

Background: LysP plays an important role in transport of lysine in bacteria.
Results: Binding of lysine to purified STM2200 was investigated using ITC; key residues involved in binding are predicted by structure modeling and verified experimentally.
Conclusion: l-Lysine binds to STM2200 selectively, and important residues were identified.
Significance: Our findings provide a functional characterization of a lysine transporter.
Keywords: Amino Acid Transport, Homology Modeling, Isothermal Titration Calorimetry, Protein Purification, Site-directed Mutagenesis, APC Superfamily, Quantitative l-Lysine Binding, Secondary Active Transporter
Abstract
Membrane proteins of the amino acid-polyamine-organocation (APC) superfamily transport amino acids and amines across membranes and play an important role in the regulation of cellular processes. We report the heterologous production of the LysP-related transporter STM2200 from Salmonella typhimurium in Escherichia coli, its purification, and functional characterization. STM2200 is assumed to be a proton-dependent APC transporter of l-lysine. The functional interaction between basic amino acids and STM2200 was investigated by thermoanalytical methods, i.e. differential scanning and isothermal titration calorimetry. Binding of l-lysine to STM2200 in its solubilized monomer form is entropy-driven. It is characterized by a dissociation constant of 40 μm at pH 5.9 and is highly selective; no evidence was found for the binding of l-arginine, l-ornithine, l-2,4-diaminobutyric acid, and l-alanine. d-Lysine is bound 45 times more weakly than its l-chiral form. We thus postulate that STM2200 functions as a specific transport protein. Based on the crystal structure of ApcT (Shaffer, P. L., Goehring, A., Shankaranarayanan, A., and Gouaux, E. (2009) Science 325, 1010–1014), a proton-dependent amino acid transporter of the APC superfamily, a homology model of STM2200 was created. Docking studies allowed identification of possible ligand binding sites. The resulting predictions indicated that Glu-222 and Arg-395 of STM2200 are markedly involved in ligand binding, whereas Lys-163 is suggested to be of structural and functional relevance. Selected variants of STM2200 where these three amino acid residues were substituted using single site-directed mutagenesis showed no evidence for l-lysine binding by isothermal titration calorimetry, which confirmed the predictions. Molecular aspects of the observed ligand specificity are discussed.
Introduction
The amino acid-polyamine-organocation (APC)2 superfamily is one of the largest families of secondary active transporters for amino acids. These physiologically important transporters are found in bacteria, archaea, yeast, fungi, unicellular eukaryotes, plants, and animals and hence are ubiquitous in nature (2). The APC superfamily members include transport proteins functioning as solute-cation symporters or solute-solute antiporters (2–5). The size of corresponding polypeptide chains varies between 350 and 800 amino acid residues. According to topology predictions (6, 7), most of these proteins have 12 (only a few of them possess 10 or 14) transmembrane helices (TMHs). In addition, sensors and transporters of amino acids in Saccharomyces cerevisiae can be considered as APC transporter homologues (8). In mammals, the APC transporters play a pivotal role in amino acid transport-related processes including redox imbalance (9), neuroadaptation (10), macrophage activation and proliferation (11), growth and survival of cancer cells (12), and primary inherited cystinuria and lysinuric protein intolerance (13–15). Various amino acid transporters including a serine transporter of the Na+/Ser symporter family (16); AroP-PheP, a tryptophan transporter of the aromatic amino acid transport family (17); AdiC, an arginine-agmatine transporter (18–20); ApcT, a proton-dependent broad specificity amino acid transporter (1) of the APC superfamily; the Na+-independent neutral amino acid transporter (21); and the glutamate and aspartate transporter of the excitatory amino acid transporter family (22, 23) have been studied using Escherichia coli. However, lysine transport has received little attention. One important member of the APC superfamily is the lysine-specific and predicted proton-dependent permease and transporter LysP from E. coli; its gene has been cloned and expressed previously (24). In addition, its regulation has been studied (24, 25). The integral membrane protein STM2200 is related to LysP. STM2200 contains 489 amino acid residues and is predicted to possess 12 TMHs (TransportDB). Here, we produced and purified STM2200 from Salmonella typhimurium LT2 using the E. coli TOP10 strain. Subsequent to the development of suitable methods for isolation, detergent solubilization, and purification, thermoanalytical methods such as differential scanning calorimetry (DSC) and isothermal titration calorimetry (ITC) were applied to characterize the thermal stability of purified STM2200 in its solubilized form and its binding affinity for basic amino acids. Selective binding of l-lysine was observed by ITC. The investigation of the oligomeric state indicated that STM2200 exists as a monomer in β-d-dodecyl maltoside (β-DDM) micelles.
An understanding of the mechanism of action of STM2200 requires detailed structural information. Because the atomic structure of STM2200 is not available, structural aspects could only be investigated on the basis of its sequence by homology modeling making use of the recently reported crystal structure of ApcT (1) in an inward-open conformation (1). Docking studies were used to identify possible ligand/substrate binding sites, which are discussed and illustrated.
EXPERIMENTAL PROCEDURES
Materials
β-DDM was purchased from Glycon Chemicals. The site-directed mutagenesis kit used was purchased from Agilent Technologies. The BCA kit was from Pierce. pBAD plasmid was purchased from Invitrogen. Ni-NTA beads were from Qiagen. Carbenicillin was from Carl Roth. Superdex 200 PC 3.2/30 was obtained from GE Healthcare. Complete protease inhibitor mixture was from Roche Applied Science, lysozyme was from Applichem, and Benzonase was obtained from Merck KGaA. d-Lysine (with a content of 0.1% l-lysine) was purchased from Bachem. l-2,4-Diminobutyric acid was from Fluka. All other reagents, l-arginine, l-lysine, l-ornithine, l-alanine, and l-arabinose were purchased from Sigma.
Bacterial Strains and Expression Vectors
The E. coli strain DH5α (Invitrogen) was used for cloning, the E. coli strain TOP10 (Invitrogen) was used for heterologous protein production, and the E. coli strain XL1-Blue (Agilent Technologies) was used for site-directed mutagenesis. For heterologous protein production in E. coli, we used the pBAD vector, which was modified in the multiple cloning site to yield the recombinant protein fused with a tobacco etch virus protease cleavage site and a C-terminal decahistidine tag (His tag) and named pBADA2 (see Fig. 2A). For co-expression and co-purification experiments in oligomerization studies, the gene of interest was cloned into a modified pBAD vector named pBAD-CM (see Fig. 2A). This vector is modified for insertion of two copies of the same gene in such a way that the first copy codes for a protein with a C-terminal hexa-His tag and the second copy codes for a protein with a C-terminal Strep-I tag.
FIGURE 2.
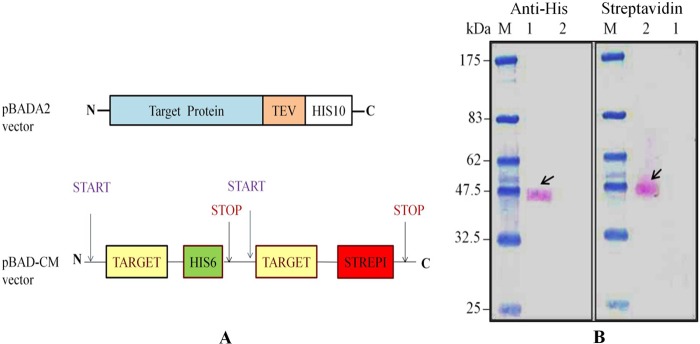
A, schematic representation of different vector constructs. The pBADA2 vector encodes the target protein followed by a tobacco etch virus (TEV) protease cleavage site and a deca-His tag at the C terminus. The pBAD-CM vector is modified in a way in which the target gene can be cloned two times, one time separately with an added hexa-His tag coding sequence and another time with an added Strep-I tag coding sequence. B, oligomerization study of STM2200 using a pBAD-CM vector. The Western blot of co-purified proteins was developed separately with an anti-His antibody and a streptavidin, both coupled to alkaline phosphatase. Lane M, marker; lane 1, His tag-purified protein; lane 2, Strep-I tag-purified protein.
Cloning of Genes
The coding sequences of targets of interest were obtained from the transporter protein analysis database (TransportDB) and used for primer design. The gene encoding the target protein was amplified by PCR using Phusion DNA polymerase (Finnzymes, Finland) with genomic DNA from S. typhimurium (a kind gift from A. Kahnert and S. H. E. Kaufmann, Max Planck Institute of Infection Biology, Berlin, Germany) and primers (Invitrogen) containing at least 15 gene-specific nucleotides. The resulting PCR products were cloned into the respective vectors according to the manufacturers' instructions. Cloning into the pBAD-CM vector required a two-step procedure. The first step involved introduction of a copy of the gene to be expressed as a fusion protein with a C-terminal hexa-His tag. The resulting construct was then used for a second cloning step in which the same gene was inserted into a second cloning site to be expressed as a C-terminal Strep-I tag fusion. Both tags were used to detect and purify the protein produced.
Site-directed Mutagenesis
The QuikChange II site-directed mutagenesis kit was used to introduce point mutations, replace amino acids, and delete or insert amino acids. This kit did not require specialized vectors, unique restriction sites, multiple transformations, or in vitro methylation treatment steps. The mutants were prepared using reagents supplied in the kit as suggested in the manufacturer's instructions.
Heterologous Protein Production and Detection
The expression constructs pBADA2 and pBAD-CM were transformed into TOP10 cells. Transformants were selected on lysogeny broth agar plates containing 50 μg/ml carbenicillin. A single colony was used for inoculation; inoculum of 50 μl from overnight culture was transferred into 10 ml of fresh lysogeny broth containing 50 μg/ml carbenicillin at 37 °C, grown until the A600/cm (Optical density measurement at 600 nm by using 1 cm pathlength cuvettes) reached a value of 0.5. Subsequently, the culture was induced with 0.02% l-arabinose and incubated at 37 °C for 3 h. When the suspension reached an A600/cm of 2, the cells were harvested by centrifugation and resuspended in 70 μl of lysis buffer consisting of 20 mm HEPES-NaOH, 100 mm NaCl, 10 mm MgSO4 pH 7.5, 1× Complete protease inhibitor mixture (Roche Applied Science), 1 mg/ml lysozyme (Applichem, Darmstadt), 1 unit of Benzonase (Merck KGaA). After 20 min of incubation at room temperature, 5 μl of 10% SDS and 25 μl of 4× SDS-PAGE loading buffer were added to the mixture. Samples of 20 μl were separated by 10% SDS-PAGE and analyzed by Western blot using anti-His alkaline phosphatase antibody (Sigma) according to the manufacturer's instructions.
Isolation of Membrane and Protein Purification
Expression vectors pBADA2 and pBAD-CM containing STM2200 were freshly transformed into E. coli TOP10 cells, and single colonies after growth on agar plates were inoculated into lysogeny broth medium. The overnight inocula were used for inoculation (50 ml of inoculum for 2 liters of medium in 5-liter flasks). The culture was incubated at 37 °C at 180 rpm agitation until the A600/cm reached a value of 0.5–0.7. The production of the protein was induced by addition of 0.02% l-arabinose at 37 °C for 3 h. Cells were harvested after centrifugation and suspended in 100 ml of lysis buffer (20 mm HEPES-NaOH, 100 mm NaCl, 2 mm EDTA, 2 mm of PMSF, pH 7.5). The cells were disrupted by passing them through an M-110LA microfluidizer (Microfluidics, Newton, MA) under ice at 10,000 p.s.i. four times. The cell debris was removed by centrifugation at 6500 × g at 4 °C for 30 min. Membrane vesicles were collected from the lysate supernatant after centrifugation at 145,000 × g at 4 °C for 90 min. The pelleted vesicles were resuspended in 20 mm HEPES-NaOH, 100 mm NaCl, pH 7.5. The total protein content in the membrane fraction was analyzed by BCA assay (Pierce) according to the manufacturer's instructions; the final concentration was adjusted to 10 mg/ml. Samples were aliquoted and stored at −80 °C.
The membranes of the aliquots were solubilized at room temperature in solubilization buffer 20 mm HEPES-NaOH, 100 mm NaCl, 2 mm PMSF (GERBU, Gailberg, Germany), 1% (w/v) β-DDM, pH 7.5 and incubated at 4 °C with slow agitation for 1 h. The insoluble membrane material was removed by ultracentrifugation at 170,000 × g at 4 °C for 1 h. The supernatant was passed through a column containing a 1 ml of Ni-NTA matrix (Qiagen, Hilden, Germany) pre-equilibrated with 20 mm HEPES-NaOH, 300 mm NaCl, 0.025% (w/v) β-DDM, pH 7.5. Subsequent to a washing step with 30 ml of 20 mm HEPES-NaOH, 300 mm NaCl, 0.025% (w/v) β-DDM, 40 mm imidazole-HCl, pH 7.5, the protein was eluted with 4 ml of 20 mm MES-NaOH, 300 mm NaCl, 0.025% (w/v) β-DDM, 350 mm imidazole-HCl, pH 5.9.
For the oligomerization studies, an affinity purification using Strep-I tag was carried out, and the streptavidin column was prepared according to published protocols (26). The membrane solubilizate was passed through the 1-ml column at a flow rate of 0.5 ml/min at 4 °C two times. Nonspecifically bound protein was removed by washing the column with 25 column volumes of 20 mm HEPES-NaOH, 300 mm NaCl, 0.025% (w/v) β-DDM, pH 7.5. Bound protein was eluted with 20 mm MES-NaOH, 300 mm NaCl, 0.025% (w/v) β-DDM, pH 5.9 containing 2 mm desthiobiotin (Sigma) using 4 × column volumes. Subsequently, all the protein fractions were analyzed by SDS-PAGE and subsequent Western blotting using anti-His antibody conjugated with alkaline phosphatase (Sigma) and streptavidin conjugated with alkaline phosphatase (Amersham Biosciences). Analytical chromatography was performed to investigate the homogeneity of the purified protein using a Superdex 200 PC 3.2/3.0 column in a SMART system (GE Healthcare).
Determination of Protein Concentration
Protein content during purification and the subsequent concentration of STM2200 and its variants was determined by BCA protein assay (Pierce) according to the manufacturer's instructions.
Differential Scanning Calorimetry
The DSC measurements were performed using a VP-Capillary DSC system (MicroCal, Inc., Northampton, MA) equipped with tantalum 61 cells, each with an active volume of 137 μl. Protein samples were concentrated to 2.5–3 mg/ml in 20 mm MES-NaOH, 150 mm NaCl, 0.025% (w/v) β-DDM, pH 5.9; this buffer was used as a reference medium. Both sample and reference solutions were filtered using a 0.2-μm filter. Scanning was performed at a rate of 90 °C/h and a N2 pressure of 4.5 bars after an initial 15 min of equilibration at a starting temperature of 10 °C. Data were analyzed using Origin 7.0 software (Microcal Origin); thermograms were corrected by subtraction of buffer-only blank scans.
Isothermal Titration Calorimetry
ITC measurements were carried out at 20 °C with a VP-ITC calorimeter (GE Healthcare, MicroCal, Inc.) consisting of a 1.45-ml reaction cell. STM2200 in 20 mm MES-NaOH, 150 mm NaCl, 0.025% (w/v) β-DDM, pH 5.9 at concentrations ranging between 15 and 30 μm was titrated with ligands under conditions where the sum of the ionic strengths of ligand and NaCl was always 150 mm. After an initial 4-μl injection, all subsequent ligand additions consisted of 5 μl every 240–300 s. Stirring was at 307 rpm. For evaluation, the corresponding heat changes of titrant dilution in the absence of protein were generally subtracted from the resulting signals in its presence. Evaluation was carried out using the software of the manufacturer (MicroCal Origin) based on a single site binding model.
Whereas binding of l- and d-lysine to STM2200 was detectable and the corresponding affinities could be determined (see Fig. 3 and Table 2), no evidence for binding was observed in the same medium for l-ornithine, l-2,4-diaminobutyric acid, and l-alanine up to total ligand concentrations of 6.1, 2.1, and 6.1 mm, respectively. In addition, again no clear evidence for l-lysine binding was observed for the STM2200 variants, i.e. R395M, E222L, K163A, and K163L, which were titrated up to total ligand concentrations of 2.0 mm in the cell.
FIGURE 3.

Titration calorimetry of STM2200 at 20 °C. A, l-lysine titration of 30 μm STM2200 in 20 mm MES-NaOH, 0.025% (w/v) β-DDM (buffer B) containing 150 mm NaCl, pH 5.9 with 1 × 4 μl and 22 × 5.0 μl of 5 mm l-lysine in buffer B containing 145 mm NaCl, pH 5.9 and corresponding reference titration (R; shifted by an offset) in the absence of protein. d-Lysine titration of 30 μm STM2200 in buffer B containing 150 mm NaCl, pH 5.9 with 1 × 4 μl and 22 × 5.0 μl of 5 mm d-lysine in buffer B containing 145 mm NaCl, pH 5.9 (d-Lys). B, heat changes (Δq) shown as a concentration ratio between l-lysine and STM2200. Data are presented under “Results,” and mean values are presented in Table 2. C, d-lysine titration of 32 μm STM2200 in buffer B containing 150 mm NaCl, pH 5.9 with 1 × 4 μl and 22 × 5.0 μl of 50 mm d-lysine in buffer B containing 100 mm NaCl, pH 5.9 and corresponding reference titration (R; shifted by an offset) in the absence of protein. D, heat changes (Δq) shown as a concentration ratio between d-lysine and STM2200. Data are presented under “Results,” and mean values are presented in Table 2.
TABLE 2.
Thermodynamic parameters for ligand binding to STM2200 and its variants in 20 mm MES-NaOH, 0.025% (w/v) β-DDM, 150 mm NaCl, pH 5.9 at 20 °C
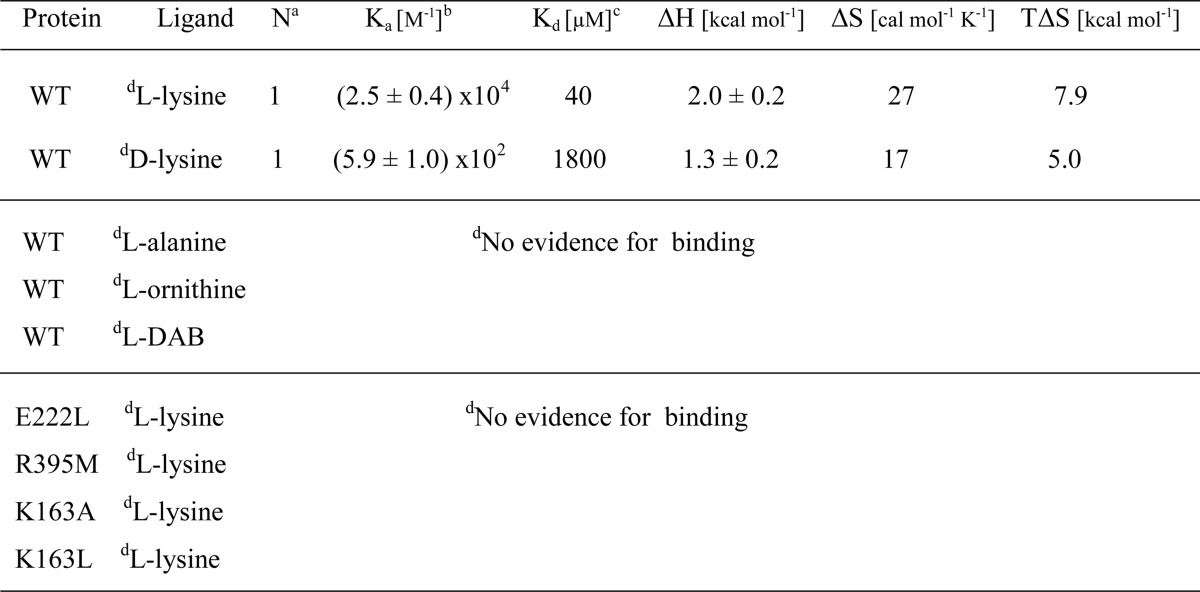
a Stoichiometric coefficient.
b Ka, association constant.
c Kd, dissociation constant; Kd = 1/Ka.
d Details are given under “Experimental Procedures” and “Results.”
Homology Modeling
A homology model of STM2200 was constructed by using the Prime module in Maestro (Prime 1.6; Schrödinger, Portland, OR). The backbone coordinates for the core of the homology model were built based on the 2.35-Å resolution crystal structure of apo-ApcT (1), a proton-coupled broad specificity amino acid transporter (Protein Data Bank code 3GIA), as template.
The sequence of STM2200 from S. typhimurium was taken from NCBI Protein Database accession number NP_461145.1. An initial secondary structure prediction was carried out using the PSIPRED Protein Structure Prediction Server and the Prime module in Maestro (Schrödinger). The HHpred web server identified apo-ApcT, a proton-coupled broad specificity amino acid transporter and an arginine/agmatine antiporter (1), as the closest relative to known structures with E-values of 6.5e−48 and 5.8e−47 and HHSearch scores of 393.0 and 386.5, respectively.
The refined STM2200 homology model was subjected to a series of validation analyses for consistency and reliability. The overall sequence identity between STM2200 and ApcT is only 19%. The stereochemical quality of the model was analyzed using the MolProbity server (27). It provides an all-atom contact analysis identifying any steric problems within the molecule and uses high accuracy Ramachandran and rotamer distributions to check the main chain and side chains for conformational outliers. In general, the STM2200 model scores were similar to those of the crystallographic structure of ApcT. The distribution of the ψ/φ angles was well within the allowed regions. The majority (87.7%) of all residues were found in the favored regions of the plot, and almost all (98.2%) residues were found in the allowed regions. The corresponding percentages for the ApcT template structure were 97.7 and 100.0. Several steric clashes and rotamer outliers were identified and fixed. Manual alteration of problematic ψ/φ angles along the entire protein backbone was performed during the refinement phase of the model construction. Comparing this result with similar ones for template crystal structures, we confirmed that the backbone conformations of the model structure are, by this criterion, as reliable as those of the template structure.
A validation check of the energetic properties of the homology model was performed using the ProSA web server (28). ProSA analysis of the STM2200 homology model revealed a z-score value of −4.6 compared with the value of −6.25 for the ApcT template that is in the range of native conformations of the template.
Docking
Structures of the ligands were taken from the PubChem Substance database (CID 389 for l-ornithine, CID 470 for l-2,4-diaminobutyric acid, CID 5950 for l-alanine, CID 5962 for l-lysine, and CID 57449 for d-lysine) as sdf files and prepared for docking using the LigPrep utility (Schrödinger) with Epik (Schrödinger) to define protonation and tautomeric states at pH 7.0 ± 2.0. The STM2200 model was prepared using the Protein Preparation Wizard utilities (Schrödinger) in Maestro with the default options to assign the charge states of ionizable residues, add hydrogens, and carry out a highly constrained minimization. Flexible docking calculations were performed with Glide XP 5.0 (Grid-based Ligand Docking from Energetics, Schrödinger) following the fragment-based procedure to increase the number of energetically favorable poses to a value of 1000. The scoring function Gscore was used to rank all poses. After docking, the best poses were minimized within the protein model. This minimization was performed using conjugate gradient minimization (0.05 convergence criteria), the optimized potentials for liquid simulations all-atoms (OPLS-AA) force field (29, 30), and the generalized Born/surface are (GB/SA) continuum water model (31). Protein residues that fell within 10 Å of the binding sites were allowed to optimize during minimization.
RESULTS
Production and Purification of STM2200
The vector pBAD in the A2 version (coding for a C-terminal deca-His tag preceded by a tobacco etch virus cleavage site) containing STM2200 in combination with the arabinose promoter had shown the largest production among all tested vectors used in small scale productions in whole cells. Three hours of induction with 0.02% (w/v) arabinose was found to be optimal. For protein identification, the whole cell lysates were subjected to SDS-PAGE and subsequent Western blot using an alkaline phosphatase-conjugated anti-polyhistidine antibody. The protein band of STM2200 was found just below the 47.5-kDa marker protein range in the Western blot (Fig. 1A). For the isolation of STM2200, membranes were prepared and solubilized in 1% (w/v) β-DDM. After ultracentrifugation, the resulting solubilizate was loaded onto a Ni-NTA matrix for protein purification. Subsequently, the protein was subjected to analytical size exclusion chromatography to determine its homogeneity and stability under different experimental conditions. The protein showed a single and narrow peak indicating its homogeneity in 20 mm MES-NaOH, 150 mm NaCl in 0.025% (w/v) β-DDM, pH 5.9 at 4 °C (Fig. 1D) and the presence of a single band on Coomassie-stained SDS-polyacrylamide gels indicating that the protein was at least 95% pure (Fig. 1C). Our procedure yielded ∼0.8 mg of purified protein/liter of lysogeny broth culture.
FIGURE 1.

A, Western blot showing the production of STM2200 in pBADA2 vector in whole cell lysates using an alkaline phosphatase-conjugated anti-His antibody. Lane M, marker; lane 1, control expression without induction; lane 2, expression after 3 h of induction with 0.02% (w/v) l-arabinose; lane 3, expression after 4 h of induction with 0.02% (w/v) l-arabinose. B, Western blot of small scale production of STM2200 and its four variants, i.e. K163A, K163L, R395M, and E222L. STM2200 and its variants were produced in small scale. Whole cell lysates were prepared, and equal amounts of total protein were subjected to SDS-PAGE and subsequent Western blotting using anti-His antibodies. Lane M, protein marker; lane 1, uninduced STM2200 as a negative control; lane 2, induced STM2200 as a positive control; lane 3, uninduced K163A; lane 4, induced K163A; lane 5, uninduced K163L; lane 6, induced K163L; lane 7, uninduced R395M; lane 8, induced R395M; lane 9, uninduced E222L; lane 10, induced E222L. C, Coomassie-stained 10% SDS-polyacrylamide gel shows purified STM2200. D, analytical gel filtration profile of purified STM2200 using a Superdex 200 column in a SMART system in buffer containing 20 mm MES-NaOH, 150 mm NaCl, 0.025% (w/v) β-DDM, pH 5.92.
Oligomerization Studies by Co-expression and Co-purification
For co-expression and co-purification, the STM2200 gene was cloned into the pBAD-CM vector (Fig. 2A). To apply the co-purification method, it is essential that the same protein can be alternatively expressed with two different tags, a hexa-His tag and a Strep-I tag, in the same cells. To purify the protein solubilized by β-DDM, two separate and different chromatographic procedures were applied, one using a Ni-NTA matrix to separate the His-tagged protein and the other using a streptavidin column for the Strep-I-tagged protein. The eluted fractions were analyzed on a Western blot (Fig. 2B) by using alkaline phosphatase coupled to His tag- and Strep-I tag-specific antibodies. If the protein is present as an oligomer, the protein purified using the Ni-NTA matrix should be detectable with alkaline phosphatase-conjugated streptavidin. The Western blot showed the presence of STM2200 purified by the Ni-NTA matrix detected by an anti-polyhistidine alkaline phosphatase-conjugated antibody. When purified with the streptavidin column, STM2200 was only detectable with alkaline phosphatase-conjugated streptavidin. This result indicates that STM2200 is present as a monomer. Moreover, the purified protein samples containing the different tags were subjected to analytical gel filtration to investigate their homogeneity. Both chromatograms showed a single narrow peak characteristic of homogeneous preparations.
Thermoanalytical Ligand Binding Studies
DSC is used to study thermal stability, unfolding, and binding of biomolecules (32, 33). STM2200 at a concentration of 50 μm in 20 mm MES-NaOH, 150 mm NaCl, 0.025% (w/v) β-DDM, pH 5.9 exhibited an endothermic transition with a transition temperature (Tm) of 43.5 °C (ΔH and full width of transition are given in Table 1). In addition, the thermal stability of the protein was analyzed upon addition of the putative ligands l-lysine and l-arginine. In the presence of 1 mm l-lysine, a marked increase in the Tm was observed that is indicative of ligand binding. The resulting transition temperature was 50.5 °C with only a slightly higher ΔH value (data in Table 1; no thermograms shown; data represent mean values from three experiments). No significant change was observed in the presence of l-arginine.
TABLE 1.
Differential scanning calorimetry of STM2200 (50 μm) and in the presence of ligands in 20 mm MES-NaOH, 150 mm NaCl, 0.025% β-DDM (w/v), pH 5.9
The full width at half-maximum height is around 11 °C. Details are given under “Results.”
| Ligand | Ligand conc. | Tm | ΔH |
|---|---|---|---|
| mm | °C | kcal mol−1 | |
| No | 0 | 43.5 ± 1.0 | 42 ± 8 |
| l-Lysine | 1.0 | 50.5 ± 1.0 | 49 ± 9 |
| l-Arginine | 1.0 | 44.5 ± 1.0 | 39 ± 8 |
For a quantitative analysis of ligand binding, isothermal titration calorimetry was applied. Because of the comparatively low transition temperature of STM2200 and its fairly broad thermal denaturation transition, all titrations were carried out at 20 °C to avoid minor amounts of protein denaturation during titrations. Fig. 3A shows a characteristic titration of 30 μm STM2200 with 5 mm l-lysine in 20 mm MES-NaOH, 145 mm NaCl, 0.025% (w/v) β-DDM, pH 5.9 together with the corresponding reference titration, which is characterized by an almost negligible exothermic ligand dilution process. Binding of l-lysine was clearly observed and is characterized by an endothermic process. The evaluation shown in Fig. 3B is based on a single site binding model. The stoichiometric coefficient N obtained from the fit procedure varied around 1. Because no precise value was obtainable under our conditions, a fixed value of N = 1.0 was used for all evaluations. This is consistent with suggestions derived from the modeling and our reported STM2200 properties. The value for the dissociation constant resulting from the fit according to the single site model in Fig. 3B was 28 μm, and a ΔH value of 2.1 kcal/mol was obtained from this experiment. An identical titration with d-lysine (Fig. 3A) showed only a minor signal increase compared with the reference titration and thus does not provide evidence for binding under the chosen conditions. If, however, the titration was carried out with considerably higher d-lysine concentrations such as 50 mm (see Fig. 3C), then again an endothermic ligand binding process could be observed. Under the new conditions, the reference titration showed an exothermic ligand dilution process. The dissociation constant resulting from the corresponding evaluation according to the single site model (Fig. 3D) was 2.0 mm, and the ΔH value was 1.2 kcal/mol. Evidently, the lower ΔH value is consistent with the lower signal intensity observed for the d-lysine titration. The resulting mean values of thermodynamic parameters were obtained from six titrations with l- and four titrations with d-lysine and are given in Table 2. The TΔS and ΔS values were calculated from the mean values. As a result, the ligand binding site of STM2200 exhibits a marked chiral selection in favor of l-lysine.
To obtain information about properties of the STM2200 ligand binding site, titrations were carried out with substitutes of l-lysine again bearing a positive charge but exhibiting shorter side chains such as l-ornithine and l-2,4-diaminobutyric acid. In addition, the zwitterionic l-alanine was investigated. Under our experimental conditions, no binding could be observed for these three compounds even if the positively charged side chain of l-lysine was shortened only by one CH2 group (l-ornithine). Nevertheless, an extremely weak affinity of these ligands cannot be excluded. Depending on the chosen titration conditions at 20 °C in the medium given above, estimated lower limits of dissociation constants for a possible binding were about 2.5 mm for l-ornithine, 1 mm for l-2,4-diaminobutyric acid, and 2.5 mm for l-alanine provided a ΔH value of at least ∼1 kcal/mol can be expected.
A structural evaluation of the ligand binding site would be of great interest. However, because no crystals suitable for x-ray analysis could be obtained, a modeling study based on the structure of the related transporter protein ApcT was carried out.
Homology Modeling
The homology model of STM2200 represents a conformation of the transporter in the occluded, inward facing state. Its 489 amino acid residues can be arranged in 12 transmembrane helices as shown in Fig. 4B. The model contains an inverted structural symmetry motif within the first 10 TMHs, organized in two repeating units, followed by the two additional TMHs, 11 and 12. The protein fold and pseudo-symmetry of STM2200 model are similar to that of LeuT from Aquifex aeolicus (LeuTAa) (34). Similarly to the apo-ApcT structure, it adopts a substrate-free, inward facing occluded conformation after substrate release, and it exhibits closure of the intracellular thin gate that shields the binding pocket from the intracellularly oriented cavity. This apo conformation is most similar to that of the substrate-bound, occluded conformation of the BetP structure (35).
FIGURE 4.

Illustration of homology model of STM2200 obtained in this work based on the crystal structure of the inward facing apo-ApcT transporter from Methanocaldococcus jannaschii (Protein Data Bank code 3GIA). A, internal cavity represented as red-dotted surface. Hydrophobic residues are colored in blue, and polar residues are in red. Amino acids are represented by single amino acid letter code. B, the overall architecture of the STM2200 homology model viewed as a cross-eyed stereopair viewed parallel to the membrane. d-Lys is bound to the structure and shown as ball-and-stick and colored by heteroatoms. TMHs are shown as ribbons and are numbered 1–12 (distances are mentioned under “Results”). C, schematic illustration of binding of l-lysine (blue) and d-lysine (red) to STM2200 including partial dehydration upon binding. ter, terminus; aq, aqueous.
Docking of Lysine and Model Compounds to the Homology Model
Analysis of the STM2200 homology model structure using ICM-Pro (36) predicts a large closed cavity at the approximate midpoint of the lipid bilayer, revealing the presence of the polar, aromatic, and aliphatic cavity-lining residues Met-61, Val-62, Ile-65, Met-212, Gly-215, Phe-216, Phe-218, Ala-385, Trp-386, Gly-388, Ile-389, Ile-391, Ser-392, Ile-431, Thr-432, Gln-219, Gly-220, Thr-221, Glu-222, Arg-395, Asn-105, and Ser-392 (for amino acid single letter codes, see Fig. 4A). This negatively charged cavity was assessed to be large enough to accommodate the substrates and, therefore, might be the ligand binding site.
To provide evidence for this finding and to identify ligand binding site(s), we applied fragment-based Glide (XP) ligand docking calculations (Schrödinger) with the STM2200 homology model. Five different ligands were tested in docking experiments: l-lysine, d-lysine, l-ornithine, l-2,4-diaminobutyric acid, and l-alanine. The results of the calculations show that the ligand binding pocket is located at the approximate center of the membrane and is identical for all tested ligands. It is formed by the residues specified above predicted by our ICM-Pro calculations.
Of the five tested ligands, d-lysine showed the best binding scoring function, i.e. lowest glide value. It demonstrates ionic interactions with the carboxyl group of Glu-222 and the guanidino group of Arg-395. The binding distances were 1.6 and 2.0 Å, respectively. Binding was further improved by a hydrogen bond between the carbonyl group of Gly-388 and the ligand as illustrated schematically in Fig. 4B. However, although l-lysine binding to STM2200 was shown by ITC experiments in this study, the scoring function (glide value) was half that of d-lysine. Similarly to d-lysine, it formed ionic interactions with the carboxyl group of Glu-222 and the guanidino group of Arg-395 as well as hydrogen bonds with the carbonyl oxygen atoms of Gly-215 and Gly-388 (Fig. 4B) with binding distances of 1.9 and 2.6 Å, respectively. These results showed that electrostatic interactions were stronger when tested in the presence of l-lysine with the highest electrostatic score value. It was further seen that Arg-395 and Glu-222 residues, present in the substrate binding sites, form ionic interactions with all the tested substrate carboxyl and amino groups, respectively. As these key residues are important in formation of salt bridges, it was hypothesized that mutations at Arg-395 and Glu-222 to non-polar and uncharged residues could result in inhibition or loss of binding or transport of l-lysine or other possible substrates. Therefore, the variants R395M and E222L were made and characterized.
On the other hand, it was also found by sequence alignment that Lys-158 of apo-ApcT was homologous to Lys-163 of STM2200. It has been shown that Lys-158 of apo-ApcT is located in a pocket that can be structurally aligned to the binding site Na2 in LeuT (37). It was hypothesized that Lys-163 could play a similar role in STM2200, so mutation at this position might abolish ion binding within the protein and hence inhibit its transport or binding activity. To test this hypothesis, K163L and K163A variants were also produced and purified to test the binding of l-lysine by using ITC to elucidate the role of this residue in STM2200.
Preparation of Site-directed Variants of STM2200 and ITC Binding Studies
STM2200 was subjected to site-directed mutagenesis to generate four different variants, i.e. R395M, E222L, K163L, and K163A, using a site-directed mutagenesis kit. The correctness of all variants was verified by sequencing. Subsequently, the plasmids for each variant were transformed into TOP10 cells and subjected to whole cell lysate expression screening analyzed by Western blots (Fig. 1B). After large scale production of each variant, membrane preparation was carried out. The membranes of all variants were solubilized using 1% (w/v) β-DDM as performed for wild type STM2200. Subsequently, all variants were purified using affinity chromatography on a Ni-NTA matrix using a batch method. In addition, the affinity-purified variants were subjected to preparative scale size exclusion chromatography to test their homogeneity and purity. All variants showed a similar purity level when compared with the wild type. However, their yield was lower. All variants showed a single narrow peak, characteristic of a homogeneous preparation, according to the gel filtration profile.
To characterize our docking predictions experimentally, the four purified variants were subjected to ITC tests for l-lysine binding as carried out for wild type. No affinity of l-lysine was found with any of the variants when titrated with 5 and 20 mm l-lysine as described above. These results confirm the prediction that Glu-222 and Arg-395 play an important role in lysine binding. In addition, K163A and K163L variants also did not show evidence for significant binding of l-lysine under the same conditions. However, an extremely weak affinity of the investigated variants with dissociation constants larger than 6.0 mm cannot be excluded unless the corresponding enthalpy changes were very small.
DISCUSSION
Properties of STM2200
Oligomerization Studies of STM2200
For this purpose, co-expression and co-purification studies were performed in the pBAD-CM vector in E. coli under conditions where the target protein was expressed twice with two different tags, one possessing a His tag and the other one having a Strep-I tag (38), followed by purification and tag-specific detection. The result indicated that the protein is present as monomer (Fig. 2B). This observation is in agreement with ApcT, which was also shown to be monomeric (1).
Thermoanalytical Studies with STM2200
The expected ability of STM2200 to bind l-lysine could be demonstrated by DSC and was characterized with a 9 °C increase of the transition temperature (see Table 1). This observation indicates that l-lysine binding stabilizes STM2200.
To characterize quantitatively the binding of l-lysine and some related ligands to STM2200, calorimetric titrations were carried out (Fig. 3) and evaluated under conditions where one ligand is bound per STM2200 monomer. The binding of l-lysine, characterized by a dissociation constant of ∼40 μm (Table 2), is of intermediate affinity and is driven by the comparatively large ΔS value of 27 cal mol−1 K−1. The TΔS value overcompensates for the small but positive enthalpy change of 2.0 kcal/mol. The observed binding affinity is within the reported ranges of ligand binding to APC transporters (19, 39, 40). Upon the much weaker binding of d-lysine, again an endothermic process could be observed. The corresponding dissociation constant is 1.8 mm, which is thus about 45 times larger than that of l-lysine. The ΔH value for d-lysine binding is markedly smaller than that of its chiral antipode. This result is consistent with the smaller signal intensity observed in the case of d-lysine titrations (Fig. 3C).
Under the experimental conditions suitable for l-lysine titrations, no heat changes of significant intensity were observed with related compounds such as l-ornithine, l-2,4-diaminobutyric acid, and l-alanine. Thus, no evidence for binding of these amino acids was observed, although an extremely weak binding of these compounds cannot be excluded.
Our results indicate that l-lysine binding to STM2200 is selective, representing a necessary condition for the expected transmembrane transport by this protein. Assuming a one-step binding equilibrium with a formation rate constant of 107–108 m−1 s−1, a dissociation rate in the millisecond time range would result. Such a property is consistent with an efficient membrane transport process under conditions where ligand dissociation is rate-limiting. A higher binding affinity would result in a slower and consequently less efficient dissociation step.
Structural Aspects and Mutagenesis of STM2200
Because there is no crystal structure of STM2200 available, a detailed molecular interpretation of the thermodynamic parameters related to l- and d-lysine binding (see Table 2) on the basis of known structural parameters is not possible at present. However, to obtain structural information concerning ligand binding, homology modeling based on the related transport protein ApcT was performed.
Homology Modeling and Docking of STM2200
The crystal structures of two transporters of the APC family including AdiC, an arginine-agmatine antiporter, and ApcT, a broad specificity amino acid transporter, were published recently (1, 19). ApcT is related (∼19% sequence identity) to STM2200 and was used to develop a homology model (Fig. 4B). The model, comprising 489 amino acid residues and 12 transmembrane helices, corresponds to the occluded, inward facing state of ApcT. It predicts a large, closed cavity in the approximate midpoint of the lipid bilayer that consists of different polar, aromatic, and aliphatic residues in various positions (Fig. 4A). This cavity is large enough to accommodate the substrates and hence is likely to represent the ligand binding site. The results of the docking study indicate that l-lysine can form ionic interactions with Arg-395 and Glu-222 residues of STM2200 that are considered to represent residues for ligand binding. Concerning STM2200, it was hypothesized that mutations at Arg-395 and Glu-222 to uncharged residues could result in the loss of l-lysine binding by STM2200. To test this hypothesis, the R395M and E222L variants were prepared and used for binding studies using titration calorimetry. These variants did not bind l-lysine as expected. These results are consistent with our hypothesis.
The charge-compensating residues Arg-395 and Glu-222 interact with the carboxylate and α-amino group of l- and d-lysine, respectively, leading to salt bridges stabilized by hydrogen bonds (Fig. 4C); the corresponding bond distances range between 1.7 and 2.4 Å. In their study of LysP (24), the authors already referred to the importance of conserved glutamate and arginine residues at various positions and predicted their role in binding of l-lysine in LysP of E. coli.
Besides the two key residues Arg-395 and Glu-222, the results of the docking study suggest that the binding of l-lysine to STM2200 is further stabilized by additional hydrogen bonds between the α- and ϵ-ammonium group of l-lysine and the carbonyl groups of Gly-388 and Gly-215, respectively (Fig. 4C). The hydrogen bond between Gly-215 and the indicated ϵ-ammonium group is missing in the case of d-lysine binding, which could be the reason for the weaker binding affinity of this chiral form. The docking study suggests bond distances between 2.0 and 2.6 Å.
In addition to l- and d-lysine, the analogues l-ornithine, l-2,4-diaminobutyric acid, and l-alanine were also investigated in the modeling and docking studies. Arg-395 and Glu-222 are involved in the binding of the two positively charged ligand analogues to STM2200. Otherwise, the binding behavior differed from that of l-lysine. Thus, it is not surprising that no ligand binding could be detected by titration calorimetry for the three studied analogues. This result, which is attributed to the mode of binding, is consistent with the results of the docking investigation.
Besides the residues mentioned above, homology modeling also focused on Lys-163 of STM2200; this residue was found to be homologous to Lys-158 of apo-ApcT on the basis of sequence alignment. In ApcT, Lys-158 is located in a pocket that can be structurally aligned to binding site Na2 in LeuT (1, 37). Na+ in LeuT as well as the protonated side chain of Lys-158 in ApcT is assumed to induce the formation of a conformation that is essential to the function as membrane transporter (1). Consequently, it was hypothesized that Lys-163 in its protonated form could play a similar role in STM2200 and that its substitution by a non-polar and uncharged amino acid could abolish l-lysine binding and probably also transport. This hypothesis was also tested experimentally by carrying out calorimetric l-lysine titrations with the STM2200 variants K163L and K163A. Because no evidence for l-lysine binding could be observed under our chosen conditions, the allosteric importance of Lys-163 for ligand binding could be proposed, which confirms the hypothesis mentioned above. This result is also in agreement with earlier published studies on the lysine-specific permease of E. coli, LysP (24), where similar predictions were made on the basis of a sequence alignment of conserved residues but had not been verified experimentally.
Thermodynamics of Ligand Binding and Structural Aspects
The suggested mode of binding between STM2200 and l- or d-lysine allows a qualitative interpretation of the resulting thermodynamic parameters (Table 2). The formation of two salt bridges stabilized by strong hydrogen bonds between the α-ammonium and carboxylate group of both chiral ligands with both oxygen atoms of the Glu-222 carboxylate residue and the guanidinium group of Arg-395 (Fig. 4C), respectively, is expected to lead to a marked negative enthalpy change. This feature is expected to enable enthalpy-driven high affinity ligand binding. Moreover, the formation of two additional, less strong hydrogen bonds between the main chain amide carbonyl groups of Gly-388 and Gly-215 and the α- and ϵ-ammonium groups of l-lysine (Fig. 4C), respectively, should even increase the negative enthalpy contribution, thus supporting high affinity ligand binding even more. The fact that the docking study did not provide evidence for hydrogen bond formation between Gly-215 and the ϵ-ammonium group of d-lysine does not alter our qualitative argumentation significantly. Our expectation outlined before, however, is not consistent with the experimentally observed positive enthalpy changes for l- and d-Lys binding (Table 2). We thus conclude that the expected negative enthalpy contribution must be overcompensated by a larger positive contribution, leading to the experimentally observed positive enthalpy change. Positive enthalpy contributions can result from the formation of hydrophobic interactions for example. However, the current modeling study did not suggest evidence for hydrophobic contacts upon binding. It is thus likely that another process must be involved in ligand binding that is responsible for the predicted large, positive enthalpy contribution.
As a prerequisite of l- or d-lysine binding, the solvation of the ligand itself as well as of the amino acid side chain and main chain carbonyl groups interacting with both ligands must be changed markedly (see Fig. 4C). The necessary desolvation can only be achieved by the energy requiring breaking of numerous hydrogen bonds between water and involved groups of ligand and protein. This would lead to a positive enthalpy contribution. If this positive contribution exceeds the originally expected negative contribution, a positive enthalpy change will result, which is in agreement with the experimental data (Table 2). The release of numerous bound water molecules upon the described, at least partial desolvation during ligand binding should also lead to a marked positive entropy change, which is also consistent with the experimental data (Table 2). A conformational change such as a contraction of the binding cavity of the protein upon ligand binding could also lead to a marked positive enthalpy change. However, this possibility is not considered here because the cavity conformation was kept fixed for our docking studies. In summary, the partial desolvation of ligand and binding cavity induced by the interaction between ligands and the preorganized binding site is assumed to represent the entropy-driven driving force for l- and d-lysine binding.
The experimental data show that binding of d-lysine is almost 45 times weaker than that of l-lysine. According to the described docking study, we assume that the ϵ-ammonium group of d-lysine in contrast to that of l-lysine is not interacting with the amide carbonyl group of Gly-215 (Fig. 4C).Therefore, the solvation of these two groups is expected to remain essentially unchanged upon binding, leading to a diminished positive enthalpy and entropy contribution. The free energy change, responsible for a 45 times weaker binding of d-lysine, corresponds to RT × ln 45 where 45 is the ratio of the association constants and is about 2.2 kcal/mol. This value characterizing the energetic difference between l- and d-lysine binding corresponds to almost one-half of the hydrogen bond energy between an uncharged donor and acceptor in the absence of solvent. The weakening of the affinity for d- compared with l-lysine could thus be due to the loss of not more than one hydrogen bond as expected by the reported docking study. Therefore, the molecular prediction of the chiral selectivity between l- and d-lysine binding resulting from the modeling and docking study (illustrated schematically in Fig. 4C) is qualitatively consistent with the obtained thermodynamic data (Table 2).
In conclusion, l-lysine is bound selectively to STM2200, characterized by a dissociation constant of 40 μm. The binding is entropy-driven and mainly attributed to a partial desolvation of the ligand and binding cavity. The combination of a biophysical study with site-directed mutagenesis derived from an extended homology model and docking study led to the proposal that Glu-222, Arg-395, and Gly-388 are the key residues that directly interact with l- and d-lysine. Gly-215 appears to interact only with l-lysine and is suggested to be relevant for the 45 times stronger preference of the l- over the d-form. In contrast to this, Lys-163 in its protonated form is thought to be necessary for adopting a suitable conformation enabling l- and d-lysine binding.
Acknowledgments
We thank Cornelia Münke for excellent technical assistance, Helga Husmann for helping in graphical representation of a figure, and Prof. Friedemann Schneider for helpful discussions.
This work was supported by the European Membrane Protein Consortium (EMeP), the Cluster of Excellence Frankfurt (“Macromolecular complexes”), Deutsche Forschungsgemeinschaft Grant SFB807 (Transport and Communication across biological membranes), and Max-Planck-Gesellschaft.
- APC
- amino acid-polyamine-organocation
- BCA
- bicinchoninic acid
- β-DDM
- n-dodecyl-β-d-maltoside
- DSC
- differential scanning calorimetry
- ITC
- isothermal titration calorimetry
- LysP
- lysine-specific permease
- Tm
- transition temperature
- TMH
- transmembrane helix
- Ni-NTA
- nickel-nitrilotriacetic acid.
REFERENCES
- 1. Shaffer P. L., Goehring A., Shankaranarayanan A., Gouaux E. (2009) Structure and mechanism of a Na+-independent amino acid transporter. Science 325, 1010–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Saier M. H. (2000) Families of transmembrane transporters selective for amino acids and their derivatives. Microbiology 146, 1775–1795 [DOI] [PubMed] [Google Scholar]
- 3. Closs E. I., Albritton L. M., Kim J. W., Cunningham J. M. (1993) Identification of a low affinity, high capacity transporter of cationic amino acids in mouse liver. J. Biol. Chem. 268, 7538–7544 [PubMed] [Google Scholar]
- 4. Sophianopoulou V., Diallinas G. (1995) Amino acid transporters of lower eukaryotes: regulation, structure and topogenesis. FEMS Microbiol. Rev. 16, 53–75 [DOI] [PubMed] [Google Scholar]
- 5. Kashiwagi K., Shibuya S., Tomitori H., Kuraishi A., Igarashi K. (1997) Excretion and uptake of putrescine by the PotE protein in Escherichia coli. J. Biol. Chem. 272, 6318–6323 [DOI] [PubMed] [Google Scholar]
- 6. Cosgriff A. J., Pittard A. J. (1997) A topological model for the general aromatic amino acid permease, AroP, of Escherichia coli. J. Bacteriol. 179, 3317–3323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jack D. L., Paulsen I. T., Saier M. H. (2000) The amino acid/polyamine/organocation (APC) superfamily of transporters specific for amino acids, polyamines and organocations. Microbiology 146, 1797–1814 [DOI] [PubMed] [Google Scholar]
- 8. Forsberg H., Ljungdahl P. O. (2001) Sensors of extracellular nutrients in Saccharomyces cerevisiae. Curr. Genet. 40, 91–109 [DOI] [PubMed] [Google Scholar]
- 9. Sato H., Shiiya A., Kimata M., Maebara K., Tamba M., Sakakura Y., Makino N., Sugiyama F., Yagami K., Moriguchi T., Takahashi S., Bannai S. (2005) Redox imbalance in cystine/glutamate transporter-deficient mice. J. Biol. Chem. 280, 37423–37429 [DOI] [PubMed] [Google Scholar]
- 10. Baker D. A., McFarland K., Lake R. W., Shen H., Tang X. C., Toda S., Kalivas P. W. (2003) Neuroadaptations in cystine-glutamate exchange underlie cocaine relapse. Nat. Neurosci. 6, 743–749 [DOI] [PubMed] [Google Scholar]
- 11. Yeramian A., Martin L., Serrat N., Arpa L., Soler C., Bertran J., McLeod C., Palacín M., Modolell M., Lloberas J., Celada A. (2006) Arginine transport via cationic amino acid transporter 2 plays a critical regulatory role in classical or alternative activation of macrophages. J. Immunol. 176, 5918–5924 [DOI] [PubMed] [Google Scholar]
- 12. Fuchs B. C., Bode B. P. (2005) Amino acid transporters ASCT2 and LAT1 in cancer: partners in crime? Semin. Cancer Biol. 15, 254–266 [DOI] [PubMed] [Google Scholar]
- 13. Calonge M. J., Gasparini P., Chillarón J., Chillón M., Gallucci M., Rousaud F., Zelante L., Testar X., Dallapiccola B., Di Silverio F., Barceló P., Estivill X., Zorzano A., Nunes V., Palacín M. (1994) Cystinuria caused by mutations in rBAT, a gene involved in the transport of cystine. Nat. Genet. 6, 420–425 [DOI] [PubMed] [Google Scholar]
- 14. Torrents D., Mykkänen J., Pineda M., Feliubadaló L., Estévez R., de Cid R., Sanjurjo P., Zorzano A., Nunes V., Huoponen K., Reinikainen A., Simell O., Savontaus M. L., Aula P., Palacín M. (1999) Identification of SLC7A7, encoding y+LAT-1, as the lysinuric protein intolerance gene. Nat. Genet. 21, 293–296 [DOI] [PubMed] [Google Scholar]
- 15. Borsani G., Bassi M. T., Sperandeo M. P., De Grandi A., Buoninconti A., Riboni M., Manzoni M., Incerti B., Pepe A., Andria G., Ballabio A., Sebastio G. (1999) SLC7A7, encoding a putative permease-related protein, is mutated in patients with lysinuric protein intolerance. Nat. Genet. 21, 297–301 [DOI] [PubMed] [Google Scholar]
- 16. Kim Y. M., Ogawa W., Tamai E., Kuroda T., Mizushima T., Tsuchiya T. (2002) Purification, reconstitution, and characterization of Na+/serine symporter, SstT, of Escherichia coli. J. Biochem. 132, 71–76 [DOI] [PubMed] [Google Scholar]
- 17. Cosgriff A. J., Brasier G., Pi J., Dogovski C., Sarsero J. P., Pittard A. J. (2000) A study of AroP-PheP chimeric proteins and identification of a residue involved in tryptophan transport. J. Bacteriol. 182, 2207–2217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fang Y., Jayaram H., Shane T., Kolmakova-Partensky L., Wu F., Williams C., Xiong Y., Miller C. (2009) Structure of a prokaryotic virtual proton pump at 3.2 Å resolution. Nature 460, 1040–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gao X., Lu F., Zhou L., Dang S., Sun L., Li X., Wang J., Shi Y. (2009) Structure and mechanism of an amino acid antiporter. Science 324, 1565–1568 [DOI] [PubMed] [Google Scholar]
- 20. Kowalczyk L., Ratera M., Paladino A., Bartoccioni P., Errasti-Murugarren E., Valencia E., Portella G., Bial S., Zorzano A., Fita I., Orozco M., Carpena X., Vázquez-Ibar J. L., Palacín M. (2011) Molecular basis of substrate-induced permeation by an amino acid antiporter. Proc. Natl. Acad. Sci. U.S.A. 108, 3935–3940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fukasawa Y., Segawa H., Kim J. Y., Chairoungdua A., Kim D. K., Matsuo H., Cha S. H., Endou H., Kanai Y. (2000) Identification and characterization of a Na+-independent neutral amino acid transporter that associates with the 4F2 heavy chain and exhibits substrate selectivity for small neutral d- and l-amino acids. J. Biol. Chem. 275, 9690–9698 [DOI] [PubMed] [Google Scholar]
- 22. Yernool D., Boudker O., Jin Y., Gouaux E. (2004) Structure of a glutamate transporter homologue from Pyrococcus horikoshii. Nature 431, 811–818 [DOI] [PubMed] [Google Scholar]
- 23. Raunser S., Appel M., Ganea C., Geldmacher-Kaufer U., Fendler K., Kühlbrandt W. (2006) Structure and function of prokaryotic glutamate transporters from Escherichia coli and Pyrococcus horikoshii. Biochemistry 45, 12796–12805 [DOI] [PubMed] [Google Scholar]
- 24. Steffes C., Ellis J., Wu J., Rosen B. P. (1992) The lysP gene encodes the lysine-specific permease. J. Bacteriol. 174, 3242–3249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ruiz J., Haneburger I., Jung K. (2011) Identification of ArgP and Lrp as transcriptional regulators of lysP, the gene encoding the specific lysine permease of Escherichia coli. J. Bacteriol. 193, 2536–2548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schmidt T. G., Skerra A. (1994) One-step affinity purification of bacterially produced proteins by means of the “Strep tag” and immobilized recombinant core streptavidin. J. Chromatogr. A 676, 337–345 [DOI] [PubMed] [Google Scholar]
- 27. Davis I. W., Leaver-Fay A., Chen V. B., Block J. N., Kapral G. J., Wang X., Murray L. W., Arendall W. B., 3rd, Snoeyink J., Richardson J. S., Richardson D. C. (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wiederstein M., Sippl M. J. (2007) ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 35, W407–W410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jorgensen W. L., Maxwell D. S., Tirado-Rivers J. (1996) Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 118, 11225–11236 [Google Scholar]
- 30. Jorgensen W. L., Tirado-Rivers J. (1988) The OPLS potential functions for proteins. Energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 110, 1657–1666 [DOI] [PubMed] [Google Scholar]
- 31. Still W. C., Tempczyk A., Hawley R., Hendrickson T. (1990) Semianalytical treatments of solvation for molecular mechanics and dynamics. J. Am. Chem. Soc. 112, 6127–6129 [Google Scholar]
- 32. Bruylants G., Wouters J., Michaux C. (2005) Differential scanning calorimetry in life science: thermodynamics, stability, molecular recognition and application in drug design. Curr. Med. Chem. 12, 2011–2020 [DOI] [PubMed] [Google Scholar]
- 33. Privalov P. L., Dragan A. I. (2007) Microcalorimetry of biological macromolecules. Biophys. Chem. 126, 16–24 [DOI] [PubMed] [Google Scholar]
- 34. Weyand S., Shimamura T., Yajima S., Suzuki S., Mirza O., Krusong K., Carpenter E. P., Rutherford N. G., Hadden J. M., O'Reilly J., Ma P., Saidijam M., Patching S. G., Hope R. J., Norbertczak H. T., Roach P. C., Iwata S., Henderson P. J., Cameron A. D. (2008) Structure and molecular mechanism of a nucleobase-cation-symport-1 family transporter. Science 322, 709–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ressl S., Terwisscha van Scheltinga A. C., Vonrhein C., Ott V., Ziegler C. (2009) Molecular basis of transport and regulation in the Na+/betaine symporter BetP. Nature 458, 47–52 [DOI] [PubMed] [Google Scholar]
- 36. Abagyan R., Totrov M., Kuznetsov D. (1994) ICM—a new method for protein modeling and design: applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 15, 488–506 [Google Scholar]
- 37. Yamashita A., Singh S. K., Kawate T., Jin Y., Gouaux E. (2005) Crystal structure of a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporters. Nature 437, 215–223 [DOI] [PubMed] [Google Scholar]
- 38. Goswami D., Kaur J., Surade S., Grell E., Michel H. (2012) Heterologous production and functional and thermodynamic characterization of cation diffusion facilitator (CDF) transporters of mesophilic and hyperthermophilic origin. Biol. Chem. 393, 617–629 [DOI] [PubMed] [Google Scholar]
- 39. Fang Y., Kolmakova-Partensky L., Miller C. (2007) A bacterial arginine-agmatine exchange transporter involved in extreme acid resistance. J. Biol. Chem. 282, 176–182 [DOI] [PubMed] [Google Scholar]
- 40. Vangelatos I., Vlachakis D., Sophianopoulou V., Diallinas G. (2009) Modelling and mutational evidence identify the substrate binding site and functional elements in APC amino acid transporters. Mol. Membr. Biol. 26, 356–370 [DOI] [PubMed] [Google Scholar]