

Abstract
Toll-like receptor (TLR) ligands induce dendritic cell (DC) maturation. During this process cells initiate proteolytic degradation of internalized protein antigens into peptides that complex with MHC II, and simultaneously increase expression of costimulatory molecules and of cytokines such as IL-6, IL-12 and IL-23. In these ways, TLR-activated DCs are able to activate naïve Th cells and initiate Th1 and Th17 responses, and TLR-ligands thus serve as adjuvants for these types of responses. In contrast, products from helminth parasites generally do not activate DCs, and act as adjuvants for Th2 response induction. We have explored the underlying basis for this form of adjuvanticity. We show that exposure of DCs to soluble antigens from the eggs of the helminth parasite Schistosoma mansoni (SEA) leads to the induction of proteolysis of internalized antigen. This occurs in the absence of significant induction of costimulatory molecule expression or production of proinflammatory cytokines. SEA-induced antigen processing occurs independently of MyD88 or Trif, but is significantly attenuated by inhibition of p38, but not ERK, signaling. In DCs exposed to SEA, ligation of CD40 provides a necessary second signal that stimulates costimulatory molecule expression, allowing DCs to mature into capable antigen presenting cells. Collectively, the data demonstrate the existence of a MyD88/Trif-independent, p38-dependent pathway of antigen processing in DCs, which is uncoupled from conventional DC maturation and is associated with induction of Th2 type immune responses.
Keywords: Dendritic cells, Th1/Th2 cells, Parasitic-Helminth, Antigen Presentation/Processing, Inflammation
Introduction
The immune system has evolved to recognize pathogen-inherent signals and respond accordingly. Much of the responsibility for sensing primary infection, and for assessing the nature of the threat lies with dendritic cells (DCs). These cells, through expression of a panel of pattern recognition receptors such as Toll like receptors (TLRs), are able to identify and respond to foreign organisms by initiating a sequence of events that link innate and adaptive immunity. Specifically, they: 1) induce inflammation by producing cytokines and chemokines; 2) internalize material (antigen) from the pathogen and proteolytically process it for the presentation of peptide epitopes, complexed with MHCII molecules to T cells, 3) upregulate expression of MHCII and costimulatory molecules, and 4) migrate to T cell rich areas of lymphoid organs and activate T cells. These events are best understood from the study of DCs activated by TLR-ligands, which stimulate the breadth of events classically associated with DC activation and maturation. It is the ability to activate DCs through TLRs that imbues extracts or molecules isolated from pathogens with adjuvanticity, the ability to strongly induce primary immune responses. In certain cases, dominant antigenicity (the property of being the target of an immune response) and adjuvanticity are the properties of individual molecules (1), but adjuvants generally possess the ability to facilitate the induction of immune responses to molecules that are introduced to the immune system alongside them.
We and others have shown that SEA, a soluble extract of the egg life stage of the helminth parasite, Schistosoma mansoni, is a potent inducer of T cell responses, and does so independently of any added adjuvant (2). Indeed, SEA acts as an adjuvant itself, promoting T cell responses to unrelated antigens (3). In contrast to DCs activated by TLR ligands, which tend to promote Th1 or Th17 responses, DCs activated by SEA, or by other helminth-derived products, strongly promote Th2 responses (4, 5). The basis for SEA's adjuvanticity and antigenicity has remained unclear, since unlike DCs activated by TLR ligands (6-9), DCs exposed to SEA remain phenotypically immature, as assessed by costimulatory molecule expression and cytokine production. Thus, superficially, SEA-stimulated DCs appear immature, and would therefore be expected to induce tolerance rather than adaptive immunity.
Although TLR-induced expression of costimulatory molecules during DC maturation has been regarded as essential for effective antigen presentation, it has become clear that the failure of immature DCs to effectively prime naïve T cells may also reflect their inability to efficiently process antigen and thus generate antigenic peptide/MHCII complexes. TLR ligation in immature DCs leads to a process of phagosome maturation, whereby the lysosomal pathway is sufficiently acidified to allow the controlled degradation of newly endocytosed antigen into peptide fragments suitable for binding MHC class II molecules (10). Moreover, there is a requirement for TLR signaling for the selection of antigen for processing and presentation (11, 12). Thus, a model has emerged in which DC maturation is coupled to antigen processing, and as such, the ability of DCs to prime naïve T cells is regulated by the capacity of the stimulating antigen to induce a program of antigen processing and presentation (13). On the basis of these findings, we hypothesized that DC maturation could be uncoupled from antigen processing, and that SEA might promote the induction of a maturation program that permits antigen processing and presentation in the absence of induced cytokine production and costimulation molecule expression commonly associated with DC maturation. Therefore, we undertook studies to investigate antigen processing by SEA-DC, and thus, gain a better understanding of how SEA acts as such a potent Th2 inducing DC antigen/adjuvant. Our data demonstrate that a Th2-inducing pathogen-derived product, SEA, induces the maturation of DCs into cells with high proteolytic capacity, without concomitant phenotypic maturation.
Materials and Methods
Mice and reagents
Six- to 12 week old Balb/c, C57BL/6, CD40-/- and Trif-/- mice were from The Jackson Laboratory (Bar Harbor, ME), CBA mice were from Taconic (Hudson, NY), MyD88-/- mice were a gift from L.Turka and Y. Choi (University of Pennsylvania) and BALB/c 4get mice were a gift from M. Mohrs (Trudeau Institute, NY) and bred at the University of Pennsylvania. Balb/c DO.11.10 and C57BL/6 OTII mice were bred and maintained under specific pathogen-free conditions at the University of Pennsylvania. DC activation was induced by 100 ng/ml LPS (Escherichia coli serotype 0111:B4; Sigma-Aldrich, St. Louis, MO), 1 μg/ml CpG (MWG Biotech, High Point, NC), or 10 μg/ml Poly(I:C) (Sigma-Aldrich). SEA was prepared aseptically, as previously described (4), and used at 50 μg/ml. Aseptically collected and sterile-filtered (0.2-μm pore size) hen egg white (which is ∼50% OVA) was used as OVA to stimulate DO.11.10 and OTII cells (14); this antigen was assumed to be endotoxin free; a conclusion supported by its inability to activate DCs (not shown). Horseradish peroxidase (HRP) (Sigma-Aldrich) was cleaned of contaminating LPS by Endobind column purification (Profos, Germany) and considered LPS free as demonstrated by LAL assay (Sigma) and failure to induce DC maturation (MHC class II and CD86 upregulation) and IL-12 production.
Bone marrow-derived dendritic cell generation
Bone marrow cells were aseptically collected from the femurs of mice and 1×105 cells/well were seeded into 48 well non-tissue culture treated plates (Corning). Cells were cultured for 6 days in RPMI 1640 (Sigma) supplemented with 5% heat-inactivated and filtered FCS (Hyclone), 2 mM L-glutamine (Life Technologies, Gathersburg, MD), 100 U/ml penicillin plus 100 μg/ml streptomycin (Life Technologies), and 50 μM 2-ME (Sigma), in the presence of 10 ng/ml GM-CSF (PeproTech, Rocky Hill, NJ). At day 3, 250 μl of culture supernatant was removed and replaced with 250 μl of fresh supplemented medium containing 10 ng/ml GM-CSF.
DC/T cell co-cultures
DCs were grown in the same conditions are described above, with the exception of being seeded at 200 μl of 2×106 cells/ml in round bottomed 98 well plates. On day 6, DCs were pulsed with 300 μg/ml OVA for 1 hr prior to extensive washing to remove excess OVA, before culture for 18 hrs in the presence or absence of SEA (50 μg/ml) or LPS (100 ng/ml) in final volume of 100 μl. DCs were then cultured with 2×105 CFSE (Molecular Probes) -labeled DO.11.10 CD4+ T cells, purified by negative selection using MACS (Mitenyi Biotech), in complete T cell medium in a final volume of 200 μl. After 3 days, cells were restimulated with PMA (80 ng/ml), ionomycin (800 ng/ml; Sigma-Aldrich) and brefeldin A (10 μg/ml; BD Pharmingen) for 4 hrs and prepared for intracellular staining by Cytofix/Cytoperm kit according to the manufacturers instructions (BD Pharmingen). Cells were then stained for IL-4, IFNγ and IL-2 using specific Abs (BD Pharmingen), prior to acquisition.
Antigen processing assay
Day 6 DCs were pulsed with 250 ng/ml LPS-depleted HRP for 1hr at 37°C. Cells were then extensively washed with warm medium to remove any excess HRP. If required, DCs were then incubated with anti-CD40 (1μg/ml: Alexis biochemicals), p38 inhibitors SB203580, SB202190, SB220025 and PD169316 (10 μM; Calbiochem), or the ERK inhibitor U0126 (10 μM; Calbiochem), which inhibits MEK-1 and MEK-2, which are direct upstream activators of ERK, for 1hr, prior to stimulation with either SEA (50 μg/ml), LPS (100 ng/ml), CpG (1 μg/ml) or PolyI:C (10 μg/ml; Amersham) for 19hrs. For the no chase time point, DCs were harvested before addition of stimuli. In order to determine the level of HRP activity in DCs at “no chase” and 19 hrs, cells were first washed with warm PBS and lysed with lysis buffer (1% TritonX-100, 20 mM HEPES, 10% glycerol and 150 mM NaCl) for 30 min on ice. A standard curve of HRP was prepared and the concentration of intact HRP within DC lysates was calculated by adding TMB substrate and subsequent reading at 450 nm. In order to determine the maturation of DCs, cells and supernatants were harvested at 19hr instead and assessed for cytokine production by ELISA and surface phenotype by flow cytometry.
Characterization of DC cytokine production and surface phenotype
Cytokine ELISAs for IL-12p40 production were performed using paired mAb in combination with recombinant cytokine standard (BD Pharmingen), as described (4). Expression of MHCII and CD86 was quantified using specific Abs purchased from BD Pharmingen.
Western Blotting
Cells were solubilized with lysis buffer (0.1% Triton X-100, 20 mM HEPES, pH 7.5, 10% glyercol, 150 mM NaCl, and 1 mM DTT), supplemented with 1mM sodium orthovanadate, 10mM sodium fluoride and protease inhibitor cocktail (Sigma-Aldrich). Equivalent amounts of cellular extracts prepared in 4× NuPage sample buffer (Invitrogen Life Technologies, Carlsbad, CA) were resolved on Bis-Tris NuPage gels (Invitrogen Life Technologies) and transferred to Immobilon membrane (Millipore, Billerica, MA). Blots were incubated with primary Ab against phosphorylated p38 and ERK1/2 (Cell Signaling Technology, Beverly, MA), HRP (Jackson ImmunoResearch Laboratories), detected using secondary HRP-conjugated Ab (Cell Signaling Technology) and visualized by the ECL system (Amersham Pharmacia Biotech, Piscataway, NJ).
Staining of lysosomes
Day 6 DCs were stimulated with SEA or LPS for 1hr, prior to the addition of 10 uM lysosensor blue (Molecular probes) for 1hr at 37°C. Cells were then harvested, washed with PBS, mounted onto Superfrost/Plus microscope slides (Fisher) and photographed using a Leica DMIRB microscope and a DC500 camera (Leica).
Immunizations
Wild type and Trif-/- DCs were pulsed o/n without or with SEA (50 μg/ml), washed and then injected i.p. into naïve mice (5 × 105 per mouse in 0.5 ml of PBS). One week later, splenocytes were recovered and cultured in vivo with or without SEA (50 μg/ml) exactly as described previously (4, 5). Three days later culture supernatants were recovered and cytokine levels were measured using mAb-based ELISAs and reagents from BD Pharmingen.
Statistical Analysis
Student's t test was used to calculate the significance of differences between means.
Results
SEA acts on DCs as an adjuvant to induce the development of Th2 cells from naive CD4+ T cell in vitro
Previous reports have shown that SEA can act as an adjuvant to promote Th2 responses both in vivo and in vitro. We sought to quantitate this effect using Ova-specific DO.11.10 cells activated by DCs that had been pulsed with Ova in the presence or absence of SEA. We found that DCs pulsed with Ova and SEA promoted a robust DO.11.10 CD4+ T cell proliferative response, equivalent in some experiments to that seen with TLR-activated (Fig. 1). The adjuvanticity of SEA resulted in a Th2-biased response, which was in contrast to the Th1 response induced by TLR-ligand matured DCs (Fig. 1, and data not shown). These data confirm and expand on previous findings (4, 5, 15, 16).
Figure 1.
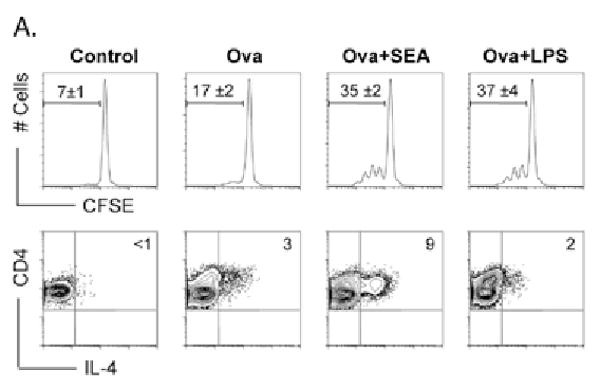
SEA from S. mansoni eggs demonstrate potent Th2 adjuvanticity in vitro. DCs were pulsed with ova for 1hr prior to washing and subsequent maturation with either SEA or LPS overnight. DCs were then co-cultured with CFSE labeled naïve CD4+ purified DO.11.10 T cells. After 3 days, cells were harvested by assessed for proliferation, activation and Th2 development by intracellular IL-4 staining A. SEA-DC induces T proliferation and acquisition of a Th2 phenotype as demonstrated by increased IL-4 staining. Data are mean ± SEM from at least 3 individual experiments.
SEA induces antigen degradation that is uncoupled from DC maturation
Studies of DCs stimulated through TLRs have indicated that for antigen to be processed for presentation, a maturation signal is required. Previous studies examining the induction of cytokine production and increased costimulatory molecule expression, rather than antigen degradation, suggest that SEA does not induce DC maturation (4, 17). However. SEA is clearly recognized by DCs, since beads coated with SEA are more rapidly and efficiently internalized than are beads coated with ovalbumin (a foreign protein from a non-pathogen-source), or LPS, a TLR-ligand (data not shown). Here we ask specifically whether incubation of immature DCs with SEA results in an increased ability to process internalized antigen. To this end, we pulsed DCs with LPS-depleted horseradish peroxidase for 1 hr at 37°C, washed, and then chased with HRP-free medium in the presence of SEA or LPS. After 19 hr, processing was assessed by measuring HRP enzymatic activity in cell extracts, and comparing loss of activity in stimulated versus unstimulated cells, as previously described (10) (Fig. 2A). We found that approximately 50% of the HRP initially internalized by DCs was lost following LPS stimulation, whereas nearly all of the HRP remained accountable for in DCs that received no microbial stimuli (Fig. 2B). Strikingly, HRP levels in DCs stimulated with SEA were comparable to those in LPS-stimulated DCs (Fig. 2B). Degradation did not occur in cells maintained at 4°C overnight (Fig. 2C). The direct addition of SEA to HRP in the extracellular environment, at either neutral or acidic pH, did not result in loss of HRP activity (data not shown). SEA- and LPS-induced degradation of HRP was evident as the appearance of a major degradation product at approximately 28 kDa on Western blots of HRP-pulsed DCs (Fig. 2D). In contrast, immature DCs demonstrated little proteolytic potential. Importantly, unlike the case for DCs stimulated with LPS, all of the observed HRP degradation in SEA-DCs occurred in the absence of upregulation of MHC class II and CD86 (Fig. 2E) or production of IL-12p40 (Fig. 2F), confirming that neither HRP itself nor SEA was inducing TLR-like DC activation. Thus, SEA induces antigen proteolysis but this process is uncoupled from changes in DCs that are conventionally associated with maturation.
Figure 2.
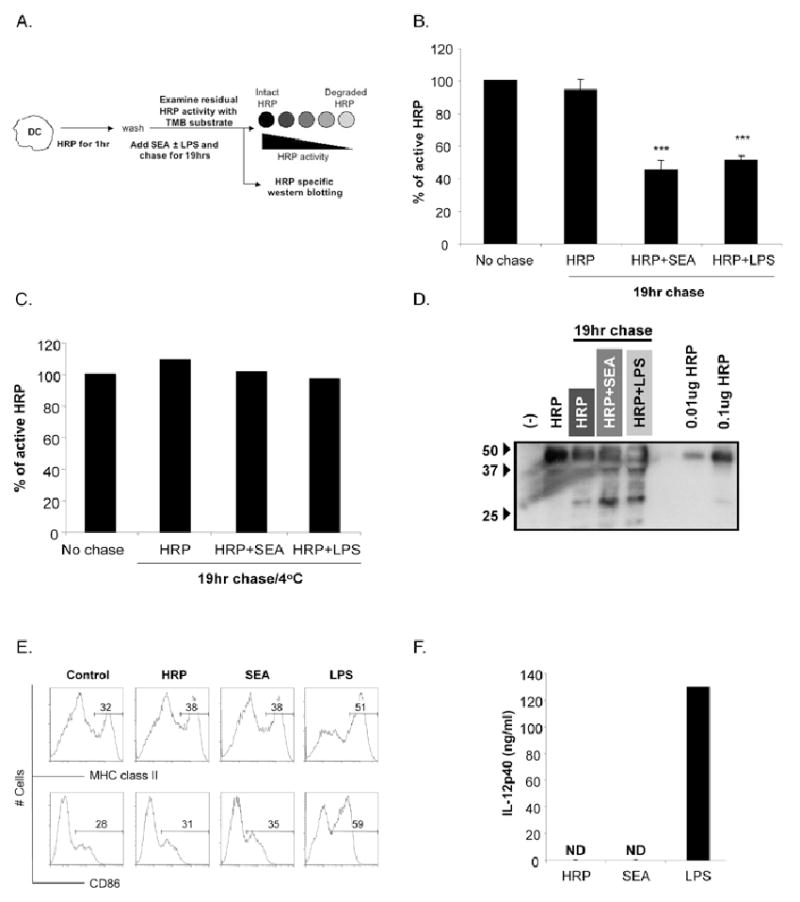
Activation of immature DCs with SEA induces antigen processing in the absence of phenotypic maturation. A. Protocol for assaying antigen processing in DCs following pulsing with HRP. DCs were pulsed with LPS-depleted HRP for 1hr prior to extensive washing to remove excess extracellular HRP. Cells were then either harvested to calculate initial (no chase) activity of HRP, or chased in the presence of SEA or LPS for 19hrs before being harvested to determine intact HRP activity (19hr chase). The percentage of intact HRP remaining after 19hrs was calculated by comparing the level of HRP activity in immature (HRP only), SEA or LPS cells after 19hr chase to initial no chase HRP activity. B. Percentage of HRP activity remaining in immature DCs (HRP), SEA and LPS DCs after 19hrs chase. C. Percentage of HRP activity remaining in immature DCs (HRP), SEA and LPS DCs after 19hrs chase at 4°C. D. After 19hr chase, extracts from HRP-pulsed-immature, SEA and LPS activated DCs were electrophoretically separated, blotted and probed with Ab specific for HRP. Cells were pulsed with HRP for 1hr as described above and subsequently cultured for 19hrs in the presence of medium (HRP), SEA or LPS. Cells and supernatant were harvested after 19hrs and analyzed for surface expression of MHC class II and CD86 and IL-12p40 by ELISA. Significant differences by Student's t test (*** p<0.05), where data points represent mean value ± SEM of data from seven experiments.
Antigen proteolysis is linked to a drop in the intralysomal pH from ∼5.6 in resting DCs to ∼4.6 following TLR-ligation (10), which approximates the pH-optima of lysosomal proteases and thus facilitates efficient proteolysis of lysosomal contents (18). Therefore, we investigated whether the activation of DCs with SEA promoted a drop in lysosomal pH that would be consistent with observed changes in antigen degradation. Lysosome acidification was assessed with fluorescent microscopy using lysosensor probes. These probes are acidotrophic agents that accumulate in acidic organelles as the result of protonation, which leads to a pH dependent increase in fluorescent intensity. The data show that compared to immature DCs (pH∼5.6), in which little lysosensor staining can be observed, DCs activated with SEA demonstrated increased intralysosomal acidification to similar levels detected in LPS activation DCs (pH∼4.6) (Fig. 3). Thus, activation of DCs with SEA leads to phagosome maturation and a decrease in lysosomal pH, which has previously been shown to be an essential requirement for DC-mediated antigen degradation (10).
Figure 3.
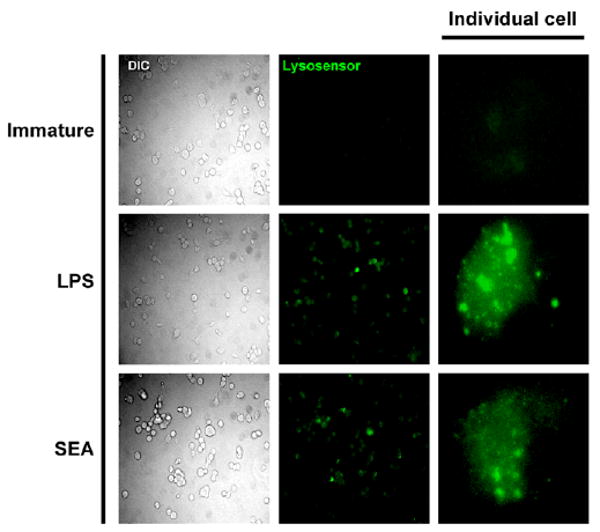
Activation of immature DCs with SEA results in lysosomal acidification. A. Immature day 6 DCs were stimulated with either medium alone (immature), LPS of SEA for 1 hr prior to the addition of lysosensor blue. Cells were then cultured in the presence of lysosensor dye for an additional 1hr before harvesting and visualization of lysosensor fluorescence by fluorescent microscopy. Immature DCs demonstrate little lysosensor fluorescence, whereas the presence of acidification of lysosomes in both SEA and LPS activated cells is indicated by positive lysosensor staining. Each image is representative of >20 individual cells from 3 or more different field of view. The study was repeated twice with similar results.
CD40 signaling regulates the ability of SEA-DC to present antigen
CD40 signaling in DCs has been shown to play an important role in the induction of Th2 immune responses in vivo (5, 19). Since SEA-activated DCs are characterized by an immature phenotype (4), which is believed to be insufficient for naïve CD4+ T cell activation (20-23), we hypothesized that CD40 ligation by CD154 following the initial interaction between SEA-DC and CD4+ T cells may provide the necessary additional signal to upregulate costimulatory molecule and MHC class II expression to allow T cell priming. Therefore, we directly investigated whether CD40 stimulation modulates the phenotype of SEA-DCs by culturing SEA or TLR-activated DCs in the presence of agonistic anti-CD40 Abs. Consistent with previous reports (4, 24), CD40 signaling in LPS or CpG activated DCs resulted in augmented production of IL-12p40, whereas SEA-activated DCs failed to make IL-12p40 with or without CD40 stimulation (Fig. 4A). We next examined whether CD40 stimulation of SEA-DCs augmented their ability to process and present antigen by investigating antigen degradation and MHC class II and CD86 expression. We found modest effects of CD40 stimulation on the ability of SEA activated DCs to further process antigen (Fig. 4B), and to increase expression of MHCII and CD86 (Fig. 4C). In contrast, these parameters were unaffected by CD40 stimulation in TLR-activated DCs, which appeared to be maximally activated. To determine the functional significance of these CD40 stimulated changes for the ability of DCs to activate naïve CD4 T cells, DCs generated from WT or CD40-/- mice were pulsed with Ova for 1 hr prior, plus or minus SEA or a TLR-ligand for a further 24 hr, and then co-cultured with CFSE labeled DO.11.10 CD4+ T cells. As expected, DCs pulsed with Ova plus SEA were more capable of activating naïve CD4+ T cells than were DCs pulsed with SEA alone (Fig. 4D). However, the adjuvant effect of SEA was highly CD40-dependent, since CD40-/- DCs pulsed with Ova and SEA failed to induce T cell proliferation (Fig. 4D). In contrast, the ability of TLR-activated DCs to induce T cell proliferation was only marginally affected by the lack of CD40 (Fig. 4D). Therefore, CD40 signaling plays a role in positively regulating the ability of SEA-activated DCs to present antigen, in the absence of proinflammatory cytokine production.
Figure 4.
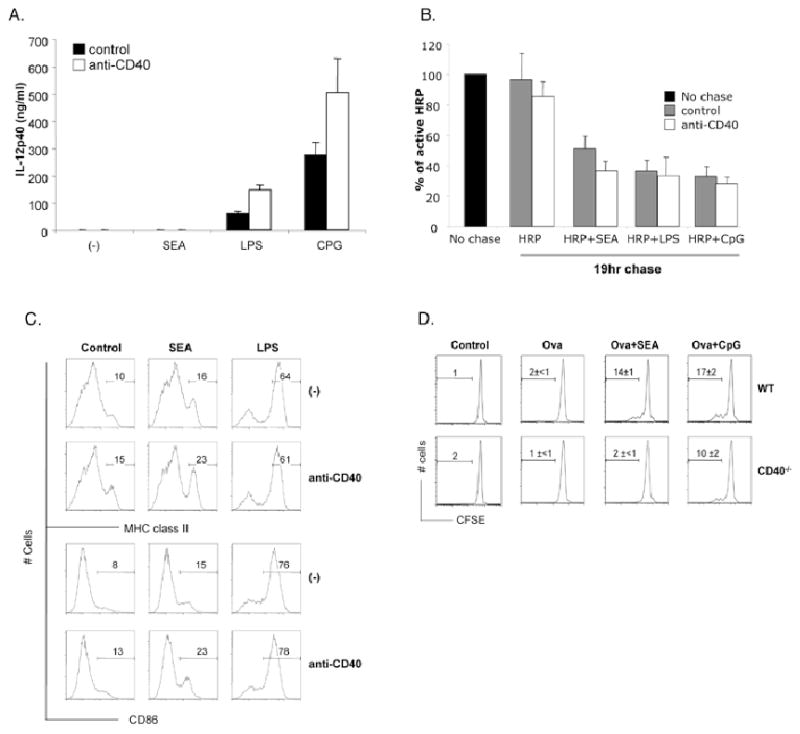
CD40 signaling in SEA DCs augments their capacity as APCs. Day six DCs were stimulated with medium alone (-), SEA, LPS or CpG with or without addition of agonistic anti-CD40 Ab for 19hrs. Cells and supernatant were then harvested and assessed for A. IL-12p40 production by ELISA. B. Antigen processing capacity as previously described. C. Surface expression of MHC class II and CD86. D. Day six WT or CD40-/- DCs were pulsed with ovalbumin protein for 1hr prior to extensive washing to remove extracellular protein. Cells were then stimulated for 19hr with SEA before being co-cultured with CFSE labeled naïve CD4+ purified DO.11.10 T cells. After 3 days, cells were harvested by assessed for proliferation by CFSE dilution. Data are mean ± SEM from at least 3 individual experiments
SEA-induced antigen processing occurs in the absence of MyD88 and Trif signaling
Recent studies have demonstrated the importance of MyD88 signaling for the generation of antigenic peptides from antigen containing TLR-ligands (11). However there is evidence to suggest that the antigenicity and adjuvanticity of SEA are TLR-independent (16). To explore directly whether SEA-induced antigen processing is MyD88-independent we generated immature DCs from MyD88-/- mice and examined antigen processing after differential maturation with TLR-ligands or SEA. As anticipated the level of HRP degraded by MyD88-/- DCs in response to SEA was comparable to that of WT DCs following activation with SEA (Fig. 5A). Thus, the ability of SEA to activate DCs and induce a pathway of antigen processing occurs in a MyD88-independent manner. Stimulation of DCs with CpG was only able to induce processing of HRP and costimulatory molecule upregulation (Fig. 5A and data not shown) in WT DCs, confirming not only the known essential role of MyD88 for CpG-induced costimulatory molecule upregulation in DCs, but that MyD88 is also essential for CpG induced antigen processing.
Figure 5.
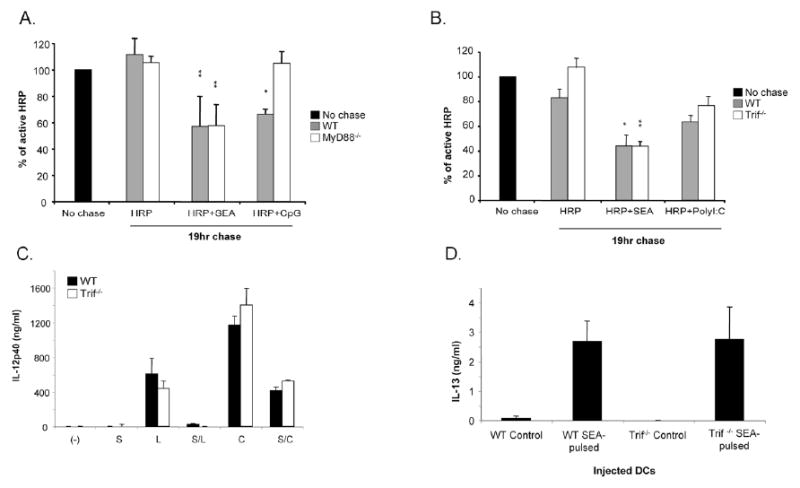
SEA induced antigen processing occurs independently of MyD88 and Trif. DCs were generated from MyD88-/- and Trif-/- mice and grown for 6 days. Cells were then pulsed with HRP as previously described prior to 19hrs chase in the presence of either medium alone, SEA, CpG or PolyI:C. The levels of intact HRP remaining after 19hr was assessed allowing the antigen processing capacity of A. MyD88-/- DCs and B. Trif-/- DCs to be determined. Significant differences by Student's t test (* p<0.05, ** p<0.01), where data points represent mean value ± SD of data from three experiments. C. The role of Trif was investigated further by examining the ability of SEA to inhibit LPS or CpG induced IL-12p40 production in its absence. WT and Trif-/- DCs were pulsed with LPS (L) or CpG (C) alone or in combination with SEA (S), or with SEA alone, and 24hr later IL-12p40 levels in culture supernatants we measured by ELISA. The suppression of TLRligand induced cytokine production by SEA is in each case significant by Student's t test (p<0.05). D. Trif-/- DCs were tested for their ability to induce Th2 responses. Splenocytes from mice immunized as indicated in the X-axis were restimulated with SEA, and levels of IL-13, as an indicator of Th2 response development, were measured by ELISA. Control DCs were not pulsed with SEA. Data points represent mean values from 3 individual mice/group ± SD.
Some reports have implicated TLR3 in the host response to schistosome egg antigen (25). TLR3-induced signaling is MyD88-independent and Trif-dependent (26, 27). To explore the possibility that SEA induced antigen degradation is Trif-dependent, we characterized the antigen-processing capacity of DCs derived from Trif-/- mice. Our data failed to reveal a role for Trif in this process (Fig. 5B). For these experiments we used the TLR3 ligand PolyI:C as a positive control, but this activation signal did not strongly induce processing even in WT mice. To further examine the role of Trif in SEA-mediated processes we examined the ability of SEA to suppress TLR-induced IL-12 production in Trif-/- DCs (a characteristic property of SEA that correlates with its ability to promote Th2 responses (15, 17); Fig. 5C), and the ability of Trif-/- DCs to induce SEA-specific Th2 responses in vivo (Fig. 5D). Our findings revealed no differences between WT and Trif-/- DCs in either of these regards, indicating that major immunologic effects of SEA are Trif-independent.
Together, these data suggest that SEA activates an antigen processing program in DCs independently of TLR signaling.
SEA-induced antigen processing requires p38 signaling
In a search for a molecular mediator of SEA-induced antigen processing we re-assessed previous data from the laboratory (17) and noted that p38 is phosphorylated following exposure of DCs to this helminth extract. This is intriguing since p38 regulates key aspects of antigen processing, including endocytotic transport and phagosome maturation (12, 28, 29). To confirm the effect of SEA on p38, we stimulated DCs with SEA or the TLR-ligand LPS and at times thereafter harvested cells for Western blot analysis of p38 phosphorylation. We were able to detect phosphorylated p38 at 10 and 30 minutes following SEA exposure (Fig. 6A). In comparison, LPS induced stronger phosphorylation of p38 that was still evident at 1 h (Fig. 6A). We next assessed whether p38 plays a role in SEA-induced antigen-processing. DCs were loaded with HRP and washed, but prior to a 19 hr chase in the presence of SEA or LPS, the cells were pre-incubated with a panel of p38 inhibitors (SB203580, SB202190 or PD169316) for 1 hr. All inhibitors were used at 10 μM and did not alter cell viability (data not shown). In these experiments, immature DCs contained approximately 60-70% of the HRP that they had internalized 19 hr earlier (Fig. 6B). As expected, SEA induced an increase in antigen processing (Fig. 6B), but this effect was significantly diminished by the inhibition of p38 (Fig. 6B,C). In these experiments, baseline antigen degradation in the absence of SEA was also diminished by the presence of p38 inhibitors (data not shown), indicating a fundamental role for this kinase in antigen processing. Previous work has indicated that SEA also induces ERK activation in DCs (17). We confirmed this result here, but found that inhibition of this event had no effect on SEA-induced antigen processing (Supplementary Fig. 1).
Figure 6.
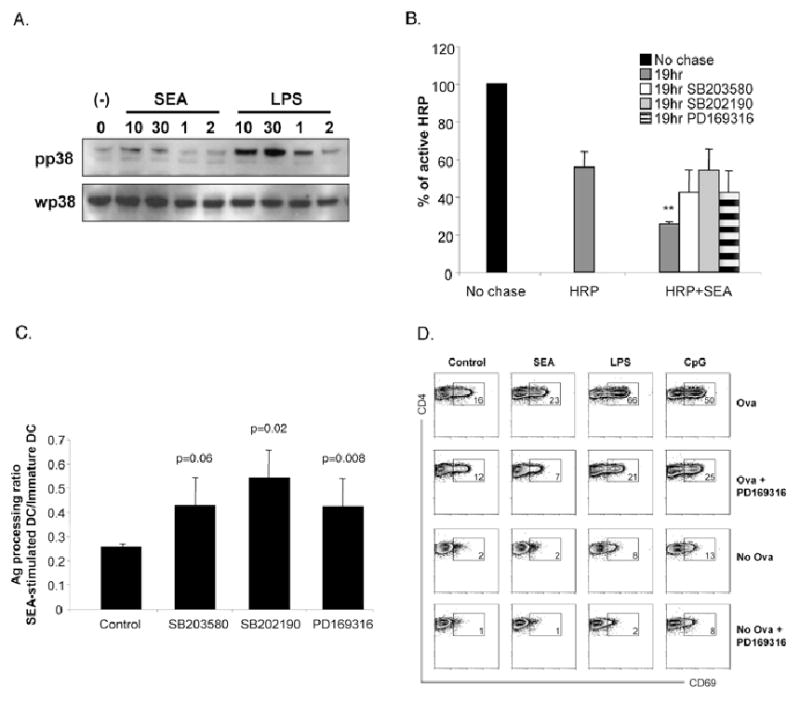
SEA induced processing occurs in a p38-dependent manner. A. Extracts from DCs pulsed for 10min, 30min, 1hr or 2hr with SEA or LPS were electrophoretically separated, blotted, probed with Ab specific for phosphorylated p38 and to serve as a loading control, reprobed with Ab specific for unphosphorylated p38. B. In order to determine if p38 signaling was required for SEA-mediated antigen-processing, DCs were pulsed with HRP for 1hr, washed and cultured in the presence of p38 specific inhibitors SB203580, SB202190 or PD169316 for 1hr prior to activation with SEA. After 19hrs, DCs were harvested as described earlier for assessment of antigen processing capacity. C. In order control for effects of inhibitors on basal antigen -processing levels, the ratio between the percentage of active HRP remaining in SEA-activated and immature-DCs was calculated for each inhibitor treated group. Significant differences by Student's t test (** p<0.01), where data points represent mean value ± SD of data from three experiments. D. The effects of p38 inhibition on the ability of SEA to induce antigen processing and presentation by DCs was assessed by inhibiting Ova-pulsed DCs with the p38-specific inhibitor PD 169316 for 1hr prior to overnight stimulation with SEA or TLR ligands, LPS or CpG. DCs were then co-cultured with CFSE labeled naïve CD4+ purified DO.11.10 T cells. After 3 days, cells were harvested by assessed for proliferation by CFSE. Data are representative of 2 individual experiments.
Finally, we examined whether the adjuvanticity of SEA is p38-dependent. For these experiments, we pulsed DCs with Ova for 1 hr prior to the addition of the PD 169316, which is likely to be the most specific of the pharmacologic inhibitors of p38 activity (14). One hour later, DCs were stimulated with SEA and incubated overnight, washed and co-cultured with DO.11.10 CD4+ T cells for 6 hr, after which CD69 expression as an indicator of T cell activation, was measured. We focused on CD69 expression since we were concerned that longer term assays to monitor T cell activation (such as the measurement of proliferation, for example), would allow the effects of the p38 inhibitor to wear off. As expected, CD4 T cells activated by SEA-conditioned DCs upregulated expression of CD69 expression to a point that was intermediate between that measured on T cells activated by control Ova pulsed DCs or DCs pulsed with ligands plus Ova (Fig. 6D). When p38 signaling was inhibited, SEA-induced antigen processing and presentation was attenuated by greater than 3-fold (Fig. 6D); p38 inhibition also affected antigen presentation by immature and TLR-activated DCs (Fig. 6D). Similar results were observed with other p38 inhibitors SB 203580 and SB 202190 (data not shown). Collectively, our data suggest that SEA induces a p38 dependent signal that promotes antigen-processing and presentation by DCs.
Discussion
Schistosome eggs, and SEA, an extract from them, are strongly antigenic, inducing notably Th2-polarized responses in the absence of adjuvant. Our previous work has shown that DCs exposed to SEA retain an immature phenotype and fail to produce inflammatory cytokines (4, 15, 17). DCs with this phenotype are generally considered to be tolerogenic (30), yet SEA-pulsed DCs induce SEA-specific Th2 responses when injected into mice (4). These findings indicate to us that: 1) to induce Th2 responses in the absence of adjuvant, SEA needs to interact directly only with DCs, and not with other cells in the immune system; 2) the interaction between SEA and DCs is likely to lead to changes in DC biology that facilitate productive antigen presentation. In considering the latter, we undertook studies to directly assess whether SEA, like TLR ligands (10), induces proteolytic processing of antigen within DCs. We show that exposure to SEA induces DCs to process antigen via a MyD88/Trif-indendent but p38-dependent pathway. As a result, SEA-conditioned antigen -pulsed DCs are better at activating naïve CD4 T cells than are DCs pulsed with antigen alone. The uncoupling of SEA-induced antigen processing from conventional DC maturation results in an environment that lacks cytokine promoters of Th1 or Th17 development, and partially as a result of this, we believe, a Th2 response emerges. This process relies on CD40/CD0L interactions, which we postulate play an important role in providing additional activation signals to DCs that are not provided by SEA. Finally, we show that coating beads with SEA enhances their ability to be taken up by DCs, suggesting that part of the inherent antigenicity of SEA might be linked to its preferential recognition by these important antigen -presenting cells.
SEA consists of a mixture of glyco-conjugates (31), which can be recognized by C-type lectins including DC-SIGN and the macrophage galactose-type lectin (MGL) (32, 33). A recent report has shown that recognition of SEA by these lectins on human DCs leads to targeting of SEA into MHC class II+ LAMP+ compartments (34). Consequently, C-type lectin-mediated recognition of SEA by DCs has the potential to promote efficient internalization and delivery of SEA to a compartment where antigen processing and generation of antigenic peptides suitable for MHCII loading would be favored. Targeting of exogenous antigen to endocytic receptors such as C-type lectins has been shown to lead to more efficient generation of MHC II:peptide complexes and activation of antigen -specific CD4+ T cells (35-38). The ligation of a C-type lectin by SEA fits with the fact that SEA, like C-type lectin-initiated signaling, can inhibit TLR-induced DC activation (17, 39, 40).
We found that CD40 signaling in SEA pulsed DCs results in modestly increased antigen processing, but more significantly, increased expression of MHC class II and CD86 in the absence of proinflammatory cytokine production. The finding that SEA-stimulated DCs require CD40 to induce T cell proliferation in vitro indicates that CD40 signaling plays an essential role in inducing full antigen -presentation abilities in these cells. Our data are consistent with previous reports the cross-linking CD40 on DC results in increased expression of peptide:MHC class II complexes and enhances T cell priming (41). Moreover, CD40-mediated interactions have been shown to be important for upregulation of costimulatory molecule expression in human monocytes and DCs (42, 43), and studies in CD40-/- and CD154-/- deficient mice have shown that this pathway is important for initiating signaling cascades required for effective T cell priming (44). Our previous findings that CD40-/- DCs fail to initiate SEA-specific Th2 responses (5, 19), imply that SEA-pulsed DCs need additional CD40-mediated signaling to present sufficient antigen for optimum T cell priming and Th2 development. Similarly, defective Th2 development has been demonstrated in other systems where CD40 signaling is deficient (45-47) whereas for promotion of Th1 immune responses, CD40-mediated interactions are redundant (48, 49).
Recently, studies by Blander and Medzhitov have proposed that TLRs allow DCs to decipher the information encoded by microbial pathogens, and thus initialize a program of phagosome intrinsic maturation that selects these antigen for effective presentation to T cells (11, 12). This differs from SEA-mediated activation of antigen-processing in DCs, since our data indicate that SEA influences antigen-processing in a more global manner. A possible reason for this discrepancy may be inherent to the nature of the stimulating antigen being examined. The phagosome intrinsic pathway of antigen -processing was demonstrated by using particulate antigen (whole bacteria or TLR-coated latex beads) which are internalized via phagocytosis, whereas, soluble antigen, such as SEA, are internalized via endocytic pathways. Therefore, it is feasible that in contrast to phagocytosis, receptors that mediate alternative modes of internalization, such as endocytosis, may have the ability to signal from the cell surface in a way that modulates the characteristics of other intracellular compartments. For example, DC-SIGN has been show to mediate signals from the cell surface that are involved in the transport of internalized HIV particles to infectious synapses in DCs. Importantly, this occurred in the absence of co-localization between HIV and DC-SIGN, and suggests that DC-SIGN might act as a sensor on the cell surface which can modulate intracellular compartments (50). Consistent with this, previous studies have already demonstrated that C-type lectins (DC-SIGN and Dectin-1) have the potential to modulate intracellular signals within DCs (51, 52). Therefore, we suggest that SEA, signaling via C-type lectins, or another as yet unidentified receptor, could potentially signal from the cell surface to enhance the proteolytic capacity of intracellular vesicles that need not necessarily contain SEA.
Using MyD88-/- and Trif-/- DCs, we found that that SEA activates a TLR-independent pathway of inducible antigen processing in DCs. The TLR-independent nature of SEA's inherent antigenicity is confirmed by the finding that both MyD88-/- and Trif-/- -SEA-pulsed DCs are capable of promoting SEA-specific Th2 immune responses in vivo (data not shown). Numerous studies have demonstrated that p38 signaling can regulate aspects of the endocytic system, and thus highlight the potential for p38-dependent regulation of DC antigen processing. For example, signaling via p38 has been shown to modulate endocytic traffic by regulating the activity of guanyl-nucleotide dissociation inhibitor (GDI) on Rab proteins (28). We found that SEA-mediated antigen processing and presentation are significantly attenuated by specific inhibition of p38 (but not ERK – Supplementary Fig. 1) signaling. Thus, SEA induces a TLR-independent, p38 dependent pathway of inducible processing in DCs. Potentially, activation of p38 signaling by SEA may lead to phosphorylation of GDI, which converts exhausted GDP-bound Rab5 to active GTP-Rab5, promoting increased fusion between endocytic vesicles and eventual delivery of antigen to degradative compartments. Consistent with this possibility, in macrophages TLR-induced fusion between phagosomes and lysosomes and eventual antigen degradation is impaired following inhibition of p38 signaling (12). There is precedent for C-type lectins initiating p38 signaling in specific situations (53, 54), Thus, C-type lectin-mediated recognition of SEA could potentially induce a p38-dependent program in which antigen-processing is uncoupled from cytokine production and other events normally associated with maturation.
The data presented here demonstrate that SEA, a product derived from the helminth parasite S. mansoni, induces an unconventional type of DC activation in which antigen-processing is uncoupled from conventional maturation. Understanding the mechanisms that allow DCs to present antigen without initiating inflammation may provide opportunities for developing new defined adjuvants for clinical use.
Supplementary Material
Supplementary Figure 1. SEA induced processing occurs in an ERK-independent manner. A. Extracts from DCs pulsed for 10min, 30min, 1hr or 2hr with SEA or LPS were electrophoretically separated, blotted, probed with Ab specific for phosphorylated ERK and to serve as a loading control, reprobed with Ab specific for unphosphorylated ERK. B. In order to determine if ERK signaling was required for SEA-mediated antigen-processing, DCs were pulsed with HRP for 1hr, washed and cultured in the presence of an ERK inhibitor, UO126 for 1hr prior to activation with SEA. After 19hrs, DCs were harvested for assessment of antigen processing capacity.
Acknowledgments
We thank C.J. Krawczyk for insightful comments and E. Jung for excellent technical assistance.
This work was supported by the National Institutes of Health (AI53825 to EJP).
References
- 1.Yarovinsky F, Kanzler H, Hieny S, Coffman RL, Sher A. Toll-like receptor recognition regulates immunodominance in an antimicrobial CD4+ T cell response. Immunity. 2006;25:655–664. doi: 10.1016/j.immuni.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 2.Pearce EJ. Priming of the immune response by schistosome eggs. Parasite Immunol. 2005;27:265–270. doi: 10.1111/j.1365-3024.2005.00765.x. [DOI] [PubMed] [Google Scholar]
- 3.Okano M, Satoskar AR, Nishizaki K, Abe M, Harn DA., Jr Induction of Th2 responses and IgE is largely due to carbohydrates functioning as adjuvants on Schistosoma mansoni egg antigens. J Immunol. 1999;163:6712–6717. [PubMed] [Google Scholar]
- 4.MacDonald AS, Straw AD, Bauman B, Pearce EJ. CD8-dendritic cell activation status plays an integral role in influencing Th2 response development. J Immunol. 2001;167:1982–1988. doi: 10.4049/jimmunol.167.4.1982. [DOI] [PubMed] [Google Scholar]
- 5.MacDonald AS, Straw AD, Dalton NM, Pearce EJ. Cutting edge: Th2 response induction by dendritic cells: a role for CD40. J Immunol. 2002;168:537–540. doi: 10.4049/jimmunol.168.2.537. [DOI] [PubMed] [Google Scholar]
- 6.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 7.Tsuji S, Matsumoto M, Takeuchi O, Akira S, Azuma I, Hayashi A, Toyoshima K, Seya T. Maturation of human dendritic cells by cell wall skeleton of Mycobacterium bovis bacillus Calmette-Guerin: involvement of toll-like receptors. Infect Immun. 2000;68:6883–6890. doi: 10.1128/iai.68.12.6883-6890.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hertz C, Kiertscher S, Godowski P, Bouis D, Norgard M, Roth M, Modlin R. Microbial lipopeptides stimulate dendritic cell maturation via toll-like receptor 2. J Immunol. 2001;166:2444–2450. doi: 10.4049/jimmunol.166.4.2444. [DOI] [PubMed] [Google Scholar]
- 9.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 10.Trombetta ES, Ebersold M, Garrett W, Pypaert M, Mellman I. Activation of lysosomal function during dendritic cell maturation. Science. 2003;299:1400–1403. doi: 10.1126/science.1080106. [DOI] [PubMed] [Google Scholar]
- 11.Blander JM, Medzhitov R. Toll-dependent selection of microbial antigens for presentation by dendritic cells. Nature. 2006;440:808–812. doi: 10.1038/nature04596. [DOI] [PubMed] [Google Scholar]
- 12.Blander JM, Medzhitov R. Regulation of phagosome maturation by signals from toll-like receptors. Science. 2004;304:1014–1018. doi: 10.1126/science.1096158. [DOI] [PubMed] [Google Scholar]
- 13.Blander JM. Signalling and phagocytosis in the orchestration of host defence. Cell Microbiol. 2007;9:290–299. doi: 10.1111/j.1462-5822.2006.00864.x. [DOI] [PubMed] [Google Scholar]
- 14.Khaled AR, Moor AN, Li A, Kim K, Ferris DK, Muegge K, Fisher RJ, Fliegel L, Durum SK. Trophic factor withdrawal: p38 mitogen-activated protein kinase activates NHE1, which induces intracellular alkalinization. Mol Cell Biol. 2001;21:7545–7557. doi: 10.1128/MCB.21.22.7545-7557.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cervi L, MacDonald AS, Kane C, Dzierszinski F, Pearce EJ. Cutting edge: dendritic cells copulsed with microbial and helminth antigens undergo modified maturation, segregate the antigens to distinct intracellular compartments, and concurrently induce microbe-specific Th1 and helminth-specific Th2 responses. J Immunol. 2004;172:2016–2020. doi: 10.4049/jimmunol.172.4.2016. [DOI] [PubMed] [Google Scholar]
- 16.Jankovic D, Kullberg MC, Caspar P, Sher A. Parasite-induced Th2 polarization is associated with down-regulated dendritic cell responsiveness to Th1 stimuli and a transient delay in T lymphocyte cycling. J Immunol. 2004;173:2419–2427. doi: 10.4049/jimmunol.173.4.2419. [DOI] [PubMed] [Google Scholar]
- 17.Kane CM, Cervi L, Sun J, McKee AS, Masek KS, Shapira S, Hunter CA, Pearce EJ. Helminth antigens modulate TLR-initiated dendritic cell activation. J Immunol. 2004;173:7454–7461. doi: 10.4049/jimmunol.173.12.7454. [DOI] [PubMed] [Google Scholar]
- 18.De Duve C, Wattiaux R. Functions of lysosomes. Annu Rev Physiol. 1966;28:435–492. doi: 10.1146/annurev.ph.28.030166.002251. [DOI] [PubMed] [Google Scholar]
- 19.MacDonald AS, Patton EA, La Flamme AC, Araujo MI, Huxtable CR, Bauman B, Pearce EJ. Impaired Th2 development and increased mortality during Schistosoma mansoni infection in the absence of CD40/CD154 interaction. J Immunol. 2002;168:4643–4649. doi: 10.4049/jimmunol.168.9.4643. [DOI] [PubMed] [Google Scholar]
- 20.Schuler G, Steinman RM. Murine epidermal Langerhans cells mature into potent immunostimulatory dendritic cells in vitro. J Exp Med. 1985;161:526–546. doi: 10.1084/jem.161.3.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gimmi CD, Freeman GJ, Gribben JG, Gray G, Nadler LM. Human T-cell clonal anergy is induced by antigen presentation in the absence of B7 costimulation. Proc Natl Acad Sci U S A. 1993;90:6586–6590. doi: 10.1073/pnas.90.14.6586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schwartz RH. Models of T cell anergy: is there a common molecular mechanism? J Exp Med. 1996;184:1–8. doi: 10.1084/jem.184.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Steinman RM, Hawiger D, Liu K, Bonifaz L, Bonnyay D, Mahnke K, Iyoda T, Ravetch J, Dhodapkar M, Inaba K, Nussenzweig M. Dendritic cell function in vivo during the steady state: a role in peripheral tolerance. Ann N Y Acad Sci. 2003;987:15–25. doi: 10.1111/j.1749-6632.2003.tb06029.x. [DOI] [PubMed] [Google Scholar]
- 24.Schulz O, Edwards AD, Schito M, Aliberti J, Manickasingham S, Sher A, Reis e Sousa C. CD40 triggering of heterodimeric IL-12 p70 production by dendritic cells in vivo requires a microbial priming signal. Immunity. 2000;13:453–462. doi: 10.1016/s1074-7613(00)00045-5. [DOI] [PubMed] [Google Scholar]
- 25.Aksoy E, Zouain CS, Vanhoutte F, Fontaine J, Pavelka N, Thieblemont N, Willems F, Ricciardi-Castagnoli P, Goldman M, Capron M, Ryffel B, Trottein F. Double-stranded RNAs from the helminth parasite Schistosoma activate TLR3 in dendritic cells. J Biol Chem. 2005;280:277–283. doi: 10.1074/jbc.M411223200. [DOI] [PubMed] [Google Scholar]
- 26.Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301:640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 27.Jiang Z, Mak TW, Sen G, Li X. Toll-like receptor 3-mediated activation of NF-kappaB and IRF3 diverges at Toll-IL-1 receptor domain-containing adapter inducing IFN-beta. Proc Natl Acad Sci U S A. 2004;101:3533–3538. doi: 10.1073/pnas.0308496101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cavalli V, Vilbois F, Corti M, Marcote MJ, Tamura K, Karin M, Arkinstall S, Gruenberg J. The stress-induced MAP kinase p38 regulates endocytic trafficking via the GDI:Rab5 complex. Mol Cell. 2001;7:421–432. doi: 10.1016/s1097-2765(01)00189-7. [DOI] [PubMed] [Google Scholar]
- 29.West MA, Wallin RP, Matthews SP, Svensson HG, Zaru R, Ljunggren HG, Prescott AR, Watts C. Enhanced dendritic cell antigen capture via toll-like receptor-induced actin remodeling. Science. 2004;305:1153–1157. doi: 10.1126/science.1099153. [DOI] [PubMed] [Google Scholar]
- 30.Steinman RM, Nussenzweig MC. Avoiding horror autotoxicus: the importance of dendritic cells in peripheral T cell tolerance. Proc Natl Acad Sci U S A. 2002;99:351–358. doi: 10.1073/pnas.231606698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hokke CH, Yazdanbakhsh M. Schistosome glycans and innate immunity. Parasite Immunol. 2005;27:257–264. doi: 10.1111/j.1365-3024.2005.00781.x. [DOI] [PubMed] [Google Scholar]
- 32.van Die I, van Vliet SJ, Nyame AK, Cummings RD, Bank CM, Appelmelk B, Geijtenbeek TB, van Kooyk Y. The dendritic cell-specific C-type lectin DC-SIGN is a receptor for Schistosoma mansoni egg antigens and recognizes the glycan antigen Lewis x. Glycobiology. 2003;13:471–478. doi: 10.1093/glycob/cwg052. [DOI] [PubMed] [Google Scholar]
- 33.van Vliet SJ, van Liempt E, Saeland E, Aarnoudse CA, Appelmelk B, Irimura T, Geijtenbeek TB, Blixt O, Alvarez R, van Die I, van Kooyk Y. Carbohydrate profiling reveals a distinctive role for the C-type lectin MGL in the recognition of helminth parasites and tumor antigens by dendritic cells. Int Immunol. 2005;17:661–669. doi: 10.1093/intimm/dxh246. [DOI] [PubMed] [Google Scholar]
- 34.van Liempt E, van Vliet SJ, Engering A, Garcia Vallejo JJ, Bank CM, Sanchez-Hernandez M, van Kooyk Y, van Die I. Schistosoma mansoni soluble egg antigens are internalized by human dendritic cells through multiple C-type lectins and suppress TLR-induced dendritic cell activation. Mol Immunol. 2007;44:2605–2615. doi: 10.1016/j.molimm.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 35.Engering AJ, Cella M, Fluitsma D, Brockhaus M, Hoefsmit EC, Lanzavecchia A, Pieters J. The mannose receptor functions as a high capacity and broad specificity antigen receptor in human dendritic cells. Eur J Immunol. 1997;27:2417–2425. doi: 10.1002/eji.1830270941. [DOI] [PubMed] [Google Scholar]
- 36.Tan MC, Mommaas AM, Drijfhout JW, Jordens R, Onderwater JJ, Verwoerd D, Mulder AA, van der Heiden AN, Scheidegger D, Oomen LC, Ottenhoff TH, Tulp A, Neefjes JJ, Koning F. Mannose receptor-mediated uptake of antigens strongly enhances HLA class II-restricted antigen presentation by cultured dendritic cells. Eur J Immunol. 1997;27:2426–2435. doi: 10.1002/eji.1830270942. [DOI] [PubMed] [Google Scholar]
- 37.Mahnke K, Guo M, Lee S, Sepulveda H, Swain SL, Nussenzweig M, Steinman RM. The dendritic cell receptor for endocytosis, DEC-205, can recycle and enhance antigen presentation via major histocompatibility complex class II-positive lysosomal compartments. J Cell Biol. 2000;151:673–684. doi: 10.1083/jcb.151.3.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Engering A, Geijtenbeek TB, van Vliet SJ, Wijers M, van Liempt E, Demaurex N, Lanzavecchia A, Fransen J, Figdor CG, Piguet V, van Kooyk Y. The dendritic cell-specific adhesion receptor DC-SIGN internalizes antigen for presentation to T cells. J Immunol. 2002;168:2118–2126. doi: 10.4049/jimmunol.168.5.2118. [DOI] [PubMed] [Google Scholar]
- 39.Chieppa M, Bianchi G, Doni A, Del Prete A, Sironi M, Laskarin G, Monti P, Piemonti L, Biondi A, Mantovani A, Introna M, Allavena P. Cross-linking of the mannose receptor on monocyte-derived dendritic cells activates an anti-inflammatory immunosuppressive program. J Immunol. 2003;171:4552–4560. doi: 10.4049/jimmunol.171.9.4552. [DOI] [PubMed] [Google Scholar]
- 40.Bergman MP, Engering A, Smits HH, van Vliet SJ, van Bodegraven AA, Wirth HP, Kapsenberg ML, Vandenbroucke-Grauls CM, van Kooyk Y, Appelmelk BJ. Helicobacter pylori modulates the T helper cell 1/T helper cell 2 balance through phase-variable interaction between lipopolysaccharide and DC-SIGN. J Exp Med. 2004;200:979–990. doi: 10.1084/jem.20041061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Manickasingham S, Reis e Sousa C. Microbial and T cell-derived stimuli regulate antigen presentation by dendritic cells in vivo. J Immunol. 2000;165:5027–5034. doi: 10.4049/jimmunol.165.9.5027. [DOI] [PubMed] [Google Scholar]
- 42.Peguet-Navarro J, Dalbiez-Gauthier C, Rattis FM, Van Kooten C, Banchereau J, Schmitt D. Functional expression of CD40 antigen on human epidermal Langerhans cells. J Immunol. 1995;155:4241–4247. [PubMed] [Google Scholar]
- 43.Cella M, Scheidegger D, Palmer-Lehmann K, Lane P, Lanzavecchia A, Alber G. Ligation of CD40 on dendritic cells triggers production of high levels of interleukin-12 and enhances T cell stimulatory capacity: T-T help via APC activation. J Exp Med. 1996;184:747–752. doi: 10.1084/jem.184.2.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grewal IS, Xu J, Flavell RA. Impairment of antigen-specific T-cell priming in mice lacking CD40 ligand. Nature. 1995;378:617–620. doi: 10.1038/378617a0. [DOI] [PubMed] [Google Scholar]
- 45.Poudrier J, van Essen D, Morales-Alcelay S, Leanderson T, Bergthorsdottir S, Gray D. CD40 ligand signals optimize T helper cell cytokine production: role in Th2 development and induction of germinal centers. Eur J Immunol. 1998;28:3371–3383. doi: 10.1002/(SICI)1521-4141(199810)28:10<3371::AID-IMMU3371>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 46.Nierkens S, van Helden P, Bol M, Bleumink R, van Kooten P, Ramdien-Murli S, Boon L, Pieters R. Selective requirement for CD40-CD154 in drug-induced type 1 versus type 2 responses to trinitrophenyl-ovalbumin. J Immunol. 2002;168:3747–3754. doi: 10.4049/jimmunol.168.8.3747. [DOI] [PubMed] [Google Scholar]
- 47.Khan WI, Motomura Y, Blennerhassett PA, Kanbayashi H, Varghese AK, El-Sharkawy RT, Gauldie J, Collins SM. Disruption of CD40-CD40 ligand pathway inhibits the development of intestinal muscle hypercontractility and protective immunity in nematode infection. Am J Physiol Gastrointest Liver Physiol. 2005;288:G15–22. doi: 10.1152/ajpgi.00159.2004. [DOI] [PubMed] [Google Scholar]
- 48.Campos-Neto A, Ovendale P, Bement T, Koppi TA, Fanslow WC, Rossi MA, Alderson MR. CD40 ligand is not essential for the development of cell-mediated immunity and resistance to Mycobacterium tuberculosis. J Immunol. 1998;160:2037–2041. [PubMed] [Google Scholar]
- 49.Reis e Sousa C, Hieny S, Scharton-Kersten T, Jankovic D, Charest H, Germain RN, Sher A. In vivo microbial stimulation induces rapid CD40 ligand-independent production of interleukin 12 by dendritic cells and their redistribution to T cell areas. J Exp Med. 1997;186:1819–1829. doi: 10.1084/jem.186.11.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arrighi JF, Pion M, Garcia E, Escola JM, van Kooyk Y, Geijtenbeek TB, Piguet V. DC-SIGN-mediated infectious synapse formation enhances X4 HIV-1 transmission from dendritic cells to T cells. J Exp Med. 2004;200:1279–1288. doi: 10.1084/jem.20041356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Geijtenbeek TB, Van Vliet SJ, Koppel EA, Sanchez-Hernandez M, Vandenbroucke-Grauls CM, Appelmelk B, Van Kooyk Y. Mycobacteria target DC-SIGN to suppress dendritic cell function. J Exp Med. 2003;197:7–17. doi: 10.1084/jem.20021229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gantner BN, Simmons RM, Canavera SJ, Akira S, Underhill DM. Collaborative induction of inflammatory responses by dectin-1 and Toll-like receptor 2. J Exp Med. 2003;197:1107–1117. doi: 10.1084/jem.20021787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Robinson MJ, Sancho D, Slack EC, LeibundGut-Landmann S, Reis e Sousa C. Myeloid C-type lectins in innate immunity. Nat Immunol. 2006;7:1258–1265. doi: 10.1038/ni1417. [DOI] [PubMed] [Google Scholar]
- 54.Chen CH, Floyd H, Olson NE, Magaletti D, Li C, Draves K, Clark EA. Dendritic-cell-associated C-type lectin 2 (DCAL-2) alters dendritic-cell maturation and cytokine production. Blood. 2006;107:1459–1467. doi: 10.1182/blood-2005-08-3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. SEA induced processing occurs in an ERK-independent manner. A. Extracts from DCs pulsed for 10min, 30min, 1hr or 2hr with SEA or LPS were electrophoretically separated, blotted, probed with Ab specific for phosphorylated ERK and to serve as a loading control, reprobed with Ab specific for unphosphorylated ERK. B. In order to determine if ERK signaling was required for SEA-mediated antigen-processing, DCs were pulsed with HRP for 1hr, washed and cultured in the presence of an ERK inhibitor, UO126 for 1hr prior to activation with SEA. After 19hrs, DCs were harvested for assessment of antigen processing capacity.