

Abstract
The endoplasmic reticulum (ER) is a central organelle for protein biosynthesis, folding, and traffic. Perturbations in ER homeostasis create a condition termed ER stress and lead to activation of the complex signaling cascade called the unfolded protein response (UPR). Recent studies have documented that the UPR coordinates multiple signaling pathways and controls various physiologies in cells and the whole organism. Furthermore, unresolved ER stress has been implicated in a variety of metabolic disorders, such as obesity and type 2 diabetes. Therefore, intervening in ER stress and modulating signaling components of the UPR would provide promising therapeutics for the treatment of human metabolic diseases.
Keywords: Diabetes, ER Stress, Metabolic Diseases, Obesity, Unfolded Protein Response
Introduction
The endoplasmic reticulum (ER)2 is a membrane-bound and structurally intricate organelle present in all eukaryotic cells and is the major place for the synthesis of secretory and membrane proteins along with membrane lipids and internal calcium storage (1). Newly synthesized proteins are modified and folded into their native structure within the ER lumen, which is tightly monitored by the ER quality control machinery. Protein synthesis in the ER is dynamically adjusted in coordination with the physiological status of cells. When the folding capacity of the ER fails to accommodate the load of unfolded proteins, ER homeostasis is perturbed to a condition referred to as “ER stress.” In response to ER stress, an adaptive mechanism termed the unfolded protein response (UPR) is implemented to re-establish homeostasis in the ER. The early role of UPR signaling is to increase expression of proteins that are involved in the ER folding machinery to enhance protein folding and also to attenuate general protein translation to reduce the load in the ER. Additionally, terminally misfolded proteins are translocated from the ER to the cytoplasm and then degraded by the proteasome, which is known as ER-associated degradation. However, UPR signaling would initiate cell death pathways when ER homeostasis is not recovered for prolonged periods of time.
Three branches of the UPR have been characterized in metazoans, and they are mediated by ER-located transmembrane proteins: IRE1 (inositol-requiring protein-1), PERK (protein kinase RNA-like ER kinase), and ATF6 (activating transcription factor-6) (Fig. 1). It is noteworthy that there is cross-talk between the UPR and other signaling pathways, such as inflammatory and metabolic signaling, and such profound orchestration is crucial for maintaining proper ER function and physiological homeostasis of cells. Furthermore, an increasing number of studies have shown that UPR signaling and ER stress are associated with pathophysiological and metabolic changes, including obesity, type 2 diabetes, atherosclerosis, heart and liver diseases, and neurodegenerative disorders (1). In this minireview, we summarize the current understanding of the molecular mechanism of IRE1, PERK, and ATF6 signaling; their integrated networks with other signaling pathways; and also the role of ER stress and UPR signaling in various metabolic disorders.
FIGURE 1.

UPR signaling and its cross-talk mediated by IRE1-XBP1, PERK and ATF6. Upon ER stress, the dissociation of GRP78 from the lumenal domain of IRE1, PERK, and ATF6 initiates UPR signaling. The direct binding of unfolded proteins to IRE1 also activates IRE1-mediated UPR. Active IRE1 selectively splices XBP1 mRNA to produce an active transcription factor (XBP1s) or down-regulates mRNAs or microRNAs (RIDD). Activated PERK phosphorylates eIF2α to attenuate global protein translation while selectively augmenting the translation of ATF4 and ATF5. An active form of ATF6 is generated upon cleavage by S1P and S2P after translocation to the Golgi from the ER. There are different cross-talks between UPR signaling components and other signal transduction pathways, such as inflammatory pathways (JNK, NF-κB, p38MAPK, and the NLRP3 inflammasome), apoptotic machinery (Bax, Bak, and BI-1), and metabolism (p85α/β, FoxO1, CRTC2, and SREBP-2). ERAD, ER-associated degradation.
IRE1 and XBP1 (X-box-binding Protein-1) Signaling Networks and Metabolic Regulation of Lipid and Glucose Homeostasis
IRE1 is the first identified signaling component of the UPR and is evolutionarily conserved from yeast to metazoans (2). There are two isoforms of IRE1 in mammals, IRE1α and IRE1β. IRE1α is ubiquitously expressed (3), whereas IRE1β is found primarily in the intestine and lung (4, 5). The role of IRE1α in the mediation of UPR signaling has been clearly established, but the role of IRE1β in the UPR is not yet clear. IRE1 is a type I ER-resident transmembrane protein consisting of an ER lumenal domain (sensing protein-folding status) and a cytoplasmic region containing a serine/threonine kinase domain and an RNase domain (2). Under normal conditions without ER stress, IRE1α exists as a monomer associated with GRP78 (glucose-regulated protein of 78 kDa) (6); however, upon ER stress, IRE1 forms a homodimer and oligomers, followed by release of GRP78 (6) and/or direct interaction of unfolded proteins with its ER lumenal domain (7). Dimerization and oligomerization lead to autotransphosphorylation of IRE1 at multiple sites, including phosphorylation of Ser-724 in mammalian IRE1α (8). These phosphorylation events lead to activation of the RNase domain, which selectively excises a 26-base fragment from full-length XBP1 (unspliced XBP1) mRNA (9). Splicing of unspliced XBP1 mRNA leads to generation of spliced XBP1 (XBP1s), which encodes a highly active transcription factor called XBP1s protein. Eventually, XBP1s translocates to the nucleus and up-regulates the transcriptions of its target genes involved in protein folding, ER biogenesis, and ER-associated degradation (Fig. 1) (9). In addition to splicing XBP1 mRNA, IRE1α has been recently shown to down-regulate microRNAs and other mRNAs under conditions in which IRE1α is activated strongly and for prolonged times. This process is referred to as “regulated IRE1-dependent decay” (RIDD) (10, 11) and has been implicated in various physiologies in the pancreas (11, 12). IRE1α and XBP1 are required for embryonic development; germ-line deletion of XBP1 or IRE1α results in embryonic lethality in mice due to liver hypoplasia (13, 14) or a developmental defect of the heart (15).
IRE1α and XBP1 have been characterized as key players in diverse and complex networks, and a number of other signaling components have been shown to interact with IRE1α and XBP1s (Fig. 1). For example, during ER stress, IRE1α plays a role in activation of inflammatory signaling pathways, including JNK and NF-κB. Although the mechanism of IRE1α in NF-κB activation remains to be clarified, IRE1α has been demonstrated to activate the JNK pathway through its physical association with TRAF2 (TNF receptor-associated factor-2) (16). JIK (JNK inhibitory kinase), ASK1 (apoptosis signaling kinase-1) and AIP1 (ASK1-interacting protein-1) have been further identified to regulate the IRE1α-JNK pathway (17–19). In addition, a genetic study in Caenorhabditis elegans proposed inflammatory p38MAPK as a positive regulator of IRE1-XBP1 signaling (20). Our group recently demonstrated that p38MAPK enhances XBP1s activity in mice via increased mRNA stability and nuclear transport of XBP1s protein, which is mediated through direct phosphorylation of XBP1s at Thr-48 and Ser-61 (21). IRE1α also activates the NLRP3 inflammasome by enhanced expression of TXNIP (thioredoxin-interacting protein) (12, 22). Furthermore, several studies have revealed that molecules involved in cell death signaling could regulate IRE1α function. Pro-apoptotic Bax and Bak are required for IRE1α activity upon ER stress via their physical association (23), whereas BI-1 (Bax inhibitor-1) inhibits IRE1α function (24). Conversely, IRE1 has been proposed to contribute to cell death through various mechanisms, including ASK1-JNK (18), caspase-12 (17), RIDD (11), and TXNIP (12, 22). Also, certain molecules that are involved in metabolic signaling have been shown to modulate IRE1 and XBP1 activity. For example, glucagon and protein kinase A, which function contrary to insulin receptor signaling in the liver, have been proposed to activate IRE1α (25). Furthermore, PTP1B (protein-tyrosine phosphatase 1B), a negative regulator of insulin and leptin receptor signaling, has been reported to be required for full activation of IRE1α upon ER stress (26). By contrast, we demonstrated that insulin receptor signaling positively regulates XBP1s activity (27) via physical interaction of the regulatory subunits of PI3K (p85α/β) with XBP1s. Interaction of p85α/β with XBP1s facilitates nuclear transportation of XBP1s (27, 28). Furthermore, XBP1s was shown to directly interact with FoxO1 (Forkhead box protein O1) and lead to its degradation (29), which suggests that XBP1s has functions other than regulating gene expression as a transcription factor.
IRE1 and XBP1s and their cross-talk with other signaling pathways have been suggested to play crucial roles in glucose and lipid metabolism. IRE1-null cells are more responsive to insulin compared with wild-type cells (30). When IRE1α activity was suppressed by RNAi-mediated silencing or by BI-1 overexpression in the liver, hepatic gluconeogenesis was reduced, and glucose tolerance was improved in obese and diabetic mice (25, 31). In contrast, haploinsufficiency of XBP1 creates ER stress and leads to development of insulin resistance and glucose intolerance upon high fat diet feeding (30). In obese and diabetic mice, XBP1s fails to translocate to the nucleus in the liver due to impaired association with p85α/β and reduced p38MAPK activity (21, 27). Ectopic expression of XBP1s in the liver via adenovirus restores glucose tolerance and insulin sensitivity in diabetic mice by suppressing hepatic FoxO1 activity and gluconeogenesis (21, 29). Furthermore, liver-specific deletion of XBP1 in adult mice at early high fat diet feeding exacerbates glucose intolerance and insulin resistance (29). In addition to the liver, pancreatic XBP1 was also shown to play a role in glucose homeostasis; β cell-specific deletion of XBP1 in the pancreas decreased insulin processing and secretion from islets, resulting in modest hyperglycemia and glucose intolerance (32). Seemingly different roles of IRE1α and XBP1s in glucose metabolism may stem from distinctive functions of IRE1α and XBP1s as exemplified by RIDD or JNK activation by IRE1α and inhibition of FoxO1 by XBP1s.
Several recent works have also uncovered the functional role of IRE1α in lipid homeostasis. Hepatic suppression of IRE1α activity via BI-1 led to development of hepatic steatosis upon high fat diet feeding while decreasing the levels of circulating cholesterol and triglycerides (TGs) (31). Similarly, liver-specific deletion of IRE1α lowered plasma lipid contents but prompted hepatosteatosis possibly due to decreased TG secretion (33). All of the above demonstrate that IRE1α and XBP1s are crucial regulators of glucose and lipid metabolism in tissues such as the liver and pancreas.
PERK Plays Important Roles in β Cell Function
PERK is a type I ER transmembrane protein, and like ATF6, the evolution of PERK started in metazoans (34). The cytoplasmic kinase domain of PERK shares homology with other eIF2α kinases, such as GCN2 (general control non-depressible-2), HRI (heme-regulated inhibitor), and PKR (34). The ER lumenal domain is structurally homologous and functionally interchangeable with the IRE1α lumenal domain, implying a similar stress-sensing mechanism between PERK and IRE1α (6). GRP78 is associated with the PERK monomer, and its dissociation from PERK upon ER stress leads to homodimerization, autotransphosphorylation, and activation of the kinase domain. Consequently, activated PERK phosphorylates eIF2α at Ser-51, leading to suppression of the assembly of the ribosomal complex and global protein translation, thus reducing protein load in the ER (Fig. 1) (35). However, PERK and eIF2α phosphorylation selectively facilitates translation of certain transcription factors, such as ATF4 and ATF5; ATF4 increases transcription of genes involved in amino acid metabolism, the oxidative stress response, and apoptosis (36), and ATF5 enhances the transcription of TXNIP (22). PERK mediates ER stress-induced apoptosis, and the pro-apoptotic transcription factor CHOP (C/EBP homologous protein) has been proposed as a major mediator of PERK-induced apoptosis (37). In addition, the PERK pathway has been reported to activate inflammatory signaling; for instance, the PERK-eIF2α axis activates the NF-κB pathway upon ER stress by suppressing IκBα (inhibitor of κBα) independently of ATF4 and IκBα phosphorylation (38, 39). Furthermore, the PERK pathway also activates the NLRP3 inflammasome via ATF5-mediated TXNIP expression (22).
Several animal studies have consistently suggested that the PERK pathway is crucial in pancreatic β cell function. PERK-null mice develop growth retardation and metabolic dysfunction, notably hyperglycemia due to loss of pancreatic islets (40, 41), and such islet dysfunctions were not reported in mice lacking GCN2, HRI, or PKR (42). The importance of pancreatic PERK in glucose homeostasis has been further supported by the fact that pancreas-specific or endocrine pancreas-specific deletion of PERK also leads to β cell loss and hyperglycemia (41, 43). Moreover, when PERK expression was reintroduced in β cells of PERK-null mice, the mice became euglycemic and displayed normal β cell mass (43). PERK has also been found to be important in human islet function; Wolcott-Rallison syndrome, which is a rare genetic disease in humans caused by mutations of PERK, shows notably similar pathologies as in mouse models, including growth retardation, skeletal dysplasia and early-onset diabetes by non-autoimmune-mediated β cell destruction (44). The eIF2α S51A mutation blocks translational inhibition by eIF2α kinases, and homozygous eIF2α S51A mice show a deficiency in pancreatic β cells as do PERK-null mice but die immediately after birth (45). Heterozygous eIF2α S51A mice are viable and do not show any β cell loss on a high fat diet but still become glucose-intolerant, resulting from reduced insulin folding and secretion (46). In addition, preventing ER stress-induced β cell death has been shown to be protective under various diabetic conditions in mice; CHOP-null mice maintain better glucose homeostasis than wild-type mice as a result of protection from β cell loss in diet-induced or genetic obesity models and in an ER stress-induced type I diabetes model (Akita mice) (47, 48).
ATF6 and Regulated Proteolysis: A Role in Glucose Metabolism
ATF6 is a type II ER transmembrane protein and belongs to the family of bZIP transcription factors, which also includes CREBH (cAMP-responsive element-binding protein H). The cytoplasmic domain of ATF6 is a transcription factor, and the ER lumenal domain senses the perturbations in the ER (49). ATF6 is retained in the ER membrane via its physical association with GRP78 under normal conditions without ER stress. Upon development of perturbations in the ER lumen, the dissociation of GRP78 allows ATF6 to translocate to the Golgi (50), where it is cleaved by S1P (site-1 protease) and S2P in a similar manner as SREBP (sterol regulatory element-binding protein) proteins. Subsequently, the N-terminal domain (cytoplasmic part) is liberated (51). The released N-terminal ATF6, which is the active transcription factor, translocates to the nucleus and up-regulates the transcription of target genes, including XBP1 (Fig. 1) (49). Several cross-talks between ATF6 and other signaling pathways have been reported; for example, p38MAPK has been shown to directly phosphorylate ATF6 and to increase its transcriptional activity (52). Additionally, PGC-1α serves as a coactivator of ATF6 and enhances ATF6 activity in muscle (53). In the liver, CRTC2 (CREB-regulated transcription coactivator-2) augments ATF6 transcriptional activity through their physical association, but conversely, ATF6 suppresses CRTC2 activity as a coactivator of CREB (54). Heterodimerization between ATF6 and XBP1s enhances their transcriptional activity (55). Furthermore, ATF6 has been shown to suppress the transcriptional activity of SREBP-2 (56).
There are two isoforms of ATF6 (ATF6α and ATF6β), which are redundant in their roles in the UPR, and mice deficient in either isoform are born normal (55). However, deletion of both isoforms together leads to embryonic lethality, suggesting that ATF6 is also crucial in early development (55). ATF6 has been suggested to have a role in glucose metabolism in the liver and pancreas. When whole body Atf6α−/− mice are fed high fat diets or crossed with agouti yellow mice, they develop early and more severe glucose intolerance, most likely due to reduced insulin content in the pancreatic β cells (57). Hepatic ATF6α has been demonstrated to attenuate CRTC2 activity and gluconeogenesis (54), and overexpression of ATF6α in the livers of obese and diabetic mice reduces blood glucose and improves glucose tolerance, whereas reduced ATF6α expression via RNAi increases blood glucose levels in lean mice (54). Another ER membrane-bound bZIP transcription factor, CREBH, the expression of which is limited to the liver and small intestine (58, 59), is activated by S1P- and S2P-mediated cleavage in the Golgi upon ER stress (59). However, unlike ATF6, CREBH has shown to activate different sets of hepatic genes in the acute phase response (59), iron metabolism (60), gluconeogenesis (61), and TG metabolism (58). CREBH expression in the liver is increased in obese and diabetic mice, and hepatic overexpression of CREBH raises blood glucose levels, whereas RNAi-mediated depletion of CREBH in the liver reduces blood glucose through direct modulation of gluconeogenic gene transcription (61). In addition, CREBH-deficient mice show hypertriglyceridemia due to reduced TG clearance (58).
ER Stress and Leptin and Insulin Resistance
Obesity leads to many debilitating diseases, including insulin resistance and type 2 diabetes, and has become a prevailing disease in the United States and other countries worldwide. In the United States, 69% of the adult population is overweight, and 36% is obese (62). Leptin is an adipocyte-derived hormone that suppresses appetite and increases energy expenditure through its action on the CNS. Despite high circulating levels of leptin, leptin cannot exert its anti-obesity effects due to the presence of leptin resistance in the obese population. We have previously shown that increased hypothalamic ER stress in obese mice is one of the main mechanisms leading to development of leptin resistance (63). Indeed, pharmacologically induced ER stress suppresses leptin signaling in the hypothalamus and increases food intake and body weight in mice (63). Also, neuronal XBP1 deletion creates hypothalamic ER stress and leptin resistance and subsequently promotes weight gain upon high fat diet feeding through increased food intake and decreased energy expenditure (63). Chemical chaperones, agents that can reduce ER stress, such as 4-phenylbutyric acid (4-PBA) and tauroursodeoxycholic acid (TUDCA), alleviate hypothalamic ER stress and restore leptin responsivity in high fat diet-fed mice, in turn reducing food consumption and body weight (63).
Obesity leads to insulin resistance in the brain, liver, and adipose tissues (64, 65). Insulin is produced and secreted from β cells in the pancreas in response to nutrients. Central insulin signaling mainly regulates food intake and body weight (66), whereas peripheral insulin receptor signaling primarily regulates blood glucose levels by suppressing gluconeogenesis in the liver and enhancing glucose uptake in the muscle and adipose tissue and adjusts peripheral storage and breakdown of fat, glycogen, and proteins (65). ER stress in the brain also contributes to central insulin resistance along with leptin resistance; central administration of insulin does not suppress food intake when acute ER stress is induced in the brain (67). Peripherally, increased ER stress in the liver and adipose tissues of obese mice has been shown to cause insulin resistance and ultimately type 2 diabetes (30). Indeed, when heterozygous XBP1-null mice are fed a high fat diet, they display augmented ER stress in the liver and adipose tissues and become more obese, insulin-resistant, and glucose-intolerant compared with wild-type mice (30). Also, peripheral administration of chemical chaperones alleviates ER stress in the liver and adipose tissues and in turn improves insulin sensitivity and glucose homeostasis in obese and diabetic mice (68). Furthermore, 4-PBA and TUDCA have been demonstrated to improve insulin sensitivity in human obese subjects, suggesting ER stress and the UPR as potential therapeutic targets for human metabolic diseases (69, 70).
Various pathologies in obesity have been proposed to contribute to development of ER stress (Fig. 2); for example, cholesterol and free fatty acids such as palmitate induce ER stress possibly via increased reactive oxygen species and ER Ca2+ depletion from SERCA (sarco/endoplasmic reticulum calcium ATPase) dysfunction (71, 72). Indeed, diminished SERCA expression and activity were observed in the livers and macrophages of obese and insulin-resistant mice, which also have higher level of ER stress (73, 74). Moreover, when SERCA activity in the liver was restored, ER stress was ameliorated, glucose homeostasis and insulin sensitivity were improved, and hepatic lipogenesis and TG accumulation were reduced in obese and diabetic mice (73). In addition, chronic activation of mTORC1 has been reported in the muscles, adipose tissues, and livers of obese animals, and tuberous sclerosis complex deficiency and subsequent activation of mTORC1 were shown to create ER stress and suppress insulin signaling (75). In contrast, inhibition of mTORC1 by rapamycin reduces ER stress and improves insulin sensitivity (76). Recently, decreased XBP1s nuclear translocation and activity due to reduced association with p85α/β and decreased phosphorylation by p38MAPK in the livers of obese mice were shown to play important roles in the development of ER stress in obesity (21, 27).
FIGURE 2.
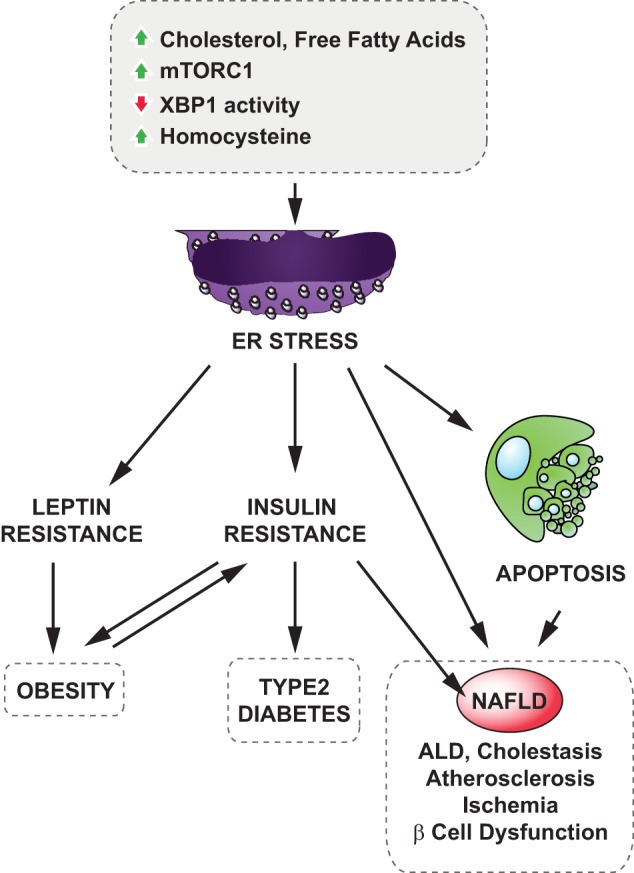
ER stress and metabolic disorders. Various stimuli such as increased loads of cholesterol or free fatty acids or chronic activation of mTORC1 activity, hyperhomocysteinemia, and decreased XBP1s activity were shown to cause ER stress under obesity conditions. ER stress leads to the development of obesity through hypothalamic leptin and insulin resistance and contributes to type 2 diabetes via inducing peripheral insulin resistance and increasing hepatic gluconeogenesis. Furthermore, ER stress has been suggested to be involved in various pathologies of metabolic liver diseases (NAFLD, ALD, and cholestasis) and cardiovascular diseases (atherosclerosis and ischemia).
Liver Diseases and ER Stress
Nonalcoholic fatty liver disease (NAFLD) and alcoholic fatty liver disease (ALD) are major liver diseases worldwide, encompassing a range of hepatic disorders from steatosis to cirrhosis. There has been a concerning increase in the incidence of NAFLD due to the obesity epidemic. Increased ER stress has been shown to contribute to the development of NAFLD under obesity conditions. Progression of NAFLD is closely associated with increased ER stress and activated UPR signaling (30). ER stress signaling in hepatocytes was demonstrated to promote lipogenesis through SREBP-1c activation (77). Livers deficient in ATF6α or IRE1α are prone to developing steatosis upon acute ER stress or high fat diet feeding (57, 78, 79). Moreover, chemical chaperone administration or hepatic overexpression of GRP78 or SERCA2b mitigates ER stress and reduces lipogenesis in liver, thus decreasing the risk of developing hepatic steatosis (68, 73, 77). ALD animal models also display augmented hepatic ER stress and increased rates of apoptosis due partly to increased circulating homocysteine, which is suggested to induce ER stress through interference with proper disulfide bond formation and protein folding (80). When circulating homocysteine and ER stress were attenuated or CHOP was depleted, mice were protected from alcohol-induced liver injury (80, 81). Aberrant retention of bile acids in the liver, a condition termed cholestasis, creates toxicity and leads to liver injury, fibrosis, and cirrhosis. Bile acids have been proposed to induce ER stress and cell death in hepatocytes; activated UPR signaling was observed in the livers of mouse models of cholestasis (82, 83). In addition, whole body CHOP deficiency protects hepatocytes from cholestasis-induced liver damage (83).
Vascular Diseases: Atherosclerosis and Ischemia
Atherosclerosis, which is the primary cause of heart disease and stroke, initially emerges from subendothelial accumulation of apolipoprotein B lipoproteins, cholesterol, and TGs, which then leads to the recruitment of monocytes and subsequent differentiation into macrophages. Further lipid uptake and oxidation along with inflammation transform these macrophages into foam cells and also increase the apoptosis of macrophages, which eventually creates necrotic cores if the growth of lesions is unresolved (84). Increased UPR has been observed in vascular endothelial cells, smooth muscle cells, and macrophages in atherosclerotic lesions (72, 85). Free fatty acids, oxidized lipids, cholesterol, and hyperhomocysteinemia have been proposed to raise ER stress in endothelial cells and macrophages during the progression of atherosclerosis (72, 86, 87). The apoptotic cell death of macrophages is considered a major pathological event in later stages of atherosclerosis, and ER stress has been suggested as a major inducer of apoptosis through the IRE1α-JNK pathway and CHOP (72, 88). Indeed, CHOP-null macrophages are protected from cholesterol-induced cytotoxicity (72), and CHOP deficiency reduces the atherosclerotic lesion size and cell death (89).
Atherosclerosis may induce ischemia, in which oxygen and nutrition supply to tissues and organs via blood is restricted, leading to stroke and heart attack (90). Ischemic conditions such as glucose deprivation have been shown to compromise protein folding and induce ER stress in cultured cells (90). Accordingly, increased ER stress, CHOP expression, and cell death have been observed in the hippocampus and cardiac myocytes after ischemia (91, 92). Non-lethal ischemic preconditioning, which increases GRP78 expression, protects the mouse brain from apoptosis after ischemia (92), and CHOP depletion alleviates ischemic neuronal apoptosis in mice (93). Similarly, cardiac overexpression of ATF6 or CHOP deletion protects mice from heart injury after ischemia/reperfusion (91, 94).
Therapeutic Potential for Treatment of Metabolic Disorders by Modulating ER Stress and UPR
ER stress and UPR signaling have been shown to be involved in the pathology of various diseases, including metabolic disorders. Pharmacological approaches to manipulate them as a treatment for human diseases have been pursued recently. For example, the Food and Drug Administration (FDA) in the United States approved two 26 S proteasome inhibitors, bortezomib (Velcade) and carfilzomib (Kyprolis), for the treatment of multiple myeloma. Both of these drugs lead to the development of higher levels of ER stress in multiple myeloma cells and induce cell death (95). For metabolic disorders, the chemical chaperones 4-PBA and TUDCA were demonstrated to improve insulin signaling and glucose homeostasis in in vivo mouse models and in human obese and insulin-resistant subjects via ameliorating ER stress (68–70).
There have also been recent efforts to develop drugs targeting a specific arm of the UPR. For example, several chemicals have been demonstrated to modulate the RNase activity of IRE1 by directly engaging the cytoplasmic part of IRE1, including the RNase and kinase domains (96, 97), and some of them have been proposed as potential treatments for multiple myeloma (96). Also, PERK-specific kinase inhibitors have been explored (98–100) and shown to inhibit tumor growth in xenograft mouse models (99) and to prevent neurodegeneration in a mouse model of prion disease (100).
In this sense, different approaches to modulate ER stress could be used for the treatment of different diseases. Although reducing ER stress in obesity could have therapeutic potential in insulin resistance and type 2 diabetes, further inducing ER stress could help to fight against cancers.
Conclusions
IRE1, the first signaling arm of the UPR, was identified 2 decades ago; since then, there has been enormous progress in understanding how the UPR components function in response to ER stress at the molecular level. Also, recent work has revealed crucial links between ER stress and different components of metabolic syndrome. Considering that each UPR component contributes diverse metabolic phenotypes, it is important to understand how an individual component of the UPR and its cross-talk with other signaling pathways are involved in the pathogenesis of metabolic diseases. Furthermore, developing specific therapeutics targeting the individual UPR components would be beneficial for the treatment of various metabolic disorders.
Acknowledgments
We thank Sang Won Park and Isin Cakir for critical reading and discussion of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 DK081009 and R01 DK098496 (to U. O.). This work was also supported by American Diabetes Association Career Development Grant 7-09-CD-10 (to U. O.).
- ER
- endoplasmic reticulum
- UPR
- unfolded protein response
- XBP1s
- spliced XBP1
- RIDD
- regulated IRE1-dependent decay
- TG
- triglyceride
- 4-PBA
- 4-phenylbutyric acid
- TUDCA
- tauroursodeoxycholic acid
- NAFLD
- nonalcoholic fatty liver disease
- ALD
- alcoholic fatty liver disease.
REFERENCES
- 1. Park S. W., Ozcan U. (2013) Potential for therapeutic manipulation of the UPR in disease. Semin. Immunopathol. 35, 351–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cox J. S., Shamu C. E., Walter P. (1993) Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 73, 1197–1206 [DOI] [PubMed] [Google Scholar]
- 3. Tirasophon W., Welihinda A. A., Kaufman R. J. (1998) A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 12, 1812–1824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bertolotti A., Wang X., Novoa I., Jungreis R., Schlessinger K., Cho J. H., West A. B., Ron D. (2001) Increased sensitivity to dextran sodium sulfate colitis in IRE1β-deficient mice. J. Clin. Invest. 107, 585–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Martino M. B., Jones L., Brighton B., Ehre C., Abdulah L., Davis C. W., Ron D., O'Neal W. K., Ribeiro C. M. (2013) The ER stress transducer IRE1β is required for airway epithelial mucin production. Mucosal Immunol. 6, 639–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bertolotti A., Zhang Y., Hendershot L. M., Harding H. P., Ron D. (2000) Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2, 326–332 [DOI] [PubMed] [Google Scholar]
- 7. Gardner B. M., Walter P. (2011) Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science 333, 1891–1894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lee K. P., Dey M., Neculai D., Cao C., Dever T. E., Sicheri F. (2008) Structure of the dual enzyme Ire1 reveals the basis for catalysis and regulation in nonconventional RNA splicing. Cell 132, 89–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yoshida H., Matsui T., Yamamoto A., Okada T., Mori K. (2001) XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891 [DOI] [PubMed] [Google Scholar]
- 10. Hollien J., Lin J. H., Li H., Stevens N., Walter P., Weissman J. S. (2009) Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 186, 323–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Han D., Lerner A. G., Vande Walle L., Upton J. P., Xu W., Hagen A., Backes B. J., Oakes S. A., Papa F. R. (2009) IRE1α kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 138, 562–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lerner A. G., Upton J. P., Praveen P. V., Ghosh R., Nakagawa Y., Igbaria A., Shen S., Nguyen V., Backes B. J., Heiman M., Heintz N., Greengard P., Hui S., Tang Q., Trusina A., Oakes S. A., Papa F. R. (2012) IRE1α induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 16, 250–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang K., Wong H. N., Song B., Miller C. N., Scheuner D., Kaufman R. J. (2005) The unfolded protein response sensor IRE1α is required at 2 distinct steps in B cell lymphopoiesis. J. Clin. Invest. 115, 268–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Reimold A. M., Etkin A., Clauss I., Perkins A., Friend D. S., Zhang J., Horton H. F., Scott A., Orkin S. H., Byrne M. C., Grusby M. J., Glimcher L. H. (2000) An essential role in liver development for transcription factor XBP-1. Genes Dev. 14, 152–157 [PMC free article] [PubMed] [Google Scholar]
- 15. Masaki T., Yoshida M., Noguchi S. (1999) Targeted disruption of CRE-binding factor TREB5 gene leads to cellular necrosis in cardiac myocytes at the embryonic stage. Biochem. Biophys. Res. Commun. 261, 350–356 [DOI] [PubMed] [Google Scholar]
- 16. Urano F., Wang X., Bertolotti A., Zhang Y., Chung P., Harding H. P., Ron D. (2000) Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287, 664–666 [DOI] [PubMed] [Google Scholar]
- 17. Yoneda T., Imaizumi K., Oono K., Yui D., Gomi F., Katayama T., Tohyama M. (2001) Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J. Biol. Chem. 276, 13935–13940 [DOI] [PubMed] [Google Scholar]
- 18. Nishitoh H., Matsuzawa A., Tobiume K., Saegusa K., Takeda K., Inoue K., Hori S., Kakizuka A., Ichijo H. (2002) ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 16, 1345–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Luo D., He Y., Zhang H., Yu L., Chen H., Xu Z., Tang S., Urano F., Min W. (2008) AIP1 is critical in transducing IRE1-mediated endoplasmic reticulum stress response. J. Biol. Chem. 283, 11905–11912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Richardson C. E., Kooistra T., Kim D. H. (2010) An essential role for XBP-1 in host protection against immune activation in C. elegans. Nature 463, 1092–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lee J., Sun C., Zhou Y., Lee J., Gokalp D., Herrema H., Park S. W., Davis R. J., Ozcan U. (2011) p38 MAPK-mediated regulation of Xbp1s is crucial for glucose homeostasis. Nat. Med. 17, 1251–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Oslowski C. M., Hara T., O'Sullivan-Murphy B., Kanekura K., Lu S., Hara M., Ishigaki S., Zhu L. J., Hayashi E., Hui S. T., Greiner D., Kaufman R. J., Bortell R., Urano F. (2012) Thioredoxin-interacting protein mediates ER stress-induced β cell death through initiation of the inflammasome. Cell Metab. 16, 265–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hetz C., Bernasconi P., Fisher J., Lee A. H., Bassik M. C., Antonsson B., Brandt G. S., Iwakoshi N. N., Schinzel A., Glimcher L. H., Korsmeyer S. J. (2006) Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1α. Science 312, 572–576 [DOI] [PubMed] [Google Scholar]
- 24. Lisbona F., Rojas-Rivera D., Thielen P., Zamorano S., Todd D., Martinon F., Glavic A., Kress C., Lin J. H., Walter P., Reed J. C., Glimcher L. H., Hetz C. (2009) BAX inhibitor-1 is a negative regulator of the ER stress sensor IRE1α. Mol. Cell 33, 679–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mao T., Shao M., Qiu Y., Huang J., Zhang Y., Song B., Wang Q., Jiang L., Liu Y., Han J. D., Cao P., Li J., Gao X., Rui L., Qi L., Li W., Liu Y. (2011) PKA phosphorylation couples hepatic inositol-requiring enzyme 1α to glucagon signaling in glucose metabolism. Proc. Natl. Acad. Sci. U.S.A. 108, 15852–15857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gu F., Nguyên D. T., Stuible M., Dubé N., Tremblay M. L., Chevet E. (2004) Protein-tyrosine phosphatase 1B potentiates IRE1 signaling during endoplasmic reticulum stress. J. Biol. Chem. 279, 49689–49693 [DOI] [PubMed] [Google Scholar]
- 27. Park S. W., Zhou Y., Lee J., Lu A., Sun C., Chung J., Ueki K., Ozcan U. (2010) The regulatory subunits of PI3K, p85α and p85β, interact with XBP-1 and increase its nuclear translocation. Nat. Med. 16, 429–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Winnay J. N., Boucher J., Mori M. A., Ueki K., Kahn C. R. (2010) A regulatory subunit of phosphoinositide 3-kinase increases the nuclear accumulation of X-box-binding protein-1 to modulate the unfolded protein response. Nat. Med. 16, 438–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhou Y., Lee J., Reno C. M., Sun C., Park S. W., Chung J., Lee J., Fisher S. J., White M. F., Biddinger S. B., Ozcan U. (2011) Regulation of glucose homeostasis through a XBP-1-FoxO1 interaction. Nat. Med. 17, 356–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ozcan U., Cao Q., Yilmaz E., Lee A. H., Iwakoshi N. N., Ozdelen E., Tuncman G., Görgün C., Glimcher L. H., Hotamisligil G. S. (2004) Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306, 457–461 [DOI] [PubMed] [Google Scholar]
- 31. Bailly-Maitre B., Belgardt B. F., Jordan S. D., Coornaert B., von Freyend M. J., Kleinridders A., Mauer J., Cuddy M., Kress C. L., Willmes D., Essig M., Hampel B., Protzer U., Reed J. C., Brüning J. C. (2010) Hepatic Bax inhibitor-1 inhibits IRE1α and protects from obesity-associated insulin resistance and glucose intolerance. J. Biol. Chem. 285, 6198–6207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lee A. H., Heidtman K., Hotamisligil G. S., Glimcher L. H. (2011) Dual and opposing roles of the unfolded protein response regulated by IRE1α and XBP1 in proinsulin processing and insulin secretion. Proc. Natl. Acad. Sci. U.S.A. 108, 8885–8890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang S., Chen Z., Lam V., Han J., Hassler J., Finck B. N., Davidson N. O., Kaufman R. J. (2012) IRE1α-XBP1s induces PDI expression to increase MTP activity for hepatic VLDL assembly and lipid homeostasis. Cell Metab. 16, 473–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Harding H. P., Zhang Y., Ron D. (1999) Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397, 271–274 [DOI] [PubMed] [Google Scholar]
- 35. Ma K., Vattem K. M., Wek R. C. (2002) Dimerization and release of molecular chaperone inhibition facilitate activation of eukaryotic initiation factor-2 kinase in response to endoplasmic reticulum stress. J. Biol. Chem. 277, 18728–18735 [DOI] [PubMed] [Google Scholar]
- 36. Wek R. C., Cavener D. R. (2007) Translational control and the unfolded protein response. Antioxid. Redox Signal. 9, 2357–2371 [DOI] [PubMed] [Google Scholar]
- 37. Zinszner H., Kuroda M., Wang X., Batchvarova N., Lightfoot R. T., Remotti H., Stevens J. L., Ron D. (1998) CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 12, 982–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Deng J., Lu P. D., Zhang Y., Scheuner D., Kaufman R. J., Sonenberg N., Harding H. P., Ron D. (2004) Translational repression mediates activation of nuclear factor κB by phosphorylated translation initiation factor 2. Mol. Cell. Biol. 24, 10161–10168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jiang H. Y., Wek S. A., McGrath B. C., Scheuner D., Kaufman R. J., Cavener D. R., Wek R. C. (2003) Phosphorylation of the α subunit of eukaryotic initiation factor 2 is required for activation of NF-κB in response to diverse cellular stresses. Mol. Cell. Biol. 23, 5651–5663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Harding H. P., Zeng H., Zhang Y., Jungries R., Chung P., Plesken H., Sabatini D. D., Ron D. (2001) Diabetes mellitus and exocrine pancreatic dysfunction in Perk−/− mice reveals a role for translational control in secretory cell survival. Mol. Cell 7, 1153–1163 [DOI] [PubMed] [Google Scholar]
- 41. Gao Y., Sartori D. J., Li C., Yu Q. C., Kushner J. A., Simon M. C., Diehl J. A. (2012) PERK is required in the adult pancreas and is essential for maintenance of glucose homeostasis. Mol. Cell. Biol. 32, 5129–5139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Marciniak S. J., Ron D. (2006) Endoplasmic reticulum stress signaling in disease. Physiol. Rev. 86, 1133–1149 [DOI] [PubMed] [Google Scholar]
- 43. Zhang W., Feng D., Li Y., Iida K., McGrath B., Cavener D. R. (2006) PERK EIF2AK3 control of pancreatic β cell differentiation and proliferation is required for postnatal glucose homeostasis. Cell Metab. 4, 491–497 [DOI] [PubMed] [Google Scholar]
- 44. Delépine M., Nicolino M., Barrett T., Golamaully M., Lathrop G. M., Julier C. (2000) EIF2AK3, encoding translation initiation factor 2-α kinase 3, is mutated in patients with Wolcott-Rallison syndrome. Nat. Genet. 25, 406–409 [DOI] [PubMed] [Google Scholar]
- 45. Scheuner D., Song B., McEwen E., Liu C., Laybutt R., Gillespie P., Saunders T., Bonner-Weir S., Kaufman R. J. (2001) Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol. Cell 7, 1165–1176 [DOI] [PubMed] [Google Scholar]
- 46. Scheuner D., Vander Mierde D., Song B., Flamez D., Creemers J. W., Tsukamoto K., Ribick M., Schuit F. C., Kaufman R. J. (2005) Control of mRNA translation preserves endoplasmic reticulum function in β cells and maintains glucose homeostasis. Nat. Med. 11, 757–764 [DOI] [PubMed] [Google Scholar]
- 47. Song B., Scheuner D., Ron D., Pennathur S., Kaufman R. J. (2008) Chop deletion reduces oxidative stress, improves β cell function, and promotes cell survival in multiple mouse models of diabetes. J. Clin. Invest. 118, 3378–3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Oyadomari S., Koizumi A., Takeda K., Gotoh T., Akira S., Araki E., Mori M. (2002) Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J. Clin. Invest. 109, 525–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Haze K., Yoshida H., Yanagi H., Yura T., Mori K. (1999) Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 10, 3787–3799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shen J., Chen X., Hendershot L., Prywes R. (2002) ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell 3, 99–111 [DOI] [PubMed] [Google Scholar]
- 51. Ye J., Rawson R. B., Komuro R., Chen X., Davé U. P., Prywes R., Brown M. S., Goldstein J. L. (2000) ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 6, 1355–1364 [DOI] [PubMed] [Google Scholar]
- 52. Thuerauf D. J., Arnold N. D., Zechner D., Hanford D. S., DeMartin K. M., McDonough P. M., Prywes R., Glembotski C. C. (1998) p38 mitogen-activated protein kinase mediates the transcriptional induction of the atrial natriuretic factor gene through a serum response element. A potential role for the transcription factor ATF6. J. Biol. Chem. 273, 20636–20643 [DOI] [PubMed] [Google Scholar]
- 53. Wu J., Ruas J. L., Estall J. L., Rasbach K. A., Choi J. H., Ye L., Boström P., Tyra H. M., Crawford R. W., Campbell K. P., Rutkowski D. T., Kaufman R. J., Spiegelman B. M. (2011) The unfolded protein response mediates adaptation to exercise in skeletal muscle through a PGC-1α/ATF6α complex. Cell Metab. 13, 160–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wang Y., Vera L., Fischer W. H., Montminy M. (2009) The CREB coactivator CRTC2 links hepatic ER stress and fasting gluconeogenesis. Nature 460, 534–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yamamoto K., Sato T., Matsui T., Sato M., Okada T., Yoshida H., Harada A., Mori K. (2007) Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6α and XBP1. Dev. Cell 13, 365–376 [DOI] [PubMed] [Google Scholar]
- 56. Zeng L., Lu M., Mori K., Luo S., Lee A. S., Zhu Y., Shyy J. Y. (2004) ATF6 modulates SREBP2-mediated lipogenesis. EMBO J. 23, 950–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Usui M., Yamaguchi S., Tanji Y., Tominaga R., Ishigaki Y., Fukumoto M., Katagiri H., Mori K., Oka Y., Ishihara H. (2012) Atf6α-null mice are glucose intolerant due to pancreatic β-cell failure on a high-fat diet but partially resistant to diet-induced insulin resistance. Metabolism 61, 1118–1128 [DOI] [PubMed] [Google Scholar]
- 58. Lee J. H., Giannikopoulos P., Duncan S. A., Wang J., Johansen C. T., Brown J. D., Plutzky J., Hegele R. A., Glimcher L. H., Lee A. H. (2011) The transcription factor cyclic AMP-responsive element-binding protein H regulates triglyceride metabolism. Nat. Med. 17, 812–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhang K., Shen X., Wu J., Sakaki K., Saunders T., Rutkowski D. T., Back S. H., Kaufman R. J. (2006) Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell 124, 587–599 [DOI] [PubMed] [Google Scholar]
- 60. Vecchi C., Montosi G., Zhang K., Lamberti I., Duncan S. A., Kaufman R. J., Pietrangelo A. (2009) ER stress controls iron metabolism through induction of hepcidin. Science 325, 877–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lee M. W., Chanda D., Yang J., Oh H., Kim S. S., Yoon Y. S., Hong S., Park K. G., Lee I. K., Choi C. S., Hanson R. W., Choi H. S., Koo S. H. (2010) Regulation of hepatic gluconeogenesis by an ER-bound transcription factor, CREBH. Cell Metab. 11, 331–339 [DOI] [PubMed] [Google Scholar]
- 62. Flegal K. M., Carroll M. D., Kit B. K., Ogden C. L. (2012) Prevalence of obesity and trends in the distribution of body mass index among US adults, 1999–2010. JAMA 307, 491–497 [DOI] [PubMed] [Google Scholar]
- 63. Ozcan L., Ergin A. S., Lu A., Chung J., Sarkar S., Nie D., Myers M. G., Jr., Ozcan U. (2009) Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab. 9, 35–51 [DOI] [PubMed] [Google Scholar]
- 64. Clegg D. J., Gotoh K., Kemp C., Wortman M. D., Benoit S. C., Brown L. M., D'Alessio D., Tso P., Seeley R. J., Woods S. C. (2011) Consumption of a high-fat diet induces central insulin resistance independent of adiposity. Physiol. Behav. 103, 10–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Biddinger S. B., Kahn C. R. (2006) From mice to men: insights into the insulin resistance syndromes. Annu. Rev. Physiol. 68, 123–158 [DOI] [PubMed] [Google Scholar]
- 66. Plum L., Schubert M., Brüning J. C. (2005) The role of insulin receptor signaling in the brain. Trends Endocrinol. Metab. 16, 59–65 [DOI] [PubMed] [Google Scholar]
- 67. Won J. C., Jang P. G., Namkoong C., Koh E. H., Kim S. K., Park J. Y., Lee K. U., Kim M. S. (2009) Central administration of an endoplasmic reticulum stress inducer inhibits the anorexigenic effects of leptin and insulin. Obesity 17, 1861–1865 [DOI] [PubMed] [Google Scholar]
- 68. Ozcan U., Yilmaz E., Ozcan L., Furuhashi M., Vaillancourt E., Smith R. O., Görgün C. Z., Hotamisligil G. S. (2006) Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 313, 1137–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kars M., Yang L., Gregor M. F., Mohammed B. S., Pietka T. A., Finck B. N., Patterson B. W., Horton J. D., Mittendorfer B., Hotamisligil G. S., Klein S. (2010) Tauroursodeoxycholic acid may improve liver and muscle but not adipose tissue insulin sensitivity in obese men and women. Diabetes 59, 1899–1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Xiao C., Giacca A., Lewis G. F. (2011) Sodium phenylbutyrate, a drug with known capacity to reduce endoplasmic reticulum stress, partially alleviates lipid-induced insulin resistance and β-cell dysfunction in humans. Diabetes 60, 918–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Cunha D. A., Hekerman P., Ladrière L., Bazarra-Castro A., Ortis F., Wakeham M. C., Moore F., Rasschaert J., Cardozo A. K., Bellomo E., Overbergh L., Mathieu C., Lupi R., Hai T., Herchuelz A., Marchetti P., Rutter G. A., Eizirik D. L., Cnop M. (2008) Initiation and execution of lipotoxic ER stress in pancreatic β-cells. J. Cell Sci. 121, 2308–2318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Feng B., Yao P. M., Li Y., Devlin C. M., Zhang D., Harding H. P., Sweeney M., Rong J. X., Kuriakose G., Fisher E. A., Marks A. R., Ron D., Tabas I. (2003) The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat. Cell Biol. 5, 781–792 [DOI] [PubMed] [Google Scholar]
- 73. Park S. W., Zhou Y., Lee J., Ozcan U. (2010) Sarco(endo)plasmic reticulum Ca2+-ATPase 2b is a major regulator of endoplasmic reticulum stress and glucose homeostasis in obesity. Proc. Natl. Acad. Sci. U.S.A. 107, 19320–19325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Liang C. P., Han S., Li G., Tabas I., Tall A. R. (2012) Impaired MEK signaling and SERCA expression promote ER stress and apoptosis in insulin-resistant macrophages and are reversed by exenatide treatment. Diabetes 61, 2609–2620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Laplante M., Sabatini D. M. (2012) mTOR signaling in growth control and disease. Cell 149, 274–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ozcan U., Ozcan L., Yilmaz E., Düvel K., Sahin M., Manning B. D., Hotamisligil G. S. (2008) Loss of the tuberous sclerosis complex tumor suppressors triggers the unfolded protein response to regulate insulin signaling and apoptosis. Mol. Cell 29, 541–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kammoun H. L., Chabanon H., Hainault I., Luquet S., Magnan C., Koike T., Ferré P., Foufelle F. (2009) GRP78 expression inhibits insulin and ER stress-induced SREBP-1c activation and reduces hepatic steatosis in mice. J. Clin. Invest. 119, 1201–1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Zhang K., Wang S., Malhotra J., Hassler J. R., Back S. H., Wang G., Chang L., Xu W., Miao H., Leonardi R., Chen Y. E., Jackowski S., Kaufman R. J. (2011) The unfolded protein response transducer IRE1α prevents ER stress-induced hepatic steatosis. EMBO J. 30, 1357–1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Rutkowski D. T., Wu J., Back S. H., Callaghan M. U., Ferris S. P., Iqbal J., Clark R., Miao H., Hassler J. R., Fornek J., Katze M. G., Hussain M. M., Song B., Swathirajan J., Wang J., Yau G. D., Kaufman R. J. (2008) UPR pathways combine to prevent hepatic steatosis caused by ER stress-mediated suppression of transcriptional master regulators. Dev. Cell 15, 829–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ji C., Kaplowitz N. (2003) Betaine decreases hyperhomocysteinemia, endoplasmic reticulum stress, and liver injury in alcohol-fed mice. Gastroenterology 124, 1488–1499 [DOI] [PubMed] [Google Scholar]
- 81. Ji C., Mehrian-Shai R., Chan C., Hsu Y. H., Kaplowitz N. (2005) Role of CHOP in hepatic apoptosis in the murine model of intragastric ethanol feeding. Alcohol. Clin. Exp. Res. 29, 1496–1503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Bochkis I. M., Rubins N. E., White P., Furth E. E., Friedman J. R., Kaestner K. H. (2008) Hepatocyte-specific ablation of Foxa2 alters bile acid homeostasis and results in endoplasmic reticulum stress. Nat. Med. 14, 828–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Tamaki N., Hatano E., Taura K., Tada M., Kodama Y., Nitta T., Iwaisako K., Seo S., Nakajima A., Ikai I., Uemoto S. (2008) CHOP deficiency attenuates cholestasis-induced liver fibrosis by reduction of hepatocyte injury. Am. J. Physiol. Gastrointest. Liver Physiol. 294, G498–G505 [DOI] [PubMed] [Google Scholar]
- 84. Moore K. J., Tabas I. (2011) Macrophages in the pathogenesis of atherosclerosis. Cell 145, 341–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Thorp E., Iwawaki T., Miura M., Tabas I. (2011) A reporter for tracking the UPR in vivo reveals patterns of temporal and cellular stress during atherosclerotic progression. J. Lipid Res. 52, 1033–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Lu Y., Qian L., Zhang Q., Chen B., Gui L., Huang D., Chen G., Chen L. (2013) Palmitate induces apoptosis in mouse aortic endothelial cells and endothelial dysfunction in mice fed high-calorie and high-cholesterol diets. Life Sci. 92, 1165–1173 [DOI] [PubMed] [Google Scholar]
- 87. Kokame K., Kato H., Miyata T. (1996) Homocysteine-respondent genes in vascular endothelial cells identified by differential display analysis. GRP78/BiP and novel genes. J. Biol. Chem. 271, 29659–29665 [DOI] [PubMed] [Google Scholar]
- 88. Li F., Guo Y., Sun S., Jiang X., Tang B., Wang Q., Wang L. (2008) Free cholesterol-induced macrophage apoptosis is mediated by inositol-requiring enzyme 1 α-regulated activation of Jun N-terminal kinase. Acta Biochim. Biophys. Sin. 40, 226–234 [DOI] [PubMed] [Google Scholar]
- 89. Thorp E., Li G., Seimon T. A., Kuriakose G., Ron D., Tabas I. (2009) Reduced apoptosis and plaque necrosis in advanced atherosclerotic lesions of Apoe−/− and Ldlr−/− mice lacking CHOP. Cell Metab. 9, 474–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Glembotski C. C. (2008) The role of the unfolded protein response in the heart. J. Mol. Cell. Cardiol. 44, 453–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Martindale J. J., Fernandez R., Thuerauf D., Whittaker R., Gude N., Sussman M. A., Glembotski C. C. (2006) Endoplasmic reticulum stress gene induction and protection from ischemia/reperfusion injury in the hearts of transgenic mice with a tamoxifen-regulated form of ATF6. Circ. Res. 98, 1186–1193 [DOI] [PubMed] [Google Scholar]
- 92. Hayashi T., Saito A., Okuno S., Ferrand-Drake M., Chan P. H. (2003) Induction of GRP78 by ischemic preconditioning reduces endoplasmic reticulum stress and prevents delayed neuronal cell death. J. Cereb. Blood Flow Metab. 23, 949–961 [DOI] [PubMed] [Google Scholar]
- 93. Tajiri S., Oyadomari S., Yano S., Morioka M., Gotoh T., Hamada J. I., Ushio Y., Mori M. (2004) Ischemia-induced neuronal cell death is mediated by the endoplasmic reticulum stress pathway involving CHOP. Cell Death Differ. 11, 403–415 [DOI] [PubMed] [Google Scholar]
- 94. Fu H. Y., Okada K., Liao Y., Tsukamoto O., Isomura T., Asai M., Sawada T., Okuda K., Asano Y., Sanada S., Asanuma H., Asakura M., Takashima S., Komuro I., Kitakaze M., Minamino T. (2010) Ablation of C/EBP homologous protein attenuates endoplasmic reticulum-mediated apoptosis and cardiac dysfunction induced by pressure overload. Circulation 122, 361–369 [DOI] [PubMed] [Google Scholar]
- 95. Goldberg A. L. (2012) Development of proteasome inhibitors as research tools and cancer drugs. J. Cell Biol. 199, 583–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Mimura N., Fulciniti M., Gorgun G., Tai Y. T., Cirstea D., Santo L., Hu Y., Fabre C., Minami J., Ohguchi H., Kiziltepe T., Ikeda H., Kawano Y., French M., Blumenthal M., Tam V., Kertesz N. L., Malyankar U. M., Hokenson M., Pham T., Zeng Q., Patterson J. B., Richardson P. G., Munshi N. C., Anderson K. C. (2012) Blockade of XBP1 splicing by inhibition of IRE1α is a promising therapeutic option in multiple myeloma. Blood 119, 5772–5781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Cross B. C., Bond P. J., Sadowski P. G., Jha B. K., Zak J., Goodman J. M., Silverman R. H., Neubert T. A., Baxendale I. R., Ron D., Harding H. P. (2012) The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc. Natl. Acad. Sci. U.S.A. 109, E869–E878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Harding H. P., Zyryanova A. F., Ron D. (2012) Uncoupling proteostasis and development in vitro with a small molecule inhibitor of the pancreatic endoplasmic reticulum kinase, PERK. J. Biol. Chem. 287, 44338–44344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Atkins C., Liu Q., Minthorn E., Zhang S. Y., Figueroa D. J., Moss K., Stanley T. B., Sanders B., Goetz A., Gaul N., Choudhry A. E., Alsaid H., Jucker B. M., Axten J. M., Kumar R. (2013) Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. 73, 1993–2002 [DOI] [PubMed] [Google Scholar]
- 100. Moreno J. A., Halliday M., Molloy C., Radford H., Verity N., Axten J. M., Ortori C. A., Willis A. E., Fischer P. M., Barrett D. A., Mallucci G. R. (2013) Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci. Transl. Med. 5, 206ra138. [DOI] [PubMed] [Google Scholar]