

Summary
Given the association between mutational load and cancer, the observation that genetic aberrations are frequently found in human pluripotent stem cells (hPSCs) is of concern. Prior studies in human induced pluripotent stem cells (hiPSCs) have shown that deletions and regions of loss of heterozygosity (LOH) tend to arise during reprogramming and early culture, whereas duplications more frequently occur during long-term culture. For the corresponding experiments in human embryonic stem cells (hESCs), we studied two sets of hESC lines: one including the corresponding parental DNA and the other generated from single blastomeres from four sibling embryos. Here, we show that genetic aberrations observed in hESCs can originate during preimplantation embryo development and/or early derivation. These early aberrations are mainly deletions and LOH, whereas aberrations arising during long-term culture of hESCs are more frequently duplications. Our results highlight the importance of close monitoring of genomic integrity and the development of improved methods for derivation and culture of hPSCs.
Introduction
Human embryonic stem cells (hESCs) are derived from the inner cell mass (ICM) cells of blastocyst stage embryos (Itskovitz-Eldor et al., 2000; Reubinoff et al., 2000; Thomson et al., 1998). These cells appear to be immortal and can be maintained and propagated in culture in an undifferentiated state essentially forever, without losing their ability to proliferate. In addition, they have the potential to develop into all three embryonic germ layers and all of their differentiated derivatives, both in vivo and in vitro (reviewed in Eiges and Benvenisty, 2002). In vivo, nonmalignant tumors (teratomas) are formed when hESCs are injected into immunodeficient mice. These tumors also demonstrate the wide developmental potential of hESCs, with the presence of derivatives of all embryonic germ layers. The vast self-renewal and differentiation capacities of hESCs make them potential sources of large quantities of differentiated cells for drug screening and cell therapy. One of the greatest concerns for the clinical use of hESCs or their derivatives is the element of safety, and of all the safety issues, tumorigenicity is of the highest priority (Ben-David and Benvenisty, 2011; Fox, 2008). As such, extensive research into the biology of stem cells and in-depth preclinical studies, especially those on safety, should be pursued in order to maximize their potential benefits while minimizing the risks of hESCs for application in regenerative medicine.
The link between stem cells and cancer cells is supported by increasing evidence that cancer can develop from stem cells, and that many cell signaling pathways essential for normal development are dysregulated in cancer (Catalina et al., 2009). In addition, genetic and epigenetic instability have been strongly associated with various types of cancer; thus, it is reasonable to assume that evidence of such instability is undesirable in cell preparations intended for clinical use. Catalina et al. (2009) have shown that hESCs continue to maintain overall genetic stability even after 70 passages (P) in culture, when grown on human feeder cells and passaged by mechanical splitting. This study evaluated genetic stability using G-banding karyotype analysis, which was confirmed by fluorescence in situ hybridization, comparative genomic hybridization (CGH), and SKY analyses (a high-resolution molecular cytogenetic tool). However, it has been shown that other culture conditions, including feederfree conditions and enzymatic splitting that are commonly used for the maintenance of undifferentiated hESCs, are associated with accumulation of genetic and epigenetic alterations with bulk passaging and extended time in culture (Baker et al., 2007; Imreh et al., 2006; Maitra et al., 2005). These genetic changes may lead to quantitative differences in gene expression. Some of the most frequent chromosomal changes observed in hESCs, such as trisomies of chromosomes 12 and 17, are similar to those seen in malignant germ cell tumors (Draper et al., 2004; Mitalipova et al., 2005).
Subchromosomal abnormalities have been detected in hESCs by CGH and SNP genotyping (Lefort et al., 2008; Närvä et al., 2010; Spits et al., 2008). Our lab (Laurent et al., 2011) recently identified recurrent duplications in specific regions of the genome in hESCs acquired with time and passaging in culture. Similarly, other groups (Ben-David et al., 2010; Hussein et al., 2011; Amps et al., 2011; Mayshar et al., 2010) identified several aberrations resulting from time in culture. The most frequently observed duplication included the region of chromosome 12 that contains pluripotency- and proliferation-associated genes, including NANOG. These findings are a cause for concern because overexpression of NANOG has been shown to increase proliferation (Darr et al., 2006). These reports on instability of hESCs and human induced pluripotent stem cells (hiPSCs) focused on alterations detectable over the course of long-term culture, implying that some late-passage hESC lines may be unsuitable for therapeutic purposes. However, genetic changes appear to be present in some hESCs even at very early passages, raising the possibility of genetic instability that occurs already during the derivation process. These results highlight the need to map the full range of common genetic aberrations in hESCs, in order to define the conditions that promote/repress genomic instability in these cells, as well as to differentiate between aberrations that lead to phenotypic changes which are “harmful” and those that are “harmless.” In order to develop strategies to prevent the accumulation of genetic alterations in hESCs, we must first identify when during the lifetime of the cultures they are most likely to occur.
hiPSCs are easy to generate and present a good system for determining whether a given alteration is a new event or similar to the parental source cells that were used for their generation (Laurent et al., 2011). However, the selective pressures imposed by the reprogramming process itself may significantly contribute to aneuploidy in hiPSCs (Ben-David et al., 2010; Laurent et al., 2011). Therefore, the goal of this study was to distinguish between the frequency and type of genetic alterations that arise during different stages of hESC derivation and culture, specifically during the period between gametogenesis and early passage of the established hESC line (including gametogenesis, fertilization, preimplantation embryo development, derivation of the line, and its early passage) and during long-term culture. This information will point to key aspects of pluripotent stem cell generation and culture that require optimization.
We took advantage of our access to two unique sets of genetically related samples, together with high-resolution genomic analysis in order to determine at a high level of precision when a specific genetic change has occurred –during gametogenesis, embryogenesis, derivation, or culture. The first genetically related samples include early and high-passage samples (collected 30–42 passages after the early-passage samples) from three hESC lines, which we derived (the preimplantation-genetic-diagnosis [PGD]-derived hESC lines), and for which we have parental DNA from the blastocyst donors. These related samples enabled us to explore the origins of the identified copy number variations, whether they were inherited or arose de novo during early embryogenesis or derivation, as well as to determine the maternal and paternal contributions to the observed genetic changes. The three PGD-derived hESC lines were generated from embryos that were deemed unsuitable for reproductive purposes because they were identified by PGD to carry single gene mutations for severe genetic disorders (Saethre-Chotzen Syndrome for Lis04_Twist, Duchenne Muscular Dystrophy for Lis12_DM, and Familial Adenomatous Polyposis for Lis25_FAP); these PGD-derived hESC lines are not intended to be used for therapy, but rather as models to understand disease and hESC biology (Ben-Yosef et al., 2008; Biancotti et al., 2010; Marteyn et al., 2011; Niclis et al., 2009; Telias et al., 2013) and, to our knowledge, are unique in the availability of parental DNA, which allows comparisons between the hESC and parental genomes. The second set of samples includes genetically related dizygotic and monozygotic twin blastomere-derived hESC lines (Chung et al., 2008; Ilic et al., 2009) that enabled us to distinguish between changes that occurred during gametogenesis, embryogenesis, and derivation. Identification of when aberrations occur will enable future research using these models to determine optimal embryo culture, hESC derivation, and culture conditions that maximize their genetic stability.
Results
In order to dissect when during the process of embryogenesis and hESC derivation genetic aberrations are arising, we analyzed samples from two unique sets of samples (Figure 1). First, we assessed the genetic stability of early-passage (passage 11–20) and high-passage (passage 44–60) samples from three hESC lines, compared to DNA from parental blood samples (Figure 1). These three hESC lines were derived from embryos carrying single-gene mutations identified by PGD and are termed the “PGD-derived hESC lines.” Analysis of DNA from the parental, early-passage hESC, and high-passage hESC samples allowed us to definitively identify de novo duplications and deletions in the hESC lines, to determine whether they occurred during the period between gametogenesis/preimplantation embryo development and hESC derivation or during long-term passage of the hESC lines, and to identify the parent of origin of the duplicated and retained alleles, respectively. In addition,we were able to distinguish between regions ofhomozygosity that were the result of loss of one parental allele followed by duplication of the remaining allele from the other parent and those that were likely acquired through normal Mendelian inheritance of homozygous alleles in cases where both parents shared the same allele. Second, we analyzed early-passage (passage 11–16) samples from nine “twin blastomere hESC lines,” which were derived using single blastomeres isolated from four sibling embryos (Figure 1, inset). One embryo yielded four lines, another embryo yielded three lines, and two embryos yielded one line each. Analysis of these monozygotic and dizygotic twin lines enabled us to determine whether the copy number variations were likely to be inherited or arising de novo, and to distinguish whether the de novo variations arose during the period encompassing gametogenesis, preimplantation embryo development, or hESC derivation and early passage.
Figure 1. hESC Lines and Parental DNA Samples Used in This Study.
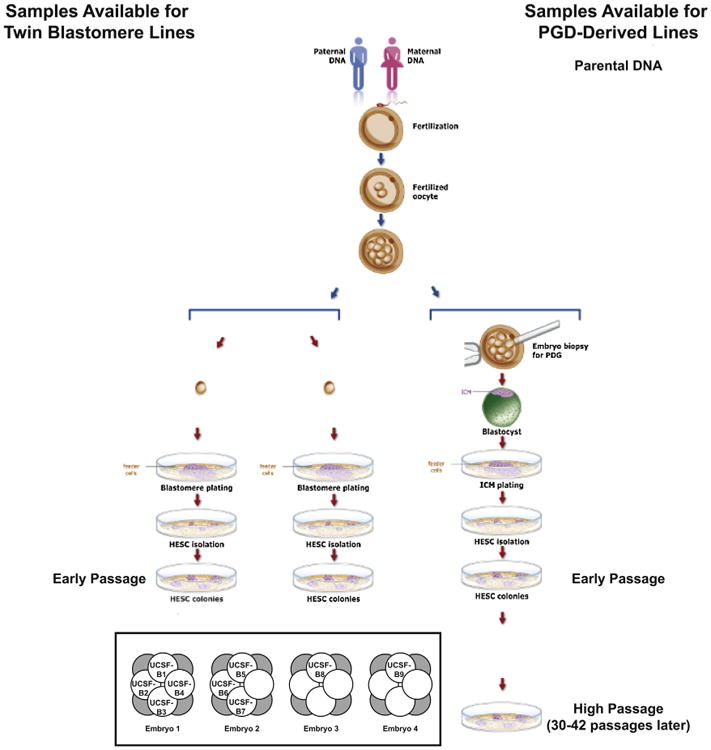
Schematic diagram summarizing the samples collected for analysis relating to the PGD-derived hESC lines. (inset) Diagram showing relationships among the nine twin blastomere (UCSF-B) hESC lines. Embryos 1, 2, 3, and 4 are dyzygotic twins; UCSF-B1, -B2, -B3, and -B4, as well as UCSF-B5, -B6, and -B7 are monozygotic twin lines. See also Figures S4, S5, and Table S1.
All samples were analyzed by high-resolution SNP genotyping using a microarray platform interrogating over 1 million SNPs (the Illumina Omni1 BeadChip). Replicate error analysis was performed to verify the relationships among the samples (Table S1). A replicate error value of ∼80% indicates that a pair of samples are not related to each other, whereas a value of ∼85%– 90% indicates a parent-child or full-sibling relationship, and a value of >99% indicates a self or identical/monozygotic twin relationship. As expected, these results confirmed the father-mother-child trios for the PGD-derived hESC lines (Table S1), the full-sibling relationships among the four embryos and the identical twin relationships among the hESCs lines derived from different blastomeres of the same embryo (Table S1) for the twin blastomere hESC lines. Copy number variations (CNVs) were identified using CNV Partition 3.1.6 (Illumina), visual inspection of the BAF and LogR Ratio plots, and/or karyotyping (see Experimental Procedures; Extended Experimental Procedures for further details).
Analysis of PGD-Derived hESC Lines
Early and high-passage samples were analyzed by karyotyping. These results revealed no karyotypic abnormalities in the early and high Lis04_Twist1 and Lis25_FAP samples and the early-passage Lis12_DM1 sample. Only the high-passage Lis12_DM1 sample was shown to have a karyotypic abnormality, consisting of a pericentric inversion on chromosome 7 and a whole chromosome duplication of chromosome 17 (Figure 2A).
Figure 2. Results for the PGD-Derived hESC Lines.
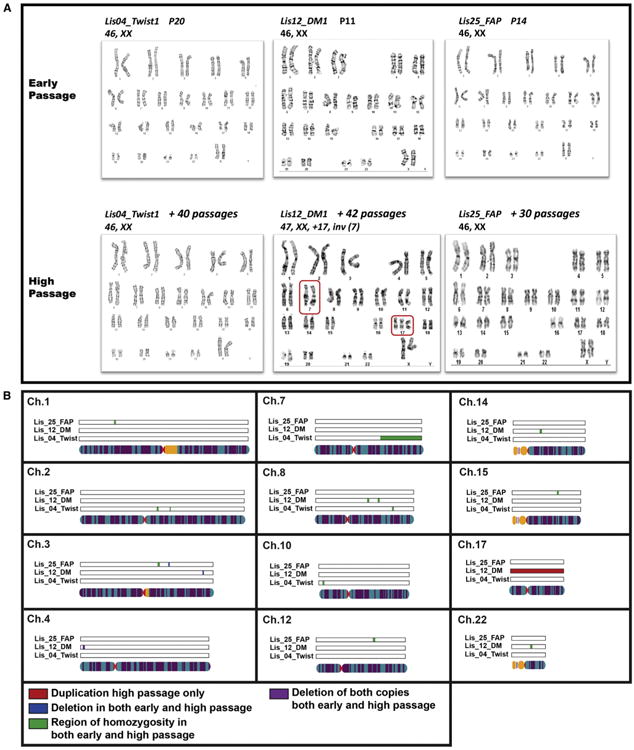
(A) Karyotypes at early and high passage for the PGD-derived hESC lines. The inversion in chromosome 7 and the duplication of chromosome 17 in the high-passage sample of the Lis12_DM hESC line are highlighted in the red boxes.
(B) CNVs that were not present in the parental DNA samples identified by SNP Genotyping analysis in the PGD-derived hESC lines. Duplications are shown in red, deletions in blue, and regions of homozygosity in green.
The SNP genotyping analysis of the same samples was able to detect the duplication of chromosome 17, but not the pericentric inversion on chromosome 7, in the high-passage Lis12_DM1 sample (Figures 2B and S1; Table S2). The SNP genotyping analysis also identified deletions and regions of homozygosity that cannot be detected by karyotyping (Figures 2B and S1; Table S2). All of the novel deletions and regions of homozygosity were present in both the early- and high-passage samples. The BAF and LogR ratio plots for the two largest identified genomic aberrations are shown in Figure 3 and confirm that the region of homozygosity on chromosome 7 of the Lis04_Twist line was present in both the early and high-passage samples (Figure 3A), and the duplication of chromosome 17 in the Lis12_DM1 line was present in the high-passage sample, but not in the early-passage sample (Figure 3B).
Figure 3. Representative BAF and LogR Ratio Plots.

(A and B) Data illustrating a region of homozygosity on chromosome 7 in the early and high-passage samples of the Lis04_Twist hESC line (A) and a duplication of chromosome 17 in the high-passage sample of the Lis12_DM hESC line (B).
(C) B allele frequency data analysis showing the paternal origin of the duplicated chromosome 17 in the high-passage Lis12_DM1 culture.
We then wished to determine the parent of origin of the duplicated chromosome 17 in the Lis12_DM1 high-passage sample. Some SNPs on chromosome 17 were homozygous in both parental genomes and discordant between parental genomes for the Lis12_DM1 hESC line (i.e., AA in the maternal and BB in the paternal genome, or BB in the maternal and AA in the paternal genome). For these SNPs, we plotted the BAF values for the maternal, paternal, and high-passage hESC samples, rank ordered according to the BAF value in the hESC sample. We reasoned that the BAF for the hESC sample should be closer to the BAF of the parent of origin of the duplicated chromosome. For example, if the maternal genotype is AA, the paternal genotype is BB, and the paternal allele is duplicated, then the hESC sample genotype should be ABB, making the hESC BAF closer to the paternal BAF. In this way, we were able to determine that the duplicated chromosome 17 in the Lis12_DM1 high-passage sample was of paternal origin (Figure 3C).
Regions of homozygosity can occur either from loss of heterozygosity, in which one parental allele is lost and the other is duplicated, or from normal Mendelian inheritance of a series of alleles that are the same on the inherited maternal and paternal chromosomes. In order to distinguish between these two alternatives, we further analyzed the SNP genotyping data and performed short tandem repeat (STR) marker analysis for three regions of homozygosity detected from the SNP genotyping data. STRs, also known as microsatellites, consist of variable numbers of repeats of 2–6 bp sequences, which are highly polymorphic. See Table S3 for details on the polymorphic markers used. Our SNP genotyping results demonstrated that, for the large region of homozygosity on chromosome 7 in the Lis04_Twist hESC line (encompassing ∼60 Mb), all of the homozygous alleles in the hESC line were present in the paternal sample, but many were not found in the maternal sample. Our finding suggests that this region of homozygosity arose from loss of the maternal allele and duplication of the paternal allele. This conclusion was confirmed by the polymorphic marker analysis, which showed that not only was the Lis04_Twist line homozygous for all tested markers, but that these markers could only have been inherited from the paternal genome (Figure 4A; Table S3). In contrast, our SNP genotyping results for the small regions of homozygosity on chromosome 8 in the Lis12_DM and on chromosome 12 in the Lis25_FAP lines (both approximately 1 Mb in extent) showed that the alleles present in the hESC lines could have originated from either parent or both parents. For these regions of homozygosity, the STR analysis showed that the hESC lines were heterozygous for at least one marker, supporting the conclusion that one set of homozygous alleles was inherited from each parent (Figures 4B and 4C). High frequencies of regions of homozygosity are more common in populations that arose from small founder group, such as the Ashkenazi Jewish population from which these hESC lines were derived. For the Lis12_DM line, the observed alleles at each marker locus could have been inherited from either parent (Figure 4B), but, for the Lis25_FAP line, the parent of origin for the three heterozygous marker loci could be definitively assigned (Figure 4C).
Figure 4. SNP Genotyping and STR Marker Analysis Data for Regions of Homozygosity.

For the SNP genotyping analysis, the A alleles are shown in purple, and the B alleles in pink. For the STR analyses, the amplicon sizes (numbers of repeats) for each repeat sequence are shown. Maternal alleles are shown in red, paternal alleles are blue, and the parental allele(s) of origin are indicated in bold typeface.
(A) Lis04_Twist, chr7.
(B) Lis12_DM, chr8.
(C) Lis25_FAP, chr12.
Therefore, of the 13 identified regions of homozygosity, the parent of origin could be identified from the SNP genotyping data only for the largest one (60 Mb) located on chromosome 7 in the Lis04_Twist line. This validated region of LOH was present in both the early and high-passage samples and likely arose during preimplantation embryo development, derivation, or early hESC culture. However, this was the only one out of the three regions tested by STR analysis to be validated as an LOH event. We conclude that the majority of small (∼1 Mb) regions of homozygosity identified in early-passage hESC lines by SNP genotyping analysis result from normal biparental inheritance of highly homozygous alleles.
Because we previously observed that CNV analysis from SNP genotyping data can result in a high percentage of false-positive calls (Laurent et al., 2011), we performed qPCR validation using TaqMan assays specifically designed to measure copy number on the three deletion calls identified in the PGD-derived hESC lines and found that two out of three deletions were confirmed (Lis12 chr3 single-copy deletion, Lis12 chr4 loss of both copies were confirmed). The Lis25 chr3 single-copy deletion was not confirmed and was thus concluded to be a false-positive deletion call from SNP genotyping (Table 1; Figure S2). From the SNP genotyping data, we were able to determine that the retained alleles for the confirmed single-copy deletion on chromosome 3 in the Lis12_DM line was of maternal origin (Table S4).
Table 1. Summary of SNP Genotyping, Karyotyping, STR Analysis, and CNV qPCR Validation for the PGD-Derived hESC Lines.
| hESC Line | Chr | Position | Early | High | Detection | Validation | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP Genotyping | Karyotyping | Short Tandem Repeat Testing | qPCR CNV assay | ||||||||||||
| CNV | Interpretation | Inh | No. of SNPs in Region | No. of Markers | STR Results | Probe Used | Ratio (versus Diploid Control) | CNV qPCR Results | Conclusion | ||||||
| Lis04_ Twist | 2 | 114925151–119741942 | + | + | 2 | ROH | UTD | 1,372 | ND | None | NA | None | NA | NA | ROH |
| 2 | 134033864–137339476 | + | + | 2 | ROH | UTD | 1,062 | ND | None | NA | None | NA | NA | ROH | |
| 7 | 98674852–158812247 | + | + | 2 | Large ROH | Pat | 19,577 | ND | 4 | Homz, Pat | None | NA | NA | Large LOH, Pat, gam-early pass | |
| 8 | 111210397–112355790 | + | + | 2 | ROH | UTD | 180 | ND | None | NA | None | NA | NA | ROH | |
| 10 | 9044003–10289903 | + | + | 2 | ROH | UTD | 425 | ND | None | NA | None | NA | NA | ROH | |
| Lis12_ DM | 3 | 177373492–177393468 | + | + | 1 | 1-copy | Mat | 15 | ND | None | NA | Hs03228327 | 0.55 | 1 -copy | 1 -copy del, Mat, gam -early pass |
| 4 | 9820707–9843332 | + | + | 0 | 0-copy | Loss of both parental copies | 96 | ND | None | NA | Hs03239014 | Undetectable | 0-copy | 0-copy del, gam - early pass | |
| 7 | p13-q11.2 | - | + | 2 | ND | ND | Peri-inv | None | NA | None | NA | NA | Peri, long-term | ||
| 8 | 85878049–86910517 | + | + | 2 | ROH | UTD | 181 | ND | None | NA | None | NA | NA | ROH. | |
| 8 | 100005461–101295619 | + | + | 2 | ROH | UTD | 191 | ND | 6 | Homz, nl inh | None | NA | NA | ROH, nl inh | |
| 14 | 39412328–40455980 | + | + | 2 | ROH | UTD | 259 | ND | None | NA | None | NA | NA | ROH | |
| 17 | Whole chromosome | - | + | 3 | Dup | Pat | 29,097 | Tri17 | None | NA | None | NA | NA | Dup, long-term | |
| 22 | 26529417–27788788 | + | + | 2 | ROH | UTD | 262 | ND | None | NA | None | NA | NA | ROH | |
| Lis25_FAP | 1 | 51610325–52868170 | + | + | 2 | ROH | UTD | 301 | ND | None | NA | Not done | NA | NA | ROH |
| 3 | 121951424–123356123 | + | + | 2 | ROH | UTD | 344 | ND | None | NA | Not done | NA | NA | ROH | |
| 3 | 133103999–133195707 | + | + | 1 | 1-copy | UTD | 45 | ND | None | NA | Hs04750306 Hs01541029 | 0.976, 1.019 | 2-copies | False-positive | |
| 12 | 86694771–87755618 | + | + | 2 | ROH | UTD | 229 | ND | 4 | Hetz, nl inh | Not done | NA | NA | ROH, nl inh | |
| 15 | 70195123–71415221 | + | + | 2 | ROH | UTD | 276 | ND | None | NA | not done | NA | NA | ROH | |
Early, early passage; High, high passage; Interp, interpretation of the SNP genotyping CNV call; Inh, inheritance pattern of regions of CNV inferred from the SNP genotyping data; ROH, region of homozygosity; CNV, CNV value; Inh, inheritance; One copy, single-copy deletion; Zero copy, zero copy deletion; ND, not detected; Dup, duplication; UTD, unable to determine parent of origin; Mat, both alleles are of maternal origin (ROH); Pat, both alleles are of paternal origin (ROH) or duplicated allele is of paternal origin; loss of both, loss of both parental alleles; NA, not applicable; Peri-inv, pericentromeric inversion; Tri17, trisomy 17; Homz, homozygous; Hetz, heterozygous; nl inh, normal inheritance; gam-early pass, occurred during the period from gametogenesis to early passage, long-term, occurred during long-term culture; Probe used, catalog number of the CNV qPCR assay obtained from Life Technologies; Undet, undetectable. See also Figures S4, S5, and Tables S3 and S4.
A summary of the SNP genotyping, karyotype, STR marker analysis, and CNV qPCR results is shown in Table 1. Overall, we saw that the two validated deletions and the validated region of LOH were present in both the early and high-passage cultures, suggesting that they arose during preimplantation embryo development, derivation, or early passage of the hESC lines. None of these aberrations were detectable by karyotyping. We investigated whether these deletions included genes for which a decrease in expression might result in a selective advantage in culture, but neither the Lis12_DM deletion on chromosome 3 nor the Lis12_DM deletion on chromosome 4 encompassed any identified genes.
The only identified genetic aberrations in the PGD-derived hESC lines that arose during long-term passage (were absent in the early-passage samples and present in the high-passage samples) were the large pericentromeric inversion on chromosome 7 and the duplication of the entire chromosome 17, both in the same line (Lis12_DM).
Analysis of the Twin Blastomere hESC Lines
Analysis of the nine twin blastomere lines was focused on distinguishing between genetic aberrations that arose during early preimplantation development and those that arose during derivation and early culture of the hESC lines, and therefore these lines were analyzed at early passage only. The twin blastomere hESC lines were derived from four sibling embryos (embryo 1, 2, 3, and 4) from the same parents, with lines UCSF-B1, -B2, -B3, and -B4 derived from embryo 1, UCSF-B5, -B6, and -B7 from embryo 2, UCSF-B8 from embryo 3, and UCSF-B9 from embryo 4 (Figure 1, inset).
Even though parental DNAs for the twin blastomere lines were not available for analysis, the study of monozygotic (Lines UCSF-B1, -B2, -B3, and -B4 and lines UCSF-B5, -B6, and -B7) and dizygotic twin blastomere hESC lines enabled us to infer whether observed variants were inherited or arose de novo. Furthermore, the sets of monozygotic twin lines derived from the same embryo enabled us to distinguish between changes that occurred during gametogenesis/embryogenesis and those that occurred during hESC derivation/early culture.
The copy number variations detected by SNP genotyping analysis of the twin blastomere lines are shown in Figure 5A (see also Table S5). If a given variant was present in all of the monozygotic lines derived from the same embryo, it is shown once. In cases in which there was a discrepancy among the lines derived from the same embryo (e.g., for the chromosome 12 duplication present in UCSF-B7 and absent in UCSF-B5 and -B6), both the normal and duplicated variants are shown and annotated. A total of 34 CNVs were present in lines from more than one sibling embryo (Table S6; e.g., aqua boxes in Figure 5A) and therefore were likely to be inherited. Twelve sub-chromosomal variants were observed in all of the lines from one embryo (i.e., all of the monozygotic twin lines from a given embryo; Table S6; e.g., green boxes in Figure 5A). These variants, therefore, must have arisen prior to the 8-cell stage; they could have been either inherited or created de novo during gametogenesis or the first two cleavage cycles. To determine whether these variants were most likely inherited or created de novo, we examined the frequency with which each of these variants was observed in the Database of Genomic Variants (DGV). In Table S6, we determined that, if the variant was present in only one embryo and was either new (not seen in DGV) or had a frequency of ≤1% in DGV (a commonly used definition for a “rare” allele), we then concluded that the variant was de novo. We used population frequency information from DGV because parental DNA was not available for the twin blastomere lines, and we fully appreciate that our threshold is arbitrary and the accuracy of our approach is limited by the small numbers of data points for many copy number variations present in DGV.
Figure 5. CNVs Identified Using SNP Genotyping in the Twin Blastomere Lines.

(A) Summary of CNVs called in the twin blastomere lines. If a given variant was present in all of the lines derived from the same embryo, they are displayed together. In cases in which there was a discrepancy among the lines derived from the same embryo, all variants are shown. Duplications are shown in red, deletions in blue, regions of homozygosity in green, and an indeterminate region (either duplication or deletion, present in a subpopulation of cells) in purple. Examples of variants present in two or more embryos are in aqua boxes, examples of variants present in all lines from only one embryo are in green boxes, and the two aberrations that are found in only one of multiple lines from a given embryo are in the red boxes. Only chromosomes for which at least one variant was detected are shown.
(B) Representative BAF and LogR Ratio plots illustrating the chromosome 20 aberration in one line from Embryo 1 and the chromosome 12 duplication in one line from Embryo 2.
There was also a duplication of chromosome 12 in UCSF-B9, which we judged to be a de novo aberration that arose during in vitro passage of the hESC line, as an earlier passage of this line had been karyotyped with a normal diploid result (data not shown). Finally, we identified two events that were present in only one line from a set of monozygotic lines (Table S6 and, e.g., red boxes in Figure 5A). We therefore concluded that these events were likely generated de novo during the derivation process itself or during early culture. These two events were (1) the duplication of chromosome 12 found in UCSF-B7, but absent from its monozygotic triplet lines UCSF-B5 and -B6; and (2) the aberration of chromosome 20 that was present in UCSF-B1 but absent in its monozygotic quadruplet lines UCSF-B2, -B3, and B4. Because the duplication of chromosome 12 was not present in karyotype results from a separate culture of the UCSF-B7 line, we concluded that this duplication arose during in vitro culture. The BAF and LRR plots for chromosome 20 in the UCSF-B1 hESC line (Figure 5B) were consistent with an aberration affecting the entire chromosome in some, but not all, cells in the analyzed sample. In mosaic cases, such as this, it is difficult to ascertain from the SNP genotyping data whether the aberration is a deletion or a duplication, because the distribution of BAF values could be consistent with either type of abnormality, and the change in the LRR is too small to allow confident discrimination between the two possibilities. We noticed that the BAF and LRR plots appeared slightly different for the q arm and the p arm of chromosome 20, with the spread in the BAF being slightly larger and the LRR being slightly lower for the p arm, suggesting that there might be a deletion of the p arm and a duplication of the q arm (Figure 5B). We attempted to determine the copy number for the q arm using qPCR, but the result was indeterminate, likely due to the mosaicism in the sample. The karyotype, which was performed on the same culture as the SNP genotyping and collected one passage earlier, proved to be the most informative assay for this aberration, revealing a mosaic aberration of chromosome 20 in 13 out of 20 spreads (data not shown). By G-banding, the aberration was determined to be an isochromosome 20q [46XY,i(20)(q10)], which essentially results in three copies of 20q and one copy of 20p, and is consistent with the SNP genotyping findings.
Given the discordance for the chromosome 12 and chromosome 20 aberrations among monozygotic twin blastomere lines derived from the same embryo, we can be certain that these variants arose after fertilization. The BAF and LRR plots illustrate that these findings were indeed seen in only one of the twin blastomere lines from these two embryos (UCSF-B1 chr20 and UCSF-B7 chr12, Figure 5B).
From the results of the analysis of all of the twin blastomere hESC lines, we conclude that the majority of the detected variants were likely inherited, with a smaller number of variants that were either inherited or acquired during gametogenesis or the first few cell divisions, and only two aberrations that definitively arose after fertilization.
A whole chromosome duplication of chromosome 12 was identified in twin blastomere lines from two different embryos, the UCSF-B7 line from embryo 2 and the UCSF-B9 line from embryo 4. It is known that chromosome 12 duplications are quite common in human pluripotent stem cell cultures and are thought to confer a selective advantage in culture (Draper et al., 2004; Laurent et al., 2011). To determine the parent of origin for these duplicated chromosomes, we compared the BAFs of each of the nonhomozygous loci in the UCSF-B7 and -B9 samples (defined in this analysis by a BAF between 0.2 and 0.8 in both samples) and found that the BAFs did not overlap. These results indicated that the duplicated copies of chromosome 12 in the UCSF-B7 and UCSF-B9 were inherited from the mother for one line and the father for the other line (Figure S3).
Having two embryos with duplications of the same chromosome allowed us to build the full haplotypes for chromosome 12 for the four sibling embryos, as well as most of the haplotypes for the two parents, even though parental DNAs were not available for these lines (Figure 6; Table S7; see Experimental Procedures and Extended Experimental Procedures for details). This analysis revealed that the duplicated chr12s in the UCSF-B7 and UCSF-B9 lines were not from the same parent. Moreover, the observed number and pattern of crossing-over events make it likely that the duplicated chromosome 12 in UCSF-B7 is of maternal origin, whereas the duplicated chromosome in UCSF-B9 is of paternal origin (Buard and de Massy, 2007; Lee et al., 2011).
Figure 6. Summary of Inheritance of Parental Haplotypes in the Four Embryos, which Were the Source of the Twin Blastomere hESC Lines.

Data from one representative hESC line are shown for each embryo. See also Table S7.
A subset of deletions in the twin blastomere hESC lines was subjected to validation by qPCR (Figure S4). These experiments validated a zero-copy deletion on chromosome 1 present in all lines from embryo 1, and a one-copy deletion on chromosome 6 present in all lines from embryo 2. A one-copy deletion on chromosome 3 in all lines from embryo 1 was not confirmed by the qPCR and is therefore likely a false-positive call from the SNP genotyping data. The CNV qPCR results were inconclusive for the aberration of chromosome 20 called in UCSF-B1 from the SNP genotyping data, likely because only a subpopulation of cells carried the isochromosome 20 detected by karyotyping. These results, as well as a summary of the SNP genotyping and probability calculation results, are listed in Table S6.
Discussion
This study presents results that directly answer questions pertaining to the timing and parental origin of genetic aberrations in hESCs. In order to answer fundamental questions about the type and timing of mutational events occurring during the process of embryogenesis, hESC derivation, and culture of hESC lines, we studied two unique sets of hESC lines using a variety of cytogenetic and molecular techniques, including karyotyping, high-resolution SNP genotyping (with copy number variation and haplotype analyses), STR analysis, and qPCR. The first set consisted of three “trios,” each of which comprised one PGD-derived hESC line and DNA from its two parents, which enabled both definitive identification of de novo mutations, and determination of the parent-of-origin of duplicated and deleted alleles. The second set consisted of nine hESC lines derived from single blastomeres isolated from four embryos from the same parents. The fact that there were both monozygotic and dizygotic “twin” hESC lines in this set allowed us to determine the timing of mutational events in relation to key developmental/biological steps.
Due to the inability to access to the parental DNA for most established hESC lines, previous studies on the genetic stability of hESCs have not been able to definitively identify genetic aberrations that have been acquired before and during early stages of hESC derivation. For hiPSCs, for which the source differentiated cell cultures can be easily studied, it has been demonstrated that most deletions and regions of homozygosity were detected soon after reprogramming, whereas duplications tended to appear over time in culture (Laurent et al., 2011; Mayshar et al., 2010). However, it is not appropriate to generalize these findings in hiPSCs to hESCs, given the significant differences in the processes of hiPSC reprogramming and hESC derivation, which could be reasonably expected to impose quite different mutagenic and selective pressures on the cultures.
This study has enabled us to demonstrate that the two novel deletions and the single region of LOH in the PGD-derived hESC lines were identified in both the early and high-passage samples, and the single detected duplication in these lines was present in a high-passage sample only (Table 1). Taken together, the previous reports on hiPSCs and results from the PGD-derived hESC lines presented in this study suggest that deletions and regions of LOH are generated at least in part from events that are common to the derivation and early culture of both hiPSCs and hESCs (e.g., the selective pressures of clonal or low-density culture). Therefore, they cannot be attributed solely to manipulations that are specific to either reprogramming (such as overexpression of reprogramming transcription factors or viral transduction) or hESC derivation (such as embryo culture).
All of the duplications and deletions and four of the regions of homozygosity identified by the SNP analysis were subjected to validation by STR analysis and/or qPCR CNV analysis. The availability of parental DNA for the PGD-derived lines allowed us to use the SNP genotyping data to determine the parent-of-origin of the chromosomes involved in one out of one duplication events, one out of three deletion events called from the CNV analysis, and one out of 13 regions of homozygosity. The other two deletions called from the CNV analyses were a loss of both parental copies and a call that failed to validate by qPCR analysis. In addition, we found that variants for which the parent of origin could be determined from the SNP genotyping data were confirmed by the validation studies, whereas variants for which the parent of origin could not be inferred from the SNP genotyping data failed to validate (Table 1). The validated deletions and region of LOH were paternal in one case and maternal in one case, suggesting that there is not a strong parent-of-origin bias, although the number of observations is too small to draw statistically significant conclusions. The results from the PGD-derived hESC analysis determined that novel deletions and regions of homozygosity arise in the period encompassing preimplantation embryo development and early-passage hESC culture.
We used the twin blastomere hESC data to classify variants as inherited (present in two or more embryos as represented by dizygotic twin lines), likely inherited (present in only one embryo with frequency >1% in DGV), likely de novo (present in only one embryo and frequency <1% in DGV), or de novo (discordant among blastomeres from the same embryo as represented by monozygotic twin lines).
The results from the twin blastomere hESC lines showed that for the embryos from which more than one monozygotic twin line was derived (embryos 1 and 2), all of the likely de novo deletions and regions of homozygosity were observed in all monozygotic twin lines. Considering these results together, we conclude that a significant subset of the novel deletions and regions of homozygosity seen in hESCs originate during the first two cleavage cycles of the preimplantation embryo. This conclusion may appear to be a curious coincidence in the context of published results indicating that deletions and regions of homozygosity tend to arise also during reprogramming and early culture of hiPSCs (Hussein et al., 2011; Laurent et al., 2011). Of note, two other studies published at around the same time did not show the same trend; however, one study used gene expression rather than DNA analysis to detect CNVs (Mayshar et al., 2010), whereas the other observed that some variants appeared with time in culture and others disappeared, resulting in no net gain or loss of observed variants with time in culture (Amps et al., 2011). However, it should be remembered that the preimplantation embryos used for generation of hESCs are cultured in vitro and therefore may be subject to selective or mutagenic pressures that are similar to those imposed by re-programming. There is growing independent evidence that more than half of human in vitro fertilization (IVF) embryos, which are the sources of cells used for the derivation of hESC lines, contain aneuploid cells, and mosaicism is a frequently occurring phenomenon during this preimplantation period (Barbash-Hazan et al., 2009; Mertzanidou et al., 2013a, 2013b; Vanneste et al., 2009; Wells and Delhanty, 2000). These data can explain some of the aberrations observed in the early-passage hESCs.
Although parental DNA could not be obtained for the twin blastomere lines, the fortuitous occurrence of chromosome 12 duplications in two twin blastomere lines from two different embryos allowed us to define the complete chromosome 12 haplotypes for the four UCSF-B embryos, as well as the nearly complete haplotypes for the parental chromosome 12 s, including sites of meiotic crossing over. These results revealed that the duplicated chromosome 12 s in the UCSF-B7 and UCSF-B9 lines originated from different parents, and that the frequency and location of crossing-over events for the two parents was markedly different, consistent with a recent publication (Lee et al., 2011), and allowing us to infer that parent A was likely paternal and parent B was likely maternal.
This study demonstrates the complementarity of the different molecular and bioinformatic techniques used. Although CNV analysis of SNP genotyping data was able to detect many small duplications and deletions, as well as regions of homozygosity, which were not detected by karyotyping, a large chromosomal rearrangement was detected by karyotyping, but not by SNP genotyping. Haplotype analysis of the SNP genotyping data was able to identify the parent-of-origin for several variants in the PGD-derived lines, as well as to infer the parental contributions to the duplicated and nonduplicated copies of chromosome 12 in the twin blastomere lines. STR and qPCR analyses served as orthogonal assays for validation of findings from the SNP genotyping data.
The valuable sample sets used in this study allowed us to define and trace the origins of mutational events that occur during critical steps in the development and derivation of embryonic stem cell lines, including gametogenesis, the first few cell divisions, and the derivation process, and show that genetic aberrations can occur at every step. Our results demonstrate proof-of-concept for several data analysis strategies, aimed at determining the timing and parent-of-origin of mutational events, and indicate that even more might be learned from analysis of more extensive pedigrees (for which parental DNAs are available for sets of monozygotic twin and sibling hESC lines) using ultra-deep whole-genome DNA sequencing, which, in a single analysis platform, would allow the identification of copy number variations, indels, point mutations, and rearrangements.
Taken in the context of previously published reports, our current findings suggest that deletions and regions of LOH frequently occur during derivation and early culture of both hESCs and hiPSCs, whereas duplications more commonly arise over long-term culture. Indeed, there were no de novo duplications observed at early passage in any of the PGD or twin blastomere lines. Of the four large duplications observed in this study, we have evidence that all occurred after derivation of the hESC line. The fact that additional deletions and regions of homozygosity were not observed to arise during long-term culture in either the PGD lines in this study, or in the iPSC lines in a previously published study (Laurent et al., 2011), suggests that conditions specific to the derivation process itself promote the occurrence and selection of these types of aberrations. Because the culture conditions were held constant for all passages of the lines examined in both studies, we infer that it is more likely that the derivation process was the source of the deletions and regions of homozygosity. Moreover, in prior studies from our lab and others, some deletions and regions of homozygosity detected in early-passage hiPSC cultures were not seen in later passages of the same hiPSC lines (Hussein et al., 2011; Laurent et al., 2011), suggesting that these aberrations may be subject to negative selection during long-term culture.
Overall, our results highlight the importance of close monitoring of genomic integrity and the development of improved methods and reagents for derivation and culture of human pluripotent stem cell cultures to minimize selection for genetically abnormal cells and ensure the safety of pluripotent stem cell-derived cells for clinical use. The similarity in the temporal pattern of aberrations in hESCs and hiPSCs suggests that the identification of factors that protect against the occurrence of deletions/regions of homozygosity during reprogramming might be useful also in preserving genetic stability during preim-plantation embryo culture and hESC derivation. This may be important given that the scarcity of starting material for hESC derivation prevents large-scale hESC derivation studies from being performed.
Experimental Procedures
In Vitro Fertilization, Donors, and Ethics Approval
PGD-Derived hESC Lines
The use of spare IVF-derived embryos following PGD for the generation and research of hESCs was approved by the National Ethics Committee and is in accordance with the guidelines released by the Bioethics Advisory Committee of the Israel Academy of Sciences and Humanities (Ruth Arnon et al., 2001).
Twin Blastomere hESC Lines
Embryos were produced by IVF for clinical purposes, and surplus frozen embryos were obtained with full informed consent and used in compliance with Advanced Cell Technology's Ethics Advisory Board and Institutional Review Board.
hESC Derivation Protocol
The PGD-derived hESC lines were derived from the ICM of blastocyst stage embryos (days 6–8 postfertilization), and twin blastomere hESC lines were derived from single blastomeres isolated from the 8-cell stage (day 3 postfertilization) (Figure 1).
PGD-Derived hESC Lines
Derivation of hESC lines from blastocysts following PGD was carried out using established protocols and as we previously described (Frumkin et al., 2010).
Twin Blastomere hESC Lines
hESC lines were successfully established from single blastomeres from four different 8-cell stage embryos. hESC lines derived from single blastomeres isolated from the same embryo are considered to be monozygotic twin hESC lines. Embryo 1 produced four monozygotic twin lines (UCSF-B1, -2, -3, and -4), embryo 2 produced three lines (UCSF-B5, -6, and -7), embryo 3 produced one line (UCSF-B8), and embryo 4 produced one line (UCSF-B9). Because all four embryos were donated by the same couple, hESC lines from different embryos are dizygotic twin lines.
The UCSF-B lines were derived under a protocol approved by the University of California's Committee on Human Research, and embryos were obtained through UCSF IVF Tissue Bank from donors undergoing IVF who gave informed consent.
The lines were derived according to the method described by Chung et al. (2008).
hESC Culture
hESCs were cultured on inactivated MEFs and in hESC medium. The medium was changed on a daily basis, and the cells were propagated until they reached the passage desired for the study. hESCs at early passage (passage 10–20) and high passage (30–40 passages later) were expanded to 100 mm Petri dishes, physically separated from the feeder cells and subjected to DNA extraction.
Phenotypic Characterization of hESC Lines
The hESC lines included in this study have been characterized for self-renewal ability, expression of undifferentiated pluripotent stem cell-specific markers, karyotype, and pluripotent potential by forming embryoid bodies in vitro or by teratoma induction in vivo. Characterization for the Lis04_Twist and Lis12_DM lines was reported in Frumkin et al. (2010); in addition, we include in this report representative images of immunofluorescent staining of the Lis12_DM line in Figure S4. Characterization for the Lis25_FAP line is shown in Figure S5. Full characterization for the twin blastomere lines is being reported in a separate manuscript (T. Zdravkovic, K.L. Nazor, N. Larocque, M. Gormley, M. Donne, N. Hunkapillar, G. Giritharan, H.S. Bernstein, G. Wei, M. Hebrok, X. Zeng, O. Genbacev, A. Mattis, M.T. McMaster, A. Krtolica, D. Valbuena, C. Simon, L.C. Laurent, J.F. Loring, and S. Fisher, unpublished data).
Parental DNA
The parental DNA isolated from blood samples of the maternal and paternal donors of the blastocysts for the PGD-derived hESC lines were analyzed and compared to the corresponding hESC lines (Figure 1).
DNA Extraction
DNA was extracted from parental blood and hESC samples using the DNeasy Blood and Tissue Genomic DNA Purification Kit (QIAGEN), following the manufacturer's instructions.
SNP Genotyping and Copy Number Variation Analysis
SNP Genotyping was performed on the Omni1 BeadChip (Illumina), which interrogates 1 million SNPs across the genome. Two micrograms of genomic DNA was amplified and labeled according to the manufacturer's instructions. The labeled product were hybridized to the array and subsequently scanned with an Iscan (Illumina). For the SNP genotyping, we performed data cleaning, filtering, SNP calling, and replicate error analysis with GenomeStudio. CNVPartition v 3.1.6 is the CNV-calling algorithm used for calling the aberrations in the samples.
Validation of Copy Number Aberrations and Single Base-Pair Mutations by qPCR
qPCR CNV assays were performed according to the manufacturer's instructions (Life Technologies). Samples of the WA09 hESC line and the HDF51 fibroblast line were used as diploid controls.
Short Tandem Repeat Analysis
For each region of homozygosity identified by SNP genotyping analysis, four to seven sets of STR markers were selected (Table S4). We used only informative markers that were heterozygous in at least one of the parents. This enabled us to determine the parental contribution. PCR conditions were as previously shown for single-cell polymorphic marker analysis (Malcov et al., 2007).
Statistical Analysis of SNP Genotyping Data
Replicate error analysis was performed in GenomeStudio and refers to the fraction of genotypes that are identical for each pair of samples tested.
The B allele frequencies and LogR ratios were calculated in GenomeStudio using the standard cluster files provided by the manufacturer.
For the PGD-derived hESC lines, the SNP genotyping data were used to determine the origin of the copy number alterations. In the case of duplications, the extra allele was identified from the B allele frequency (BAF) data by (1) filtering out homozygous SNPs by removing loci for which the BAF was <0.1 or >0.9 in the hESC line; (2) removing indeterminate SNPs with BAFs between 0.4 and 0.6; and (3) determining the duplicated allele to be A if the BAF was <0.4 and B if the BAF was >0.6. In the case of deletions, the remaining alleles were simply the genotype values called in GenomeStudio. For the parental allele assignments, we used the genotype values provided by GenomeStudio.
Haplotype analysis of the twin blastomere lines for chromosome 12 was performed on SNPs for which at least one sibling twin blastomere hESC line was heterozygous (i.e., had a BAF between 0.1 and 0.9). The extra chromosome 12 alleles for the lines with duplications, UCSF-B7 and UCSF-B9, were determined in the same way the extra chromosome 17 alleles were identified in the Lis12_DM line, except that the availability of several closely related lines allowed us to determine the identities of the duplicated alleles even when the BAFs in the duplicated hESC lines were close to 0.5. By iteratively comparing the chr12 genotypes for UCSF-B1, -B7, -B8, and -B9, we were able to infer the chromosome 12 haplotypes for each UCSF-B line, as well as both of the parents (Figure 6 and Table S7).
Supplementary Material
Acknowledgments
The laboratory of Susan Fisher (UCSF, including Tamara Zdravkovic and Olga Genbacev) made the blastomere-derived lines available to the authors. The derivation and characterization of these lines were supported by grants from the California Institute for Regenerative Medicine (RL1-00648 and RC1-00113). We thank Jeanne F. Loring and Candace Lynch for running the genomic DNA from the hESC cultures on SNP genotyping microarrays, supported by CIRM TR-1250. We thank Dr. Sagit Peleg and Veronica Gold for performing the PGD analysis of patients and the haplotype analysis for the PGD-HESC lines, Tsvia Frumkin for the karyotype analysis and skillful assistance in the stem cell research lab, and the embryologists of the Racine IVF lab (Ariela Carmon, Tanya Cohen, Tamar Shwartz, and Nava Mei-Raz) for their skillful assistance. We thank Sigalit Siso from Tel Aviv Medical Center for the graphics. This research was partly funded by a grant from the Recanaty Foundation, Tel Aviv University. F.S.B. was supported by a California Institute for Regenerative Medicine (CIRM) Bridges grant to the California State University, Channel Islands. F.S.B. and L.C.L. were supported by The Hartwell Foundation. L.C.L. was supported by NIH K12 HD001259.
Footnotes
Accession Numbers: The GEO accession number for the SNP genotyping data reported in this paper is GSE49452.
Supplemental Information: Supplemental Information includes Extended Experimental Procedures, five figures, and seven tables and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2013.08.009.
References
- Amps K, Andrews PW, Anyfantis G, Armstrong L, Avery S, Baharvand H, Baker J, Baker D, Munoz MB, Beil S, et al. Screening ethnically diverse human embryonic stem cells identifies a chromosome 20 minimal amplicon conferring growth advantage. Nat Biotechnol. 2011;29:1132–1144. doi: 10.1038/nbt.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DE, Harrison NJ, Maltby E, Smith K, Moore HD, Shaw PJ, Heath PR, Holden H, Andrews PW. Adaptation to culture of human embryonic stem cells and oncogenesis in vivo. Nat Biotechnol. 2007;25:207–215. doi: 10.1038/nbt1285. [DOI] [PubMed] [Google Scholar]
- Barbash-Hazan S, Frumkin T, Malcov M, Yaron Y, Cohen T, Azem F, Amit A, Ben-Yosef D. Preimplantation aneuploid embryos undergo self-correction in correlation with their developmental potential. Fertil Steril. 2009;92:890–896. doi: 10.1016/j.fertnstert.2008.07.1761. [DOI] [PubMed] [Google Scholar]
- Ben-David U, Benvenisty N. The tumorigenicity of human embryonic and induced pluripotent stem cells. Nat Rev Cancer. 2011;11:268–277. doi: 10.1038/nrc3034. [DOI] [PubMed] [Google Scholar]
- Ben-David U, Benvenisty N, Mayshar Y. Genetic instability in human induced pluripotent stem cells: classification of causes and possible safeguards. Cell Cycle. 2010;9:4603–4604. doi: 10.4161/cc.9.23.14094. [DOI] [PubMed] [Google Scholar]
- Ben-Yosef D, Malcov M, Eiges R. PGD-derived human embryonic stem cell linesas a powerful tool for the study of human genetic disorders. Mol Cell Endocrinol. 2008;282:153–158. doi: 10.1016/j.mce.2007.11.010. [DOI] [PubMed] [Google Scholar]
- Biancotti JC, Narwani K, Buehler N, Mandefro B, Golan-Lev T, Yanuka O, Clark A, Hill D, Benvenisty N, Lavon N. Human embryonic stem cells as models for aneuploid chromosomal syndromes. Stem Cells. 2010;28:1530–1540. doi: 10.1002/stem.483. [DOI] [PubMed] [Google Scholar]
- Buard J, de Massy B. Playing hide and seek with mammalian meiotic crossover hotspots. Trends Genet. 2007;23:301–309. doi: 10.1016/j.tig.2007.03.014. [DOI] [PubMed] [Google Scholar]
- Catalina P, Bueno C, Montes R, Nieto A, Ligero G, Sanchez L, Jara M, Rasillo A, Orfao A, Cigudosa J, et al. Genetic stability of human embryonic stem cells: a first-step toward the development of potential hESC-based systems for modeling childhood leukemia. Leuk Res. 2009;33:980–990. doi: 10.1016/j.leukres.2008.08.028. [DOI] [PubMed] [Google Scholar]
- Chung Y, Klimanskaya I, Becker S, Li T, Maserati M, Lu SJ, Zdrav-kovic T, Ilic D, Genbacev O, Fisher S, et al. Human embryonic stem cell lines generated without embryo destruction. Cell Stem Cell. 2008;2:113–117. doi: 10.1016/j.stem.2007.12.013. [DOI] [PubMed] [Google Scholar]
- Darr H, Mayshar Y, Benvenisty N. Overexpression of NANOG in human ES cells enables feeder-free growth while inducing primitive ectoderm features. Development. 2006;133:1193–1201. doi: 10.1242/dev.02286. [DOI] [PubMed] [Google Scholar]
- Draper JS, Smith K, Gokhale P, Moore HD, Maltby E, Johnson J, Meisner L, Zwaka TP, Thomson JA, Andrews PW. Recurrent gain of chromosomes 17q and 12 in cultured human embryonic stem cells. Nat Biotechnol. 2004;22:53–54. doi: 10.1038/nbt922. [DOI] [PubMed] [Google Scholar]
- Eiges R, Benvenisty N. A molecular view on pluripotent stem cells. FEBS Lett. 2002;529:135–141. doi: 10.1016/s0014-5793(02)03191-5. [DOI] [PubMed] [Google Scholar]
- Fox JL. FDA scrutinizes human stem cell therapies. Nat Biotechnol. 2008;26:598–599. doi: 10.1038/nbt0608-598. [DOI] [PubMed] [Google Scholar]
- Frumkin T, Malcov M, Telias M, Gold V, Schwartz T, Azem F, Amit A, Yaron Y, Ben-Yosef D. Human embryonic stem cells carrying mutations for severe genetic disorders. In Vitro Cell Dev Biol Anim. 2010;46:327–336. doi: 10.1007/s11626-010-9275-5. [DOI] [PubMed] [Google Scholar]
- Hussein SM, Batada NN, Vuoristo S, Ching RW, Autio R, Närvä E, Ng S, Sourour M, Hämäläinen R, Olsson C, et al. Copy number variation and selection during reprogramming to pluripotency. Nature. 2011;471:58–62. doi: 10.1038/nature09871. [DOI] [PubMed] [Google Scholar]
- Ilic D, Giritharan G, Zdravkovic T, Caceres E, Genbacev O, Fisher SJ, Krtolica A. Derivationof human embryonic stem cell lines from bio-psied blastomeres on human feeders with minimal exposure to xenomaterials. Stem Cells Dev. 2009;18:1343–1350. doi: 10.1089/scd.2008.0416. [DOI] [PubMed] [Google Scholar]
- Imreh MP, Gertow K, Cedervall J, Unger C, Holmberg K, Szöke K, Csöregh L, Fried G, Dilber S, Blennow E, Ahrlund-Richter L. Invitro culture conditions favoring selectionof chromosomalabnormal-ities in human ES cells. J Cell Biochem. 2006;99:508–516. doi: 10.1002/jcb.20897. [DOI] [PubMed] [Google Scholar]
- Itskovitz-Eldor J, Schuldiner M, Karsenti D, Eden A, Yanuka O, Amit M, Soreq H, Benvenisty N. Differentiation of human embryonic stem cells into embryoid bodies compromising the three embryonic germ layers. Mol Med. 2000;6:88–95. [PMC free article] [PubMed] [Google Scholar]
- Laurent LC, Ulitsky I, Slavin I, Tran H, Schork A, Morey R, Lynch C, Harness JV, Lee S, Barrero MJ, et al. Dynamic changes in the copy number of pluripotency and cell proliferation genes in human ESCs and iPSCs during reprogramming and time in culture. Cell Stem Cell. 2011;8:106–118. doi: 10.1016/j.stem.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Chao A, Chen CH, Chou T, Wang SY, Wang TH. Analysis of human meiotic recombination events with a parent-sibling tracing approach. BMC Genomics. 2011;12:434. doi: 10.1186/1471-2164-12-434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefort N, Feyeux M, Bas C, Féraud O, Bennaceur-Griscelli A, Tachdjian G, Peschanski M, Perrier AL. Human embryonic stem cells reveal recurrent genomic instability at 20q11.21. Nat Biotechnol. 2008;26:1364–1366. doi: 10.1038/nbt.1509. [DOI] [PubMed] [Google Scholar]
- Maitra A, Arking DE, Shivapurkar N, Ikeda M, Stastny V, Kassauei K, Sui G, Cutler DJ, Liu Y, Brimble SN, et al. Genomic alterations in cultured human embryonic stem cells. Nat Genet. 2005;37:1099–1103. doi: 10.1038/ng1631. [DOI] [PubMed] [Google Scholar]
- Marteyn A, Maury Y, Gauthier MM, Lecuyer C, Vernet R, Denis JA, Pietu G, Peschanski M, Martinat C. Mutant human embryonic stem cells reveal neurite and synapse formation defects in type 1 myotonic dystrophy. Cell Stem Cell. 2011;8:434–444. doi: 10.1016/j.stem.2011.02.004. [DOI] [PubMed] [Google Scholar]
- Mayshar Y, Ben-David U, Lavon N, Biancotti JC, Yakir B, Clark AT, Plath K, Lowry WE, Benvenisty N. Identification and classification of chromosomal aberrations in human induced pluripotent stem cells. Cell Stem Cell. 2010;7:521–531. doi: 10.1016/j.stem.2010.07.017. [DOI] [PubMed] [Google Scholar]
- Mertzanidou A, Spits C, Nguyen HT, Van de Velde H, Sermon K. Evolution of aneuploidy up to Day 4 of human preimplantation development. Hum Reprod. 2013a;28:1716–1724. doi: 10.1093/humrep/det079. [DOI] [PubMed] [Google Scholar]
- Mertzanidou A, Wilton L, Cheng J, Spits C, Vanneste E, Moreau Y, Ver-meesch JR, Sermon K. Microarray analysis reveals abnormal chromosomal complements in over 70% of 14 normally developing human embryos. Hum Reprod. 2013b;28:256–264. doi: 10.1093/humrep/des362. [DOI] [PubMed] [Google Scholar]
- Mitalipova MM, Rao RR, Hoyer DM, Johnson JA, Meisner LF, Jones KL, Dalton S, Stice SL. Preserving the genetic integrity of human embryonic stem cells. Nat Biotechnol. 2005;23:19–20. doi: 10.1038/nbt0105-19. [DOI] [PubMed] [Google Scholar]
- Närvä E, Autio R, Rahkonen N, Kong L, Harrison N, Kitsberg D, Borghese L, Itskovitz-Eldor J, Rasool O, Dvorak P, et al. High-resolution DNA analysis of human embryonic stem cell lines reveals culture-induced copy number changes and loss of heterozygosity. Nat Biotechnol. 2010;28:371–377. doi: 10.1038/nbt.1615. [DOI] [PubMed] [Google Scholar]
- Niclis J, Trounson AO, Dottori M, Ellisdon A, Bottomley SP, Verlinsky Y, Cram D. Human embryonic stem cell models of Huntington disease. Reprod Biomed Online. 2009;19:106–113. doi: 10.1016/s1472-6483(10)60053-3. [DOI] [PubMed] [Google Scholar]
- Reubinoff BE, Pera MF, Fong CY, Trounson A, Bongso A. Embryonic stem cell lines from human blastocysts: somatic differentiation in vitro. Nat Biotechnol. 2000;18:399–404. doi: 10.1038/74447. [DOI] [PubMed] [Google Scholar]
- Ruth Arnon MR, Ben-Or G, Berman-Schmidt S, Heyd D, Halperin M, Ishay R, Kasher A, Levy-Lahad E, Keynan A, Soreq H, et al. The use of embryonic stem cells for therapeutic research. 2001 http://bioethicsacademyacil.
- Spits C, Mateizel I, Geens M, Mertzanidou A, Staessen C, Vandeskelde Y, Van der Elst J, Liebaers I, Sermon K. Recurrent chromosomal abnormalities in human embryonic stem cells. Nat Biotechnol. 2008;26:1361–1363. doi: 10.1038/nbt.1510. [DOI] [PubMed] [Google Scholar]
- Telias M, Segal M, Ben-Yosef D. Neural differentiation of Fragile X human Embryonic Stem Cells reveals abnormal patterns of development despite successful neurogenesis. Dev Biol. 2013;374:32–45. doi: 10.1016/j.ydbio.2012.11.031. [DOI] [PubMed] [Google Scholar]
- Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- Vanneste E, Voet T, Le Caignec C, Ampe M, Konings P, Melotte C, Debrock S, Amyere M, Vikkula M, Schuit F, et al. Chromosome instability is common in human cleavage-stage embryos. Nat Med. 2009;15:577–583. doi: 10.1038/nm.1924. [DOI] [PubMed] [Google Scholar]
- Wells D, Delhanty JD. Comprehensive chromosomal analysis of human preimplantation embryos using whole genome amplification and single cell comparative genomic hybridization. Mol Hum Reprod. 2000;6:1055–1062. doi: 10.1093/molehr/6.11.1055. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.