

Background: The gap junction protein Cx43 is implicated in maintaining anti-oxidative defense in astrocytes.
Results: In contrast to hypoxia/reoxygenation, oxidative stress induced by H2O2 triggers more astrocytic death in the absence of Cx43 channels.
Conclusion: Gap junction intercellular communication is required for Cx43-mediated resistance to H2O2.
Significance: An altered Cx43 phosphorylation state in response to cellular stress may be critical for Cx43-mediated cell death or recovery.
Keywords: Astrocytes, Connexin, Gap Junctions, Hydrogen Peroxide, Hypoxia, Hemichannels, Hypoxia/Reoxygenation
Abstract
Oxidative stress induced by reactive oxygen species (ROS) is associated with various neurological disorders including aging, neurodegenerative diseases, as well as traumatic and ischemic insults. Astrocytes have an important role in the anti-oxidative defense in the brain. The gap junction protein connexin43 (Cx43) forms intercellular channels as well as hemichannels in astrocytes. In the present study, we investigated the contribution of Cx43 to astrocytic death induced by the ROS hydrogen peroxide (H2O2) and the mechanism by which Cx43 exerts its effects. Lack of Cx43 expression or blockage of Cx43 channels resulted in increased ROS-induced astrocytic death, supporting a cell protective effect of functional Cx43 channels. H2O2 transiently increased hemichannel activity, but reduced gap junction intercellular communication (GJIC). GJIC in wild-type astrocytes recovered after 7 h, but was absent in Cx43 knock-out astrocytes. Blockage of Cx43 hemichannels incompletely inhibited H2O2-induced hemichannel activity, indicating the presence of other hemichannel proteins. Panx1, which is predicted to be a major hemichannel contributor in astrocytes, did not appear to have any cell protective effect from H2O2 insults. Our data suggest that GJIC is important for Cx43-mediated ROS resistance. In contrast to hypoxia/reoxygenation, H2O2 treatment decreased the ratio of the hypophosphorylated isoform to total Cx43 level. Cx43 has been reported to promote astrocytic death induced by hypoxia/reoxygenation. We therefore speculate the increase in Cx43 dephosphorylation may account for the facilitation of astrocytic death. Our findings suggest that the role of Cx43 in response to cellular stress is dependent on the activation of signaling pathways leading to alteration of Cx43 phosphorylation states.
Introduction
Astrocytes are the most abundant non-neuronal cell type in the central nervous system (1). They play an essential role in adult brain homeostasis, including glutamate uptake, potassium ion buffering, nutrient support, and antioxidant protection for neurons (2–5). The gap junction protein connexin43 (Cx43)2 is highly expressed in astrocytes and is crucial for maintaining their normal function (6, 7). Cx43 can form gap junction channels as well as hemichannels. Gap junctions allow the passive intercellular diffusion of small molecules, such as glutamate, glutathione, glucose, ATP, cAMP, IP3, and ions (Ca2+, Na+, K+) (8, 9). A single gap junction channel consists of two opposing channels, called hemichannels, which are made of six connexin proteins (10, 11). Hemichannels predominantly exist in a closed state under normal physiological conditions, mainly due to ambient levels of Ca2+ (12, 13). However, various cellular stress conditions, such as hypoxia/reoxygenation (H/R) and metabolic starvation, have been reported to cause the opening of hemichannels in cultured astrocytes (14, 15).
Oxidative stress is a result of the imbalance between reactive oxygen species (ROS) production and antioxidant activity. Astrocytes are the center of the brain's defense from oxidative stress as they maintain high intracellular concentration of antioxidant molecules (16). However, oxidative stress can interfere with astrocyte function depending on the severity of the insults (17, 18). Knockdown of Cx43 in cortical astrocytes is reported to increase cell death induced by the ROS hydrogen peroxide (H2O2) (19). However, the mechanism underlying this Cx43-mediated ROS resistance in astrocytes has not been investigated, although several lines of evidence indicate the involvement of Cx43 gap junctions or hemichannels in H2O2-mediated cell death in epithelial cells and osteocytes (20–23). In addition, whether Cx43 plays a protective or detrimental role in response to ROS-induced oxidative stress is still under debate (21–23). In the present study, we employed two different systems, one using Cx43−/− astrocytes and another with Cx43 channel blockers, to carefully examine the importance of Cx43 expression and channel activity in H2O2-induced astrocytic death. Previous studies reported that phosphorylation of Cx43 and its channel activity are altered during hypoxia and reoxygenation in astrocytes, leading to cell death (14, 15, 24–27). Therefore, we investigated whether H2O2 generated by hypoxia and reoxygenation (28, 29) can induce those changes in Cx43. Here, we demonstrated that Cx43 oppositely modulates H2O2- and H/R-induced cell death in astrocytes and that these distinct effects of Cx43 are correlated with differential regulation of Cx43 phosphorylation and spatial distribution.
EXPERIMENTAL PROCEDURES
Animals
Wild-type (WT) mice (C67BL) were obtained from homozygous mating. Cx43−/− mice were generated from crossing Cx43+/− mice. Complete knock-out of Cx43 is neonatal lethal due to abnormal heart development (30). Therefore, Cx43−/− astrocytes were isolated from embryonic day 20 (E20) mice. Mice of either sex used for all experiments were maintained in an animal facility for 12 h light/dark cycle and provided food and water ad libitum. All breeding and animal procedures were approved by The University of British Columbia Animal Care Committee and performed in accordance with the guidelines established by the Canadian Council on Animal Care.
Astrocyte Culture
WT astrocytes were isolated from early postnatal (P0-P1) cortices. Cx43−/− astrocytes were isolated from E20 brains. Each brain from littermates of heterozygous mating was processed separately and Cx43−/− mice were characterized by gross right ventricle morphologic abnormalities (31). The absence of astrocytic Cx43 was confirmed by immunostaining. Astrocytes were prepared from mouse cortices as previously described (32). Briefly, dissected cortices were triturated in DMEM (Sigma-Aldrich). The cell suspension was passed through a 70 μm cell filter strainer and then seeded into flasks (2 cortices/T75 flask). Culture media (DMEM supplemented with 10% FBS, 10 units/ml penicillin, and 10 μg/ml streptomycin) was replaced 3 days after plating and every second day thereafter. Primary astrocytes reached subconfluence at 7–8 days in vitro. Subconfluent cells were vigorously shaken to remove cells loosely attached to the astrocyte monolayer (mainly oligodendrocytes). Astrocytes were then harvested with trypsin-EDTA (Invitrogen) and frozen in freezing medium (DMEM, 10% FBS, and 8% DMSO). Frozen astrocytes were thawed and plated on glass coverslips coated with poly-l-ornithine (0.01% solution, Sigma-Aldrich) or culture dishes. Cultures were maintained for 5–7 days prior to experiments. All experiments were carried out on confluent astrocytes and performed independently at least three times. Astrocytes isolated from different breeding pairs were used for each set of experiments.
Hydrogen Peroxide and Gap Junction Blocker Treatment
Astrocytes were washed once with serum-free medium and then subjected to indicated concentrations of H2O2 (Sigma-Aldrich) in the same medium for 45 min. Cells were then washed once and maintained in fresh culture medium for recovery. The control group was treated the same way except for the addition of H2O2.
To study the effect of gap junction blockers on H2O2-induced cell death, gap junction blockers carbenoxelone (100 μm, Sigma-Aldrich) and 18-α-glycyrrhetinic acid (100 μm, Sigma-Aldrich) were added 30 min before and during H2O2 treatment, and in the recovery medium.
Cell Death Analysis
Cell death by loss of membrane integrity was evaluated using the dye rhodamine B dextran (Rdex) as previously described (15). Cultured astrocytes were incubated with 100 μm Rdex (10 kDa, Invitrogen) for 3 min followed by five washes with phosphate-buffered saline (PBS). Total number of cells was determined by nuclear staining with 1 μm Hoechst 33342 (Invitrogen). The staining was immediately detected by microscopy using a Zeiss Axioplan2 fluorescence microscope (Carl Zeiss). The number of cells positive for Rdex was evaluated.
In other experiments, cell death was accessed using the dye propidium iodine (PI) (33). Briefly, astrocytes were exposed to PI (40 μg/ml, Sigma-Aldrich) for 10 min. Cells were then fixed with 4% paraformaldehyde for 15 min, washed with PBS, and mounted using Prolong Gold reagent with DAPI (Molecular Probes). PI positive round red nuclei and condensed blue nuclei were considered dead cells. Pictures were taken from random fields using a Zeiss Axioplan2 fluorescence microscope. Nuclei were counted using ImageJ with a pixel value of 100.
Dye Coupling Measurement
To evaluate gap junction coupling, astrocytes were grown on 35 mm culture dishes to 100% confluence. After medium was removed, a mixture of gap junction-permeable carboxyfluorescein (0.1%) and gap junction-impermeable Rdex (0.1%) in serum free medium was added, and cells were then scraped with a surgical blade. Two minutes later, the excess dye was washed off with serum-free medium. Pictures were taken after 10 min incubation using a Zeiss Axioplan2 fluorescence microscope. The gap junction blocker carbenoxolone was added to prevent further coupling. The distance carboxyfluorescein traveled was measured from the scrape line to the point the fluorescence intensity reduced to 1.5× the background intensity using ImageJ software.
Ethidium Bromide Uptake Measurement
To assess hemichannel activity, astrocytes grown on poly-l-ornithine coated coverslips were exposed to 5 μm ethidium bromide in Locke's solution (in mm: 154 NaCl, 5.4 KCl, 1.8 CaCl2, 1 MgCl2, 10 HEPES pH 7.4) supplemented with 5 mm glucose. For time lapse imaging, fluorescent signal was recorded every 30 s using a Zeiss Axioplan2 fluorescence microscope. To determine the changes in slope, regression lines were fitted to points before and after various treatments using Microsoft Excel, and mean values of the slopes were compared.
Western Blot Analysis
Cells were washed with cold PBS and then lysed in RIPA buffer (50 mm Tris, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1% Triton X-100, 0.5% NaDOC, and 0.1% SDS) supplemented with protease and phosphatase inhibitors. Cell homogenates were incubated on ice for 30 min and then centrifuged at 16,000 × g for 15 min. The resulting supernatants were collected and assayed for protein concentration using BCA protein assay reagent (Thermo Scientific). To examine the expression level of Cx43, 20–30 μg of protein was loaded on SDS-PAGE and transferred to PVDF membranes. The membranes were blocked with 5% skim milk and treated with rabbit anti-Cx43 (1:5,000, Sigma-Aldrich) and mouse anti-GAPDH (1:5000, HyTest) antibodies. To determine the expression level of Panx1, the membranes were incubated with rabbit anti-Panx1 antibody (1:5,000, a gift from Dr. D. Laird, University of Western Ontario, Canada). The secondary antibodies were goat anti-rabbit and anti-mouse horseradish-peroxidase conjugated (Sigma-Aldrich). The intensity of the bands was quantified using Quantity-One software (Bio-Rad).
Immunocytochemistry
Cells were fixed with 4% paraformaldehyde for 15 min at room temperature, rinsed three times with PBS, and permeabilized in 0.2% Triton X-100 (Sigma-Aldrich) for 10 min. Cells were blocked in 2% bovine serum albumin (Invitrogen) in PBS for 1 h, then incubated with rabbit polyclonal anti-Cx43 (1:2000, Sigma-Aldrich) primary antibody overnight at 4 °C or for 1 h at room temperature. After incubation with primary antibody, cells were washed three times with PBS and then incubated with secondary antibody, goat anti-rabbit antibody conjugated to Alexa Fluor 488 (1:1000, Molecular Probe). Following the incubation, cells were washed three times with PBS and mounted using Prolong Gold reagent with DAPI. All images were captured using a Zeiss Axioplan2 fluorescence microscope.
To evaluate the average size of Cx43 plaques, the images captured at 40× magnification were used. Briefly, each image was converted into a binary image and the average plaque areas were determined by the “Analyze particles” function in the ImageJ software.
Hypoxia/Reoxygenation Protocol
Astrocytes were subjected to hypoxia for 4 h in Locke's solution inside a chamber in an incubator. The chamber was purged with a CO2/N2 (5%/95%) flow. The oxygen concentration inside the chamber was kept between 0.3 to 0.5%. After hypoxia, cells were maintained in culture medium and returned to an incubator containing a CO2/air atmosphere (5%/95%) environment. For normoxic control, cells were maintained in the same solution under normoxic conditions for 4 h. All other procedures were the same as described in the hypoxia experiments.
Assessment of Mitochondrial Respiration
Astrocytes in 24-well plate were incubated with 0.5 mg/ml MTT (Sigma-Aldrich) in Hank's balanced salt solution (Invitrogen) for 30 min at 37 °C. Formazan crystals generated by living cells were dissolved in 0.5 ml of dimethyl sulfoxide (Sigma-Aldrich). Color formation was determined by measuring the optical density at 562 nm.
Statistical Analysis
Data are expressed as the average ± S.E. and analyzed using Student's t test to evaluate the significant between groups. p < 0.05 is considered significant (*), and p < 0.01 is considered highly significant (**).
RESULTS
Functional Channels Contribute to Cx43-mediated ROS Resistance in Cortical Astrocytes
It has been previously reported that siRNA knockdown of Cx43 leads to an increase in H2O2-induced apoptosis in primary astrocytes, almost 2-fold over the scrambled siRNA control (19). However, whether Cx43 hemichannels and/or gap junction channels contribute to the anti-apoptotic effect of astrocytic Cx43 has not been investigated. Therefore, we first determined whether complete lack of Cx43 in astrocytes further enhances H2O2 toxicity. Secondly, we asked whether functional Cx43 channels in astrocytes are essential for their resistance to H2O2. Wild-type (WT) and Cx43−/− astrocytes were exposed to 0.7 mm and 1.4 mm H2O2. This compound is unstable and decomposes rapidly over time to undetectable levels after 1 h in cell culture medium (34). To obtain the maximum effect, we treated cells with H2O2 in serum-free medium for 45 min. The medium was then removed and replaced with fresh culture medium to prevent H2O2 from reacting with other components of the medium and allow cells to recover. H2O2 is reported to induce cell death in astrocytes by both necrotic and apoptotic pathways, 39 ± 10% and 26 ± 14% of the total number of cells, respectively (35). In our study, cell death was analyzed using the dye Rdex, an indicator of loss of membrane integrity, 24 h post-treatment. No dead cells were detected after exposing WT cells to 0.7 mm H2O2 (Fig. 1, A and B). However, astrocytes lacking Cx43 exhibited 13 ± 3% cell death at this concentration. With 1.4 mm H2O2, only 20 ± 3% of WT astrocytes died while the majority of Cx43−/− cells (63 ± 5%) were positive for Rdex. This result indicates that Cx43-deficient astrocytes are more sensitive to H2O2 toxicity. To determine whether functional Cx43 channels are important for protection against H2O2, astrocytes were exposed to the Cx43 channel blockers carbenoxelone (Cbx) or 18-α-glycyrrhetinic acid (18-αGA), which have been previously shown to inhibit both astrocytic Cx43 gap junction coupling and hemichannel activity (36). Astrocytes were left untreated (control) or treated with 1.2 mm H2O2. Cx43 channel blockers were added 30 min before, during, and after treatment (Fig. 1, C and D). Cells that were positive for the membrane impermeable dye PI or had condensed round nuclei were considered dead cells. WT astrocytes exhibited 12 ± 2% cell death when treated with H2O2. There was a 3.3- or 2.8-fold increase in ROS-induced cell death in the presence of Cbx or 18-αGA, respectively, over the vehicle control. Altogether, this finding suggests that channel activity of Cx43 is important for cell survival in response to H2O2.
FIGURE 1.

Connexin43 channels protect astrocytes from hydrogen peroxide toxicity. A, WT and Cx43−/− astrocytes were exposed to 0.7 mm or 1.4 mm H2O2 for 45 min in serum-free medium and then maintained in fresh culture medium for up to 24 h. Live cells were incubated with Rdex and Hoechst. Representative micrographs show H2O2-induced cell death by loss of membrane integrity (uptake of Rdex, red). Total cells were identified by staining with Hoechst (blue). Scale bar: 50 μm. B, quantification of cell death by Rdex staining method. Data represent average ± S.E. (n = 3). C, WT astrocytes were treated with 1.2 mm H2O2 in the absence (vehicle) or presence of channel blockers carbenoxolone (Cbx) and 18-α-glycyrrhetinic acid (18-αGA). Cells were labeled with PI (red) and DAPI (blue) at 24 h post-treatment. Scale bar: 50 μm. D, cell death was quantified (red round nuclei and condensed nuclei), as shown in the graph. Data represent average ± S.E. (n = 3). *, p < 0.05 and **, p < 0.01.
Hydrogen Peroxide Treatment Leads to Reduced Cx43 Gap Junction Intercellular Communication in Astrocytes
Cx43 can function as a gap junction channel or as a hemichannel. First, we examined whether H2O2 treatment affects Cx43 GJIC in astrocytes. We employed two fluorescent dyes, carboxyfluorescein (376 Da) and Rdex (10 kDa), in a scrape loading assay. Cx43 channels are permeable to carboxyfluorescein but not to Rdex. WT and Cx43−/− cells were either untreated (control) or treated with 0.7 mm H2O2 for 45 min; gap junction activity was assessed immediately or after 3, 7, and 24 h recovery in fresh media. In agreement with previous findings (37, 38), lack of Cx43 prevented cultured astrocytes from transferring dye to neighboring cells, as dye could not be detected beyond the injured cells at the scratch boundary (Fig. 2A). Dye travel distance in WT astrocytes was measured and is shown in Fig. 2B. WT untreated astrocytes exhibited active GJIC (170 ± 9 μm), while treatment of astrocytes with H2O2 for 45 min resulted in less GJIC, which is evident from the shorter dye travel distance (125 ± 8 μm) (Fig. 2B, 0 h). Following the 3 h recovery, the extent of gap junction coupling in treated cells was still reduced (Fig. 2, A and B). However, GJIC had recovered to a level comparable to control cells by 7 h after treatment (Fig. 2B), suggesting that H2O2 treatment leads to transient Cx43 gap junction uncoupling in cortical astrocytes. The recovery of GJIC may be due to an increased level of newly synthesized Cx43 that has yet to be phosphorylated, or altered activities of kinases and phosphatases, all of which contribute to an increased level of hypophosphorylated protein that we observed in Fig. 4.
FIGURE 2.
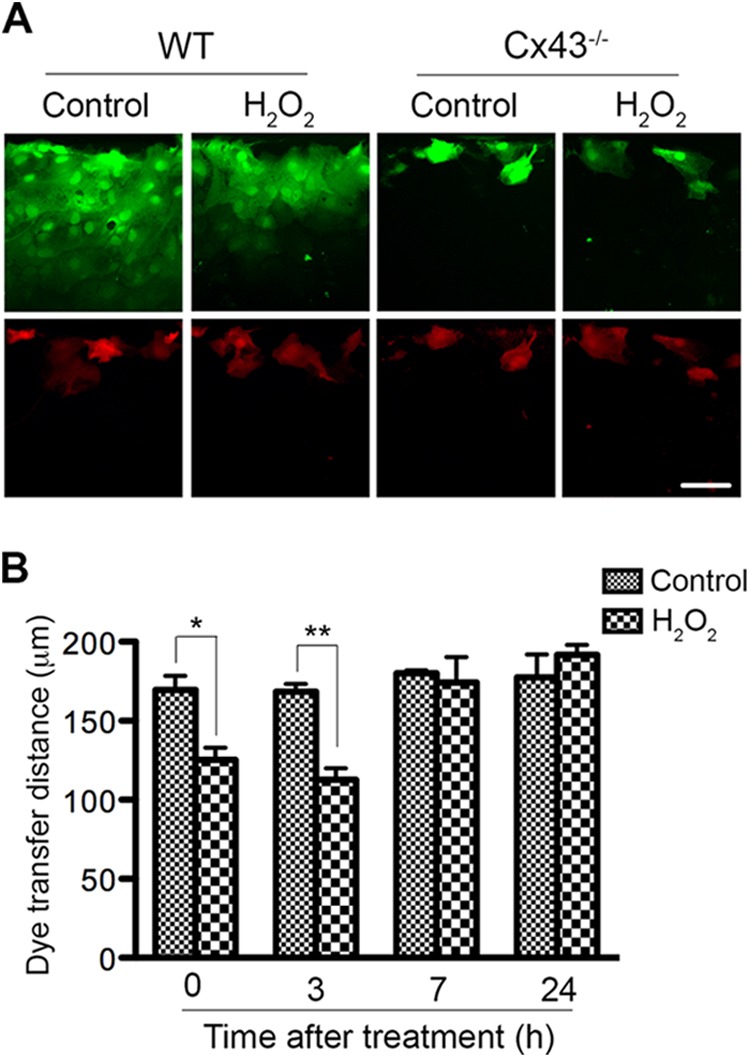
Hydrogen peroxide treatment transiently decreases astrocytic Cx43 gap junction coupling. WT and Cx43−/− astrocytes were left untreated (control) or exposed to 0.7 mm H2O2 for 45 min and then cells were maintained in fresh culture medium for recovery. Gap junction coupling was measured at 0, 3, 7, and 24 h post-treatment using a scrape loading assay. A, representative micrographs show carboxyfluorescein (green) and Rdex (red) after scrape loading at 3 h after treatment. Scale bar: 50 μm. B, distance of dye transfer in the control and H2O2-treated WT cells was quantified, as shown in the graph. Data represent average ± S.E. (n = 3). *, p < 0.05 and **, p < 0.01.
FIGURE 4.
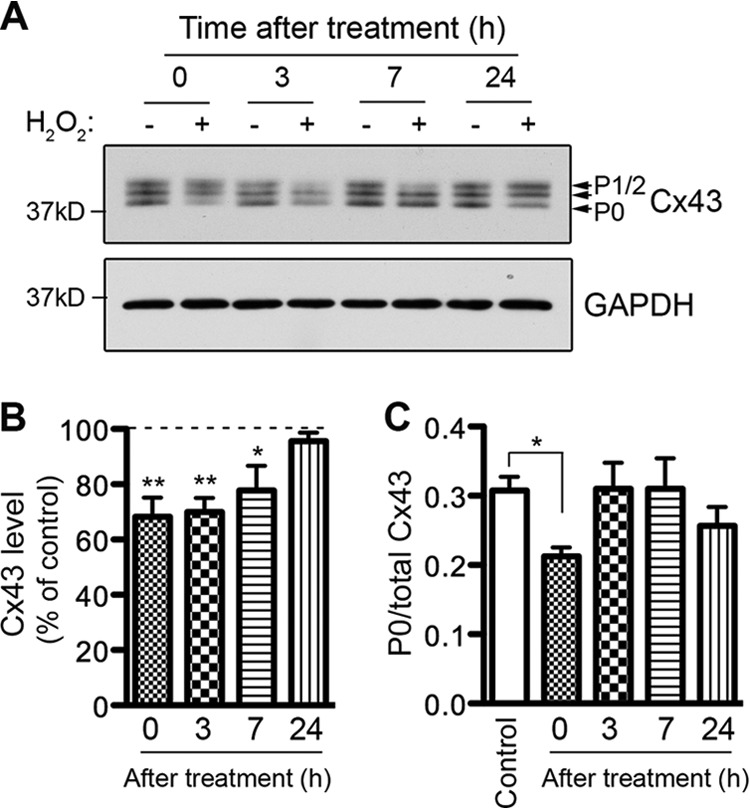
Effect of hydrogen peroxide treatment on Cx43 level and phosphorylation in astrocytes. WT astrocytes were exposed to 0.7 mm H2O2 for 45 min in serum-free medium and then maintained in fresh culture medium for recovery. A, cells were harvested at 0, 3, 7, and 24 h post-treatment (+). Control cells (−) were maintained in the same condition without H2O2 and harvested at the same time. Lysates prepared from these astrocytes were immunoprobed with anti-Cx43 antibody (top panel) and anti-GAPDH antibody (bottom panel). The results were quantified and shown in the graphs. B, bars represent the amount of Cx43 protein as a percentage of the loading control, GAPDH. The control samples were set at 100% and the treated samples were expressed relative to the control samples. Data represent average ± S.E. (n = 4). C, bars represent the ratio of the P0 form to total Cx43 in the untreated (control) and treated samples. Data represent average ± S.E. (n = 4). *, p < 0.05 and **, p < 0.01.
Hydrogen Peroxide Treatment Results in Enhanced Cx43 Hemichannel Activity in Astrocytes
Cx43 hemichannels are usually closed under normal physiological conditions (12, 13). Opening of hemichannels is regulated by various physiological and pathological conditions (14, 39–41). A previous study showed that oxidative stress induced by H2O2 leads to membrane depolarization and hemichannel opening in epithelial cells (21). We asked whether H2O2 causes the opening of hemichannels, especially Cx43 hemichannels, in primary astrocytes by measuring the uptake rate of the hemichannel-permeable ion ethidium (Etd) using time lapse imaging. Representative pictures demonstrate Etd uptake before (Fig. 3A, untreated) and 15 min after addition of H2O2 (Fig. 3A, H2O2). Quantification of uptake rate is shown in Fig. 3B. The uptake rate prior to the addition of H2O2 was minimal and set at 100%. We observed a 3.4-fold increase in the Etd uptake rate by WT astrocytes when 0.7 mm H2O2 was added. This is similar to Etd uptake rate triggered by the removal of extracellular Ca2+/Mg2+ (Fig. 3A). The hemichannel stimulating effect of H2O2 was reduced, but not completely abolished, in Cx43-deficient astrocytes. Similarly, there was a decrease in Etd uptake when Cx43 channel blockers, Gap 26 and Cbx, were applied. WT cells preincubated with the antioxidant glutathione abolished the H2O2-induced increase in Etd uptake. The opening of hemichannels is, however, transient as we did not observe an increase in Etd uptake after 45 min treatment with H2O2 (Fig. 3, C and D, 0 h).
FIGURE 3.
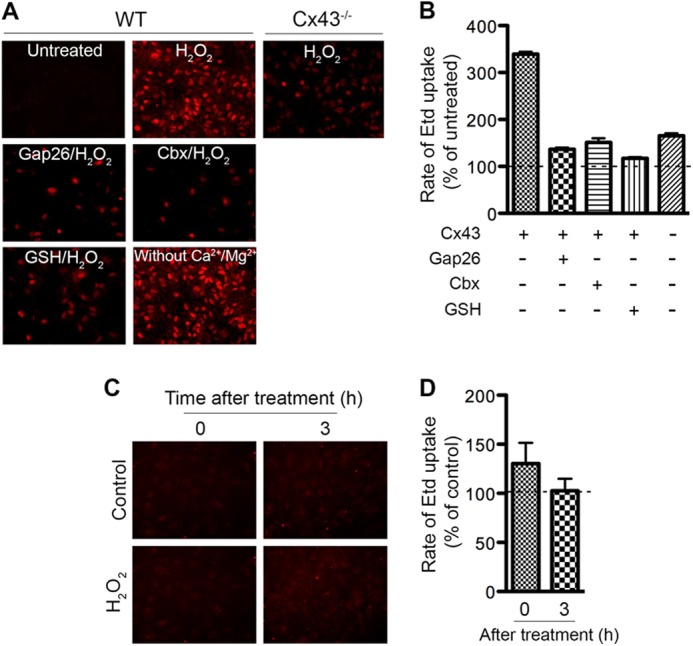
Hydrogen peroxide treatment increases astrocytic Cx43 hemichannel activity. A, Etd uptake was recorded before (untreated) and after addition of H2O2 using time-lapse imaging. Representative micrographs show Etd uptake (red) by WT and Cx43−/− astrocytes. Gap junction blockers Gap26 (200 μm) and Cbx (100 μm), and the reducing agent glutathione (GSH, 1 mm) were added simultaneously with Etd. B, Rate of Etd uptake was determined and normalized to the untreated cells as shown in the graph. Etd uptake before addition of H2O2 was set at 100%. C, astrocytes were left untreated (control) or treated (H2O2) with H2O2 for 45 min. Etd uptake was recorded right after the removal of H2O2 (0 h) or at 3 h after treatment (3 h). Representative micrographs show Etd uptake. D, rate of Etd uptake in H2O2-treated astrocytes was determined and normalized to the untreated astrocytes. Data represent average ± S.E.
Cx43 Expression, Phosphorylation, and Distribution Is Altered in Response to Hydrogen Peroxide
It is well-established that Cx43 has a short half-life of l.5 to 5 h (42–44). Thus, regulation of Cx43 assembly and turnover are important for its physiological and pathological functions (45). In addition, it is reported that Cx43 contains multiple phosphorylation sites, which influence gap junction assembly, channel gating, and half-life (46, 47). As an initial step in understanding the mechanism that regulates Cx43-mediated ROS resistance, we investigated the expression and phosphorylation of Cx43 as well as its spatial distribution in WT astrocytes in response to ROS treatment.
Astrocytes were treated with 0.7 mm H2O2 for 45 min in serum-free medium and then left to recover in culture medium. Cells were harvested immediately after treatment (0 h) or at 3, 7, and 24 h post-treatment. Untreated (control) cells were maintained in the same condition and harvested at the same time. In control cells, we detected three different bands when probing with anti-Cx43 antibody (which recognizes total Cx43): P0, P1, and P2, of which P0 is the isoform with the fastest mobility on SDS-PAGE gels (Fig. 4A). These isoforms represent different phosphorylation states of Cx43 (48). H2O2-treated astrocytes showed a 32% reduction in Cx43 level immediately after 45 min of treatment (Fig. 4B, 0 h). Cx43 levels remained low at 3 h and 7 h post-treatment. At 24 h after treatment, the Cx43 level returned back to the basal level. Since H2O2 induced junctional uncoupling in astrocytes, we investigated whether Cx43 phosphorylation is a mechanism that regulates astrocytic Cx43 GJIC in response to H2O2. Cx43 can be phosphorylated on multiple sites by various protein kinases and dephopshorylated by phosphatases PP1 and PP2A (46, 47). The P0 isoform is conventionally classified as the non-phosphorylated form (49), however, a later study suggested that the P0 isoform can be phosphorylated (50), therefore the P0 isoform is referred to as the hypophosphorylated isoform. The ratio of the P0 isoform to total Cx43 level was analyzed and shown in Fig. 4C. We observed a 30% decrease in the ratio after 45 min of treatment (Fig. 4C, 0 h). Since the level of Cx43 is also reduced at this time, our results suggest that the P0 isoform of Cx43 is dramatically reduced after 45 min of treatment. Interestingly, the ratio increased back to a level comparable to the control at 3 h and 7 h post-treatment although total Cx43 is still low compared with untreated cells (Fig. 4, B and C). It is not clear whether the decrease in P0 isoform is due to increased conversion to the phosphorylated P1 and P2 isoforms, however, we did not detect any significant difference in the ratio of P1/2 level to total Cx43 level (data not shown). At 24 h, the level of P0 as well as the total Cx43 level has recovered. In summary, H2O2 treatment leads to a decrease in total Cx43 expression and a transient reduction in the ratio of the hypophosphorylated form to total Cx43 level in astrocytes. To determine whether the changes in Cx43 phosphorylation can translate into alterations in gap junction organization, astrocytes were immunostained for Cx43. A previous study reported that Cx43 in confluent primary astrocytes is distributed between astrocytes at the contact areas in a pattern known as gap junction plaques (38). Similarly, we found that prior to H2O2 treatment, the majority of Cx43 was arranged into large gap junction plaques, evident as intense punctate labeling (Fig. 5A, Control). After 45 min of treatment, the number of large plaques was reduced (Fig. 5A, 0 h). At 1 h post-treatment, the intense punctate structure disappeared. This phenomenon was also observed at 3 h and 5 h post-treatment. By 24 h, large gap junction plaques had reappeared. The average size of Cx43 plaques was quantified by ImageJ and is shown in Fig. 5B. The average size of Cx43 positive plaques was reduced at 1–5 h post-treatment, but recovered by 24 h. The reduction in Cx43 plaque size is caused by a decrease in the number of larger plaques and an increase in the number of smaller plaques (data not shown). Altogether, this evidence suggests that H2O2 decreases Cx43 levels and transiently reduces the ratio of the hypophosphorylated isoform to total Cx43 level. Interestingly, there is a correlation between reduced Cx43 plaque size and GJIC (Figs. 2 and 5).
FIGURE 5.
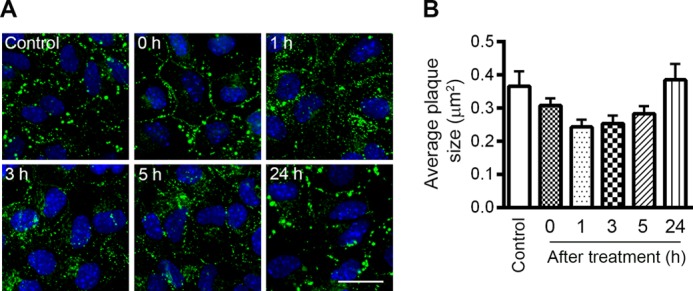
Effect of hydrogen peroxide treatment on Cx43 distribution in astrocytes. WT astrocytes were exposed to 0.7 mm H2O2 for 45 min in serum-free medium and then maintained in fresh culture medium for recovery. A, WT astrocytes were fixed before treatment (control) and at 0, 1, 3, 5, and 24 h after treatment. Fixed cells were immunostained for Cx43 (green) and DAPI (blue). Scale bar: 20 μm. B, average size of Cx43 plaques was quantified, as shown in the graph. Data represent average ± S.E.
Panx1 Channels Do Not Contribute to Hydrogen Peroxide-induced Astrocyte Death
It is reported that Panx1 is a major hemichannel contributor and does not have gap junction activity in cultured astrocytes (51–54). Our results showed that application of Cx43-specific pharmacological agents incompletely blocked the ROS-induced increase in astrocyte membrane permeability as measured by Etd uptake, suggesting that Panx1 may also mediate the increase in Etd uptake in response to H2O2 treatment. First, we asked whether the level of Panx1 is altered in Cx43−/− astrocytes. We detected two major bands in lysates from WT and Cx43−/− astrocytes but not in lysates from Panx-1−/− astrocytes, indicating that the two bands are Panx1 (Fig. 6A). Previous studies showed that Panx1 is N-glycosylated, therefore, these bands most likely represent the degree of glycosylation (55, 56). We did not detect any difference in Panx1 level between WT and Cx43−/− cells at any investigated time points, suggesting elevated ROS-mediated cell death in Cx43−/− astrocytes is not due to altered Panx1 expression. This is consistent with an earlier study (51). Interestingly, Panx1 levels significantly increased 24 h after treatment in both WT and Cx43−/− astrocytes. To investigate whether the elevated Panx1 protein level is critical for the recovery of astrocytes in response to ROS, we evaluated H2O2-induced cell death in Panx1−/− astrocytes. WT and Panx1−/− astrocytes were treated with 0.7 mm or 1.4 mm H2O2. Cell death was analyzed using the dye Rdex as described earlier. Lack of Panx1 did not result in any changes in H2O2-induced cell death (Fig. 6B). This finding suggests that Panx1 does not have a major role in astrocytic death induced by ROS.
FIGURE 6.
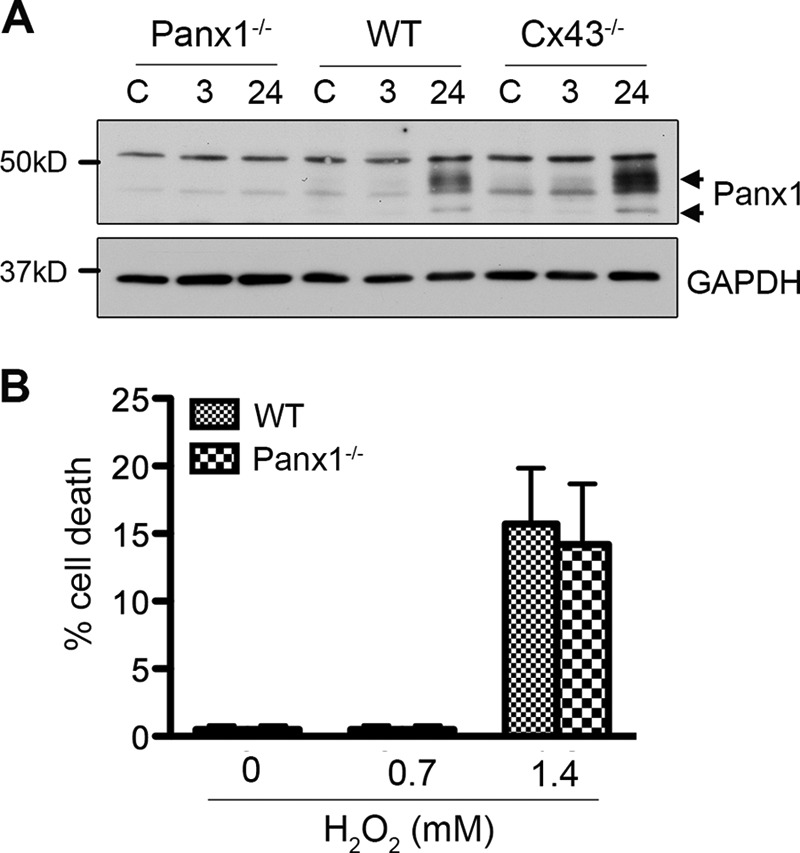
Panx1 expression and function in response to hydrogen peroxide treatment in astrocytes. WT, Cx43−/−, and Panx1−/− astrocytes were exposed to 0.7 mm H2O2 for 45 min in serum-free medium and then maintained in fresh culture medium for recovery. A, astrocytes were harvested before treatment (C) and at 3 h, and 24 h post-treatment. Lysates prepared from these cells were immunoprobed with anti-Panx1 and anti-GAPDH antibodies. B, WT and Panx1−/− astrocytes were exposed to 0.7 mm or 1.4 mm H2O2. Cell death was analyzed by Rdex staining method. Data represent average ± S.E. (n = 3).
Distinct Alterations in Cx43 Phosphorylation and Distribution in Response to Hypoxia/Reoxygenation Cause Astrocytic Death
ROS have been long thought to be a critical mediator of brain damage related to H/R (28, 29). In stroke, hypoxia and reoxygenation are important components of ischemia and reperfusion, respectively. In contrast to the cell protective effect of Cx43 in response to H2O2, Cx43 has been implicated in promoting astrocytic death in response to H/R (15). Therefore, we characterized the modulatory mechanisms that may explain these opposite effects of Cx43 in astrocytes. WT and Cx43−/− astrocytes underwent hypoxia for 4 h and then were reoxygenated in culture medium for 20 h. Control cells were maintained in the same solution but under normal oxygen conditions. First, we examined the contribution of Cx43 in H/R-induced cell death. Astrocytes lacking Cx43 showed no reduction of cell viability after H/R, whereas WT astrocytes exhibited a 30 ± 7% reduction in cell viability (Fig. 7A). This result agrees with a previous study showing that Cx43 hemichannel activity is associated with H/R-induced cell death (15). To identify the reason underlying the opposite effect whereby Cx43 prevents H2O2-induced cell death but promotes H/R-mediated cell death in astrocytes, we examined Cx43 expression and distribution in WT astrocytes. Interestingly, 4 h of hypoxia caused a marked increase in Cx43 immunostaining at the plasma membrane, and a complete disappearance of Cx43 punctate structures (Fig. 7B, 0 h, upper panel). This is distinct from H2O2-induced subcellular changes of Cx43 plaques (Fig. 5). After 1 h of reoxygenation, Cx43 was redistributed but the punctate structures were still missing. By 20 h of reoxygenation Cx43 returned to its normal (normoxic condition) distribution pattern as punctate plaques (Fig. 7B, 20 h). Next, we investigated Cx43 expression and phosphorylation. Similar to H2O2 treatment, 4 h of hypoxia caused a reduction in Cx43 expression (Fig. 7, C and D). In contrast to H2O2, hypoxia treatment resulted in a 2-fold increase in the ratio of the P0 isoform to total Cx43 level (Fig. 7E). After 1 h of reoxygenation, this ratio was reduced to a 1.4-fold increase, suggesting that Cx43 P0 level was decreased. Cx43 expression and phosphorylation was completely restored to control levels after 20 h of reoxygenation in culture medium. In summary, hypoxia causes the disappearance of punctuate gap junction plaques and a marked increase in the hypophosphorylated isoform of Cx43.
FIGURE 7.

Effect of hypoxia/reoxygenation on astrocytic Cx43 level, phosphorylation, and distribution. WT astrocytes were subjected to hypoxia or maintained under normoxic conditions (normoxia) in Locke's solution for 4 h and then reoxygenated in fresh culture medium. A, cell survival was determined by MTT assay at 20 h after reoxygenation. The bars represent astrocytic survival calculated as percentage of the control under normoxia. Data represent average ± S.E. (n = 3). B, cells were fixed after 4 h hypoxia or 4 h normoxia (0 h), and 1 h and 20 h after reoxygenation. Fixed cells were immunostained for Cx43 (green) and DAPI (blue). Scale bar: 20 μm. C, cells were harvested before (Control), after 4 h hypoxia or 4 h normoxia (0 h), and at 1 h and 20 h after reoxygenation. Lysates prepared from these astrocytes were immunoprobed with anti-Cx43 antibody (top panel), and anti-GAPDH antibody (bottom panel). D, bars represent the amount of Cx43 protein as a percentage of the loading control, GAPDH. The control samples (harvested before hypoxia) were set at 100%, and the samples harvested after hypoxia and reoxygenation were expressed relative to the control samples. Data represent average ± S.E. (n = 3). E, bars represent the ratio of the P0 isoform to total Cx43 before (control) and after 4 h hypoxia (0 h) and 1 and 20 h after reoxygenation. Data represent average ± S.E. (n = 3). *, p < 0.05 and **, p < 0.01.
DISCUSSION
The role for Cx43 channels in H2O2-induced cell death has been studied in several cell types, including epithelial cells and osteocytes, with mixed results. Some studies suggest a cell protective role whereas others propose a cell destructive role (21–23). Moreover, whether Cx43 hemichannels and/or gap junctions are important for H2O2-mediated cell death is still under debate. The involvement of Cx43 in H2O2-mediated astrocytic death has been previously reported (19, 35). Knockdown of Cx43 sensitized primary rat brain astrocytes to H2O2 (19). However, inhibition of Cx43 GJIC by pharmacological agents did not interfere with H2O2-induced cell death in rat striatum astrocytes (35). In this study, we have demonstrated that Cx43 channel activity is crucial for Cx43-mediated ROS resistance of cortical astrocytes as astrocytes lacking Cx43 or blockage of Cx43 channels show similar elevated cell death induced by H2O2.
Next, we addressed the mechanism by which astrocytic Cx43 exerts its effects. Cx43 can form gap junction channels and hemichannels in astrocytes. Here, we show that H2O2 causes an increase in hemichannel activity but a reduction in GJIC mediated by Cx43. Enhanced hemichannel activity could result from an increase in opening probability of hemichannels and/or an increase in the number of hemichannels in the plasma membrane. The immediate increase in the dye uptake after the addition of H2O2 suggests that H2O2 causes hemichannel opening in astrocytes. In addition, we detected a reduction in Cx43 average plaque size which is associated with an increase in the number of smaller Cx43 plaques and a reduction in the number of larger plaques in astrocytes after H2O2 treatment. H2O2-induced increases in Cx43 hemichannel activity and cell surface expression were reported in osteocytes (23). Thus, it is likely that the H2O2-induced increase in Cx43 hemichannel activity is also associated with an increase in the number of Cx43 hemichannels at the cell surface in astrocytes. However, the effect is transient as hemichannels returned to a closed state soon after the removal of H2O2. In contrast to the effect on hemichannels, H2O2 caused uncoupling of gap junctions in cultured cortical astrocytes. This is in agreement with various studies done in epithelial cells and other cell types (20, 23, 57–61) but inconsistent with the finding from rat striatum astrocytes (35). These authors pointed out that H2O2-induced increase in GJIC in astrocytes is very atypical as endogenous GJIC is typically high in astrocytes (35). This difference may explain why Cx43 in striatum astrocytes exhibits distinct effects on H2O2-induced cell death. Unlike hemichannels, the effect of H2O2 on GJIC is prolonged as GJIC returned to control levels only observed after 7 h post-treatment. Astrocytes lacking Cx43 did not exhibit coupling, suggesting that Cx43 is solely responsible for GJIC in astrocytes in culture under the conditions used in this study. Therefore, the reduction in coupling observed in WT astrocytes treated with H2O2 as measured by dye transfer is mainly due to Cx43. This is supported by the finding that there is a significant change in the average size of Cx43 plaques and Cx43 level in response to ROS treatment in WT astrocytes. We detected a reduction in the number of large Cx43 gap junction plaques (62), suggesting that reduced Cx43 levels after H2O2 treatment could be a result of increased degradation of Cx43 gap junctions (46, 63). The phosphorylation of Cx43 has been linked to its internalization and degradation (46, 64). In this study, we show that Cx43 phosphorylation state is altered after H2O2 treatment, suggesting that changes in phosphorylation of Cx43 may contribute to reduced number of Cx43 gap junctions.
Our findings suggest that H2O2 treatment alters Cx43 GJIC as well as hemichannel activity in cortical astrocytes, leading to ROS resistance. If hemichannels contributed to ROS resistance, the opening of hemichannels would be expected to benefit the cells. However, a previous study suggested that ROS can enter cells via opened hemichannels, indicating the detrimental effect of hemichannels (21). To dissect hemichannel function from GJIC, we studied the role of Panx1 in H2O2-induced cell death. It has been reported that Panx1 is the major substrate of hemichannels and does not form functional gap junction in cultured astrocytes (51–54). Here, we reported that knock-out of Panx1 in cultured astrocytes did not prevent H2O2-mediated cell death. It is worth noting that H2O2-induced hemichannel opening is very transient, as short as 45 min, whereas cell death appeared at much later time point. Altogether, Panx1 as well as Cx43 hemichannels do not appear to play an important role in astrocytic death induced by H2O2. This suggests that GJIC is a major contributor to ROS resistance mediated by Cx43.
It is interesting to note the distinct effects of Cx43 in oxidative stress and H/R pathways. This is unexpected because ROS including H2O2 are also produced during H/R and thought to contribute to H/R-induced cell death (28, 29). This suggests that H/R may stimulate additional pathways besides ROS-mediated signaling, leading to distinctively different modulation of Cx43. Previous studies reported an immediate change in Cx43 expression and distribution at the plasma membrane after hypoxia (15, 24, 65). Here, we demonstrated that after 4 h hypoxia, intense punctate structures disappear and non-punctate Cx43 localizes over the entire plasma membrane, which was correlated with a marked increase in the P0 isoform of Cx43. This phenomenon was not observed in response to ROS. Interestingly, the non-punctate membrane staining of Cx43 in hypoxia treated cells are very similar to astrocytes harboring a G60S mutation at its extracellular loop (66). This mutant also shows increased hemichannel activity, indicating that a redistribution of Cx43 may be correlated with the reciprocal increase of hemichannel and decrease of gap junction activity. Indeed, hypoxia and reoxygenation increases hemichannel activity and decreases GJIC in astrocytes (14, 15, 24). In addition, Orellana et al. (2010) reported that blockage of Cx43 hemichannels during reoxygenation can prevent astrocytic death caused by H/R (15). This suggests that a prolonged increase in Cx43 hemichannel activity is the main cause of astrocytic death induced by H/R.
In summary, our finding suggests that the controversial role of Cx43 may be attributed to the extent of dephosphorylation that results in varying degrees of hemichannel enhancement and GJIC inhibition. If hypoxia-induced redistribution of Cx43 is correlated with marked dephosphorylation resulting in sustained hemichannel opening compared with H2O2-treated cells, the cellular damage by the GJIC mediated role of Cx43 may be irreversible. In contrast, the damage caused by transient opening of Cx43 channels in H2O2-treated cells is reversed by the recovery of GJIC, and therefore Cx43 has a beneficial role.
This work was supported by a grant-in-aid from the Heart and Stroke Foundation of Canada (to C. C. N.) and the Fondo Nacional de Desarrollo Científico y Tecnologico Grant FONDECYT 3120006 (to J. L. V.) of Chile. Funding was partly provided through the CIHR Team Grant on ”Vascular Cognitive Impairment: Animal Models of Co-morbidity.”
- Cx43
- connexin43
- ROS
- reactive oxygen species
- Cbx
- carbenoxelone
- 18-αGA
- 18-α-glycyrrhetinic acid
- Etd
- ethidium.
REFERENCES
- 1. Tower D. B., Young O. M. (1973) The activities of butyrylcholinesterase and carbonic anhydrase, the rate of anaerobic glycolysis, and the question of a constant density of glial cells in cerebral cortices of various mammalian species from mouse to whale. J. Neurochem. 20, 269–278 [DOI] [PubMed] [Google Scholar]
- 2. Walz W. (2000) Role of astrocytes in the clearance of excess extracellular potassium. Neurochem. Int. 36, 291–300 [DOI] [PubMed] [Google Scholar]
- 3. Maragakis N. J., Rothstein J. D. (2001) Glutamate transporters in neurologic disease. Arch. Neurol. 58, 365–370 [DOI] [PubMed] [Google Scholar]
- 4. Pekny M., Nilsson M. (2005) Astrocyte activation and reactive gliosis. Glia 50, 427–434 [DOI] [PubMed] [Google Scholar]
- 5. Bélanger M., Allaman I., Magistretti P. J. (2011) Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell. Metab. 14, 724–738 [DOI] [PubMed] [Google Scholar]
- 6. Dermietzel R., Spray D. C. (1993) Gap junctions in the brain: where, what type, how many and why? Trends Neurosci. 16, 186–192 [DOI] [PubMed] [Google Scholar]
- 7. Nagy J. I., Rash J. E. (2000) Connexins and gap junctions of astrocytes and oligodendrocytes in the CNS. Brain Res. Brain Res. Rev. 32, 29–44 [DOI] [PubMed] [Google Scholar]
- 8. Alexander D. B., Goldberg G. S. (2003) Transfer of biologically important molecules between cells through gap junction channels. Curr. Med. Chem. 10, 2045–2058 [DOI] [PubMed] [Google Scholar]
- 9. Sáez J. C., Berthoud V. M., Brañes M. C., Martínez A. D., Beyer E. C. (2003) Plasma membrane channels formed by connexins: their regulation and functions. Physiol. Rev. 83, 1359–1400 [DOI] [PubMed] [Google Scholar]
- 10. Foote C. I., Zhou L., Zhu X., Nicholson B. J. (1998) The pattern of disulfide linkages in the extracellular loop regions of connexin 32 suggests a model for the docking interface of gap junctions. J. Cell Biol. 140, 1187–1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Unger V. M., Kumar N. M., Gilula N. B., Yeager M. (1999) Three-dimensional structure of a recombinant gap junction membrane channel. Science 283, 1176–1180 [DOI] [PubMed] [Google Scholar]
- 12. Li H., Liu T. F., Lazrak A., Peracchia C., Goldberg G. S., Lampe P. D., Johnson R. G. (1996) Properties and regulation of gap junctional hemichannels in the plasma membranes of cultured cells. J. Cell Biol. 134, 1019–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Quist A. P., Rhee S. K., Lin H., Lal R. (2000) Physiological role of gap-junctional hemichannels. Extracellular calcium-dependent isosmotic volume regulation. J. Cell Biol. 148, 1063–1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Contreras J. E., Sánchez H. A., Eugenin E. A., Speidel D., Theis M., Willecke K., Bukauskas F. F., Bennett M. V., Sáez J. C. (2002) Metabolic inhibition induces opening of unapposed connexin 43 gap junction hemichannels and reduces gap junctional communication in cortical astrocytes in culture. Proc. Natl. Acad. Sci. U.S.A. 99, 495–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Orellana J. A., Hernández D. E., Ezan P., Velarde V., Bennett M. V., Giaume C., Sáez J. C. (2010) Hypoxia in high glucose followed by reoxygenation in normal glucose reduces the viability of cortical astrocytes through increased permeability of connexin 43 hemichannels. Glia 58, 329–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wilson J. X. (1997) Antioxidant defense of the brain: a role for astrocytes. Can. J. Physiol. Pharmacol. 75, 1149–1163 [PubMed] [Google Scholar]
- 17. Chen Y., Chan P. H., Swanson R. A. (2001) Astrocytes overexpressing Cu,Zn superoxide dismutase have increased resistance to oxidative injury. Glia 33, 343–347 [DOI] [PubMed] [Google Scholar]
- 18. Choi J. H., Kim D. H., Yun I. J., Chang J. H., Chun B. G., Choi S. H. (2007) Zaprinast inhibits hydrogen peroxide-induced lysosomal destabilization and cell death in astrocytes. Eur. J. Pharmacol. 571, 106–115 [DOI] [PubMed] [Google Scholar]
- 19. Giardina S. F., Mikami M., Goubaeva F., Yang J. (2007) Connexin 43 confers resistance to hydrogen peroxide-mediated apoptosis. Biochem. Biophys. Res. Commun. 362, 747–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Upham B. L., Kang K. S., Cho H. Y., Trosko J. E. (1997) Hydrogen peroxide inhibits gap junctional intercellular communication in glutathione sufficient but not glutathione deficient cells. Carcinogenesis 18, 37–42 [DOI] [PubMed] [Google Scholar]
- 21. Ramachandran S., Xie L. H., John S. A., Subramaniam S., Lal R. (2007) A novel role for connexin hemichannel in oxidative stress and smoking-induced cell injury. PLoS One 2, e712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hutnik C. M., Pocrnich C. E., Liu H., Laird D. W., Shao Q. (2008) The protective effect of functional connexin43 channels on a human epithelial cell line exposed to oxidative stress. Invest. Ophthalmol. Vis. Sci. 49, 800–806 [DOI] [PubMed] [Google Scholar]
- 23. Kar R., Riquelme M. A., Werner S., Jiang J. X. (2013) Connexin 43 channels protect osteocytes against oxidative stress-induced cell death. J. Bone Miner. Res. 28, 1611–1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li W., Hertzberg E. L., Spray D. C. (2005) Regulation of connexin43-protein binding in astrocytes in response to chemical ischemia/hypoxia. J. Biol. Chem. 280, 7941–7948 [DOI] [PubMed] [Google Scholar]
- 25. Martínez A. D., Sáez J. C. (2000) Regulation of astrocyte gap junctions by hypoxia-reoxygenation. Brain Res. Brain Res. Rev. 32, 250–258 [DOI] [PubMed] [Google Scholar]
- 26. Retamal M. A., Cortés C. J., Reuss L., Bennett M. V., Sáez J. C. (2006) S-nitrosylation and permeation through connexin 43 hemichannels in astrocytes: induction by oxidant stress and reversal by reducing agents. Proc. Natl. Acad. Sci. U.S.A. 103, 4475–4480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lin J. H., Lou N., Kang N., Takano T., Hu F., Han X., Xu Q., Lovatt D., Torres A., Willecke K., Yang J., Kang J., Nedergaard M. (2008) A central role of connexin 43 in hypoxic preconditioning. J. Neurosci. 28, 681–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Armstead W. M., Mirro R., Busija D. W., Leffler C. W. (1988) Postischemic generation of superoxide anion by newborn pig brain. Am. J. Physiol. 255, H401–403 [DOI] [PubMed] [Google Scholar]
- 29. Flamm E. S., Demopoulos H. B., Seligman M. L., Poser R. G., Ransohoff J. (1978) Free radicals in cerebral ischemia. Stroke 9, 445–447 [DOI] [PubMed] [Google Scholar]
- 30. Reaume A. G., de Sousa P. A., Kulkarni S., Langille B. L., Zhu D., Davies T. C., Juneja S. C., Kidder G. M., Rossant J. (1995) Cardiac malformation in neonatal mice lacking connexin43. Science 267, 1831–1834 [DOI] [PubMed] [Google Scholar]
- 31. Liu S., Liu F., Schneider A. E., St Amand T., Epstein J. A., Gutstein D. E. (2006) Distinct cardiac malformations caused by absence of connexin 43 in the neural crest and in the non-crest neural tube. Development 133, 2063–2073 [DOI] [PubMed] [Google Scholar]
- 32. Ozog M. A., Bechberger J. F., Naus C. C. (2002) Ciliary neurotrophic factor (CNTF) in combination with its soluble receptor (CNTFRα) increases connexin43 expression and suppresses growth of C6 glioma cells. Cancer Res. 62, 3544–3548 [PubMed] [Google Scholar]
- 33. Danilov C. A., Fiskum G. (2008) Hyperoxia promotes astrocyte cell death after oxygen and glucose deprivation. Glia 56, 801–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fan X., Hussien R., Brooks G. A. (2010) H2O2-induced mitochondrial fragmentation in C2C12 myocytes. Free Radic. Biol. Med. 49, 1646–1654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rouach N., Calvo C. F., Duquennoy H., Glowinski J., Giaume C. (2004) Hydrogen peroxide increases gap junctional communication and induces astrocyte toxicity: regulation by brain macrophages. Glia 45, 28–38 [DOI] [PubMed] [Google Scholar]
- 36. Ye Z. C., Wyeth M. S., Baltan-Tekkok S., Ransom B. R. (2003) Functional hemichannels in astrocytes: a novel mechanism of glutamate release. J. Neurosci. 23, 3588–3596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dermietzel R., Hertberg E. L., Kessler J. A., Spray D. C. (1991) Gap junctions between cultured astrocytes: immunocytochemical, molecular, and electrophysiological analysis. J. Neurosci. 11, 1421–1432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Giaume C., Fromaget C., el Aoumari A., Cordier J., Glowinski J., Gros D. (1991) Gap junctions in cultured astrocytes: single-channel currents and characterization of channel-forming protein. Neuron 6, 133–143 [DOI] [PubMed] [Google Scholar]
- 39. John S. A., Kondo R., Wang S. Y., Goldhaber J. I., Weiss J. N. (1999) Connexin-43 hemichannels opened by metabolic inhibition. J. Biol. Chem. 274, 236–240 [DOI] [PubMed] [Google Scholar]
- 40. Bruzzone S., Guida L., Zocchi E., Franco L., De Flora A. (2001) Connexin 43 hemi channels mediate Ca2+-regulated transmembrane NAD+ fluxes in intact cells. FASEB J. 15, 10–12 [DOI] [PubMed] [Google Scholar]
- 41. Stout C. E., Costantin J. L., Naus C. C., Charles A. C. (2002) Intercellular calcium signaling in astrocytes via ATP release through connexin hemichannels. J. Biol. Chem. 277, 10482–10488 [DOI] [PubMed] [Google Scholar]
- 42. Laird D. W., Puranam K. L., Revel J. P. (1991) Turnover and phosphorylation dynamics of connexin43 gap junction protein in cultured cardiac myocytes. Biochem. J. 273, 67–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Laing J. G., Beyer E. C. (1995) The gap junction protein connexin43 is degraded via the ubiquitin proteasome pathway. J. Biol. Chem. 270, 26399–26403 [DOI] [PubMed] [Google Scholar]
- 44. Beardslee M. A., Laing J. G., Beyer E. C., Saffitz J. E. (1998) Rapid turnover of connexin43 in the adult rat heart. Circ. Res. 83, 629–635 [DOI] [PubMed] [Google Scholar]
- 45. Laird D. W. (2006) Life cycle of connexins in health and disease. Biochem. J. 394, 527–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Laird D. W. (2005) Connexin phosphorylation as a regulatory event linked to gap junction internalization and degradation. Biochim. Biophys. Acta 1711, 172–182 [DOI] [PubMed] [Google Scholar]
- 47. Solan J. L., Lampe P. D. (2007) Key connexin 43 phosphorylation events regulate the gap junction life cycle. J. Membr. Biol. 217, 35–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Musil L. S., Beyer E. C., Goodenough D. A. (1990) Expression of the gap junction protein connexin43 in embryonic chick lens: molecular cloning, ultrastructural localization, and post-translational phosphorylation. J. Membr. Biol. 116, 163–175 [DOI] [PubMed] [Google Scholar]
- 49. Musil L. S., Cunningham B. A., Edelman G. M., Goodenough D. A. (1990) Differential phosphorylation of the gap junction protein connexin43 in junctional communication-competent and -deficient cell lines. J. Cell Biol. 111, 2077–2088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Solan J. L., Fry M. D., TenBroek E. M., Lampe P. D. (2003) Connexin43 phosphorylation at S368 is acute during S and G2/M and in response to protein kinase C activation. J. Cell Sci. 116, 2203–2211 [DOI] [PubMed] [Google Scholar]
- 51. Iglesias R., Dahl G., Qiu F., Spray D. C., Scemes E. (2009) Pannexin 1: the molecular substrate of astrocyte “hemichannels”. J. Neurosci. 29, 7092–7097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sosinsky G. E., Boassa D., Dermietzel R., Duffy H. S., Laird D. W., MacVicar B., Naus C. C., Penuela S., Scemes E., Spray D. C., Thompson R. J., Zhao H. B., Dahl G. (2011) Pannexin channels are not gap junction hemichannels. Channels 5, 193–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Scemes E. (2012) Nature of plasmalemmal functional “hemichannels”. Biochim. Biophys. Acta 1818, 1880–1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dahl G., Locovei S. (2006) Pannexin: to gap or not to gap, is that a question? IUBMB Life 58, 409–419 [DOI] [PubMed] [Google Scholar]
- 55. Boassa D., Ambrosi C., Qiu F., Dahl G., Gaietta G., Sosinsky G. (2007) Pannexin1 channels contain a glycosylation site that targets the hexamer to the plasma membrane. J. Biol. Chem. 282, 31733–31743 [DOI] [PubMed] [Google Scholar]
- 56. Penuela S., Bhalla R., Gong X. Q., Cowan K. N., Celetti S. J., Cowan B. J., Bai D., Shao Q., Laird D. W. (2007) Pannexin 1 and pannexin 3 are glycoproteins that exhibit many distinct characteristics from the connexin family of gap junction proteins. J. Cell Sci. 120, 3772–3783 [DOI] [PubMed] [Google Scholar]
- 57. Hu J., Cotgreave I. A. (1995) Glutathione depletion potentiates 12-O-tetradecanoyl phorbol-13-acetate(TPA)-induced inhibition of gap junctional intercellular communication in WB-F344 rat liver epithelial cells: relationship to intracellular oxidative stress. Chem. Biol. Interact. 95, 291–307 [DOI] [PubMed] [Google Scholar]
- 58. Kuo M. L., Jee S. H., Chou M. H., Ueng T. H. (1998) Involvement of oxidative stress in motorcycle exhaust particle-induced DNA damage and inhibition of intercellular communication. Mutat. Res. 413, 143–150 [DOI] [PubMed] [Google Scholar]
- 59. Kang K. S., Kang B. C., Lee B. J., Che J. H., Li G. X., Trosko J. E., Lee Y. S. (2000) Preventive effect of epicatechin and ginsenoside Rb2 on the inhibition of gap junctional intercellular communication by TPA and H2O2. Cancer Lett. 152, 97–106 [DOI] [PubMed] [Google Scholar]
- 60. Todt I., Ngezahayo A., Ernst A., Kolb H. A. (2001) Hydrogen peroxide inhibits gap junctional coupling and modulates intracellular free calcium in cochlear Hensen cells. J. Membr. Biol. 181, 107–114 [DOI] [PubMed] [Google Scholar]
- 61. Lee D. E., Kang N. J., Lee K. M., Lee B. K., Kim J. H., Lee K. W., Lee H. J. (2010) Cocoa polyphenols attenuate hydrogen peroxide-induced inhibition of gap-junction intercellular communication by blocking phosphorylation of connexin 43 via the MEK/ERK signaling pathway. J. Nutr. Biochem. 21, 680–686 [DOI] [PubMed] [Google Scholar]
- 62. Bukauskas F. F., Jordan K., Bukauskiene A., Bennett M. V., Lampe P. D., Laird D. W., Verselis V. K. (2000) Clustering of connexin 43-enhanced green fluorescent protein gap junction channels and functional coupling in living cells. Proc. Natl. Acad. Sci. U.S.A. 97, 2556–2561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Salameh A. (2006) Life cycle of connexins: regulation of connexin synthesis and degradation. Adv. Cardiol. 42, 57–70 [DOI] [PubMed] [Google Scholar]
- 64. Segretain D., Falk M. M. (2004) Regulation of connexin biosynthesis, assembly, gap junction formation, and removal. Biochim. Biophys. Acta 1662, 3–21 [DOI] [PubMed] [Google Scholar]
- 65. Li W. E., Nagy J. I. (2000) Connexin43 phosphorylation state and intercellular communication in cultured astrocytes following hypoxia and protein phosphatase inhibition. Eur. J. Neurosci. 12, 2644–2650 [DOI] [PubMed] [Google Scholar]
- 66. Kozoriz M. G., Lai S., Vega J. L., Sáez J. C., Sin W. C., Bechberger J. F., Naus C. C. (2013) Cerebral ischemic injury is enhanced in a model of oculodentodigital dysplasia. Neuropharmacology 75, 549–556 [DOI] [PubMed] [Google Scholar]