

Abstract
Starting in ancient China and Greece, arsenic-containing compounds have been used in the treatment of disease for over 3000 years. They were used for a variety of diseases in the 20th century, including parasitic and sexually transmitted illnesses. A resurgence of interest in the therapeutic application of arsenicals has been driven by the discovery that low doses of a 1% aqueous solution of arsenic trioxide (i.e. arsenous acid) leads to complete remission of certain types of leukemia. Since FDA approval of arsenic trioxide (As2O3) for treatment of acute promyelocytic leukemia (APL) in 2000, it has become a front line therapy in this indication. There are currently over 100 active clinical trials involving inorganic arsenic or organoarsenic compounds registered with the FDA for the treatment of cancers. New generations of inorganic and organometallic arsenic compounds with enhanced activity or targeted cytotoxicity are being developed to overcome some of the shortcomings of arsenic therapeutics, namely short plasma half-lives and narrow therapeutic window.
Arsenic has a rich history in medicine, but it has not always been administered with the intention of benefiting the recipient. Arsenic trioxide (As2O3, ATO), a white powder which dissolves readily in alkaline solution to form a stable, odorless, and tasteless solution of arsenous acid, was widely known as ‘the king of poisons/the poison of kings’.1 Arsenic poisoning mimics the hemorrhagic gastroenteritis symptomatic with cholera, allowed arsenic trioxide to be used as an acute poison. The well-known toxicity of As2O3 that led to applications as a rat poison in times of plague2 did not prevent our predecessors from exploring other applications. Members of European royal courts used lower doses of arsenic to achieve the rosy lips and pale skin considered a sign of good breeding1. Mountaineers in the Austrian and Swiss Alps reportedly consumed small doses of arsenic on a regular basis in order to improve stamina and ward off disease.3 While it seems a high risk for little benefit, the key to many profound lifesaving pharmacological applications of arsenic trioxide are perhaps rooted in the Renaissance dictum attributed to Paracelsus, namely that it is the dose that determines the poison. In the course of this Forum, we discuss the evidence of carefully controlled administration of a variety of forms of AsIII for treatment of cancer. We focus first on the history and development of low dose treatments with the inorganic drug arsenous acid, and it subsequent development into Trisenox, into a front line, FDA-approved treatment for some types of blood cancers. The aqueous reaction chemistry and some of the physiological targets of this drug, which surprisingly can act to promote the reversion of a cancer cell to a normal phenotype, will also be discussed. Given the clear efficacy of arsenicals for specific cancer types, given that several of these agents clear rapidly and do not accumulate in the body, we argue that deeper investigation of the pharmacology of new complexes and nano-scale formulations that target the delivery and release of this metalloid are well warranted.
Some of the earliest records of direct applications of arsenic to treat disease are found in traditional Chinese medicine where it was used as a devitalizing agent prior to dental work. Hippocrates and other ancient Greek physicians used arsenic as an escharotic, a substance that destroys tissue and produces a thick black scab known as an eschar, to treat skin and breast cancers.4 Two other common mineral forms of arsenic have also been major components in other traditional medicines in China for more than 1500 years: realgar, tetra-arsenic tetrasulfide (As4S4) and orpiment, diarsenic trisulfide (As2S3) (Figure 1). Following the success of arsenic trioxide, recently both As4S4 and As2S3 have been used in clinical trials in China for treatment of APL.5 Arsenic became popular in western medicine in the 18th century, when Thomas Fowler patented a solution of potassium arsenite to be administered orally.6 Known as Fowler's Solution, it saw a wide variety of uses for over 150 years including Hodgkin's disease, leukemia, asthma, pemphigus, psoriasis, and eczema.
Figure 1.
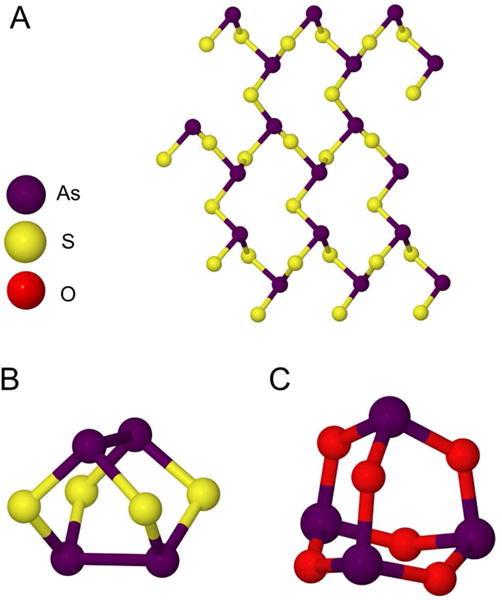
Structure of major arsenic ores. A) Orpiment. B) Realgar. C) Arsenic trioxide or “white arsenic”. 143, 144
Paul Ehrlich won the Nobel Prize in 1908 for his studies of immunity and the development of small molecule drugs targeting cellular receptors. This work lead to the development of his “magic bullet” hypothesis, namely that a properly designed molecule would bind to a receptor present on the syphilis-causing pathogen (the gram negative spirochete T. pallidum) and enable the specific delivery of a toxin to that organism. He first identified Salvarsan in 1907, but it was not commonly used until 1911 for the treatment of syphilis.7 Prepared from the reduction of nitro-oxyphenyl arsonic acid, Salvarsan was used to treat syphilis and other diseases until penicillin became widely available during World War II. The structure of Salvarsan was the subject of some controversy for many years, since Elrich and others originally determined that Salvarsan had dimeric structure akin to azobenzene (Figure 2). Recent mass spectrometric data suggests that the compound is a mixture of the trimer (Figure 2) and pentamer (Figure 2) in solution.8
Figure 2.
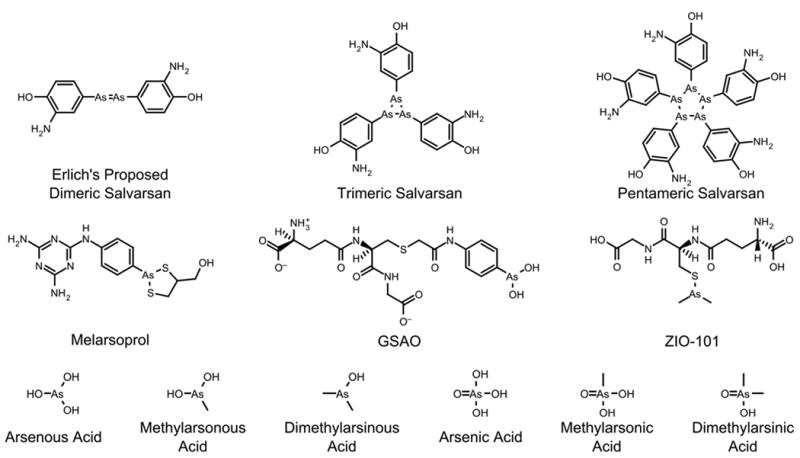
Structures of relevant arsenic compounds.
Acute Promyelocytic Leukemia (APL)
Aside from a few specific indications, arsenic had fallen out of wide use in western medicine by the mid-20th century due to the aforementioned toxicity at high doses. However, in 1971, a group of Chinese physicians tested the efficacy of a 1% solution of arsenic trioxide in patients with leukemia. The responses of the cohort were mixed; however, the group of patients that presented with Acute Promeylocytic Leukemia had drastically better response to arsenic trioxide than other patients9
Up to this point, the greatest clinical success of arsenic trioxide has been in the treatment of hematological cancers, most notably in acute promyelocytic leukemia (APL) which is a subtype of acute myeloid leukemia (AML).10 APL was first identified as a distinct clinical entity in 1957 by a Norwegian physician, Hillestad, which he categorized as one of the most malignant forms of acute leukemia.10 APL diagnosis is characterized by an accumulation of promyelocytes in the blood and bone marrow which can lead to severe bleeding tendencies with a rapid course of only a few weeks. On a molecular level, APL is characterized by a balanced reciprocal translocation between chromosomes 15 and 17, t(15;17)(q22;q21), which results in a fusion of the promyelocytic leukemia (PML) gene with the retinoic acid receptor α (RARα) gene. The resulting PML-RARα fusion protein is the essential driver in ≥98% of APL patients and acts as a constitutive transcriptional repressor of normal RARα signaling. This results in a differentiation block for promyelocytes maturing to functional granulocytes.10
In the early 1970s it was shown that AML leukemic cells were particularly sensitive to a combination chemotherapy treatment of daunorubicin, a member of the anthracycline family that are thought to be DNA-intercalators, and cytarabine (Ara-C), a pyrimidine nucleoside analog that inhibits DNA polymerase during the synthesis phase of the cell cycle. While an early 1973 study showed this chemotherapy treatment resulted in 33% complete remission (CR) of APL patients,11 more recent studies in the 1990s showed CR rates at 80-90%.12 This chemotherapy regiment successfully inhibited the proliferation of leukemogenic cells but is not curative and the majority of patients eventually relapsed. Lacking better alternatives the regiment remained the standard of care for twenty years until the discovery and adoption of all-trans retinoic acid (ATRA) therapy. As a single agent ATRA has demonstrated effective but often short-lasting remission for the treatment of APL; however, when ATRA is administered in combination with anthracycline-based chemotherapy (most commonly daunorubicin or idarubicin) the complete remission rate stays equally high and the disease-free survival rate is drastically improved. Long-term results from a randomized trial run by the European APL group showed that ATRA in combination with chemotherapy (daunorubicin and cytarabine) resulted in 96% complete remission and a 10-year event-free survival rate of 76%.13 The study also concluded that following complete remission, a prolonged maintenance treatment (up to two years) of intermittent ATRA and chemotherapy helped to reduce the rate of relapse.13
In many cases of refractory or relapsed APL the disease eventually becomes resistant to ATRA-therapy. It is in this salvage setting that arsenic trioxide was originally utilized for the treatment of refractory, ATRA-resistant APL.12 As mentioned previously, arsenic has been utilized in traditional Chinese medicines for thousands of years, and in the early 1970s a group of physicians from Harbin Medical University in China identified arsenic as an active ingredient from a traditional Chinese therapy for leukemia leading them to use arsenic trioxide as a therapy for APL.14 Results from the first moderately sized trials of arsenic trioxide for the treatment of APL came out of Harbin Medical University and the Shanghai Institute of Hematology in the mid-1990s and reported impressive rates of complete remission and overall survival for both newly diagnosed and relapsed APL.14 The success of arsenic trioxide in China led to these trials being repeated worldwide with much success.15 While arsenic was originally reserved for refractory APL, in 2010 the North American Leukemia Intergroup published the results of a trial testing arsenic trioxide as consolidation treatment for APL patients in their first remission. In this study adding two five-week consolidation courses of arsenic trioxide at 0.15 mg/kg/day via intravenous infusion for five days a week increased the three-year disease-free survival rate to 90% while the control group not treated with arsenic trioxide had a rate of 70% disease-free survival.16 Additional studies have shown that arsenic trioxide is the single most effective agent for treating APL. One study using single-agent arsenic trioxide therapy as a front-line treatment reported in 2011 that of 197 patients with newly-diagnosed APL, 85.8% achieved complete remission with a 5-year disease-free survival rate of 66.7%.17
Every study mentioned so far has used an intravenous formulation of As2O3 that, while very effective and generally well-tolerated, requires daily infusions, often for ten weeks, and has resulted in some reports of cardiotoxicity with QTc prolongation measured by electrocardiogram. Given the demonstrated benefit of ATO in the treatment of APL, a group in Hong Kong has developed an oral formulation of ATO that can be self-administered in the outpatient setting.18 Several studies by Au et al. have found that orally administered ATO is well absorbed with bioavailability of up to 95% of an equivalent dose of intravenous ATO. The slower oral absorption of ATO also led to a reduction in peak plasma arsenic levels which was associated with lower levels of cardiotoxicity.18, 19 The success of arsenic as an effective, economical, and easily-administered therapy for APL has warranted testing in a number of other cancers which will be discussed later in this Forum.
Toxicity and Reaction Chemistry of Arsenic(III)
The toxicity of arsenic has been widely observed and is likely better known in popular culture than the rich therapeutic history of arsenic. Acute arsenic poisoning has symptoms similar to many other diseases, including vomiting, diarrhea, bloody urine, and eventual death. These common disease-like symptoms led to the wide use of arsenic as a poison. During the rush to World War I arsenic was developed into a chemical weapon. Methyldichloroarsine was used to great effect by the German army, and the United States developed, but never deployed, 2-chloroethenylarsonous dichloride, also known as lewisite. 20
The salts of arsenous acid, such as sodium arsenite (NaAsO2), potassium arsenite (KAsO2), calcium arsenite (CaHAsO3), copper arsenite (CuHAsO3, Scheele's green), copper acetoarsenite (3Cu(AsO2)2·Cu(O2CCH3)2, Paris green), and lead arsenite (PbHAsO3), and the salts of arsenic acid, such as calcium arsenate (Ca3(AsO4)2), and lead arsenate (Pb3(AsO4)2) are also poisonous. They have been used as anticancer agents (sodium arsenite and potassium arsenite), in preserving hides, in the manufacturer of soap and antiseptics, and in viticulture as insecticides, weed killers, germicide and rodenticides.21, 22
Aside from its more infamous uses as an acute poison, arsenic is sometimes present in high concentrations in ground water, resulting in the exposure of entire populations, depending upon the local geologic strata.23 Natural groundwater contamination is the most common source of chronic arsenic exposure though it may also be encountered through dietary sources as well as anthropogenic sources such as mining, smelting, and agricultural runoff.24 The most common side effect of chronic arsenic exposure is hyperpigmentation of the skin which, according to the US EPA, typically begins after 6 months to 3 years of high contamination levels of 0.04 mg/kg/day or 5 to 15 years of low contamination levels of 0.01 mg/kg/day.25 More serious side effects of chronic exposure include hyperkeratosis of the palms and feet which often leads to skin lesions that are thought to increase incidence of squamous cell skin cancer. Bladder, lung, liver, kidney, and prostate cancers have also been linked to chronic arsenic exposure as well as leukopenia, peripheral vascular diseases (Blackfoot disease), anemia, neuropathy, encephalopathy, and diabetes. While many of these diseases have been linked to chronic arsenic exposure, researchers have had difficulty determining dose dependence due to complications involved with estimating total arsenic exposure from drinking water for various populations.26 Arsenic also may be acutely toxic at much larger doses and is most often observed with severe gastrointestinal distress (vomiting, abdominal cramps, and diarrhea), heart dysrhythmias, headaches, and kidney and/or liver failure.27
The introduction of arsenic trioxide in the treatment of APL and other cancers naturally led to concerns about its acute and chronic toxicities. This was addressed by the development of a very low dose administered daily over a month long course of treatment. A common dosing schedule for arsenic trioxide therapy for APL is two courses of 0.15 mg ATO per kilogram of patient bodyweight daily for five days a week for five weeks with a two week rest between courses.16 With this dosing, ATO is generally well-tolerated with commonly reported side effects of differentiation syndrome (APL-specific effect associated with elevated white blood cell counts), headache, elevated liver enzymes, and cardiac QTc prolongation and arrhythmias. Most of these side effects are low-grade and reversible after halting lowering or halting ATO therapy.15 In a follow-up study of APL patients in remission after receiving ATO therapy, urine analysis for arsenic showed that urine arsenic concentrations in patients who were off ATO treatment for at least 24 months were only slightly above that of healthy controls and were well below safety limit recommendations by the US Agency for Toxic Substances and Disease Registry and the Ministry of Health of China.28 A second follow-up study of APL patients who had received ATO therapy at least 24 months prior confirmed these findings of low arsenic retention through the analysis of hair and nails of patients.29
As mentioned elsewhere in this article, arsenic trioxide is the most effective single agent therapy for APL. The recent release of 10-year follow-up studies on APL patients receiving ATO therapy has shown that ATO increases the rate of event free survival and also has little/no long term side effects.28 The use of ATO in induction/consolidation therapy of APL may allow for the reduction of anthracycline chemotherapy which is known to have much more serious and longer lasting side effects. Currently orally administered formulations of arsenic trioxide are being studied and seem to have a similar efficacy as IV-administered ATO while reducing acute toxicities (especially QTc prolongation) from the lower peak plasma arsenic levels encountered with slower oral absorption of ATO.18
Arsenic trioxide, when dissolved in water at neutral pH forms arsenous acid, As(OH)3 (Figure 2). This species is neutral at physiological pH (pKa1 = 9.3, pKa2 = 13.5, pKa3 = 14.0).30 The aqueous form of arsenic(V) oxide is arsenic acid, H3AsO4 (Figure 2), and in physiological conditions, it exists as hydrogen arsenate and dihydrogen arsenate (pKa1 = 2.19, pKa2 = 6.94, pKa3=11.5).31
In all cases, exposure to trivalent arsenic (AsIII) is more toxic than exposure to its pentavalent form (AsV). It is thought that the increased toxicity of AsIII compared to AsV is due to the increased affinity of AsIII for sulfur ligands.32 Arsenic in groundwater is typically inorganic arsenite forms of AsIII while dietary arsenic tends to be AsV derivatives such as dimethylarsinic acid or arsenobetaine (major component of dietary arsenic found in seafood). The greatest contribution to total urinary arsenic levels is dimethylarsinic acid.33
Organic arsenic compounds have long been used for treating various disease, with their greatest drawbacks being toxicity. Melarsoprol (Figure 2) was developed in the late 1940s and is still used for standard-of-care treatment of African Trypanosomiasis, commonly known as sleeping sickness.34 It is used for more advanced stages of the disease, due to fatal reactive encephalopathy in 5-10% of patients treated with melarsoprol. Other organoarsenicals, such as salvarsan, display dose-limiting acute toxicity as well as chronic toxicities, and have fallen out of use for safer alternatives.
Arsenate as a phosphate analog
Arsenate has long been known to participate in similar reactions as phosphates. Recently there was a controversial report in the literature that arsenate had been shown to replace phosphate along the DNA backbone in an extremophile bacterial strain. Subsequent analyses in other laboratories have demonstrated that there is no detectable arsenic in the DNA of these bacteria,35 and that these organisms require phosphate as an essential nutrient for growth.36
Arsenate (AsO43-) is known to participate in certain cellular processes in place of phosphate (PO43-), particularly phosphorylation reactions.37 When arsenate is substituted for phosphate during glucose phosphorylation, it results in the rapid decomposition of glucose-1-arsenate.38 Arsenate and other transition metal oxoanions, such as vanadate, are potent inhibitors of many phosphatase enzymes.39 This action is thought to be the result of the increased stability of a metalloester intermediate compared to the native phosphoester intermediate. Other enzymes are not inhibited by arsenate or vanadate, and can perform these reactions on substrates that have arsenate or vanadate in place of the normal phosphate.40, 41
Despite the potential disruption of these metabolic pathways, there is little evidence that arsenate is responsible for the majority of the symptoms of acute arsenic toxicity. First, the intracellular concentrations required to inhibit these enzymes is high (10-4 M)39 and not achievable in patients without acute arsenic toxicity. Second, the phosphate concentration in the cell (10-100 mM)42 is high enough that even a toxic dose of arsenic to the cell would not be enough to displace phosphate from these essential metabolic pathways.
Arsenic-thiol Thermodynamics
Arsenic exerts its effect in cells by interacting with proteins and peptides, typically through the sulfhydryl groups of cysteine residues. These interactions are energetically favored but have heats of reaction that make the interactions rapid and reversible. One important ligand that arsenic binds is glutathione. Glutathione (GSH) is a tripeptide molecule (γ-L-glutamyl-L-cysteinyl glycine) that is responsible for protecting the cell from reactive oxygen species generated by normal cellular metabolism.43
The interaction of arsenous acid with glutathione occurs in a stepwise manner, where each hydroxyl ligand is displaced by the nucleophilic sulfur ligand.44 The interaction of the arsenic center with a single sulfur ligand has the smallest heat of reaction, with each additional interaction more exothermic (ΔH1=-2.5 kcal mol-1, ΔH2= -3.1 kcal mol-1, ΔH3= -33.1 kcal mol-1). This is likely due to the increased electronic degeneracy of these interactions. The formation of the tris-glutathione arsenite, As(GS)3, species is spontaneous at pH 7.4 at 37°C (ΔG = -8.8 kcal mol-1).44 Scott et al. isolated and characterized an arsenite complex of glutathione as As(GS)3 by mass spectrometry.45 Delnomdedieu et al. found that glutathione reduces arsenate to arsenite and forms a As(GS)3 complex (Figure 3).46
Figure 3.
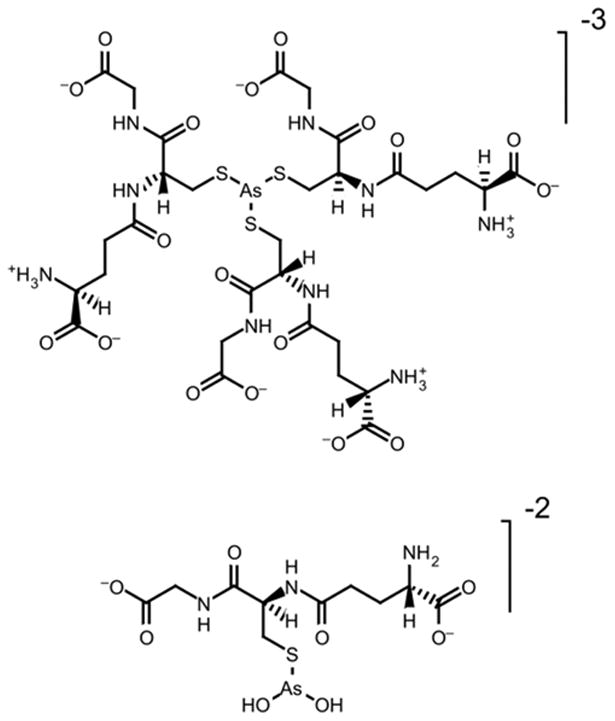
Proposed structure of arsenic-glutathione complexes.46
The formation constants of the cysteine (Cys) complexes [As(Cys)3], logK = 29.84(6), and [As(Cys)(OH)2]-, logK = 12.01(9), and of the glutathione complexes [As(GS)3]3-, logK = 32.0(6), and [As(GS)(OH)2]2-, logK = 10(3) were calculated from potentiometric and spectroscopic data.47 Spectroscopic data clearly indicate that the arsenite binding site is S-atom of cysteine. At pH 7.0-7.5 with arsenic and glutathione concentrations of 5 mM and 15 mM, respectively, approximately 70% of the arsenic species present are [As(GS)3]3-.47 The other major species in this range are As(OH)3 and [As(GS)(OH)2]2-.
Methylated arsenic also interacts with sulfur ligands, albeit with higher affinity than inorganic arsenous acid.44, 48 The formation of the bis-glutathione methyl arsenite species is slightly more favored than the tris-glutathione arsenite (ΔG = -10.1 kcal mol-1). The increased reactivity of methylarsenous acid with sulfur ligands is thought to be the basis for the increased toxicity of this species.49 The thermodynamics of arsenic-protein interactions are less well understood, however, examining the interactions of arsenites with thiol ligands provides some insight into the strengths of these interactions. Much like the monothiol case of glutathione, these interactions are energetically favored; however, dithiol interactions are more energetically stable than monothiol interactions due to the chelate effect.44
Cellular Mechanisms for Arsenic Transport and Detoxification
At elevated temperatures and in membranes with large amounts of cholesterol, As(OH)3 is able to rapidly cross lipid membranes, likely due to the increased fluidity of the membranes.50 It is likely that arsenic acid enters cells by co-opting a phosphate transporter.51 Arsenous acid is also able to cross cell membranes by utilizing the neutral species transporter aquaglyceroporin 9 (AQP9).51, 52 Aquaglyceroporins are members of the major intrinsic protein (MIP) superfamily and are responsible for the transport of glycerol and other neutral solutes into the cells. Another member of this superfamily is aquaporin which regulates water transport in cells.53 The transport of neutral molecules into and out of cells by aquaglyceroporins is typically a regulated response to metabolic cues.54 Arsenic has been shown to utilize AQP9 and AQP7 channels as a pathway for cellular uptake.52 In leukemia cells, the presence of these transporters, particularly AQP9, confers sensitivity to arsenous acid as well as potassium antimonyl tartarate, another metalloid compound.55, 56 The expression of AQP9 in Xenopus laevis oocytes increases arsenous acid uptake 40 fold over 90 seconds.52 AQP7, in contrast, only increases arsenous acid uptake by 10 fold in the same assay. 52
The expression of AQP9 has been demonstrated to confer arsenic sensitivity to leukemia cell lines in vitro.56 When AQP9 expression is induced by DNA transfection, the arsenic concentration required for 50% cell growth inhibition (IC50) is reduced by 3.8 fold.56 The effect is similar when AQP9 expression is induced pharmacologically using Vitamin D pretreatment; the cells are rendered 2.5 fold more sensitive to arsenic treatment.57 This effect is shown in a panel of 11 leukemia cell lines, where AQP9 expression correlates with directly with arsenic uptake and sensitivity. Interestingly, AQP9 is most highly expressed in acute promeylocytic leukemia (APL). 57
Arsenic can be present in groundwater to widely varying levels depending on local mineral strata. Organisms exposed to moderate arsenic levels have developed specialized systems for detoxifying and removing arsenic from cells.51 In bacteria, a specialized system called the Ars operon is controlled by the ArsR protein in response to arsenic exposure and begins exporting arsenic from the cell by first reducing arsenates to arsenites and then utilizing a specialized arsenite transporter.51
In mammalian cells, the metabolism of arsenic involves export from the cell and metabolic detoxification of the arsenic. Metabolic processing of inorganic arsenic(III) is carried out by a series of methylation reactions. These reactions involve the reaction of arsenous acid with arsenic(III) methyltransferase (AS3MT).58-61 AS3MT uses S-adenosyl methionine as a methyl donor in this reaction.58 This protein catalyzes both the methylation of arsenous acid as well as methylarsonous acid in the presence of adequate reducing and methyl equivalents.60, 61 It is not entirely clear if the product of methylation is methylarsonous acid or methylarsonic acid, however, examination of a bacterial S-adenosyl methionine methyltransferase, ArsM, indicates that the arsenic center remains arsenic(III) during the reaction.62 There are three cysteines in the protein that are important for catalytic activity, two likely participate directly in arsenic binding, and the third is involved in the methylation of arsenous acid,60 but not methylarsonous acid.62 The position of the arsenic center is such that the arsenic lone electron pair is positioned towards the methyl group of S-adensosyl methionine, facilitating methylation via an SN2 reaction.62 Further crystallographic, EXAFS, or other physical examination of the active site is required to produce a more precise mechanism of this enzyme.
Glutathione also plays a role in the direct export of arsenous acid from the cell.63 Glutathione complexes with metal ions are substrates for the multi-drug resistance export proteins (MRP1-10) that are found in cells.64 These proteins are responsible for exporting a diverse range of ionic substrates. Arsenic-glutathione complexes (Figure 3) are substrates for MRP1,63, 64 and high levels of expression of MRP1 is associated with arsenic resistance in cancer cells.65 Glutathione-S-transferase pi (GST-pi) is responsible for catalyzing the reaction of arsenic with glutathione and enabling the subsequent export of the arsenic-glutathione complex.65 Depleting intracellular glutathione by inhibiting its synthesis sensitizes cells to arsenous acid and other arsenic compounds.66-68 High expression levels of GST-pi results in resistance to arsenous acid.65
Targets of Arsenous Acid Activity
Due to the large concentration of thiols in the cell, with the majority of them in the reduced state, arsenic has a wide array of targets. The majority of the proteins that bind arsenic have spatially close cysteine resides that bind the arsenic through the sulfhydryl side chain. In cancer cells, there are a more limited set of interactions that are thought to exert the anti-tumor effect of arsenous acid.
Metabolic proteins
Arsenous acid acts to increase the levels of reactive oxygen species in cells; however, there is little evidence that oxidizing equivalents originate from the reduction of arsenous acid to metallic arsenic or arsine. Arsenic is known, however, to interact with a mitochondrial membrane protein complex, called the mitochondrial membrane transition pore complex.69 As arsenic binds to this pore complex, it initiates the collapse of the mitochondrial membrane potential and the release of cytochrome c from the mitochondria into the cytoplasm of the cell. 70, 71 Other researchers have demonstrated that arsenous acid will bind to enzymes involved in glucose metabolism.72
Proteins regulating intracellular redox potentials
Another class of proteins affected by intracellular arsenic is involved in redox balance and antioxidant pathways.6, 73 These proteins have redox active sulfhydryl groups that detoxify reactive oxygen species produced by normal mitochondrial function. Arsenic acts on these proteins by binding to the thiol groups and removing their ability to bind other nucleophiles, such as superoxide, hydroxide radicals, or oxide radicals.
Thioredoxin is an oxidoreductase that contains a dithiol active site. It performs a number of signaling and homeostasis activities related to cellular oxidation state and can also modulate the levels of adventitious reactive oxygen species.74 The dithiol active site is maintained in the reduced state by another enzyme, thioredoxin reductase. Thioredoxin reductase is a NADPH-dependent flavoenzyme that utilizes an active site composed of a redox-active cysteine pair and a redox-active selenocysteine-cysteine pair.75 Arsenic containing compounds interact strongly with selenocysteine residues in proteins.76 Arsenous acid and methylarsonous acid inhibits enzyme activity which prevents the reduction of the disulfide form of thioredoxin and may result in the accumulation of oxidized proteins within the cell.77 Mass spectroscopy of peptide fragments of thioredoxin reductase reveal that arsenic binds to both redox-active sites within the active site, preventing normal oxidation and reduction of the disulfide and the selenosulfide bonds.78
The effect of arsenic on the cell cycle is thought to be mediated through the stabilization of microtubules.79-82 In cells treated with arsenic, a portion of cells halt normal cell cycling in the mitosis phase due to the stabilization of tubulin required for progression through the normal cell cycle.82 This effect is similar to paclitaxel; however, in combination the two drugs behave antagonistically which suggests that arsenic may be a useful agent in patients with taxane-resistant tumors. 79
Arsenic interactions with mutant proteins in Leukemia: PML-RARα
The key driver to leukemogenesis in APL is the PML-RARα fusion protein resulting from the reciprocal t(15;17) translocation. This translocation recruits histone deacetylases and other transcription repressors that inhibit normal RARα signaling which regulates myeloid differentiation. In APL, the disrupted RARα signaling prevents promyelocytes from differentiating to mature granulocytes. While the normal function of PML has not been fully elucidated, it appears to function as a tumor suppressor protein and has been tightly associated with p53 regulation of important cell processes like differentiation, senescence, and apoptosis.83 PML is also fascinating in its localization to highly dynamic yet discrete nuclear structures known as PML nuclear bodies (NBs) which appear to play a role in stress response by regulating post-translational modification of a number of proteins. The PML- RARα fusion in APL disrupts the formation of NBs in the nucleus though treatment with arsenic trioxide reverses this abnormality. 84 Arsenic trioxide has been shown to induce apoptosis at high concentrations (1-2 μM) and promote cell differentiation at low concentrations (0.25-0.5 μM).14
The PML protein contains the RBCC motif characterized by an N-terminal RING finger motif, which is a cysteine rich type of zinc finger domain, two cysteine rich B box zinc finger domains, and a coiled coil domain which mediates homodimer formation.85 The abundance of cysteine-containing zinc-binding sites in PML led researchers to hypothesize that arsenic may directly bind to the PML protein. Using a biotin-arsenic pull-down affinity assay fluorescent organic arsenic compounds, several groups have shown that arsenic co-localizes and directly binds to both wild-type PML and the PML- RARα fusion proteins.86, 87 Zhang et al. used the purified PML RING peptide containing C3HC4 (aa 57-91) zinc finger for arsenic-binding analysis and with MALDI-TOF reported that one PML RING molecule was capable of binding two arsenic atoms. Near-ultraviolet absorbance spectroscopy showed the formation of arsenic-sulfur bonds while X-ray absorption spectra showed the local structures of the trivalent arsenics which were each coordinated by three cysteines in ZF1 and ZF2 of PML RING.87 Upon binding to arsenic, PML RING is thought to undergo a conformational change that results in oligomerization, likely due to intermolecular As-S bonds. Arsenic-binding also promotes sumoylation by UBC9 which then recruits the 11S proteasome for degradation.88 While these reports of in vitro arsenic activity strongly suggest a mechanism for arsenic-induced PML redistribution and degradation, it is unclear what effects the strongly reducing nature of the nucleus would have on these As-S and S-S interactions inside the cell.
Ongoing Clinical Trials
This Forum article is not intended to be an exhaustive review on the clinical use of arsenic in modern medicine, for that the reader should turn to a number of thorough reviews referenced here, especially those of Tallman,12, 89 Chen,14, 88 de Thé,9 and Dilda.90Table 1 below summarizes much of the available data that has been published from the 129 clinical trials registered with the FDA.
Table 1.
Clinical trials for arsenical drugs in cancer registered with the Food and Drug Administration. For more details see http://www.clinicaltrials.gov
| Malignancy | Number of Trials | Treatment | Comments | Ref |
|---|---|---|---|---|
| Hematological | 86 | |||
|
| ||||
| Acute Promyelocytic Leukemia (APL) | 18 | ATO most commonly used for induction of refractory APL or as part of combination therapy (induction and/or consolidation)for newly-diagnosed APL | Most effective single-agent therapy; very effective with no major side effects | 15,16,92-94 |
| Multiple Myeloma | 16 | ATO monotherapy or in combination with ascorbic acid and/or thalidomide; ZIO-101 | Moderate activity and reasonably well tolerated; NR | 95-98 |
| Myelodysplastic Syndromes (MDS) | 13 | ATO monotherapy or in combination with chemotherapy | Moderate activity and reasonably well tolerated | 99-102 |
| Lymphoma | 11 | ATO monotherapy or in combination with other agents | Partial and/or transient response with variable toxicity | 103,104 |
| Acute Myeloid Leukemia (AML) | 8 | ATO monotherapy | Variable response | 105-109 |
| Chronic Myeloid Leukemia (CML) | 6 | ATO monotherapy or in combination with imatinib | Partial response and well-tolerated | 110 |
| Solid Tumors | 25 | |||
|
| ||||
| Glioma | 5 | ATO and radiotherapy | Effect on tumor vasculature; well-tolerated | 111,112 |
| Various Advanced Solid | 3 | ATO; ZIO-101; GSAO | NR; Early evidence of clinical activity; NR | 91 |
| Liver | 2 | ATO monotherapy | Not active | 113 |
| Lung | 2 | ATO monotherapy | NR | |
| Skin | 2 | ATO monotherapy | Well-tolerated; not active | 114 |
| Breast | 1 | ATO monotherapy | NR | |
| Colorectal | 1 | ATO and 5-Fluorouracil | Moderate activity in refractory cases | 115 |
| Pancreatic | 1 | ATO monotherapy | Not active | 116 |
| Cervical | 1 | ATO monotherapy | NR | |
| Prostate | 1 | ATO monotherapy | Decreased PSA markers | 117 |
| Kidney | 1 | ATO monotherapy | No response | 118 |
There are currently 129 clinical trials registered with the FDA at www.clinicaltrials.gov that deal with arsenic. Of these 129 clinical trials, 107 trials incorporate arsenic trioxide or arsenic-based drugs as therapeutics for the treatment of cancer. Of particular interest are ZIO-101 (Darinaparsin) and GSAO (Figure 2), two novel organoarsenical drugs that are under evaluation for the treatment of solid tumors.91 The cancer-related trials can subdivided into three groups: leukemias (75 trials), lymphomas (11 trials), and other cancers, mostly solid tumors (25 trials). There were several trials that dealt with more than one of these subgroups. Clearly hematological malignancies have been the major focus of clinical arsenic research with the largest number of trials involving acute promyelocytic leukemia APL (18), multiple myeloma (16), myelodysplastic syndromes (13), non-APL acute myeloid leukemia (8), and chronic myeloid leukemia (6). In solid tumors, arsenic is has been used in glioblastoma (5) mostly as a radiosensitizer for radiation therapy.
The majority of the remaining trials (19 of 22) focused on chronic arsenic exposure, typically from groundwater contamination or occupational hazards while one ongoing trial is testing arsenic trioxide for the treatment of lupus.
Shortcomings of Arsenic Trioxide as an Anti-cancer Agent
Despite the strong efficacy of arsenic in hematological malignancies, free arsenic trioxide has a number of serious limitations. The primary limitation to wide use of arsenic trioxide as an anti-cancer agent is the systemic toxicity associated with large amounts of arsenic in the blood. Low doses of arsenic trioxide work well for treating hematological malignancies; however, solid tumors remain a treatment challenge. In the animal model, there have been numerous studies performed in breast, ovarian, and other cancers with positive results.90 Unfortunately these successes have not translated into clinical success in human patients. This is likely due to the rapid renal clearance of arsenic metabolites which limits the concentration of arsenic that can be delivered to a tumor site.
Nanoparticulate Arsenic Compounds Designed to Improve Arsenic Delivery to Tumors
Novel arsenic sulfide preparations for the treatment of cancer
Recently, researchers have begun investigating arsenic sulfide compounds for their anti-tumor properties.119, 120 In addition to new clinical trials taking place in China,5 new compounds and formulations are also being developed. Wang and coworkers reported the first synthesis of realgar (As4S4) quantum dots produced from bulk realgar, and produced a stabilized colloidal formulation of these nanoparticles using ethylenediamine and acetic acid.121 Further work has produced a hydrogel comprised of these realgar nanoparticles (Figure 6). The hydrogel formulation is pH responsive and has improved anti-tumor cell activity compared to the stabilized realgar nanoparticles.122
Figure 6.
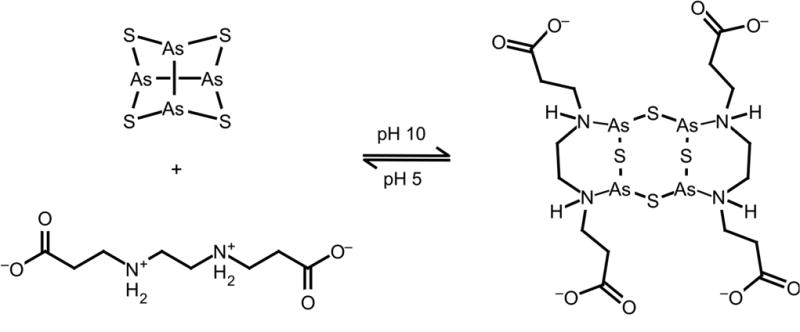
Arsenic sulfide nanogel assembly is driven by coordination of the arsenic centers by nitrogen ligands and hydrogen bonds between the chelator molecules.122
Arsenic nanobins show improved anti-tumor efficacy compared to arsenous acid
In order to extend the clinical utility of arsenic trioxide, our laboratory has developed a novel method for stably encapsulating arsenic trioxide inside nano-scale liposomes called nanobins.123-129 The nanobins utilize a metal-ion gradient loading mechanism that relies on the reaction of arsenous acid with a divalent metal ion such as nickel(II), cobalt(II)123 or platinum(II).125
The preparation of the nanobins proceeds by first dissolving lipids and cholesterol in chloroform. The lipid formulation is intended to be stable in the plasma and long-circulating, and so poly(ethylene glycol) and cholesterol are included. The lipids are dissolved at a molar ratio of 0.51/0.45/0.04 1,2-distearoyl-sn-glycero-3-phosphocholine/cholesterol/ 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000]. The chloroform is then removed by rotary evaporation to form a lipid film. After overnight drying under high vacuum, the lipids are hydrated in a metal acetate, M(OAc)2 solution, typically nickel(II) acetate or cis-diaqua-diammine platinum(II) acetate. The lipid solution is then subjected to high-pressure extrusion to size the nanobins, after which any external metal solution is removed and replaced by buffer, using either size-exclusion chromatography or tangential flow filtration. Arsenic trioxide solution is then added to the exterior of the nanobins, and is allowed to load for up to 12 hours at elevated temperature. Arsenic loading is driven by the complexation and precipitation of arsenic with the metal ions inside the nanobins, and the efflux of protonated acetic acid from the nanobin (Figure 7). The complexation of arsenic with metal salts is required for stability, otherwise encapsulated arsenic will leak rapidly from the liposome.130
Figure 7.
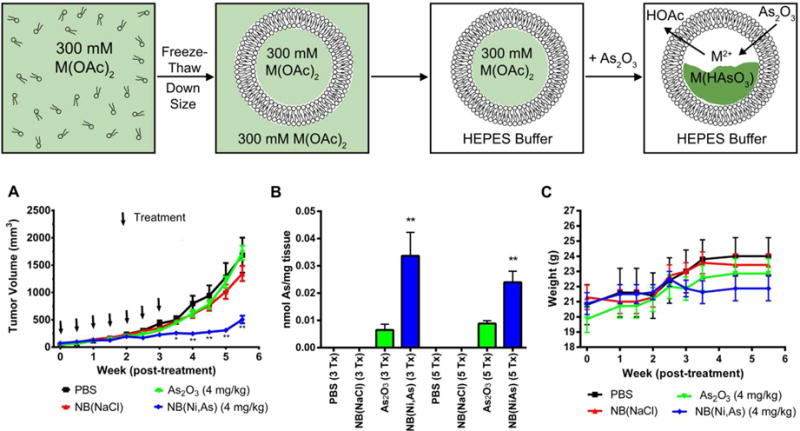
Preparation and use of arsenic nanobins. Top: Synthesis scheme of arsenic nanobins. M = Ni2+, [Pt(NH3)2(OH2)2]2+ A)Tumor volume plot of human triple-negative orthotopic xenograft tumors implanted in the 4th mammary fat pad of female nude mice showing decreased tumor volume in nanobin treated mice vs. mice treated with arsenic trioxide. B) Nanobin treatment increases total amount of arsenic in the tumor compared to arsenic trioxide treatment. C) Stable animal weight during treatment indicates all treatments were well tolerated.126
This loading mechanism is capable of concentrating arsenic inside the liposome to > 270mM as a solid, crystalline payload. If this amount of arsenic were to be dissolved in the volume of a mammalian cell, the resulting concentration would be on the micromolar scale, very likely resulting in cell death. The presence of a lipid bilayer surrounding the arsenic payload greatly attenuates the toxicity of the arsenic, reducing the toxicity 3-4 fold in cell culture experiments compared to arsenic trioxide.124-126
The nanobins have been shown to improve the anti-tumor efficacy of arsenic trioxide in a mouse model of breast cancer.126 In this model, triple-negative breast cancer cells were implanted in the 4th mammary fat pad of nude mice and allowed to grow for two weeks. The animals were then treated with equivalent amounts of arsenic trioxide or nickel-arsenic nanobins [NB(Ni,As)]. The nanobin treatment group showed durable reduction in tumor growth compared to the arsenic trioxide group, which showed no statistically significant difference from the untreated control groups. This improved efficacy was due to the increased tumor accumulation of arsenic where the nanobins were able to deliver 2-3 times more arsenic to the tumors. The nanobins also reduced the arsenic clearance rate from the plasma by over 300-fold, greatly increasing the exposure of the tumor to the drug.
The nanobin formulation is analogous to liposomal doxorubicin, sold as Doxil (Janssen) or Lipodox (Sun Pharma Global FZE) in the United States, wherein doxorubicin is stably encapsulated inside a liposome using a pH gradient created by encapsulated ammonium sulfate. Our laboratory uses similar processing technology to manufacture the nanobins, which ensures consistent and homogeneous particle size and drug loading
These nanobins are stable, with a greater than six month shelf-life, likely due to the presence of a direct arsenic-metal bond. Previous attempts to encapsulate arsenous acid had very short shelf lives, because the arsenic would easily leak out of the vesicles.131 In the case of the diaqua-cisplatin arsenic nanobins, the arsenic reacts directly with the platinum center, resulting in a platinum-arsenic bond length of 2.32 Å.125 This structure was elucidated by EXAFS, and has spurred further research in our group into these arsenic-platinum bonds.
Coordination Chemistry of Arsenous Acid and Design of New Pt-As Anticancer Agents
Platinum compounds with arsenous acid were not known in the literature until above mentioned nanobins were formulated. The coordination chemistry of arsenous acid is not well understood since only two complexes of arsenous acid, one with nickel and one with palladium, have been isolated and characterized by X-ray so far. The complex of arsenous acid with nickel(II) is a heterometallic supramolecular adduct with cucurbit[6]uril (cuc), [W3(Ni(As(OH)3)S4(H2O)8Cl])·cuc·13H2O, in which arsenous acid is coordinated through arsenic(III) acting as a Lewis base (Figure 8).132, 133 In this complex, the arsenic center has trigonal pyramidal geometry with the NiII-AsIII bond length of 2.225(4) Å. In the arsenous acid complex with palladium, [Mo3(Pd(As(OH)3)S4(H2O)6Cl3]2Cl2·C36H36N24O12·19H2O, the geometry around the arsenic center is more tetrahedral with O-As-Pd bond angle of 113.8° and O-As-O bond angle of 104.7°.134 The observed Pd-As bond length is 2.368(3) Å.
Figure 8.
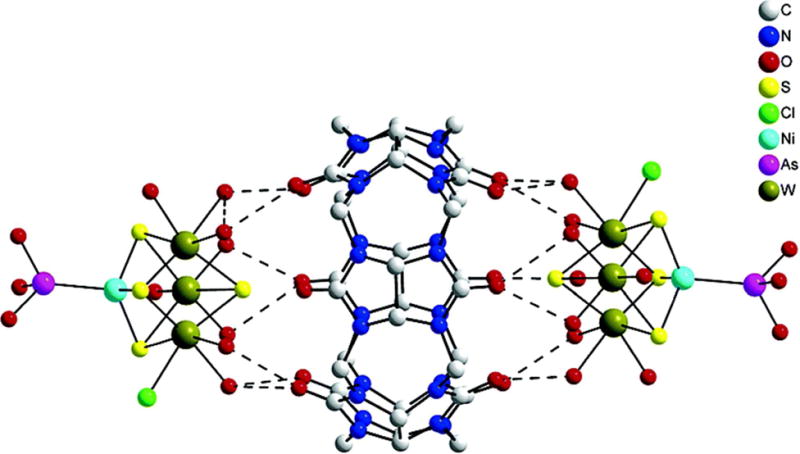
Structure of supramolecular complex including arsenic-nickel coordination.132
A new synthetic approach to combine platinum and aqueous form of arsenic trioxide in one molecular compound has been recently discovered.135 The new complexes were synthesized from cisplatin and arsenic trioxide in water-nitrile mixture. In arsenoplatin-1 (1) (Figure 9) platinum forms expected square planar geometry, but arsenic(III) forms trigonal bipyramidal geometry with arsenic(III) acting unexpectedly as a Lewis base and as a Lewis acid simultaneously.135 Multiple interactions of arsenic(III) are partly responsible for the very short PtII-AsIII bonds in these complexes which are in 2.2687(4)- 2.2732 (3) Å range. The shortest bond obtained is just 0.0012 Å longer than the shortest PtII-AsIII bond found in one organoarsenical compound according the CSD. The PtII-AsIII bond in nanobins is 2.3 Å on the basis of EXAFS. In arsenoplatins this bond is even shorter reflecting the stability of PtII-AsIII core in arsenoplatins.
Figure 9.
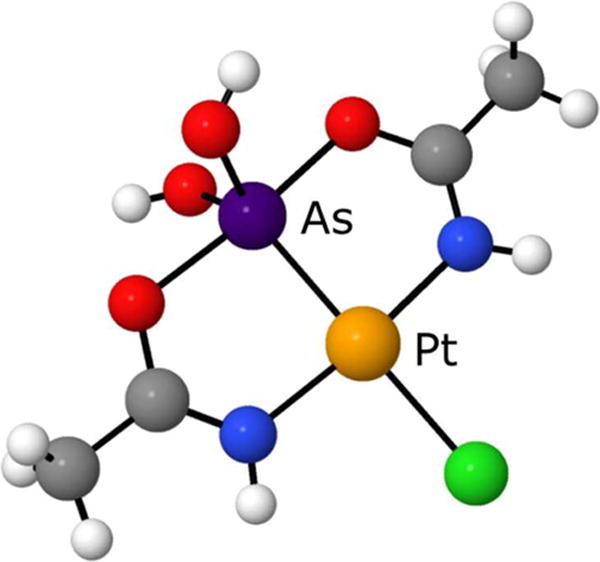
Structure of Arsenoplatin 1 based on single-crystal X-ray crystallography. Unlabeled atoms are colored according to the key in Figure 8.135
Stability of the PtII-AsIII core is confirmed in substitution reactions in aqueous solution. Substitution of chloride ion in arsenoplatin-1 with thiocyanate ion in water occurs immediately at room temperature, and the addition of AgNO3 to facilitate substitution is not necessary (Scheme 1). 135
Scheme 1.
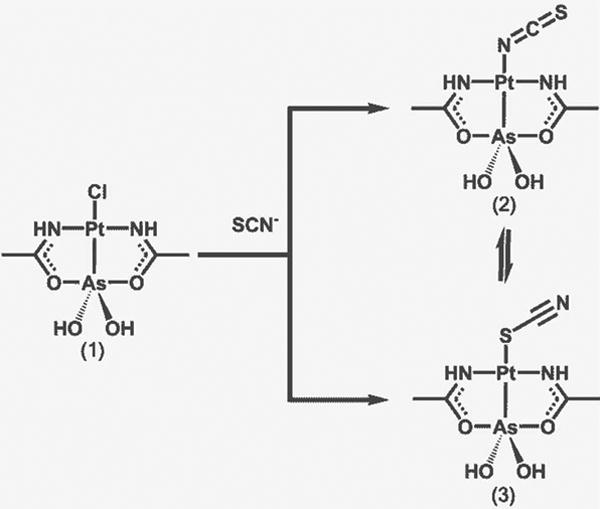
Stability of the As-Pt bond in Arsenoplatin 1. The strong trans effect of As enables rapid displacement of the chloride ligand with thiocyanate. Both the thiocyanate and isothiocyanate species are present in DMSO.135
The rapid substitution is likely driven by the strong trans effect of the arsenic moiety. Obtained SCN-derivate undergoes isomerization in DMSO solution, and the equilibrium mixture contains 64 ± 1.2% of S-isomer and 36 ± 1.5% of N isomer. In these substitution/isomerization reactions the PtII-AsIII core remains intact, confirmed on the basis of a single crystal X-ray structure of the SCN- derivate. The stability of the core is caused by arsenic(III) forming multiple interactions; with platinum(II) acting as a Lewis base, and with oxygen atoms from acetylamido moieties acting as a Lewis acid, which leads to the formation of two chelate rings in the molecular structure. Arsenoplatins are stable in saline solutions. These complexes are soluble in DMSO, methanol, and partially in water.135
The idea behind the synthesis of arsenoplatins was to design a new type of compounds containing both arsenic(III) and platinum(II), which would have potential to overcome resistance obstacles.136-138 When the cytotoxicity of arsenoplatins was investigated against various cancer cell lines, we found that in some cell lines arsenoplatin compounds were more cytotoxic than cisplatin. In the case of ovarian cisplatin resistant A2780CP cell line arsenoplatin-1 was two times more cytotoxic than cisplatin with the resistant factor (RF) of 1.1, where RF < 2 denotes non cross- resistance.139-142
The anticancer efficacy of arsenoplatins may be the result of the strong trans effect of arsenic moiety combined with the trans stereochemistry of N-atoms at the platinum center. The new coordination mode of arsenous acid discovered in our lab will trace a path for the syntheses of new small molecular compounds, derivates of the parent compounds, with different structural motifs and with different modes of action. Investigation of arsenoplatin compounds in vivo is an ongoing project.
Outlook for Arsenic in Therapeutic Agents
The complex nature of arsenic's mechanism of action suggests that there will be many uses of arsenic in medicine in the future. The development of delivery vehicles or molecules that will prevent systemic toxicity while delivering efficacious doses of arsenic to disease sites is a promising method that may help expand the medical use of this imporant metalloid. Previously, numerous organic arsenic compounds were developed to reduce toxicity and improve efficacy, with mixed results. In light of these new inorganic arsenic-metal compounds, perhaps arsenoplatins and other coordination compounds represent a new class of arsenic drugs that will prove useful for arsenic therapy in the future.
Figure 4.
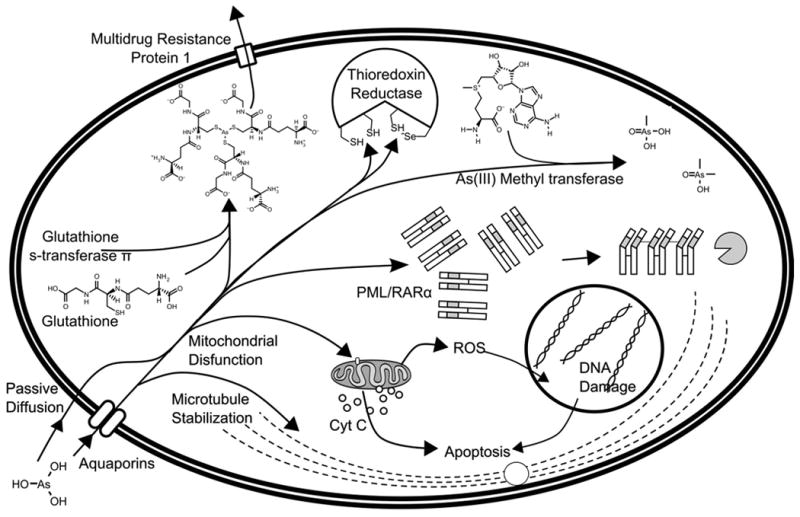
Proposed mechanisms of action for arsenic trioxide. Arsenous acid enters mammalian cells either by passive diffusion, or by the neutral solute transporter proteins in the aquaporin family.51, 52, 55, 56 It then binds to the numerous thiol moieties present in the cell, particularly glutathione, the interaction of which is catalyzed by glutathione s-transferase.65 Arsenous acid can bind to microtubules, preventing depolymerization and halting the cell cycle.79 Arsenic also binds to the active site of thioredoxin reductase, which in turn causes the buildup of oxidized, mis-folded proteins in the cell.78 In acute promyelocytic leukemia, arsenic binds to a zinc finger fusion protein, PML/RARα, which causes aggregation and degradation of the protein.86, 87 Arsenic can be metabolized into methylarsonic acid or dimethylarsinic acid.62 Arsenic interacts with mitochondrial proteins to collapse the mitochondrial membrane potential, produce reactive oxygen species (ROS), release cytochrome c into the cytoplasm and initiate apoptosis.73
Figure 5.
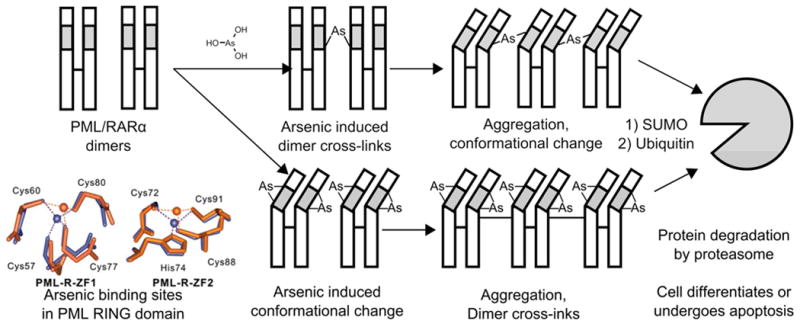
Mechanism of PML/RARα degradation. It is unclear in which order these processes occur, however, arsenous acid may induce crosslinking between PML/RARα dimers which results in a conformational change, or it may induce a conformational change in PML/RARα dimers that causes aggregation. The fate of the aggregated PML/RARα is degradation in the proteasome, which results in cell death or differentiation. Inset, the change of coordination geometry when arsenic is bound to the RING domain of PML (orange) compared to the zinc binding (blue).84, 86, 87
Acknowledgments
The authors would like to thank Emily Que and Richard Ahn for their insights and expertise while writing of this article.
Funding Sources: This work was supported by the NCI Center for Nanotechnology Platform Partnerships U01CA151461-01. PLH and EPS were partially supported by the Biotechenology Cluster Training Program at Northwestern University. PLH was also supported by NSF GRFP DGE-0824162.
Footnotes
Author Contributions: The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Abbreviations
- 1.Parascandola J. King of Poisons: A History of Arsenic. 2012:134–134. [Google Scholar]
- 2.Konkola K. J Hist Med Allied Sci. 1992;47:186–209. doi: 10.1093/jhmas/47.2.186. [DOI] [PubMed] [Google Scholar]
- 3.Przygoda G, Feldmann Jr, Cullen WR. Appl Organomet Chem. 2001;15:457–462. [Google Scholar]
- 4.Haller JS. Pharmacy in History. 1975;17:87–100. [PubMed] [Google Scholar]
- 5.Lu DP, Wang Q. Int J Hematol. 2002;76(Suppl 1):316–8. doi: 10.1007/BF03165273. [DOI] [PubMed] [Google Scholar]
- 6.Jungwirth U, Kowol CR, Keppler BK, Hartinger CG, Berger W, Heffeter P. Antioxidants & redox signaling. 2011;00 doi: 10.1089/ars.2010.3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ehrlich P, Bertheim A. Ber Dtsch Chem Ges. 1912;45:756–766. [Google Scholar]
- 8.Lloyd NC, Morgan HW, Nicholson BK, Ronimus RS. Angew Chem Int Ed. 2005;44:941–4. doi: 10.1002/anie.200461471. [DOI] [PubMed] [Google Scholar]
- 9.Zhu J, Chen Z, Lallemand-Breitenbach V, de Thé H. Nat Rev Cancer. 2002;2:1–9. doi: 10.1038/nrc887. [DOI] [PubMed] [Google Scholar]
- 10.Degos L. Br J Haematol. 2003:539–553. doi: 10.1046/j.1365-2141.2003.04460.x. [DOI] [PubMed] [Google Scholar]
- 11.Crowther D, Powles RL, Bateman CJ, Beard ME, Gauci CL, Wrigley PF, Malpas JS, Fairley GH, Scott RB. Br Med J. 1973;1:131–7. doi: 10.1136/bmj.1.5846.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tallman MS. Blood. 2002;99:759–767. doi: 10.1182/blood.v99.3.759. [DOI] [PubMed] [Google Scholar]
- 13.Adès L, Guerci A, Raffoux E, Sanz M, Chevallier P, Lapusan S, Recher C, Thomas X, Rayon C, Castaigne S, Tournilhac O, de Botton S, Ifrah N, Cahn JY, Solary E, Gardin C, Fegeux N, Bordessoule D, Ferrant A, Meyer-Monard S, Vey N, Dombret H, Degos L, Chevret S, Fenaux P. Blood. 2010;115:1690–6. doi: 10.1182/blood-2009-07-233387. [DOI] [PubMed] [Google Scholar]
- 14.Wang ZY, Chen Z. Blood. 2008;111:2505–15. doi: 10.1182/blood-2007-07-102798. [DOI] [PubMed] [Google Scholar]
- 15.Soignet SL, Frankel SR, Douer D, Tallman MS, Kantarjian H, Calleja E, Stone RM, Kalaycio M, Scheinberg Da, Steinherz P, Sievers EL, Coutré S, Dahlberg S, Ellison R, Warrell RP. J Clin Oncol. 2001;19:3852–60. doi: 10.1200/JCO.2001.19.18.3852. [DOI] [PubMed] [Google Scholar]
- 16.Powell BL, Moser B, Stock W, Gallagher RE, Willman CL, Stone RM, Rowe JM, Coutre S, Feusner JH, Gregory J, Couban S, Appelbaum FR, Tallman MS, Larson RA. Blood. 2010;116:3751–7. doi: 10.1182/blood-2010-02-269621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ghavamzadeh A, Alimoghaddam K, Rostami S, Ghaffari SH, Jahani M, Iravani M, Mousavi SA, Bahar B, Jalili M. J Clin Oncol. 2011;29:2753–7. doi: 10.1200/JCO.2010.32.2107. [DOI] [PubMed] [Google Scholar]
- 18.Au WY, Kumana CR, Lee HKK, Lin SY, Liu H, Yeung DYM, Lau JSM, Kwong YL. Blood. 2011;118:6535–43. doi: 10.1182/blood-2011-05-354530. [DOI] [PubMed] [Google Scholar]
- 19.Altman JK, Tallman MS. Blood. 2011;118:6477–8. doi: 10.1182/blood-2011-11-386557. [DOI] [PubMed] [Google Scholar]
- 20.Noort D, Benschop HP, Black RM. Toxicol Appl Pharmacol. 2002;184:116–126. [PubMed] [Google Scholar]
- 21.The Merck Index. 1983;10th [Google Scholar]
- 22.Columbia Encyclopedia. 2004;6th [Google Scholar]
- 23.Korte NE, Fernando Q. Crit Rev Environ Control. 1991;21:1–39. [Google Scholar]
- 24.Ferguson JF, Gavis J. Water Res. 1972;6:1259–1274. [Google Scholar]
- 25.Environmental Protection, A. Fed Regist. 2001;66:6975–7066. [Google Scholar]
- 26.National Research, C. Arsenic in Drinking Water: 2001 Update. The National Academies Press; 2001. [PubMed] [Google Scholar]
- 27.Agency for Toxic, S; Disease, R. Toxicological Profile for Arsenic. 2007 [PubMed] [Google Scholar]
- 28.Hu J, Liu YF, Wu CF, Xu F, Shen ZX, Zhu YM, Li JM, Tang W, Zhao WL, Wu W, Sun HP, Chen QS, Chen B, Zhou GB, Zelent A, Waxman S, Wang ZY, Chen SJ, Chen Z. Proc Natl Acad Sci U S A. 2009;106:3342–7. doi: 10.1073/pnas.0813280106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mathews V, George B, Chendamarai E, Lakshmi KM, Desire S, Balasubramanian P, Viswabandya A, Thirugnanam R, Abraham A, Shaji RV, Srivastava A, Chandy M. J Clin Oncol. 2010;28:3866–71. doi: 10.1200/JCO.2010.28.5031. [DOI] [PubMed] [Google Scholar]
- 30.Ni Dhubhghaill O, Sadler P. Bioinorganic Chemistry. Vol. 78. Springer Berlin Heidelberg; 1991. The structure and reactivity of arsenic compounds: Biological activity and drug design; pp. 129–190. [Google Scholar]
- 31.Sellers P, Sunner S, Wadso I. Acta Chem Scand. 1964;18:202–206. [Google Scholar]
- 32.Hirano S, Cui X, Li S, Kanno S, Kobayashi Y, Hayakawa T, Shraim A. Arch Toxicol. 2003;77:305–12. doi: 10.1007/s00204-003-0447-x. [DOI] [PubMed] [Google Scholar]
- 33.Caldwell KL, Jones RL, Verdon CP, Jarrett JM, Caudill SP, Osterloh JD. J Exposure Sci Environ Epidemiol. 2009;19:59–68. doi: 10.1038/jes.2008.32. [DOI] [PubMed] [Google Scholar]
- 34.Soignet SL, Maslak P, Wang ZG, Jhanwar S, Calleja E, Dardashti LJ, Corso D, DeBlasio A, Gabrilove J, Scheinberg DA, Pandolfi PP, Warrel RP. N Engl J Med. 1998;339:1341–1348. doi: 10.1056/NEJM199811053391901. [DOI] [PubMed] [Google Scholar]
- 35.Reaves ML, Sinha S, Rabinowitz JD, Kruglyak L, Redfield RJ. Science (New York, NY) 2012;337:470–3. doi: 10.1126/science.1219861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Erb TJ, Kiefer P, Hattendorf B, Günther D, Vorholt Ja. Science (New York, NY) 2012;337:467–70. doi: 10.1126/science.1218455. [DOI] [PubMed] [Google Scholar]
- 37.Hughes MF. Toxicology letters. 2002;133:1–16. doi: 10.1016/s0378-4274(02)00084-x. [DOI] [PubMed] [Google Scholar]
- 38.Doudoroff M, Barker HA, Hassid WZ. Journal of Biological Chemistry. 1947:147–150. [PubMed] [Google Scholar]
- 39.VanEtten RL, Waymack PP, Rehkop DM. Journal of the American Chemical Society. 1974;96:6782–6785. doi: 10.1021/ja00828a053. [DOI] [PubMed] [Google Scholar]
- 40.Drueckhammer DG, Durrwachter JR, Pederson RL, Crans DC, Daniels L, Wong CH. J Org Chem. 1989;54:70–77. [Google Scholar]
- 41.Crans DC, Simone CM, Blanchard JS. J Am Chem Soc. 1992;114:4926–4928. [Google Scholar]
- 42.Auesukaree C, Homma T, Tochio H, Shirakawa M, Kaneko Y, Harashima S. J Biol Chem. 2004;279:17289–17294. doi: 10.1074/jbc.M312202200. [DOI] [PubMed] [Google Scholar]
- 43.Reipa V. Bioelectrochemistry. 2004;65:47–9. doi: 10.1016/j.bioelechem.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 44.Spuches AM, Kruszyna HG, Rich AM, Wilcox DE. Inorg Chem. 2005;44:2964–72. doi: 10.1021/ic048694q. [DOI] [PubMed] [Google Scholar]
- 45.Scott N, Hatlelid KM, MacKenzie NE, Carter DE. Chem Res Toxicol. 1993;6:102–6. doi: 10.1021/tx00031a016. [DOI] [PubMed] [Google Scholar]
- 46.Delnomdedieu M, Basti MM, Otvos JD, Thomas DJ. Chem Biol Interact. 1994;90:139–155. doi: 10.1016/0009-2797(94)90099-x. [DOI] [PubMed] [Google Scholar]
- 47.Rey Na, Howarth OW, Pereira-Maia EC. J Inorg Biochem. 2004;98:1151–9. doi: 10.1016/j.jinorgbio.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 48.Percy AJ, Gailer J. Bioinorg Chem Appl. 2008;2008:539082–539082. doi: 10.1155/2008/539082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Petrick JS, Ayala-Fierro F, Cullen WR, Carter DE, Vasken Aposhian H. Toxicol Appl Pharmacol. 2000;163:203–7. doi: 10.1006/taap.1999.8872. [DOI] [PubMed] [Google Scholar]
- 50.Gier JD, Mandersloot JG, Deenen LLMV. Biochim Biophys Acta. 1968;150:666–675. doi: 10.1016/0005-2736(68)90056-4. [DOI] [PubMed] [Google Scholar]
- 51.Yang HC, Fu HL, Lin YF, Rosen BP. Chapter Twelve - Pathways of Arsenic Uptake and Efflux. In: José MA, Svetlana L, editors. Current Topics in Membranes. Vol. 69. Academic Press; 2012. pp. 325–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu Z, Carbrey JM, Agre P, Rosen BP. Biochem Biophys Res Commun. 2004;316:1178–85. doi: 10.1016/j.bbrc.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 53.Tsukaguchi H, Shayakul C. J Biol Chem. 1998;273:24737–24743. doi: 10.1074/jbc.273.38.24737. [DOI] [PubMed] [Google Scholar]
- 54.Carbrey JM, Gorelick-Feldman Da, Kozono D, Praetorius J, Nielsen S, Agre P. Proc Natl Acad Sci U S A. 2003;100:2945–50. doi: 10.1073/pnas.0437994100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu Z, Shen J, Carbrey JM, Mukhopadhyay R, Agre P, Rosen BP. Proc Natl Acad Sci U S A. 2002;99:6053–8. doi: 10.1073/pnas.092131899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bhattacharjee H, Carbrey J, Rosen BP, Mukhopadhyay R. Biochem Biophys Res Commun. 2004;322:836–41. doi: 10.1016/j.bbrc.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 57.Leung J, Pang A, Yuen WH, Kwong YL, Tse EWC. Blood. 2007;109:740–6. doi: 10.1182/blood-2006-04-019588. [DOI] [PubMed] [Google Scholar]
- 58.Lin S, Shi Q, Nix FB, Styblo M, Beck Ma, Herbin-Davis KM, Hall LL, Simeonsson JB, Thomas DJ. J Biol Chem. 2002;277:10795–803. doi: 10.1074/jbc.M110246200. [DOI] [PubMed] [Google Scholar]
- 59.Wood TC, Salavagionne OE, Mukherjee B, Wang L, Klumpp AF, Thomae Ba, Eckloff BW, Schaid DJ, Wieben ED, Weinshilboum RM. J Biol Chem. 2006;281:7364–73. doi: 10.1074/jbc.M512227200. [DOI] [PubMed] [Google Scholar]
- 60.Song X, Geng Z, Li X, Zhao Q, Hu X, Zhang X, Wang Z. Biochimie. 2011;93:369–75. doi: 10.1016/j.biochi.2010.10.010. [DOI] [PubMed] [Google Scholar]
- 61.Wang S, Li X, Song X, Geng Z, Hu X, Wang Z. The Journal of biological chemistry. 2012;287:38790–9. doi: 10.1074/jbc.M112.368050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ajees aA, Marapakala K, Packianathan C, Sankaran B, Rosen BP. Biochemistry. 2012;51:5476–85. doi: 10.1021/bi3004632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Leslie EM, Haimeur A, Waalkes MP. J Biol Chem. 2004;279:32700–8. doi: 10.1074/jbc.M404912200. [DOI] [PubMed] [Google Scholar]
- 64.Zaman GJ, Lankelma J, van Tellingen O, Beijnen J, Dekker H, Paulusma C, Oude Elferink RP, Baas F, Borst P. Proc Natl Acad Sci U S A. 1995;92:7690–4. doi: 10.1073/pnas.92.17.7690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu J, Chen H, Miller DS, Saavedra JE, Keefer LK, Johnson DR, Klaassen CD, Waalkes MP. Mol Pharmacol. 2001;60:302–9. doi: 10.1124/mol.60.2.302. [DOI] [PubMed] [Google Scholar]
- 66.Yang CH, Kuo ML, Chen JC, Chen YC. Br J Cancer. 1999;81:796–9. doi: 10.1038/sj.bjc.6690766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Davison K, Côté S, Mader S, Miller WH. Leukemia. 2003;17:931–40. doi: 10.1038/sj.leu.2402876. [DOI] [PubMed] [Google Scholar]
- 68.Dai J, Weinberg RS, Waxman S, Jing Y. Blood. 1999;93:268–77. [PubMed] [Google Scholar]
- 69.Larochette N, Decaudin D, Jacoto tE, Brenner C, Marzo I, Susin Sa, Zamzami N, Xie Z, Reed J, Kroemer G. Experimental cell research. 1999;249:413–21. doi: 10.1006/excr.1999.4519. [DOI] [PubMed] [Google Scholar]
- 70.Zheng Y, Shi Y, Tian C, Jiang C, Jin H, Chen J, Almasan A, Tang H, Chen Q. Oncogene. 2004;23:1239–47. doi: 10.1038/sj.onc.1207205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cai X, Shen YL, Zhu Q, Jia PM, Yu Y. Leukemia. 2000;14:262–270. doi: 10.1038/sj.leu.2401650. [DOI] [PubMed] [Google Scholar]
- 72.Zhang X, Yang F, Shim JY, Kirk KL, Anderson DE, Chen X. Cancer Letters. 2007;255:95–106. doi: 10.1016/j.canlet.2007.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Platanias LC. Journal of Biological Chemistry. 2009;284:18583–18587. doi: 10.1074/jbc.R900003200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nordberg J, Arnér ESJ. Free Radical Biology and Medicine. 2001;31:1287–1312. doi: 10.1016/s0891-5849(01)00724-9. [DOI] [PubMed] [Google Scholar]
- 75.Zhong L, Arnér ES, Holmgren A. Proc Natl Acad Sci U S A. 2000;97:5854–9. doi: 10.1073/pnas.100114897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Johansson L, Chen C, Thorell JO. Nat Methods. 2004;1:1–6. doi: 10.1038/nmeth707. [DOI] [PubMed] [Google Scholar]
- 77.Lin S, Del Razo LM, Styblo M, Wang C, Cullen WR, Thomas DJ. Chem Res Toxicol. 2001;14:305–11. doi: 10.1021/tx0001878. [DOI] [PubMed] [Google Scholar]
- 78.Lu J, Chew EH, Holmgren A. Proc Natl Acad Sci U S A. 2007;104:12288–93. doi: 10.1073/pnas.0701549104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ling YH, Jiang JD, Holland JF, Perez-Soler R. Mol Pharmacol. 2002;62:529–38. doi: 10.1124/mol.62.3.529. [DOI] [PubMed] [Google Scholar]
- 80.Li YM, Broome JD. Cancer Res. 1999:776–780. [PubMed] [Google Scholar]
- 81.Ma DC, Sun YH, Chang KZ, Ma XF, Huang SL, Bai YH, Kang J, Liu YG, Chu JJ. Eur J Haematol. 1998;61:27–35. doi: 10.1111/j.1600-0609.1998.tb01057.x. [DOI] [PubMed] [Google Scholar]
- 82.Perkins C, Kim CN, Fang G, Bhalla KN. Blood. 2000:1014–1022. [PubMed] [Google Scholar]
- 83.de Stanchina E, Querido E, Narita M, Davuluri RV, Pandolfi PP, Ferbeyre G, Lowe SW. Mol Cell. 2004;13:523–35. doi: 10.1016/s1097-2765(04)00062-0. [DOI] [PubMed] [Google Scholar]
- 84.Lallemand-Breitenbach V, de Thé H. Cold Spring Harbor Perspect Biol. 2010;2:a000661–a000661. doi: 10.1101/cshperspect.a000661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jensen K, Shiels C, Freemont PS. Oncogene. 2001;20:7223–33. doi: 10.1038/sj.onc.1204765. [DOI] [PubMed] [Google Scholar]
- 86.Jeanne M, Lallemand-Breitenbach V, Ferhi O, Koken M, Le Bras M, Duffort S, Peres L, Berthier C, Soilihi H, Raught B, de Thé H. Cancer cell. 2010;18:88–98. doi: 10.1016/j.ccr.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 87.Zhang XW, Yan XJ, Zhou ZR, Yang FF, Wu ZY, Sun HB, Liang WX, Song AX, Lallemand-Breitenbach V, Jeanne M, Zhang QY, Yang HY, Huang QH, Zhou GB, Tong JH, Zhang Y, Wu JH, Hu HY, de Thé H, Chen SJ, Chen Z. Science. 2010;328:240–3. doi: 10.1126/science.1183424. [DOI] [PubMed] [Google Scholar]
- 88.Chen SJ, Zhou GB, Zhang XW, Mao JH, de Thé H, Chen Z. Blood. 2011;117:6425–37. doi: 10.1182/blood-2010-11-283598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Douer D, Tallman MS. J Clin Oncol. 2005;23:2396–410. doi: 10.1200/JCO.2005.10.217. [DOI] [PubMed] [Google Scholar]
- 90.Dilda PJ, Hogg PJ. Cancer Treat Rev. 2007;33:542–64. doi: 10.1016/j.ctrv.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 91.Camacho LH, Hong DS, Gutierrez C, Vertovsek S, Tannir N, Parker CA, Purdom MA, Lewis J, Gale RP, Kurzrock R. Phase-1 Trial of ZIO-101, a novel organic arsenic in patients with advanced cancers. 2006:13041–13041. [Google Scholar]
- 92.Aribi A, Kantarjian HM, Estey EM, Koller Ca, Thomas Da, Kornblau SM, Faderl SH, Laddie NM, Garcia-Manero G, Cortes JE. Cancer. 2007;109:1355–9. doi: 10.1002/cncr.22524. [DOI] [PubMed] [Google Scholar]
- 93.Estey E, Garcia-Manero G, Ferrajoli A, Faderl S, Verstovsek S, Jones D, Kantarjian H. Blood. 2006;107:3469–73. doi: 10.1182/blood-2005-10-4006. [DOI] [PubMed] [Google Scholar]
- 94.Tsimberidou AM, Kantarjian HM, Garcia-Manero G, Koller C, Jones DM, Keating MJ, Estey E. Clinical outcomes and rates of molecular remission with all-trans retinoic acid (ATRA) and arsenic trioxide (As2O3) combination therapy in newly diagnosed acute promyelocytic leukemia (APL) 2006:6503–6503. [Google Scholar]
- 95.Berenson JR, Boccia R, Siegel D, Bozdech M, Bessudo A, Stadtmauer E, Talisman Pomeroy J, Steis R, Flam M, Lutzky J, Jilani S, Volk J, Wong SF, Moss R, Patel R, Ferretti D, Russell K, Louie R, Yeh HS, Swift RA. Br J Haematol. 2006;135:174–83. doi: 10.1111/j.1365-2141.2006.06280.x. [DOI] [PubMed] [Google Scholar]
- 96.Hussein MA, Saleh M, Ravandi F, Mason J, Rifkin RM, Ellison R. Br J Haematol. 2004;125:470–6. doi: 10.1111/j.1365-2141.2004.04941.x. [DOI] [PubMed] [Google Scholar]
- 97.Munshi NC, Tricot G, Desikan R, Badros a, Zangari M, Toor a, Morris C, Anaissie E, Barlogie B. Leukemia. 2002;16:1835–7. doi: 10.1038/sj.leu.2402599. [DOI] [PubMed] [Google Scholar]
- 98.Qazilbash MH, Saliba RM, Nieto Y, Parikh G, Pelosini M, Khan FB, Jones RB, Hosing C, Mendoza F, Weber DM, Wang M, Popat U, Alousi A, Anderlini P, Champlin RE, Giralt S. Biol Blood Marrow Transplant. 2008;14:1401–7. doi: 10.1016/j.bbmt.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Batista WM, Hineman L, Seneviratne L, Bagdasarian B, Wada JK, Aboulafia D, Malpass T, Gota CH. Phase I/II study of arsenic trioxide (ATO) in combination with cytosine arabinoside (ARA-C) in patients with myelodysplastic syndromes (MDS) 2006:16525–16525. [Google Scholar]
- 100.Raza A, Buonamici S, Lisak L, Tahir S, Li D, Imran M, Chaudary NI, Pervaiz H, Gallegos JA, Alvi MI, Mumtaz M, Gezer S, Venugopal P, Reddy P, Galili N, Candoni A, Singer J, Nucifora G. Leuk Res. 2004;28:791–803. doi: 10.1016/j.leukres.2003.11.018. [DOI] [PubMed] [Google Scholar]
- 101.Schiller GJ, Slack J, Hainsworth JD, Mason J, Saleh M, Rizzieri D, Douer D, List AF. J Clin Oncol. 2006;24:2456–64. doi: 10.1200/JCO.2005.03.7903. [DOI] [PubMed] [Google Scholar]
- 102.Stroh A, Carter TH, Hohl RJ, Zehr P, Ritchie JM. Pilot Study of Lovastatin in Patients with Myelodysplastic Syndrome (MDS) Receiving Arsenic Trioxide (Trisenox) Therapy. 2005:4928–4928. [Google Scholar]
- 103.Chang JE, Voorhees PM, Kolesar JM, Ahuja HG, Sanchez FA, Rodriguez GA, Kim K, Werndli J, Bailey HH, Kahl BS. Hematol Oncol. 2009;27:11–16. doi: 10.1002/hon.870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ravandi F, van Besien K. Leukemia. 2003;17:271–2. doi: 10.1038/sj.leu.2402735. [DOI] [PubMed] [Google Scholar]
- 105.Douer D, Watkins K, Louie R, Weitz IC, Mohrbacher A, Mark L, Levine AM. Treatment of non-APL acute myelogenous leukemia with intravenous arsenic trioxide plus ascorbic acid. 2006:1959–1959. [Google Scholar]
- 106.Kornblau SM, Jackson CE, Worthing A, Faderl S, Beran M, Fayad L, Ravandi-Kashani F, Bothakur G, Gale R, Verstovsek S. A phase I trial of a novel organic arsenic S-dimethylarsino-glutathione (ZIO-101) in hematological malignancies. 2006:16503–16503. [Google Scholar]
- 107.Parmar S, Rundhaugen LM, Boehlke L, Riley M, Nabhan C, Raji A, Frater JL, Tallman MS. Leuk Res. 2004;28:909–19. doi: 10.1016/j.leukres.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 108.Roboz GJ, Ritchie EK, Curcio T, Provenzano J, Carlin R, Samuel M, Wittenberg B, Mazumdar M, Christos PJ, Mathew S, Allen-Bard S, Feldman EJ. Cancer. 2008;113:2504–11. doi: 10.1002/cncr.23855. [DOI] [PubMed] [Google Scholar]
- 109.Wetzler M, Andrews C, Ford La, Tighe S, Barcos M, Sait SNJ, Block AW, Nowak NJ, Baer MR, Wang ES, Baumann H. Cancer. 2011;117:4861–8. doi: 10.1002/cncr.26097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ravandi-Kashani F, Ridgeway J, Nishimura S, Agarwal M, Feldman L, Salvado A, Kovak MR, Hoffman R. Pilot study of combination of imatinib mesylate and trisenox (As2O3) in patients with accelerated (AP) and blast phase (BP) CML. 2003:314–314. [Google Scholar]
- 111.Grimm SA, Marymont M, Chandler JP, Muro K, Newman SB, Levy RM, Jovanovic B, McCarthy K, Raizer JJ. J Neurooncol. 2012;110:237–43. doi: 10.1007/s11060-012-0957-6. [DOI] [PubMed] [Google Scholar]
- 112.Ryu S, Crocker I, Stieber V, Ye X, Fisher JD, Grossman S. Perfusion and diffusion MRI in the assessment of the antivascular effect of arsenic trioxide combined with radiotherapy for glioblastoma multiforme: NABTT phase I study. 2005 [Google Scholar]
- 113.Lin CC, Hsu C, Hsu CH, Hsu WL, Cheng AL, Yang CH. Invest New Drugs. 2007;25:77–84. doi: 10.1007/s10637-006-9004-9. [DOI] [PubMed] [Google Scholar]
- 114.Kim KB, Bedikian AY, Camacho LH, Papadopoulos NE, McCullough C. Cancer. 2005;104:1687–92. doi: 10.1002/cncr.21386. [DOI] [PubMed] [Google Scholar]
- 115.Ardalan B, Subbarayan PR, Ramos Y, Gonzalez M, Fernandez A, Mezentsev D, Reis I, Duncan R, Podolsky L, Lee K, Lima M, Ganjei-Azar P. Clin Cancer Res. 2010;16:3019–27. doi: 10.1158/1078-0432.CCR-09-2590. [DOI] [PubMed] [Google Scholar]
- 116.Kindler HL, Aklilu M, Nattam S, Vokes EE. Am J Clin Oncol. 2008;31:553–6. doi: 10.1097/COC.0b013e318178e4cd. [DOI] [PubMed] [Google Scholar]
- 117.Gallagher RE, Ferrari A, Kaubisch A, Makower D, Stein C, Rajdev L, Gucalp R, Wadler S, Mandeli J, Sarta C. Arsenic trioxide (ATO) in metastatic hormone-refractory prostate cancer (HRPC): Results of phase II trial T99-0077. 2004:4638–4638. [Google Scholar]
- 118.Vuky J, Yu R, Schwartz L, Motzer RJ. Invest New Drugs. 2002;20:327–30. doi: 10.1023/a:1016270206374. [DOI] [PubMed] [Google Scholar]
- 119.Du YH, Ho PC. Cancer Chemother Pharmacol. 2001;47:481–490. doi: 10.1007/s002800100278. [DOI] [PubMed] [Google Scholar]
- 120.Wu J, Shao Y, Liu J, Chen G, Ho PC. J Ethnopharmacol. 2011;135:595–602. doi: 10.1016/j.jep.2011.03.071. [DOI] [PubMed] [Google Scholar]
- 121.Wang J, Lin M, Zhang T, Yan Y, Ho PC, Xu QH, Loh KP. J Am Chem Soc. 2008;130:11596–7. doi: 10.1021/ja804436w. [DOI] [PubMed] [Google Scholar]
- 122.Wang J, Loh KP, Wang Z, Yan Y, Zhong Y, Xu QH, Ho PC. Angew Chem Int Ed. 2009;48:6282–5. doi: 10.1002/anie.200900586. [DOI] [PubMed] [Google Scholar]
- 123.Chen H, MacDonald RC, Li S, Krett NL, Rosen ST, O'Halloran TV. J Am Chem Soc. 2006;128:13348–9. doi: 10.1021/ja064864h. [DOI] [PubMed] [Google Scholar]
- 124.Chen H, Ahn R, Van den Bossche J, Thompson DH, O'Halloran TV. Mol Cancer Ther. 2009;8:1955–63. doi: 10.1158/1535-7163.MCT-09-0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Chen H, Pazicni S, Krett NL, Ahn RW, Penner-Hahn J, Rosen ST, O'Halloran TV. Angew Chem Int Ed. 2009;121:9459–9463. doi: 10.1002/anie.200903655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ahn RW, Chen F, Chen H, Stern ST, Clogston JD, Patri AK, Raja MR, Swindell EP, Parimi V, Cryns VL, O'Halloran TV. Clin Cancer Res. 2010:3607–3617. doi: 10.1158/1078-0432.CCR-10-0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lee Sm, Lee Os, Halloran TVO, Schatz GC, Nguyen ST. ACS Nano. 2011:3961–3969. doi: 10.1021/nn200478m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ahn RW, Barrett SL, Raja MR, Jozefik JK, Spaho L, Chen H, Bally MB, Mazar AP, Avram MJ, Winter JN, Gordon LI, Shea LD, O'Halloran TV, Woodruff TK. PloS one. 2013;8:e58491–e58491. doi: 10.1371/journal.pone.0058491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Winter ND, Murphy RKJ, O'Halloran TV, Schatz GC. J Liposome Res. 2011;21:106–115. doi: 10.3109/08982104.2010.483597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kallinteri P, Fatouros D, Klepetsanis P, Antimisiaris SG. J Liposome Res. 2004;14:27–38. doi: 10.1081/lpr-120039661. [DOI] [PubMed] [Google Scholar]
- 131.Kallinteri P, Fatouros D, Klepetsanis P, Antimisiaris SG. Journal of liposome research. 2004;14:27–38. doi: 10.1081/lpr-120039661. [DOI] [PubMed] [Google Scholar]
- 132.Hernandez-Molina R, Sokolov MN, Clausen M, Clegg W. Inorg Chem. 2006;45:10567–75. doi: 10.1021/ic061146n. [DOI] [PubMed] [Google Scholar]
- 133.Sokolov MN, Virovets AV, Dybtsev DN, Chubarova EV, Fedin VP, Fenske D, Mo HOM. Inorg Chem. 2001:4816–4817. doi: 10.1021/ic015520p. [DOI] [PubMed] [Google Scholar]
- 134.Phadnis PP, Jain VK, Klein A, Schurr T, Kaim W. New J Chem. 2003;27:1584–1584. [Google Scholar]
- 135.Miodragovic´ ÐUQ, Kurutz JA, Stern JW, Ahn CL, Kandela RW, Mazar I, O'Halloran TV. Angew Chemie Int Ed. 2013 doi: 10.1002/anie.201303251. Angew Chem Int Ed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Du YH, Ho P. Cancer Chemother Pharmacol. 2001;47:481–490. doi: 10.1007/s002800100278. [DOI] [PubMed] [Google Scholar]
- 137.Helm CW, States JC. J Ovarian Res. 2009;2:2–2. doi: 10.1186/1757-2215-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Jungwirth U, Kowol CR, Keppler BK, Hartinger CG, Berger W, Heffeter P. Antioxid Redox Signaling. 2011;00 doi: 10.1089/ars.2010.3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kido Y, Khokhar AR, Siddik ZH. Anti-Cancer Drugs. 1993 doi: 10.1097/00001813-199304000-00018. [DOI] [PubMed] [Google Scholar]
- 140.Fricker SP. Dalton Trans. 2007:4903–17. doi: 10.1039/b705551j. [DOI] [PubMed] [Google Scholar]
- 141.Helleman J, Burger H, Hamelers IHL, Boersma AWM, Anton IP, Kroon MD, Stoter G, Nooter K. Cancer Biol Ther. 2006;5:943–949. doi: 10.4161/cbt.5.8.2876. [DOI] [PubMed] [Google Scholar]
- 142.Intini FP, Pellicani RZ, Boccarelli A, Sasanelli R, Coluccia M, Natile G. Eur J Inorg Chem. 2008;2008:4555–4561. [Google Scholar]
- 143.Ballirano P, Maras A. Z Kristallogr. 2002;217:177–178. [Google Scholar]
- 144.Mullen DJE, Nowacki W. Z Kristallogr. 1972;136:48–65. [Google Scholar]