

Abstract
Interleukin-33 (IL-33) is a member of the IL-1 cytokine family. It predominantly induces type 2 immune responses and thus is protective against atherosclerosis and nematode infections but contributes to allergic airway inflammation. Interleukin-33 also plays a pivotal role in the development of many autoimmune diseases through mechanisms that are still not fully understood. In this review, we focus on the recent advances in understanding of the expression and function of IL-33 in some autoimmune disorders, aiming to provide insight into its potential role in disease development.
Keywords: autoimmune diseases, inflammation, interleukin-33, ST2
Introduction
Interleukin-33 (IL-33) was originally recognized as a nuclear factor of high endothelial venules.1 In 2005 it was identified, via a sequence database search for genes homologous to the IL-1 family, as the ligand for the orphan molecule ST2.2 In contrast to other IL-1 family members such as IL-1 and IL-18, which require caspase-1 for their activation, full-length IL-33 released via cell necrosis is, in fact, biologically active, whereas its cleavage by caspases during apoptotic cell death destroys its bioactivity.3,4 IL-33 can be classified as an alarmin because it is released into the extracellular space following cell damage or tissue injury and acts as an endogenous danger signal by sending out warning signals to alert neighbouring cells and tissues.5 However, in their study determining the sub-cellular localization of IL-33 and tracking its intracellular mobility and extracellular release, Kakkar et al.6 demonstrated that IL-33 is localized simultaneously to nuclear euchromatin and membrane-bound cytoplasmic vesicles, and is secreted by living cells to carry out its extracellular functions without the need for cellular necrosis.
IL-33 is constitutively expressed by tissue barrier cells such as the epithelial and endothelial cells of many organs,2,7 but it is also expressed by some innate immune cells such as macrophages and dendritic cells.2,8,9 IL-33 possesses dual roles both as a traditional extracellular cytokine and as an intracellular nuclear factor with transcriptional regulatory properties.10,11 The pathophysiological role of IL-33 as a nuclear factor is not fully understood but a recent report suggests that full-length nuclear IL-33 sequesters nuclear factor-κB (NF-κB) and reduces NF-κB-triggered gene expression to dampen pro-inflammatory signalling.12
IL-33 exerts its classical cytokine biological effects via binding to a heterodimer receptor complex consisting of ST2 and the IL-1 receptor accessory protein (IL-1RAcP),13 and activating the signalling pathway of MyD88 and NF-κB.5 In addition to the membrane form of ST2, soluble ST2 (sST2) is produced as a result of alternative splicing and acts as a decoy receptor; binding of IL-33 to sST2 results in the inhibition of the IL-33/ST2 pathway. ST2 is expressed by many resting or activated immune cells including T helper type 2 (Th2) cells, mast cells, basophils, macrophages, dendritic cells, CD8 T cells and B cells.2,14–16 IL-33 predominantly induces type 2 immune responses via activation of dendritic cells that drive the differentiation of naive lymphocytes into Th2 cells.17 IL-33 also acts on Th2 cells, nuocytes18 and B1 cells19 to produce IL-5 and IL-13, but not the archetypal Th2 cytokine IL-4.2,20 Furthermore, IL-33 polarizes macrophages towards an alternatively activated phenotype,21 enhances cytotoxic T-lymphocyte clonal expansion and the antiviral function of these cells.16 While IL-33 has also been shown to synergize with IgE in mast cell activation and degranulation,22 long-term exposure to IL-33 renders mast cells refractory to FcεRI stimulation.23
In addition to immune cells, ST2 is also expressed by the endothelium, epithelium cells10,24–26 and fibroblasts7 in many tissues and organs, suggesting the involvement of the IL-33/ST2 pathway in the pathogenesis of a large number of diseases. Whereas the expression of ST2 in specific tissue and organ cells such as adipocytes, osteoblasts, cardiac myocytes and neuronal cells7 indicates that IL-33 also has tissue-specific functions contributing to disease development in these organs. Indeed, emerging evidence suggests that IL-33 has important but also pleiotropic effects in many immune-mediated diseases. It activates immune cells to produce Th2 cytokines such as IL-5 and IL-1320,27 and is often associated with type 2 immune responses that exacerbate allergic airway inflammation,20,21,27–32 confer resistance to nematode infection33 and protect against cardiovascular diseases.34–36 However, IL-33 is also able to induce inflammatory hypernociception,37 a hallmark of pro-inflammatory activity, and is a pro-inflammatory cytokine in collagen-induced arthritis (CIA).38 Thus it is likely that IL-33 can play either pro- or anti- inflammatory roles depending on the specific disease and immune context.
This review therefore aims to bring together current findings from clinical research and models of autoimmune disease to highlight and better understand the role of IL-33. Although we acknowledge that there are limitations to translating research in animal models to human conditions, valuable insights on immune mechanisms that are difficult to explore in humans may be gained.
IL-33 in rheumatoid arthritis
A large body of research suggests that IL-33 is heavily involved in the pathogenesis of rheumatoid arthritis (RA). Early studies using in situ hybridization showed that IL-33 is detected in the blood vessels of RA synovium tissues.10 We and others further confirmed that both IL-33 and ST2 are present in the lining layer and in the interstitial sub-lining layer of tissues from RA patients.38,39 Although resting primary synovial fibroblasts from RA patients express little or no IL-33, the expression level is dramatically up-regulated following the stimulation of pro-inflammatory cytokines such as tumour necrosis factor-α (TNF-α) and IL-1β.38 The above observations indicate that the IL-33/ST2 pathway may contribute to the pathogenesis of joint inflammation and destruction.
Some recent studies have attempted to evaluate the levels of IL-33 and ST2 in serum and synovial fluid (SF) in RA patients with the aim of identifying potential biomarkers for the disease. Robust data from different laboratories indicate that the levels of IL-33 and ST2 in sera and SF samples are significantly increased in RA patients, particularly those with active disease, compared with healthy controls, and osteoarthritis and psoriatic arthritis patients.40–43 Furthermore, higher levels of IL-33 are observed in SF than serum samples of the same RA patients.44 Xiangyang et al.40 further observed that the higher levels of IL-33 detected in the sera of RA patients are positively correlated with disease severity, rheumatic factor and the Modified Sharp Score that was used to evaluate bone erosion; the authors therefore suggest that IL-33 may contribute to RA pathogenesis partly through bone erosion. However, as IL-33 is an inhibitor of bone resorption,45 it can also be speculated that IL-33 might be secreted as a result, rather than being the cause, of bone erosion after joint inflammation. Our own investigations confirmed elevated serum and SF levels of IL-33 in RA patients, although no correlation was found between IL-33 concentration and acute-phase inflammation reactant or the score of the Disease Activity Index.46 Interestingly, we observed that the serum IL-33 concentration correlated with the production of IgM and RA-related auto-antibodies including rheumatic factor and anti-citrullinated protein antibodies, suggesting a more complex and indirect link between IL-33 and RA inflammation involving mast cells and B cells.46 This link is further supported by the strong correlation between serum IL-33 levels and RA treatment as IL-33 levels are decreased in patients after receiving etanercept treatment.47 Similarly, RA patients who responded to treatment with a TNF inhibitor also showed a reduction in serum IL-33.42,46 These important observations suggest that the measurement of IL-33 levels in serum could be a useful marker of RA severity and for monitoring the effect of RA treatment.
Increasing evidence supports a role for the IL-33/ST2 pathway in the pathology associated with chronic articular inflammation. A few years before IL-33 was identified, Leung et al.48 reported that IL-33 decoy receptor sST2-Fc was able to reduce disease severity in CIA mice. The protective effect is also obtained after administering anti-ST2 blocking antibody at the onset of disease, and the attenuated CIA is associated with reduced joint destruction and a marked decrease of interferon-γ (IFN-γ) and IL-17 production by the draining lymph node cells.39 The role of IL-33 as a pro-inflammatory cytokine in arthritis disease was further confirmed by our own reports that exogenous recombinant IL-33 (rIL-33) exacerbated disease severity in CIA or autoantibody-induced arthritis (AIA) mice, whereas ST2-deficient mice developed attenuated CIA or AIA together with marked reduction of pro-inflammatory cytokine production (IL-17, TNF-α and IFN-γ).38,49 We further identified that ST2-expressing mast cells, which are abundant in RA synovium tissues, are the likely cells mediating IL-33 function in RA. We demonstrated that rIL-33 treatment exacerbates CIA or AIA only in ST2-deficient mice engrafted with mast cells from wild-type but not from ST2-deficient mice. It is therefore likely that IL-33 primarily derived from synovial fibroblasts contributes to synovial pathology by activating mast cells to degranulate and produce pro-inflammatory cytokines. It should be noted, however, that extrapolating data based on exogenous IL-33 treatment to form conclusions regarding endogenous IL-33 may be misleading. A recent study50 using IL-33-deficient mice reported that endogenous IL-33 was not required for the development of joint inflammation in AIA. Interestingly, in good agreement with our findings,49 ST2-deficient mice in that study were found to have reduced arthritis severity,50 which could indicate an IL-33-independent effect of ST2, though this requires further investigation.
Taken together, the above data suggest that the IL-33/ST2 pathway is critical in promoting arthritogenic inflammation (Fig. 1) and is a novel therapeutic target for RA disease. However, any treatment involving this pathway will need to be closely monitored as IL-33 also protects bone through inhibiting osteoclast differentiation as shown in in vitro culture.45 In addition, osteoclastogenesis was confirmed to be inhibited in transgenic mice over-expressing IL-33.51
Figure 1.
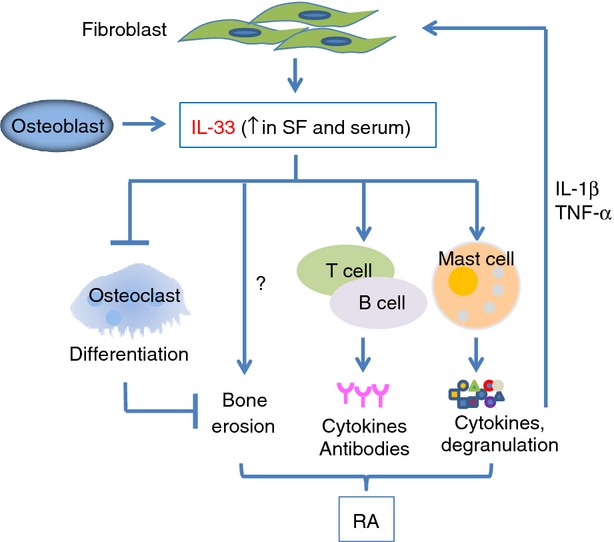
Schematic of the role of interleukin-33 (IL-33) in the development of rheumatoid arthritis (RA). IL-33 released by synovial fibroblasts under inflammatory conditions [detected in the synovial fluid (SF) and serum samples of RA patients] binds to ST2 on immune cells such as mast cells and lymphocytes (Th2 and B cells) and activates these cells to release inflammatory molecules and antibodies contributing to the pathology of the disease. Pro-inflammatory cytokines released by immune cells such as IL-1β and TNF-α can activate fibroblast cells creating a positive feedback system and thus releasing more IL-33. Meanwhile, expression of IL-33 is also increased during osteoblast differentiation and IL-33 is able to inhibit bone absorption by blocking osteoclast formation, therefore protecting bone in RA. In this setting, IL-33 might be secreted as a result, rather than as a cause, of bone erosion.
IL-33 in multiple sclerosis
The role of IL-33 in the development of immune-mediated central nervous system (CNS) diseases such as multiple sclerosis (MS) has gained extra attention because of the extremely high level of IL-33 mRNA expression in the brain and spinal cord,2 suggesting that IL-33/ST2 may have CNS-specific functions in addition to a role as an immune-mediator. Indeed IL-33 is associated with Alzheimer's disease,52 and genetic variants of IL-33 affect disease susceptibility.53 Some recent studies have focused on the expression and function of IL-33 in CNS cells. While pathogen-associated molecular patterns can significantly increase IL-33 expression in cultured CNS glia cells, IL-33 levels and activity are also dramatically increased in the brains of mice infected with Theiler's murine encephalomyelitis virus.54 Subsequent reports confirmed that IL-33 is mainly expressed by astrocytes in murine spinal cord55,56 and in human CNS tissues,57 indicating that IL-33-producing astrocytes may act as a potentially critical regulator of innate immune responses in the CNS. However, less is known about the target CNS cells that IL-33 binds to and activates. We previously observed that ST2 protein is abundantly expressed in murine spinal cord tissues.55 Others using primary CNS cell culture suggested that mRNA of ST2, IL-1RAcP and sST2 were detected in microglia and astrocytes, and only IL-1RAcP was present in neuronal cells.56 Further studies defining the precise cellular location of IL-33 receptor in CNS will help to elucidate the functions of the IL-33/ST2 pathway in CNS homeostasis and disease development.
The importance of IL-33 in MS development was first indicated by the significant up-regulation of IL-33 and/or ST2 in the spinal cord tissues from experimental autoimmune encephalomyelitis (EAE) mice compared with naive ones.55,58 This phenomenon was then confirmed in humans: the levels of IL-33 were significantly elevated in normal-appearing white matter and plaque areas in the CNS of MS patients when compared with the white matter of normal controls.57 In addition, a more than fourfold increase of IL-33 was observed in the sera of patients with relapsing–remitting MS (untreated for over 2 months) compared with healthy controls. Consistent with the serum data, IL-33 levels were also significantly increased in freshly prepared peripheral leucocytes (mRNA) as well as in the supernatants of stimulated lymphocytes and macrophages of these patients.57 Interestingly, both plasma IL-33 level and its mRNA expression in peripheral leucocytes are suppressed in MS patients when they receive a 3-month treatment of recombinant IL-1β-1α.57 As such, IL-33 could be a potential biomarker of MS disease activity and for monitoring clinical therapies.
Despite the emerging evidence suggesting a role for IL-33 in CNS inflammation, functional data as to whether IL-33 is beneficial or detrimental to MS disease is less clear. Research in animal models for MS disease has provided an insight into the function of IL-33 in MS pathogenesis. Mice that were IL-33-deficient developed EAE normally, indicating that IL-33 is not essential for the pathogenesis of EAE.59 However, we demonstrated that ST2-deficient mice developed exacerbated EAE, and that rIL-33 appears to exert protective effects when administered to EAE mice after disease onset.55 The protection is accompanied by a reduced production of IL-17 and IFN-γ but increased IL-5 and IL-13 by lymphocytes of peripheral lymphoid organs, together with a shift of macrophage function towards the alternative (M2) phenotype,55 a characteristic function of IL-33 that was also observed in airway and adipose tissue inflammation.21,60 In fact, growing insight into IL-33 function in CNS inflammation can also be found in other reports, which suggest that ST2 gene deletion alters polarization of antigen-presenting cells, and subsequently leads to the development of highly pathogenic T-helper cell subsets thus contributing to CNS pathology.61,62 However, contradictory findings were reported that rIL-33 exacerbates the disease course of EAE and anti-IL-33 antibody delays the onset and inhibits the severity of EAE.58 The reason for the discrepancies in the above findings is currently not known. However, in our investigation, we observed that the administration time of exogenous rIL-33 is critical to its function in EAE development. While delayed administration of IL-33 attenuates EAE, treatment before disease onset exacerbates EAE development (H.-R. Jiang, F.Y. Liew, unpublished data). In addition to its role as an immunomodulatory cytokine, IL-33 is also a potent endothelial activator. It increases endothelial permeability with reduced vascular endothelial-cadherin-facilitated cell–cell junctions in vitro and induces vascular leakage in mouse skin.25 As the breakdown of the blood–brain barrier is an essential step for the subsequent CNS inflammation, it is tempting to speculate that early administration of IL-33 may contribute to MS and EAE inflammation via disrupting the blood–brain barrier in addition to modulating the immune system. Delayed treatment with IL-33 may also be more important in its currently undefined role in the CNS compartment.
Although the precise pathophysiological function of the IL-33/ST2 axis in MS and other CNS diseases remains to be determined, current evidence supports a role for IL-33 in autoimmune MS development (Fig. 2). It is to be hoped that IL-33 may be a potential biomarker for MS development and treatment, and become a potential target/reagent in the future treatment of MS.
Figure 2.

Schematic of the role of interleukin-33 (IL-33) in the development of multiple sclerosis (MS). MS is a disease which involves both the immune and the central nervous systems (CNS). In the immune system, IL-33 levels in serum samples of MS patients are elevated, suggesting active immune responses in the periphery. IL-33 promotes vascular permeability and may be involved in the disruption of the blood–brain barrier (a) thus facilitating the infiltration of immune cells to the CNS. Peripheral IL-33 can also activate multiple immune cells such as dendritic cells (DC), macrophages (M) and lymphocytes to induce type 2 immune responses, which then down-regulate systemic inflammation (b). In the CNS, IL-33 may be released by the CNS-resident cells such as astrocytes, under inflammatory conditions/cell death and subsequently it activates CNS cells and/or infiltrating immune cells via binding to its ST2 receptor. These activated cells then produce important cytokines and antibodies that play important roles in CNS damage and repair, e.g. demyelination or remyelination, and thus modulate MS development (c). IL-33 may also have a direct effect on oligodendrocytes and myelin damage/repair in CNS (d).
IL-33 in inflammatory bowel disease
In an early study characterizing the expression and role of IL-33 in organs and cells, Schmitz et al. reported that IL-33 mRNA is found in the stomach, and observed that IL-33 induces severe pathological changes in the digestive tract including infiltration of immune cells, epithelial hyperplasia, goblet cell hypertrophy and increased mucus in the lumen.2 In fact, a recent genotyping study has revealed an association between genetic variants of genes of IL-33 and ST2 and susceptibility to autoimmune inflammatory bowel diseases (IBD) including ulcerative colitis (UC) and Crohn's disease (CD).63 Some recent studies have subsequently focused on the expression and potential role of IL-33 in UC and CD development.
Carrier et al. were the first to report that endothelial cells of CD are a major source of IL-33 in chronically inflamed tissues.10 However, some other reports have suggested that IL-33 may play a potential role in UC but not CD. Interleukin-33 is produced by gut epithelial cells and lamina propria mononuclear cells, and the expression is highly up-regulated in UC samples and correlates with disease activity.64–66 Such results were not observed in CD patients. However, the correlation between serum IL-33 level and UC activity appears to be inconclusive. While Beltran et al.65 and others67 reported that IL-33 is barely detected in the serum of all UC and CD patients, Pastorelli et al.66 observed elevated cleaved IL-33 in the serum of the IBD patients. The above disagreement may suggest that different forms of IL-33 exist in tissues and they potentially indicate the different mechanisms of IL-33 functions in disease. Similar to IL-33, sST2 expression in the colonic mucosa was mainly observed in UC rather than CD patients or healthy controls.65 In contrast to the cellular source of IL-33, ST2 is increased in lamina propria mononuclear cells but is absent/decreased in the epithelial cells in inflamed UC colons. In addition there is less controversy about the increased serum levels of sST2 in both active UC and CD patients, although the levels in CD are still about fivefold lower than that in UC patients.68 Similar to the findings in RA and MS, the levels of circulating IL-33 and sST2 in UC patients also appear to correlate with disease severity and are modulated by treatment with infliximab (anti-TNF).66 Thus it is reasonable to propose that the IL-33/ST2 axis is important in orchestrating inflammation associated with UC but not CD. Why IL-33/ST2 is less obviously involved with CD is unclear but may be due to the fact that CD is predominantly mediated by Th1/Th17 cells whereas UC is Th2 mediated69,70 and IL-33 is a strong inducer of type 2 immune responses.71
To understand the function and underlying mechanisms of IL-33 in IBD, several groups have investigated the role of IL-33 in IBD development in animals. Elevated IL-33 is observed in tissues of mice with trinitrobenzene sulphonic acid-induced colitis and rIL-33 significantly ameliorates the clinical symptoms of colitis and colonic tissue damage, together with enhanced Th2 type immune responses and Foxp3+ T regulatory cell functions.72 A similar protective role for IL-33 was also reported in a chronic dextran sodium sulphate (DSS) -induced colitis model when exogenous IL-33 reduced disease severity by modulating Th1 inflammation and induced a shift to Th2-associated cytokine production.73 Aberrant mucosal immune responses to intestinal microbiota are believed to be the cause of IBD74 and interestingly, translocation of bacteria across the gut epithelium is reduced by IL-33 treatment in the chronic DSS-induced colitis model.73 However, in acute DSS colitis, which is predominantly mediated by innate immune responses, IL-33-deficient mice displayed greater viability than wild-type controls during the early stage of colitis suggesting that IL-33 is important for the induction of mucosal inflammation at the onset of DSS colitis.59 Interestingly, these mice also have delayed recovery of body weight after changing from DSS water to normal plain drinking water. The authors suggested that the delayed local inflammation in IL-33-deficient mice resulted in delayed resolution of tissue damage. However, the data may support a novel unrecognized function of IL-33 in mucosal healing and wound repair in IBD diseases,75,76 therefore indicating a more complicated role for IL-33 in IBD in addition to being an immune modulator.
Current evidence clearly shows that the up-regulation of IL-33 and ST2 in the inflamed mucosa tissues of IBD is a good indicator of active inflammation, and it is more specific for UC than CD. These findings may lead to exciting potential therapeutic strategies for UC patients if we are able to better understand the dichotomous function of IL-33 in mucosal inflammation and IBD development.
IL-33 in diabetes
IL-33 may play a role in pancreatic diseases as it is constitutively expressed by murine pancreas77 and activates pancreatic stellate cells.78 Indeed, IL-33 has a protective role in acute pancreatitis, and serum sST2 levels correlate with disease severity.77 Surprisingly, there is very little published information about the role of IL-33 in both insulin-dependent (type 1 diabetes, T1D) and non-insulin-dependent (type 2 diabetes, T2D) diabetes.
It is well recognized that the IL-33/ST2 signalling pathway is beneficial in cardiovascular disease and obesity through enhancing Th2 cytokine production in adipocytes and adipose tissues and reducing the expression of adipogenic and metabolic genes.79 Treatment with rIL-33 exerts protective effects in genetically obese diabetic (ob/ob) mice with reduced adiposity and improves glucose and insulin tolerance60 via polarizing T cells and macrophages towards type 2 immune responses. The above evidence suggests that manipulating IL-33 expression may be a useful therapeutic strategy for treating or preventing T2D in obese patients. In fact, high levels of sST2 are detected in T2D patients compared with healthy controls, and the levels are even higher in those with left ventricular diastolic dysfunction.80 Furthermore, sST2 levels are positively and independently correlated with glycaemic controls in T2D and the authors proposed that chronic inflammation most likely establishes the basis of increased sST2 levels. In a cross-sectional study, Miller et al. examined whether sST2 concentration in serum correlates with classic and novel markers of cardiovascular and diabetic risk factors.81 They concluded that sST2 levels are related principally to the markers associated with diabetes and ectopic fat, indicating a potential role for the IL-33/ST2 pathway in diabetes.
The function and mechanisms by which the IL-33 signalling pathway operates in diabetes are still waiting to be characterized. In a murine model of T1D induced by multiple doses of streptozotocin, ST2 gene deletion enhances diabetes susceptibility in the disease-resistant BALB/c strain, confirmed by the levels of glycaemia, glycosuria, the number of infiltrating cells and β cell loss.82 The authors proposed that the IL-33/ST2 pathway exerts a protective effect in T1D, possibly through balancing the Th1/Th17 and Th2 responses as the clinical severity of the disease is accompanied by enhanced mRNA expression of TNF-α, IFN-γ and IL-17 in the pancreatic lymph nodes. Using the same animal model, others have reported that the myocardial levels of IL-33 are decreased compared with controls, and the reduction enhances the sensitivity of the myocardium to ischaemia/reperfusion-induced injury through mechanisms of activating protein kinase C βII.83 Treatment with exogenous rIL-33, on the other hand, attenuates ischaemia/reperfusion-induced injury. In general, the above findings point to an important protective role for IL-33 in diabetes, and the disease-related heart complications.
IL-33 in systemic lupus erythematosus
It was reported over a decade ago that ST2 protein was identified in the serum of some systemic lupus erythematosus (SLE), RA and other rheumatic disease patients.84 However, information regarding the role of the IL-33/ST2 signalling pathway in SLE is still limited. A recent report from Hong Kong confirmed that the serum levels of sST2 are significantly increased in patients with active SLE compared with patients with inactive disease and normal controls,85 and they correlate significantly with disease severity, anti-dsDNA antibody and prednisolone dosage and negatively with C3. A parallel study demonstrated that serum IL-33 levels in Chinese SLE patients are also significantly up-regulated relative to healthy controls,86 but lower than that of RA patients. Further investigation into the association of IL-33 levels with various SLE-associated laboratory tests indicates that IL-33 plays a role in the acute phase of SLE, but a role in the subsequent course of the disease is unlikely.86 In addition, IL-33 may exert biological effects on erythrocytes and platelets or their precursors in SLE. As all the patients in the above studies are of Far Eastern origin, it will be interesting and important to confirm the findings in western SLE populations. Nevertheless the above observations point to a possible role for the IL-33/ST2 pathway in the development of SLE. However, a lack of published data in the field indicates the need for further investigation into SLE disease models to determine the precise functions and immune mechanisms of IL-33 and ST2 in this context.
IL-33 in systemic sclerosis
Interleukin-33 was recently found to be related to systemic sclerosis (SSc). Both IL-33 and ST2 are abnormally expressed in the affected skin and visceral organs of SSc patients.87 In fact, the expression levels of IL-33 and ST2 in tissues are either up-regulated or down-regulated depending on the disease stage. IL-33 is lost in most endothelial cells of affected organs in early but not late SSc tissue samples, whereas the expression of ST2 in infiltrating immune cells is enhanced in early SSc but weakened in late SSc. The authors propose that upon endothelial cell activation/damage, IL-33 may be mobilized from these cells to signal through ST2 in key profibrotic players such as inflammatory/immune cells and fibroblasts/myofibroblasts. Further analysis of IL-33 concentrations in serum demonstrated that the levels are elevated in SSc patients and correlate with the extent of skin sclerosis (higher levels in patients with diffuse cutaneous SSc than those with limited cutaneous SSc) and the severity of pulmonary fibrosis.88 The increased serum IL-33 levels are also indicative of vascular involvement in SSc development.89 Hence IL-33 is likely to play a role in cutaneous and pulmonary fibrosis in SSc patients. To explore how IL-33 contributes to SSc development, a recent study in mice has demonstrated that subcutaneous injection of IL-33 results in the development of cutaneous fibrosis, which is similar to that seen in SSc patients confirming that IL-33 is a profibrotic mediator.90 In addition, it appears that the development of IL-33-induced skin fibrosis was dependent on IL-13, predominantly being produced by eosinophils, while IL-4 and mast cells were not required.90
IL-33 in other autoimmune diseases
Interleukin-33 is also linked to the development of many other autoimmune disorders an example being atopic dermatitis. Combined stimulation of TNF-α and IFN-γ efficiently induces IL-33 production by dermal fibroblasts and keratinocytes.91 In the murine model of atopic dermatitis, increased expression of IL-33 and ST2 are also observed in skin lesions after allergen or staphylococcal enterotoxin B exposure, and the expression can be suppressed by topical Tacrolimus treatment.91 Further investigation suggests that IL-33 influences atopic dermatitis susceptibility by significantly down-regulating human β-defensin 2 expression in keratinocytes and in acute eczematous reactions.92 Elevated levels of IL-33 are also reported in some less common autoimmune diseases such as ankylosing spondylitis, amyotrophic lateral sclerosis and Behçet's disease. The IL-33 levels are increased in the sera of ankylosing spondylitis patients when compared with healthy controls, and positively correlate with disease activity and TNF-α and IL-17 levels.93 In contrast, the levels of serum IL-33 are significantly lower in amyotrophic lateral sclerosis patients compared with healthy controls, while the levels of sST2 are significantly higher and reflect inflammation in amyotrophic lateral sclerosis.94 Increased expression of IL-33 has also been reported in serum, peripheral blood mononuclear cells and skin lesions of Behçet's patients, with a correlation to other cytokines such as IL-6, IL-17 and active status of the disease,95 suggesting that IL-33 could be used as a biomarker for active Behçet's disease.
Conclusions
This review has summarized the current evidence for a role of IL-33 in various autoimmune diseases (Table 1). The correlations, both positive and negative, with the levels of IL-33 and its receptor ST2 in tissue and serum with disease severities and following treatments suggest that IL-33 and/or ST2 may represent potential biomarkers in monitoring autoimmune disease severity and subsequent efficacy of clinical treatment. The importance of the IL-33/ST2 signalling pathway in the development of many autoimmune diseases indicates that modulating this pathway may have potential therapeutic benefits for the patients. However, much remains to be elucidated regarding the mechanisms that activate cells to express and secrete IL-33 in each organ and the precise functions and underlying mechanisms of the IL-33/ST2 axis in these systemic and organ-specific autoimmune diseases.
Table 1.
IL-33 expression and function in some autoimmune diseases
| Disease | IL-33 and ST2 expression in tissues (Refs) | Function of IL-33 (Refs) |
|---|---|---|
| RA | IL-33↑ and ST2↑ in synovium, SF and serum40–43 Serum IL-33 level correlates with RA severity40 Serum IL-33 level correlates with RA-related autoantibodies46 IL-33↓ in RA serum after treatment45–47 | Harmful in RA but protects bones sST2 attenuates CIA48 ST2 blocking antibody attenuates CIA39 rIL-33 exacerbates CIA38 and AIA49 IL-33 inhibits osteoclastogenesis45,51 ST2 deficiency attenuates CIA or AIA38,49,50 |
| MS | IL-33↑ in serum, PBMCs and CNS lesion of MS patients57 IL-33↑ and ST2↑ in EAE spinal cord55,58 IL-33↓ in MS serum after treatment57 | Contradictory rIL-33 attenuates EAE55 ST2 deficiency exacerbates EAE55,62 IL-33 deficient mice develop normal EAE59 rIL-33 exacerbates EAE and IL-33 antibody inhibits EAE58 |
| IBD | IL-33↑ in UC but not CD mucosa, and its level correlates with UC activity64–66 Cleaved (not full–length) IL-33↑ in UC serum65–67 sST2↑ in UC mucosa and serum65,68 Serum IL-33↓ and sST2↓ after treatment66 | Protective? rIL-33 ameliorates trinitrobenzene sulphonic acid-induced colitis72 rIL-33 attenuates chronic DSS colitis73 IL-33 deficient mice have delayed acute colitis and delayed recovery59 IL-33 promotes mucosal healing75,76 |
| Diabetes | IL-33 is expressed by murine pancreas77 sST2↑ in serum of T2D80 sST2 level correlates with markers of diabetic and cardiovascular risk factor81 | Protective ST2 deficiency enhances T1D susceptibility in mice82 rIL-33 attenuates T1D and related heart disease83 |
| SLE | IL-33↑ and sST2↑ in serum84–86 Serum sST2 correlate with severity85 | Not clear |
Acknowledgments
Financial support: Dr Karen Fairlie-Clarke is supported by a grant from Medical Research Scotland.
Glossary
Abbreviations
- AIA
autoantibody-induced arthritis
- CD
Crohn's disease
- CIA
collagen-induced arthritis
- CNS
central nervous system
- DM
diabetes mellitus
- DSS colitis
dextran sodium sulphate-induced colitis
- EAE
experimental autoimmune encephalomyelitis
- IBD
inflammatory bowel diseases
- IFN-γ
interferon-γ
- IL
interleukin
- IL-1RAcP
IL-1 receptor accessory protein
- M2
alternative macrophages
- MS
multiple sclerosis
- NF-κB
nuclear factor-κB
- RA
rheumatoid arthritis
- SF
synovial fluid
- SLE
systemic lupus erythematosus
- SSc
systemic sclerosis
- sST2
soluble ST2
- T1D
type 1 DM
- T2D
type 2 DM
- Th2
T helper type 2
- TNBS
trinitrobenzene sulphonic acid
- TNF-α
tumour necrosis factor-α
- UC
ulcerative colitis
Disclosures
The authors declare that there are no conflicts of interest.
References
- 1.Baekkevold ES, Roussigne M, Yamanaka T, et al. Molecular characterization of NF-HEV, a nuclear factor preferentially expressed in human high endothelial venules. Am J Pathol. 2003;163:69–79. doi: 10.1016/S0002-9440(10)63631-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schmitz J, Owyang A, Oldham E, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–90. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 3.Cayrol C, Girard JP. The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1. Proc Natl Acad Sci U S A. 2009;106:9021–6. doi: 10.1073/pnas.0812690106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luthi AU, Cullen SP, McNeela EA, et al. Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity. 2009;31:84–98. doi: 10.1016/j.immuni.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 5.Liew FY, Pitman NI, McInnes IB. Disease-associated functions of IL-33: the new kid in the IL-1 family. Nat Rev Immunol. 2010;10:103–10. doi: 10.1038/nri2692. [DOI] [PubMed] [Google Scholar]
- 6.Kakkar R, Hei H, Dobner S, Lee RT. Interleukin 33 as a mechanically responsive cytokine secreted by living cells. J Biol Chem. 2012;287:6941–8. doi: 10.1074/jbc.M111.298703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mirchandani AS, Salmond RJ, Liew FY. Interleukin-33 and the function of innate lymphoid cells. Trends Immunol. 2012;33:389–96. doi: 10.1016/j.it.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 8.Ohno T, Oboki K, Kajiwara N, et al. Caspase-1, caspase-8, and calpain are dispensable for IL-33 release by macrophages. J Immunol. 2009;183:7890–7. doi: 10.4049/jimmunol.0802449. [DOI] [PubMed] [Google Scholar]
- 9.Talabot-Ayer D, Calo N, Vigne S, Lamacchia C, Gabay C, Palmer G. The mouse interleukin (Il)33 gene is expressed in a cell type- and stimulus-dependent manner from two alternative promoters. J Leukoc Biol. 2012;91:119–25. doi: 10.1189/jlb.0811425. [DOI] [PubMed] [Google Scholar]
- 10.Carriere V, Roussel L, Ortega N, Lacorre DA, Americh L, Aguilar L, Bouche G, Girard JP. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc Natl Acad Sci U S A. 2007;104:282–7. doi: 10.1073/pnas.0606854104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roussel L, Erard M, Cayrol C, Girard J-P. Molecular mimicry between IL-33 and KSHV for attachment to chromatin through the H2A-H2B acidic pocket. EMBO Rep. 2008;9:1006–12. doi: 10.1038/embor.2008.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ali S, Mohs A, Thomas M, Klare J, Ross R, Schmitz ML, Martin MU. The dual function cytokine IL-33 interacts with the transcription factor NF-κB to dampen NF-κB-stimulated gene transcription. J Immunol. 2011;187:1609–16. doi: 10.4049/jimmunol.1003080. [DOI] [PubMed] [Google Scholar]
- 13.Ali S, Huber M, Kollewe C, Bischoff SC, Falk W, Martin MU. IL-1 receptor accessory protein is essential for IL-33-induced activation of T lymphocytes and mast cells. Proc Natl Acad Sci U S A. 2007;104:18660–5. doi: 10.1073/pnas.0705939104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Allakhverdi Z, Smith DE, Comeau MR, Delespesse G. Cutting edge: the ST2 ligand IL-33 potently activates and drives maturation of human mast cells. J Immunol. 2007;179:2051–4. doi: 10.4049/jimmunol.179.4.2051. [DOI] [PubMed] [Google Scholar]
- 15.Smithgall MD, Comeau MR, Yoon BR, Kaufman D, Armitage R, Smith DE. IL-33 amplifies both Th1- and Th2-type responses through its activity on human basophils, allergen-reactive Th2 cells, iNKT and NK cells. Int Immunol. 2008;20:1019–30. doi: 10.1093/intimm/dxn060. [DOI] [PubMed] [Google Scholar]
- 16.Bonilla WV, Frohlich A, Senn K, et al. The alarmin interleukin-33 drives protective antiviral CD8 T cell responses. Science. 2012;335:984–9. doi: 10.1126/science.1215418. [DOI] [PubMed] [Google Scholar]
- 17.Besnard AG, Togbe D, Guillou N, Erard F, Quesniaux V, Ryffel B. IL-33-activated dendritic cells are critical for allergic airway inflammation. Eur J Immunol. 2011;41:1675–86. doi: 10.1002/eji.201041033. [DOI] [PubMed] [Google Scholar]
- 18.Barlow JL, Peel S, Fox J, et al. IL-33 is more potent than IL-25 in provoking IL-13-producing nuocytes (type 2 innate lymphoid cells) and airway contraction. J Allergy Clin Immunol. 2013;132:933–41. doi: 10.1016/j.jaci.2013.05.012. [DOI] [PubMed] [Google Scholar]
- 19.Komai-Koma M, Gilchrist DS, McKenzie ANJ, Goodyear CS, Xu D, Liew FY. IL-33 activates B1 cells and exacerbates contact sensitivity. J Immunol. 2011;186:2584–91. doi: 10.4049/jimmunol.1002103. [DOI] [PubMed] [Google Scholar]
- 20.Kurowska-Stolarska M, Kewin P, Murphy G, et al. IL-33 induces antigen-specific IL-5+ T cells and promotes allergic-induced airway inflammation independent of IL-4. J Immunol. 2008;181:4780–90. doi: 10.4049/jimmunol.181.7.4780. [DOI] [PubMed] [Google Scholar]
- 21.Kurowska-Stolarska M, Stolarski B, Kewin P, et al. IL-33 amplifies the polarization of alternatively activated macrophages that contribute to airway inflammation. J Immunol. 2009;183:6469–77. doi: 10.4049/jimmunol.0901575. [DOI] [PubMed] [Google Scholar]
- 22.Silver MR, Margulis A, Wood N, Goldman SJ, Kasaian M, Chaudhary D. IL-33 synergizes with IgE-dependent and IgE-independent agents to promote mast cell and basophil activation. Inflamm Res. 2010;59:207–18. doi: 10.1007/s00011-009-0088-5. [DOI] [PubMed] [Google Scholar]
- 23.Jung MY, Smrz D, Desai A, et al. IL-33 induces a hyporesponsive phenotype in human and mouse mast cells. J Immunol. 2013;190:531–8. doi: 10.4049/jimmunol.1201576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aoki S, Hayakawa M, Ozaki H, Takezako N, Obata H, Ibaraki N, Tsuru T, Tominaga S, Yanagisawa K. ST2 gene expression is proliferation-dependent and its ligand, IL-33, induces inflammatory reaction in endothelial cells. Mol Cell Biochem. 2010;335:75–81. doi: 10.1007/s11010-009-0244-9. [DOI] [PubMed] [Google Scholar]
- 25.Choi YS, Choi HJ, Min JK, Pyun BJ, Maeng YS, Park H, Kim J, Kim YM, Kwon YG. Interleukin-33 induces angiogenesis and vascular permeability through ST2/TRAF6-mediated endothelial nitric oxide production. Blood. 2009;114:3117–26. doi: 10.1182/blood-2009-02-203372. [DOI] [PubMed] [Google Scholar]
- 26.Matsuda A, Okayama Y, Terai N, et al. The role of interleukin-33 in chronic allergic conjunctivitis. Invest Ophthalmol Vis Sci. 2009;50:4646–52. doi: 10.1167/iovs.08-3365. [DOI] [PubMed] [Google Scholar]
- 27.Stolarski B, Kurowska-Stolarska M, Kewin P, Xu D, Liew FY. IL-33 exacerbates eosinophil-mediated airway inflammation. J Immunol. 2010;185:3472–80. doi: 10.4049/jimmunol.1000730. [DOI] [PubMed] [Google Scholar]
- 28.Lloyd CM. IL-33 family members and asthma – bridging innate and adaptive immune responses. Curr Opin Immunol. 2010;22:800–6. doi: 10.1016/j.coi.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kondo Y, Yoshimoto T, Yasuda K, et al. Administration of IL-33 induces airway hyperresponsiveness and goblet cell hyperplasia in the lungs in the absence of adaptive immune system. Int Immunol. 2008;20:791–800. doi: 10.1093/intimm/dxn037. [DOI] [PubMed] [Google Scholar]
- 30.Ohno T, Morita H, Arae K, Matsumoto K, Nakae S. Interleukin-33 in allergy. Allergy. 2012;67:1203–14. doi: 10.1111/all.12004. [DOI] [PubMed] [Google Scholar]
- 31.Borish L, Steinke JW. Interleukin-33 in asthma: how big of a role does it play? Curr Allergy Asthma Rep. 2011;11:7–11. doi: 10.1007/s11882-010-0153-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Salmond RJ, Mirchandani AS, Besnard A-G, Bain CC, Thomson NC, Liew FY. IL-33 induces innate lymphoid cell-mediated airway inflammation by activating mammalian target of rapamycin. J Allergy Clin Immunol. 2012;130(1159–66):e6. doi: 10.1016/j.jaci.2012.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Humphreys NE, Xu D, Hepworth MR, Liew FY, Grencis RK. IL-33, a potent inducer of adaptive immunity to intestinal nematodes. J Immunol. 2008;180:2443–9. doi: 10.4049/jimmunol.180.4.2443. [DOI] [PubMed] [Google Scholar]
- 34.Kakkar R, Lee RT. The IL-33/ST2 pathway: therapeutic target and novel biomarker. Nat Rev Drug Discov. 2008;7:827–40. doi: 10.1038/nrd2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miller AM, Xu D, Asquith DL, et al. IL-33 reduces the development of atherosclerosis. J Exp Med. 2008;205:339–46. doi: 10.1084/jem.20071868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miller AM, Liew FY. The IL-33/ST2 pathway – a new therapeutic target in cardiovascular disease. Pharmacol Ther. 2011;131:179–86. doi: 10.1016/j.pharmthera.2011.02.005. [DOI] [PubMed] [Google Scholar]
- 37.Verri WA, Jr, Guerrero AT, Fukada SY, et al. IL-33 mediates antigen-induced cutaneous and articular hypernociception in mice. Proc Natl Acad Sci U S A. 2008;105:2723–8. doi: 10.1073/pnas.0712116105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu D, Jiang HR, Kewin P, et al. IL-33 exacerbates antigen-induced arthritis by activating mast cells. Proc Natl Acad Sci U S A. 2008;105:10913–8. doi: 10.1073/pnas.0801898105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Palmer G, Talabot-Ayer D, Lamacchia C, et al. Inhibition of interleukin-33 signaling attenuates the severity of experimental arthritis. Arthritis Rheum. 2009;60:738–49. doi: 10.1002/art.24305. [DOI] [PubMed] [Google Scholar]
- 40.Xiangyang Z, Lutian Y, Lin Z, Liping X, Hui S, Jing L. Increased levels of interleukin-33 associated with bone erosion and interstitial lung diseases in patients with rheumatoid arthritis. Cytokine. 2012;58:6–9. doi: 10.1016/j.cyto.2011.12.010. [DOI] [PubMed] [Google Scholar]
- 41.Hong YS, Moon SJ, Joo YB, et al. Measurement of interleukin-33 (IL-33) and IL-33 receptors (sST2 and ST2L) in patients with rheumatoid arthritis. J Korean Med Sci. 2011;26:1132–9. doi: 10.3346/jkms.2011.26.9.1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Matsuyama Y, Okazaki H, Hoshino M, et al. Sustained elevation of interleukin-33 in sera and synovial fluids from patients with rheumatoid arthritis non-responsive to anti-tumor necrosis factor: possible association with persistent IL-1β signaling and a poor clinical response. Rheumatol Int. 2012;32:1397–401. doi: 10.1007/s00296-011-1854-6. [DOI] [PubMed] [Google Scholar]
- 43.Talabot-Ayer D, McKee T, Gindre P, Bas S, Baeten DL, Gabay C, Palmer G. Distinct serum and synovial fluid interleukin (IL)-33 levels in rheumatoid arthritis, psoriatic arthritis and osteoarthritis. Joint Bone Spine. 2012;79:32–7. doi: 10.1016/j.jbspin.2011.02.011. [DOI] [PubMed] [Google Scholar]
- 44.Matsuyama Y, Okazaki H, Tamemoto H, et al. Increased levels of interleukin 33 in sera and synovial fluid from patients with active rheumatoid arthritis. J Rheumatol. 2010;37:18–25. doi: 10.3899/jrheum.090492. [DOI] [PubMed] [Google Scholar]
- 45.Schulze J, Bickert T, Beil FT, et al. Interleukin-33 is expressed in differentiated osteoblasts and blocks osteoclast formation from bone marrow precursor cells. J Bone Miner Res. 2011;26:704–17. doi: 10.1002/jbmr.269. [DOI] [PubMed] [Google Scholar]
- 46.Mu R, Huang HQ, Li YH, Li C, Ye H, Li ZG. Elevated serum interleukin 33 is associated with autoantibody production in patients with rheumatoid arthritis. J Rheumatol. 2010;37:2006–13. doi: 10.3899/jrheum.100184. [DOI] [PubMed] [Google Scholar]
- 47.Kageyama Y, Torikai E, Tsujimura K, Kobayashi M. Involvement of IL-33 in the pathogenesis of rheumatoid arthritis: the effect of etanercept on the serum levels of IL-33. Mod Rheumatol. 2012;22:89–93. doi: 10.1007/s10165-011-0480-1. [DOI] [PubMed] [Google Scholar]
- 48.Leung BP, Xu D, Culshaw S, McInnes IB, Liew FY. A novel therapy of murine collagen-induced arthritis with soluble T1/ST2. J Immunol. 2004;173:145–50. doi: 10.4049/jimmunol.173.1.145. [DOI] [PubMed] [Google Scholar]
- 49.Xu D, Jiang HR, Li Y, et al. IL-33 exacerbates autoantibody-induced arthritis. J Immunol. 2010;184:2620–6. doi: 10.4049/jimmunol.0902685. [DOI] [PubMed] [Google Scholar]
- 50.Martin P, Talabot-Ayer D, Seemayer CA, et al. Disease severity in K/BxN serum transfer-induced arthritis is not affected by IL-33 deficiency. Arthritis Res Ther. 2013;15:R13. doi: 10.1186/ar4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Keller J, Catala-Lehnen P, Wintges K, et al. Transgenic over-expression of interleukin-33 in osteoblasts results in decreased osteoclastogenesis. Biochem Biophys Res Commun. 2012;417:217–22. doi: 10.1016/j.bbrc.2011.11.088. [DOI] [PubMed] [Google Scholar]
- 52.Chapuis J, Hot D, Hansmannel F, et al. Transcriptomic and genetic studies identify IL-33 as a candidate gene for Alzheimer's disease. Mol Psychiatry. 2009;14:1004–16. doi: 10.1038/mp.2009.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yu JT, Song JH, Wang ND, et al. Implication of IL-33 gene polymorphism in Chinese patients with Alzheimer's disease. Neurobiol Aging. 2012;33(1014):e11–4. doi: 10.1016/j.neurobiolaging.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 54.Hudson CA, Christophi GP, Gruber RC, Wilmore JR, Lawrence DA, Massa PT. Induction of IL-33 expression and activity in central nervous system glia. J Leukoc Biol. 2008;84:631–43. doi: 10.1189/jlb.1207830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jiang HR, Milovanovic M, Allan D, et al. IL-33 attenuates experimental autoimmune encephalomyelitis by suppressing IL-17 and IFN-γ production and inducing alternatively-activated macrophages. Eur J Immunol. 2012;42:1804–14. doi: 10.1002/eji.201141947. [DOI] [PubMed] [Google Scholar]
- 56.Yasuoka S, Kawanokuchi J, Parajuli B, et al. Production and functions of IL-33 in the central nervous system. Brain Res. 2011;1385:8–17. doi: 10.1016/j.brainres.2011.02.045. [DOI] [PubMed] [Google Scholar]
- 57.Christophi GP, Gruber RC, Panos M, Christophi RL, Jubelt B, Massa PT. Interleukin-33 upregulation in peripheral leukocytes and CNS of multiple sclerosis patients. Clin Immunol. 2012;142:308–19. doi: 10.1016/j.clim.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li M, Li Y, Liu X, Gao X, Wang Y. IL-33 blockade suppresses the development of experimental autoimmune encephalomyelitis in C57BL/6 mice. J Neuroimmunol. 2012;247:25–31. doi: 10.1016/j.jneuroim.2012.03.016. [DOI] [PubMed] [Google Scholar]
- 59.Oboki K, Ohno T, Kajiwara N, et al. IL-33 is a crucial amplifier of innate rather than acquired immunity. Proc Natl Acad Sci U S A. 2010;107:18581–6. doi: 10.1073/pnas.1003059107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miller AM, Asquith DL, Hueber AJ, et al. Interleukin-33 induces protective effects in adipose tissue inflammation during obesity in mice. Circ Res. 2010;107:650–8. doi: 10.1161/CIRCRESAHA.110.218867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Milovanovic M, Volarevic V, Radosavljevic G, Jovanovic I, Pejnovic N, Arsenijevic N, Lukic ML. IL-33/ST2 axis in inflammation and immunopathology. Immunol Res. 2012;52:89–99. doi: 10.1007/s12026-012-8283-9. [DOI] [PubMed] [Google Scholar]
- 62.Milovanovic M, Volarevic V, Ljujic B, Radosavljevic G, Jovanovic I, Arsenijevic N, Lukic ML. Deletion of IL-33R (ST2) abrogates resistance to EAE in BALB/C mice by enhancing polarization of APC to inflammatory phenotype. PLoS ONE. 2012;7:e45225. doi: 10.1371/journal.pone.0045225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Latiano A, Palmieri O, Pastorelli L, et al. Associations between genetic polymorphisms in IL-33, IL1R1 and risk for inflammatory bowel disease. PLoS ONE. 2013;8:e62144. doi: 10.1371/journal.pone.0062144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Seidelin JB, Bjerrum JT, Coskun M, Widjaya B, Vainer B, Nielsen OH. IL-33 is upregulated in colonocytes of ulcerative colitis. Immunol Lett. 2010;128:80–5. doi: 10.1016/j.imlet.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 65.Beltran CJ, Nunez LE, Diaz-Jimenez D, et al. Characterization of the novel ST2/IL-33 system in patients with inflammatory bowel disease. Inflamm Bowel Dis. 2010;16:1097–107. doi: 10.1002/ibd.21175. [DOI] [PubMed] [Google Scholar]
- 66.Pastorelli L, Garg RR, Hoang SB, et al. Epithelial-derived IL-33 and its receptor ST2 are dysregulated in ulcerative colitis and in experimental Th1/Th2 driven enteritis. Proc Natl Acad Sci U S A. 2010;107:8017–22. doi: 10.1073/pnas.0912678107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ajdukovic J, Tonkic A, Salamunic I, Hozo I, Simunic M, Bonacin D. Interleukins IL-33 and IL-17/IL-17A in patients with ulcerative colitis. Hepatogastroenterology. 2010;57:1442–4. [PubMed] [Google Scholar]
- 68.Diaz-Jimenez D, Nunez LE, Beltran CJ, et al. Soluble ST2: a new and promising activity marker in ulcerative colitis. World J Gastroenterol. 2011;17:2181–90. doi: 10.3748/wjg.v17.i17.2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fuss IJ, Neurath M, Boirivant M, Klein JS, de la Motte C, Strong SA, Fiocchi C, Strober W. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn's disease LP cells manifest increased secretion of IFN-γ, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J Immunol. 1996;157:1261–70. [PubMed] [Google Scholar]
- 70.Brand S. Crohn's disease: Th1, Th17 or both? The change of a paradigm: new immunological and genetic insights implicate Th17 cells in the pathogenesis of Crohn's disease. Gut. 2009;58:1152–67. doi: 10.1136/gut.2008.163667. [DOI] [PubMed] [Google Scholar]
- 71.Liew FY. IL-33: a Janus cytokine. Ann Rheum Dis. 2012;71(Suppl. 2):i101–4. doi: 10.1136/annrheumdis-2011-200589. [DOI] [PubMed] [Google Scholar]
- 72.Duan L, Chen J, Zhang H, et al. Interleukin-33 ameliorates experimental colitis through promoting Th2/Foxp3 regulatory T-cell responses in mice. Mol Med. 2012;18:753–61. doi: 10.2119/molmed.2011.00428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Grobeta P, Doser K, Falk W, Obermeier F, Hofmann C. IL-33 attenuates development and perpetuation of chronic intestinal inflammation. Inflamm Bowel Dis. 2012;18:1900–9. doi: 10.1002/ibd.22900. [DOI] [PubMed] [Google Scholar]
- 74.Baumgart DC, Carding SR. Inflammatory bowel disease: cause and immunobiology. Lancet. 2007;369:1627–40. doi: 10.1016/S0140-6736(07)60750-8. [DOI] [PubMed] [Google Scholar]
- 75.Sponheim J, Pollheimer J, Olsen T, et al. Inflammatory bowel disease-associated interleukin-33 is preferentially expressed in ulceration-associated myofibroblasts. Am J Pathol. 2010;177:2804–15. doi: 10.2353/ajpath.2010.100378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lopetuso LR, Scaldaferri F, Pizarro TT. Emerging role of the interleukin (IL)-33/ST2 axis in gut mucosal wound healing and fibrosis. Fibrogenesis Tissue Repair. 2012;5:18. doi: 10.1186/1755-1536-5-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ouziel R, Gustot T, Moreno C, et al. The ST2 pathway is involved in acute pancreatitis: a translational study in humans and mice. Am J Pathol. 2012;180:2330–9. doi: 10.1016/j.ajpath.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Masamune A, Watanabe T, Kikuta K, Satoh K, Kanno A, Shimosegawa T. Nuclear expression of interleukin-33 in pancreatic stellate cells. Am J Physiol Gastrointest Liver Physiol. 2010;299:G821–32. doi: 10.1152/ajpgi.00178.2010. [DOI] [PubMed] [Google Scholar]
- 79.Miller AM. Role of IL-33 in inflammation and disease. J Inflamm. 2011;8:22. doi: 10.1186/1476-9255-8-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fousteris E, Melidonis A, Panoutsopoulos G, et al. Toll/interleukin-1 receptor member ST2 exhibits higher soluble levels in type 2 diabetes, especially when accompanied with left ventricular diastolic dysfunction. Cardiovasc Diabetol. 2011;10:101. doi: 10.1186/1475-2840-10-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Miller AM, Purves D, McConnachie A, et al. Soluble ST2 associates with diabetes but not established cardiovascular risk factors: a new inflammatory pathway of relevance to diabetes? PLoS ONE. 2012;7:e47830. doi: 10.1371/journal.pone.0047830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zdravkovic N, Shahin A, Arsenijevic N, Lukic ML, Mensah-Brown EP. Regulatory T cells and ST2 signaling control diabetes induction with multiple low doses of streptozotocin. Mol Immunol. 2009;47:28–36. doi: 10.1016/j.molimm.2008.12.023. [DOI] [PubMed] [Google Scholar]
- 83.Rui T, Zhang J, Xu X, Yao Y, Kao R, Martin CM. Reduction in IL-33 expression exaggerates ischaemia/reperfusion-induced myocardial injury in mice with diabetes mellitus. Cardiovasc Res. 2012;94:370–8. doi: 10.1093/cvr/cvs015. [DOI] [PubMed] [Google Scholar]
- 84.Kuroiwa K, Arai T, Okazaki H, Minota S, Tominaga S. Identification of human ST2 protein in the sera of patients with autoimmune diseases. Biochem Biophys Res Commun. 2001;284:1104–8. doi: 10.1006/bbrc.2001.5090. [DOI] [PubMed] [Google Scholar]
- 85.Mok MY, Huang FP, Ip WK, Lo Y, Wong FY, Chan EY, Lam KF, Xu D. Serum levels of IL-33 and soluble ST2 and their association with disease activity in systemic lupus erythematosus. Rheumatology (Oxford) 2010;49:520–7. doi: 10.1093/rheumatology/kep402. [DOI] [PubMed] [Google Scholar]
- 86.Yang Z, Liang Y, Xi W, Li C, Zhong R. Association of increased serum IL-33 levels with clinical and laboratory characteristics of systemic lupus erythematosus in Chinese population. Clin Exp Med. 2011;11:75–80. doi: 10.1007/s10238-010-0115-4. [DOI] [PubMed] [Google Scholar]
- 87.Manetti M, Ibba-Manneschi L, Liakouli V, et al. The IL1-like cytokine IL33 and its receptor ST2 are abnormally expressed in the affected skin and visceral organs of patients with systemic sclerosis. Ann Rheum Dis. 2010;69:598–605. doi: 10.1136/ard.2009.119321. [DOI] [PubMed] [Google Scholar]
- 88.Yanaba K, Yoshizaki A, Asano Y, Kadono T, Sato S. Serum IL-33 levels are raised in patients with systemic sclerosis: association with extent of skin sclerosis and severity of pulmonary fibrosis. Clin Rheumatol. 2011;30:825–30. doi: 10.1007/s10067-011-1686-5. [DOI] [PubMed] [Google Scholar]
- 89.Terras S, Opitz E, Moritz RK, Hoxtermann S, Gambichler T, Kreuter A. Increased serum IL-33 levels may indicate vascular involvement in systemic sclerosis. Ann Rheum Dis. 2012;72:144–5. doi: 10.1136/annrheumdis-2012-201553. [DOI] [PubMed] [Google Scholar]
- 90.Rankin AL, Mumm JB, Murphy E, et al. IL-33 induces IL-13-dependent cutaneous fibrosis. J Immunol. 2010;184:1526–35. doi: 10.4049/jimmunol.0903306. [DOI] [PubMed] [Google Scholar]
- 91.Savinko T, Matikainen S, Saarialho-Kere U, et al. IL-33 and ST2 in atopic dermatitis: expression profiles and modulation by triggering factors. J Invest Dermatol. 2012;132:1392–400. doi: 10.1038/jid.2011.446. [DOI] [PubMed] [Google Scholar]
- 92.Alase A, Seltmann J, Werfel T, Wittmann M. Interleukin-33 modulates the expression of human β-defensin 2 in human primary keratinocytes and may influence the susceptibility to bacterial superinfection in acute atopic dermatitis. Br J Dermatol. 2012;167:1386–9. doi: 10.1111/j.1365-2133.2012.11140.x. [DOI] [PubMed] [Google Scholar]
- 93.Han GW, Zeng LW, Liang CX, Cheng BL, Yu BS, Li HM, Zeng FF, Liu SY. Serum levels of IL-33 is increased in patients with ankylosing spondylitis. Clin Rheumatol. 2011;30:1583–8. doi: 10.1007/s10067-011-1843-x. [DOI] [PubMed] [Google Scholar]
- 94.Lin CY, Pfluger CM, Henderson RD, McCombe PA. Reduced levels of interleukin 33 and increased levels of soluble ST2 in subjects with amyotrophic lateral sclerosis. J Neuroimmunol. 2012;249:93–5. doi: 10.1016/j.jneuroim.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 95.Hamzaoui K, Kaabachi W, Fazaa B, Zakraoui L, Mili Boussen I, Haj Sassi F. Serum IL-33 levels and skin mRNA expression in Behcet's disease. Clin Exp Rheumatol. 2013;31(Suppl. 77):6–14. [PubMed] [Google Scholar]