

Abstract
Tuberculosis (TB) remains a major global health problem accounting for millions of deaths annually. Approximately one-third of the world's population is infected with the causative agent Mycobacterium tuberculosis. The onset of an adaptive immune response to M. tuberculosis is delayed compared with other microbial infections. This delay permits bacterial growth and dissemination. The precise mechanism(s) responsible for this delay have remained obscure. T-cell activation is preceded by dendritic cell (DC) migration from infected lungs to local lymph nodes and synapsis with T cells. We hypothesized that M. tuberculosis may impede the ability of DCs to reach lymph nodes and initiate an adaptive immune response. We used primary human DCs to determine the effect of M. tuberculosis on expression of heterodimeric integrins involved in cellular adhesion and migration. We also evaluated the ability of infected DCs to adhere to and migrate through lung endothelial cells, which is necessary to reach lymph nodes. We show by flow cytometry and confocal microscopy that M. tuberculosis-infected DCs exhibit a significant reduction in surface expression of the β2 (CD18) integrin. Distribution of integrin β2 is also markedly altered in M. tuberculosis-infected DCs. A corresponding reduction in the αL (CD11a) and αM (CD11b) subunits that associate with integrin β2 was also observed. Consistent with reduced integrin surface expression, we show a significant reduction in adherence to lung endothelial cell monolayers and migration towards lymphatic chemokines when DCs are infected with M. tuberculosis. These findings suggest that M. tuberculosis modulates DC adhesion and migration to increase the time required to initiate an adaptive immune response.
Keywords: CD18, dendritic cells, integrins, migration, mycobacteria
Introduction
Tuberculosis (TB) remains a formidable infectious disease accounting for approximately 2 million deaths worldwide each year.1 Estimates of infection indicate that one-third of the world's population has been infected with the causative agent, Mycobacterium tuberculosis.1 The host immune response is typically sufficient to contain the infection but seldom able to eradicate the bacterium. Nearly 5–10% of infections result in active TB while many result in a latent state, which can later reactivate depending on host immune status.2 In humans, development of adaptive immunity to M. tuberculosis is measured as a response to the tuberculin skin test and evidence shows a 5–6-week period following exposure before acquired immunity develops.3 In mice, approximately 18–20 days are required before antigen-specific T cells arrive in the infected lungs (reviewed in ref. 4). Compared with other microbial infections this is substantially longer and allows for expansion of the mycobacterial population.4–12 Detailed mechanisms that underlie the delayed acquired immune response to M. tuberculosis have remained obscure.
Mycobacterium tuberculosis is spread by aerosol route and is first encountered by alveolar phagocytes such as macrophages and dendritic cells (DCs). DCs can internalize M. tuberculosis and M. tuberculosis-derived antigens through numerous cell surface receptors. These include complement receptors (CR), mannose receptor, and DC-SIGN (reviewed in ref. 13). Although evidence suggests that DC-SIGN may be the primary receptor for mycobacterial uptake by DCs.14 Subsequently the DC undergoes maturation and up-regulates chemokine receptor 7 (CCR7) expression. This allows for directed migration to secondary lymphoid organs in response to a gradient of CCL19 and CCL21 chemokines.15,16 Other coordinated cellular events involve increased expression of T-cell co-stimulatory and adhesion molecules.17,18 The latter aid in adhesion to endothelial cells and migration through afferent lymphatics to regional lymph nodes.
Integrins are transmembrane heterodimers that consist of a unique α subunit and a shared β subunit, and are indispensable for proper immune function. The β1 family consists of a diverse group of integrins. Among the most notable adhesion integrins is very late antigen 4 (VLA-4; CD49d/CD29: α4/β1), a ligand for vascular cell adhesion molecule-1 and fibronectin expressed by vascular endothelium. In addition, the well characterized β2 family consists of 4 different α subunits that share a common β2 (CD18) subunit. Most notable are LFA-1 (CD11a/CD18 or αL/β2), Mac-1/CR3 (CD11b/CD18 or αM/β2) and CR4 (CD11c/CD18 or αX/β2). They are all expressed by DCs and are involved in cell-cell adhesion and T-cell activation.19 In the context of TB, integrins have been found to be absolutely required for host control of infection. Survival is substantially reduced in mice deficient for CD11a and CD18.20 Moreover, CD11a knock-out mice are impaired in their ability to control M. tuberculosis and exhibit decreased numbers of effector T cells in the lungs compared with wild-type mice.20
The process of DC attachment to endothelial cells and subsequent migration is mediated by members of the β1 and β2 integrin family of adhesion molecules. Blocking antibodies to CD18, CD11a, CD11b and VLA-4 have been reported to inhibit DC adhesion to and transmigration through endothelial cells.21,22 Similarly, blocking antibodies to their cognate ligands intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 block DC adhesion to and transmigration across lymphatic endothelium.23 In addition, LFA-1 on DCs is involved in regulating the duration of contact with T cells, allowing the enhancement of T helper type 1 cell proliferation.24–26
The effect of M. tuberculosis on primary human DC integrin expression has not been explored. Given the prominent role of these integrins in DC migration, adhesion to vascular endothelium, and interactions with T cells, we investigated the influence of M. tuberculosis infection on integrin expression in primary human DCs. Our results demonstrate that DC exposure to M. tuberculosis results in a decrease in α and β integrin subunit expression with impaired DC adherence to endothelial cells and migration toward a chemokine gradient. We further suggest that this could influence the delayed onset of an adaptive immune response during TB.
Materials and methods
Cell culture
Monocytes were isolated from buffy coats purchased from the New York Blood Center, (New York, NY) by sequential Ficoll (Amersham Biosciences, Piscataway, NJ) and OptiPrep™ (Sigma, St Louis, MO) density gradient centrifugations as described previously.27 Eligible donors were 16 years of age or older, weighed at least 110 lb (50 kg), and were in good physical health. The donor samples were anonymous and de-identifed. Monocytes were washed once with PBS and resuspended in RPMI-1640 (Thermo Scientific, Waltham, MA) supplemented with 25 mm HEPES and 2 mm l-glutamine. Monocytes were seeded on 24-well tissue culture plates (1 × 106 cells/well) and allowed to adhere for 1 hr before washing twice with PBS to remove non-adherent cells. Adherent monocytes were then immediately cultured in RPMI-1640 supplemented with 10% human serum AB, 25 mm HEPES, 2 mm l-glutamine, 0·1 mm non-essential amino acids, 1 mm sodium pyruvate, 0·05 mm 2-mercaptoethanol and recombinant human interleukin-4 (IL-4) and granulocyte–macrophage colony-stimulating factor (GM-CSF; Gemini Bioproducts, West Sacramento, CA) were added at 1600 U and 290 U per 106 cells, respectively. Cells were cultured for 7 days at 37° with 5% CO2. On day 7 cells were considered immature DC and harvested with PBS that contained 4 mg/ml lidocaine and 5 mm EDTA for 15 min at 37°. Monocytes cultured in this manner routinely yield a DC population that is heterogeneous in size and shape, MHC IIhigh, CD86high, CD14−, CD80+, CD1b+, CD40high, CD11chigh and DC-SIGN+ (CD 209; Fig. 1). Harvested DCs were pelleted and washed with PBS. DCs were then cultured in medium described above without IL-4 and GM-CSF (i.e. infection medium) for downstream assays.
Figure 1.
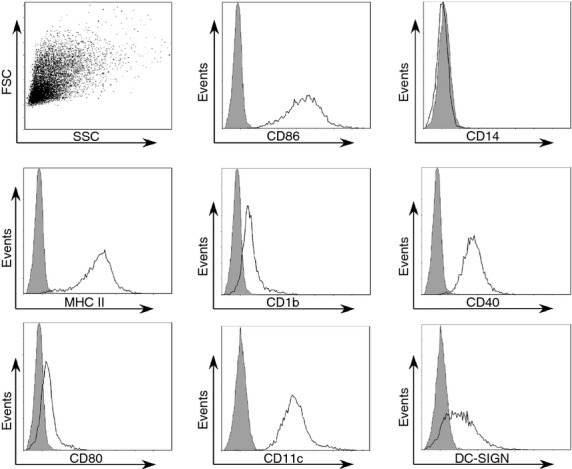
Culture of adherent monocytes with interleukin-4 (IL-4) and granulocyte–macrophage colony-stimulating factor (GM-CSF) results in a characteristic dendritic cell (DC) phenotype. Monocytes were isolated from human peripheral blood by sequential density gradient centrifugations followed by adhesion in serum-free medium. Adherent monocytes were then washed twice with PBS and cultured in medium containing IL-4 and GM-CSF for 7 days. After 7 days, cells were harvested and immunolabelled with monoclonal antibodies against the indicated molecule or isotype control and analysed by flow cytometry. A representative scatter plot and histogram overlays are shown; grey = isotype control.
Primary blood DCs were isolated by immunomagnetic selection from frozen human peripheral blood mononuclear cells with a blood DC isolation kit (Miltenyi Biotech, Bergisch Gladbach, Germany) following the instructions provided by the manufacturer. Blood DCs were then cultured in infection medium with or without M. tuberculosis infection.
Mycobacterium strains, culture conditions, cell infections and bead treatment
Mycobacterium tuberculosis strain mc27000 was described before and generously provided by Dr William Jacobs (Albert Einstein College of Medicine) and grown in Middlebrook 7H9 broth supplemented with albumin, dextrose and pantothenate (24 μg/ml) at 37° with 5% CO2.28,29 For DC infection, M. tuberculosis were washed with PBS to remove residual broth, suspended in infection medium (as described above) at the indicated multiplicity of infection (MOI), and passaged through a 27-gauge needle. Determination of the actual MOI was performed by plating the inoculum on Middlebrook 7H10 agar plates supplemented with oleic acid, albumin, dextrose and pantothenate (24 μg/ml) and grown at 37° with 5% CO2 for 3 weeks. Irradiated M. tuberculosis H37Rv was obtained from BEI Resources (Manassas, VA). For bead treatments, 10 μl of latex beads (Sigma) were suspended in 1 ml of infection medium followed by two washes with PBS and again suspended in 1 ml infection medium. This preparation of beads was diluted 1000-fold in the DC culture to yield a ratio of two beads per cell.
Flow cytometry
For cell surface protein expression analysis, 2 × 105 DCs were seeded on 24-well low binding plates (Nalge Nunc, Rochester, NY) and infected or subjected to 2 μm latex beads (Sigma) at a quantity comparable to the MOI. DC were harvested as described above and suspended in 0·1 ml of flow cytometry staining buffer (R&D Systems, Minneapolis, MN). Fc receptors were blocked with human FcR blocking reagent (Miltenyi Biotec) according to the supplier recommendation for 30 min at room temperature with gentle shaking. Cells were then incubated with the specific monoclonal antibody indicated or isotype control for 30 min at room temperature with gentle shaking. All monoclonal antibodies were purchased from Ebiosciences (San Diego, CA) and used according to the manufacturer's recommendation. Monocloncal antibodies used were as follows: mouse anti-CD18 conjugated to phycoerythrin (PE), mouse anti-CD11a PE-conjugated, mouse anti-CD11b PE-conjugated, mouse anti-CD29 PE-conjugated, mouse anti-CD14 FITC-conjugated, mouse anti-CD1b FITC-conjugated, mouse anti-CD80/86 PE-conjugated, mouse anti-CD40 PE-conjugated, mouse anti-MHCII PE-conjugated, and mouse anti-CD54 PE-conjugated. The DCs were then washed once with 1 ml flow cytometry staining buffer, pelleted and fixed in 0·5 ml of 4% paraformaldehyde. A minimum of 5000 cells per sample were analysed by flow cytometry on a FC 500 (Beckman Coulter, Brea, CA).
RNA isolation and quantitative RT-PCR
RNA was extracted and isolated using PureZOL (Bio-Rad, Hercules, CA) following the manufacturer's instructions. The RNA was reverse transcribed using an iScript cDNA synthesis kit (Bio-Rad). The resulting cDNA was diluted 20-fold and real-time PCR was performed using iQ SYBR Green Supermix and an iQ5 thermal cycler (Bio-Rad) with 40 cycles of amplification. Samples were analysed in duplicate. Gene expression was normalized to that of GAPDH and expressed relative to untreated controls using the 2−ΔΔCt method. Oligonucleotide primer sets used were as follows: GAPDH, 5′-CAGCCGCATCTTCTTTTG-3′ (forward), 5′-GCAACAATATCCACTTTACCA-3′ (reverse), integrin β2 (itgb2), 5′-CACCTACGACTCCTTCTGC-3′ (forward), 5′-TGACAAACGACTGCTCCTG-3′ (reverse), integrin αL (itgal), 5′-CCTCTTCCATGTTCAGCCTC-3′ (forward), 5′-TTCTCATACACCACGTCAACC-3′ (reverse), and integrin αM (itgam), 5′-CACGGATGGAGAAAAGTTTGG-3′ (forward), 5-TGGATGCGATGGTATTAAGCT-3′ (reverse).
Confocal microscopy
DCs (1 × 105) were seeded on glass coverslips in 24-well plates in infection medium and either left uninfected or infected with a 5 μm Syto-9 (Life Technologies, Carlsbad, CA) -stained log-phase culture of M. tuberculosis at an MOI of ∼ 2 for 48 hr. At that time the medium was removed and cells were washed with PBS followed by fixation with 350 μl of 4% paraformaldehyde for 30 min at room temperature. Cells were then washed thrice with 0·5 ml PBS that contained 0·2% BSA (PB solution) for 30 min. Next, mouse anti-CD18, mouse anti-CD11a or CD11b (Santa Cruz Biotechnology, Santa Cruz, CA) or isotype control diluted in 150 μl PB solution was added for 2 hr with gentle shaking in the dark. The DCs were then washed thrice with PB solution and 150 μl of goat anti-mouse AlexaFlour 568-conjugated secondary antibody (Invitrogen, Carlsbad, CA) was added for 1 hr with shaking in the dark. Following three washes with PBS, coverslips were mounted on microscope slides with ProLong® Gold mounting solution (Life Technologies, Grand Island, NY). Confocal microscopy was performed on a Zeiss LSM 510 Meta.
DC/Endothelial cell adhesion assay
Human microvascular lung endothelial cells (HMVLEC) were used and cultured to confluency following the manufacturer's instructions (Lonza, Basel, Switzerland). HMVLEC were harvested with a trypsin/EDTA solution (ThermoScientific), washed with PBS, and seeded at a density of 105 per well in a 96-well plate in 0·2 ml of infection medium. After 24 hr, HMVLEC were activated to express adhesion molecules by 16 hr culture with TNF-α (20 ng/ml). All wells were blocked to reduce non-specific DC adhesion with HEPES-buffered RPMI-1640 that contained 0·5% BSA for 90 min. Untreated, M. tuberculosis-infected (MOI ∼ 2), lipopolysaccharide (LPS) -treated, or latex bead-treated (as above) DCs were stained with 1 ml of a 5 μm Syto-61 (Molecular Probes, Eugene, OR) solution in HEPES-buffered saline (HBS) for 30 min with gentle shaking. DCs were then washed twice with 1 ml of HBS, suspended in HEPES-buffered RPMI-1640, and 1 × 105 were overlaid onto HMVLEC. DCs were allowed to adhere for 30 min, then all wells were washed thrice with 0·1 ml of room temperature HEPES-buffered RPMI-1640 to remove non-adherent cells. The cultures were then fixed with 4% paraformaldehyde and fluorescence was measured with a Synergy plate reader (BioTek, Winooski, VT) at an excitation of 620 nm and emission of 645 nm. Each treatment was performed in duplicate. This was repeated with three independent donors for LPS and six for all other DC treatments.
Transendothelial migration assay
DC migration was assayed using the Cytoselect™ Leukocyte Transmigration Assay kit from Cell Biolabs, Inc. (San Diego, CA) following the instructions provided by the manufacturer. Briefly, HMVLEC (1 × 105) were seeded on 0·3-μm porous inserts for 48 hr for monolayer formation. TNF-α (20 ng/ml) was used to activate HMVLEC to expression adhesion molecules for 16 hr. DCs (2 × 105) that were cultured in medium alone, stimulated with LPS (200 ng/ml) or infected with M. tuberculosis (MOI ∼ 2) for 48 hr were fluorescently stained and 1 × 105 were overlaid. The lymphatic chemokines CCL19 and CCL21 (100 ng/ml each) were added to the lower chamber of the well, and cells were incubated for 24 hr. Migrated cells were then lysed, and fluorescence was measured with a Synergy plate reader using an excitation of 480 nm and emission of 520 nm. Each treatment was performed in duplicate. This was repeated with three independent donors.
Statistical anlaysis
Student's t-test was used as indicated to determine statistical significance in the 95% confidence interval.
Results
Human dendritic cells infected with M. tuberculosis express reduced levels of cell surface integrins
Surface expression levels of the β1 and β2 family of integrins have been shown to be unchanged during maturation of human DCs in response to LPS.30 We wanted to examine the effect of M. tuberculosis infection on human DC integrin expression. First, to confirm our DC differentiation technique, cells were harvested and immunolabelled against the indicated molecules and analysed by flow cytometry. The phenotype of the monocyte-derived DCs cultured as described is demonstrated in Fig. 1. Notably, these cells are CD86high, MHC class IIhigh, CD11chigh, CD1b+ and CD14−. To begin, we first investigated surface levels of the β chains that are common among a number of distinct heterodimers. DCs were infected with M. tuberculosis (MOI ∼ 2) for 48 hr, immunolabelled for surface integrins, and analysed by flow cytometry. CD18 surface expression was significantly decreased when DCs were infected with M. tuberculosis (Fig. 2a,b). The mean fluorescent intensity from eight independent donors was reduced by approximately twofold during infection (P = 0·033, Fig. 2a). The expression of CD18 was distributed in a bimodal manner for some blood donors; this was observed in both infected and control cultures. To investigate the possibility that reduced surface levels of CD18 were mediated by a decrease in CD18 (itgb2) transcription upon infection with M. tuberculosis, quantitative RT-PCR was performed. We observed an approximate two-fold mean decrease in CD18 transcripts over four independent donors that was not statistically significant compared with uninfected DCs (data not shown). We also evaluated CD29 (β1) integrin surface expression. Although there was a modest decrease in the surface expression of CD29 during infection, the findings did not reach statistical significance (Fig. 2c,d). To determine if changes in surface protein expression were limited to integrins, the expression of ICAM-1 (CD54) was evaluated on infected DCs. In contrast to CD18 and CD29, ICAM-1 expression was significantly increased on M. tuberculosis-infected DCs (P = 0·012, Fig. 2e,f). This is consistent with previous reports.31,32
Figure 2.
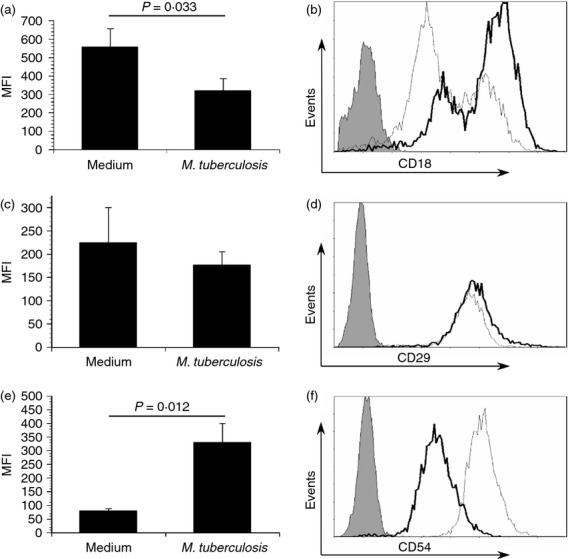
Mycobacterium tuberculosis infection results in a reduction of cell surface integrin expression. Primary human dendritic cells (DCs) were cultured in medium alone or infected with M. tuberculosis (multiplicity of infection ∼ 2), harvested, immunolabelled for CD18 (a, b), CD29 (c, d), or CD54 (e, f) and analysed by flow cytometry. The mean fluorescent intensity (MFI) ± standard error for eight (a) or three independent donors (c and e) along with a representative histogram overlay (b, d, and f; grey = isotype control; thin line = infected; bold black line = medium alone) for each molecule is displayed. Statistical significance in the 95% confidence interval was determined by Student's t-test.
Integrin subunits cell surface distribution and expression are altered following M. tuberculosis infection
Figure 2 indicates a specific change in surface expression of CD18 on human DCs during infection by M. tuberculosis. To further confirm this result and examine cellular distribution upon infection with M. tuberculosis, CD18, CD11a and CD11b surface expression were evaluated by confocal microscopy. Consistent with flow cytometric data, we observed that uninfected DCs expressed higher levels of all three integrin subunits (Fig. 3). In addition, we observed a redistribution of CD18 during infection. Rather than diffuse localization throughout the cell in the uninfected cells, CD18 was located primarily at the cell periphery in M. tuberculosis-infected DCs (Fig. 3). The levels of CD11b were also reduced during infection with some redistribution. In contrast, CD11a surface expression was reduced when DCs were M. tuberculosis-infected with more intermediate redistribution compared with CD11b (Fig. 3). CD18 has been shown to be recruited to podosomes and focal adhesions where an actin–integrin linkage is required to maintain stability.30 In the absence of the ability to regulate actin organization, CD18 has been shown to be located at the cell periphery with the consequence of a reduced ability to adhere to ICAM-1.30 By comparison, this suggests that the distribution of CD18 is important to its function in adherence and motility. Importantly in our experiments, mycobacteria induce a redistribution that is also accompanied by a loss of cell surface protein levels.
Figure 3.
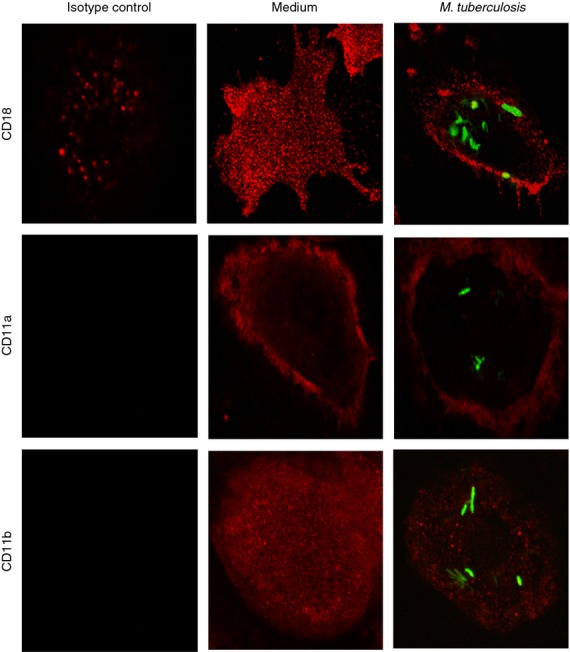
Cell surface distribution and expression of integrin subunits is altered by Mycobacterium tuberculosis infection. To visually assess CD18, CD11a and CD11b expression and distribution, primary human dendritic cells were infected with syto-61 (green)-stained M. tuberculosis. Integrin subunits were immunolabelled (red) and analysed by confocal microscopy. The expression of all three integrin subunits is reduced upon M. tuberculosis infection. CD18 and CD11a are redistributed from a uniform pattern in medium alone to the cell periphery in M. tuberculosis-infected cells. CD11b exhibits a uniform cellular distribution in either condition. A representative of three independent donors demonstrating similar results is shown.
Reduced CD18 expression is mediated by M. tuberculosis-derived antigen and maintained over time
To determine if live bacteria are required for the reduction in CD18 levels, we exposed DCs to irradiated M. tuberculosis (uvi M. tuberculosis ) at an MOI ∼ 2 for 48 hr. Irradiated M. tuberculosis mediate a comparable reduction in cell surface CD18 to live mycobacteria (Fig. 4a). As a control for phagocytosis, we included 2-μm latex beads in DC cultures for 48 hr and evaluated CD18 levels as before. Latex beads are ideal because the number supplied does not change as would be the case with other faster-growing bacteria, and they remove inflammatory signalling as a compounding factor. Treatment with beads did not lead to a reduction of CD18 surface levels that was comparable to M. tuberculosis (Fig. 4a). Cumulatively, this suggests that M. tuberculosis-derived antigens are responsible for the observed change in CD18 expression; neither bacterial replication nor secretion are required. Moreover, CD18 is a component of CR3 used for internalization of M. tuberculosis by phagocytes. As CD18 expression was limited 48 hr after infection, we considered the possibility that CD18 surface levels may be reduced as a consequence of mycobacterial internalization. CD18 surface levels were not strongly influenced by increasing numbers of bacteria in a linear fashion, which would be expected if this was purely a result of phagocytosis (Fig. 4b). To further evaluate the reduction kinetics and determine if CD18 surface expression returns to levels observed with uninfected DCs at later time points, CD18 expression was evaluated at 48-hr intervals over a period of 6 days. Although there is some increase in CD18 over time with uninfected cells, the reduction in CD18 expression was maintained over 6 days in the M. tuberculosis-infected cultures (Fig. 4c). Cumulatively, these data suggest an M. tuberculosis-driven event and indicate that internalization of complement receptors during M. tuberculosis phagocytosis is not responsible for the observed reduction in CD18 expression.
Figure 4.
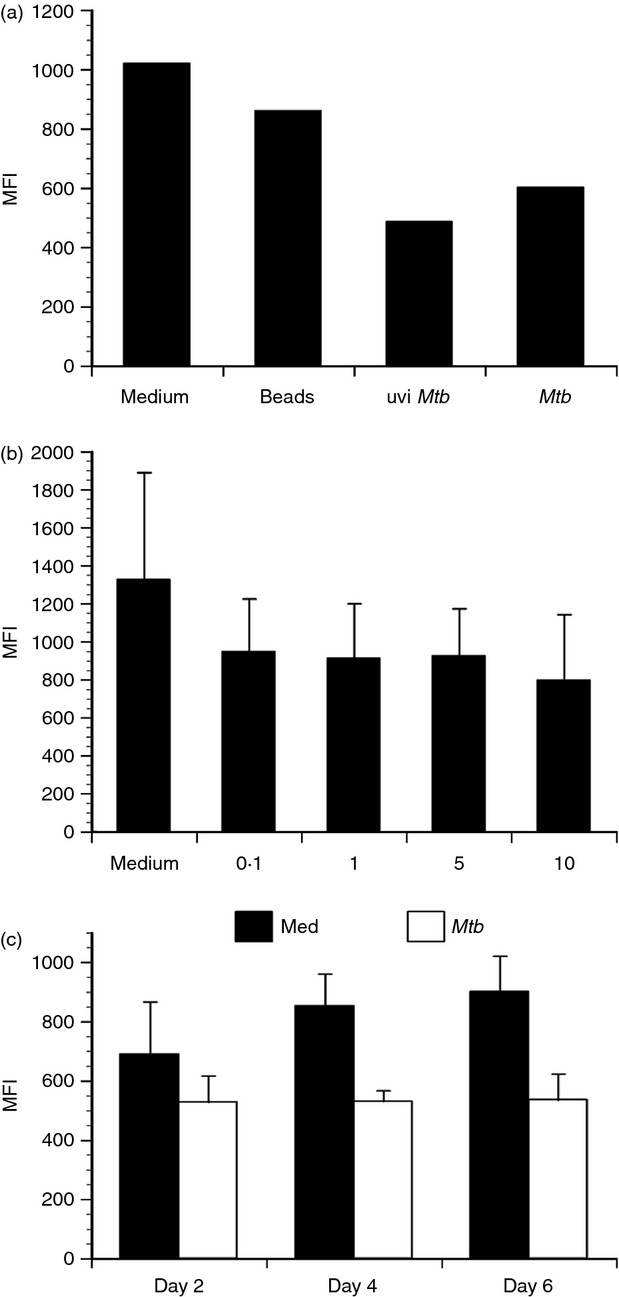
CD18 expression decrease is mediated by M. tuberculosis-derived antigen and maintained over time. Primary human dendritic cells (DCs) were treated as indicated and CD18 cell surface expression evaluated by flow cytometry. (a) A representative experiment of three performed with different donors displaying the mean fluorescence intensity (MFI) of CD18 in response to fluorescent beads, ultraviolet-inactivated M. tuberculosis (uviMtb), and live M. tuberculosis (Mtb) at a multiplicity of infection (MOI) ∼ 2. (b) CD18 MFI ± standard error in response to increasing numbers of M. tuberculosis (MOI ∼ 0·1–10) for three combined experiments with separate donors. (c) CD18 MFI ± standard error for DCs cultured in in medium alone or infected with M. tuberculosis (MOI ∼ 2) over 6 days. The data represent three combined experiments performed with different donors.
M. tuberculosis infection results in a reduction of cell surface-expressed integrins
As we observed a reduction in CD18 expression, we also investigated the effect of M. tuberculosis infection on two α subunits that pair with CD18 to form integrin heterodimers LFA-1 and Mac-1, which are commonly involved in DC adhesion and biology. To do this, DCs were immunolabelled for CD18 in parallel with each α subunit. Mycobacterium tuberculosis infection resulted in reduced surface expression of both α subunits CD11a and CD11b, respectively (Fig. 5); however, neither achieved statistical significance. Reduced surface levels of CD11a or CD11b were not the product of meaningful changes in gene expression observed over three independent donors (data not shown).
Figure 5.

Mycobacterium tuberculosis infection results in a reduction of cell surface-expressed integrin heterodimers and individual α subunits. Dendritic cells (DCs) were cultured in medium alone or infected with M. tuberculosis (multiplicity of infection ∼ 2), harvested, and double-immunolabelled for either CD18/CD11a (LFA-1) or CD18/CD11b (Mac-1) and analysed by flow cytometry. Representative scatter plots of LFA-1 on medium alone cells (a) or M. tuberculosis-infected (b) are shown. Representative scatter plots of Mac-1 on medium alone cells (d) and M. tuberculosis-infected (e) are shown. Overlaid histogram representations of the same data sets highlight the changes in a CD11a (c) or CD11b (f) surface expression. (g) The mean fluorescent intensity (MFI) ± standard error for CD11a (αL) and CD11b (αM), with five combined experiments performed with independent donors, are shown. Representative histogram overlays for CD11a (c) and CD11b (f) are shown; grey = isotype control; thin line = infected; bold black line = medium alone.
To confirm our findings using primary blood DCs, these cells were isolated from peripheral blood mononuclear cells by immunomagnetic selection. Blood DCs were then either left in cell culture medium or infected with M. tuberculosis at an MOI ∼ 2 for 48 hr. They were then immunolabelled for CD18 in parallel with each α subunit and analysed by flow cytometry. Similar to monocyte-derived DCs, blood DCs exhibited reductions in both LFA-1 (Fig. 6a–c) and Mac-1 (Fig. 6d–f) after infection with M. tuberculosis, respectively. The decrease was more substantial for Mac-1, which exhibited a more than twofold decrease following infection.
Figure 6.
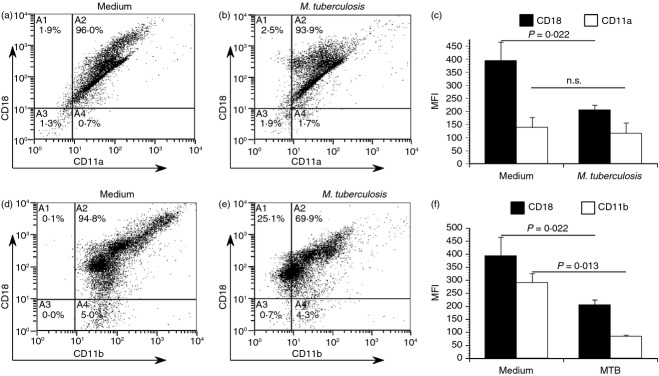
Mycobacterium tuberculosis infected primary blood dendritic cells (DCs) exhibit reduced cell surface expression of both LFA-1 and Mac-1. To confirm our previous findings on blood DC, these cells were isolated from peripheral blood mononuclear cells by immunomagnetic selection. Blood DC were then either left in cell culture medium or infected with M. tuberculosis at a multiplicity of infection ∼ 2 for 48 hr. Cells were then immunolabelled for either LFA-1 (CD18/CD11a, a–c) or Mac-1 (CD18/CD11b, d–f) and analysed by flow cytometry. Scatter plots of a representative donor are shown, and the mean fluorescence intensity (MFI) ± standard error for two donors. Statistical significance in the 95% confidence interval was determined by a Student's t-test.
M. tuberculosis-infected DCs exhibit a reduced ability to adhere to lung endothelial cells and migrate toward a chemokine gradient
Migration of DCs from infected lung to local lymph nodes is preceded by adhesion to and transmigration through lymphatic endothelia. To determine the functional consequence of limited integrin expression on the surface of infected DCs, we measured DC adherence to primary HMVLEC. DCs were left untreated, infected with M. tuberculosis, or treated with LPS or latex beads for 48 hr. Latex beads were used as a control to determine if cells that have phagocytosed a non-stimulating agent are generally less adherent. DCs were then harvested, labelled with Syto-61 and cultured with endothelial cells for 30 min. Non-adherent DCs were removed by washing and the fluorescence associated with adhered DCs was quantified. Unstimulated DCs bound efficiently to HMVLEC and this was not altered by uptake of latex beads. LPS-stimulated DCs bound as efficiently to HMVLEC as bead-treated DCs (Fig. 7a). However, M. tuberculosis-infected DCs (MOI ∼ 2) exhibited a significant reduction (P < 0·0001) in their ability to adhere to HMVLEC.
Figure 7.
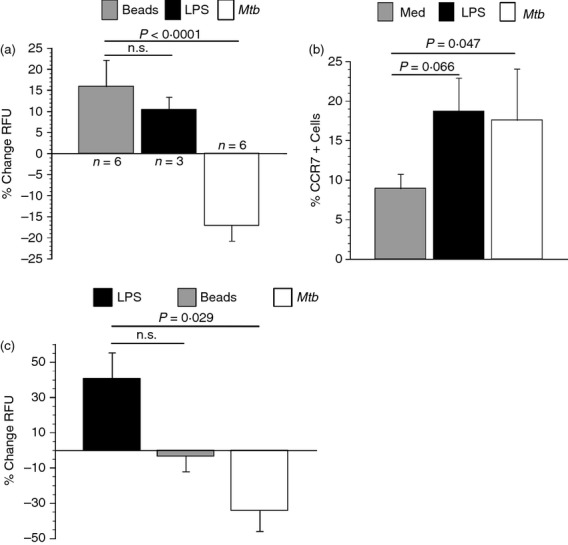
Mycobacterium tuberculosis infected dendritic cells (DCs) exhibit a reduced ability to adhere to lung endothelial cells and migrate toward lymphatic chemokines. (a) Human microvascular lung endothelial cells (HMVLEC) were seeded in 96-well plates and activated to express adhesion molecules with tumour necrosis factor-α (TNF-α; 20 ng/ml). DCs were cultured in medium alone, infected with M. tuberculosis, treated with lipopolysaccharide (LPS; 200 ng/ml) or latex beads for 48 hr. The DCs were then harvested, syto-61-stained and placed over the HMVLEC for 30 min. Fluorescence indicative of DC adherence was measured by spectrophotometry. Data are expressed as average per cent decrease in relative fluorescent units (RFU) ± standard error relative to medium alone. Data represent three separate donors for LPS and six for all other DC treatments. Each treatment contained a replicate. Statistical significance for the 95% confidence interval was determined by a Student's t-test. (b) CCR7 on resting (medium), LPS-stimulated (200 ng/ml) and M. tuberculosis-infected (multiplicity of infection ∼ 2) DCs was measured by flow cytometry. Per cent CCR7-positive cells over four independent blood donors is shown. (c) HMVLEC were seeded on porous inserts and cultured for 48 hr. TNF-α (20 ng/ml) was added to induce expression of adhesion molecules overnight. DCs (1 × 105) that were cultured in medium alone, LPS-treated (200 ng/ml), or infected with M. tuberculosis (multiplicity of infection ∼ 2) were fluorescently stained and overlaid. The lymphatic chemokines CCL19 and CCL21 (100 ng/ml) were added to the lower chamber of the well. The cultures were incubated for 24 hr. Replicate well-readings were taken for each DC treatment and the data are expressed as the mean per cent change in relative fluorescent units (RFU) ± standard error relative to medium alone. Data represent three experiments carried out with separate donors. Statistical significance in the 95% confidence interval was determined by a Student's t-test; n.s. = not significant.
Upon firm adhesion to lymphatic endothelial cells, DCs traverse the endothelium and enter the afferent lymphatic system for migration to local lymph nodes. CCR7 facilitates DC migration to lymph nodes. We observed a significant increase (P = 0·047) in CCR7-expressing DCs upon M. tuberculosis infection relative to untreated DCs (Fig. 7b). Lipopolysaccharide-treated DCs exhibited a similar increase in CCR7. To test the ability of M. tuberculosis-infected DCs to migrate through lung endothelial cells, we used an in vitro migration assay. The DCs were treated with LPS as a positive control, latex beads as negative control, infected with M. tuberculosis (MOI ∼ 2), or left untreated for 48 hr. The DCs were then placed over a monolayer of activated HMVLEC (1 × 105) and allowed to migrate for 24 hr toward CCL19 and CCL21 (lymphatic-produced chemokines) placed in the lower chamber of the well. The M. tuberculosis-infected DCs exhibit a substantial reduction (P = 0·029) in their ability to migrate through HMVLEC and towards lymphatic chemokines compared with the LPS-positive control (Fig. 7c). As expected, latex bead-treated cells did not migrate.
Discussion
Dendritic cells in peripheral tissues sample the microenvironment for antigens and upon recognition migrate through afferent lymphatics to regional lymph nodes. The DCs can then present antigens to T cells and in this way serve as the link between innate and adaptive immunity. Numerous studies in vivo have estimated that migration is typically complete in 24 hr.33–35 The onset of adaptive immunity during TB suggests that this process is prolonged with DCs following mycobacterial infection compared with other pathogens.36–39 In this study we show that DCs infected with M. tuberculosis express reduced levels of cell surface integrins that contain CD18. This impacts their adhesion to lymphatic endothelial cells and the ability to migrate through a lung endothelial monolayer toward chemokines in vitro.
Surface levels of CD29, CD18 and α chain partners have previously been shown to be unaffected by DC maturation.30 We wanted to determine if their expression was altered as a consequence of infection by M. tuberculosis. To the best of our knowledge there are no reports that have examined integrin expression on primary human DCs infected by M. tuberculosis. There have been numerous studies conducted with macrophages, monocytes and murine DCs that report increases in integrin expression during M. tuberculosis infection. In one study, human monocyte-derived macrophages exhibited increased expression of LFA-1 that augmented cellular adhesion.40 Similarly, Mac-1 cell surface expression was increased on murine macrophages upon infection with M. tuberculosis in vitro.41 Moreover, it was shown that murine lung DCs increased cell surface expression of both CD11a and CD11b in response to M. tuberculosis after 24 hr.42 In contrast, our findings demonstrate that both monocyte-derived and blood DCs infected with M. tuberculosis exhibit reduced expression of surface molecules that facilitate adherence to and transmigration through endothelial cells. This does not suggest a motile phenotype capable of initiating a prompt effector T-cell response. The modulation of adhesive and migratory molecules on DCs may alter/disrupt the trafficking of these cells and contribute to the delayed onset of adaptive immunity to M. tuberculosis in vivo.
Flow cytometric analysis of cell surface integrins sometimes revealed a bimodal expression pattern, although this was not observed with all donors. Similarly, others have reported bimodal integrin expression on various cell types including DCs.43–45 It is unclear why this may be in some cases but there does not appear to be a relationship to M. tuberculosis infection. Confocal microscopy showed a redistribution of CD18, CD11a and CD11b when DCs are infected with M. tuberculosis; this was not as pronounced for CD11a. Rather than demonstrating a uniform distribution throughout the cell as observed in the unstimulated group, M. tuberculosis-infected DCs exhibited a reduced level of all three integrin subunits examined with surface expression, which was localized mainly to the cell periphery for CD18 and CD11a. Although still reduced, CD11b distribution was not as clearly localized to the cell periphery as the other two subunits. In a previous report by Burns et al.30 that examined integrin distribution in LPS-matured DCs, CD18 and CD11b associated with actin-rich podosomes that were shown in a separate report to assemble and disassemble during DC maturation.46 However, in the absence of the Wiskott–Aldrich syndrome protein that contributes to actin organization, CD18 is localized at the cell periphery in a manner similar to what is reported here in infected DCs.30 This may suggest an influence of the actin cytoskeleton during M. tuberculosis infection. Additionally and unique to M. tuberculosis-infected DCs is the reduction of integrin surface levels. In contrast, a fourfold increase in ICAM-1 surface expression on M. tuberculosis-infected DCs suggests a specific influence on integrins involved in interactions with endothelial cells and DC migration.
A reduction in surface-expressed CD18-containing integrins would be expected to impact adherence and migration. Antibodies specific for CD18 significantly reduce adherence of human DCs to ICAM-1.30 Therefore, we wanted to evaluate if there was a functional consequence to limited integrin surface expression during infection of DCs by M. tuberculosis. We report a significant reduction in the ability of M. tuberculosis-infected DCs to adhere to lung endothelial cells. Latex beads and LPS-stimulation were used in parallel, which resulted in an increase in cellular adhesion, as opposed to decreased adhesion in M. tuberculosis-infected DCs. This indicates that M. tuberculosis-infection, and not phagocytosis or general activation, are responsible for the decrease in CD18 that impacts adhesiveness of DCs to endothelial cells. Consistent with reduced adhesion to lung endothelial cells, we report a significant reduction in the ability of M. tuberculosis-infected DCs to migrate toward the lymphatic chemokines CCL19/CCL21. Dendritic cells responded and migrated when treated with LPS, suggesting that M. tuberculosis infection of DCs mediates the reduced migratory capacity. Under the same conditions, we observed a comparable increase in CCR7-expressing DCs between LPS-treated and M. tuberculosis–infected cells. This indicates that they possess the ability to respond to the above chemokines and migrate accordingly; further highlighting that this inhibition is exclusive to M. tuberculosis and is suggestive of a mechanism independent of CCR7. This is in contrast to reports of mycobacteria inhibiting chemotaxis of DCs through a block in the up-regulation of CCR7.47,48 Our data also suggest a mechanism by which a loss of surface-expressed integrins impacts migration. In line with this, several studies have shown that specific blocking of integrins resulted in drastic reductions in the ability of DCs to adhere and migrate through endothelial cells.21,22 In further support of impaired DC migration during tuberculosis, another study also found that M. tuberculosis-infected murine DCs exhibited an impaired ability to migrate using an in vitro model similar to the one used in this study.49 Given that adhesion to and migration through endothelial cells is a prerequisite to reach lymph nodes, this could be a mechanism leading to prolonged presence of M. tuberculosis-infected DCs at the site of infection. This could contribute to a high rate of bacterial growth in the lungs and help to explain the delayed onset of an adaptive immune response that characterizes TB. However, in vivo assays will be needed to further substantiate these ideas.
There are several hypotheses that attempt to account for the delayed adaptive immune response during TB. Evidence in mice indicates that migration of DCs from the infected lung to lymph nodes takes at least 8–9 days.36 Studies have shown that DCs reach lymph nodes in much shorter time frames following uptake and activation by other stimuli.12,33 One hypothesis has been that there is insufficient antigen to mount an immune response. Wolf et al.50 examined this in vivo by increasing the M. tuberculosis inoculum per mouse and examined proliferation of adoptively transferred, transgenic T cells. They found that even the highest inoculum did not account for earlier T-cell proliferation. This indicates that the delay seen between infection and CD4+ T-cell activation is not solely attributed to the low number of bacteria, and further implies that slow bacterial growth does not explain the slow immune response to M. tuberculosis. T-cell proliferation is preceded by DC migration/transport of live M. tuberculosis to draining lymph nodes and synapsis with T cells50,51 – further highlighting an inhibition of DC migration to lymph nodes as a bottleneck in initiation of adaptive responses. Additionally, the authors provide information that M. tuberculosis may directly target DC migration and inhibit antigen presentation in vivo.51 The authors also report that the limiting step in the development of the adaptive immune response is attributed to a delay in the initial activation of CD4+ T cells. In line with this, a recent report found that mannose-capped lioparabinomannan (a M. tuberculosis cell wall component) preferentially inhibited murine and human T helper type 1 cell migration toward the lymph node egress signal sphingosine-1-phosphate.52 A recent report directly monitored DC migration into the mediastinal lymph nodes rather than activation of T cells as a measure of DCs arrival in mice receiving intratracheal delivery of either M. tuberculosis or purified protein derivative. The authors observed disparate patterns in the migration timing of DCs to the mediastinal lymph node.53 Dendritic cells from purified protein derivative-treated mice were detectable and peaked much earlier (7 days) in the mediastinal lymph node whereas DCs from M. tuberculosis-infected mice were not detectable early and did not peak until much later (21 days). The authors suggested that this may be a mechanism whereby M. tuberculosis evades an early expansion of effector T cells. Our data support those observations and provide a mechanistic explanation. Collectively, this indicates that M. tuberculosis is capable of interfering with the migration of multiple subsets of leucocytes to ultimately delay/subvert the onset of a timely adaptive immune response.
In conclusion, we report for the first time a reduction in cell surface integrin expression on primary human DCs infected with M. tuberculosis. It is clear that a robust host T-cell response is mounted following infection54 as manifested in the granuloma. However, as mentioned previously, the time taken for DCs to reach T-cell-rich zones in the lymph nodes is delayed and the mechanisms responsible have remained unknown. Our data suggest that the modulation of adhesion molecules, adherence to lung endothelial cells, and impaired migration by M. tuberculosis-infected DCs could account for the delayed activation and presence of T cells at the site of pulmonary TB. Measures aimed at combating the M. tuberculosis-mediated influence on CD18 integrin expression may promote an earlier and more efficient immune response with the hope of limiting early exponential increase in the bacterial burden.
Acknowledgments
This work was supported by institutional funds supplied by the University of South Carolina School of Medicine and National Institutes of Health grant HL093300.
Glossary
Abbreviations
- BSA
bovine serum albumin
- CCL-19
chemokine (C-C motif) ligand-19
- CCL-21
chemokine (C-C motif) ligand-21
- CR
complement receptor
- CCR7
chemokine receptor 7
- DC
dendritic cell
- DC-SIGN
dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin
- GM-CSF
granulocyte–macrophage colony-stimulating factor
- HMVLEC
human microvascular lung endothelial cell
- ICAM-1
intercellular adhesion molecule-1
- LFA-1
lymphocyte function-associated antigen-1
- LPS
lipopolysaccharide
- Mac-1
macrophage-1 antigen
- MOI
multiplicity of infection
- PE
phycoerythrin
- TB
tuberculosis
- VLA-4
very late antigen-4
Authorship
LLR contributed to the design of the experiments and was responsible for the performance of experiments, data analysis and manuscript preparation. CMR conceived the study idea, designed experiments, and contributed equally to the preparation of the manuscript.
Disclosures
The authors declare that they have no competing interests.
References
- 1.World Health Organization. Global Tuberculosis Control Report. Geneva, Switzerland: World Health Organization; 2011. http://www.who.int/tb/publications/global_report/2011/gtbr11_full.pdf. [Google Scholar]
- 2.Marino S, Pawar S, Fuller C, Reinhart T, Flynn JL, Kirschner DE. Dendritic cell trafficking and antigen presentation in the human immune response to Mycobacterium tuberculosis. J Immunol. 2004;173:494–506. doi: 10.4049/jimmunol.173.1.494. [DOI] [PubMed] [Google Scholar]
- 3.Wallgren A. The time-table of tuberculosis. Tubercle. 1948;29:245–51. doi: 10.1016/s0041-3879(48)80033-4. [DOI] [PubMed] [Google Scholar]
- 4.Cooper AM. Cell-mediated immune responses in tuberculosis. Annu Rev Immunol. 2009;27:393–422. doi: 10.1146/annurev.immunol.021908.132703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baron SD, Singh R, Metzger DW. Inactivated Francisella tularensis live vaccine strain protects against respiratory tularemia by intranasal vaccination in an immunoglobulin A-dependent fashion. Infect Immun. 2007;75:2152–62. doi: 10.1128/IAI.01606-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chakravarty S, Cockburn IA, Kuk S, Overstreet MG, Zavala F. CD8+ T lymphocytes protective against malaria liver stages are primed in skin-draining lymph nodes. Nat Med. 2007;13:1035–41. doi: 10.1038/nm1628. [DOI] [PubMed] [Google Scholar]
- 7.Kursar M, Bonhagen K, Kohler A, Mittrucker HW. Organ-specific CD4+ T cell response during Listeria monocytogenes infection. J Immmunol. 2002;168:6382–7. doi: 10.4049/jimmunol.168.12.6382. [DOI] [PubMed] [Google Scholar]
- 8.Lira R, Doherty M, Modi G, Sacks D. Evolution of lesion formation, parasitic load, immune response, and reservoir potential in c57bl/6 mice following high and low-dose challenge with Leishmania major. Infect Immun. 2000;68:5176–82. doi: 10.1128/iai.68.9.5176-5182.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McSorley SJ, Asch S, Costalonga M, Reinhardt RL, Jenkins MK. Tracking Salmonella-specific CD4 T cells in vivo reveals a local mucosal response to a disseminated infection. Immunity. 2002;16:365–77. doi: 10.1016/s1074-7613(02)00289-3. [DOI] [PubMed] [Google Scholar]
- 10.Moskophidis D, Kioussis D. Contribution of virus-specific CD8+ cytotoxic T cells to virus clearance or pathologic manifestations of influenza virus infection in a T cell receptor transgenic mouse model. J Exp Med. 1998;188:223–32. doi: 10.1084/jem.188.2.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Srinivasan A, Foley J, Ravindran R, McSorley SJ. Low-dose Salmonella infection evades activation of flagellin-specific CD4 T cells. J Immunol. 2004;173:4091–9. doi: 10.4049/jimmunol.173.6.4091. [DOI] [PubMed] [Google Scholar]
- 12.Grayson MH, Ramos MS, Rohlfing MM, Kitchens R, Wang HD, Gould A, Agapov E, Holtman MJ. Controls for lung dendritic cell maturation and migration during respiratory viral infection. J Immunol. 2007;179:1438–48. doi: 10.4049/jimmunol.179.3.1438. [DOI] [PubMed] [Google Scholar]
- 13.Herrmann JL, Lagrange PH. Dendritic cells and Mycobacterium tuberculosis: which is the Trojan horse? Pathol Biol. 2006;53:35–40. doi: 10.1016/j.patbio.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 14.Tailleux L, Schwartz O, Herrmann JL, Pivert E, Jackson M, Amara A. DC-SIGN is the major Mycobacterium tuberculosis receptor on human dendritic cells. J Exp Med. 2003;197:121–7. doi: 10.1084/jem.20021468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Randolph GJ. Dendritic cell migration to lymph nodes: cytokines, chemokines, and lipid mediators. Semin Immunol. 2001;13:267–74. doi: 10.1006/smim.2001.0322. [DOI] [PubMed] [Google Scholar]
- 16.Riol-Blanco L, Sanchez N, Torres A, Tejedor A, Narumiya S, Corbi A, Mateos P, Rodriguez-Fernandez J. The chemokine receptor CCR7 activates in dendritic cells two signaling molecules that independently regulate chemotaxis and migratory speed. J Immunol. 2005;174:4070–80. doi: 10.4049/jimmunol.174.7.4070. [DOI] [PubMed] [Google Scholar]
- 17.Hanekom WA, Mendillo M, Manca C, Haslett P, Siddiqui M, Barry C, Kaplan G. Mycobacterium tuberculosis inhibits maturation of human monocyte-derived dendritic cells in vitro. J Infect Dis. 2003;188:257–66. doi: 10.1086/376451. [DOI] [PubMed] [Google Scholar]
- 18.Puig-Kroger A, Sanz-Rodriquez F, Longo N, Sanchez-Mateos P, Botella L, Teixido J, Bernabeu C, Corbi AL. Maturation-dependent expression and function of the CD49d integrin on monocyte-derived human dendritic cells. J Immunol. 2000;165:4338–45. doi: 10.4049/jimmunol.165.8.4338. [DOI] [PubMed] [Google Scholar]
- 19.Ammon C, Meyer P, Schwarzfischer L, Krause SW, Anderseen R, Kreutz M. Comparative analysis of integrin expression on monocyte-derived macrophages and monocyte-derived dendritic cells. Immunology. 2000;100:364–9. doi: 10.1046/j.1365-2567.2000.00056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ghosh S, Chackerian AA, Parker CM, Ballantyne CM, Behar SM. The LFA-1 adhesion molecule is required for protective immunity during pulmonary Mycobacterium tuberculosis infection. J Immunol. 2006;176:4914–22. doi: 10.4049/jimmunol.176.8.4914. [DOI] [PubMed] [Google Scholar]
- 21.D'Amico G, Bianchi G, Bernasconi S, Bersani L, Piemonti L, Sozzani S, Mantovani A, Allavena P. Adhesion, transendothelial migration, and reverse transmigration of in vitro cultured dendritic cells. Blood. 1998;92:207–14. [PubMed] [Google Scholar]
- 22.De la Rosa G, Longo N, Rodriguez-Fernandez J, Puig-Kroger A, Pineda A, Corbi AL, Sanzhez-Mateos P. Migration of human blood dendritic cells across endothelial cell monolayers: adhesion molecules and chemokines involved in subset-specific transmigration. J Leukoc Biol. 2003;73:639–49. doi: 10.1189/jlb.1002516. [DOI] [PubMed] [Google Scholar]
- 23.Johnson LA, Clasper S, Holt AP, Lalor PF, Baban D, Jackson DG. An inflammation-induced mechanism for leukocyte transmigration across lymphatic vessel endothelium. J Exp Med. 2006;203:2763–77. doi: 10.1084/jem.20051759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Balkow S, Heinz S, Schmidbauer P, Kolanus W, Holzmann B, Grabbe S, Laschinger M. LFA-1 activity state on dendritic cells regulates contact duration with T-cells and promotes T-cell priming. Blood. 2010;116:1885–94. doi: 10.1182/blood-2009-05-224428. [DOI] [PubMed] [Google Scholar]
- 25.Smits HH, de Jong EC, Schuitemaker JHN, Geijenbeek TBH, van Kooyk Y, Kapsenberg ML, Wierenga EA. Intercellular adhesion molecule-1/LFA-1 ligation favors human Th1 development. J Immunol. 2002;168:1710–6. doi: 10.4049/jimmunol.168.4.1710. [DOI] [PubMed] [Google Scholar]
- 26.Varga G, Nippe N, Balkow S, et al. LFA-1 contributes to signal 1 of T-cell activation and to the production of Th1 cytokines. J Invest Dermatol. 2010;130:1005–12. doi: 10.1038/jid.2009.398. [DOI] [PubMed] [Google Scholar]
- 27.Carlson PE, Carroll JA, O'Dee DM, Nau GJ. Modulation of virulence factors in Francisella tularensis determines human macrophage responses. Microb Pathog. 2007;42:204–14. doi: 10.1016/j.micpath.2007.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sambandamurthy VK, Derrick SC, Hsu T, Chen B, Larsen MH. Mycobacterium tuberculosis ΔRD1 ΔpanCD: a safe and limited replicating mutant strain that protects immunocompetent and immunocompromised mice against experimental tuberculosis. Vaccine. 2006;24:6309–20. doi: 10.1016/j.vaccine.2006.05.097. [DOI] [PubMed] [Google Scholar]
- 29.Sambandamurthy VS, Wang X, Chen B, Russell R, Collins F. A pantothenate auxotroph of Mycobacterium tuberculosis is highly attenuated and protects mice against tuberculosis. Nat Med. 2002;8:1171–4. doi: 10.1038/nm765. [DOI] [PubMed] [Google Scholar]
- 30.Burns S, Hardy S, Buddle J, Yong K, Jones G, Thrasher A. Maturation of DC is associated with changes in motile characteristics and adherence. Cell Mot Cytoskel. 2004;57:118–32. doi: 10.1002/cm.10163. [DOI] [PubMed] [Google Scholar]
- 31.Henderson R, Watkins S, Flynn JL. Activation of human dendritic cells following infection with Mycobacterium tuberculosis. J Immunol. 1997;159:635–43. [PubMed] [Google Scholar]
- 32.Mihret A, Mamo G, Tafesse M, Hailu A, Parida S. Dendritic cells activate and mature after infection with Mycobacterium tuberculosis. BMC Res Note. 2011;4:247–53. doi: 10.1186/1756-0500-4-247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Macatonia SE, Knight SC, Edwards AJ, Fryer P. Localization of antigen on lymph node dendritic cells after exposure to the contact sensitizer fluorescein isothiocyanate. Functional and morphological studies. J Exp Med. 1987;166:1654–67. doi: 10.1084/jem.166.6.1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kupiec-Weglinski JW, Austyn JM, Morris JP. Migration patterns of dendritic cells in the mouse. Traffic from the blood, and T cell-dependent and independent entry to lymphoid tissues. J Exp Med. 1988;176:632–45. doi: 10.1084/jem.167.2.632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cumberbatch M, Kimber I. Dermal tumor necrosis factor-α induces dendritic migration to draining lymph nodes, and possibly provides one stimulus for Langerhans' cell migration. Immunology. 1992;75:257–63. [PMC free article] [PubMed] [Google Scholar]
- 36.Reiley WW, Calayag MD, Wittmer ST, et al. ESAT-6 specific CD4 T cell responses to aerosol Mycobacterium tuberculosis infection are initiated in the mediastinal lymph nodes. Proc Natl Acad Sci USA. 2008;105:10961–6. doi: 10.1073/pnas.0801496105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roman E, Miller E, Harmsen A, Wiley J, von Adrian U, Huston G, Swain S. CD4 effector T cell subsets in the response to influenza: heterogeneity, migration, and function. J Exp Med. 2004;196:957–68. doi: 10.1084/jem.20021052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chakerian AA, Alt JM, Perera TV, Behar SM. Dissemination of Mycobacterium tuberculosis is influenced by host factors and precedes the initiation of T-cell immunity. Infect Immun. 2002;70:4501–9. doi: 10.1128/IAI.70.8.4501-4509.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Flynn KJ. Virus-specific CD8+ T cells in primary and secondary influenza pneumonia. Immunity. 1998;8:683–91. doi: 10.1016/s1074-7613(00)80573-7. [DOI] [PubMed] [Google Scholar]
- 40.Desjardin LE, Kaufman TM, Potts B, Kutzbach B, Yi H, Schlesinger LS. Mycobacterium tuberculosis-infected human macrophages exhibit enhanced cellular adhesion with increased expression of LFA-1 and ICAM-1 and reduced expression and/or function of complement receptors, FcγRII and the mannose receptor. Microbiology. 2002;148:3161–71. doi: 10.1099/00221287-148-10-3161. [DOI] [PubMed] [Google Scholar]
- 41.Ghosh S, Saxena RK. Early effect of Mycobacterium tuberculosis infection on Mac-1 and ICAM-1 expression on mouse peritoneal macrophages. Exp Mol Med. 2004;36:387–95. doi: 10.1038/emm.2004.51. [DOI] [PubMed] [Google Scholar]
- 42.Gonzalez-Juarrero M, Orme IM. Characterization of murine lung dendritic cells infected with Mycobacterium tuberculosis. Infect Immun. 2001;69:1127–33. doi: 10.1128/IAI.69.2.1127-1133.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li A, Simmons P, Kaur P. Identification and isolation of candidate human keratinocyte stem cells based on cell surface phenotype. Proc Natl Acad Sci USA. 1998;95:3902–7. doi: 10.1073/pnas.95.7.3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Andreason S, Thomsen A, Christensen J. Expression and functional importance of collagen-binding integrins α1β1 and α2β1, on virus activated T cells. J. Immunol. 2003;171:2804–11. doi: 10.4049/jimmunol.171.6.2804. [DOI] [PubMed] [Google Scholar]
- 45.Tjomsland V, Ellegard R, Kjolhede P, Larsson M. Blocking of integrins inhibits HIV-1 infection of human cervical mucosa immune cells with free and complement-opsonized virions. Eur J Immunol. 2013;43:1–12. doi: 10.1002/eji.201243257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.West MA, Wallin RP, Matthews SP, Svensson HG, Zaru R, Ljunggren HG, Prescott AR, Watts C. Enhanced dendritic cell antigen capture via toll-like receptor-induced acting remodeling. Science. 2004;305:1153–7. doi: 10.1126/science.1099153. [DOI] [PubMed] [Google Scholar]
- 47.Rajashree P, Supriya P, Das SD. Differential migration of human monocyte-derived dendritic cells after infection with prevalent clinical strains of Mycobacterium tuberculosis. Immunobiology. 2008;213:567–75. doi: 10.1016/j.imbio.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 48.Sanarico N, Colone A, Grassi M, Mariani F. Different transcriptional profiles of human monocyte-derived dendritic cells infected with distinct strains of Mycobacterium tuberculosis and Mycobacterium bovis Bacillus Calmette–Guérin. Clin Dev Immunol. 2011;2011 doi: 10.1155/2011/741051. Article ID 741051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Blomgran R, Ernst JD. Lung neutrophils facilitate activation of naïve antigen-specific CD4+ T cells during Mycobacterium tuberculosis infection. J Immunol. 2011;186:7110–9. doi: 10.4049/jimmunol.1100001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wolf J, Desvignes L, Linas B, Banaiee N, Tamura T, Takatsu K, Ernst JD. Initiation of the adaptive immune response to Mycobacterium tuberculosis depends on antigen production in the local lymph node, not the lungs. J Exp Med. 2008;205:105–15. doi: 10.1084/jem.20071367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wolf AJ, Linas B, Tervejo-Nunez G, Kincaid E, Tumara T, Takatsu K, Ernst JD. Mycobacterium tuberculosis infects dendritic cells with high frequency and impairs their function in vivo. J Immunol. 2007;179:2509–19. doi: 10.4049/jimmunol.179.4.2509. [DOI] [PubMed] [Google Scholar]
- 52.Richmond J, Lee J, Green D, Kornfeld H, Cruikshank W. Mannose-capped lipoarabinomannan from Mycobacterium tuberculosis preferentially inhibits sphingosine-1-phosphate-induced migration of Th1 cells. J Immunol. 2012;189:5886–95. doi: 10.4049/jimmunol.1103092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Garcia-Romo G, Pedroza-Gonzalez A, Flores-Romo L. Mycobacterium tuberculosis manipulates pulmonary APCs subverting early immune responses. Immunobiology. 2013;218:393–401. doi: 10.1016/j.imbio.2012.05.022. [DOI] [PubMed] [Google Scholar]
- 54.Lopez-Ramierz GM, Rom W, Ciotoli C, Martiniuk F, Cronstein B, Reibman J. Mycobacterium tuberculosis alters expression of adhesion molecules on monocytic cells. Infect Immun. 1994;62:2515–20. doi: 10.1128/iai.62.6.2515-2520.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]