

Abstract
Background
As severe alcohol intoxication impairs memory function, a high concentration of ethanol (60 mM) acutely inhibits long-term potentiation (LTP), a cellular model of learning and memory, in rat hippocampal slices. Neurosteroids are involved in this LTP inhibition. We recently reported that the inhibitory effects of 60 mM ethanol are blocked by 4-methylpyrazole (4MP), an inhibitor of alcohol dehydrogenase, suggesting that acetaldehyde locally generated within the hippocampus participates in LTP inhibition.
Aim
We investigated whether acetaldehyde generated by ethanol metabolism contributes to neurosteroidogenesis and LTP inhibition.
Results
Like 60 mM ethanol, we found that exogenous acetaldehyde enhanced neurosteroid immunostaining in CA1 pyramidal neurons, and that augmented neurosteroid immunostaining by high ethanol alone was blocked by 4MP but not by inhibitors of other ethanol metabolism pathways. The inhibitory effects of 60 mM ethanol on LTP were mimicked by a lower concentration of ethanol (20 mM) plus acetaldehyde (60 μM), although neither agent alone was effective at these concentrations, suggesting that 60 mM ethanol inhibits LTP via multiple actions, one of which involves acetaldehyde and the other of which requires only 20 mM ethanol. The effects of ethanol and acetaldehyde on neurosteroid staining and LTP were overcome by inhibition of neurosteroid synthesis and by blockade of N-methyl-D-aspartate receptors (NMDARs).
Conclusion
These observations indicate that acetaldehyde generated by local ethanol metabolism within the hippocampus serves as a signal for neurosteroid synthesis in pyramidal neurons, and participates in the synaptic dysfunction associated with severe alcohol intoxication.
Keywords: Class III ADH, allopregnanolone, binge drinking, blackout, finasteride, hangover
INTRODUCTION
Severe alcohol intoxication can result in periods of dense anterograde amnesia, referred to as memory “blackouts” 1. Alcohol is thought to induce this amnesia by inhibiting long-term potentiation (LTP), a form of synaptic plasticity associated with memory processing2, and in the CA1 region of rat hippocampal slices, administration of 60 mM ethanol acutely inhibits LTP induction3–5. Because ethanol-induced neurosteroidogenesis in hippocampal CA1 pyramidal neurons6 contributes to LTP inhibition4,5, it appears that GABAergic neurosteroids are involved in blackouts. The acute effects of ethanol on neurosteroid synthesis and LTP inhibition require high concentrations with no effect at 20 mM ethanol, but full effects at 60 mM5. Interestingly, 20 mM ethanol attenuates LTP when administered with the neurosteroid, allopregnanolone (alloP)4. Thus, we hypothesize that ethanol has dual actions on LTP: one effect occurs at lower concentrations and the other requires high concentrations that exceed the threshold for neurosteroid synthesis. We further hypothesize that accumulation of ethanol metabolites may contribute to neurosteroidogenesis and represents an effect of ethanol that involves high concentrations.
Consistent with this, we recently found that 4-methylpyrazole (4MP), an inhibitor of alcohol dehydrogenase (ADH), overcomes the inhibitory effects of 60 mM ethanol on LTP induction in hippocampal slices, indicating that local metabolism of ethanol to acetaldehyde contributes to LTP inhibition7. The subtype of ADH responsible for the effects of 4MP is uncertain, although ADH3, a member of the ADH family8,9 with low ethanol affinity10, is expressed in hippocampal neurons11,12, suggesting a possible role in the region. Acetaldehyde may also be generated de novo in the CNS by other mechanisms including catalase and the cytochrome P450 system, and, in cultured astrocytes and rat brain homogenates, accumulates in the presence of ethanol13–15.
In this study, we found that in the presence of exogenous acetaldehyde even 20 mM ethanol inhibits LTP induction, although neither agent alone altered LTP at the concentrations used. Thus, in the presence of acetaldehyde, a low concentration of ethanol is sufficient to prevent synaptic plasticity. Coupled with the effects of 4MP noted above, these findings support the idea that locally generated acetaldehyde is one contributor to the ability of high concentrations of ethanol to inhibit LTP. Based on the role that GABAergic neurosteroids play in ethanol-mediated LTP inhibition, we sought to test the hypothesis that metabolism of ethanol to acetaldehyde contributes to local neurosteroidogenesis in the hippocampus.
MATERIALS AND METHODS
Animals
Male Sprague-Dawley albino rats were used for all studies16. Protocols for animal use were approved by the Washington University Animal Studies Committee in accordance with the NIH guidelines for care and use of laboratory animals.
Immunohistochemistry
Immunostaining was performed using antiserum against 5α-reduced neurosteroids This antibody primarily recognizes alloP and has been characterized previously for use in tissue sections16,17. In hippocampal slices, neurosteroid immunoreactivity is markedly diminished by exogenous alloP and completely blocked by the 5α-reductase inhibitor, finasteride16.
Immunostaining methods are described in detail elsewhere16. Briefly, 500 μm hippocampal slices were prepared from postnatal day 30–32 male rats under isoflurane anesthesia. After dissection, slices were cut from the septal hippocampus with a rotary slicer in artificial cerebrospinal fluid (ACSF) containing (in mM): 124 NaCl, 5 KCl, 2 MgSO4, 2CaCl2, 1.25 NaH2PO4, 22 NaHCO3, 10 glucose, bubbled with 95% O2 / 5% CO2 at 4–6 °C.
After recovering for at least one hour at 30oC, slices used for immunohistochemistry were initially screened by electrophysiology to verify robust physiological responses to synaptic stimulation5,16 and were incubated with various reagents in separate 10 ml beakers. Following drug treatment, slices were fixed in 4% paraformaldehyde in phosphate buffered saline (PBS) for 30 min then washed with PBS and incubated in blocking solution (1% donkey serum/PBS) for 2 h at 25ºC. Slices were incubated with an antibody raised in sheep against 5α-reduced neurosteroids diluted 1:2500 in 1% donkey serum/PBS for 48h at 4ºC then rinsed with PBS and incubated with secondary antibody for 2 h at 25ºC. Alexa Flour 488 donkey anti-sheep IgG (diluted 1:500) was used to visualize neurosteroids.
Confocal images were obtained using a 60X oil-immersion objective (1.4 N.A.), a C1 laser scanning confocal microscope with a 488 nm laser and Z-C1 software (Nikon Instruments, Melville, NY). All parameters (1021 × 1024 resolution, Z-stack step size 0.40 μm, 1.92 μs dwell time, small pinhole) were kept constant within an experiment. Digital images were analyzed and the average intensity of the tissue was calculated from three consecutive planes at the same depth (40–60 μm) from the sample surface within an experiment using MetaMorph software (Universal Imaging Corporation, Downingtown, PA). Similar quantification has been performed by other investigators18–20.
Electrophysiological recording in vitro
For electrophysiology, hippocampal slices were transferred to a submerged recording chamber with continuous bath perfusion of ACSF at 2 ml/min at 30°C. Extracellular recordings were obtained from the apical dendritic layer of the CA1 region elicited with 0.1 ms constant current pulses through a bipolar stimulating electrode placed in stratum radiatum. During an experiment, excitatory postsynaptic potentials (EPSPs) were monitored using a half-maximal stimulus based on a baseline input-output curve. After establishing a stable baseline, LTP was induced by applying a single 100 Hz × 1 s high frequency stimulus (HFS) using the same intensity stimulus as used for monitoring. An input-output curve was repeated 60 min following HFS for statistical comparisons of changes in EPSP slopes at half-maximal intensity. Signals were digitized and analyzed using the PCLAMP software (Axon Instruments, Union City, CA). All chemicals were used at concentrations that did not suppress baseline EPSPs. Because 30 min administration of 0.3 mM sodium azide suppressed EPSPs (49.8 ± 3.8%, n = 3), we used this agent at 0.1 mM.
Materials
Finasteride and neurosteroids were obtained from Steraloids (Newport, RI). D-APV was purchased from Tocris (St. Louis, MO). All other chemicals were purchased from Sigma (St. Louis, MO). Picrotoxin (PTX) was dissolved in ethanol as a 10 mM stock solution. One μM PTX in ethanol sustains EPSPs but augments the second population spike evoked by paired stimuli at an interval of 21 msec. The antibody against 5α-reduced neurosteroids was purchased from Dr. Robert Purdy, University of California-San Diego. Alexa Flour 488 was purchased from Invitrogen (Carlsbad, CA).
Data analysis and statistics
All data are expressed as mean ± s.e.m with baseline responses normalized to 100%. For multiple comparisons, analysis of variance followed by post hoc Holm-Sidak test was employed. For comparisons between two groups Student’s t-test was used. P-values of less than 0.05 were considered statistically significant.
RESULTS
Acetaldehyde and neurosteroidogenesis
To determine whether acetaldehyde mimics the effects of ethanol on neurosteroidogenesis, we examined neurosteroid immunostaining in the CA1 region using a previously characterized antibody against alloP and other 5α-reduced neurosteroids16,17. Acetaldehyde increased neurosteroid staining in pyramidal neurons in a concentration-dependent fashion, with significant effects following 15 min administration of 60 μM (277.1 ± 36.8% vs. control, n = 5, *P < 0.001), but not 6 μM acetaldehyde (100.6 ± 6.1% vs. control, n = 5). At 20 μM, acetaldehyde had marginal effects on immunostaining (129.4 ± 5.3% vs. control, n = 5) (Fig. 1a,b). The enhanced staining by 60 μM acetaldehyde was blocked completely by 1 μM finasteride (100.5 ± 3.6% vs. control, n = 5), a 5α-reductase inhibitor that blocks neurosteroid synthesis, and by 50 μM D-2-amino-5-phosphonovalerate (D-APV) (103.1 ± 3.3%, n = 5) (Fig. 1c,d), a competitive NMDAR antagonist.
Figure 1.

Acetaldehyde enhances neurosteroidogenesis via activation of NMDARs and 5α-reductase. A, Immunostaining against 5α-reduced neurosteroids is observed in cell bodies of CA1 pyramidal neurons in naïve hippocampal slices (left panel). The neurosteroid staining is enhanced by 15 min incubation with 60 μM acetaldehyde (second) but not by 20 μM or 6 μM acetaldehyde (third and fourth panels). B, Summary of immunostaining studies shows fluorescence intensity (arbitrary units) as mean ± s.e.m. C, Immunostaining against 5α-reduced neurosteroids in the CA1 area (left) is not enhanced by 20 mM ethanol alone (second panel). The enhancement by 60 μM acetaldehyde (third panel) is blocked by 1μM finasteride (fourth panel) and 50 μM D-APV (right panel). D, Summary of immunostaining studies shows fluorescence intensity (arbitrary units) as mean ± s.e.m. Calibration bar: 25 μm. P-values are calculated by Holm-Sidek post-hoc test. n = 5. * P < 0.001.
Because the ADH inhibitor, 4 MP, overcomes the effects of 60 mM ethanol on LTP induction16, we hypothesized that ethanol enhances immunostaining via local metabolism to acetaldehyde. Indeed, the enhanced neurosteroid staining by 60 mM ethanol was completely attenuated by 1 mM 4MP (105.4 ± 3.3% vs. control, n = 5, P < 0.001), although 4MP alone had no effect on baseline neurosteroid levels (Fig. 2).
Figure 2.

Involvement of ADH in neurosteroidogenesis. A, Immunostaining against 5α-reduced neurosteroids in the CA1 area (left panel) is enhanced by 60 mM ethanol (second panel). 4MP (1 mM), an ADH inhibitor, blocks the effects of ethanol (third) but does not alter basal staining (fourth panel). Calibration bar: 25 μm. B, Summary of immunohistological studies shows fluorescence intensity as mean ± s.e.m (n = 5). P value was calculated by Holm-Sidak post-hoc method. * P < 0.001.
Based on the proposed role of catalase in brain ethanol metabolism21,22, we examined whether catalase inhibition blocks the enhancement of immunostaining by 60 mM ethanol. In the presence of 100 μM sodium azide, a catalase inhibitor23, 60 mM ethanol still enhanced neurosteroid immunostaining in the CA1 region (289.6 ± 29.5%, n = 5) (Fig. 3).
Figure 3.
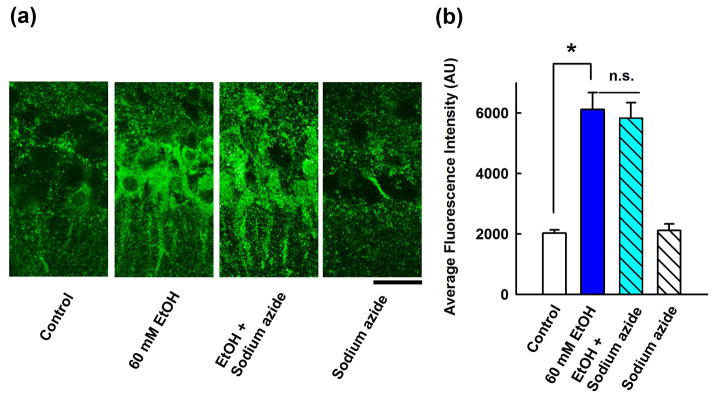
Effects of a catalase inhibitor on neurosteroidogenesis. A, Immunostaining against 5α-reduced neurosteroids in the CA1 area (left panel) is enhanced by 60 mM ethanol (second). Sodium azide has no effect on ethanol (third) and does not alter basal staining (fourth panel). Calibration bar: 25 μm. B, Summary of immunohistological studies with sodium azide shows fluorescence intensity as mean ± s.e.m (n = 5). P value was calculated by Holm-Sidak post-hoc method. * P < 0.001.
Ethanol can also be metabolized in brain by the cytochrome P450 enzyme, CYP2E121,24. Similar to sodium azide, we found that 3 mM allyl sulfide, an inhibitor of CYP2E122, failed to block the enhancement of neurosteroid immunostaining produced by 60 mM ethanol (295.0 ± 29.4% vs. control, n = 5) (Fig. 4).
Figure 4.
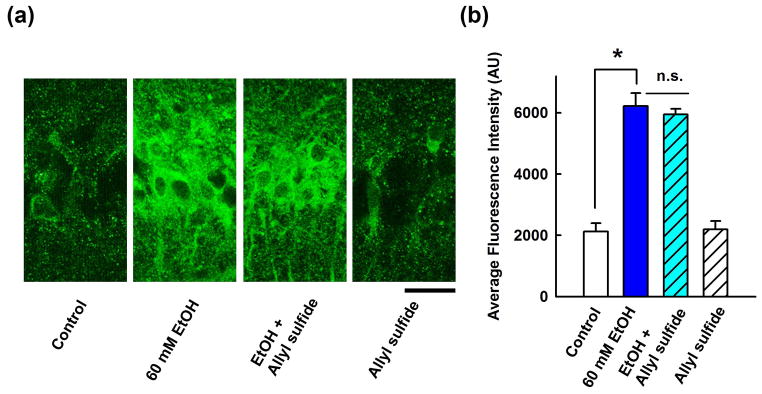
Effects of a CYP2E1 inhibitor on neurosteroidogenesis. A, Immunostaining against 5α-reduced neurosteroids in the CA1 area (left) is enhanced by 60 mM ethanol (second panel) and ethanol’s effects are not blocked by allyl sulfide, a CYP2E1 inhibitor (third panel). Allyl sulfide alone had no effect on basal staining (fourth panel). Calibration bar: 25 μm. B, Summary of immunohistological studies with allyl sulfide shows fluorescence intensity as mean ± s.e.m.
Effects of acetaldehyde on LTP
As previously reported4,5, LTP in the CA1 region of rat hippocampal slices was blocked acutely by 60 mM but not by 20 mM ethanol administered for 15 min prior to and during a single 100 Hz × 1 s HFS (EPSP change; 153.6 ± 3.0% in control slices, n = 6, 144.2 ± 5.2% at 20 mM ethanol, n = 5, 102.1 ± 2.5% at 60 mM ethanol, n = 6, measured 60 min after HFS compared to baseline with pre-HFS EPSPs normalized to 100%; P < 0.01 at 60 mM vs. control LTP). Because alcohol metabolizing enzymes are found in the hippocampus12, we hypothesized that high levels of ethanol might be converted to acetaldehyde and that acetaldehyde would then contribute to ethanol’s actions. To test this, we first examined the effects of acetaldehyde on LTP alone and in combination with ethanol. LTP was dose-dependently inhibited by acetaldehyde administered for 15 min prior to high frequency stimulation (HFS) (EPSP change; 150.5 ± 3.6%, n=6, 116.5 ± 1.3%, n = 5, 111.1 ± 7.8%, n = 5, 108.5 ± 5.4%, n = 5, of baseline at 60 μM, 600 μM, 1 mM and 6 mM acetaldehyde, respectively; P < 0.01 at 600 μM and above vs. control LTP) (Fig. 5a). These results indicate that acetaldehyde alone alters LTP only at very high concentrations that are unlikely to be achieved in vivo.
Figure 5.

Effects of acetaldehyde and ethanol on LTP induction. A, In control slices, LTP is readily induced (green triangles) by a single 100 Hz × 1 s high frequency stimulation (HFS, arrow). Fifteen min administration of acetaldehyde (AcH, hatched bar) at 0.6 mM (white diamonds), 1 mM (white squares) or 6 mM (white triangles) inhibits LTP. At 60 μM, acetaldehyde did not alter LTP induction (white circles). B, LTP is not blocked by 20 mM ethanol (EtOH) (blue bar, blue circles), but LTP is blocked by a combination of 60 μM acetaldehyde (white bar) plus 20 mM ethanol (aqua squares). White circles show effects of 60 μM acetaldehyde alone for comparison. C, The inhibition of LTP by 20 mM ethanol plus 60 μM acetaldehyde is overcome by continuous administration of 1 μM picrotoxin (PTX). Traces depict EPSPs before (dashed lines) and 60 min after HFS (solid lines). Solid lines in color match symbols in the graphs. Scale; 1mV, 5 msec.
Although 20 mM ethanol alone or 60 μM acetaldehyde alone did not inhibit LTP, the combination markedly dampened LTP induction (EPSP change: 107.5 ± 3.5% of baseline, n = 5; P < 0.01 vs. control) (Fig. 5b). A combination of 20 mM ethanol with 20 μM acetaldehyde also attenuated LTP (EPSP change: 127.4 ± 3.5% of baseline, n = 5; P < 0.01), though a combination of 20 mM ethanol plus 6 μM acetaldehyde had no significant effect (EPSP change: 139.8 ± 5.0% of baseline, n = 5).
We recently reported that inhibition of LTP by 60 mM ethanol is overcome by 1 mM 4MP7. This finding indicates that locally generated acetaldehyde resulting from the metabolism of high concentrations of ethanol plays a pivotal role in LTP inhibition. However in the present study, 1 mM 4 MP failed to promote LTP induction in the presence of 60 μM acetaldehyde plus 20 mM ethanol (EPSP slope; 95.9 ± 4.5 % of baseline, n = 5) (Fig. 5c), an observation that is consistent with 4MP acting as an ADH inhibitor.
We previously found that picrotoxin (PTX), a non-competitive GABAAR antagonist, also overcomes the effects of 60 mM ethanol on LTP induction3. Similarly, 1 μM PTX, a low concentration that does not alter LTP magnitude in control slices4 overcame the combination of 20 mM ethanol plus 60 μM acetaldehyde (EPSP slope: 145.2 ± 5.2% of baseline, n = 5; P < 0.01 compared to ethanol + acetaldehyde) (Fig. 5d), suggesting the involvement of GABAARs.
Although 60 mM ethanol is a partial NMDA receptor (NMDAR) antagonist3, its effects on LTP are prevented by complete NMDAR blockade during the time that slices are exposed to ethanol5. Consistent with this, complete NMDAR inhibition also blocks the effects of acetaldehyde plus ethanol. Similar to 60 mM ethanol alone3, LTP inhibition by 20 mM ethanol plus 60 μM acetaldehyde persisted for several hours following washout of these two agents (EPSP change following 3 successive HFSs delivered at 30 min intervals after washout: 103.3 ± 7.7% of baseline, measured 60 min after the third HFS; n = 3; P < 0.01)(Fig. 6a). In the following study, therefore, HFS was delivered after washout of APV with ethanol and acetaldehyde. Using a paradigm in which 20 μM acetaldehyde and 20 mM ethanol were administered in combination for 15 min and then washed out 30 min prior to HFS, we found that LTP was inhibited (EPSP change; 106.8 ± 1.9% of baseline, n = 5; P < 0.01 vs. control) (Fig. 6b). However, coadministration of 50 μM D-APV with ethanol plus acetaldehyde overcame the inhibitory effects on LTP (EPSP change: 156.5 ± 8.9% of baseline, n = 5; P < 0.01 compared to ethanol + acetaldehyde) (Fig. 6b). We have also shown that inhibitory effects of 60 mM ethanol on LTP are blocked by finasteride4,5. Consistent with this, 1 μM finasteride, a concentration that had no effect on LTP alone4,5,16 overcame the inhibitory effects of 20 mM ethanol plus 60 μM acetaldehyde (EPSP change: 153.8 ± 10.1% of baseline, n = 5; P < 0.01) (Fig. 6c).
Figure 6.

LTP inhibition by a combination of 20 mM ethanol and 60 μM acetaldehyde involves activation of NMDARs and 5α-reductase. A, LTP inhibition by 20 mM ethanol plus 60 μM acetaldehyde( blue and white bars) persists long following washout of these two agents. Three successive HFSs delivered at 30 min intervals after washout failed to induce LTP (aqua circles). B, Fifteen min administration of 20 mM ethanol plus 60 μM acetaldehyde inhibits LTP induction 30 min after washout (aqua circles). Co-administration of D-APV (50 μM, pink bar), with ethanol and AcH overcame LTP inhibition (purple diamonds). C, Thirty min administration of 1 μM finasteride (gray bar) also overcomes the inhibitory effects of 20 mM ethanol plus 60 μM acetaldehyde on LTP. Traces depict EPSPs before (dashed lines) and 60 min after HFS (solid lines). Solid lines in color match symbols in the graphs. Scale; 1mV, 5 msec.
Results of these LTP studies are summarized as histograms (Fig. 7).
Figure 7.

Summaries of LTP studies. A, LTP induction is dose-dependently inhibited by both ethanol and acetaldehyde. B, LTP induction in the presence of 20 mM ethanol is dese-dependently inhibited by acetaldehyde. LTP inhibition by 20 mM ethanol plus 60 μM acetaldehyde is overcome by 4MP, an ADH inhibitor, and PTX, a GABAR antagonist. C, HFS delivered 30 min after washout of 20 mM ethanol plus 60 μM acetaldehyde fails to induce LTP. APV, an NMDAR antagonist, administered with ethanol and acetaldehyde allows LTP induction in the same paradigm. * P < 0.01.
DISCUSSION
The recent finding that 4MP overcomes LTP inhibition by 60 mM ethanol7 raised a question whether 60 mM ethanol directly enhances neurosteroid synthesis or acts via acetaldehyde. In the present experiments, we examined whether the effects of high acute ethanol involve production of acetaldehyde locally in the hippocampus. Roles for acetaldehyde in ethanol’s effects have been observed previously but remain controversial21,22. Importantly, systemic acetaldehyde does not mimic ethanol’s effects on brain alloP levels22, suggesting that systematically produced acetaldehyde is unlikely to contribute to ethanol-induced neurosteroidogenesis in brain26,27. However, other evidence suggests a role for brain ethanol metabolism in modulating neuronal function13,14,23, raising the possibility that acetaldehyde generated locally within the brain may be involved in ethanol-induced neuronal changes. There are several classes of ADH for alcohol metabolism: Class I (containing α, β and γ isoenzymes) and II (π ADH) are mostly in the liver and Class IV (σ and μ ADH) is in the upper digestive tract, whereas Class III (χ ADH) are found in all tissues studied28, Mori et al. have shown that ADH3, a member of the ADH family8,9, is expressed in the hippocampus11, and although this ADH variant has low affinity for ethanol, recent studies suggest a role in alcohol metabolism10. Importantly, ADH3 is expressed in pyramidal neurons12, raising the possibility that high concentrations of ethanol will result in acetaldehyde production within pyramidal neurons. Thus, if acetaldehyde acts locally within brain regions where it is produced, then measurements of acetaldehyde levels in plasma or cerebrospinal fluid may not accurately reflect its contributions to synaptic or behavioral changes29. Because 20 mM ethanol alone does not enhance neurosteroid immunostaining5, neurosteroidogenesis appears to be induced acutely only by high levels of ethanol through accumulation of acetaldehyde from regional ethanol metabolism (Fig. 8-1). Again, the low affinity of Class III ADH3 for ethanol10, may explain why high levels of ethanol are required for neurosteroidogenesis and LTP inhibition.
Figure 8.

A Scheme shows possible mechanisms for ethanol to inhibit LTP. 1. Only High concentrations of ethanol are metabolized into acetaldehyde via group III ADH. However, ADH can be activated by other substrates. 2. Acetaldehyde activates NMDARs through unknown fashion. 3. Activation of NMDARs activates TSPO resulting in synthesis of neurosteroids including alloP. This process is blocked by Finasteride. 4. Neurosteroids activate GABARs. 5. Activation of GABARs synergetically inhibits LTP induction with other various stressful conditions. These conditions include ethanol exposure even if the concentrations are moderate.
In our present study, we found that exogenous acetaldehyde alone, at micromolar concentrations, enhances immunostaining against 5α-reduced steroids in CA1 pyramidal neurons in a finasteride-sensitive fashion. This facilitated staining is blocked by APV (current study) and mimicked by low concentrations of NMDA5, indicating that NMDAR activation contributes to acetaldehyde-mediated neurosteroidogenesis just as it does with high concentrations of ethanol (Fig. 8-2,3). We also found that enhanced neurosteroid immunostaining by 60 mM ethanol is blocked by 4MP, suggesting that generation of acetaldehyde in pyramidal neurons via ADH is a mediator of these actions.
Moderate concentrations of ethanol inhibit LTP when combined with micromolar acetaldehyde, though neither agent alone inhibits LTP at the effective concentrations. Similar to 60 mM ethanol, the effects of 20 mM ethanol plus 60 μM acetaldehyde on LTP in vitro persist for several hours after washout3 and are blocked by inhibitors of GABAARs, NMDARs and 5α reductase4,5(Fig. 8-2,3,4), suggesting that the postulated dual actions of 60 mM ethanol are mimicked by moderate ethanol plus lower micromolar acetaldehyde (Fig. 8-5). Our finding that acetaldehyde does not inhibit LTP at concentrations below 0.1 mM indicates that acetaldehyde alone is unlikely to be the sole mediator of impaired synaptic plasticity. However, a combination of 20 mM ethanol and 60 μM acetaldehyde persistently inhibits LTP (Fig. 5b), indicating that moderate levels of ethanol may impair cognitive function if acetaldehyde accumulates in the brain. Furthermore, it is possible that other factors that increase neurosteroid production can induce synaptic and cognitive dysfunction in the presence of moderate levels of ethanol. These other factors could include behavioral stressors or concomitant use of neurosteroidogenic medications such as benzodiazepines16. Moreover, our results have experimental implications and suggest that conditions that trigger acute neurosteroid production can alter LTP in vitro when as little as 0.1 % ethanol (~16 mM) is added as a drug-solvent.
Of note, our results indicate that the acetaldehyde contributing to LTP inhibition and neurosteroid formation can be formed locally within the hippocampus and do not require systemic production. This strongly suggests that binge drinking and its associated high ethanol concentrations may induce regional brain acetaldehyde formation independent of peripheral acetaldehyde levels.
In summary, we show that regionally formed acetaldehyde promotes neurosteroid synthesis in hippocampal pyramidal neurons, and participates in the synaptic dysfunction associated with acute high concentrations of alcohol.
Acknowledgments
This work was supported by grants AA017413, MH07791 and GM47969 from the National Institutes of Health, and the Bantly Foundation.
Abbreviations
- 4MP
4-methylpyrazole
- ACSF
artificial cerebrospinal fluid
- ADH
alcohol dehydrogenase
- alloP
allopregnanolone
- APV
2-amino-5-phosphonovalerate
- EPSP
excitatory postsynaptic potential
- HFS
high frequency stimulus
- LTP
long-term potentiation
- NMDA
N-methyl-D-aspartic acid
- NMDAR
NMDA receptor
- PBS
phosphate buffered saline
- PTX
Picrotoxin
Footnotes
DISCLOSURE
CFZ serves on the Scientific Advisory Board of Sage Therapeutics. The authors have no other competing interests to declare.
References
- 1.White M. What happened? Alcohol, memory blackouts and the brain. Alcohol Res Health. 2003;27:186–196. [PMC free article] [PubMed] [Google Scholar]
- 2.Bliss TVP, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 3.Izumi Y, Nagashima K, Murayama K, Zorumski CF. Acute effects of ethanol on hippocampal long-term potentiation and long-term depression are mediated by different mechanisms. Neuroscience. 2005;136:509–517. doi: 10.1016/j.neuroscience.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 4.Izumi Y, Murayama K, Tokuda K, Krishnan K, Covey DF, Zorumski CF. GABAergic neurosteroids mediate the effects of ethanol on long-term potentiation in rat hippocampal slices. Eur J Neurosci. 2007;26:1881–1888. doi: 10.1111/j.1460-9568.2007.05809.x. [DOI] [PubMed] [Google Scholar]
- 5.Tokuda K, Izumi Y, Zorumski CF. Ethanol enhances neurosteroidogenesis in hippocampal pyramidal neurons by paradoxical NMDA receptor activation. J Neurosci. 2011;31:9905–9909. doi: 10.1523/JNEUROSCI.1660-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sanna E, Talani G, Busonero F, et al. Brain steroidogenesis mediates ethanol modulation of GABAA receptor activity in rat hippocampus. J Neurosci. 2004;24:6521–6530. doi: 10.1523/JNEUROSCI.0075-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tokuda K, Izumi Y, Zorumski CF. Locally-generated acetaldehyde is involved in ethanol-mediated LTP inhibition in the hippocampus. Neurosci Lett. 2013;537:40–43. doi: 10.1016/j.neulet.2013.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parés X, Vallee BL. New human liver alcohol dehydrogenase forms with unique kinetic characteristics. Biochem Biophys Res Commun. 1081;98:122–130. doi: 10.1016/0006-291x(81)91878-7. [DOI] [PubMed] [Google Scholar]
- 9.Haseba T, Hirakawa K, Tomita Y, Watanabe T. Characterization of high Km alcohol dehydrogenase from mouse liver. In: Hirai H, editor. Electrophoresis ‘83. Berlin and New York: Walter de Gruyter & Co; 1984. pp. 393–400. [Google Scholar]
- 10.Haseba T, Ohno Y. A new view of alcohol metabolism and alcoholism--role of the high-Km Class III alcohol dehydrogenase (ADH3) Int J Environ Res Public Health. 2010;7:1076–1092. doi: 10.3390/ijerph7031076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mori O, Haseba T, Kameyama K, et al. Histological distribution of class III alcohol dehydrogenase in human brain. Brain Res. 2000;852:186–190. doi: 10.1016/s0006-8993(99)02201-5. [DOI] [PubMed] [Google Scholar]
- 12.Galter D, Carmine A, Buervenich S, Duester G, Olson L. Distribution of class I, III and IV alcohol dehydrogenase mRNAs in the adult rat, mouse and human brain. Eur J Biochem. 2003;270:1316–1326. doi: 10.1046/j.1432-1033.2003.03502.x. [DOI] [PubMed] [Google Scholar]
- 13.Aragon CM, Rogan F, Amit Z. Ethanol metabolism in rat brain homogenates by a catalase-H2O2 system. Biochem Pharmacol. 1992;44:93–98. doi: 10.1016/0006-2952(92)90042-h. [DOI] [PubMed] [Google Scholar]
- 14.Gill K, Menez JF, Lucas D, Deitrich RA. Enzymatic production of acetaldehyde from ethanol in rat brain tissue. Alcohol Clin Exp Res. 1992;16:910–915. doi: 10.1111/j.1530-0277.1992.tb01892.x. [DOI] [PubMed] [Google Scholar]
- 15.Eysseric H, Gonthier B, Soubeyran A, Bessard G, Saxod R, Barret L. Characterization of the production of acetaldehyde by astrocytes in culture after ethanol exposure. Alcohol Clin Exp Res. 1997;21:1018–1023. [PubMed] [Google Scholar]
- 16.Tokuda K, O’Dell KA, Izumi Y, Zorumski CF. Midazolam inhibits hippocampal long-term potentiation and learning through dual central and peripheral benzodiazepine receptor activation and neurosteroidogenesis. J Neurosci. 2010;30:16788–16795. doi: 10.1523/JNEUROSCI.4101-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saalmann YB, Kirkcaldie MT, Waldron S, Calford MB. Cellular distribution of the GABAA receptor-modulating 3α-hydroxy, 5α-reduced pregnane steroids in the adult rat brain. J Neuroendocrinol. 2007;19:272–284. doi: 10.1111/j.1365-2826.2006.01527.x. [DOI] [PubMed] [Google Scholar]
- 18.Dudek SM, Fields RD. Somatic action potentials are sufficient for late-phase LTP- related cell signaling. Proc Natl Acad Sci U S A. 2002;99:3962–3967. doi: 10.1073/pnas.062510599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thompson KR, Otis KO, Chen DY, Zhao Y, O’Dell TJ, Martin KC. Synapse to nucleus signaling during long-term synaptic plasticity; a role for the classical active nuclear import pathway. Neuron. 2004;44:997–1009. doi: 10.1016/j.neuron.2004.11.025. [DOI] [PubMed] [Google Scholar]
- 20.Birbach A, Verkuyl JM, Matus A. Reversible, activity-dependent targeting of profilin to neuronal nuclei. Exp Cell Res. 2006;312:2279–2287. doi: 10.1016/j.yexcr.2006.03.026. [DOI] [PubMed] [Google Scholar]
- 21.Quertemont E, Tambour S, Tirelli The role of acetaldehyde in the neurobehavioral effects of ethanol: a comprehensive review of animal studies. Prog Neurobiol. 2005;75:247–274. doi: 10.1016/j.pneurobio.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 22.Correa M, Salamone JD, Segovia KN, et al. Piecing together the puzzle of acetaldehyde as a neuroactive agent. Neurosci Biobehav Rev. 2012;36:404–430. doi: 10.1016/j.neubiorev.2011.07.009. [DOI] [PubMed] [Google Scholar]
- 23.Zimatkin SM, Pronko SP, Vasiliou V, Gonzalez FJ, Deitrich RA. Enzymatic mechanisms of ethanol oxidation in the brain. Alcohol Clin Exp Res. 2006;30:1500–1505. doi: 10.1111/j.1530-0277.2006.00181.x. [DOI] [PubMed] [Google Scholar]
- 24.Ronis MJ, Korourian S, Blackburn ML, Badeaux J, Badger TM. The role of ethanol metabolism in development of alcoholic steatohepatitis in the rat. Alcohol. 2010;44:157–169. doi: 10.1016/j.alcohol.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boyd KN, O’Buckley TK, Morrow AL. Role of acetaldehyde in ethanol-induced elevation of the neuroactive steroid 3α-hydroxy-5α-pregnan-20-one in rats. Alcohol Clin Exp Res. 2008;32:1774–1781. doi: 10.1111/j.1530-0277.2008.00762.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quertemont E, Tambour S, Bernaerts P, Zimatkin SM, Tirelli E. Behavioral characterization of acetaldehyde in C57BL/6J mice: locomotor, hypnotic, anxiolytic and amnesic effects. Psychopharmacology (Berl) 2004;177:84–92. doi: 10.1007/s00213-004-1911-x. [DOI] [PubMed] [Google Scholar]
- 27.Deng XS, Deitrich RA. Putative role of brain acetaldehyde in ethanol addiction. Curr Drug Abuse Rev. 2008;1:3–8. doi: 10.2174/1874473710801010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crabb DW, Matsumoto M, Chang D, You M. Overview of the role of alcohol dehydrogenase and aldehyde dehydrogenase and their variants in the genesis of alcohol-related pathology. Proc Nutr Soc. 2004;63:49–63. doi: 10.1079/pns2003327. [DOI] [PubMed] [Google Scholar]
- 29.Kim SW, Bae KY, Shin HY, et al. The role of acetaldehyde in human psychomotor function: a double-blind placebo-controlled crossover study. Biol Psychiatry. 2010;67:840–845. doi: 10.1016/j.biopsych.2009.10.005. [DOI] [PubMed] [Google Scholar]