

Background: Multiheme proteins have crucial roles in diverse nitrogen cycle processes.
Results: The kustc1061 octaheme protein from anaerobic ammonium-oxidizing (anammox) bacteria specifically oxidizes hydroxylamine to NO.
Conclusion: Enzyme specificity derives from subtle amino acid changes near the P460 catalytic heme.
Significance: The presence of kustc1061 homologs in anammox and other bacteria enables the detoxification of hydroxylamine, thereby generating NO for respiratory purposes.
Keywords: Enzyme Kinetics, Enzyme Purification, Nitric Oxide, Nitrogen Metabolism, Protein Structure, Reactive Nitrogen Species, X-ray Crystallography, Anaerobic Ammonium Oxidation, Hydroxylamine, Octaheme Proteins
Abstract
Nitric oxide is an important molecule in all domains of life with significant biological functions in both pro- and eukaryotes. Anaerobic ammonium-oxidizing (anammox) bacteria that contribute substantially to the release of fixed nitrogen into the atmosphere use the oxidizing power of NO to activate inert ammonium into hydrazine (N2H4). Here, we describe an enzyme from the anammox bacterium Kuenenia stuttgartiensis that uses a novel pathway to make NO from hydroxylamine. This new enzyme is related to octaheme hydroxylamine oxidoreductase, a key protein in aerobic ammonium-oxidizing bacteria. By a multiphasic approach including the determination of the crystal structure of the K. stuttgartiensis enzyme at 1.8 Å resolution and refinement and reassessment of the hydroxylamine oxidoreductase structure from Nitrosomonas europaea, both in the presence and absence of their substrates, we propose a model for NO formation by the K. stuttgartiensis enzyme. Our results expand the understanding of the functions that the widespread family of octaheme proteins have.
Introduction
Nitric oxide has a multitude of roles in cell-cell communication, in microbial defense, and as a metabolite in nitrogen cycle microorganisms, where it is an intermediate in the reduction and oxidation of inorganic nitrogen compounds (1–7). Anaerobic ammonium-oxidizing (anammox) bacteria use the oxidizing power of NO to activate inert ammonium into hydrazine (N2H4) (8). In all these processes, NO is synthesized by specific enzymes. Here, we describe an enzyme previously (8) identified as kustc1061 from the anammox bacterium Kuenenia stuttgartiensis that makes NO from hydroxylamine (NH2OH) (Equation 1). This enzyme is related to hydroxylamine oxidoreductase (HAO)5 of aerobic ammonium-oxidizing bacteria (9). In aerobic ammonium-oxidizing bacteria, HAO catalyzes the oxidation of hydroxylamine to nitrite (NO2−), the second step in the aerobic oxidation of ammonium (Equation 2). Several properties of this enzyme have been elucidated including the determination of a crystal structure of the Nitrosomonas europaea protein (NeHAO) (9–13). Each subunit of the homotrimeric NeHAO harbors eight c-type hemes for catalysis and electron transfer, which are involved in the oxidation of its substrate. A distinguishing feature of NeHAO is the covalent attachment of the catalytic heme (fourth) of one subunit to a tyrosine residue (Tyr491) of the adjacent subunit in the trimer (11, 13).
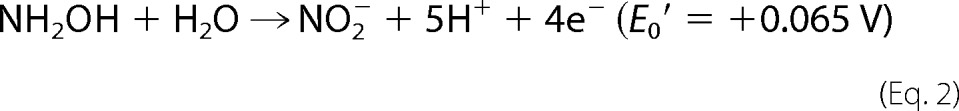 |
HAO-like proteins are not unique to aerobic ammonium-oxidizing bacteria. A BlastP search reveals hundreds of octaheme proteins from a wide range of microorganisms that are homologous to NeHAO. Anammox bacteria, such as K. stuttgartiensis, which were shown to contribute substantially to nitrogen release into the atmosphere (14), harbor no less than 10 divergent HAO paralogs. In fact, one of these (kustc0694 in K. stuttgartiensis) is dedicated to the oxidation of hydrazine, the N2-forming reaction in these organisms (8). Octaheme proteins from other organisms reduce nitrite to ammonium (15, 16). Apparently, various types of HAO are capable of taking the nitrogen atom through all its oxidation states between −3 (ammonium) and +3 (nitrite), both in oxidative and reductive directions. The presence of the tyrosine-dependent covalent linkage between different subunits seems to be a distinctive feature in governing reactions into an oxidative direction (17). These diverse catalytic potentials could very well make HAO-like proteins central but thus far overlooked catalysts in biogeochemical nitrogen cycle processes. In addition to the primary question of which reactions are performed by various HAO-related proteins, an even more intriguing issue is how these variants are tuned to their specific functions.
Presently, it is unknown which structural features determine the differences in reaction specificities of HAO-like proteins. Furthermore, these differences cannot be explained by sequence analyses and structural modeling, which are hampered by the lack of structural and biochemical information. To address these questions, we studied the catalytic and redox properties of the HAO-like protein kustc1061 of K. stuttgartiensis. Further, we determined the high resolution structure of both the as-isolated kustc1061 and crystals soaked with substrates. In parallel and independent from a recent refinement (12, 13), we reassessed the structure of NeHAO not only in the absence but also in presence of substrates. The results of this study shed light on determinants in a widespread group of homologs about which very little is known at the biochemical level.
EXPERIMENTAL PROCEDURES
Enzyme Purification and Identification
Kustc1061 was purified from a K. stuttgartiensis enrichment culture (∼95% pure) that was grown continuously as planktonic cells in a 10-liter membrane bioreactor using methods described by Kartal et al. (18). Purification typically used 4 liters of cells (A600 = ∼1) that were collected by centrifugation (8,000 × g, 15 min). This and all following steps, except for FPLC, were done at 4 °C. Pelleted cells were resuspended in 5 ml of 20 mm potassium Pi buffer (pH 7) and broken by three subsequent passages through a French pressure cell operating at 138 MPa. The lysate was incubated with 1% deoxycholate (w/v) on a rotating incubator (20 rpm) for 1 h. After incubation, cell debris was removed by centrifugation (3,000 × g, 15 min), and the supernatant was subjected to ultracentrifugation (150,000 × g, 1 h) in a Discovery 100 ultracentrifuge equipped with a T-1270 rotor (Sorvall, Newton, CT) to pellet cell membranes. The deep dark red supernatant represented the crude extract. Kustc1061 was brought to homogeneity in a two-step FPLC procedure using an Äkta purifier (GE Healthcare). Columns were eluted at 2 ml min−1; the eluate was monitored at 280 nm and collected in 2-ml fractions. In the first step, crude extract was applied to a column packed with 30 ml of Q Sepharose XL (GE Healthcare) and equilibrated with two column volumes of 20 mm Tris-HCl buffer (pH 8). After washing the column with two column volumes of this Tris-HCl buffer, a hydroxylamine-oxidizing fraction (20 ml) was eluted by isocratic washing with 200 mm NaCl in the Tris-HCl buffer. This fraction was desalted in the above potassium Pi buffer and concentrated to 5 ml using 100-kDa molecular mass cutoff spin filters (Vivaspin 20; Sartorius Stedim Biotech, Aubagne, France). The concentrated fraction was loaded onto 10-ml column packed with Ceramic Hydroxyapatite (Bio-Rad) that had been equilibrated by washing with five column volumes of potassium Pi buffer. After application of the sample, the column was eluted with a 120-ml linear gradient of 20–500 mm potassium Pi (pH 7). Hydroxylamine oxidizing activity eluted as a prominent bright red symmetrical peak at 80 mm potassium Pi. This peak (12 ml) was collected, desalted in 20 mm potassium Pi buffer (pH 7), and concentrated as above to ∼2.8 mg of protein ml−1. Purity was checked by nondenaturating PAGE and SDS-PAGE (19). The identity of kustc1061 was established by MALDI-TOF analysis (see below). By the procedure outlined, kustc1061 was purified 5.3-fold in 31% yield. Both the yield and purification factor will likely be underestimates because crude extract contained substantial hydroxylamine oxidizing activity derived from hydrazine synthase (8).
N. europaea ATCC 19718 was cultured, and NeHAO was purified essentially as described by Hooper and Nason (20). 110 liters of a N. europaea culture were grown at room temperature in mineral medium (ATCC medium 2265) in 20-liter carboys, which were vigorously sparged with air. The pH of the medium was regularly adjusted to 8 by adding 20% (w/v) Na2CO3. The cells were harvested after 6 days (12 g of wet weight), frozen in liquid nitrogen, and stored at −80 °C. The cells were resuspended in 50 mm Tris-HCl (pH 7.5; buffer A) and disrupted by sonication, and the cell-free extract was fractionated by ammonium sulfate precipitation followed by subsequent DEAE-Sepharose (Merck) and ceramic hydroxyapatite (Bio-Rad) liquid chromatography. After exchange to buffer A using a PD-10 desalting column (GE Healthcare), the 35–70% ammonium sulfate fraction was loaded onto a 30-ml DEAE-Sepharose column, which was eluted with a linear gradient of 0–400 mm NaCl in buffer A. The red fractions eluting at 200 mm NaCl were combined, buffer-exchanged to 20 mm potassium Pi buffer (pH 7.5), and loaded onto a 5-ml hydroxyapatite column, which was eluted with a gradient of 20–500 mm potassium phosphate (pH 7.5). NeHAO and Ne1300 co-eluted at ∼250 mm potassium phosphate. These fractions were concentrated and buffer-exchanged to 25 mm HEPES-KOH (pH 7.5) containing 25 mm KCl. The identity of the proteins was confirmed by MALDI-TOF mass spectrometry after tryptic digest from SDS-PAGE gel slices.
Analytical Ultracentrifugation
Sedimentation velocity and equilibrium ultracentrifugation was performed in a Beckman XL-I Proteomelab ultracentrifuge, using 1.2-cm path length cells at 20 °C. The protein was dissolved in 25 mm HEPES-KOH (pH 7.5), 25 mm KCl to an absorbance at 280 nm of less than 0.5 (for 1.2 cm path length). The speeds used were 9,000 and 12,000 rpm in an An60Ti rotor. Equilibrium data were analyzed using SEDFIT (21).
Spectrophotometric Enzyme Assays
By routine, reactions were followed at 37 °C by measuring the reduction of bovine cytochrome c at 550 nm (Δϵ550 = 19,600 m−1 cm−1) (22) in a Cary 50 spectrophotometer (Agilent, Santa Clara, CA). Reaction rates were determined from the initial linear portion of the progress curves employing the Cary 50 software package. For enzyme kinetics, initial reaction rates were fitted by nonlinear regression analysis (Origin 8.5.1; OriginLab Corporation, Northampton, MA) applying Michaelis-Menten equations.
Reaction mixtures (0.4 ml) in potassium Pi buffer contained 50 μm cytochrome c and an appropriate amount of enzyme. After following the absorption at 550 nm for 1 min, reactions were started by the addition of substrate (hydroxylamine or hydrazine) in the requested concentration from 100 μm anoxic stock solutions. Potential inhibitors (90 μm NO, 5 mm phenyl hydrazine) were added prior to the enzyme. An NO stock solution (0.9 mm) was prepared by sparging anoxic potassium Pi buffer with He/NO (50%:50%, v/v) in a butyl-rubber capped serum vial for 10 min. For assays in the direction of reduced cytochrome c oxidation, a stock solution was prepared by mixing 50 μm cytochrome c with 20 μm ascorbic acid, giving 40 μm reduced cytochrome c.
Nitrite reduction with reduced methyl viologen as electron donor was followed by recording methyl viologen oxidation at 600 nm (ϵ600 = 13,700 m−1 cm−1) (23). Reduced methyl viologen monocation radical was made by zinc reduction of a 1 mm stock solution of the oxidized compound (24). The preparation of reduced methyl viologen and of the assay mixtures and subsequent enzymatic reactions were carried out in an anaerobic glove box (97% cytochrome, 3% H2; O2 < 0.2 ppm). Reduced methyl viologen was added to an A600 = 1 (73 μm), and reactions were started by the addition of enzyme.
Spectrofluorometric NO Measurements
Nitric oxide production was measured by two different fluorescence-based methods. Fluorescence was recorded in 4-ml rubber-stoppered fluorescence cuvettes (Hellma, Müllheim, Germany) in a Cary Eclipse fluorometer (Agilent) set at a slit width of 5 nm and high photomultiplier voltage. Calibration was performed in the under-mentioned reaction mixtures without enzyme containing 0–13.5 μm of NO.
The first method used the Cu(II) complex of FL2E (2-{4,5-bis[(6-(2-ethoxy-2-oxoethoxy)-2-methylquinolin-8-ylamino)methyl]-6-hydroxy-3-oxo-3H-xanthen-9-yl}benzoic acid) (25) supplied in the NO-ON nitric oxide sensing kit (Strem Chemicals, Newburyport, MA) as the trapping agent. Cu(II)FL2E reacts specifically with NO in a 1:2 stoichiometry to form a fluorescent nitrosylated derivative. Assays were performed anaerobically at room temperature. Reaction mixtures (4 ml) were prepared in the anaerobic glove box and contained 8 μm hydroxylamine, 50 μm cytochrome c, 0.5% (v/v) Me2SO, and 10 μm Cu(II)FL2E in 20 mm potassium Pi buffer (pH 7). After recording fluorescence (excitation wavelength, 480 nm; emission wavelength, 519 nm) for 2 min, the reaction was started by adding enzyme.
The second method took advantage of the NO- and oxygen-dependent formation of nitrosylated 4-amino-5-methylamino-2′,7′-difluorofluorescein (Calbiochem) (26, 27). In this case, assays were performed aerobically. Reaction mixtures (4 ml) contained 2 μm hydroxylamine, 50 μm cytochrome c, 0.1% (v/v) Me2SO, and 8.25 μm 4-amino-5-methylamino-2′,7′-difluorofluorescein in 20 mm potassium Pi buffer (pH 7). Reactions were recorded at an excitation wavelength of 495 nm and an emission wavelength of 518 nm and were started by the addition of 0.6 μg of enzyme.
Other Analytical Methods
(15N-Labeled) gaseous nitrogen compounds (NO, NO2, N2O, and N2) were determined by GC-MS analysis of head space samples as described previously (8). Here, reactions were performed in 3-ml Exetainers (Labco, High Wycombe, UK) closed with rubber stoppers. Reaction mixtures (2 ml) containing either 10 μm [15N]hydroxylamine (99% pure; Isotec, Miamisburg, OH) or 7.5 μm double-(15N) labeled hydrazine (98% pure; Cambridge Isotope Laboratories, Cambridge, UK) were prepared in the anaerobic glove box. Ammonium and nitrite were measured colorimetrically according to established methods (28). Protein concentrations were measured with the Bio-Rad protein assay, based on the method of Bradford (29), using bovine serum albumin as a standard.
The identity of proteins was established by MALDI-TOF analysis of the trypsin-cleaved protein prepared from the gels as detailed elsewhere (8, 30). Spectra were evaluated by the Mascot Peptide Mass Fingerprint search program (Matrix Science, London, UK) against the K. stuttgartiensis and the N. europaea protein databases. In case of kustc1061, 12 of 43 predicted peptides were retrieved (Mowse identification score, >50) (supplemental Fig. S1).
Electronic Absorbance Spectra
UV-visible spectra were measured in 1-ml quartz cuvettes (path length, 1 cm). Samples contained 80 μg ml−1 of as-isolated fully oxidized kustc1061 in potassium Pi buffer. Spectra of the reduced kustc1061 were recorded immediately after adding few crystals of sodium dithionite to the solution containing oxidized enzyme. Spectra in the presence of substrates were made after preincubating oxidized or reduced kustc1061 solution with 100 μm of the compound to be tested (5 mm for phenyl hydrazine). NO (final concentration, 90 μm) was added from a stock solution prepared as described above.
Spectroelectrochemistry
Heme reduction potentials were measured through changes in the electronic absorbance of kustc1061 that had been adsorbed on an optically transparent SnO2 electrode and then equilibrated at 4 °C at a series of defined potentials following the protocol previously published (31). The electrode was composed of a mesoporous, nanocrystalline layer of SnO2 coated onto a glass slide (32). The electrode (∼0.7-cm2 geometric area) was covered with 5 μl of 3.5 mg ml−1 kustc1061 in potassium Pi buffer and incubated on ice for 10 min. After removal of excess protein solution, the electrode was rinsed with 50 mm MOPS buffer (pH 7) and placed in an anaerobic 3-ml quartz cuvette (path length, 1 cm) filled with 50 mm MOPS buffer (pH 7). A platinum wire and an AgCl-coated silver wire served as counter and reference (E0′ = +400 mV versus standard hydrogen electrode) electrodes, respectively. The cuvette containing the kustc1061-coated SnO2 electrode was placed in a spectrophotometer (V650; Jasco Analytical Instruments, Easton, MD) and connected to a potentiostat (PGSTAT20; Autolab, Utrecht, The Netherlands). To minimize the spectral contribution from scattering by the SnO2 particles, a cuvette containing a SnO2 electrode that had not been exposed to kustc1061 was placed in the reference beam of the spectrometer during measurements. UV-visible spectra were recorded after setting the potential applied to the kustc1061-coated electrode to the desired value and allowing the protein to equilibrate with that potential. Voltage-dependent spectral changes were fully reproducible for at least five cycles between −600 and +400 mV (20 mV steps) in reductive and oxidative directions. Changes in absorbance at 551 and 418 nm derived from the heme c α and Soret bands, respectively, and at 468 nm of the P460 chromophore were normalized and evaluated by Nernstian curve fitting using Microsoft Excel and applying least square minimizing.
Protein Crystallization and Crystal Handling
Kustc1061 was concentrated by ultrafiltration to A280,1 cm ≈ 20 (A409,1 cm ≈ 95) in 25 mm HEPES-KOH buffer (pH 7.5), containing 25 mm KCl. Needle-shaped, hexagonal crystals of kustc1061 grew from hanging drops equilibrated against 1.2 m ammonium sulfate, 0.1 m sodium phosphate buffer (pH 7.4). These crystals were soaked in reservoir solution containing 25% (v/v) ethylene glycol and 10 mm HgCl2 before flash-cooling in liquid nitrogen. Another, cubic crystal form diffracting to much higher resolution grew from 1.3 m ammonium sulfate, 0.05–0.1 m sodium phosphate buffer (pH 7.4), and the detergent additive cyclohexylbutanoyl-N-hydroxyethylglucamide. These crystals were cryoprotected in reservoir solution with 25% (v/v) ethylene glycol or 30% (w/v) sucrose and then flash-cooled in liquid nitrogen.
NeHAO (A280,1 cm ≈ 37; A409,1 cm ≈ 133) was crystallized essentially as described by Cedervall et al. (12, 13). Thin oval plates of up to 200-μm longest dimension grew at 20 °C under Al's oil (a 1:1 mixture of paraffin oil and silicon oil) in batch crystallization using 42% (v/v) PEG400 and 50 mm KNO3 in 100 mm MES/NaOH buffer (pH 7.5), after 2 weeks. When flashed-cooled in their mother liquor, the space group was P212121 with a = 142 Å, b = 143 Å, c = 214 Å. These crystals could also be dehydrated by increasing the PEG400 concentration to 65% (v/v) in 5% steps, resulting in space group P21212 with a = 140 Å, b = 141 Å, c = 106 Å. A large improvement in diffraction quality was observed upon annealing (33, 34) the flash-cooled crystals by interrupting the cryostream once for 15 s.
Data Collection and Structure Solution
Diffraction data from kustc1061 were collected at the X10SA Beamline of the Swiss Light Source (Villigen, CH) at 100 K and processed with XDS (35, 36). Statistics are reported in Table 1. A highly redundant, 3.5 Å resolution SAD data set was collected from a hexagonal crystal at a wavelength of 1.7384 Å, in which AutoSHARP (37) detected seven heavy atom sites per asymmetric unit using SHELXD (38). Phasing and density modification using SOLOMON (39) resulted in an excellent electron density map into which an initial model was built, which was then refined against a 2.6 Å resolution data set from a hexagonal crystal collected at a wavelength of 1.000 Å. After several rounds of rebuilding in COOT (40) and refinement with REFMAC (41) and PHENIX (42), a model with excellent geometry and R factors was obtained (Table 1). This model was used to phase the high resolution data obtained from the cubic crystal form by molecular replacement with PHASER (43). Here, too, repeated rounds of rebuilding in COOT (40) and refinement with REFMAC (41) resulted in an excellent model with good R factors (Table 1). Geometric parameters for the modified heme co-factor were obtained from semiempirical calculations at the PM-3 level carried out using HyperChem 8.0 (Hypercube Inc., Gainesville, FL). Crystals of protein-ligand complexes were obtained by soaking cubic crystals in artificial mother liquor containing the respective compounds (100 mm hydroxylammonium chloride, 10 min; 5 mm hydrazinium sulfate, 10 min; and 50 mm phenyl hydrazine, 30 min) followed by flash-cooling in liquid nitrogen. Soaking with NO was done by incubating crystals in thoroughly degassed and cytochrome-saturated buffers, and cryoprotectants were placed inside an anaerobic glove box for 15 min at room temperature with ∼4 mm of the NO donor 1-(N,N-diethylamine)diazen-1-ium-1,2-diolate.
TABLE 1.
Data collection, phasing, and refinement statistics
The values in parentheses are for highest resolution shell.
| KustC1061 native HgCl2 Fe SAD | KustC1061 native initial model | KustC1061 native high resolution (4N4J) | KustC1061 NH2OH soak (4N4K) | KustC1061 N2H4 soak (4N4L) | KustC1061 phenyl hydrazine soak (4N4M) | Native NeHAO (4N4N) | NeHAO NH2OH soak (4N4O) | |
|---|---|---|---|---|---|---|---|---|
| Data collection | ||||||||
| Space group | P63 | P63 | P213 | P213 | P213 | P213 | P21212 | P21212 |
| Cell dimensions | ||||||||
| a, b, c (Å) | 96.6, 96.6, 118.0 | 96.4, 96.4, 118.0 | 130.0, 130.0, 130.0 | 130.1, 130.1, 130.1 | 130.0, 130.0, 130.0 | 130.3, 130.3, 130.3 | 140.7, 142.0, 106.7 | 140.1,142.0, 106.5 |
| α, β, γ (°) | 90, 90, 120 | 90, 90, 120 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| Wavelength | 1.7384 | 1.0000 | 0.9785 | 1.0000 | 0.9785 | 1.0000 | 0.9786 | 0.9778 |
| Resolution (Å) | 20.0-3.5 (3.6-3.5) | 30.0-2.60 (2.7-2.6) | 30.0-1.8 (1.9-1.8) | 30.0-2.2 (2.3-2.2) | 30.0-1.9 (2.0-1.9) | 30.0-2.1 (2.2-2.1) | 50-2.2 (2.3-2.2) | 50-2.5 (2.7-2.5) |
| Rmerge | 0.083 (0.151) | 0.081 (0.479) | 0.084 (0.387) | 0.076 (0.356) | 0.100 (0.464) | 0.072 (0.455) | 0.102 (0.588) | 0.124 (0.541) |
| I/σI | 37.0 (21.7) | 19.7 (5.0) | 16.8 (4.8) | 17.3 (4.9) | 19.9 (5.4) | 18.0 (3.9) | 13.6 (3.1) | 11.0 (3.1) |
| Completeness (%) | 99.9 (100) | 99.9 (100) | 100.0 (100.0) | 99.9 (100.0) | 100.0 (99.9) | 99.9 (100.0) | 99.5 (99.3) | 99.3 (99.3) |
| Redundancy | 20.4 (20.2) | 7.2 (7.4) | 7.5 (7.6) | 6.6 (6.9) | 13.4 (13.7) | 18.0 (3.9) | 5.6 (5.5) | 4.5 (4.5) |
| Refinement | ||||||||
| Resolution (Å) | 46-1.8 | 46-2.2 | 46-1.9 | 46-2.1 | 50-2.2 | 50-2.5 | ||
| No. reflections | 64,440 | 35,645 | 54,927 | 41,094 | 98,124 | 68,039 | ||
| Rwork/Rfree | 0.143/0.157 | 0.154/0.191 | 0.148/0.163 | 0.165/0.202 | 0.179/0.213 | 0.224/0.273 | ||
| No. atoms | ||||||||
| Protein | 3,979 | 3,968 | 3,962 | 3,930 | 13,323 | 13,314 | ||
| Ligand/ion | 344 (8 heme) | 344 (8 heme) | 344 (8 heme) | 344 (8 heme) | 1034 (24 heme) | 1034 (24 heme) | ||
| 20 (4 PO43−) | 15 (3 PO43−) | 20 (4 PO43−) | 20 (4 PO43−) | 5 (PO43−) | 5 (PO43−) | |||
| 58 (4 C- HEGA-10) | 12 (3 EG) | 28 (7 EG) | 28 (7 EG) | 1 (1 K+) | 36 (2 PEG) | |||
| 2 (H2NOH) | 12 (C-HEGA-10) | 6 (1 C6H5) | 6 (3 NH2OH) | |||||
| 2 (H2NNH2) | 2 (2 K+) | |||||||
| Water | 472 | 306 | 417 | 268 | 1385 | 993 | ||
| B-factors | ||||||||
| Protein | 15.2 | 26.8 | 17.9 | 28.9 | 28.9 | 33.4 | ||
| Ligand/ion | 17.1 | 21.9 | 17.6 | 23.5 | 22.7 | 51.0 | ||
| Water | 21.8 | 33.7 | 24.6 | 33.1 | 34.7 | 32.3 | ||
| Root mean square deviations | ||||||||
| Bond lengths (Å) | 0.007 | 0.008 | 0.008 | 0.008 | 0.007 | 0.006 | ||
| Bond angles (°) | 1.086 | 0.961 | 1.086 | 1.042 | 1.025 | 0.917 | ||
NeHAO crystals of either crystal form were annealed as described above, and native data were collected at PETRA-III using a Pilatus 6 m detector at 100 K and processed with XDS (35, 36). Molecular replacement with PHASER (43) using the structure from Igarashi et al. (11) (Protein Data Bank 1FGJ) as the search model. During iterative rebuilding in COOT (40) and refinement with REFMAC (41), an extra polypeptide chain became visible in the electron density maps. This protein was confirmed to be Ne1300 by peptide mass fingerprinting and was built accordingly. A ligand-coordinated structure of NeHAO was obtained by performing the dehydration protocol in the presence of 100 mm hydroxylamine. Data were collected at the X10SA Beamline of the Swiss Light Source (Villigen, CH) at 100 K. Difference electron density maps clearly displayed extra density connected to the position where a water molecule is bound in the native structure, clearly showing that a larger molecule had bound to the P460 co-factor.
Bioinformatics
Protein sequence homology searches were performed against nonredundant protein databases using the BlastP program (44) at the NCBI website. N-terminal signal cleavage sites were predicted with SignalP 4.1 (45). Multiple amino acid sequence alignments were made with the ClustalW program (46) at the EMBL-EBI website. All structural figures were prepared in PyMOL (47).
RESULTS
Kustc1061 Is a Specific NO-forming Hydroxylamine Oxidase
Previously, we purified an abundant HAO-like protein from K. stuttgartiensis and identified this as kustc1061 (8). This protein performed the three-electron oxidation of hydroxylamine, resulting in the stoichiometric formation of NO and three molecules of reduced cytochrome c (Fig. 1, A–D, and Equation 1). In contrast and as expected, the purified NeHAO catalyzed a four-electron oxidation of hydroxylamine in accordance with Equation 2 (Fig. 1A). Hydroxylamine oxidation by kustc1061 followed Michaelis-Menten kinetics (apparent Km = 4.4 ± 0.9 μm, Vmax = 4.8 ± 0.2 μmol min−1 mg protein−1). NO in concentrations up to 80 μm did not have any effect on this reaction. Further, kustc1061 was also able to catalyze the four-electron oxidation of hydrazine to N2 (Fig. 1, A and E), albeit with lower affinity and maximum rate (apparent Km = 54 ± 3.3 μm, Vmax = 1.60 ± 0.05 μmol min−1 mg protein−1), i.e., with a 34-fold lower catalytic efficiency (kcat/Km) (Table 2). Kustc1061 was capable of nitrite reduction with an artificial reductant, reduced methyl viologen, but only with a very low rate (0.18 μmol min−1 mg protein−1), which is also comparable to nitrite reduction by NeHAO (Table 2). In contrast to what was reported for NeHAO (48), we could not measure any hydroxylamine disproportionation to ammonium and NO, nitrite, N2O, or N2. These findings strongly suggested that the oxidation of hydroxylamine to NO was the distinctive physiological reaction of kustc1061. The kustc1061 catalytic constants were in the same range as those reported for not well defined, homologous enzymes of the anammox bacteria Brocadia anammoxidans and strain KSU-1 (Table 2) (49, 50). NeHAO oxidized hydroxylamine and hydrazine at 6- and 9-fold higher maximal rates, respectively.
FIGURE 1.

Hydroxylamine and hydrazine oxidation by kustc1061 and NeHAO. A, cytochrome c reduction (50 μm) catalyzed by kustc1061 (200 ng) in the presence of 1.5 μm hydroxylamine (blue) or 1.5 μm hydrazine (red). The oxidation of hydroxylamine (1.5 μm) coupled to the reduction of cytochrome c (50 μm) by NeHAO (30 ng) is represented by the green curve. Reactions were followed and quantified by measuring the increase in the absorption at 550 nm (Δϵ550 = 19,600 m−1 cm−1) as the result of cytochrome c reduction. B, hydroxylamine oxidation (9 μm) by 1.4 μg of kustc1061 (blue) and hydrazine oxidation (9 μm) by 20 μg of kustc1061 (red) recorded by measuring the reduction of cytochrome c at 550 nm (Δϵ550 = 19,600 m−1 cm−1, right axis). C, NO production from hydroxylamine (8 μm) by kustc1061 (150 ng) in the presence of 50 μm cytochrome c. NO formation was followed by recording the increase in fluorescence as the result of the formation of the nitrosylated derivative of Cu(II)FL2E and was quantified from a calibration curve prepared from known NO stock solutions. D, hydroxylamine oxidation (2 μm) recorded by the increase in fluorescence resulting from the formation of a nitrosylated derivative of the NO-probe 4-amino-5-methylamino-2′,7′-difluorofluorescein (black) and cytochrome c reduction under the same conditions (blue), showing a 1:3 stoichiometry of NO production versus cytochrome c reduction. E, accumulation of 30N2 during [15N]hydrazine oxidation. 15N15N (open squares) was produced upon addition of 7.5 μm H15N-15NH to 2 ml of 20 mm potassium Pi buffer containing 1.3 μg of kustc1061 and 50 μm oxidized cytochrome c. The concentration of reduced cytochrome c was determined as in A. NO in C and D was quantified from calibration curves prepared from known NO stock solutions.
TABLE 2.
Catalytic and structural properties of kustc1061 from K. stuttgartiensis, characterized orthologues from B. anammoxidans, and anammox strain KSU1 and hydroxylamine oxidoreductase from N. europaea
Kinetic values are given for mammalian cytochrome c as electron acceptor. Reduced methyl viologen was used as electron donor for NO2− reduction. NR, not reported.
| Property | K. stuttgartiensis | B. anammoxidans | KSU-1 | N. europaea |
|---|---|---|---|---|
| Vmax NH2OH (μmol min−1 mg−1) | 4.8 | 21 | 9.6 | 28.5 |
| kcat NH2OH (s−1) | 15 | 64 | 19 | 95 |
| Km NH2OH (μm) | 4.4 | 26 | 33 | 3.6 |
| kcat/Km NH2OH (s−1 μm−1) | 3.4 | 2.5 | 0.57 | 26 |
| Vmax N2H4 (μmol min−1 mg−1) | 1.6 | 1.1 | 0.54 | 14 |
| kcat N2H4 (s−1) | 4.9 | 3.4 | 1.1 | 47 |
| Km N2H4 (μm) | 54 | 18 | 25 | 4 |
| kcat/Km N2H4 (s−1 μm−1) | 0.1 | 0.19 | 0.04 | 12 |
| NO2− reduction (μmol min−1 mg−1) | 0.18a | 0.87b | NR | 6.18c |
| Total size (kDa) | 184 | 183 | 118 | 200 |
| Subunit size (kDa) | 61.5 | 58 | 53 | 67 |
| Subunit composition | α3 | α3 | α2 | α3 |
| Catalytic heme optical maximum (nm) | 468 | 468 | 468 | 463 |
| References | This paper | 49 | 50 | 20, 51, 52 |
a Specific activity with 0.1 mm nitrite.
b Specific activity with 2.5 mm nitrite.
c Vmax.
Kustc1061 Is a Multiheme Protein with a Low Redox Potential P460 Chromophore
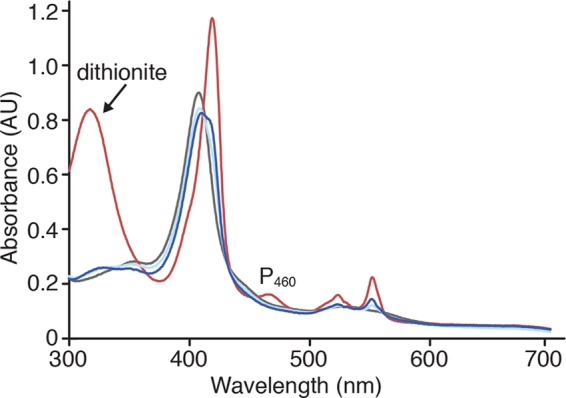
Kustc1061 was purified as a protein with a molecular mass of 184.20 kDa as determined by sedimentation equilibrium ultracentrifugation. This value was in excellent agreement with the theoretical mass of 184.16 kDa calculated for a homotrimeric gene product, after cleavage of a predicted signal peptide, harboring 24 heme c molecules in total. The resolution of the crystal structure described below confirmed the presence of three identical subunits, each binding eight c-type hemes.
The UV-visible spectrum of the as-isolated kustc1061 exhibited the characteristic features of low spin ferric c-heme, namely, a pronounced maximum at 408 nm in the Soret region and a broad feature of lower intensity between 500 and 600 nm (Fig. 2). Upon addition of dithionite, these features of the fully oxidized enzyme changed, giving rise to maxima at 418, 522, and 551 nm and at 468 nm. The maxima at 418, 522, and 551 nm were indicative of (His/His ligated) low spin ferrous c-hemes, whereas that at 468 nm was reminiscent of a ferrous heme with covalent modification by tyrosine as found for the P460 catalytic heme in NeHAO (11, 13, 53). Cross-linking of kustc1061 subunits by covalent modification of the heme was supported by the failure of the three subunits to dissociate upon denaturing SDS-PAGE (data not shown) and confirmed by x-ray crystallography (see below). Using protein film spectroelectrochemistry, the low spin His/His-ligated hemes could be reversibly reduced in seven consecutive one-electron reduction steps showing midpoint redox potentials at pH 7 (Em′) ranging from −10 to −410 mV versus the standard hydrogen electrode (Fig. 3). This potential window encompassed six Em′ values for the His/His-ligated hemes in NeHAO (53, 54). The P460 chromophore in kustc1061 displayed an Em′ of −300 ± 10 mV compared with Em′ = −260 mV for NeHAO.
FIGURE 2.
Electronic absorbance spectra of kustc1061. Spectra are the following: as-isolated (fully oxidized) kustc1061 (80 μg ml−1) (gray), dithionite-reduced kustc1061 (red), and the as-isolated enzyme incubated either with 100 μm hydroxylamine (dark blue) or with 100 μm hydrazine (light blue).
FIGURE 3.

Protein film electrochemistry of kustc1061. A, full spectra of the oxidative titration from −490 to +230 mV versus standard hydrogen electrode (SHE) in 20-mV steps. B, table showing the Em′ values (± 10 mV) calculated for the successive oxidations and reductions of the c-type hemes in kustc1061 and the comparison to those reported for NeHAO (53, 54). Numbers in parentheses refer to the corresponding hemes in NeHAO. Note that the heme c midpoint redox potentials in kustc1061 are generally lower, but the method applied does not allow the assignment to specific hemes, except for the catalytic heme 4. C, spectral details of the Soret region. D, signals at 418 nm normalized to 1 and plotted against the applied potentials. E, spectral details of the heme c-α and -β bands. F, signals at 551 nm normalized to 1 and plotted against the applied potentials. G, spectral details of the P460 region. H, signals at 468 nm normalized to 1 and plotted against the applied potentials. Squares in D, F, and H represent experimental values. Solid lines in these graphs are Nernstian fits. The data in D and F were well described by the sum of seven Nernstian contributions from isolated, single-electron (n = 1) redox centers having the Em′ values listed in B (all ± 10 mV). It was assumed that P460 spectral properties did not interfere significantly with the spectral changes of the Soret (C) and α and β (E) bands of the His/His ligated hemes; clear isosbestic points at 435 nm and at 505 and 561 nm, respectively, supported this assumption. The variation in absorbance at 468 nm had contributions from redox transformation of the P460 co-factor in addition to those of low spin His/His ligated hemes. The changes in absorbance caused by the P460 co-factor were readily identified through their larger magnitude and the appearance of a peak at low potential with an isosbestic point at 455 nm. The plot of normalized A468 nm − A455 nm was well described by a fit to the Nernst equation for an n = 1 center with Em′ = −300 mV.
When as-isolated kustc1061 was incubated with its substrate hydroxylamine, the protein became partly reduced (Fig. 2). Comparison with protein film spectroelectrochemistry results suggested the reduction of two hemes such that hydroxylamine would undergo a two-electron oxidation upon binding to the enzyme. Incubation with hydrazine also resulted in a spectrum indicative of two-electron reduction. In contrast, NO had no effect on the absorption spectrum of oxidized kustc1061, but it reoxidized the reduced enzyme completely.
Kustc1061 and NeHAO Share a Common Architecture
The high resolution x-ray crystal structure of kustc1061 was predominantly α-helical, with long helices running approximately parallel to the 3-fold symmetry axis of the trimer and surrounding a central, water-filled cavity (Figs. 4 and 5A). The subunits were linked by two covalent bonds between Tyr451 of one subunit and heme 4 of an adjacent subunit. Heme 4 was ligated by a single amino acid (His227) and could be assigned as the P460 co-factor identified by UV-visible spectroscopy. The additional seven c-type hemes had His/His ligation, so that together they readily accounted for the additional ferric and ferrous heme features observed in the UV-visible spectra.
FIGURE 4.
X-ray crystal structure of kustc1061 from K. stuttgartiensis. A, views of the kustc1061 trimer perpendicular to (left panel) and along the 3-fold symmetry axis (right panel). The three monomers are shown in different colors. The 24 heme groups are shown in red. B, the same views of the kustc1061 trimers, showing the outline of the protein and the ring-like arrangement of heme groups (red). C, stereo figure, showing a close-up of the active site of kustc1061. The P460 co-factor consists of a heme c moiety (white sticks) covalently bound in two places to the Tyr451 side chain of an adjacent protein monomer (blue). The conserved Asp262/His263 is shown, as well as His227, which coordinates the heme iron as the proximal ligand. A water molecule (red sphere) forms the distal ligand. The final, refined 2mFo − DFc electron density map calculated from 1.8 Å resolution data is overlaid at a 2 σ contour level (blue mesh). The figures were prepared in PyMOL (47).
FIGURE 5.

Views of kustc1061 and NeHAO along the 3-fold axis of the trimers. A, kustc1061, with the N-terminal domain shown in red. B, NeHAO, with the Ne1300 polypeptide shown in orange. In kustc1061, the N-terminal domain adopts the position of Ne1300 in its complex with NeHAO.
Despite a sequence identity of only 26% to NeHAO (supplemental Fig. S1), the structures of kustc1061 and NeHAO were similar and could be superimposed to a root mean square positional difference of 1.7 Å for 377 Cα atoms. This conservation also applied to the position of the heme moieties (Figs. 4–6). One may note that the same heme configuration was observed in other octaheme proteins and in pentaheme nitrite reductase (NrfA) in which the five hemes (1–5) superimpose with hemes 4–8 of the octaheme proteins (14, 15, 55, 56). However, in the trimeric kustc1061 and NeHAO, hemes were arranged in a ring. It was proposed earlier that such a ring-like arrangement of single electron carriers would enable electron transfer between the subunits, thus facilitating the extraction of two electrons from the substrate in rapid succession (54), which could also be essential for the oxidation of NH2OH to NO catalyzed by kustc1061.
FIGURE 6.

Structural similarity of the catalytic features of kust1061 and NeHAO. A, stereo view of a superposition of the active sites of kustc1061 (light gray) and NeHAO (dark gray) soaked in hydroxylamine. B, stereo view of a superposition of kustc1061 and NeHAO based on Cα-carbons, showing only the heme groups. The two proteins show a virtually identical arrangement of hemes.
Apart from this structural similarity between kustc1061 and NeHAO, we observed distinguishing differences between these two proteins, notably in their N- and C-terminal parts. Kustc1061 had a negatively charged N-terminal domain (N1; amino acids 36–80 of the translated protein) that clung to a positively charged amino acid stretch of an adjacent subunit (Fig. 5). This domain was absent in NeHAO. Instead, and in full agreement with the structure recently published by Cedervall et al. (13), the subunit interface in our native and complexed NeHAO structures was occupied by a polypeptide of 69 amino acids, which was identified as a fragment of the hypothetical protein Ne1300 (101 amino acids). However, these overall structural differences between the two proteins did not immediately explain why in kustc1061 the oxidation of NH2OH stops at NO, whereas in NeHAO the reaction continues to NO2−.
Kustc1061 and NeHAO Have Structurally Similar P460 Co-factors
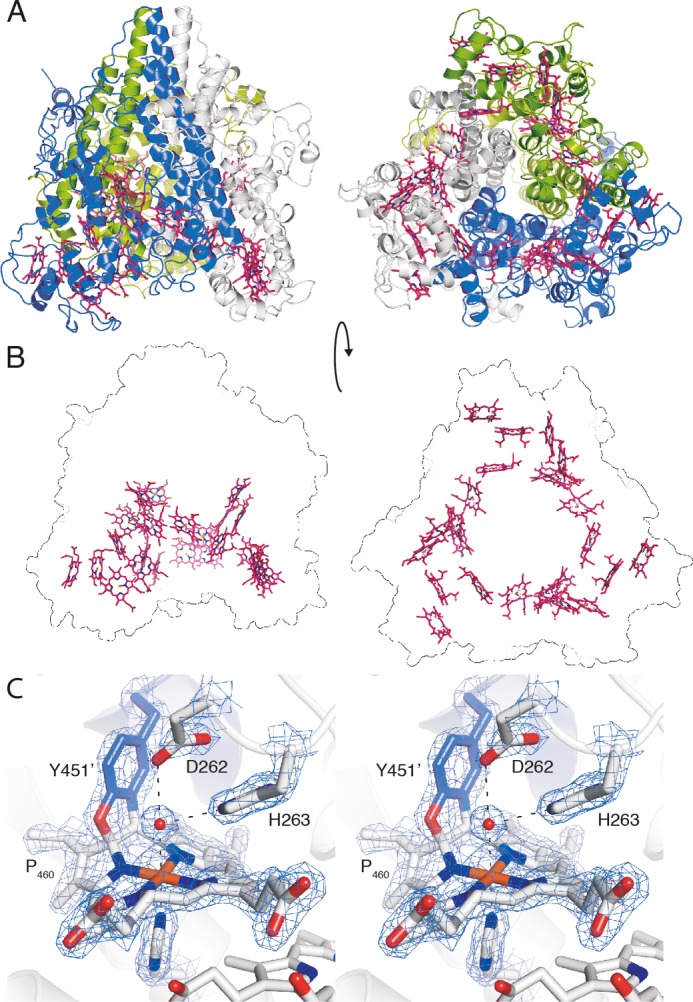
We determined kustc1061 crystal structures soaked with substrates, observing binding to heme 4 (the P460 co-factor), which was apparently the active site, as in NeHAO (11, 13). Considering the difference in reaction specificity, it was surprising that at first glance the active sites of kustc1061 and NeHAO appeared highly similar as well (Figs. 6 and 7A). In both proteins, heme 4 was covalently linked to a tyrosine from another subunit, causing the co-factor to be substantially ruffled. This ruffling was proposed to support electron abstraction from the substrate by the heme (11, 54). In the 1.8 Å resolution structure of kustc1061, we observed two covalent bonds between Tyr451 and the P460 co-factor, between a Tyr451 Cϵ and the heme meso-carbon C5, as well as between the Tyr451 Oη and the heme carbon atom C4 (Figs. 4C, 6A, 7, A–C, and 8, A–C). Although only the latter of these two was built in the previous, 2.8 Å resolution structural model of NeHAO (11), both in our higher resolution structures and in the recent refinement (2.1 Å) (13), two bonds were clearly present (Figs. 6A, 7D, and 8D), excluding the possibility that the different reaction specificities result from differences in tyrosine binding. Similarly, the direct vicinity of the substrate-binding site on the P460 co-factor did not immediately explain reaction specificities either. In as-isolated kustc1061, a water molecule was present as the sixth (distal) ligand to the iron in the P460 co-factor (Fig. 4C). Also in NeHAO, a distal water molecule was bound to the catalytic P460 co-factor, as deduced from our native (2.2 Å) and recent (2.2 Å) (13) resolution structures. This feature could not be discerned originally because of limited resolution (11).
FIGURE 7.
Stereo views of Fo − DFc electron density maps for the substrate soaks, calculated prior to the inclusion of the ligands in the model. Positive difference density (green) is contoured at 4 σ, and negative difference density (red) is contoured at −4 σ. A, kustc1061 soaked in NH2OH. B, kustc1061 soaked in hydrazine. C, kustc1061 soaked in phenyl hydrazine. D, Nitrosomonas HAO soaked in NH2OH.
FIGURE 8.

X-ray structures of the kustc1061 and NeHAO catalytic sites in crystals soaked with substrates. Close-ups of the active sites of kustc1061 soaked with hydroxylamine (A), hydrazine (B), and phenyl hydrazine (C) and of NeHAO soaked with hydroxylamine (D) are shown.
Substrates Bind to P460 in Both Kustc1061 and in NeHAO
In kustc1061, the water bound to the iron in the P460 co-factor was replaced by a larger molecule after soaking crystals with hydroxylamine (Figs. 6A, 7A, and 8A). In the resulting 2.2 Å structure of kustc1061, this compound was modeled as an NO species coordinating at an angle of ∼107° with respect to the heme-iron bond. Considering that hydroxylamine underwent a two-electron oxidation upon binding, this structure most likely represented a {FeNO}7 nitrosyl derivative, which was consistent with the observed angle between the NO molecule and the iron-nitrogen bond. Interestingly, a similarly sized molecule bound with comparable orientation to the P460 was observed in the 1.9 Å electron density maps of a hydrazine-soaked kustc1061 crystal, possibly representing an iron-bound diazene (HN = NH) (Figs. 7B and 8B). Such a diatomic molecule was not observed in crystals incubated anaerobically with the NO donor 1-(N,N-diethylamine)diazen-1-ium-1,2-diolate. In the hydroxylamine-soaked kustc1061 structure, the putative nitrosyl group bound to the P460 heme iron atom contacted the conserved aspartate (Asp262) and histidine (His263) residues. The structure of the kustc1061 hydrazine soak was very similar, except that Asp262 had rotated away from the binding site. We also determined the structure of a complex of kustc1061 with a phenyl radical at 2.1 Å resolution by soaking a crystal in phenyl hydrazine (Figs. 7C and 8C). In this structure, the loop carrying the Asp262/His263 pair moved by several Ångströms, which further suggested flexibility in this region. In our structure of NeHAO soaked with hydroxylamine, a diatomic molecule was found as well (Figs. 6A, 7D, and 8D). Again, this molecule was bound to the heme iron at an angle of 118°, and it was coordinated by the Asp291/His292 pair, which corresponded to Asp262/His263 in kustc1061. Probably, the aspartate/histidine pair served to abstract protons from hydroxylamine (Equations 1 and 2) as was proposed for the orthologous residues in NeHAO (11, 54, 57). Given the flexibility of this region observed in kustc1061 and its proximity to the central, water-filled cavity, these residues could shuttle the protons into this cavity, from where they could be transferred to the solvent. Indeed, in our native NeHAO structure, two chains of water molecules were observed leading from Asp291 to the central cavity, which could assist in the proposed proton transfer (Fig. 9).
FIGURE 9.
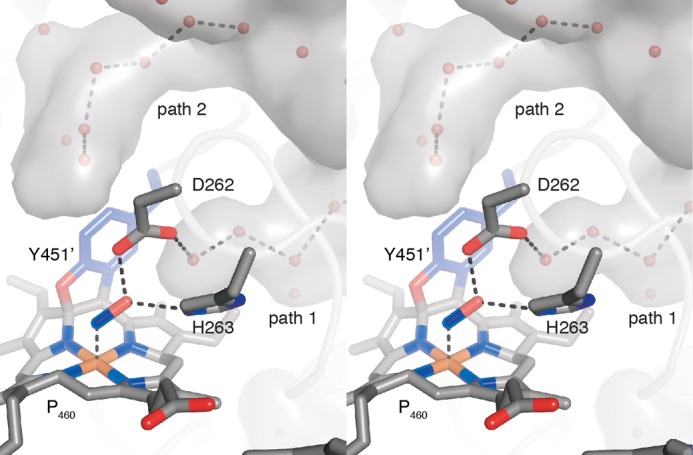
Stereo view of the water structure in the active site of NeHAO. From the central, water-filled cavity (gray surface), two water channels protrude toward the active site (paths 1 and 2). Path 1 could relay protons from the active site to the central cavity when the conserved histidine is in the conformation observed in the hydroxylamine soak. In the conformation observed in the kustc1061-hydrazine soak, path 2 could be used.
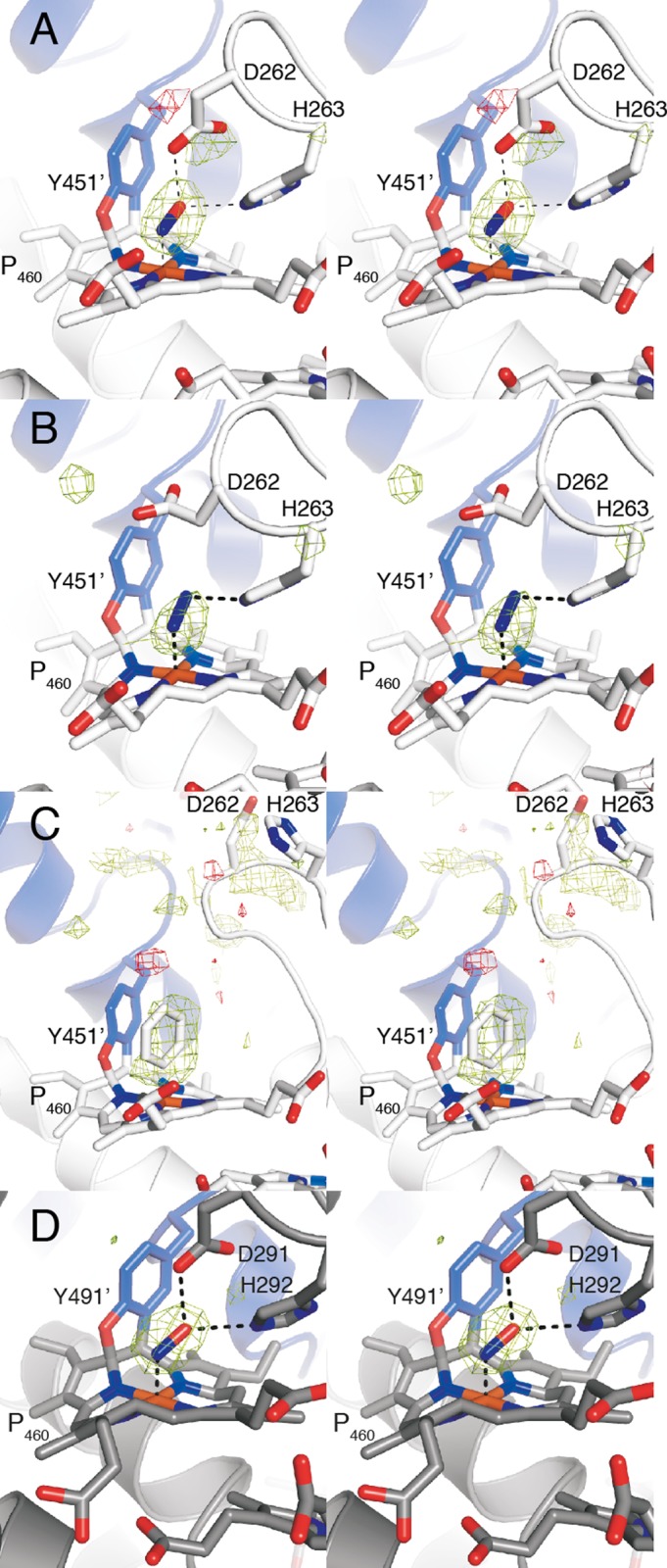
Ultimately, only one significant structural difference between the kustc1061 and NeHAO active sites was apparent. Close to the NeHAO active site, a tyrosine (Tyr358) was present at the distal side of the porphyrin ring (Fig. 10A). As judged from the amino acid sequence (supplemental Fig. S1), an orthologous tyrosine (Tyr320) was present in kustc1061. However, a two-amino acid contraction in kustc1061 resulted in Tyr320 being moved away from the active site, where it was replaced by a hydrophobic methionine (Met323) (Fig. 10B).
FIGURE 10.
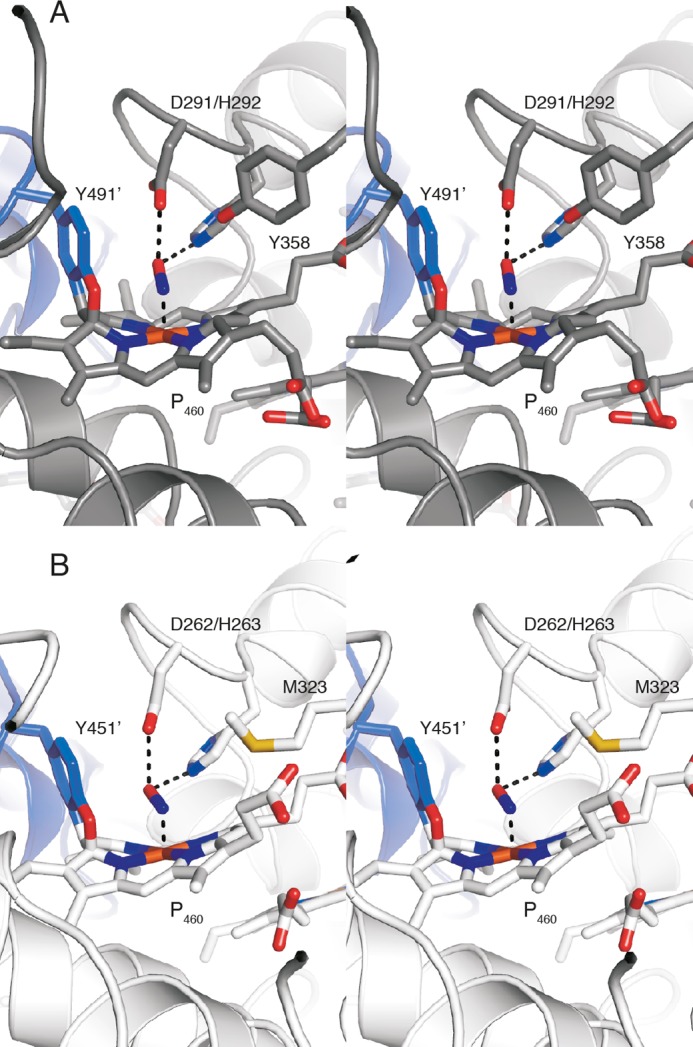
View of the active site tyrosine of NeHAO and the active site methionine of kustc1061 soaked with hydroxylamine. A, stereo view of the NeHAO active site as in Fig. 7D, rotated by ∼90° around the vertical. The conserved Tyr358 provides a possible binding site for a water molecule that can then attack the proposed {FeNO}7 species for nitrite formation. B, stereo view of the kustc1061 active site as in Fig. 7A, again after ∼90° rotation. In kustc1061, the hydrophobic Met323 takes the position of Tyr358 in NeHAO, precluding the proposed water binding.
DISCUSSION
Here, we functionally and structurally characterized the octaheme protein kustc1061 from the anammox bacterium K. stuttgartiensis. Despite the limited sequence identity, kustc1061 shared multiple characteristics with HAO from N. europaea. Both proteins were organized into covalently bound homotrimers harboring eight c-type hemes per monomer. The arrangement of these hemes, as well as their comparable midpoint redox potentials, and the presence of the P460-type catalytic centers were similar. Both kustc1061 and NeHAO use hydroxylamine as their substrate, but whereas kustc1061, and presumably many other HAO-like proteins mentioned below, oxidizes the substrate to NO, nitrite is the (major) product of NeHAO activity. This resulted in the question of what determined the reaction specificity.
Crystals both of kustc1061 and NeHAO soaked with hydroxylamine showed the presence of a diatomic molecule in the active site that could be modeled as a {FeNO}7 nitrosyl (Figs. 6A; 7, A and D; and 8, A and D). This would imply that these two proteins follow a common catalytic cycle (57) at least up to this point, involving the abstraction of two protons and two electrons (Fig. 10). Considering that these electrons have to be shuttled one at a time through the wire of His/His-ligated cytochrome c one-electron carriers, it is possible to speculate that the tyrosine bound to the catalytic heme assists in electron transfer by temporarily storing one of the electrons as a tyrosine radical. The subsequent one-electron oxidation of the {FeNO}7 nitrosyl would then yield Fe(III)-bound NO ({FeNO}6) as proposed for NeHAO in Ref. 57 (Fig. 11). Apparently, in kustc1061 this would be followed by the dissociation of NO and the rebinding of water, regenerating the resting state. Although it is known that NO is a minor product of the reaction catalyzed by NeHAO (60), its oxidation reaction has to continue to the nitrite state. To reach this state, the NeHAO reaction requires the addition of an oxygen atom, which derived from a water molecule (57), to the substrate. Comparing the active sites of kustc1061 and NeHAO, it seems that the reaction specificity relies on facilitating or preventing this step. In NeHAO, the hydrophilic Oη hydroxyl of the Tyr358 side chain is perfectly positioned to bind and orient a water molecule to attack a substrate bound to the P460 iron (Fig. 10A). Importantly, in kustc1061 this particular tyrosine was replaced by the hydrophobic Met323 (Fig. 10B), which would make the binding of a water molecule in this position unlikely, thus preventing the reaction from proceeding to nitrite.
FIGURE 11.

Proposed reaction mechanism of hydroxylamine oxidation to NO by kustc1061. The model, based on the NeHAO mechanism (57), shows the binding of hydroxylamine (H2N-OH) to the water-bound resting state (upper left), followed by subsequent oxidation of the substrate and removal of the protons that proceed through the {FeNO}7 and {FeNO}6 nitrosyl states. The model takes into account experimentally established (58, 59) and theoretically predicted (57) reaction intermediates in N-converting ferrihemoproteins, which includes the {FeNO}7 nitrosyl species observed in the present study.
Hydroxylamine is a potent inhibitor of the N2-forming reaction catalyzed by the HAO-like hydrazine dehydrogenase (kustc0694 in K. stuttgartiensis) (8). The presence of kustc1061 provides these microorganisms a means for hydroxylamine detoxification, at the same time generating NO, a substrate for hydrazine synthesis (8). Indeed, kustc1061 homologs are found in all anammox genomes sequenced so far (supplemental Fig. S1), in which close paralogs may even be observed, such as kusta0043 in K. stuttgartiensis. Furthermore, hydroxylamine is a toxic product of ammonium co-metabolism of the particulate methane monooxygenase, the key enzyme in the highly diverse an environmentally relevant methanotrophic bacteria. Many of these organisms harbor at least one HAO-like protein that has an elusive role. It was suggested that these HAO-like proteins could either oxidize hydroxylamine to nitrite or NO or alternatively reduce nitrite to NO (61, 62). Despite substantial sequence divergence, HAO-like proteins from methane oxidizers have two prominent features in common with kustc1061 and its homologs in anammox bacteria: they do contain the tyrosine involved in the covalent binding of the catalytic heme that favors oxidative reactions and lack the tyrosine present in NeHAO (Tyr358; supplemental Fig. S1), which we hypothesize to be involved in the addition of water for nitrite formation. This suggests that methanotroph HAOs are also NO producing, which would then be reduced to N2O by a respiratory cNOR-like protein that is commonly found in methane oxidizers. Through these reactions, methanotrophs might even benefit from ammonium co-metabolism, explaining the enigmatic presence of HAO-like proteins in these microorganisms.
In conclusion, here we present a detailed characterization of kustc1061 from K.stuttgartiensis including the first crystal structure of an enzyme from an anammox organism. Despite remarkable similarity to the related NeHAO enzyme, the kustc1061 enzyme converts NH2OH into NO rather than NO2− because of second shell effects steering reaction specificity so as to prevent the addition of a water-derived oxygen atom. Anammox bacteria and possibly methane oxidizers take advantage of this novel way of making NO to detoxify hydroxylamine, thereby generating electrons and substrate for respiration.
Acknowledgments
The Dortmund-Heidelberg data collection team and the staff of Beamline X10SA at the Swiss Light Source of the PSI in Villigen are acknowledged for help and facilities. Portions of this research were carried out at the light source PETRA III at DESY, a member of the Helmholtz Association. We thank Anja Burkhardt, Saravanan Panneerselvam, and Alke Meents for assistance in using Beamline P11 and Marion Gradl for assistance with MALDI-TOF analyses. T. R. M. B. is grateful to Ilme Schlichting for continuous support.
This work was supported by Netherlands Organization for Scientific Research Darwin Grants 142.16.1201 (to W. J. M.) and VENI 863.11.003 (to B. K.), by European Research Council Advanced Grant 232937 (to M. S. M. J), by the Max-Planck Society, and by UK Biotechnology and Biological Sciences Research Council Grant BB/G009228 (to S. J. M.).
The atomic coordinates and structure factors (codes 4N4J, 4N4K, 4N4L, 4N4M, 4N4N, and 4N4O) have been deposited in the Protein Data Bank (http://wwpdb.org/).

This article contains supplemental Fig. S1.
- HAO
- hydroxylamine oxidoreductase
- NeHAO
- N. europaea HAO
- FL2E
- 2-{4,5-bis[(6-(2-ethoxy-2-oxoethoxy)-2-methylquinolin-8-ylamino)methyl]-6-hydroxy-3-oxo-3H-xanthen-9-yl}benzoic acid.
REFERENCES
- 1. Bruckdorfer R. (2005) The basics about nitric oxide. Mol. Aspects Med. 26, 3–31 [DOI] [PubMed] [Google Scholar]
- 2. Hirst D. G., Robson T. (2011) Nitric oxide physiology and pathology. Methods Mol. Biol. 704, 1–13 [DOI] [PubMed] [Google Scholar]
- 3. Leitner M., Vandelle E., Gaupels F., Bellin D., Delledonne M. (2009) NO signals in the haze. Nitric oxide signalling in plant defence. Curr. Opin. Plant Biol. 12, 451–458 [DOI] [PubMed] [Google Scholar]
- 4. Wang Y., Ruby E. G. (2011) The roles of NO in microbial symbioses. Cell Microbiol. 13, 518–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lundberg J. O., Weitzberg E. (2013) Biology of nitrogen oxides in the gastrointestinal tract. Gut 62, 616–629 [DOI] [PubMed] [Google Scholar]
- 6. Einsle O., Kroneck P. M. (2004) Structural basis of denitrification. Biol. Chem. 385, 875–883 [DOI] [PubMed] [Google Scholar]
- 7. Schreiber F., Wunderlin P., Udert K. M., Wells G. F. (2012) Nitric oxide and nitrous oxide turnover in natural and engineered microbial communities. Biological pathways, chemical reactions, and novel technologies. Front. Microbiol. 3, 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kartal B., Maalcke W. J., de Almeida N. M., Cirpus I., Gloerich J., Geerts W., Op den Camp H. J., Harhangi H. R., Janssen-Megens E. M., Francoijs K.-J., Stunnenberg H. G., Keltjens J. T., Jetten M. S., Strous M. (2011) Molecular mechanism of anaerobic ammonium oxidation. Nature 479, 127–130 [DOI] [PubMed] [Google Scholar]
- 9. Hooper A. B., Vannelli T., Bergmann D. J., Arciero D. M. (1997) Enzymology of the oxidation of ammonia to nitrite by bacteria. Antonie Van Leeuwenhoek 71, 59–67 [DOI] [PubMed] [Google Scholar]
- 10. Arp D. J., Stein L. Y. (2003) Metabolism of inorganic N compounds by ammonia-oxidizing bacteria. Crit. Rev. Biochem. Mol. Biol. 38, 471–495 [DOI] [PubMed] [Google Scholar]
- 11. Igarashi N., Moriyama H., Fujiwara T., Fukumori Y., Tanaka N. (1997) The 2.8 Å structure of hydroxylamine oxidoreductase from a nitrifying chemoautotrophic bacterium, Nitrosomonas europaea. Nat. Struct. Biol. 4, 276–284 [DOI] [PubMed] [Google Scholar]
- 12. Cedervall P. E., Hooper A. B., Wilmot C. M. (2009) Crystallization and preliminary x-ray crystallographic analysis of a new crystal form of hydroxylamine oxidoreductase from Nitrosomonas europaea. Acta Crystallogr Sect. F Struct. Biol. Cryst. Commun. 65, 1296–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cedervall P. E., Hooper A, B., Wilmot C. M. (2013) Structural studies of hydroxylamine oxidoreductase reveal a unique heme cofactor and a previously unidentified interaction partner. Biochemistry 52, 6211–6218 [DOI] [PubMed] [Google Scholar]
- 14. Arrigo K. R. (2005) Marine microorganisms and global nutrient cycles. Nature 437, 349–355 [DOI] [PubMed] [Google Scholar]
- 15. Polyakov K. M., Boyko K. M., Tikhonova T. V., Slutsky A., Antipov A. N., Zvyagilskaya R. A., Popov A. N., Bourenkov G. P., Lamzin V. S., Popov V. O. (2009) High-resolution structural analysis of a novel octaheme cytochrome c nitrite reductase from the haloalkaliphilic bacterium Thioalkalivibrio nitratireducens. J. Mol. Biol. 389, 846–862 [DOI] [PubMed] [Google Scholar]
- 16. Atkinson S. J., Mowat C. G., Reid G. A., Chapman S. K. (2007) An octaheme c-type cytochrome from Shewanella oneidensis can reduce nitrite and hydroxylamine. FEBS Lett. 581, 3805–3808 [DOI] [PubMed] [Google Scholar]
- 17. Klotz M. G., Schmid M. C., Strous M., op den Camp H. J., Jetten M. S., Hooper A. B. (2008) Evolution of an octahaem cytochrome c protein family that is key to aerobic and anaerobic ammonia oxidation by bacteria. Environ. Microbiol. 10, 3150–3163 [DOI] [PubMed] [Google Scholar]
- 18. Kartal B., Geerts W., Jetten M. S. (2011) Cultivation, detection, and ecophysiology of anaerobic ammonium-oxidizing bacteria. Methods Enzymol. 486, 89–108 [DOI] [PubMed] [Google Scholar]
- 19. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 20. Hooper A. B., Nason A. (1965) Characterization of hydroxylamine-cytochrome c reductase from the chemoautotrophs Nitrosomonas europaea and Nitrosocystis oceanus. J. Biol. Chem. 240, 4044–4057 [PubMed] [Google Scholar]
- 21. Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 78, 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yonetani T. (1965) Studies on cytochrome c peroxidase. II. Stoichiometry between enzyme, H2O2, and ferrocytochrome c and enzymic determination of extinction coefficients of cytochrome c. J. Biol. Chem. 240, 4509–4514 [PubMed] [Google Scholar]
- 23. Watanabe T., Honda K. (1982) Measurement of the extinction coefficient of the methyl viologen cation radical and the efficiency by semiconductor photocatalysis. J. Phys. Chem. 86, 2617–2619 [Google Scholar]
- 24. Clarke T. A., Mills P. C., Poock S. R., Butt J. N., Cheesman M. R., Cole J. A., Hinton J. C., Hemmings A. M., Kemp G., Söderberg C. A., Spiro S., Van Wonderen J., Richardson D. J. (2008) Escherichia coli cytochrome c nitrite reductase NrfA. Methods Enzymol. 437, 63–77 [DOI] [PubMed] [Google Scholar]
- 25. McQuade L. E., Lippard S. J. (2010) Fluorescence-based nitric oxide sensing by Cu(II) complexes that can be trapped in living cells. Inorg. Chem. 49, 7464–7471 [DOI] [PubMed] [Google Scholar]
- 26. Guo F. Q., Okamoto M., Crawford N. M. (2003) Identification of a plant nitric oxide synthase gene involved in hormonal signaling. Science 302, 100–103 [DOI] [PubMed] [Google Scholar]
- 27. Nagano T. (1999) Practical methods for detection of nitric oxide. Luminescence 14, 283–290 [DOI] [PubMed] [Google Scholar]
- 28. Kartal B., Tan N. C., Van de Biezen E., Kampschreur M. J., Van Loosdrecht M. C., Jetten M. S. (2010) Effect of nitric oxide on anammox bacteria. Appl. Environ. Microbiol. 76, 6304–6306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 30. Farhoud M. H., Wessels H. J., Steenbakkers P. J., Mattijssen S., Wevers R. A., van Engelen B. G., Jetten M. S., Smeitink J. A., van den Heuvel L. P., Keltjens J. T. (2005) Protein complexes in the archaeon Methanothermobacter thermautotrophicus analyzed by blue native/SDS-PAGE and mass spectrometry. Mol. Cell. Proteomics 4, 1653–1663 [DOI] [PubMed] [Google Scholar]
- 31. Marritt S. J., Kemp G. L., Xiaoe L., Durrant J. R., Cheesman M. R., Butt J. N. (2008) Spectroelectrochemical characterization of a pentaheme cytochrome in solution and as electrocatalytically active films on nanocrystalline metal-oxide electrodes. J. Am. Chem. Soc. 130, 8588–8589 [DOI] [PubMed] [Google Scholar]
- 32. Astuti Y., Topoglidis E., Briscoe P. B., Fantuzzi A., Gilardi G., Durrant J. R. (2004) Proton-coupled electron transfer of flavodoxin immobilized on nanostructured tin dioxide electrodes. Thermodynamics versus kinetics control of protein redox function. J. Am. Chem. Soc. 126, 8001–8009 [DOI] [PubMed] [Google Scholar]
- 33. Harp J. M., Timm D. E., Bunick G. J. (1998) Macromolecular crystal annealing. Overcoming increased mosaicity associated with cryocrystallography. Acta Crystallogr. D Biol. Crystallogr. 54, 622–628 [DOI] [PubMed] [Google Scholar]
- 34. Harp J. M., Hanson B. L., Timm D. E., Bunick G. J. (1999) Macromolecular crystal annealing. Evaluation of techniques and variables. Acta Crystallogr. D Biol. Crystallogr. 55, 1329–1334 [DOI] [PubMed] [Google Scholar]
- 35. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kabsch W. (2010) Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr. 66, 133–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vonrhein C., Blanc E., Roversi P., Bricogne G. (2007) Automated structure solution with autoSHARP. Methods Mol. Biol. 364, 215–230 [DOI] [PubMed] [Google Scholar]
- 38. Schneider T. R., Sheldrick G. M. (2002) Substructure solution with SHELXD. Acta Crystallogr. D Biol. Crystallogr. 58, 1772–1779 [DOI] [PubMed] [Google Scholar]
- 39. Abrahams J. P., Leslie A. G. (1996) Methods used in the structure determination of bovine mitochondrial F1 ATPase. Acta Crystallogr. D Biol. Crystallogr. 52, 30–42 [DOI] [PubMed] [Google Scholar]
- 40. Emsley P., Cowtan K. (2004) COOT. Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 41. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 42. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX. A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. McCoy A. J., Grosse-Kunstleve R. W., Storoni L. C., Read R. J. (2005) Likelihood-enhanced fast translation functions. Acta Crystallogr. D Biol. Crystallogr. 61, 458–464 [DOI] [PubMed] [Google Scholar]
- 44. Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J. (1990) Basic local alignment search tool. J. Mol. Biol. 215, 403–410 [DOI] [PubMed] [Google Scholar]
- 45. Petersen T. N., Brunak S., von Heijne G., Nielsen H. (2011) SignalP 4.0. Discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786 [DOI] [PubMed] [Google Scholar]
- 46. Chenna R., Sugawara H., Koike T., Lopez R., Gibson T. J., Higgins D. G., Thompson J. D. (2003) Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 31, 3497–3500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. DeLano W. L. (2002) The PyMOL Molecular Graphics System. DeLano Scientific, San Carlos, CA [Google Scholar]
- 48. Pacheco A. A., McGarry J., Kostera J., Corona A. (2011) Techniques for investigating hydroxylamine disproportionation by hydroxylamine oxidoreductases. Methods Enzymol. 486, 447–463 [DOI] [PubMed] [Google Scholar]
- 49. Schalk J., de Vries S., Kuenen J. G., Jetten M. S. (2000) Involvement of a novel hydroxylamine oxidoreductase in anaerobic ammonium oxidation. Biochemistry 39, 5405–5412 [DOI] [PubMed] [Google Scholar]
- 50. Shimamura M., Nishiyama T., Shinya K., Kawahara Y., Furukawa K., Fujii T. (2008) Another multiheme protein, hydroxylamine oxidoreductase, abundantly produced in an anammox bacterium besides the hydrazine-oxidizing enzyme. J. Biosci. Bioeng. 105, 243–248 [DOI] [PubMed] [Google Scholar]
- 51. Terry K. R., Hooper A. B. (1981) Hydroxylamine oxidoreductase. A 20-heme, 200,000 molecular weight cytochrome c with unusual denaturation properties which forms a 63 000 molecular weight monomer after heme removal. Biochemistry 20, 7026–7032 [DOI] [PubMed] [Google Scholar]
- 52. Kostera J., McGarry J., Pacheco A. A. (2010) Enzymatic interconversion of ammonia and nitrite. The right tool for the job. Biochemistry 49, 8546–8553 [DOI] [PubMed] [Google Scholar]
- 53. Collins M. J., Arciero D. M., Hooper A. B. (1993) Optical spectropotentiometric resolution of the hemes of hydroxylamine oxidoreductase. Heme quantitation and pH dependence of Em. J. Biol. Chem. 268, 14655–14662 [PubMed] [Google Scholar]
- 54. Kurnikov I. V., Ratner M. A., Pacheco A. A. (2005) Redox equilibria in hydroxylamine oxidoreductase. Electrostatic control of electron redistribution in multielectron oxidative processes. Biochemistry 44, 1856–1863 [DOI] [PubMed] [Google Scholar]
- 55. Einsle O., Messerschmidt A., Stach P., Bourenkov G. P., Bartunik H. D., Huber R., Kroneck P. M. (1999) Structure of cytochrome c nitrite reductase. Nature 400, 476–480 [DOI] [PubMed] [Google Scholar]
- 56. Mowat C. G., Rothery E., Miles C. S., McIver L., Doherty M. K., Drewette K., Taylor P., Walkinshaw M. D., Chapman S. K., Reid G. A. (2004) Octaheme tetrathionate reductase is a respiratory enzyme with novel heme ligation. Nat. Struct. Mol. Biol. 11, 1023–1024 [DOI] [PubMed] [Google Scholar]
- 57. Fernández M. L., Estrin D. A., Bari S. E. (2008) Theoretical insight into the hydroxylamine oxidoreductase mechanism. J. Inorg. Biochem. 102, 1523–1530 [DOI] [PubMed] [Google Scholar]
- 58. Hoshino M., Maeda M., Konishi R., Seki H., Ford P. C. (1996) Studies on the reaction mechanism for reductive nitrosylation of ferrihemoproteins in buffer solutions. J. Am. Chem. Soc. 118, 5702–5707 [Google Scholar]
- 59. Einsle O., Messerschmidt A., Huber R., Kroneck P. M., Neese F. (2002) Mechanism of the six-electron reduction of nitrite to ammonia by cytochrome c nitrite reductase. J. Am. Chem. Soc. 124, 11737–11745 [DOI] [PubMed] [Google Scholar]
- 60. Hooper A. B., Terry K. R. (1979) Hydroxylamine oxidoreductase of Nitrosomonas. Production of nitric oxide from hydroxylamine. Biochim. Biophys. Acta 571, 12–20 [DOI] [PubMed] [Google Scholar]
- 61. Poret-Peterson A. T., Graham J. E., Gulledge J., Klotz M. G. (2008) Transcription of nitrification genes by the methane-oxidizing bacterium, Methylococcus capsulatus strain Bath. ISME J. 2, 1213–1220 [DOI] [PubMed] [Google Scholar]
- 62. Campbell M. A., Nyerges G., Kozlowski J. A., Poret-Peterson A. T., Stein L. Y., Klotz M. G. (2011) Model of the molecular basis for hydroxylamine oxidation and nitrous oxide production in methanotrophic bacteria. FEMS Microbiol. Lett. 322, 82–89 [DOI] [PubMed] [Google Scholar]