

Abstract
STAT protein species are well-known as transcription factors that regulate nuclear gene expression. Recent novel lines of research suggest new non-genomic functions of STAT5A/B and STAT6. It was discovered in human pulmonary arterial endothelial cells that STAT5A, including STAT5A-GFP, constitutively associated with the Golgi apparatus, and both STAT5A and B with the endoplasmic reticulum. Acute siRNA-mediated knockdown of STAT5A/B led to the rapid development of a dramatic cystic change in the endoplasmic reticulum (ER) characterized by deposition of the ER structural protein reticulon-4 (RTN4; also called Nogo-B) and the ER-resident GTPase atlastin-3 (ATL3) along cyst membranes and cyst-zone boundaries, accompanied by Golgi fragmentation. Functional consequences included reduced anterograde trafficking, an ER stress response (increased GRP78/BiP) and eventual mitochondrial fragmentation. This phenotype was “non-genomic” in that it was elicited in enucleated cytoplasts. In cross-immunopanning assays STAT5A and B species associated with ATL3, and the ER-lumen spacer CLIMP63 (also called cytoskeleton-associated protein 4, CKAP4) but not RTN4. From a disease significance perspective we posit that STAT5, which is known to be affected by estradiol-17β and prolactin, represents the gender-sensitive determinant in the pathogenesis of idiopathic pulmonary hypertension (IPAH), a disease which includes ER/Golgi dysfunctions but with a 2- to 4-fold higher prevalence in postpubertal women. A separate line of recent research produced evidence for the association of STAT6-GFP, but not STAT3-GFP, STAT3-DsRed, or STAT3-Flag, with mitochondria in live-cell, immunofluorescence, and immunoelectron microscopy. An N-terminal truncation of STAT6-GFP (1–459), which lacked the SH2 domain and Tyr-phosphorylation site, constitutively associated with mitochondria. Thus, the emergent new of biology STAT proteins includes non-genomic roles—structurally and functionally—in the three closely related membrane organelles consisting of the endoplasmic reticulum, Golgi apparatus, and mitochondria.
Keywords: STAT5A, STAT5B, STAT6, STAT3, nongenomic functions, endoplasmic reticulum, Golgi apparatus, mitochondria, pulmonary arterial hypertension, anterograde trafficking
Introduction
The transcriptional gene regulatory functions in the cell nucleus of the Tyr- and/or Ser-phosphorylated as well as the non-phosphorylated species of the 7 mammalian STAT proteins (STAT1, 2, 3, 4, 5A, 5B, and 6) and their alternatively spliced or proteolytically cleaved isoforms have been extensively discussed over the last two decades (reviewed in refs. 1 and 2). Thus, the mechanisms mediating the constitutive shuttling of nonphosphorylated or of phosphorylated STAT protein dimer complexes between the cytoplasm and the nucleus resulting in regulation of gene expression in the nucleus (“genomic functions”) have attracted extensive attention (reviewed in refs. 1–4). Within this context of constitutive shuttling, the common (mis)representation that STAT species “translocate” to the nucleus upon phosphorylation reflects largely a decrease in the rate of exit of phosphorylated STAT species from the nucleus of growth-factor or cytokine-stimulated cells due to increased DNA binding affinity and thus increased nuclear residence times of the phosphorylated species (reviewed in ref. 3). Strikingly, it has been clear from the very beginning of the discovery of the respective STAT proteins that the vast majority of the cellular pool of these proteins (≥85%) resided in the cytoplasm.4-6 Moreover, there have been reports that in at least some instances the majority of the cellular pool even of Tyr-phosphorylated STAT3 remained cytoplasmic in cytokine-stimulated cells in culture and in single cells in tissues in vivo.6,7 Thus, in the last few years the question of possible functions of STAT protein species in the cytoplasm itself—“non-genomic functions”—has begun to attract attention. These have included functions at the level of microtubules,8 focal adhesions,7,9 and sequestering endosomes.10-12 The present essay focuses on STAT protein involvement in the structure and function of cytoplasmic membrane organelles—endoplasmic reticulum, Golgi apparatus, and mitochondria.
In this overview we shall consider two aspects of the emerging non-genomic role of STAT protein species in mammalian cells. The major focus will be to summarize the involvement of STAT5A and STAT5B in maintaining the structure and function of the endoplasmic reticulum and Golgi apparatus (and by extension nuclear shape).13,14 Thematically, this new biology of STAT5 relates to maintaining intracellular organellar structures involved in the anterograde vesicular secretory pathway. This seems an example of biological economy in that a major transcriptional function of STAT5 species is the postpartum bulk synthesis and secretion of breast milk proteins.15,16 We shall also suggest an involvement of non-genomic STAT5 effects in the pathogenesis of pulmonary arterial hypertension—a disease with high morbidity and mortality and a clear sexual dimorphism (2- to 4-fold more prevalent in postpubertal women).13,14 We shall also relate these new findings to prior literature from 1999–2006 concerning the disruption of IL-6/caveolin-1/raft signaling and hyperactivation of PY-STAT3 in the pathogenesis of this disease.2,3,7 The second area of focus will be to evaluate recent data reporting the association of STAT3 with mitochondria,17 point to technical issues with the STAT3/mitochondria experimental data,18 and review definitive live-cell imaging evidence showing that it is STAT6-GFP which associates with mitochondria.18
Association of STAT5A with the Golgi Apparatus and Endoplasmic Reticulum
The technique of first washing cell cultures with a low digitonin-isotonic sucrose buffer to remove bulk soluble STAT proteins followed by fixation assisted in uncovering the association of endogenous STAT5A with the Golgi apparatus and centrosomes in immunofluorescence studies.13 It was observed that STAT5A immunofluorescence was constitutively associated with the Golgi apparatus and centrosomes (and abscission complexes) in cultures of primary human pulmonary arterial endothelial and smooth muscle cells (HPAEC and HPASMC respectively) (Fig. 1A).13 (These cells were used because of our long-term interest in the pathogenesis of idiopathic pulmonary hypertension [reviewed in refs. 3 and 13]). Replacing the customary Triton permeabilization after fixation with digitonin permeabilization disclosed the association of STAT5A with the endoplasmic reticulum (ER) (Fig. 1C).14 These experiments also disclosed the presence of STAT5B in the perinuclear ER sheet region.14 The Golgi and ER association was “constitutive” and did not require Tyr- or Ser-phosphorylation. In contrast STAT5A which associated with centrosomes was Ser-phosphorylated. The constitutive association of STAT5A with the Golgi apparatus was also evident upon transfection of cells with an exogenous expression vector for STAT5A-GFP (Fig. 1B).13 Mutants of STAT5A-GFP at either or both of the Ser- or Tyr-phosphorylation sites all exhibited Golgi association.
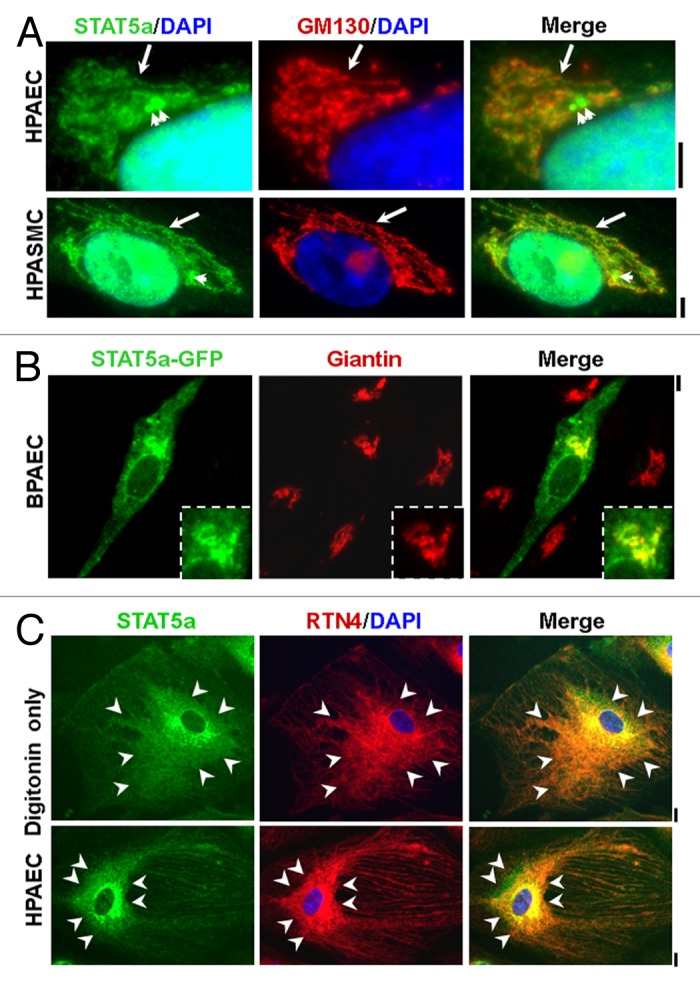
Figure 1. Association of STAT5A with the Golgi apparatus, endoplasmic reticulum and centrosomes. (A) Immunofluorescence imaging of the association of STAT5A with the Golgi tether GM130 (long arrow) and centrosomes (short arrows) in human pulmonary arterial endothelial (HPAEC) and smooth muscle cells (HPASMC) after fixation and then Triton permeabilization. Scale bars = 5 µm. (B) Fluoresence imaging of bovine PAECs transfected with a human STAT5A-GFP expression vector after fixation together with immunofluorescence imaging of the Golgi tether giantin. Scale bar = 5 µm; inset shows Golgi region at higher magnification. (C) Immunofluorescence colocalization of STAT5A with the endoplasmic reticulum (ER) structural protein RTN4/Nogo-B in HPAECs after fixation and permeabilization with only digitonin. Arrowheads demarcate the region of ER sheets; Scale bars = 10 µm. Illustrations in this composite figure were adapted from references 13 and 14.
The consequences of acute STAT5A/B knockdown using siRNAs on the structure and function of the ER-Golgi-mitochondrial axis were unexpected. In these acute knockdown experiments there was 70–80% reduction of STAT5A and STAT5B (with some crossover) in 1–2 d with the best results obtained using both siRNA preparations (Fig. 2).13 This acute knockdown of STAT5A/B led to cystic dilatation of the ER, dilatation, and fragmentation of the Golgi cisternae, lunate distortion of the nucleus and mitochondrial fragmentation in cystic ER zones in several different cell types (Fig. 3). There was a marked tubule to cyst or tubule to sheet to cyst change in the ER, with the development of juxtanuclear cysts in which the outer nuclear membrane had been pulled away from the inner nuclear membrane (thus distinguishing this phenotype from simple “ER stress” in which the nucleus remains intact). The cyst zone boundaries in the periphery of the cytoplasm were demarcated by deposition of the ER structural protein reticulon-4b (RTN4/Nogo-B) and the ER- and Golgi-resident GTPase atlastin-3 (ATL3). Remarkably, the nucleus was twisted in shape with an ice-cream scoop scalloped appearance. The concave side of the lunate nuclear deformation corresponded to a cyst due to separation of the outer and inner nuclear membranes; the convex side showed marked deposition of RTN4 (again distinguishing this phenotype from simple “ER stress”). Cells with STAT5A/B knockdown displayed decreased anterograde VSV-G trafficking, mitochondrial fragmentation and decreased mitochondrial function13,19. The observation that STAT5A/B knockdown altered the structure of cytoplasmic organelles—the ER and Golgi (and eventually mitochondria) in the present instance—was without precedent. Off-target effects of the particular STAT5A/B siRNAs used in these studies were thought unlikely to contribute to the present observations in that (1) siRNAs for STAT1, STAT3, RTN4, or ATL3 did not elicit the cystic phenotype, (2) the human-specific STAT5A/B siRNAs did not elicit a cystic phenotype in mouse embryo fibroblasts (MEFs) although murine-specific STAT5A/B siRNAs did, and (3) STAT5a/b−/− null MEFs also displayed altered ER/Golgi dynamics. Specifically, compared with early passage wt MEFs, early passage STAT5a/b−/− null MEFs responded to the stress of plating by exhibiting a flagrant cystic ER phenotype which was however transient and diminished by 12–24 h. Moreover, such STAT5a/b−/− null MEFs displayed persistent Golgi enlargement and fragmentation.13
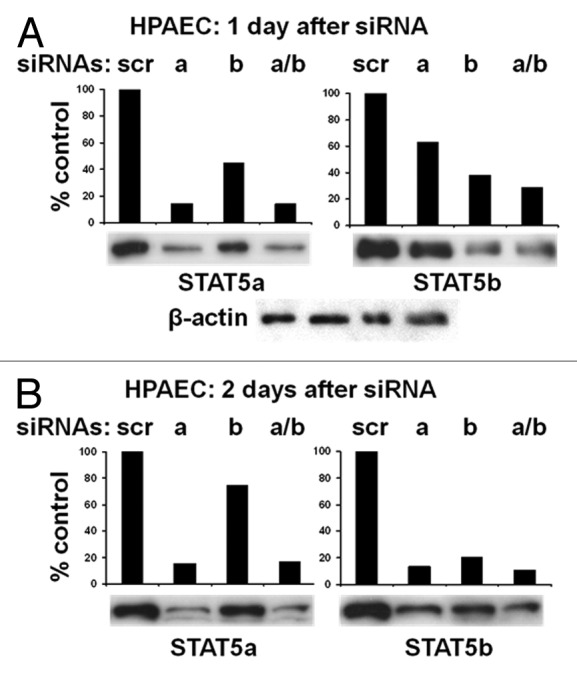
Figure 2. Acute knockdown of STAT5A or STAT5B or both in HPAECs by respective siRNAs. (A and B) The content of respective STAT5A and STAT5B proteins in cell extracts prepared from HPAECs either 1 d (A) or 2 d (B) after transfection with the indicated siRNAs was evaluated using western blotting. Equal amounts of total cellular protein (approximately 14 µg/lane) were loaded in each of the four lanes in each gel; reprobing for β-actin confirmed equivalent protein loading in (A). The densitometric quantitations are of blots immediately below each graph. Illustration was adapted from reference 13.
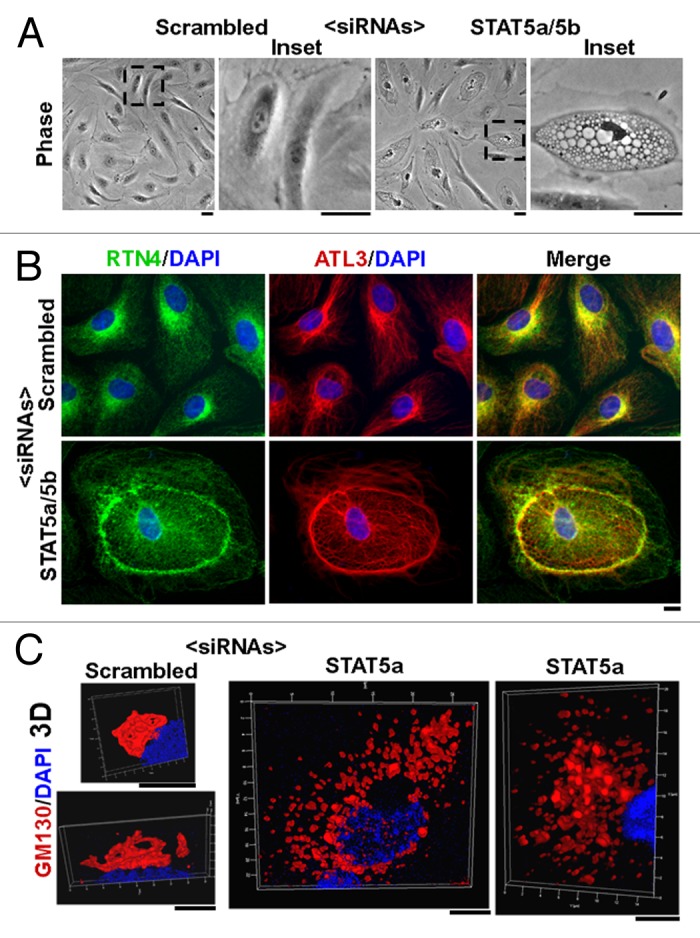
Figure 3. The cystic ER/Golgi fragmentation/lunate nucleus phenotype produced by STAT5A/B siRNAs in HPAEC cultures. (A) Phase-contrast images of cultures 18 h after transfection with respective siRNAs. Scale bars = 25 µm. (B) Immunofluorescence characterization of the cystic ER phenotype showing the cystic change and increased deposition of RTN4 and ATL3 at the cyst-zone boundaries. Scale bar = 10 µm. (C) HPAEC cultures transfected with scrambled or STAT5A siRNA were immunostained for GM130 (Golgi marker) and DAPI (nucleus marker) and imaged using z-stack data collection and rendered in 3D. Scale bars = 5 µm. Illustrations in this composite figure were adapted from references 13 and 14.
STAT5A/B Knockdown Produces a Cystic ER Phenotype in Enucleated Cytoplasts
Following the discovery of the association of STAT5A with the Golgi apparatus and the observation that acute siRNA-mediated knockdown of STAT5A/B led to an extensive cystic change in ER structure and to Golgi fragmentation,13 the question of whether manifestations of this phenotype were dependent on alterations in the transcriptional/nuclear functions of STAT5 species became a critical mechanistic question. That development of the cystic ER phenotype was unaffected by the transcriptional inhibitor 5,6-dichloro-1-β-D-ribofuranosylbenzimidazole (DRB) suggested a nongenomic basis for the development of the cystic ER/fragmented Golgi phenotype in response to STAT5A/B siRNAs.13,14 However, such inhibitor effects could be leaky. This question was addressed definitively in experiments using enucleated cytoplasts.14
The cytochalasin B-centrifugation method20 was applied to adherent cultures of primary human pulmonary arterial endothelial cells grown in 35 mm dishes to generate enucleated cytoplasts (65–75% of enucleated cells). Such cytoplasts showed a normal ER structure and a compact Golgi apparatus. It was thus possible 5 h later to expose such cultures to respective siRNAs and to monitor development of organellar changes at the single-cell level in DAPI-negative cytoplasts. Such provably nucleus-free cytoplasts developed the cystic ER/fragmented Golgi phenotype in response to acute STAT5A/B siRNA-knockdown.14 Thus, STAT5A/B knockdown modulated ER and Golgi structure independent of nuclear transcription or any other nuclear function, consistent with the direct localization of STAT5 species in the ER/Golgi.
Interactions between STAT5 Species and ER/Golgi-Resident Organellar Proteins
The question of how STAT5 species might interact with the ER/Golgi was addressed in magnetic-bead cross-immunopanning and western blotting experiments using antibodies to STAT5A and STAT5B and the organelle-associated proteins RTN4, ATL3, and CLIMP63/CKAP4 (Fig. 4).13,14 Anti-STAT3 pAb and competition with respective relevant or irrelevant peptides in the immunopanning were used as controls. The data obtained showed an association between STAT5A and STAT5B with ATL3 and CLIMP63 but not with RTN4. Since GTPase activity of atlastin species is thought to mediate the constant and rapid process of homotypic fusion cycles between ER tubules, our working hypothesis is that STAT5 may be a necessary co-factor for the ATL3 GTPase activity, and thus reduced STAT5 would lead to reduced homotypic fusion and loss of membrane curvature (i.e., development of cysts). Since we have also detected a palmitoylation motif within the amino acid sequence of STAT5A (and a weaker such motif in STAT5B), an additional/alternative possibility is the association of STAT5A with endomembranes resulting from palmitoylation.
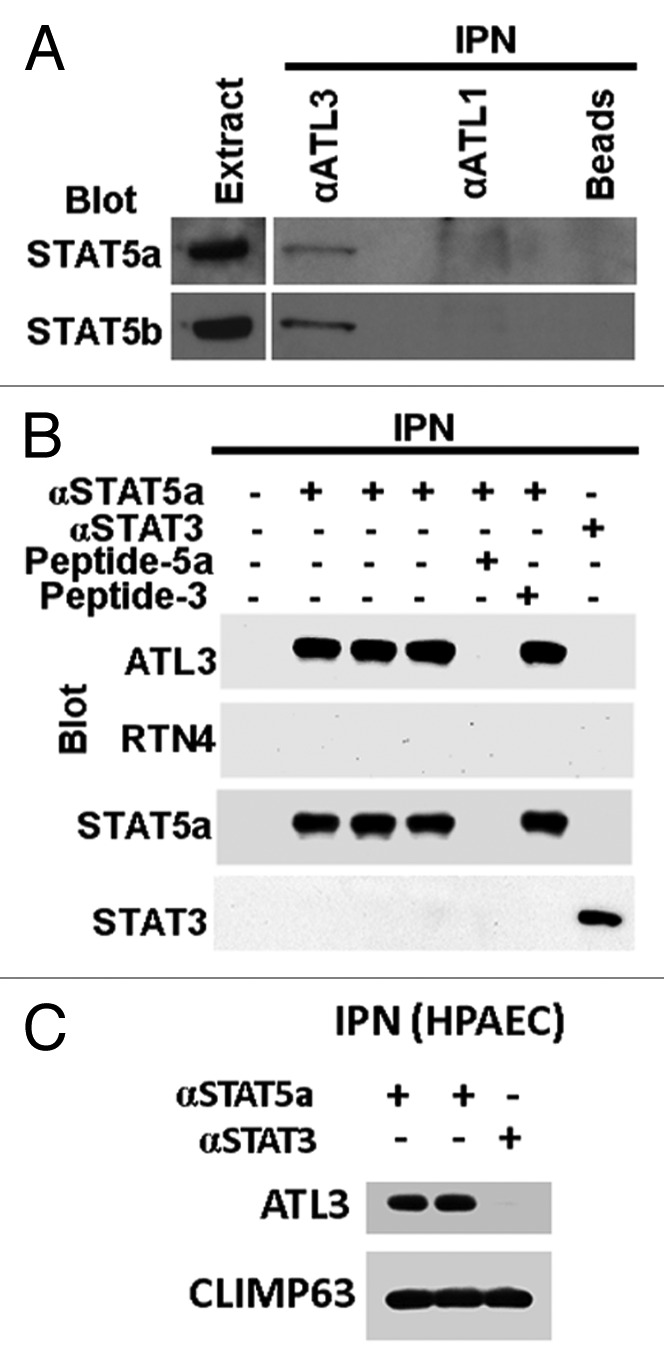
Figure 4. Association between STAT5A and STAT5B with ATL3 but not RTN4 in magnetic-bead cross-immunopanning assays and western blotting. (A) Immunopanning (IPN) of proteins from extracts of human endothelial cells (EA.hy926) was performed using anti-ATL3 or anti-ATL1 rabbit pAb as well as control beads alone (endothelial cells do not express ATL1; thus this also acts as a “negative” IgG control). The immunoisolates were western blotted for STAT5A or STAT5B as indicated. (B) Immunopanning was performed using extracts from human endothelial cells (EA.hy926 cells) using anti-STAT5A rabbit pAb in triplicate together with controls which included absence of any IgG, use of an irrelevant pAb (anti-STAT3 pAb) and prior incubation of the anti-STAT5A pAb with either relevant competing peptide (Peptide-5A) or an irrelevant competing peptide (Peptide-3). Western blotting of the immunoisolated complexes was performed as indicated. (C) Equal aliquots of HPAEC extracts were subjected to magnetic-bead immunopanning using anti-STAT5A or anti-STAT3 pAb. The immunoisolates were sequentially western blotted using Abs to ATL3 or CLIMP63 as indicated. Illustrations in this composite figure were adapted from references 13 and 14.
The STAT5A/B siRNA Knockdown Phenotype Includes Conventional “ER Stress”
There is a large amount of literature on the subject of what is colloquially called ER stress (reviewed in refs. 21–23). This phenomenon and term encompasses the response of cells to deal with an excess of imperfectly folded proteins, imperfectly glycosylated proteins, proteins whose turnover has been impeded and a variety of other “stressors.” These pathways begin in the ER/Golgi (e.g., the Ire1α, XBP1/ATF6, and PERK pathways), and mainly converge on the nucleus to affect transcriptional regulation of genes for proteins that act as chaperones and scavengers in the ER lumen (e.g., glucose-regulated protein 78 [GRP78/BiP]). The cystic phenotype produced in human pulmonary arterial endothelial cells as a result of acute STAT5A/B knockdown included the accumulation of KDEL-mCherry (a marker of ER stress) and CLIMP63-myc in the cystic lumen.14 Moreover, similar cystic cells produced in the human endothelial cell line EA.hy926 showed the marked and increased accumulation of GRP78/BiP in the ER cyst lumen (a conventional readout of ER stress). Thus it is clear that activation of conventional ER stress pathways is part of the STAT5A/B knockdown phenotype.
Implications in a Human Disease: Pulmonary Arterial Hypertension (PAH)
Insights concerning the non-genomic effects of STAT5 surfaced in studies performed using HPAECs and HPASMCs because of our long-standing interest in Golgi blockade in pulmonary arterial hypertension.24 Idiopathic pulmonary arterial hypertension (IPAH) is a relentlessly fatal disease with a sexual dimorphism in that women are more susceptible with a disease linkage to mutations in BMPR2 (reviewed in ref. 19). Histologically, lung vascular lesions in this disease have been reported to contain “plump” and “vacuolated” cells. We previously reported Golgi enlargement and fragmentation, and increased cytoplasmic dispersal of giantin in pulmonary vascular cells within IPAH lesions accompanied by defects in intracellular trafficking and hyperactivation of PY-STAT3.24-29 Strikingly, the electron microscopy (EM) data showing cystic ER dilatation in HPAECs upon STAT5A/B knockdown13 were reminiscent of the EM data of Smith and Heath30,31 from the late 1970s showing cystic dilatation of the ER in cells in a pulmonary vascular lesion in a patient with PAH as well as in PAECs in the hypoxic rat. Very recently we reported observing cystic cells with lunate distortion of the nucleus with RTN4 lining and cytoplasmically sequestered STAT5A in vascular lesions in IPAH.13 Sutendra et al.32 reported increased accumulation of RTN4/Nogo-B and activation of the ATF6 pathway in lesion tissues in IPAH. Additionally, mice with RTN4−/− deletions had reduced susceptibility to hypoxia-induced PAH. The new STAT5 data raise the question of whether a reduction in STAT5 in pulmonary vascular cells and consequent effects on anterograde trafficking might also contribute to the pathogenesis and observed ER stress in this disease.19
Indeed, given the low penetrance (10–15%) of BMPR2 mutations/deletions in causing overt disease even in familial PAH investigators have searched extensively for candidate “second-hit” genes (reviewed in ref. 19). The observation that VSV-G trafficking was significantly inhibited upon combining the functional haploinsufficiencies of STAT5A and BMPR2 (using siRNA knockdowns)19 suggested that STAT5A had the potential to be such a “second-hit” gene. Moreover it is noteworthy that STAT5 species are known to be estrogen- and prolactin-responsive15,16 and thus STAT5 biology might well underlie sexual dimorphic features of IPAH (higher prevalence in women).19 This represents an exciting new area of research in the pathogenesis of a fatal human disease.
However, STAT5 is not the only STAT protein implicated in the pathogenesis of PAH. Our interest in trafficking dysfunctions in the pathogenesis of PAH25 began with the twin discoveries in 1999 by Bhargava et al.33 of IL-6 involvement in the monocrotaline-rat model of PAH and our own understanding of the commencement of IL-6/STAT3 signaling at plasma membrane caveolin-1/rafts.34,35 We thus investigated whether there was activation of PY-STAT3 in endothelial cells in culture exposed to monocrotaline pyrolle and in the monocrotaline-rat model in vivo. We observed disruption of caveolin-1 rafts accompanied by hyperactivation of STAT3 signaling, and presented these findings already in 2003–2004.25,34,35 Lisanti and colleagues also reported similar findings relating the hyperactivation of STAT3 inversely to the loss of caveolin-1 either due to genetic deletion or in response to monocrotaline.36,37 Moreover, the latter investigators were able to block the STAT3 activation and block the disease using a caveolin-1 peptide.37 Mukhopadhyay et al.7 extended these studies to the human disease and showed remarkably that PY-STAT3 remained exclusively cytoplasmic in endothelial and smooth muscle cells in the vascular lesions in the monocrotaline–rat model and in human IPAH. It was the discovery of the inverse relationship between the loss of caveolin-1 rafts and hyperactivation of PY-STAT3 that led us to investigate defective anterograde membrane trafficking as a mechanism that might underlie the pathogenesis of PAH as well as the association of various STAT proteins with intracellular membrane organelles.24
In light of the existence of this prior literature from 1999–2006 cited above, it is incorrect for Paulin et al.38 in a review written in 2012 in this journal to state that “a role of STAT3 in PAH has been [first] suggested in 2007 […] and strengthened in the last couple of years, even leading to the conclusion that STAT3 activation might be an early event in PAH etiology, …” That conclusion was already presented in 2003–2004 from the laboratories of Mathew, Sehgal and Lisanti deriving from data going back to 1999 (see citations above). Moreover, Paulin et al.38 fail to consider the mechanism of an inverse relationship between caveolin-1/raft disruption and hyperactivation of IL-6/STAT3 signaling, and more generally changes in membrane trafficking in PAH.
Difficulties with Recent Claims of the Association of STAT3 with Mitochondria
Beginning in 1999–2000 we reported data showing the association of endogenous STAT3 in Hep3B cells with intracellular membranes including cell fractions that contained a mitochondrial marker.4,10,39-41 (These studies were performed in human Hep3B hepatocytes because this cell is a professional-going responder to IL-6 in terms of the synthesis of acute phase plasma proteins [reviewed in refs. 2–4]). However, extensive studies showed that this STAT3 was associated with plasma membrane rafts, endosomes and lysosomes.4,7,10,11,39-41 Indeed live-cell STAT3-GFP studies presented in 2007–2008 showed clearly the association of STAT3-GFP with endosomes and lysosomes but not the MitoTracker-positive mitochondria.7,11
Nevertheless, beginning in 2009 several investigators inferred the constitutive association of the transcription factor STAT3 with mitochondria in various human and murine cell types based upon observing the presence of STAT3 in mitochondria-enriched cell fractions as assayed by western blotting.17,42-44 It is noteworthy that these studies were accompanied by controls showing the absence of soluble cytosolic proteins in the mitochondria-enriched pellet but no effort was made to address contamination by other membrane organelles. It was simply stated that such pellets represented “pure” mitochondria. While, molecularly modified STAT3 carrying an engineered mitochondrial targeting sequence was reported able to modulate mitochondrial energy-generation functions,17,42 no microscopy evidence using molecular tools for the association of STAT3 with mitochondria has been forthcoming. We note that we ourselves have observed mitochondrial immunofluorescence labeling using a particular commercial rabbit serum antibody preparation designated “anti-STAT3” (including its competition by the STAT3 peptide). However, such data are open to interpretation about the composition of the polyclonal IgG in rabbit serum.
Subsequently, Cimica et al.45 also reported that exogenously expressed STAT3-GFP did not associate with MitoTracker-positive organelles in the cytoplasm of HeLa or Hep3B cells. Additionally, Phillips et al.46 reported their inability to detect any STAT3 by mass spectrometric approaches in mitochondrial fractions derived from porcine and murine heart and liver. The absence of microscopy data (STAT3-GFP fluorescence or immunogold electron microscopy) from unfractionated cells showing association of STAT3 with mitochondria remained a difficulty.
The potential functional importance of the association of a STAT-protein family member with mitochondria led us to revisit the possible association of STAT3-GFP with mitochondria using a detergent-dissection approach in adherent cell cultures (as in Fig. 1). In these experiments a low-concentration digitonin-sucrose buffer was used to remove bulk STAT proteins from the cell cytoplasm followed by fluorescence or immunofluorescence microscopy. We have remained unable to confirm the association of GFP-, DsRed-, or Flag-tagged STAT3 with mitochondria (Fig. 5).18 It has been suggested that the various tags used in these studies which were attached to the C-terminus of STAT3 might have interfered with the mitochondrial import of the STAT3 protein. This possibility, while theoretically possible, needs careful consideration in that a C-terminal GFP tag did not interfere with the mitochondrial import of STAT6 or even of an N-terminal half-truncation of STAT618 (also see below). It is now for the “STAT3 in mitochondria” proponents to provide definitive evidence for the association of endogenous STAT3 with mitochondria without resorting to post hoc explanations.
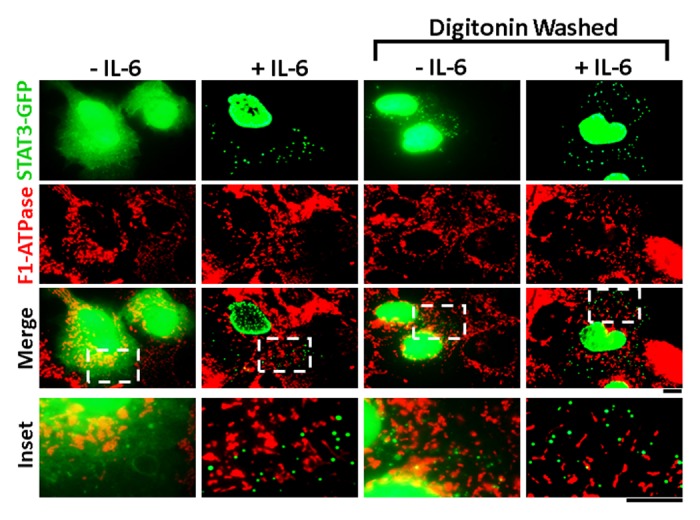
Figure 5. Lack of association of STAT3-GFP with mitochondria. Hep3B cells cultured in 6-well plates were transfected with the STAT3-GFP expression plasmid and one day later exposed to IL-6 for 30 min or left unstimulated. The cultures were then fixed or first washed 4× with the digitonin (50 µg/ml)-0.3 M sucrose buffer, then fixed followed by fluorescence imaging for GFP and immunofluorescence imaging for F1-ATPase to mark mitochondria as indicated. Insets show the boxed areas (broken white lines) at higher magnification. Scale bars = 10 µm. Illustration adapted from reference 18.
Live-Cell Imaging of the Association of STAT6-GFP with Mitochondria
The broader question of a possible association of other STAT-family of proteins with mitochondria was addressed by first using immunofluorescence assays in Hep3B and human pulmonary arterial endothelial and smooth muscle cells using commercial pAbs for all seven STAT proteins.18 Strong anti-STAT6-immunolocalization with mitochondria was apparent in fluorescence and electron microscopy assays of cells first washed with a digitonin-sucrose buffer to remove bulk soluble STAT proteins. Critically, in live-cell imaging studies, STAT6-GFP, but not N1-GFP, was observed to constitutively colocalize with MitoTracker- and tetramethylrhodamine ethyl ester-positive mitochondria, and with mitochondrial F1-ATPase when assayed by immunofluorescence after fixation18 (Fig. 6A). This association was Tyr-phosphorylation independent in that a STAT6 truncated protein (STAT61–459-GFP) which lacked the SH2 domain (517–632) and the cytokine-activated Y641 phosphorylation site also accumulated in MitoTracker-positive mitochondria in live-cell imaging (Fig. 6B). This was consistent with the unexpected discovery that anti-STAT6-immunofluorescence also associated with mitochondria in MEFs from both wild-type and the STAT6SH2-/SH2- mouse.18 MEFs from the latter mouse, which had been engineered in 1996 to be deleted in the STAT6 SH2 domain (amino acids 505–584) expressed an immune-specific ~50 kDa protein detectable in whole cell and mitochondria-enriched fractions. Taken together, these data provide the first definitive evidence of the association of any STAT-protein family member with mitochondria—that of STAT6.
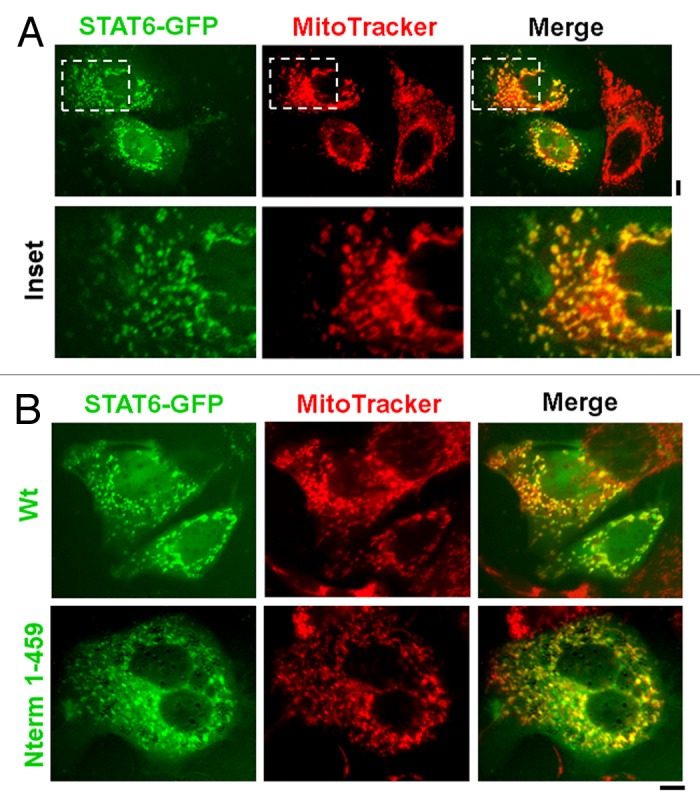
Figure 6. Live-cell imaging of STAT6-GFP and of its N-terminal truncation with mitochondria. (A) Hep3B cells in 35-mm plates were transfected with the STAT6-GFP expression plasmid and 2 d later the cultures were exposed to MitoTracker Red CMXRos at 100 nM for 15 min. After washing with PBS the live cells were imaged using a 40× water-immersion objective in green (STAT6-GFP) and red (MitoTracker). Insets depicted within each panel in the top row are shown at high magnification in the bottom row. Scale bars = 10 µm. Quantitative colocalization analyses using the Pearson’s and Costes’ plugins in Image J confirmed colocalization between STAT6-GFP and MitoTracker fluorescence at a setting of p < 0.05 in this experiment. (B) Hep3B cells in 35-mm plates were transfected with expression constructs for either the full-length or the N-terminal 1–459 truncated version of STAT6-GFP. Two days later the plates were exposed to MitoTracker Red CMXRos at 100 nM for 15 min (B) and imaged as (A). Scale bars = 10 µm. Illustrations in this composite figure were adapted from reference 18.
That proteins with major functions in the nucleus can also serve significant functions in the mitochondria is an emergent theme in cell biology. A recent example is the discovery that the nuclear telomerase complex protein TIN2 is imported into mitochondria, there processed by proteolytic cleavage to serve functions that include generation of reactive oxygen species.47 That the full-length ~100 kDa STAT6 undergoes specific proteolytic processing to a ~65 kDa fragment has already been documented.48 More recently, Chen et al.49 reported the recruitment of STAT6 to the endoplasmic reticulum in response to virus infection to serve antiviral functions independent of the Janus-activated kinases. However, inferences of the organellar association of STAT proteins and their potential nongenomic functions in the cytoplasm need careful data evaluation. Thus a recent claim of cytokine-stimulated translocation of STAT5 (including of Tyr-phosphorylated STAT5) to mitochondria in leukemic T cells exposed to IL-3 rests on inadequately controlled cell-fractionation and immunofluorescence evidence50 in that the data do not exclude the association of PY-STAT5 with cytoplasmic endosomes, as is already known for IL-6-activated PY-STAT3.10-12
STAT6 lacks an obvious mitochondrial import signal sequence. Nevertheless, the N-terminal 459 amino acids appear sufficient to mediate this mitochondrial association. That this mitochondrially-targeted fragment of STAT6 lacked the SH2 domain (amino acids 1–459) and the Y641 phosphorylation site indicates that the mitochondrial import mechanism involved is likely independent of cytokine-mediated activation of STAT6.
Comments
The field of STAT protein biology, which arose out of considerations of transcriptional regulation two decades ago, is on the cusp of a paradigm addition. While the emphasis on genomic transcriptional regulation continues to be deservedly well-placed and its importance easy to understand, reapplying simple regular-going cell biology methods with critical attention to technical issues (essentially overlooked in the decades-old rush to collect transcription-related STAT protein data) has now disclosed new aspects of the localization and functions of STAT proteins in the cytoplasm. However, with an increasing and fashionable focus on the biology of STAT proteins in the cytoplasm, it is necessary that modern-day investigators (and journal reviewers) dust off their old textbooks of cell biology methods and reacquaint themselves with some of the simple principles of cell fixation, detergent effects, and cell fractionation established by cell biologists in the 1970s and 80s so as to carry their investigations forward on a sound technical footing. The technical assumptions and approximations that entered the STAT protein field over the last two decades while pursuing the techniques of making nuclear extracts, DNA binding studies, and reporter-construct assays are not necessarily valid when evaluating basic cell biology in the cytosol and function in association with cytoplasmic organelles. Fortunately established cell biology methods already provide us with the necessary tools—however, these need to be applied with care and an understanding of their shortcomings so as not to generate a lot of noise in the literature as has been happening in the area of claims of finding assorted STAT proteins in “mitochondria” including claims of their rapid mitochondrial import accompanied by various phosphorylation events within minutes of exposing cells to respective cytokines or growth factors (some examples have been cited above). Moreover, modern-day investigators (and reviewers) would do well to familiarize themselves with earlier studies in the STAT protein field so as not to overlook easily avoided pitfalls.
The new “paradigm addition” in STAT proteins biology in recent years concerns the roles of these proteins in cytoplasmic organellar functions. These include—structurally and functionally—the axis of three closely-related membrane organelles consisting of the endoplasmic reticulum, Golgi apparatus, and mitochondria. What lies ahead is elucidating the specific details of which STAT protein does what, where, when and how in the cytoplasm—all questions clearly subject to further debate, technical dissection, and new insights.
Acknowledgments
Research in the author’s laboratory was supported in part by National Institutes of Health Research Grants R01 HL-087176 and R03 HL-114601
Glossary
Abbreviations:
- ATL3
atlastin-3
- BMPR2
bone morphogenetic protein receptor 2
- BPAEC
bovine pulmonary arterial endothelial cells
- CLIMP63
cytoskeleton linking protein 63 (also called cytoskeleton associated protein 4, CKAP4)
- DRB
5,6-dichloro-1-β-D-ribofuranosulbenzimidazole
- EM
electron microscopy
- ER
endoplasmic reticulum
- GRP78
glucose-regulated protein 78 (also called BiP)
- HPAEC
human pulmonary arterial endothelial cell
- HPASMC
human pulmonary arterial smooth muscle cell
- IPAH
idiopathic pulmonary arterial hypertension (also shortened to PAH)
- MEF
mouse embryo fibroblast
- RTN4
reticulon 4 (also called Nogo-B)
- VSV-G
vesicular stomatitis virus G glycoprotein
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/jak-stat/article/24860
References
- 1.Stark GR, Darnell JE., Jr. The JAK-STAT pathway at twenty. Immunity. 2012;36:503–14. doi: 10.1016/j.immuni.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sehgal PB, Levy DE, Hirano T. Signal Transducers and Activators of Transcription (STATs): Activation and Biology, Dordrecht, The Netherlands; Kluwer Academic Publishers, 2003:1-748 [Google Scholar]
- 3.Sehgal PB. Paradigm shifts in the cell biology of STAT signaling. Semin Cell Dev Biol. 2008;19:329–40. doi: 10.1016/j.semcdb.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ndubuisi MI, Guo GG, Fried VA, Etlinger JD, Sehgal PB. Cellular physiology of STAT3: Where’s the cytoplasmic monomer? J Biol Chem. 1999;274:25499–509. doi: 10.1074/jbc.274.36.25499. [DOI] [PubMed] [Google Scholar]
- 5.Haspel RL, Salditt-Georgieff M, Darnell JE., Jr. The rapid inactivation of nuclear tyrosine phosphorylated Stat1 depends upon a protein tyrosine phosphatase. EMBO J. 1996;15:6262–8. [PMC free article] [PubMed] [Google Scholar]
- 6.Sehgal PB, Kumar V, Guo G, Murray WC. Different patterns of regulation of Tyr-phosphorylated STAT1 and STAT3 in human hepatoma Hep3B cells by the phosphatase inhibitor orthovanadate. Arch Biochem Biophys. 2003;412:242–50. doi: 10.1016/S0003-9861(03)00050-X. [DOI] [PubMed] [Google Scholar]
- 7.Mukhopadhyay S, Shah M, Xu F, Patel K, Tuder RM, Sehgal PB. Cytoplasmic provenance of STAT3 and PY-STAT3 in the endolysosomal compartments in pulmonary arterial endothelial and smooth muscle cells: implications in pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2008;294:L449–68. doi: 10.1152/ajplung.00377.2007. [DOI] [PubMed] [Google Scholar]
- 8.Ng DC, Lin BH, Lim CP, Huang G, Zhang T, Poli V, et al. Stat3 regulates microtubules by antagonizing the depolymerization activity of stathmin. J Cell Biol. 2006;172:245–57. doi: 10.1083/jcb.200503021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Silver DL, Naora H, Liu J, Cheng W, Montell DJ. Activated signal transducer and activator of transcription (STAT) 3: localization in focal adhesions and function in ovarian cancer cell motility. Cancer Res. 2004;64:3550–8. doi: 10.1158/0008-5472.CAN-03-3959. [DOI] [PubMed] [Google Scholar]
- 10.Shah M, Patel K, Mukhopadhyay S, Xu F, Guo G, Sehgal PB. Membrane-associated STAT3 and PY-STAT3 in the cytoplasm. J Biol Chem. 2006;281:7302–8. doi: 10.1074/jbc.M508527200. [DOI] [PubMed] [Google Scholar]
- 11.Xu F, Mukhopadhyay S, Sehgal PB. Live cell imaging of interleukin-6-induced targeting of “transcription factor” STAT3 to sequestering endosomes in the cytoplasm. Am J Physiol Cell Physiol. 2007;293:C1374–82. doi: 10.1152/ajpcell.00220.2007. [DOI] [PubMed] [Google Scholar]
- 12.German CL, Sauer BM, Howe CL. The STAT3 beacon: IL-6 recurrently activates STAT 3 from endosomal structures. Exp Cell Res. 2011;317:1955–69. doi: 10.1016/j.yexcr.2011.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee JE, Yang YM, Liang FX, Gough DJ, Levy DE, Sehgal PB. Nongenomic STAT5-dependent effects on Golgi apparatus and endoplasmic reticulum structure and function. Am J Physiol Cell Physiol. 2012;302:C804–20. doi: 10.1152/ajpcell.00379.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee JE, Yang YM, Yuan H, Sehgal PB. Definitive evidence using enucleated cytoplasts for a nongenomic basis for the cystic change in endoplasmic reticulum structure caused by STAT5a/b siRNAs. Am J Physiol Cell Physiol. 2013;304:C312–23. doi: 10.1152/ajpcell.00311.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gouilleux F, Wakao H, Mundt M, Groner B. Prolactin induces phosphorylation of Tyr694 of Stat5 (MGF), a prerequisite for DNA binding and induction of transcription. EMBO J. 1994;13:4361–9. doi: 10.1002/j.1460-2075.1994.tb06756.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hennighausen L, Robinson GW. Interpretation of cytokine signaling through the transcription factors STAT5A and STAT5B. Genes Dev. 2008;22:711–21. doi: 10.1101/gad.1643908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wegrzyn J, Potla R, Chwae YJ, Sepuri NB, Zhang Q, Koeck T, et al. Function of mitochondrial Stat3 in cellular respiration. Science. 2009;323:793–7. doi: 10.1126/science.1164551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khan R, Lee JE, Yang Y-M, Liang F-X, Sehgal PB. Live-cell imaging of the association of STAT6-GFP with mitochondria. PLoS One. 2013;8:e55426. doi: 10.1371/journal.pone.0055426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang Y-M, Lane KB, Sehgal PB. Subcellular mechanisms in pulmonary arterial hypertension: combinatorial modalities that inhibit anterograde trafficking and cause BMPR2 mislocalization. Pulm Circ. 2013 doi: 10.1086/674336. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prescott DM, Myerson D, Wallace J. Enucleation of mammalian cells with cytochalasin B. Exp Cell Res. 1972;71:480–5. doi: 10.1016/0014-4827(72)90322-9. [DOI] [PubMed] [Google Scholar]
- 21.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–29. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 22.St-Pierre P, Dang T, Joshi B, Nabi IR. Peripheral endoplasmic reticulum localization of the Gp78 ubiquitin ligase activity. J Cell Sci. 2012;125:1727–37. doi: 10.1242/jcs.096396. [DOI] [PubMed] [Google Scholar]
- 23.Varadarajan S, Bampton ETW, Smalley JL, Tanaka K, Caves RE, Butterworth M, et al. A novel cellular stress response characterised by a rapid reorganisation of membranes of the endoplasmic reticulum. Cell Death Differ. 2012;19:1896–907. doi: 10.1038/cdd.2012.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sehgal PB, Lee JE. Protein trafficking dysfunctions: Role in the pathogenesis of pulmonary arterial hypertension. Pulm Circ. 2011;1:17–32. doi: 10.4103/2045-8932.78097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mathew R, Huang J, Shah M, Patel K, Gewitz M, Sehgal PB. Disruption of endothelial-cell caveolin-1α/raft scaffolding during development of monocrotaline-induced pulmonary hypertension. Circulation. 2004;110:1499–506. doi: 10.1161/01.CIR.0000141576.39579.23. [DOI] [PubMed] [Google Scholar]
- 26.Shah M, Patel K, Sehgal PB. Monocrotaline pyrrole-induced endothelial cell megalocytosis involves a Golgi blockade mechanism. Am J Physiol Cell Physiol. 2005;288:C850–62. doi: 10.1152/ajpcell.00327.2004. [DOI] [PubMed] [Google Scholar]
- 27.Sehgal PB, Mukhopadhyay S, Xu F, Patel K, Shah M. Dysfunction of Golgi tethers, SNAREs, and SNAPs in monocrotaline-induced pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1526–42. doi: 10.1152/ajplung.00463.2006. [DOI] [PubMed] [Google Scholar]
- 28.Sehgal PB, Mukhopadhyay S, Patel K, Xu F, Almodóvar S, Tuder RM, et al. Golgi dysfunction is a common feature in idiopathic human pulmonary hypertension and vascular lesions in SHIV-nef-infected macaques. Am J Physiol Lung Cell Mol Physiol. 2009;297:L729–37. doi: 10.1152/ajplung.00087.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee JE, Patel K, Almodóvar S, Tuder RM, Flores SC, Sehgal PB. Dependence of Golgi apparatus integrity on nitric oxide in vascular cells: implications in pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol. 2011;300:H1141–58. doi: 10.1152/ajpheart.00767.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith P, Heath D. Ultrastructure of hypoxic hypertensive pulmonary vascular disease. J Pathol. 1977;121:93–100. doi: 10.1002/path.1711210205. [DOI] [PubMed] [Google Scholar]
- 31.Smith P, Heath D. Electron microscopy of the plexiform lesion. Thorax. 1979;34:177–86. doi: 10.1136/thx.34.2.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sutendra G, Dromparis P, Wright P, Bonnet S, Haromy A, Hao Z, et al. The role of Nogo and the mitochondria-endoplasmic reticulum unit in pulmonary hypertension. Sci Transl Med. 2011;3:88ra55. doi: 10.1126/scitranslmed.3002194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bhargava A, Kumar A, Yuan N, Gewitz MH, Mathew R. Monocrotaline induces interleukin-6 mRNA expression in rat lungs. Heart Dis. 1999;1:126–32. [PubMed] [Google Scholar]
- 34.Sehgal PB, Shah M. Raft-STAT signaling and transcytoplasmic trafficking. In: Sehgal PB, Levy DE, Hirano T, eds. Signal Transducers and Activators of Transcription (STATs): activation and biology. Dordrecht, The Netherlands; Kluwer Academic Publishers, 2003; 247-67. [Google Scholar]
- 35.Sehgal PB. Plasma membrane rafts and chaperones in cytokine/STAT signaling. Acta Biochim Pol. 2003;50:583–94. [PubMed] [Google Scholar]
- 36.Jasmin JF, Mercier I, Hnasko R, Cheung MW, Tanowitz HB, Dupuis J, et al. Lung remodeling and pulmonary hypertension after myocardial infarction: pathogenic role of reduced caveolin expression. Cardiovasc Res. 2004;63:747–55. doi: 10.1016/j.cardiores.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 37.Jasmin JF, Mercier I, Dupuis J, Tanowitz HB, Lisanti MP. Short-term administration of a cell-permeable caveolin-1 peptide prevents the development of monocrotaline-induced pulmonary hypertension and right ventricular hypertrophy. Circulation. 2006;114:912–20. doi: 10.1161/CIRCULATIONAHA.106.634709. [DOI] [PubMed] [Google Scholar]
- 38.Paulin R, Meloche J, Bonnet S. STAT3 signaling in pulmonary arterial hypertension. JAK-STAT. 2012;1:223–33. doi: 10.4161/jkst.22366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guo GG, Patel K, Kumar V, Shah M, Fried VA, Etlinger JD, et al. Association of the chaperone glucose-regulated protein 58 (GRP58/ER-60/ERp57) with Stat3 in cytosol and plasma membrane complexes. J Interferon Cytokine Res. 2002;22:555–63. doi: 10.1089/10799900252982034. [DOI] [PubMed] [Google Scholar]
- 40.Sehgal PB, Guo GG, Shah M, Kumar V, Patel K. Cytokine signaling: STATS in plasma membrane rafts. J Biol Chem. 2002;277:12067–74. doi: 10.1074/jbc.M200018200. [DOI] [PubMed] [Google Scholar]
- 41.Shah M, Patel K, Fried VA, Sehgal PB. Interactions of STAT3 with caveolin-1 and heat shock protein 90 in plasma membrane raft and cytosolic complexes. Preservation of cytokine signaling during fever. J Biol Chem. 2002;277:45662–9. doi: 10.1074/jbc.M205935200. [DOI] [PubMed] [Google Scholar]
- 42.Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC, Levy DE. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science. 2009;324:1713–6. doi: 10.1126/science.1171721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jeong K, Kwon H, Min C, Pak Y. Modulation of the caveolin-3 localization to caveolae and STAT3 to mitochondria by catecholamine-induced cardiac hypertrophy in H9c2 cardiomyoblasts. Exp Mol Med. 2009;41:226–35. doi: 10.3858/emm.2009.41.4.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boengler K, Hilfiker-Kleiner D, Heusch G, Schulz R. Inhibition of permeability transition pore opening by mitochondrial STAT3 and its role in myocardial ischemia/reperfusion. Basic Res Cardiol. 2010;105:771–85. doi: 10.1007/s00395-010-0124-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cimica V, Chen HC, Iyer JK, Reich NC. Dynamics of the STAT3 transcription factor: nuclear import dependent on Ran and importin-β1. PLoS One. 2011;6:e20188. doi: 10.1371/journal.pone.0020188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Phillips D, Reilley MJ, Aponte AM, Wang G, Boja E, Gucek M, et al. Stoichiometry of STAT3 and mitochondrial proteins: Implications for the regulation of oxidative phosphorylation by protein-protein interactions. J Biol Chem. 2010;285:23532–6. doi: 10.1074/jbc.C110.152652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen LY, Zhang Y, Zhang Q, Li H, Luo Z, Fang H, et al. Mitochondrial localization of telomeric protein TIN2 links telomere regulation to metabolic control. Mol Cell. 2012;47:839–50. doi: 10.1016/j.molcel.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Suzuki K, Nakajima H, Kagami S, Suto A, Ikeda K, Hirose K, et al. Proteolytic processing of Stat6 signaling in mast cells as a negative regulatory mechanism. J Exp Med. 2002;196:27–38. doi: 10.1084/jem.20011682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen H, Sun H, You F, Sun W, Zhou X, Chen L, et al. Activation of STAT6 by STING is critical for antiviral innate immunity. Cell. 2011;147:436–46. doi: 10.1016/j.cell.2011.09.022. [DOI] [PubMed] [Google Scholar]
- 50.Chueh FY, Leong KF, Yu CL. Mitochondrial translocation of signal transducer and activator of transcription 5 (STAT5) in leukemic T cells and cytokine-stimulated cells. Biochem Biophys Res Commun. 2010;402:778–83. doi: 10.1016/j.bbrc.2010.10.112. [DOI] [PMC free article] [PubMed] [Google Scholar]