

Abstract
Altered carbohydrate metabolism in cancer cells was first noted by Otto Warburg more than 80 years ago. Upregulation of genes controlling the glycolytic pathway under normoxia, known as the Warburg effect, clearly differentiates malignant from non-malignant cells. The resurgence of interest in cancer metabolism aims at a better understanding of the metabolic differences between malignant and non-malignant cells and the creation of novel therapeutic and diagnostic agents exploiting these differences.
Modified d-glucose and d-mannose analogs were shown to interfere with the metabolism of their respective monosaccharide parent molecules and are potentially clinically useful anticancer and diagnostic agents.
One such agent, 2-deoxy-d-glucose (2-DG), has been extensively studied in vitro and in vivo and also clinically evaluated. Studies clearly indicate that 2-DG has a pleiotropic mechanism of action. In addition to effectively inhibiting glycolysis, 2-DG has also been shown to affect protein glycosylation. In order to better understand its molecular mechanism of action, we have designed and synthesized deuterated molecular probes to study 2-DG interference with d-glucose and d-mannose metabolism using mass spectrometry. We present here the synthesis of all desired probes: 2-deutero-d-glucose, 2-deutero-d-mannose, 6-deutero-d-glucose, 6-deutero-d-mannose, and 2-deutero-2-deoxy-d-glucose as well as their complete chemical characterization.
Keywords: 2-Deutero-d-glucose, 2-Deutero-d-mannose, 6-Deutero-d-glucose, 6-Deutero-d-mannose, 2-Deutero-2-deoxy-d-glucose, Metabolic probes
1. Introduction
Metabolism of simple monosaccharides plays an important role in tumor growth and survival. Unlike normal cells, which reserve the anaerobic metabolism of sugars for hypoxic conditions, tumor cells preferentially metabolize monosaccharides via the ancient anaerobic pathway of glycolysis regardless of ambient oxygen tension.
Given this metabolic discrepancy, modified d-glucose and d-mannose analogs have been developed to exploit the metabolism of malignant cells. These modified analogs are known to interfere with the metabolism of their respective monosaccharides, and are thus potentially clinically useful anticancer and diagnostic agents. For example, 18F-labeled 2-deoxy-2-fluoro-d-glucose (2-18FDG) is now widely used in oncology as a diagnostic and imaging agent in conjunction with positron emission tomography (PET).1
2-FDG works by exploiting the eponymous Warburg effect,2 that is the increased dependence of tumor cells on glycolysis even under normoxic conditions. Diagnostic and therapeutic opportunities stem from the fact that, in contrast to malignant cells, normal cells under normoxia will preferentially use aerobic glucose metabolism to produce energy (via the Krebs/TCA cycle), while glycolysis, the anaerobic metabolism of d-glucose, will be used only when cells are deprived of oxygen (hypoxic conditions). This provides a potentially wide therapeutic window.
The replacement of the hydroxyl at C-2 with the fluorine atom in 2-FDG does not interfere with the first step of glycolysis and allows for hexokinase-mediated phosphorylation at the C-6 hydroxyl to 2-FG 6-phosphate; however, further metabolism to d-fructose 6-phosphate is not possible. Increased accumulation of 18F-labeled 2-FDG 6-phosphate inside the highly glycolytic tumor cells can be easily detected with the use of PET.
The therapeutic option of blocking glycolysis, the primary means of energy-generating glucose metabolism in tumor cells, has been clinically explored using 2-deoxy-d-arabino-hexose (2-deoxy-d-glucose, 2-DG)3 and has led to numerous in vitro studies.4 Like 2-FDG, the lack of a hydroxyl group at C-2 in 2-DG leads to the accumulation of 2-DG 6-phosphate inside highly glycolytic tumor cells and the blockade of d-glucose metabolism. It should be noted that, for historical reasons, 2-DG has been called 2-deoxy-d-glucose, even though it is also a 2-deoxy-d-mannose and, because of sharing the same structural feature, the 2-DG can also affect d-mannose metabolism, including glycosylation processes. 2-DG has been demonstrated to affect glycosylation processes5 and induce endoplasmic reticulum stress.6
In order, to study in detail the molecular aspects of the effects of 2-DG on the metabolism of d-glucose and d-mannose using mass spectrometry, we have designed specifically deuterated probes and performed their synthesis. We present in detail chemical synthesis and characterization of 2-deutero-d-glucose (8), 2-deutero-d-mannose (13), 6-deutero-d-glucose (19), 6-deutero-d-mannose (26), and 2-deutero-2-deoxy-d-glucose (4).
2. Results and discussion
2.1. Synthesis of 2-deutero-2-deoxy-d-glucose
We proposed an efficient method for synthesis of 2-deutero-2-deoxy-d-glucose. Current efficient methods led to 2-deutero-2-deoxy-d-mannose with an axially oriented deuterium atom rather than to 2-deutero-2-deoxy-d-glucose. The first synthesis of 2-deutero-2-deoxy-d-mannose by Wong and Gray7 utilized appropriately protected ketene dithioacetal from d-glucose and its reduction with lithium aluminum deuteride, and final deprotection to 2-deutero-2-deoxy-d-mannose. Yet another method described by Lehmann and Petry8 started from fructose that, in a five-step process, led to 2-azi-2-deoxy-d-arabino-hexitol that, when irradiated at 350 nm in water gave 2-deutero-2-deoxy-d-mannose. Both methods are multistep processes and are not useful for preparing a multigram amount of final product.
Looking for the method that would be easiest to perform on a multigram scale, we decided to use commercially available and inexpensive per-O-acetylated d-glucal as the starting material. The whole process is presented in Scheme 1. The per-O-acetylated d-glucal was deacetylated in standard Zemplén condition, and without purification was per-O-benzylated. The addition of D2O in the presence of DBr in tetrahydrofuran gave 3,4,6-tri-O-benzyl-2-deutero-d-glucose (3).
Scheme 1.
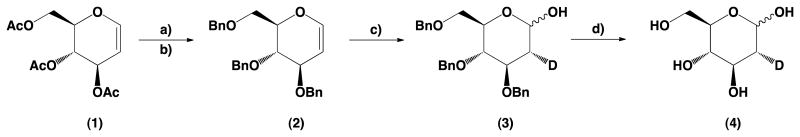
Synthesis of 2-deoxy-2-deutero-d-glucose. Reagents and conditions: (a) 1 N MeONa/MeOH; (b) NaH, BnBr, DMF; (c) DBr/D2O, THF; (d) Pd/C, H2, THF, EtOH.
Proton nuclear magnetic resonance (1H NMR) analysis demonstrated that the obtained product had only equatorial deuterium atom. Debenzylation of (3) by hydrogenation using a Paar apparatus led to the final product with a total yield of 42%. The described method may be easily scaled up, if needed.
2.2. Synthesis of 2-deutero-d-glucose
The most commonly described methods in the literature for preparation of 2-deutero-d-glucose (8) are the enzymatic process with milligram amounts of final products. The only synthetic route for preparation of (8) was described by Haines.9 Starting from methyl 3-O-benzoyl-4,6-benzylidene-α-d-glucopyranoside in a five-step process, Haines obtained milligram amounts of (8), which was per-O-acetylated and analyzed as its per-acetylated derivative. No analytical data for free (8) was included. In our approach (Scheme 2), we chose glucose as a starting material. We decided to use benzyl ethers and benzylidene acetals for protection of hydroxyl groups in positions 1, 3, 5, and 6 because that allowed us to achieve one-step deprotection of the final product. The key step in our process was to obtain benzyl α-glucoside with ketone group in position 2. Reduction of the ketone with NaBD4 led exclusively to the desired gluco product (8).
Scheme 2.

Synthesis of 2-deoxy-2-deutero-d-glucose. Reagents and conditions: (a) Dess–Martin periodinate, CH2Cl2; (b) NaBD4, CH2Cl2, MeOH; (c) Pd/C, H2.
Specifically, benzyl 3-O-benzyl-4,6-benzylidene-α-d-mannopyranoside (5) used as the starting compound was obtained using methods described in the literature. Briefly d-mannose was transformed into its benzyl glycoside,10 followed by formation of 4,6-benzylidene acetal,11 and benzylation at position 3 utilizing a stannyl ester.11 The free 2-hydroxyl group was oxidized with high yield (64%) using Dess–Martin periodinate. Reduction of such obtained ketone (6) with NaBD4 gave benzyl 3-O-benzyl-4,6-benzylidene-2-deutero-α-d-glucopyranose (7). Hydrogenation of (7) with hydrogen and palladium on carbon Degussa type allowed us to obtain high purity product in gram scale. Our approach required fewer synthetic steps than the method described by Haines. The final deprotection step was fast, with easy work-up and purification of the final compound, and may be performed in multigram scale.
2.3. Synthesis of 2-deutero-d-mannose
Our approach (Scheme 3) toward synthesis of 2-deutero-d-mannose (13) was based on the reported stereoselective reduction of β-d-arabino-hexopyranoside-2-ulose derivatives with NaBH4 compounds with manno configuration.12 Our goal was to prepare β-glycoside with a ketone group at C-2 position, with all remaining hydroxyl groups protected with benzyl ether or benzylidene acetals. We applied the Lichtenthaler synthetic process leading in two-steps from 2-acetoxy-d-glucal with good yield to respective β-ketone.13
Scheme 3.

Synthesis of 2-deutero-d-mannose. Reagents and conditions: (a) HBr/AcOH, CH2Cl2; (b) DBU, CH2Cl2; (c) NBS, EtOH, CH2Cl2; (d) BnOH, Ag2CO3, CH2Cl2; (e) NaBD4, CH2Cl2, MeOH; (f) Pd/C, H2.
The starting compound 1,2-di-O-acetyl-3,4,6-tri-O-benzyl-d-glucose (9), was prepared from per-O-benzylated-d-glucal by its cis-hydroxylation14 followed by acetylation. Compound (9) was reacted with HBr (33% solution in acetic acid) to give glycosyl bromide which upon treatment with DBU formed 2-O-acetoxy-3,4,6-tri-O-benzyl-d-glucal (10). Subsequently, using the method described by Lichtenthaler,12 2-acetoxy-d-glucal (10) was transformed into a ketone (11). Reduction of (11) using NaBD4 gave benzyl 3,4,6-tri-O-benzyl-β-d-mannopyranoside (12) with high yield (90%). Hydrogenation of (12) with hydrogen and palladium on carbon Degussa type gave the final compound, 2-deutero-d-mannose (13). Each step in the described method can be scaled up to a multigram scale.
2.4. Synthesis of 6-deutero-d-glucose
Synthesis of (S)-d-6-deutero-glucose was already described by Xu15 and used as a substrate for methyl 2,3,4-tri-O-benzyl-d-(6,6-2H2)glucopyranoside which upon Swern oxidation of 6-OH group and subsequent reduction of an aldehyde with nondeuterated (R)-(+)-Alpine-Borane gave with high optical purity an (S) isomer. As there was no need in our studies for pure (S) or (R) stereoisomers; we opted for a rather simpler high yield synthesis of 6-deutero-glucose (19) as an (S/R) mixture, as shown in Scheme 4.
Scheme 4.

Synthesis of 6-deutero-d-glucose. Reagents and conditions: (a) NaH, BnBr, DMF; (b) H2SO4, MeOH; (c) Swern oxidation; (d) NaBD4, CH2Cl2, MeOH; (e) Pd/C, H2.
To economize synthesis of 19 a mixture of α and β anomers 14 was used as a starting material. Benzyl α,β-d-glucopyranoside (14) protected with 6-O-tert-butyldimethylsilyl ether was benzylated to give compound 15 and then desilylated to benzyl 2,3,4-tri-O-benzyl-d-glucopyranoside (16). Product 16 has not been described in the literature thus for analytical purposes α and β anomers 16 were separated and characterized as 16α and 16β. Swern oxidation of the primary hydroxyl group at C-6 gave the aldehyde 17 that without separation was subjected to reduction with sodium borodeuteride to give compound 18 deuterated at position C-6. The same oxidation and reduction reactions were performed using pure anomers 16α and 16β in order to secure analytical data for the new chemical entities. Subsequent debenzylation of 18 by hydrogenation gave 6-deutero-d-glucose (19) as a mixture of (6R) and (6S) epimers.
2.5. Synthesis of 6-deutero-d-mannose
We found no evidence in the literature of a method for the synthesis of 6-deutero-d-mannose. Since we did not seek to obtain pure (R) or (S) isomer, we decided to use a strategy similar to that used for preparation of 6-deutero-d-glucose. Our approach is shown in Scheme 5.
Scheme 5.

Synthesis of 6-deutero-d-mannose. Reagents and conditions: (a) t-BuMe2SiCl, imidazole, DMF; (b) NaH, BnBr, DMF; (c) H2SO4, MeOH; (d) Swern oxidation; (e) NaBD4, CH2Cl2, MeOH; (f) Pd/C, H2.
Starting from benzyl α-d-mannopyranoside (20) protection of the 6-hydroxyl group with tert-butyldimethylsilyl was followed by benzylation of the remaining hydroxyl groups and deprotection of the position 6 to give benzyl 2,3,4-tri-O-benzyl-α-d-mannopyranoside (23). Swern oxidation of a primary hydroxyl group at C-6 gave an aldehyde (24) with a high yield as indicated by TLC. Reduction of (24) with sodium borodeuteride gave benzyl 2,3,4-tri-O-benzyl-6-deutero-α-d-mannopyranoside as a mixture of (R) and (S) isomers (25). Subsequent debenzylation of (25) by hydrogenation gave the desired 6-deutero-d-mannose (26).
3. Conclusions
To study 2-DG interference with d-glucose and d-mannose metabolism, five deuterated molecular probes, namely 2-deutero-d-glucose (8), 2-deutero-d-mannose (13), 6-deutero-d-glucose (19), 6-deutero-d-mannose (26), and 2-deutero-2-deoxy-d-glucose (4) were synthesized in gram scale using modified or novel methodology. The described synthetic procedures are practical and scalable.
4. Experimental
4.1. Materials and methods
Unless otherwise noted, reagents were purchased from commercial sources and used without further purification. DBr/D2O solution was purchased from Sigma–Aldrich and has isotopic purity of 99%+, NaBD4 was purchased from Cambridge Isotope Laboratories (D4 99%, chemical purity 90–95%). 1H NMR spectra were recorded on Bruker Avanti 300 or Bruker Avanti 500-MHz spectrometers in CDCl3 with TMS or D2O as the internal reference standard. 13C NMR spectra were recorded on a Bruker Avanti 500-MHz spectrometer in CDCl3 or DMSO-d6 with TMS as the internal reference standard or D2O. 1H and 13C signals for fully deprotected compounds were assigned based on 2D spectra: proton–proton correlation (GRADCOSY) and proton–carbon correlation (C13HSQC) and are shown in Tables 1 and 2, respectively. Mass spectrometry analysis was performed using Acuity UPLC-MS/MS (TQD-3) and AMD-604 mass spectrometers. Melting points were measured on a Büchi melting point apparatus (Büchi) and were uncorrected. Optical rotations were measured using Perkin Elmer 341 Polarimeter (Perkin Elmer). Thin–layer chromatography (TLC) was performed on an aluminum sheet coated with SilicaGel 60 F254 (EMD Millipore). Compounds were visualized on TLC plates by spraying the plates with 20% H2SO4 in EtOH and gently heating them. Silica gel column chromatography was performed with a CombiFlash Companion purification system (ISCO, Inc.) and a Biotage SP1 purification system (Biotage, LLC).
Table 1. 1H NMR data for α and β anomers of deutero-sugarsa.
| Compound | H-1 J1,2 |
H-2 J2,3 |
H-3 J3,4 |
H-4 J4,5 |
H-5 J5,6 ; J5,6 |
H-6 J6,6 |
H-6 J6,6 |
|---|---|---|---|---|---|---|---|
| 4α | 5.29 3.4 Hz | 1.70 11.9 Hz | 3.95 9.2 Hz | 3.38 9.0 Hz | 3.89−3.77 m | 3.89−3.77 m | 3.89−3.77 m |
| 4β | 4.95 9.8 Hz | 1.51 10.9 Hz | 3.77−3.70 m | 3.29 9.4 Hz | 3.43−3.36 m | 3.91 12.4 Hz | 3.77−3.70 m |
| 8α | 5.25 | – | 3.73 8.9 Hz | 3.39−3.46 m | 3.82−3.89 m | 3.81−3.74 m | 3.95−3.84 m |
| 8β | 4.66 | – | 3.50 8.8 Hz | 3.39−3.46 m | 3.51−3.46 m | 3.81−3.74 m | 3.95−3.84 m |
| 13α | 5.20 | – | 3.86 9.7 Hz | 3.68 9.5 Hz | 3.84 9.2 Hz | 3.80−3.72 m | 3.95−3.86 m |
| 13β | 4.90 | – | 3.67 9.6 Hz | 3.59 9.7 Hz | 3.40 9.2 Hz | 3.80−3.72 m | 3.95−3.86 m |
| 19α | 5.25 3.8 Hz | 3.55 9.2 Hz | 3.73 9.6 Hz | 3.45−3.39 m | 3.85 5.2 Hz | 3.83 − | -- |
| 19β | 4.66 8.0 Hz | 3.26 9.2 Hz | 3.51 9.1 Hz | 3.45−3.39 | 3.48 5.7 Hz | 3.76 − | -- |
| 26α | 5.22 | 3.99−4.00 m | 3.99 9.5 Hz | 3.69 9.7 Hz | 3.86 5.8 Hz | 3.79 − | – |
| 26β | 4.97 | 3.99−4.00 m | 3.70 9.5 Hz | 3.61 9.8 Hz | 3.42 6.2 Hz | 3.75 − | – |
All spectra recorded in D2O.
Table 2. 13C NMR spectra of deutero-sugarsa.
| Compound | C-1 | C-2 JC,D |
C-3 | C-4 | C-5 | C-6 JC,D |
|---|---|---|---|---|---|---|
| 4α | 93.36 | 36.94 19.8 Hz |
67.91 | 71.17 | 72.00 | 60.73 |
| 4β | 91.26 | 39.17 19.7 Hz |
70.42 | 70.84 | 75.99 | 60.98 |
| 8α | 95.85 | 71.05 22.0 Hz |
72.70 | 69.64 | 71.41 | 60.75 |
| 8β | 92.04 | 73.68 22.0 Hz |
75.68 | 69.59 | 75.90 | 60.60 |
| 13α | 95.50 | 71.79 21.7 Hz |
71.66 | 68.35 | 73.89 | 63.49 |
| 13β | 95.13 | 72.31 21.7 Hz |
74.47 | 68.11 | 77.64 | 62.49 |
| 19α | 92.04 | 71.43 | 72.73 | 69.61 | 71.32 | 60.42 20.2 Hz |
| 19β | 95.86 | 74.09 | 75.71 | 69.56 | 75.83 | 60.25 20.2 Hz |
| 26α | 95.50 | 72.16 | 71.72 | 68.34 | 74.52 | 62.16 23.0 Hz |
| 26β | 95.14 | 72.69 | 74.52 | 68.10 | 77.55 | 62.16 23.0 Hz |
All spectra recorded in D2O.
4.2. Benzylation–General procedure (A)
4.2.1. 3,4,6-Tri-O-benzylo-d-glucal (2)
d-Glucal (0.184 mol) was dissolved in DMF (300 mL). The obtained solution was cooled down to −20 °C and NaH (60% suspension in mineral oil) (0.83 mol, 33.2 g) was added. After 5 min, benzyl bromide (0.83 mol, 97.5 mL) was added dropwise with vigorous stirring. The reaction mixture was stirred while the temperature was allowed to rise to ambient. Progress of the reaction was monitored by the TLC method. After the reaction was completed, acetic acid (0.278 mol, 16 mL) in water (100 mL) was added dropwise. The mixture was then diluted with water (500 mL) and the product was extracted with hexanes (2 × 300 mL).
Combined organic solutions were washed with water (3 × 200 mL), and dried over anhydrous Na2SO4. The solids were filtered off and solvent was evaporated to dryness. The remaining crude product was purified by low-pressure column chromatography using hexanes–ethyl acetate gradient. Fractions containing product were pooled together and evaporated to dryness, and residual solvents were removed by additional drying with a high-vacuum oil pump, resulting in 70 g of white crystalline product (2) (yield 92%). Analytical data are consistent with the literature.16
4.2.2. 3,4,6-Tri-O-benzylo-2-deoxy-2-deutero-d-glucopyranose (3)
3,4,6-Tri-O-Benzylo-d-glucal (2) (70 g, 0.168 mol) was dissolved in THF (400 mL). A solution of DBr in D2O (48 wt %) (0.182 mol, 21 mL) was added and the reaction mixture was stirred at room temperature until all substrate disappeared (as determined by TLC). The reaction was quenched by the addition of solid Na2CO3 (91 mmol, 9.65 g) followed by water (50 mL). The mixture was stirred for an additional 15 min, and then THF was evaporated to dryness. AcOEt (500 mL) was added to the remaining oily product. Layers were separated, and the organic layer was washed with water (150 mL), then brine (150 mL), and then dried over anhydrous Na2SO4. The drying agent and solvents were removed to give a crude product that was purified by column chromatography using hexanes–ethyl acetate gradient. Fractions containing product were pooled together and evaporated to dryness, and then crystallized from the hexanes–ethyl acetate mixture, resulting in 40.2 g of white crystals of product (3) (yield 56%) as an inseparable anomeric mixture. Mp 100–101 °C. [α]d +48.1 (c = 1.06, CDCl3). HRMS calcd for C27H29DO5 [M+Na]+ m/z 458.21; found, m/z 458.42.
1H NMR (300 MHz, CDCl3): δ 7.37−7.14 (m, 15 H, H-arom), 5.39 (br s, 1H, H-1α), 4.89 (d, 1H, J = 10.9 Hz, CH2α), 4.88 (d, 1H, J = 10.9 Hz, CH2β), 4.75 (dd, 1H, J = 9.5 Hz, J = 5.9 Hz, H-1β), 4.70−4.47 (m, 5H, CH2α,β), 4.09−3.98 (m, 2H, H-3α, H-5α), 3.75−3.60 (m, 2H, H-6), 3.50 (dd, 1H, J = 9.5 Hz, J = 9.2 Hz, H-4α), 2.76 (br s, 1H, OHα), 2.27 (d, 1H, J = 5.0 Hz, OHβ), 1.67 (br d, 1H, J = 11.3 Hz, H-2α), 1.55 (dd, 1H, J = 11.4 Hz, J = 9.8 Hz, H-2β).
4.2.3. Synthesis of 2-deoxy-2-deutero-d-glucopyranose (4)–General procedure B
3,4,6-Tri-O-benzylo-2-deoxy-2-deutero-d-glucopyranose (3) (11.8 g, 27 mmol) was dissolved in the mixture of THF and methanol (1:2, v/v) (120 mL); Pd/C (10% Pd, Degussa type) (0.8 g) was added and the mixture was hydrogenated using a Paar apparatus for 25 min at room temperature under hydrogen pressure of 42 psi. After 25 min, the mixture was filtered through Celite®, solvent was evaporated to dryness and subsequently purified by column chromatography using a chloroform–methanol gradient for elution. Fractions containing product were pooled together and evaporated to dryness. The residual solvent was removed using a high-vacuum oil pump, resulting in 3.72 g of white crystals of product (4) (yield 83%); mp 117–119 °C, [α]d +47.0 (c = 1.88 H2O). HRMS calcd for C6H11DO5 [M+Na]+ m/z 188.06; found, m/z 188.05.
4.2.4. Synthesis of benzyl 3-O-benzyl-4,6-benzylidene α-d-arabino-hexos-2-ulo-pyranoside (6)
Dess–Martin periodinane (70 g, 165 mmol) was added to the solution of benzyl 3-O-benzyl-4,6-benzylidene-α-d-mannopyranoside (5) (25.9 g, 57.8 mmol) in dry CH2Cl2 (300 mL) and the obtained mixture was stirred at room temperature for 48 h (TLC control). The reaction mixture was diluted with CH2Cl2 (200 mL) and was washed with a saturated solution of K2CO3 (3 × 200 mL) and then with water, until neutral pH. The solution was dried over anhydrous Na2SO4. Inorganic salts and solvents were removed and the product was purified by column chromatography using hexanes–ethyl acetate gradient for elution. Fractions containing product were pooled together and evaporated to dryness. The residual solvent was removed using a high-vacuum oil pump to give 16.5 g of pure product (6) (yield 64%). Mp 131–133 °C lit.17 115–117 °C; [α]d +42.4 (c = 1.14, CDCl3). Lit.17 [α]d +42 (c = 1.15, CDCl3). HRMS calcd for C27H26O6 [M+Na]+ m/z 469.16; found, m/z 469.36.
1H NMR (300 MHz, CDCl3): δ 7.53 −7.24 (m, 15H, H-arom), 5.56 (s, 1H, CH benzylidene), 4.93 (d, 1H, J = 13.8 Hz, CH2), 4.77 (d, 1H, J = 11.7 Hz, CH2), 4.71 (d, 1H, J = 12.0 Hz, CH2), 4.63 (d, 1H, J = 11.7 Hz, CH2), 4.58 (d, 1H, J = 10.3 Hz, H-2), 4.25−4.18 (m, 2H, H-5, H-6), 3.89 (dd, 1H, J = 10.2 Hz, J = 9.3 Hz, H-4), 3.80 (dd, 1H, J = 9.9 Hz, J = 9.8 Hz, H-6).
4.2.5. Synthesis of benzyl 3-O-benzyl-4,6-benzylidene α-d-glucopyranoside (7)
Benzyl 3-O-benzyl-4,6-benzylidene α-d-arabino-hexos-2-ulo-pyranoside (6) (24.1 g, 54 mmol) was dissolved in a mixture of CH2Cl2 (80 mL) and methanol (100 mL). Sodium borodeuteride (2.3 g, 55 mmol) was added to the solution in 4 portions. The reaction mixture was stirred at room temperature for 20 min. Solvents were partially removed until the product started to precipitate. The resulting white crystals were filtered and washed with methanol and water, then dried to give 22.9 g of (7) (yield 94%); mp 114–115 °C; [α]d +83.3 (c = 1.09, CDCl3). HRMS calcd for C27H27DO6 [M+Na]+ m/z 472.18; found, m/z 472.53.
1H NMR (300 MHz, CDCl3): δ 7.53 − 7.23 (m, 15H, H-arom), 5.57 (s, 1H, CH benzylidene), 5.03 (s, 1H, H-1), 4.96 (d, 1H, J = 11.5 Hz, CH2), 4.81 (d, 1H, J = 11.5 Hz, CH2), 4.77 (d, 1H, J = 11.7 Hz, CH2), 4.59 (d, 1H, J = 11.7 Hz, CH2), 4.23 (dd, 1H, J = 10.0 Hz, J = 4.6 Hz, H-6), 3.88 (ddd, 1H, J = 10.1 Hz, J = 9.9 Hz, J = 4.6 Hz, H-5), 3.86 (d, 1H, J = 9.5 Hz, H-3), 3.75 (dd, 1H, J = 10.1 Hz, J = 10.2 Hz, H-6), 3.66 (dd, 1H, J = 9.4 Hz, J = 9.3 Hz, H-4), 2.27 (s, 1H, OH).
4.2.6. Synthesis of 2-deutero-d-glucose (8)
Pd(OH)2/C (20% Pd(OH)2, Degussa type) (1.1 g) was added to the solution of benzyl 3-O-benzyl-4,6-benzylidene-α-d-glucopyranoside (7) (17 g, 37.8 mmol) in THF (80 mL). The mixture was hydrogenated using a Paar apparatus for 1 h (42 psi); methanol (100 mL) was then added and the mixture was hydrogenated for an additional 12 h. The reaction mixture was filtered through Celite, the solvents were evaporated, and crude product was purified by column chromatography using a chloroform–methanol gradient for elution. Fractions containing product were pooled together and evaporated to dryness. The residual solvent was removed using a high-vacuum oil pump, resulting in 5.3 g of (8) as white crystals (yield 77%); mp 144–148 °C, [α]d +50 (c = 1.57, H2O); HRMS calcd for C6H11DO6 [M+Na]+ m/z 204.06; found, m/z 204.05.
4.2.7. Synthesis of 2-acetoxy-3,4,6-tri-O-benzyl-d-glucal (10)
The 1,2-di-O-acetyl-3,4,6-tri-O-benzyl-d-glucopyranose (9) (14.0 g, 26.2 mmol) was dissolved in CH2Cl2 (140 mL) then a solution of HBr in acetic acid (30 wt %) (9.5 mL, 52.3 mmol) was added. The reaction mixture was stirred at room temperature until all substrate was converted into glycosyl bromide (as determined by TLC). The reaction mixture was diluted with CH2Cl2 (140 mL) and washed with an ice-cold saturated solution of sodium carbonate then with water until neutral, and with brine. It was then filtered through a layer of anhydrous Na2SO4. The mixture's volume was reduced to about 140 mL and DBU (6 mL, 52.3 mmol) was added; the resulting mixture was stirred at room temperature while progress of the reaction was monitored by TLC. After the reaction was completed the reaction mixture was diluted with CH2Cl2 (100 mL) and washed with water. The organic layer was dried over anhydrous Na2SO4. The drying agent and solvent were removed and the product was purified by column chromatography using hexanes–ethyl acetate gradient for elution. The fractions containing products were combined and evaporated to dryness. The residual solvents were removed using high-vacuum oil pump to give 6.8 g of (10) as a colorless oil which solidify upon standing (yield 55%). [α]d +21.9° (c = 1.38, CDCl3) lit.18 [α]d + 24° (c = 1.6, CDCl3); HRMS calcd for C29H30O6 [M+Na]+ m/z 497.19; found, m/z 497.44.
1H NMR (300 MHz, CDCl3): δ 7.38 −7.23 (m, 15H, H-arom), 6.49 (br s, 1H, H-1), 4.57 (d, 1H, J = 11.5 Hz, CH2), 4.63 (d, 1H, J = 11.5 Hz, CH2), 4.59 (d, 1H, J = 11.5 Hz, CH2), 4.56 (s, 2H, CH2), 4.51 (d, 1H, J = 11.5 Hz, CH2), 4.44 (d, 1H, J = 5.1 Hz, H-3), 4.26−4.19 (m, 1H, H-5), 3.97 (dd, 1H, J = 7.2 Hz, J = 5.1 Hz, H-4), 3.83 (dd, 1H, J = 10.7 Hz, H-6), 3.73 (dd, 1H, J = 10.7 Hz, J = 3.5 Hz, H-6), 2.07 (s, 3H, OAc).
4.2.8. Synthesis of benzyl 3,4,6-tri-O-benzyl-β-d-arabino-hexos-2-ulo-pyranoside (11)
A mixture of 2-acetoxy-3,4,6-tri-O-benzyl-d-glucal (10) (6.6 g, 13.9 mmol), molecular sieves 3 Å, in CH2Cl2 (66 mL), and ethanol (20 mmol, 1.2 mL) was prepared, cooled down to 0 °C, and stirred for 10 min. NBS (3 g, 16.7 mmol) was added and stirring was continued until yellow color appeared. The solids were filtered off, and the filtrate was washed with ice-cold 10% solution of Na2S2O3 and then with water, and the remaining organic solution was dried over anhydrous Na2SO4. The drying agent was filtered off and the solvent was evaporated to dryness. The residue was dissolved in CH2Cl2 (25 mL) and added to a previously prepared mixture of benzyl alcohol (2.8 mL, 27.8 mmol), CH2Cl2 (50 mL), Ag2CO3 (11.5 g, 41.7 mmol), and molecular sieves 4 Å (1 g). The obtained mixture was stirred at room temperature for 1 h, and then filtered through Celite®. The volume of the mixture was reduced to ∼30 mL by evaporation. Upon addition of hexanes (200 mL) the product (11) was precipitated, filtered, washed with hexanes and dried to give 4.2 g of pure 11 (yield 56%). Mp 129–130 °C; [α]d −70.2 (c = 1.01, CDCl3). HRMS calcd for C34H34O6 [M+Na]+ m/z 561.23; found, m/z 561.52.
1H NMR (300 MHz, CDCl3): δ 7.40−7.16 (m, 20H, H-arom), 4.97 (d, 1H, J = 11.4 Hz, CH2), 4.93 (d, 1H, J = 12.0 Hz, CH2), 4.75 (s, 1H, H-1), 4.73 (d, 1H, J = 12.0 Hz, CH2), 4.62−4.52 (m, 4H, CH2), 4.19 (d, 1H, J = 8.9 Hz, H-3), 3.90 (dd, 1H, J = J = 9.0 Hz, H-4), 3.81 (ddd, 1H, J = 9.0 Hz, J = 4.9 Hz, J = 2.3 Hz, H-5), 3.76 (dd, 1H, J = 10.7 Hz, J = 2.3 Hz, H-6), 3.72 (dd, 1H, J = 10.7 Hz, J = 5.1 Hz, H-6).
4.2.9. Synthesis of benzyl 3,4,6-tri-O-benzyl-β-d-mannopyranoside (12)–General procedure C
Benzyl 3,4,6-tri-O-benzyl β-d-arabino-hexos-2-ulo-pyranoside (11) (3.5 g, 6.5 mmol) was dissolved in CH2Cl2 (15 mL), followed by addition of methanol (15 mL), then NaBD4 (1.1 g, 27 mmol) was added and the reaction mixture was stirred at room temperature, while progress of the reaction was monitored by TLC. After the reaction was completed, the reaction mixture was diluted with CH2Cl2 (75 mL), and washed with water until neutral. The crude product was purified by column chromatography using hexanes–ethyl acetate gradient for elution. Fractions containing product were combined and evaporated to dryness. The residual solvents were removed using a high-vacuum oil pump to give 3.2 g of (12) as a colorless oil (yield 90%); [α]d −53.4 (c = 1.17, CDCl3).
1H NMR (300 MHz, CDCl3): δ 20H, H-arom), 4.95 (d, 1H, J = 12.0 Hz, CH2), 4.89 (d, 1H, J = 10.9 Hz, CH2), 4.75 (d, 1H, J = 10.9 Hz, CH2), 4.70−4.52 (m, 5H, CH2), 4.45 (s, 1H, H-1), 3.88 (d, 1H, J = 9.3 Hz, J = 9.4 Hz, H-4), 3.81 (dd, 1H, J = 10.8 Hz, J = 2.2 Hz, H-6), 3.74 (dd, 1H, J = 10.8 Hz, J = 5.2 Hz, H-6), 3.54 (d, 1H, J = 9.0 Hz, H-3), 3.42 (ddd, 1H, J = 9.5 Hz, J = 5.2 Hz, J = 2.2 Hz, H-5), 2.45 (s, 1H, OH)
4.2.10. Synthesis of 2 β-d-mannopyranose (13)
Benzyl 3,4,6-tri-O-benzyl-β-d-mannopyranoside (12) (3 g, 5.53 mmol) was dissolved in a mixture of THF–methanol (1:1 v/v) (30 mL). Pd/C (10% Pd, Degussa type) (230 mg) was added and the mixture was hydrogenated using a Paar apparatus (42 psi H2 pressure) for 4 h. The mixture was filtered through Celite®, the filtrate was evaporated to dryness and the crude product was purified by column chromatography using chloroform–methanol gradient for elution. Fractions containing product were pooled together and evaporated to dryness. The residual solvents were removed using high-vacuum oil pump to give 0.942 g of white crystals of product (13) (yield 94%). Mp 125–129 °C, [α]d +13.5° (c = 1.75, H2O); HRMS calcd for C6H11DO6 [M+Na]+ m/z 204.06; found, m/z 204.05.
4.2.11. Synthesis of benzyl 2,3,4-tri-O-benzyl-6-tert-butyldimethylsilyl-d-glucopyranoside (15)
A solution of benzyl 6-tert-butyldimethylsilyl-d-glucopyranoside (14) (8.2 g, 21.5 mmol) in DMF (150 mL) was prepared and cooled to −15 °C. NaH (60% suspension in mineral oil) (3.1 g, 77.7 mmol) was added and the mixture was stirred for 5 min, then benzyl bromide (9.2 mL, 77.7 mmol) was added dropwise. The mixture was stirred vigorously while temperature was allowed to rise to ambient. After the reaction was completed (TLC) the reaction mixture was cooled down to 0 °C; then acetic acid (0.755 mL, 13, 2 mmol) followed by water (200 mL) was added. The mixture was extracted with hexanes (250 mL). The hexane solution was washed with water (2 × 100 mL), then dried over anhydrous Na2SO4. Drying agent and solvent were removed and product was purified by column chromatography using hexanes–ethyl acetate gradient for elution. Fractions containing product were pooled together and evaporated to dryness, residual solvents were removed using a high-vacuum oil pump to give 13 g of (15) (yield 92%). HRMS calcd for C40H50O6Si [M+Na]+ m/z 677.33; found, m/z 677.90.
1H NMR (500 MHz, CDCl3): δ 7.42−7.24 (m, 20H, H-arom), 5.0−4.75 (m, H CH2α,β), 4.81 (d, 1H, J = 3.8 Hz, H-1α), 4.75−4.52 (m, H, CH2-α,β), 4.49 (d, 1H, J = 7.7 Hz, H-1β), 4.05 (dd, 1H, J = 9.3 Hz, H-3α), 3.89 (d, 1H, J = 10.6 Hz, H-6β), 3.84 (dd, 1H, J = 11.4 Hz, J = 4.2 Hz, H-6β), 3.77 (dd, 1H, J = 11.3 Hz, J = 3.4 Hz, H-6α), 3.62 (br d, 1H, J = 12.2 Hz, H-6α), 3.71−3.67 (m, 1H, H-5α), 3.67−3.57 (m, 2H, H-3β H-4β), 3.54 (dd, J = 9.7 Hz, J = 9.4 Hz, H-4α), 3.50 (dd, 1H, J = 9.7 Hz, J = 3.9 Hz, H-2α), 3.47 (dd, 1H, J = 9.6 Hz, J = 8.2 Hz, H-2β), 3.29 (m, H-5β), 0.92, 0.89 (2s, 9H ea, t-Bu), 0.102, 0.089, 0.051, 0.04 (4s, 3H ea, Me)
13C NMR (500 MHz, CDCl3): δ 139.06, 138.82, 138.76, 138.69, 138.61, 138.45, 137.69, 137.49 (C-arom), 128.55, 128.53, 128.51, 128.49, 128.40, 128.34, 128.31, 128.24, 128.20, 128.13, 128.04, 128.01, 127.92, 127.88, 127.84, 127.75, 127.73, 127.72, 127.66, (C-arom), 102.47 (C-1β), 95.31 (C-1α), 84.88 (C-3β), 82.74 (C-2β), 82.34 (C-3α), 80.51 (C-2α), 77.99 (C-4α), 77.82 (C-4β), 75.99 (C-5β), 75.96, 75.93, 75.21, 75.14, 75.04, 73.10, 71.18, 70.97, 68.86 (CH2Ph), 71.98 (C-5α), 62.46, 62.39 (C-6α β), 26.22, 26.12 (t-BuSi), −4.79, −4.93, −5.12, −5.16 (Me2Si).
4.2.12. Synthesis of benzyl 2,3,4-tri-O-benzyl-d-glucopyranoside (16)
Sulfuric acid (1 mL, 18.8 mmol) was added to the vigorously stirred suspension of benzyl 2,3,4-tri-O-benzyl-6-tert-butyldimethylsilyl-d-glucopyranoside (15) (10 g, 15.3 mmol) in methanol (100 mL). The reaction mixture was stirred at room temperature. After the reaction was completed (as determined by TLC), the reaction mixture was diluted with water (100 mL). A stoichiometric amount of Na2CO3 was added, and the product was extracted with ethyl acetate (250 mL). The extract was washed with water and dried over Na2SO4. The product was purified by column chromatography using hexanes–ethyl acetate gradient. Fractions containing product were pooled together and evaporated to dryness, residual solvents were removed using a high-vacuum oil pump to give 7 g of product (16) (yield 85%).
Benzyl 2,3,4-tri-O-benzyl-β-d-glucopyranoside (16β): mp 101–103 °C [α]d −8.5° (c = 1.53, CHCl3), HRMS calcd for C34H36O6 [M+Na]+ m/z 563.24; found, m/z 563.54.
1H NMR (300 MHz, CDCl3): δ 20H, H-arom), 4.99−4.60 (m, 8H, CH2), 4.57 (d, 1H, J = 7.8 Hz, H-1), 3.87 (ddd, 1H, J = 11.9 Hz, J = 5.9 Hz, J = 2.8 Hz, H-6), 3.70 (ddd, 1H, J = 11.9 Hz, J = 7.5 Hz, J = 4.7 Hz, H-6), 3.67 (dd, 1H, J = 9.0 Hz, J = 8.7 Hz, H-3), 3.57 (dd, 1H, J = 9.4 Hz, J = 9.0 Hz, H-4), 3.49 (dd, 1H, J = 8.8 Hz, J = 7.8 Hz, H-2), 3.36 (ddd, 1H, J = 9.6 Hz, J = 4.6 Hz, J = 2.8 Hz, H-5), 1.84 (dd, 1H, J = 7.5 Hz, J = 6.1 Hz, OH).
Benzyl 2,3,4-tri-O-benzyl-α-d-glucopyranoside (16α): Mp 86–87 °C [α]d +70.1° (c = 1.80, CHCl3); HRMS calcd for C34H36O6 [M+Na]+ m/z 563.24; found, m/z 563.54.
1H NMR (300 MHz, CDCl3): δ 7.42−7.24 (m, 20H, H-arom), 5.01 (d, 1H, J = 10.9 Hz, CH2), 4.89 (d, 1H, J = 11.0 Hz, CH2), 4.84 (d, 1H, J = 10.9 Hz, CH2), 4.80 (d, 1H, J = 3.7 Hz, H-1), 4.67 (dd, 3H, J = 12.2 Hz, J = 12.7 Hz, CH2), 4.56 (d, 1H, J = 11.9 Hz, CH2), 4.55 (d, 1H, J = 12.3 Hz, CH2), 4.07 (dd, 1H, J = J = 9.3 Hz, H-3), 3.75−3.60 (m, 3H, H-6, H-6, H-5), 3.54 (dd, 1H, J = 9.1 Hz, H-4), 3.50 (dd, 1H, J = 9.5 Hz, J = 3.7 Hz, H-2), 1.57 (br s, 1H, OH).
4.2.13. Synthesis of benzyl 2,3,4-tri-O-benzyl-6-deutero-d-glucopyranoside (18)
Benzyl 2,3,4-tri-O-benzyl-d-glucopyranoside (16) (5 g, 9,24 mmol) was oxidized using method reported in the literature19 and, without purification such obtained crude aldehyde (17) was subjected to the reduction with NaBD4 (general procedure C). After the reaction was completed, the reaction mixture was diluted with CH2Cl2 (100 mL) and washed with water until neutral. The crude product was purified by column chromatography using hexanes–ethyl acetate gradient for elution. Fractions containing product were combined and evaporated to dryness. The residual solvents were removed using a high-vacuum oil pump to give 4.94 g of (18) (yield 90%).
Benzyl 2,3,4-tri-O-benzyl-6-deutero-α-d-glucopyranoside (18α): Mp 86–87 °C, [α]d +70.2° (c = 1.27, CHCl3); HRMS calcd for C34H35DO6 [M+Na]+ m/z 564.25; found, m/z 564.55.
1H NMR (500 MHz, CDCl3): δ 7.42−7.24 (m, 20H, H-arom), 5.01 (d, 1H, J = 10.8 Hz, CH2), 4.88 (d, 1H, J = 11.0 Hz, CH2), 4.84 (d, 1H, J = 10.8 Hz, CH2), 4.81 (d, 1H, J = 3.5 Hz, H-1), 4.68 (d, 2H, J = 11.0 Hz, CH2), 4.64 (d, 1H, J = 11.0 Hz, CH2), 4.56 (d, 1H, J = 11.0 Hz, CH2), 4.55 (d, 1H, J = 10.8 Hz, CH2), 4.07 (dd, 1H, J = J = 9.3 Hz, H-3), 3.75−3.62 (m, 2H, H-6, H-5), 3.34 (dd, 1H, J = J = 9.3 Hz, H-4), 3.59 (dd, 1H, J = 9.7 Hz, J = 3.6 Hz, H-2), 1.54 (dd, 1H, J = 3.5 Hz, OH).
13C NMR (500 MHz, CDCl3): δ 138.95, 138.28, 138.25, 137.22, (C-arom), 128.56, 128.51, 128.58, 128.47, 128.44, 128.14, 128.00, 127.95, 127.87, 127.66 (C-arom), 95.71 (C-1), 82.04 (C-3), 80.19 (C-2), 77.58 (C-4), 75.77 (CH2), 75.15 (CH2), 73.13 (CH2), 77.11 (C-5), 69.34 (CH2), 61.52 (t, J = 21 Hz, C-6).
Benzyl 2,3,4-tri-O-benzyl-6-deutero-β-d-glucopyranoside (18β): Mp 101–103 °C, [α]d −8.6° (c = 1.42, CHCl3); HRMS calcd for C34H35DO6 [M+Na]+ m/z 564.25; found, m/z 564.55.
1H NMR (500 MHz, CDCl3): δ 7.40−7.24 (m, 20H, H-arom), 4.94 (d, 1H, J = 10.6 Hz, CH2), 4.92 (d, 1H, J = 10.2 Hz, CH2), 4.91 (d, 1H, J = 11.8 Hz, CH2), 4.86 (d, 1H, J = 11.0 Hz, CH2), 4.80 (d, 1H, J = 11.0 Hz, CH2), 4.72 (d, 1H, J = 11.0 Hz, CH2), 4.69 (d, 1H, J = 11.8 Hz, CH2), 4.63 (d, 1H, J = 11.0 Hz, CH2), 4.56 (d, 1H, J = 7.8 Hz, H-1), 3.85 (dd, 1H, J = 5.5 Hz, J = 2.5 Hz, H-6 (R or S)), 3.68 (dd, 1H, J = 9.2 Hz, J = 9.0 Hz, H-3), 3.70−3.66 (m, 1H, H-6 (R or S)), 3.57 (dd, 1H, J = 9.5 Hz, J = 9.0 Hz, H-4 (R or S)), 3.56 (dd, 1H, J = 9.43 Hz, J = 9.2 Hz, H-4 (R or S)), 3.49 (dd, 1H, J = 8.8 Hz, J = 8.1 Hz, H-2), 3.36 (dd, 1H, J = 9.6 Hz, J = 4.6 Hz, H-5), 1.81 (d, 1H, J = 7.7 Hz, OH).
13C NMR (500 MHz, CDCl3): δ 138.66, 138.46, 138.13, 137.41, (C-arom), 128.57, 128.47, 128.44, 128.21, 128.13, 128.03, 128.01, 127.98, 127.95, 127.76, 127.71 (C-arom), 102.94 (C-1), 84.66 (C-3), 82.45 (C-2), 77.70 (C-4), 75.76 (CH2), 75.19 (C-5), 75.12 (CH2), 75.05 (CH2), 71.70 (CH2), 61.80 (t, J = 20.5 Hz, C-6).
4.2.14. Synthesis of 6-deutero-d-glucopyranoside (19)
Benzyl 2,3,4-tri-O-benzyl-6-deutero-d-glucopyranoside (18) (16.5 g, 30 mmol) was debenzylated according to general procedure B. After purification, 4.11 g of (19) was obtained (yield 76%) mp 148–151 °C, [α]d +52.2° (c = 1.53, H2O).
4.2.15. Synthesis of benzyl 6-tert-butyldimethylsilyl-α-d-mannopyranoside (21)
A mixture of benzyl α-d-mannopyranoside (20) (7 g, 25.9 mmol) and imidazole (2.72 g, 40 mmol) in DMF (80 mL) was prepared and cooled to 0 °C. Tert-butyldimethylsilyl chloride (4.2 g, 28 mmol) was added and the mixture was stirred overnight while the temperature was allowed to rise to ambient. The reaction mixture was diluted with water (200 mL) and the product was extracted with ethyl acetate (3 × 70 mL). The combined organic extracts were washed with water and brine, then dried over Na2SO4. Inorganic salts were filtered off, the solvent was evaporated to dryness, and the product was purified by column chromatography using hexanes–ethyl acetate gradient for elution. Fractions containing product were combined and evaporated to dryness. The residual solvents were removed using a high-vacuum oil pump to give 8.5 g of (21) (yield 85%). Mp 68–69 °C, [α]d +56.3° (c = 1.7, CHCl3).
1H NMR (300 MHz, DMSO-d6 + D2O): δ 7.40−7.26 (m,5H, H-arom), 4.69 (d, 1H, J = 1.5 Hz, H-1), 4.66 (d, 1H, J = 11.8 Hz, CH2), 4.42 (d, 1H, J = 11.8 Hz, CH2), 3.92 (dd, 1H, J = 10.9 Hz, J = 1.3 Hz, H-3), 3.66−3.57 (m, 2H, H-2, H-4), 3.49 (dd, 1H, J = 9.0 Hz, J = 3.3 Hz, H-6), 3.46−3.40 (m, 1H, H-5), 3.33 (dd, 1H, J = 9.0 Hz, J = 9.5 Hz, H-6), 0.87 (s, 9H, t-BuSi), 0.06, 0.05 (2s, 3Hea, Me2Si).
4.2.16. Synthesis of benzyl 2,3,4-tri-O-benzyl-6-tert-butyldimethylsilyl-α-d-mannopyranoside (22)
The 6-tert-butyldimethylsilyl-α-d-mannopyranoside (21), (3.5 g, 9.1 mmol) was dissolved in DMF (50 mL). NaH (60% mineral oil suspension) (2.15 g, 54 mmol) was added and the mixture was stirred at room temperature for 10 min, then benzyl bromide (6.5 mL, 54 mmol) was added. The mixture was heated to 50 °C for 10 min. The mixture was cooled down to room temperature; then hexanes (100 mL), followed by acetic acid (1.53 mL, 26.7 mmol) in water (150 mL), were added. After 15 min of vigorous stirring layers were separated. The organic layer was washed with water, then dried over Na2SO4. Inorganic salts were filtered off, the solvent was evaporated to dryness and the product was purified by column chromatography using hexanes–ethyl acetate gradient for elution. Fractions containing product were combined and evaporated to dryness. The residual solvents were removed using a high-vacuum oil pump to give 4.56 g of (22) as a colorless oil (yield 77%). [α]d +45.0° (c = 1.56, CHCl3).
1H NMR (300 MHz, CDCl3): δ 7.38−7.24 (m, 20H, H-arom), 4.92 (d, 1H, J = 10.8 Hz, CH2), 4.91 (d, 1H, J = 1.6 Hz, H-1), 4.75−4.60 (m, 6H, CH2), 4.42 (d, 1H, J = 10.8 Hz, CH2), 3.98−3.92 (m, 2H, H-6, H-6), 3.88−3.83 (m, 2H, H-3, H-4), 3.80 (dd, 1H, J = 1.8 Hz, J = 2.5 Hz, H-2), 3.67 (ddd, 1H, J = 9.3 Hz, J = 5.8 Hz, J = 2.6 Hz, H-5), 0.90 (s, 9H, t-BuSi), 0.08, 0.07 (2s, 3Hea, Me2Si).
4.2.17. Synthesis of benzyl 2,3,4-tri-O-benzyl-α-d-mannopyranoside (23)
Sulfuric acid (1 mL, 18.8 mmol) was added to the vigorously stirred suspension of benzyl 2,3,4-tri-O-benzyl-6-tert-butyldimethylsilyl-α-d-mannopyranoside (22) (4.1 g, 6.1 mmol) in methanol (50 mL). The reaction mixture was stirred at room temperature. After the reaction was completed (as determined by TLC), the reaction mixture was diluted with water (100 mL). A stoichiometric amount of Na2CO3 was added, and product was extracted with ethyl acetate (3 × 50 mL). The combined extracts were washed with water and dried over Na2SO4. The product was purified by column chromatography using hexanes–ethyl acetate gradient. Fractions containing product were pooled together and evaporated to dryness, and the residual solvents were removed using a high-vacuum oil pump to give 3.1 g of product (23) as a colorless oil (yield 93%). [α]d +53.4° (c = 1.2, CHCl3). Lit.20 [α]d +54° (c = 0.3, CHCl3).
1H NMR (500 MHz, CDCl3): δ 7.37−7.24 (m, 20H, H-arom), 4.94 (d, 1H, J = 10.8 Hz, CH2), 4.91 (d, 1H, J = 1.1 Hz, H-1), 4.75 (d, 1H, J = 12.3 Hz, CH2), 4.69−4.60 (m, 5H, CH2), 4.43 (d, 1H, J = 12.0 Hz, CH2), 4.02−3.94 (m, 2H, H-3, H-4), 3.86 (m, 3H, H-2, H-6, H-6), 3.70 (m, 1H, H-5), 1.98 (dd, 1H, J = 6.2 Hz, OH).
13C NMR (500 MHz, CDCl3): δ 138.55, 138.48, 138.28, 137.25, (C-arom), 128.52, 128.49, 128.44, 128.16, 127.93, 127.88, 127.81, 127.78, 127.71, 127.65, (C-arom), 97.64 (C-1), 80.27 (C-3), 75.33 (CH2), 74.99 (C-2, C-4), 73.01 (CH2), 72.59 (C-5), 72.38 (CH2), 69.20 (CH2), 62.39 (C-6).
4.2.18. Synthesis of benzyl 2,3,4-tri-O-benzyl-6-deutero-α-d-mannopyranoside (25)
Benzyl 2,3,4-tri-O-benzyl-α-d-mannopyranoside (23) (3.0 g, 5.5 mmol) was oxidized according to the method reported in the literature19 and, without purification, such obtained crude aldehyde (24) was reduced with NaBD4 (general procedure C). After the reaction was completed, the reaction mixture was diluted with CH2Cl2 (100 mL) and washed with water until neutral. The crude product was purified by column chromatography using hexanes–ethyl acetate gradient for elution. Fractions containing product were combined and evaporated to dryness. The residual solvents were removed using a high-vacuum oil pump to give 2.52 g of (25) as a colorless oil (yield 84%). [α]d + 50.1° (c = 1.48, CHCl3); HRMS calcd for C34H35DO6 [M+Na]+ m/z 564.25; found, m/z 564.55.
1H NMR (500 MHz, CDCl3): δ 7.37−7.24 (m, 20H, H-arom), 4.94 (d, 1H, J = 10.8 Hz, CH2), 4.91 (br s, 1H, H-1), 4.75 (d, 1H, J = 12.3 Hz, CH2), 4.69−4.60 (m, 5H, CH2), 4.43 (d, 1H, J = 12.0 Hz, CH2), 4.02−3.94 (m, 2H, H-3, H-4), 3.82 (br s, 1H, H-2), 3.78−3.73 (m, 1H, H-6), 3.72−3.67 (m, 1H, H-5), 1.98 (d, 1H, J = 6.2 Hz, OH).
13C NMR (500 MHz, CDCl3): δ 138.67, 138.61, 138.37, 137.37, (C-arom), 128.60, 128.56, 128.53, 128.24, 128.03, 127.99, 127.97, 127.87, 127.79, 127.73, (C-arom), 97.72 (C-1), 80.36 (C-3), 75.39 (CH2), 75.11 (C-2), 75.04 (C-4), 73.08 (CH2), 72.73 (C-5), 72.44 (CH2), 69.25 (CH2), 62.05 (t, J = 20 Hz, C-6).
4.2.19. Synthesis of 6-deutero-d-mannopyranoside (26)
Benzyl 2,3,4-tri-O-benzyl-6-deutero-α-d-mannopyranoside (25) (3.4 g, 6.3 mmol) was debenzylated according to general procedure B). After purification, 0.93 g of (25) (yield 82%) was obtained; mp 122–129 °C, [α]d +14.2° (c = 1.4, H2O).
Acknowledgments
This work was supported by grants from the CERN Foundation (WP) and Viragh Foundation (WP).
We also acknowledge the NCI Cancer Center Support Grant CA016672 for the support of the NMR and Pharmacology and Analytical Facilities at MD Anderson Cancer Center.
References
- 1.(a) Gambhir SS. Nat Rev Cancer. 2002;2:683–693. doi: 10.1038/nrc882. [DOI] [PubMed] [Google Scholar]; (b) Fowler S, Amett CD, Wolf AP, et al. J Nulc Med. 1982;23:437–445. [Google Scholar]; (c) Ido T, Wan CN, Casella JS, Fowler JS, Wolf AP, Reivich M, KUHL DE. J Labelled Compd Radiopharm. 1978;14:175–182. [Google Scholar]
- 2.Warburg O, Wind F, Negelein E. J Gen Physiol. 1927;8:519–530. doi: 10.1085/jgp.8.6.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pelicano H, Martin DS, Xu RH, Huang P. Oncogene. 2006;25:4633–4646. doi: 10.1038/sj.onc.1209597. [DOI] [PubMed] [Google Scholar]
- 4.(a) Zhang DX, Deslandes E, Villedieu M, Poulain L, Duval M, Gauduchon P, Schwartz L, Icard P. Anticancer Res. 2006;26:3561–3566. [PubMed] [Google Scholar]; (b) Maschek G, Savaraj N, Priebe W, Braunschweiger P, Hamilton K, et al. Cancer Res. 2004;64:31–34. doi: 10.1158/0008-5472.can-03-3294. [DOI] [PubMed] [Google Scholar]
- 5.Datema R, Schwartz RT. Biochem J. 1979;184:113–123. doi: 10.1042/bj1840113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu SM, Kim SJ. Exp Mol Med. 2010;42(11):777–786. doi: 10.3858/emm.2010.42.11.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wong MY, Gray GR. Carbohydr Res. 1980;80:87–89. [Google Scholar]
- 8.Lehmann J, Petry S. Carbohydr Res. 1993;239:133–142. [Google Scholar]
- 9.Haines AH. Carbohydr Res. 1977;58:212–216. doi: 10.1016/s0008-6215(00)83418-7. [DOI] [PubMed] [Google Scholar]
- 10.Krohn K, Borner G, Gringard S. J Org Chem. 1994;59:6069–6074. [Google Scholar]
- 11.Ekholm FS, Polakova M, Pawlowicz AJ, Leino R. Synthesis. 2009;4:567–576. [Google Scholar]
- 12.Lichtenthaler FW, Lergenmuller M, Peters S, Varga Z. Tetrahedron: Asymmetry. 2003;14:727–736. [Google Scholar]
- 13.Lichtenthaler FW, Klaeres U, Lergenmueller M, Schwidetzky S. Synthesis. 1992:179–184. [Google Scholar]
- 14.Nicolaou KC, Mitchell HJ, Suzuki H, Rodriguez RM, Baudoin O, Fylaktakidou KC. Angew Chem, Int Ed. 1999;38:3334–3339. [PubMed] [Google Scholar]
- 15.Xu L, Price NPJ. Carbohydr Res. 2004;339:1173–1178. doi: 10.1016/j.carres.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 16.Bovin NV, Zurabyan SE, Khorlin AY. Carbohydr Res. 1981;98:25–35. [Google Scholar]
- 17.Fairbanks AJ, Sinai P. Tetrahedron Lett. 1995;36(6):893–896. [Google Scholar]
- 18.Lichtenthaler RW, Schneider-Adams T. J Org Chem. 1994;59:6728–6734. [Google Scholar]
- 19.Mancuso AJ, Huang SL, Swern D. J Org Chem. 1978;43(12):2480–2482. [Google Scholar]
- 20.Liptak A, Imre J, Harangi J, Palnanas I. Carbohydr Res. 1983;116:217–225. [Google Scholar]