

Background: TGF-β1 activates RhoA and nuclear factor-κB (NF-κB), but the activation mechanism was not clearly elucidated.
Results: IKKγ disrupts RhoA-Rho guanine nucleotide dissociation inhibitor (RhoGDI) complex, facilitating GTP binding to RhoA, resulting in IKKβ phosphorylation by ROCK.
Conclusion: IKKγ facilitates RhoA activation, which in turn activates NF-κB.
Significance: We found the new mechanism of IKKγ to activate RhoA and NF-κB by TGF-β1.
Keywords: Cell Migration, Inflammation, NF-kB Transcription Factor, Rhoa, Transforming Growth Factor Beta (TGFbeta)
Abstract
Transforming growth factor (TGF)-β1 plays several roles in a variety of cellular functions. TGF-β1 transmits its signal through Smad transcription factor-dependent and -independent pathways. It was reported that TGF-β1 activates NF-κB and RhoA, and RhoA activates NF-κB in several kinds of cells in a Smad-independent pathway. However, the activation molecular mechanism of NF-κB by RhoA upon TGF-β1 has not been clearly elucidated. We observed that RhoA-GTP level was increased by TGF-β1 in RAW264.7 cells. RhoA-GDP and RhoGDI were bound to N- and C-terminal domains of IKKγ, respectively. Purified IKKγ facilitated GTP binding to RhoA complexed with RhoGDI. Furthermore, Dbs, a guanine nucletotide exchange factor of RhoA much more enhanced GTP binding to RhoA complexed with RhoGDI in the presence of IKKγ. Indeed, si-IKKγ abolished RhoA activation in response to TGF-β1 in cells. However, TGF-β1 stimulated the release of RhoA-GTP from IKKγ and Rho-associated kinase (ROCK), an active RhoA effector protein, directly phosphorylated IKKβ in vitro, whereas TGF-β1-activated kinase 1 activated RhoA upon TGF-β1 stimulation. Taken together, our data indicate that IKKγ facilitates RhoA activation via a guanine nucletotide exchange factor, which in turn activates ROCK to phosphorylate IKKβ, leading to NF-κB activation that induced the chemokine expression and cell migration upon TGF-β1.
Introduction
TGF-β is a signal protein that regulates many cellular functions, including cell proliferation, differentiation, migration, and survival as well as development, carcinogenesis, fibrosis, wound healing, and the immune response (1). At the cell surface, the functional complex of the TGF-β family of receptors is composed of two “type I” and two “type II” transmembrane serine/threonine kinase receptors (2). In general, TGF-β signaling is classified into two categories: Smad-dependent and -independent pathways (2). In the Smad-dependent pathway, the ligand binds to the type I and II receptor complex at the cell surface and induces phosphorylation of the type II receptor. After transphosphorylation by the type II receptor, the activated type I receptor then phosphorylates R-Smads, which, in turn, form a complex with the co-Smad, Smad4. The resulting Smads complex is translocated into the nucleus where it regulates the transcription of its target genes (2). In the Smad-independent pathway, TGF-β activates a variety of kinases, including ERK (extracellular signal-regulated kinases), JNK (c-Jun N-terminal kinase), p38 MAPK (mitogen-activated protein kinase), and PI3K (phosphoinositide 3-kinase) (3).
Ras-related small GTPase Rho family plays several roles in the regulation of cellular functions such as actin filament formation, migration, cell cycling, and transcription. Similar to Ras, Rho GTPases behave as molecular switches, alternating between the active GTP-bound and inactive GDP-bound forms; these transitions are achieved by guanine nucleotide exchange factors (GEFs)2 and GTPase-activating proteins, respectively. In addition, inactive GDP-bound Rho proteins form a complex with Rho guanine nucleotide dissociation inhibitor (RhoGDI) (4). RhoA-GDP complexed with RhoGDI is not directly activated by GEFs in a cell-free system (5, 6). This suggests that another factor, referred to as GDI displacement factor (GDF) to disrupt RhoA-RhoGDI complex, is required for the activation of RhoA by GEFs. Signals from active Rho GTPases transmit to a variety of effector proteins, including Rho-associated coiled-coil forming serine/threonine kinase (ROCK), which is activated by RhoA, and p21-activated kinase, which is activated by Cdc42/Rac (7).
Nuclear factor-κB (NF-κB) is a transcription factor that controls the expression of specific target genes, including cytokines, chemokines, cell adhesion molecules, and inducible enzymes to regulate inflammation, cancer, apoptosis, and several other physiological phenomena (8). The NF-κB family consists of p65 (RelA), RelB, c-Rel, NF-κB1 (p105, precursor of p50), and NF-κB2 (p100, precursor of p52) forming hetero- or homodimers. Two principal pathways of NF-κB activation have been elucidated: the classical pathway and the alternative pathway. In the classical pathway, the IκB kinase (IKK) complex consists of two catalytic subunits, IKKα (IKK1) and IKKβ (IKK2), and a regulatory subunit, IKKγ (also referred to as NEMO (NF-κB essential modifier). When IKKβ is activated by phosphorylation, it can then phosphorylate inhibitor of NF-κB (IκB), which is bound to NF-κB. When the phosphorylated IκB is ubiquitinated and degraded, NF-κB dimer such as p65/p50, which is released from IκB, is translocated into the nucleus, where it binds to and activates the transcription of specific target genes. In the alternative pathway, the activation of IKKα homodimer by phosphorylation induces p100 processing and the nuclear translocation of the RelB/p52 dimer (9). Intriguingly, ubiquitination, as well as phosphorylation, is involved in the activation of IKK (10).
It is noteworthy that Rho subfamily small GTPases are implicated in NF-κB activation (11–15). In addition, TGF-β1 can rapidly activate Rho subfamily GTPases, including RhoA, Cdc42, and Rac1, depending on the cell lines (3, 16–18). Furthermore, TGF-β1 induces the activation of NF-κB signaling (3, 19, 20) leading to cell motility (16, 21). Although the relevance of TGF-β1, Rho GTPases, and NF-κB has been reported in a variety of cells, the molecular mechanism of the activation of NF-κB by TGF-β1 via RhoA GTPase activation in macrophages has not been well elucidated (17).
Therefore, we attempted to discover the underlying molecular mechanism how RhoA regulates NF-κB or vice versa upon TGF-β1. We found that IKK activates RhoA; IKKγ binds to the RhoA-RhoGDI complex, facilitating the activation of RhoA, likely by disrupting the RhoA-RhoGDI complex. Thereafter, active RhoA-GTP and its downstream component ROCK phosphorylates IKKβ, which in turn phosphorylates IκB and p65, thereby leading to NF-κB activation.
EXPERIMENTAL PROCEDURES
Materials
BSA, MG132, leptomycin B, isopropyl-β-d-thiogalactoside, Triton X-100, PMSF, the anti-actin antibody, 2′/3′-O-(N-methylanthraniloyl)guanosine-5′-(γ-thio) triphosphate triethylammonium salt (Mant-GTP), and CHAPS were purchased from Sigma. TGF-β1 expressed in CHO cells and active ROCK (amino acids 17–535) were purchased from R&D Systems (Minneapolis, MN). Y27632 and HA1077, inhibitors of ROCK, were purchased from Calbiochem (La Jolla, CA). FBS was purchased from Invitrogen. Protein A-agarose beads were purchased from Pierce. DMEM-F12, penicillin, and streptomycin were purchased from Lonza (Walkersville, MD). Glutathione-Sepharose 4B beads were from Amersham Biosciences. Glutathione was purchased from Elpis Biotech (Daejoen, Korea). The anti-phospho-p65 (Ser-536), anti-phospho-IKKα/β (Ser-180/Ser-181), anti-phospho-Smad3 (Ser-423/425), and phospho-TGF-β-activated kinase 1 (TAK1) antibodies were purchased from Cell Signaling Technology (Beverly, MA), and the anti-IKKγ antibody was purchased from BD Biosciences. The anti-IKKα and anti-IKKβ antibodies were purchased from Upstate Biotechnology (Waltham, MA). The anti-RhoA, anti-p65, anti-IκB, anti-Smad3, anti-phospho-IκBα (Ser-32/36), anti-phospho-myosin light chain phosphatase and anti-TAK1 antibodies, and GST-IκB were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The TAK inhibitor 5z-7-oxozeaenol was purchased from Tocris Bioscience (Ellisville, MO). BMS345541, an IKKβ inhibitor, was purchased from Calbiochem. [35S]GTPγ and [γ-32P]ATP were purchased from the Institute of Isotopes Co., Ltd. (Budapest, Hungary). Recombinant Tat-C3 toxin was purified from Escherichia coli (17). GST-Rho binding domain (RBD) of rhotekin (GST-rhotekin-RBD) beads were prepared from cultured E. coli or obtained from Pierce. Dbs from the RhoGEF assay kit was purchased from Cytoskeleton (Denver, CO). Recombinant GST-His-IKKβ expressed in Sf9 cells was purchased from Creative Biomat (Shirley, NY). The RhoA-pCDNA3.1 and RhoGDI-pCDNA3.1 constructs were purchased from the Missouri S&T cDNA Resource Center. The GST-RhoA and GST-RhoGDI constructs were prepared by subcloning the appropriate genes into a pGEX4T-1 vector using the EcoRI/XhoI sites. The pGEX4T-1-IKKγ and pET-IKKγ constructs were provided by Dr. J. Ashwell (National Cancer Institute), and RelA (p65) cFLAG-pCDNA3 was provided by Dr. S. Smale (University of California, Los Angeles) through the Addgene plasmid repository. GST-p65 was constructed by subcloning the appropriate genes into pGEX4T-1 using the EcoRI/XhoI sites.
Cell Culture, Fluorescence Microscopy, and Confocal Microscopy
The RAW264.7 (mouse macrophage) cell line was cultured (22); if necessary, TGF-β1 (5 ng/ml) was treated. HeLa cells were cultured in DMEM containing 10% FBS, 100 units/ml streptomycin, and 100 units/ml penicillin at 37 °C in 5% CO2. The cells were fixed with 4% paraformaldehyde for 10 min, neutralized with 20 mm glycine for 10 min, and then washed three times with PBS containing 0.1% Triton X-100. The samples were incubated with primary antibody (1:100) overnight at 4 °C, washed, and then incubated with the appropriate fluorescent dye-conjugated secondary antibody for 2 h at 24 °C. DAPI (1 μg/ml) was added 10 min before washing. Fluorescence was observed by fluorescence microscopy (Axiovert 200; Carl Zeiss; Göttingen, Germany) and confocal microscopy (LSM 780NLO; Carl Zeiss). RhoA was identified using an anti-RhoA antibody, which is recognized by an Alexa Fluor 488-conjugated secondary antibody (green), and IKKγ was identified by an anti-IKKγ antibody, which is recognized by an Alexa Flour 568-conjugated secondary antibody (red). Nuclei were identified with DAPI staining (blue).
Assay of Cell Migration
Migration of RAW264.7 cells was determined using a Transwell permeable support kit with polycarbonate filter (22).
Luciferase Reporter Assays
RAW264.7 cells were grown to 80% confluence in six-well plates and then transiently transfected with the pNF-κB-Luc cis reporter plasmid (Stratagene; Santa Clara, CA) by incubating with Lipofectamine 2000 (Invitrogen) or Attractene (Qiagen; Hilden, Germany) for 3 h according to the manufacturer's instructions. To calibrate the variation in transfection efficiency, the cells were co-transfected with 1 μg of pCS2+-β-galactosidase plasmids, an expression plasmid for the E. coli galactosidase gene. The transfected cells were incubated in serum-free medium for 24 h, rinsed with PBS, lysed in 1× reporter lysis buffer (Promega; Madison, WI), and the cell debris was removed by centrifugation. The relative luciferase activity of the supernatant was measured using a luminometer according to the manufacturer's instructions (Lumat LB 9057; EG & G Bertold).
Loading of GDP and GTPγS onto GTP-binding Proteins in Vitro
Cell lysates (1 μg/μl protein in 500 μl) were incubated with 10 mm EDTA, pH 8.0. Next, GTPγS or GDP was added to the cell lysates to a final concentration of 0.1 or 1 mm, respectively, and incubated at 30 °C for 30 min with constant agitation. The reaction was terminated by thoroughly mixing with MgCl2 at a final concentration of 60 mm on ice. To determine the level of RhoA-GTP, GST-rhotekin-RBD beads (23) and an EZ-Detect Rho activation kit containing GST-RBD (Pierce) were used (24).
Assay of GTP Binding to RhoA
RhoA in the absence or presence of RhoGDI or the RhoA-RhoGDI complex in buffer (10 mm HEPES, pH 7.4, 50 mm NaCl, 1 or 5 mm MgCl2, 2 or 1 mm EDTA, respectively, 1 mm DTT, 0.1% CHAPS) was incubated with [35S]GTPγ at 24 °C for 30 min in the presence of IKKγ, Dbs (a GEF of RhoA), or CHAPS. The reaction was terminated by adding ice-cold stop buffer (10 mm HEPES, pH 7.5, 50 mm NaCl, 25 mm MgCl2), filtered with a BA85 membrane (0.45 μm; Schleicher & Schuell; Dassel, Germany), and washed twice with 2 ml of stop buffer. The radioactivity on the membrane, which corresponded to [35S]GTPγ bound with RhoA, was measured with a liquid scintillation counter (Beckman LS5801) (25). Mant-GTP was incubated with RhoA in buffer (10 mm HEPES, pH 7.4, 50 mm NaCl, 1 mm MgCl2, 2 mm EDTA, 1 mm DTT, 0.1% CHAPS). Fluorescence was measured at an emission wavelength of 440 nm and an excitation wavelength of 360 nm using a fluorescence spectrophotometer (Spectra M2; Molecular Devices; Sunnyvale, CA).
Construction and Transient Transfection of a Small Hairpin RNA Targeting RhoA
A shRNA-expressing sequence for targeting RhoA mRNA was cloned into the pSUPER RNAi system (Oligoengine; Seattle, WA) (22, 26).
Immunoprecipitation
Immunoprecipitation was performed according to the previous report (17). IKKβ and IKKγ were immunoprecipitated with an anti-IKKβ antibody and an anti-IKKγ antibody, respectively.
Construction of Domains of IKKγ and Protein-protein Interaction
Human IKKγ (Addgene plasmid 11965) was provided from Addgene. Truncation domains of IKKγ were generated by PCR. The GST-IKKγ domain constructs was made by cloning a PCR-generated fragment encompassing the region from amino acids 1–43, 44–111, 1–100, 101–200, 201–305, 351–419, 101–419, and 1–419 into pGEX-4T1. EcoRI and XhoI sites were introduced into the 5′ and 3′ ends, respectively. The GST-IKKγ fusion protein was expressed in E. coli BL-21. To determine protein-protein binding, GST-IKKγ beads and the proteins were incubated in a buffer (50 mm Tris-HCl, pH 7.5, 1× PBS, 10% glycerol, and 1 μg/ml each aprotinin, leupeptin, and pepstatin A and 1 mm PMSF) for 2 h at 4 °C. After washing the beads, bound proteins were identified with Western blotting.
Purification of RhoA, RhoGDI, IKKγ, and p65
To generate the GST-RhoA, GST-RhoGDI, GST-IKKγ, and GST-p65 fusion proteins, E. coli were transformed with pGEX-4T-1 plasmids (Invitrogen) containing the RhoA, RhoGDI, IKKγ, and p65 genes, respectively. The GST tag was removed by cleavage with thrombin (27). In some experiments, His-IKKγ was purified using Ni2+-nitrilotriacetic acid beads (Novagen; Darmstadt, Germany). RhoA expressed in Sf9 cells harboring baculovirus containing cDNA encoding RhoA was purified (27). RhoA from the cytosol and membrane were noted as C-RhoA and M-RhoA, respectively. The RhoA-RhoGDI complex was prepared by mixing and incubating RhoA and RhoGDI in buffer (10 mm HEPES, pH 7.5, 50 mm NaCl, 0.1 mm EDTA, 5 mm MgCl2, 5% glycerol, 1 mm DTT, 1 mm GDP, and protease inhibitors) for 1 h at 24 °C. To remove the uncomplexed proteins (RhoA is ∼22 kDa; RhoGDI is ∼26 kDa) from RhoA-RhoGDI, which has an estimated molecular mass of 48 kDa, the solution was centrifuged using an ultrafiltration kit (Amicon Ultrafilter; cut-off size, 30 kDa). The concentrated solution was washed three times by adding 0.5 ml of dialysis buffer (10 mm HEPES, pH 7.5, 50 mm NaCl, 0.1 mm EDTA, 5 mm MgCl2, 2.5% glycerol, 1 mm DTT, and protease inhibitors). Finally, the protein complex was aliquoted and stored at −70 °C until use.
Proximity Ligation Assay
To detect the interaction between RhoA and IKK, we utilized the DuoLink in situ proximity ligation assay (Olink Bioscience; Uppsala, Sweden) according to the manufacturer's protocol.
In Vitro Kinase Assay of ROCK
Cells were lysed in a 50-μl lysis buffer (50 mm Tris-Cl, pH 7.5, 50 mm glycerophosphate, 150 mm NaCl, 10% glycerol, 1% Tween 20, 1 mm each of DTT, PMSF, NaF, and NaVO4, 1 μg/ml each of leupeptin, aprotinin, and pepstatin A) containing 10 mm MgCl2 and 25 μm ATP. To determine direct phosphorylation of IKKβ by ROCK, 100 ng of active ROCK1 (amino acids, aa 17–535) and 100 ng of recombinant IKKβ were mixed in the presence or absence of cell lysates (50 μg) and incubated for 30 min at 30 °C in 40 μl of a kinase assay buffer (10 mm HEPES, pH 7.5, 50 mm glycerophosphate, 50 mm NaCl, 10 mm MgCl2, 10 mm MnCl2, 1 mm DTT, 30 μm ATP), and samples were incubated for 30 min at 30 °C. Samples were analyzed by Western blot using IKKα (Ser-176)/IKKβ (Ser-177) and anti-phospho-IKKα (Ser-180)/IKKβ (Ser-181) antibodies.
Measurement of MIP-1α
Macrophage inflammatory protein (MIP)-1α secreted from the RAW264.7 cells in response to TGF-β1 was quantitatively determined using ELISA (R&D Systems) according to the manufacturer's instructions. RAW264.7 cells were incubated with TGF-β1 for various time periods. Reverse transcription PCR and real-time PCR for MIP-1α mRNA expression was performed according to the previous report (22).
Statistical Analysis
Data are presented as the means ± S.E. of at least three independent experiments. The Student's t test was used to compare groups using the GraphPad Prism program (San Diego, CA).
RESULTS
RhoA Interacts with the IKK Complex
TGF-β1 increased RhoA-GTP levels in a 30–60-min treatment and then decreased (Fig. 1A), which is accordance with the previous results (17). Considering the relevance between RhoA and NF-κB, we found that the amino acid sequence of IKKγ is partially identical to RhoA effector proteins, including ROCK1, protein kinase N, rhophilin, and rhotekin although the homologous regions of IKKγ are not identical with the known Rho-binding domains of the effector proteins (supplemental Fig. S1). Therefore, we examined the possible binding of RhoA to IKKγ. Immunoprecipitation of IKKγ resulted in the co-precipitation of RhoA in a resting state, whereas TGF-β1 reduced the co-precipitation of RhoA and IKKγ (Fig. 1B). In addition, RhoA was co-immunoprecipitated with IKKα/β without TGF-β1, but TGF-β1 also reduced the amount of RhoA that was co-immunoprecipitated with IKKα/β (Fig. 1B). Whereas co-immunoprecipitation of RhoA with IKKγ was reduced in 1 h treatment of TGF-β1, their interaction was recovered after 24 h (Fig. 1C). This suggests that RhoA-GDP instead of RhoA-GTP is preferentially bound to IKKγ.
FIGURE 1.

RhoA interacts with the IKK complex. A, RAW264.7 cells were incubated with 5 ng/ml TGF-β1 at 37 °C, and GTP-RhoA levels were determined. B, the cells were treated TGF-β1 for 3 h. IKKα/IKKβ and IKKγ were immunoprecipitated with an anti-IKKγ antibody for 2 h at 4 °C, and then their co-precipitation with RhoA was determined by Western blot. C, control; T, TGF-β1 treatment. C, RAW264.7 cells were incubated with TGF-β1, and IKKγ was immunoprecipitated. The bound RhoA were analyzed by Western blot. Intensity of RhoA was quantified by densitometry (lower panel). D, RAW264.7 cells were treated with TGF-β1 for 1 h, and the interaction between RhoA and IKK was visualized in situ using a proximity ligation assay (PLA). The results are given as the means ± S.E. of three independent experiments (*, p < 0.05; **, p < 0.01; ***, p < 0.001). E, HeLa cells were treated with TGF-β1 for 1 h, and RhoA (green), IKKγ (red), and nuclei (blue) were visualized. Arrows indicate RhoA localization in the membrane. F, GST-IKKγ (1 μg of protein)-Sepharose 4B beads were incubated with 0.5–4 μg of purified M-RhoA for 2 h at 4 °C, and RhoA bound to GST-IKKγ was detected by Western blot. The amount of RhoA bound to IKKγ was calculated from the control standard curve. GST-IKKγ was detected with Ponceau S staining. CON, control; IB, immunoblot.
In addition, the proximity ligation assay, which evaluates the interaction of two proteins in situ showed an interaction between RhoA and IKKγ in a resting state, but TGF-β1 markedly reduced the interaction between RhoA and IKKγ (Fig. 1D). Consistently, confocal microscopy showed the co-localization of RhoA and IKKγ in HeLa cells. However, TGF-β1 reduced the co-localization of RhoA and IKKγ; RhoA was instead translocated to the plasma membrane (Fig. 1E). Notably, RhoA was observed in both the cytosol and nucleus of HeLa cells (Fig. 1E). Furthermore, purified RhoA was bound with purified recombinant GST-IKKγ in a concentration-dependent manner (Fig. 1F), suggesting that RhoA directly interacts with IKKγ.
RhoA-GDP and RhoGDI Interact with IKKγ
To investigate whether the GDP- or GTP-bound state of RhoA affects its interaction with IKKγ, cell lysates were preincubated with GDP or GTPγS, and immunoprecipitation was performed. RhoA underwent co-immunoprecipitation with IKKγ in the presence of GDP, but the co-immunoprecipitation was markedly reduced in the presence of GTPγS (Fig. 2A). Consistently, RhoA purified from the membranous (M-RhoA) and cytosolic (C-RhoA) fractions of Sf9 insect cells directly bound to GST-IKKγ in its GDP-bound state, whereas RhoA-GTPγS rarely interacted with GST-IKKγ (Fig. 2B). The recombinant RhoA-GDP purified from E. coli was able to bind to GST-IKKγ, instead RhoA-GTPγS rarely bound to IKKγ (Fig. 2C), suggesting that prenyl group of RhoA is not essential for the binding to IKKγ.
FIGURE 2.

RhoA and RhoGDI interact with IKKγ. A, RAW264.7 cell lysates were preloaded with 0.1 mm GTPγS or 1 mm GDP for 30 min. The lysates were subjected to immunoprecipitation with an anti-IKKγ antibody and analyzed by Western blot with the anti-RhoA and anti-IKKγ antibodies. B, RhoA proteins purified from the membrane (M-RhoA) and cytosolic fractions (C-RhoA) of Sf9 cells, preloaded with 1 mm GDP or 0.1 mm GTPγS, were precipitated with purified GST-IKKγ-Sepharose 4B beads. The GST and GST-IKKγ levels were measured with Ponceau S staining. C, recombinant RhoA protein (1 μg) expressed in E. coli was preloaded with 1 mm GDP or 0.1 mm GTPγS and then precipitated with purified GST-IKKγ-Sepharose 4B beads. GST and GST-IKKγ were identified with Ponceau S staining. The data represent the means ± S.E. of three independent experiments (*, p < 0.05; **, p < 0.01; ***, p < 0.001). D, lysates of RAW264.7 cells (500 μg of protein) were preloaded with 1 mm GDP and 0.1 mm GTPγS for 30 min at 30 °C. WCL, whole cell lysate. IKKγ was immunoprecipitated using 2 μg of anti-IKKγ antibody for 2 h at 4 °C. Co-precipitated RhoGDI was detected by Western blot analysis. E, 1 μg of GST-IKKγ conjugated to glutathione beads was incubated with 0.5–4 μg of recombinant RhoGDI for 2 h at 4 °C. RhoGDI bound to IKKγ was detected by Western blot. The amount of RhoGDI bound to IKKγ was calculated from the control standard curve. GST-IKKγ was detected with Ponceau S staining. F, 1 μg of GST-IKKγ conjugated to glutathione beads (GSH) was incubated with 2 μg of recombinant RhoA (R) purified from Sf9 cell membranes, 2 μg of purified RhoGDI (G), and 2 μg of purified RhoA-RhoGDI complex (C) for 2 h at 4 °C. RhoA and RhoGDI bound to IKKγ were detected by Western blot.
Because cytosolic RhoA-GDP in a resting state was considered to form a complex with RhoGDI, we examined whether RhoGDI is also capable of binding to IKKγ. Indeed, cytosolic RhoGDI was bound to IKKγ irrespective of the presence of either GDP or GTPγS (Fig. 2D). The purified RhoGDI directly bound to GST-IKKγ/glutathione-Sepharose beads in a concentration-dependent manner (Fig. 2E). In addition, the RhoA-RhoGDI complex, as well as RhoA or RhoGDI alone, was bound to IKKγ, suggesting that a trimeric complex of RhoA-RhoGDI-IKKγ is formed in vitro (Fig. 2F).
RhoA-GDP and RhoGDI Bind to Different Domains of IKKγ
To determine RhoA- or RhoGDI-binding domains of IKKγ, GST-IKKγ fragments (aa 1–43, 44–111, 1–100, 101–200, 201–350, 351–419, and 101–419) were prepared (Fig. 3A). RhoA-GDP was bound to N- and C-terminal domains of IKKγ (aa 1–43, 1–100, and 350–419, respectively), but it was not bound to the large C-terminal domain (aa 44–419) of IKKγ. Instead, RhoGDI was bound to C-terminal domains (aa 101–419) but not to the N-terminal domain (aa 1–111) (Fig. 3B). Because zinc finger domain (aa 389–419) in C-terminal region of IKKγ was known to be a binding site of IκB (28), the competition between IκB and RhoGDI for the binding toward IKKγ was explored. High concentration of IκB prevented RhoGDI from binding to IKKγ in vitro (Fig. 3C). Consistently, co-immunoprecipitation of IKKγ with IκB increased upon TGF-β1 in the presence of MG132, an inhibitor of proteasomal degradation, and the co-immunoprecipitation of IKKγ with RhoGDI was slightly reduced in cells (Fig. 3D), suggesting that TGF-β1 stimulates IκB binding to IKKγ.
FIGURE 3.

Binding domains of IKKγ for RhoA and RhoGDI. A, the constructs of GST-IKKγ of specific regions were designed; amino acids of IKKγ domains were denoted. B, GST-IKKγ domains (0.1 μg) conjugated with glutathione beads (GSH) were incubated with RhoA (0.1 μg) preloaded with 1 mm GDP or RhoGDI (0.5 μg) for 2 h at 4 °C, and washed. Bound RhoA and RhoGDI were identified with Western blotting. C, GST-IKKγ domains (0.5 μg) conjugated with glutathione beads were incubated with RhoGDI (0.5 μg) and various concentrations of IκB (0.5–3 μg) for 2 h at 4 °C and washed. Bound RhoGDI and IκB were identified with Western blotting. D, RAW264.7 cells were pretreated with MG132 (10 μm) for 30 min and then treated 5 ng/ml TGF-β1. IKKγ was immunoprecipitated with anti-IKKγ antibody (1 μg) and then co-precipitated RhoGDI and IκB were identified with Western blotting. E, GST-IKKγ domains (0.5 μg) conjugated with glutathione beads were incubated with RhoA (0.5 μg) and various concentrations of IKKβ (0.4–1.6 μg) for 2 h at 4 °C and washed. Bound RhoA and IKKβ were identified with Western blotting. The data represent the means ± S.E. of three independent experiments (*, p < 0.05; **, p < 0.01) except for D, which are means ± ranges of two independent experiments. F, proposed diagram of the binding of the proteins to IKKγ. C-term, C-terminal; a.a, amino acid(s).
Similarly, because the N-terminal domain (aa 44–111) was known to be a binding site of IKKβ (29), the competition between RhoA-GDP and IKKβ was examined; high concentration of IKKβ did not interfere with the binding of RhoA-GDP to IKKγ (Fig. 3E), suggesting that IKKβ and RhoA-GDP do not compete each other for their binding to IKKγ. The proposed diagram of the interaction of the proteins was shown in Fig. 3F.
IKKγ Facilitates the Activation of RhoA Complexed with RhoGDI in Vitro
Herein, we inferred that IKKγ could disrupt the interaction between RhoA and RhoGDI because RhoA-GDP bound to the N-terminal domain, and RhoGDI bound to the C-terminal domain of IKKγ, respectively. To demonstrate this hypothesis, we prepared purified proteins, including RhoA, RhoGDI, a complex of RhoA-RhoGDI, and IKKγ (Fig. 4A). Next, we examined whether IKKγ is able to facilitate the incorporation of GTP into RhoA in the complex of RhoA-RhoGDI. The preliminary experiment showed that the fluorescence intensity of 2′(3′)-O-(N-methylanthraniloyl) (Mant)-GTP (30, 31) was linearly correlated with RhoA concentration, suggesting that the extent of fluorescence presents the binding of Mant-GTP to RhoA. The fluorescence intensity of Mant-GTP bound to M-RhoA (Fig. 4B) and C-RhoA (data not shown) increased in a time-dependent manner, but RhoGDI interfered with the increase in Mant-GTP binding. However, IKKγ significantly augmented the fluorescence of Mant-GTP by binding to RhoA complexed with RhoGDI (Fig. 4B). However, IKKγ could not augment the fluorescence of Mant-GTP-bound to RhoA alone (Fig. 4F), suggesting that IKKγ does not play a role as a GEF.
FIGURE 4.

IKKγ facilitates the binding of GTP to RhoA complexed with RhoGDI. A, RhoA purified from the membrane (M-RhoA) and cytosol (C-RhoA) of Sf9 cells, recombinant RhoGDI purified from E. coli, a complex of RhoA-RhoGDI and recombinant IKKγ purified from E. coli (each ∼5 μg) were subjected to SDS-PAGE and Coomassie Blue staining. B, RhoA (0.5 μm) was preincubated with RhoGDI (2 μm) for 30 min at 24 °C and then incubated with IKKγ (2 μm) for an additional 30 min. Mant-GTP (10 μm) was mixed and incubated, and the fluorescence of Mant-GTP was measured (left panel), and the relative fluorescence intensity of Mant-GTP after 30 min of incubation was plotted in a bar graph (right panel). RFU, relative fluorescence unit. C, the RhoA-RhoGDI complex was prepared after ultrafiltration to remove free RhoA and RhoGDI. The RhoA-RhoGDI complex (0.5 μm) was incubated with [35S]GTPγ (20 μm) at 24 °C for 30 min in the presence of IKKγ (2 μm), Dbs (0.5 μm), or CHAPS (1%). D, RhoA (0.1 μm) was incubated with RhoGDI (0.4 μm) for 30 min, then IKKγ (1.6 μm) for 30 min, and then Dbs (0.5 μm) at 24 °C for 30 min. Finally, [35S]GTPγ (20 μm) was added to the mixture, which was incubated at 24 °C for 30 min. E, RhoA (0.1 μm) and RhoGDI (0.4 μm) were preincubated for 30 min. Then, IKKγ of defined concentrations was added, and the samples were incubated for another 30 min. [35S]GTPγ (20 μm) was incubated for 30 min with the protein mixture. The value of 0% indicates the binding of [35S]GTPγ to RhoA complexed with RhoGDI, and 100% indicates its binding to free RhoA. F, RhoA (0.5 μm), BSA (2 μm), and IKKγ (2 μm) were incubated with Mant-GTP (10 μm), and the fluorescence of Mant-GTP was measured in a time course. G, RAW264.7 cells were transfected with si-IKKγ (100 nm) and incubated for 72 h. Then, TGF-β1 was treated to cells for 1 h. GTP-RhoA was detected in the cell lysates (500 μg of protein) using a pulldown assay and Western blot. The data are the means ± S.E. of three independent experiments (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
To confirm this result again, we measured the binding of [35S]GTPγ to RhoA. As expected, RhoGDI suppressed the binding of [35S]GTPγ to RhoA. However, IKKγ significantly increased the binding of [35S]GTPγ to RhoA complexed with RhoGDI (Fig. 4, C and D), which was in a concentration-dependent manner (Fig. 4E). At a low MgCl2 concentration, much more [35S]GTPγ was incorporated into RhoA than at a high MgCl2 concentration (Fig. 4E). Furthermore, Dbs, a GEF for RhoA (13) much more enhanced the binding of [35S]GTPγ to RhoA complexed with RhoGDI in the presence of IKKγ than in the absence of it (Fig. 4, C and D). Here, the detergent CHAPS (1%) was used to disrupt the RhoA-RhoGDI complex. CHAPS seemed to impair Dbs activity, likely by preventing Dbs from acessing RhoA. Consistently, knockdown of IKKγ by si-IKKγ blocked the induction of RhoA-GTP in cells upon TGF-β1 stimulation (Fig. 4G), suggesting that IKKγ is indispensable for RhoA activation by TGF-β1.
RhoA Is Involved in NF-κB Activation in Response to TGF-β1
We tried to ascertain the involvement of RhoA in NF-κB activation due to TGF-β1. TGF-β1 increased the NF-κB reporter gene activity in 1 h, but its activity then decreased after 12–24 h (Fig. 5A). However, both treatment with an IKKβ inhibitor (BMS34551) and transfection of an IκB super suppressor (SR, IκB S32A/S36A) abolished NF-κB activation by TGF-β1 (Fig. 5B). TGF-β1 induced IκB degradation, but this degradation was blocked by the proteasomal inhibitor MG132 (Fig. 5C), suggesting that TGF-β1 activates NF-κB by degrading IκB in the proteasome.
FIGURE 5.

TGF-β1 induces RhoA-mediated NF-κB activation. A, RAW 264.7 cells were co-transfected with a NF-κB luciferase cis-reporter construct and a pCS2+-β-galactosidase plasmid, incubated for 24 h, and then stimulated with TGF-β1. Luciferase activity was measured with a luminometer. B, cells were co-transfected with the NF-κB luciferase reporter construct and pCS2+-β-galactosidase plasmids, or each 1 μg/ml mock (M) vector and the IκBα super repressor (S32A/S36A) DNA construct (SR) and incubated for 24 h. Cells transfected with the NF-κB luciferase construct were also preincubated with DMSO or 10 μm IKKβ inhibitor (BMS345541, BMS) dissolved in DMSO for 1 h. Then, the cells were incubated with TGF-β1 for 1 h, and the NF-κB activity was determined. C, cells were pretreated with DMSO or 10 μm MG132 dissolved in DMSO for 1 h and incubated with TGF-β1 at 37 °C. IκBα and β-actin were analyzed by Western blotting. D, RAW 264.7 cells were co-transfected with the NF-κB-luciferase reporter construct, pCS2+-β-galactosidase plasmids, and 1 μg/ml mock vector (M) or an HA-tagged RhoA construct (WT, constitutively active (G14V), or dominant-negative (T19N)), incubated for 24 h, and then incubated with TGF-β1 for 1 h. Luciferase activity was determined, and RhoA expression was assessed by Western blotting using an anti-HA antibody. E, cells were co-transfected with the NF-κB-luciferase construct and pCS2+-β-galactosidase plasmids. After 24 h, the cells were incubated with 10 μg/ml Tat-C3 for 1 h and then with TGF-β1 for 1 h. The resulting luciferase activity was measured. F, cells were co-transfected with the NF-κB-luciferase reporter construct, pCS2+-β-galactosidase plasmids, and either scrambled RNA (SCR) or 2 μg/ml sh-RhoA plasmid (Sh). After 72 h, the cells were incubated with or without TGF-β1 for 1 h, and then the luciferase activity was measured. G, cells were transfected with scrambled RNA or sh-RhoA and then stimulated with TGF-β1. The phosphorylations of IκB, IKKα/β, and p65 were analyzed by Western blotting. When the control value was set to 1, the amount of RhoA expressed in the presence of sh-RhoA was 0.33 ± 0.058. The values represent the means ± S.E. of three independent experiments (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
In addition, transfection of dominant-negative RhoA (T19N) (Fig. 5D), treatment of Tat-C3 toxin (Fig. 5E), and transfection of sh-RhoA (Fig. 5F) reduced TGF-β1-induced NF-κB promoter fused luciferase activity, whereas transfection of WT RhoA and constitutively active RhoA (G14V) promoted this activity (Fig. 5D). However, TGF-β1 induced the phosphorylation of IKKα/β, IκBα, and p65. However, the depletion of RhoA by transfecting sh-RhoA into the cells inhibited the phosphorylation of IKKα/β, IκBα, and p65 (Fig. 5G). These results suggest that RhoA is essential for NF-κB activation in the TGF-β1 signaling pathway in macrophages.
ROCK Is Involved in NF-κB Activation
Furthermore, Y27632 and HA1077 (Fasudil), inhibitors of ROCK reduced the NF-κB reporter gene luciferase activities by TGF-β1 (Fig. 6A). However, Y27632 did not alter the level of phosphorylated Smad3 in response to TGF-β1 (data not shown), indicating that although TGF-β1 can stimulates the Smad pathway, RhoA/ROCK activation is not implicated in Smad activation. Moreover, Y27632 and 5z-7-oxozeaenol, a TAK1 inhibitor (32), prevented the degradation of IκB, as well as the phosphorylation of IκB, IKKα/β, and p65 by TGF-β1 (Fig. 6B), suggesting that ROCK and TAK1 are essential for NF-κB activity in response to TGF-β1. Thus, we clarified whether ROCK directly phosphorylates IKKβ. When the constitutively active form of ROCK (aa 17–535) lacking the C-terminal region of the Rho-binding domain (33) and recombinant GST-IKKβ were incubated in the presence (Fig. 6D) or in the absence of cytosol (Fig. 6C), the phosphorylated IKKβ was detected with anti-phospho-IKKβ antibodies (Ser-177 and Ser-181), suggesting that ROCK directly phosphorylates Ser-177 and Ser-181 of IKKβ. Here, ROCK activity was demonstrated by measurement of the phosphorylation of myosin light chain phosphatase using anti-phospho-myosin light chain phosphatase antibody (Fig. 6D). However, because TAK1 is required for NF-κB activation (34), we examined the involvement of TAK1 in the regulation of the activation of RhoA. A TAK1 inhibitor, 5z-7-oxozeaenol, markedly abolished a TGF-β1-dependent increase of RhoA-GTP levels (Fig. 6E), suggesting that TAK1 is involved in RhoA activation upon TGF-β1 stimulation.
FIGURE 6.

ROCK is involved in NF-κB activation. A, RAW264.7 cells were co-transfected with the NF-κB-luciferase reporter construct and the pCS2+-β-galactosidase plasmids. The transfected cells were pretreated with 10 μm Y27632 and 10 μm HA1077 for 1 h and then incubated with TGF-β1 for 1 h. Luciferase and β-galactosidase activities were measured. B, cells were pretreated with 10 μm Y27632 or 0.5 μm 5z-7-oxozeaenol (OXO) for 1 h and then with TGF-β1 for the indicated times. The phosphorylation of IκB, IKKα/β, and p65 was analyzed by Western blotting. The data represent the means ± S.E. of three independent experiments (*, p < 0.05; **, p < 0.01). C and D, GST-IKKβ (0.1 μg), ROCK (0.1 μg), with 10 mm MgCl2 and 25 μm ATP were incubated for 30 min at 30 °C in the presence (D) or absence (C) of cell lysates (50 μg), and p-IKKα/β (Ser-176/177 and Ser-180/181) and phospho-myosin light chain phosphatase (p-MYPT) were identified with Western blotting. E, RAW264.7 cells were first pretreated with DMSO or 0.5 μm 5z-7-oxozeaenol dissolved in DMSO for 1 h and then incubated with TGF-β1 for 1 h. RhoA-GTP levels were determined using a GST-rhotekin-RBD pulldown assay. C, control; T, TGF-β1. The data represent the means ± S.E. of three independent experiments (*, p < 0.05; **, p < 0.01), and the means ± range of two independent experiment for p-IKK α/β at Ser-176/177.
NF-κB Regulates Chemokine Expression
Finally, we observed that TGF-β1 induced the transcription and protein expression of the chemokine such as MIP-1α (Fig. 7, A and B) and cell migration (Fig. 7C). However, BMS-345541 (an inhibitor of IKKβ) inhibited the MIP-1α and cell migration, suggesting that NF-κB activation is involved in the cell migration upon TGF-β1 stimulation. Consistently, we recently reported that RhoA is required for the expression of MIP-1α upon TGF-β1 stimulation (17, 22).
FIGURE 7.
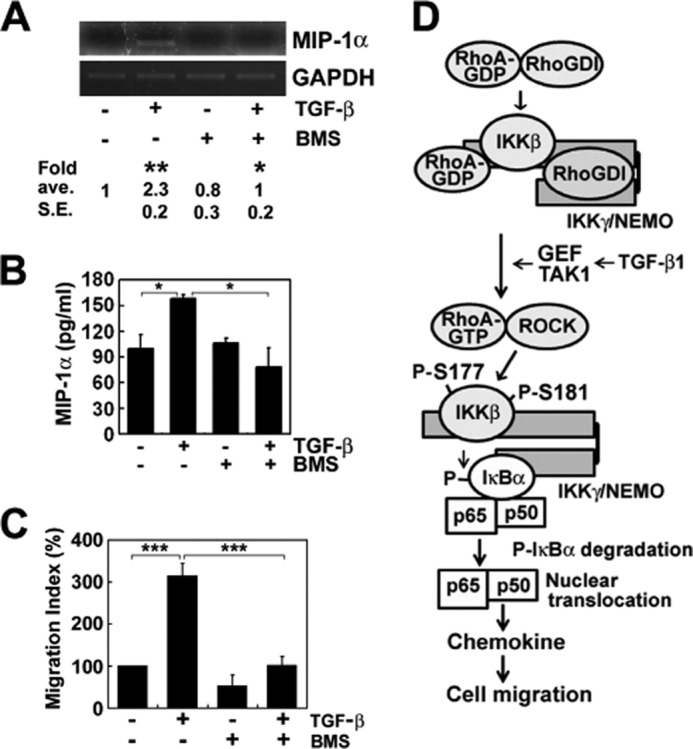
NF-κB mediates TGF-β1-induced cell migration. A, BMS345541 (BMS; IKKβ inhibitor, 10 μm) was pretreated for 1 h, TGF-β1 was treated for 1 h, and then secreted MIP-1α was determined by reverse transcription PCR. B, cells were pretreated with DMSO or 10 μm BMS345541 dissolved in DMSO for 1 h. Then, cells were treated with TGF-β1 for 6 h, and secreted MIP-1α was measured using ELISA. C, cells were preincubated with DMSO and 10 μm IKKβ inhibitor (BMS345541) for 1 h and then incubated with or without TGF-β1 for 1 h in the lower chamber, allowing cells of the upper chamber to migrate for 6 h. D, proposed scheme of the molecular mechanism to induce NF-κB activation through RhoA activation by IKKγ and in turn IKKβ phosphorylation by ROCK. The data represent the means ± S.E. of three independent experiments (*, p < 0.05; **, p < 0.01; ***, p < 0.001). ave., average.
DISCUSSION
Regulation of Transcription by RhoA
Rho proteins such as RhoA, Cdc42, and Rac1 regulate transcription via several transcription factors, including serum response factor (SRF), NF-κB, and others regulated by the kinases such as JNK1 and p38 MAPK. The substrates of these kinases include Stat3, Stat5a, ELK, PEA3, ATF2, Max, and CHOP/GADD153 (35). Among them, it has been well known that RhoA activates SRF (36). The mechanism by which RhoA activates SRF has been proposed as follows; MAL/MKL1, a member of the myocardin-related transcription factor (MRTF) is a cofactor of SRF that binds to monomeric G-actin in the cytosol but translocates to the nucleus after MAL is released from polymerized F-actin upon serum stimulation (37). However, in this study, we disclosed that the activation of NF-κB by RhoA is utterly different from SRF activation by RhoA.
RhoA-RhoGDI Complex Binds to IKKγ Allowing RhoA Activation
Generally, IKKγ provides binding sites for many proteins; 16 proteins have been reported as directly binding to IKKγ and promoting the activation of NF-κB (38). Here, we presented that RhoA-GDP and RhoGDI are also bound to IKKγ; RhoA-GDP was bound to the N-terminal domain (aa 1–43) and RhoGDI was bound to the large C-terminal domain (estimated aa 112–419) of IKKγ. The Rho-binding domain of IKKγ is not identical with the known Rho-binding domains of effector proteins, including ROCK1, protein kinase N, rhophilin, and rhotekin (supplemental Fig. S1). It is noteworthy that RhoA-GDP was also bound to small C-terminal domain (aa 351–419) of IKKγ, but not to the large C-terminal domain, suggesting that a part of C-terminal region (aa 101–350) interfere with RhoA-GDP binding to IKKγ. However, CC2 (coiled coil 2) and leucine zipper domains of IKKγ (Fig. 3A) directly interact in an anti-parallel orientation through the connecting loop containing proline (39), suggesting that N and C termini may be localized in the same direction (Fig. 3F). Interestingly, IκB binds to the C-terminal zinc finger domain (aa 389–419) (28), and IKKβ binds to the N-terminal domain of IKKγ (aa 44–111) (29). These results indicate that IKKβ and IκB bound to IKKγ might be spatially close each other (Fig. 3F). Indeed, IKKβ itself interacts with the C terminus of IκB forming a stable ternary complex with IKKγ (40). In addition, IκB competes with RhoGDI for the binding to IKKγ in vitro and in vivo upon TGF-β1 (Fig. 3, C and D, respectively). It remains unclear how the binding of IκB to IKKγ increases upon TGF-β1 with the decrease of RhoGDI. However, RhoA-binding domain may be a relatively short length of N terminus (aa 1–43), which is not overlapped with the IKKβ-binding site of IKKγ (aa 44–111); therefore, RhoA-GDP did not compete with IKKβ for the binding to IKKγ (Fig. 3E).
In conclusion, we here propose that IKKγ likely functions to activate RhoA by serving as a scaffold protein to recruit the RhoA-RhoGDI complex. It was indeed known that IKKγ recruits IκB to IKKβ as a scaffold protein, allowing IKKβ to phosphorylate IκB (28, 41). Because RhoA-GDP complexed with RhoGDI is not directly activated by GEFs (5, 6), a specific GDF to disrupt RhoA-RhoGDI complex has been accepted to be required for the activation of RhoA. Herein, we demonstrated that IKKγ could allow RhoA of RhoA-RhoGDI complex to be readily incorporated with GTP (Fig. 3). Moreover, the Dbs, a GEF of RhoA, much enhanced GTP binding to the RhoA-RhoGDI complex in the presence of IKKγ (Fig. 3), suggesting that IKKγ allows the GEF to recognize, access, or act on RhoA complexed with RhoGDI; IKKγ may play a role as a GDF.
Depending on the signaling pathway, there may be diverse GDFs that activate RhoA by a variety of specific stimuli. Indeed, several GDFs, including ezrin/radixin/moesin, Etk/Bmx, and neurotrophin receptor p75NTR, have been reported (6, 42, 43). Given that there are several GDFs, it is likely that each GDF activates RhoA in a different particular signal pathway.
In addition to GDFs, modification of RhoGDI itself releases Rho GTPases from the RhoA-RhoGDI complex; phosphorylation of RhoGDI at Tyr-156 by Src alleviates its affinity for RhoA, Rac1, and Cdc42 (44), and phosphorylation RhoGDI by PKCα leads to dissociation from RhoA (45). Because IKKγ can be also modified with proteins similar to ubiquitin (46), it is possible that a variety modification of RhoA, RhoGDI, or IKKγ actually regulates to disrupt the RhoA-RhoGDI complex in response to several stimuli in vivo. Nonetheless, it is evident that in vitro system unmodified recombinant IKKγ facilitates GTP-binding to RhoA complexed with recombinant RhoGDI without modification of these proteins by any other stimuli.
Although RhoA was reported to be activated by TGF-β1 via a GEF, including Vav2 or Net1 (18, 47), Lbc, which is also referred to as AKAP13, AKAP-Lbc, and ARHGEF13 (48) was recently reported to be associated with α-catulin, which is bound to IKK-β (49). It is likely that different types of GEFs in different cells may be involved in the activation of RhoA in response to TGF-β1.
Active RhoA/ROCK Activates NF-κB
Eventually, RhoA-GTP released from IKKγ by TGF-β1 was thought to activate ROCK. Indeed, active ROCK directly phosphorylated the recombinant GST-IKKβ at Ser-177 and Ser-181 residues in vitro (Fig. 6C). Although TGF-β1 activates TAK1 (50) and only TAK1 is generally accepted as an IKK kinase in a canonical pathway (51, 52), it remains unclear whether IKK is activated by an upstream kinase or autophosphorylation (53, 54). Here, we demonstrated that TAK1 is involved in RhoA activation upon TGF-β1 (Fig. 6E). Because ROCK and TAK1 are involved in NF-κB activation (Fig. 6B), we examined whether ROCK regulates TAK1 activity in a positive feedback manner. However, Y27632, an inhibitor of ROCK, did not influence the phosphorylation of TAK1. Consistent with this observation, neither si-ROCK1 nor si-ROCK2 had an influence on the phosphorylation of TAK1 (data not shown). These results suggest that RhoA/ROCK does not promote TAK1 activity. Although it is still ambiguous that TAK1 and/or ROCK directly activate(s) IKKβ, we propose that ROCK is able to directly phosphorylate IKKβ and TAK1 is instead involved in the activation of RhoA (Fig. 6E). However, it remains to be studied in the future how TAK1 is involved in RhoA regulation in TGF-β1 signaling. On the other hand, a long term treatment of TGF-β1 inactivates RhoA (Fig. 1A) through p190RhoGAP and cAMP/Epac/ARAP3 activation (17, 22). Subsequently, NF-κB activation was also abolished by a long term treatment of TGF-β1 (Fig. 5A).
Thus, it is a very intriguing novel finding that IKKγ facilitates RhoA activation, which in turn activates ROCK, leading to direct phosphorylation of IKKβ and subsequent activation of NF-κB. Conclusively, the RhoA and IKK complexes may regulate each other and form a positive feedback loop to activate NF-κB. Thus, we proposed the scheme of the new regulatory mechanism of NF-κB activation through RhoA activation in response to TGF-β (Fig. 7D).
Acknowledgments
We thank Dr. Ashwell for providing the pGEX-IKKγ (Addgene plasmid 11966) and pET-IKKγ (Addgene plasmid 11965) constructs and Dr. Smale for providing the RelA (p65)-pCDNA3 construct (Addgene plasmid 20012). We thank Dr. Bokoch for providing baculovirus containing RhoA and Sf9 cells. We thank Hye-Kyoung Han for construction of plasmids.
This work was supported by the Basic Science Research Program of the National Research Foundation of Korea funded by the Ministry of Education, the Ministry of Science, Information Communication Technology, and Future Planning (Grants NRF-2013R1A1A2A10006114 and MRC-2010-0009180) and Hallym University (Grant HRF-S-41).

This article contains supplemental Fig. S1.
- GEF
- guanosine nucleotide exchange factor
- GDF
- GDI displacement factor
- GDI
- guanine nucleotide dissociation inhibitor
- GTPγS
- guanosine 5′-3-O-(thio)triphosphate
- IκB
- inhibitor of NF-κB
- IKK
- IκB kinase
- Mant-GTP
- 2′/3′-O-(N-methylanthraniloyl)guanosine-5′-(γ-thio) triphosphate triethylammonium salt
- MIP
- macrophage inflammatory protein
- ROCK
- Rho-associated coiled-coil containing Ser/Thr protein kinase
- si
- small interfering
- SRF
- serum response factor
- TAK1
- TGF-β1-activated kinase 1
- RhoGDI
- Rho guanine nucleotide dissociation inhibitor
- aa
- amino acid(s)
- DMSO
- dimethyl sulfoxide.
REFERENCES
- 1. Li M. O., Wan Y. Y., Sanjabi S., Robertson A. K., Flavell R. A. (2006) Transforming growth factor-β regulation of immune responses. Annu. Rev. Immunol. 24, 99–146 [DOI] [PubMed] [Google Scholar]
- 2. Derynck R., Zhang Y. E. (2003) Smad-dependent and Smad-independent pathways in TGF-β family signaling. Nature 425, 577–584 [DOI] [PubMed] [Google Scholar]
- 3. Zhang Y. E. (2009) Non-Smad pathways in TGF-β signaling. Cell Res. 19, 128–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Olofsson B. (1999) Rho guanine dissociation inhibitors: pivotal molecules in cellular signaling. Cell Signal. 11, 545–554 [DOI] [PubMed] [Google Scholar]
- 5. Takai Y., Sasaki T., Tanaka K., Nakanishi H. (1995) Rho as a regulator of the cytoskeleton. Trends Biochem. Sci. 20, 227–231 [DOI] [PubMed] [Google Scholar]
- 6. Takahashi K., Sasaki T., Mammoto A., Takaishi K., Kameyama T., Tsukita S., Takai Y. (1997) Direct interaction of the Rho GDP dissociation inhibitor with ezrin/radixin/moesin initiates the activation of the Rho small G protein. J. Biol. Chem. 272, 23371–23375 [DOI] [PubMed] [Google Scholar]
- 7. Jaffe A. B., Hall A. (2005) GTPases: Biochemistry and Biology. Annu. Rev. Cell Dev. Biol. 21, 247–269 [DOI] [PubMed] [Google Scholar]
- 8. Yamamoto Y., Gaynor R. B. (2004) IκB kinases: key regulators of the NF-κB pathway. Trend. Biochem. Sci. 29, 72–79 [DOI] [PubMed] [Google Scholar]
- 9. Hayden M. S., Ghosh S. (2004) Signaling to NF-κB. Genes Dev. 18, 2195–2224 [DOI] [PubMed] [Google Scholar]
- 10. Chen Z. J. (2005) Ubiquitin signaling in the NF-κB pathway. Nat. Cell Biol. 7, 758–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hippenstiel S., Soeth S., Kellas B., Fuhrmann O., Seybold J., Krüll M., Eichel-Streiber C., Goebeler M., Ludwig S., Suttorp N. (2000) Rho proteins and p38-MAPK pathway are important for LPS-induced interleukin-8 expression in human endothelial cells. Blood 95, 3044–3051 [PubMed] [Google Scholar]
- 12. Perez-Moreno M., Davis M. A., Wong E., Pasolli H. A., Reynolds A. B., Fuchs E. (2006) p120-catenin mediates inflammatory response in the skin. Cell 124, 631–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cammarano M. S., Minden A. (2001) Dbl and the Rho GTPases activate NF-κB IκB kinase (IKK)-dependent and IKK-independent pathways. J. Biol. Chem. 276, 25876–25882 [DOI] [PubMed] [Google Scholar]
- 14. Montaner S., Perona R., Saniger L., Lacal J. C. (1998) Multiple signaling pathways lead to the activation of the nuclear factor kB by the Rho family of GTPases. J. Biol. Chem. 273, 12779–12785 [DOI] [PubMed] [Google Scholar]
- 15. Montaner S., Perona R., Saniger L., Lacal J. C. (1999) Activation of serum response factor by RhoA is mediated by the nuclear factor-κB and C/EBP transcription factors. J. Biol. Chem. 274, 8506–8515 [DOI] [PubMed] [Google Scholar]
- 16. Edlund S., Landström M., Heldin C. H., Aspenström P. (2002) Transforming growth factor-β-induced mobilization of actin cytoskeleton requires signaling by small GTPases Cdc42 and RhoA. Mol. Biol. Cell 13, 902–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim J. S., Kim J. G., Moon M. Y., Jeon C. Y., Won H. Y., Kim H. J., Jeon Y. J., Seo J. Y., Kim J. I., Kim J., Lee J. Y., Kim P. H., Park J. B. (2006) Transforming growth factor-β1 regulates macrophage migration via RhoA. Blood 108, 1821–1829 [DOI] [PubMed] [Google Scholar]
- 18. Papadimitriou E., Kardassis D., Moustakas A., Stournaras C. (2011) TGFβ-induced early activation of the small GTPase RhoA is Smad2/3-independent and involves Src and the guanine nucleotide exchange factor Vav2. Cell Physiol. Biochem. 28, 229–238 [DOI] [PubMed] [Google Scholar]
- 19. Arsura M., Panta G. R., Bilyeu J. D., Cavin L. G., Sovak M. A., Oliver A. A., Factor V., Heuchel R., Mercurio F., Thorgeirsson S. S., Sonenshein G. E. (2003) Transient activation of NF-κB through a TAK/IKK kinase pathway by TGF-β inhibits AP-1/SMAD signaling and apoptosis: implications in liver tumor formation. Oncogene 22, 412–425 [DOI] [PubMed] [Google Scholar]
- 20. Ishinaga H., Jono H., Lim J. H., Kweon S. M., Xu H., Ha U. H., Xu H., Koga T., Yan C., Feng X. H., Chen L. F., Li J. D. (2007) TGF-β induces p65 acetylation to enhance bacteria-induced NF-κB activation. EMBO J. 26, 1150–1162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yeh Y. Y., Chiao C. C., Kuo W. Y., Hsiao Y. C., Chen Y. J., Wei Y. Y., Lai T. H., Fong Y. C., Tang C. H. (2008) TGF-β1 increases motility and αvβ3 integrin up-regulation via PI3K, Akt and NF-κB-dependent pathway in human chondrosarcoma cells. Biochem. Pharmacol. 75, 1292–1301 [DOI] [PubMed] [Google Scholar]
- 22. Moon M. Y., Kim H. J., Kim J. G., Lee J. Y., Kim J., Kim S. C., Choi I. G., Kim P. H., Park J. B. (2013) Small GTPase Rap1 regulates cell migration through regulation of small GTPase RhoA activity in response to transforming growth factor-β1. J. Cell Physiol. 228, 2119–2126 [DOI] [PubMed] [Google Scholar]
- 23. Ren X. D., Schwartz M. A. (2000) Determination of GTP loading on Rho. Methods Enzymol. 325, 264–272 [DOI] [PubMed] [Google Scholar]
- 24. Kim J. G., Moon M. Y., Kim H. J., Li Y., Song D. K., Kim J. S., Lee J. Y., Kim J., Kim S. C., Park J. B. (2012) Ras-related GTPases Rap1 and RhoA collectively induce the phagocytosis of serum-opsonized zymosan particles in macrophage. J. Biol. Chem. 287, 5145–5155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kikuchi A., Sasaki T., Araki S., Hata Y., Takai Y. (1989) Purification and characterization from bovine cytosol of two GTPase-activating proteins specific for smg21, a GTP-binding protein having the same effector domain as c-ras p21s. J. Biol. Chem. 264, 9133–9136 [PubMed] [Google Scholar]
- 26. Shimizu S., Tahara M., Ogata S., Hashimoto K., Morishige K., Tasaka K., Murata Y. (2007) Involvement of nuclear factor-kB activation through RhoA/Rho-kinase pathway in LPS-induced IL-8 production in human cervical stromal cells. Mol. Hum. Reprod. 13, 181–187 [DOI] [PubMed] [Google Scholar]
- 27. Diebold B. A., Bokoch G. M. (2001) Molecular basis for Rac2 regulation of phagocyte NADPH oxidase. Nat. Immunol. 2, 211–215 [DOI] [PubMed] [Google Scholar]
- 28. Schröfelbauer B., Polley S., Behar M., Ghosh G., Hoffmann A. (2012) NEMO ensures signaling specificity of the pleiotropic IKKβ by directing its kinase activity toward IκBα. Mol. Cell 47, 111–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rushe M., Silvian L., Bixler S., Chen L. L., Cheung A., Bowes S., Cuervo H., Berkowitz S., Zheng T., Guckian K., Pellegrini M., Lugovskoy A. (2008) Structure of a NEMO/IKK-associating domain reveals architecture of the interaction site. Structure 16, 798–808 [DOI] [PubMed] [Google Scholar]
- 30. Hutchinson J. P., Eccleston J. F. (2000) Mechanism of nucleotide release from Rho by the GDP dissociation stimulator protein. Biochemistry 39, 11348–11359 [DOI] [PubMed] [Google Scholar]
- 31. Thanbichler M., Bock A., Goody R. S. (2000) Kinetics of the interaction of translation factor SelB from Escherichia coli with guanosine nucleotides and selenocysteine insertion sequence RNA. J. Biol. Chem. 275, 20458–20466 [DOI] [PubMed] [Google Scholar]
- 32. Ear T., Fortin C. F., Simard F. A., McDonald P. P. (2010) Constitutive association of TGF-β-activated kinase 1 with the IκB kinase complex in the nucleus and cytoplasm of human neutrophils and its impact on downstream processes. J. Immunol. 184, 3897–3906 [DOI] [PubMed] [Google Scholar]
- 33. Kishida S., Yamamoto H., Kikuchi A. (2004) Wnt-3a and Dvl induce neurite retraction by activating Rho-associated kinase. Mol. Cell Biol. 24, 4487–4501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shim J. H., Xiao C., Paschal A. E., Bailey S. T., Rao P., Hayden M. S., Lee K. Y., Bussey C., Steckel M, Tanaka N., Yamada G., Akira S., Matsumoto K., Ghosh S. (2005) TAK1, but not TAB1 or TAB2, plays an essential role in multiple signaling pathways in vivo. Genes Dev. 19, 2668–2681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Aznar S., Lacal J. C. (2001) Rho signals to cell growth and apoptosis. Cancer Lett. 165, 1–10 [DOI] [PubMed] [Google Scholar]
- 36. Hill C. S., Wynne J., Treisman R. (1995) The Rho family GTPases RhoA, Rac1, Cdc42Hs regulate transcriptional activation by SRF. Cell 81, 1159–1170 [DOI] [PubMed] [Google Scholar]
- 37. Vartiainen M. K., Guettler S., Larijani B., Treisman R. (2007) Nuclear actin regulates dynamic subcellular localization and activity of the SRF cofactor MAL. Science 316, 1749–1752 [DOI] [PubMed] [Google Scholar]
- 38. Shifera A. S. (2010) Protein-protein interactions involving IKKγ (NEMO) that promote the activation of NF-κB. J. Cell Physiol. 223, 558–561 [DOI] [PubMed] [Google Scholar]
- 39. Agou F., Traincard F., Vinolo E., Courtois G., Yamaoka S., Israël A., Véron M. (2004) The trimerization domain of Nemo is composed of the interacting CC2 and LZ coiled-coil subdomains. J. Biol. Chem. 279, 27861–27869 [DOI] [PubMed] [Google Scholar]
- 40. Xu G., Lo Y. C., Li Q., Napolitano G., Wu X., Jiang X., Dreano M., Karin M., Wu H. (2011) Crystal structure of inhibitor of κB kinase β. Nature 472, 325–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yamamoto Y., Kim D. W., Kwak Y. T., Prajapati S., Verma U., Gaynor R. B. (2001) IKKγ/NEMO facilitates the recruitment of the IκB proteins into the IκB kinase complex. J. Biol. Chem. 276, 36327–36336 [DOI] [PubMed] [Google Scholar]
- 42. Kim O., Yang J., Qiu Y. (2002) Selective activation of small GTPase RhoA by tyrosine kinase Etk through its pleckstrin homology domain. J. Biol. Chem. 277, 30066–30071 [DOI] [PubMed] [Google Scholar]
- 43. Yamashita T., Tohyama M. (2003) The p75 receptor acts as a displacement factor that releases Rho from Rho-GDI. Nat. Neurosci. 6, 461–467 [DOI] [PubMed] [Google Scholar]
- 44. DerMardirossian C., Rocklin G., Seo J. Y., Bokoch G. M. (2006) Phosphorylation of RhoGDI by Src regulates Rho GTPase binding and cytosol-membrane cycling. Mol. Biol. Cell 17, 4760–4768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dovas A., Choi Y., Yoneda A., Multhaupt H. A., Kwon S. H., Kang D., Oh E. S., Couchman J. R. (2010) Serine 34 phosphorylation of Rho guanine dissociation inhibitor (RhoGDIα) links signaling from conventional protein kinase C to RhoGTPase in cell adhesion. J. Biol. Chem. 285, 23296–23308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rahighi S., Ikeda F., Kawasaki M., Akutsu M., Suzuki N., Kato R., Kensche T., Uejima T., Bloor S., Komander D., Randow F., Wakatsuki S., Dikic I. (2009) Specific recognition of linear ubiquitin chains by NEMO is important for NF-κB activation. Cell 136, 1098–1109 [DOI] [PubMed] [Google Scholar]
- 47. Shen X., Li J., Hu P. P., Waddell D., Zhang J., Wang X. F. (2001) The activity of guanine exchange factor NET1 is essential for transforming growth factor-β-mediated stress fiber formation. J. Biol. Chem. 276, 15362–15368 [DOI] [PubMed] [Google Scholar]
- 48. Medina F., Carter A. M., Dada O., Gutowski S., Hadas J., Chen Z., Sternweis P. C. (2013) Activated RhoA is a positive feedback regulator of the Lbc family of Rho guanine nucleotide exchange factor proteins. J. Biol. Chem. 288, 11325–11333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wiesner C., Winsauer G., Resch U., Hoeth M., Schmid J. A., van Hengel J., van Roy F., Binder B. R., de Martin R. (2008) α-Catulin, Rho signaling component, can regulate NF-κB through binding to IKK-β, and confers resistance to apoptosis. Oncogene 27, 2159–2169 [DOI] [PubMed] [Google Scholar]
- 50. Yamaguchi K., Shirakabe K., Shibuya H., Irie K., Oishi I., Ueno N., Taniguchi T., Nishida E., Matsumoto K. (1995) Identification of a member of the MAPKKK family as a potential mediator of TGF-β signal transduction. Science 270, 2008–2011 [DOI] [PubMed] [Google Scholar]
- 51. Wang C., Deng L., Hong M., Akkaraju G. R., Inoue J., Chen Z. J. (2001) TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 412, 346–351 [DOI] [PubMed] [Google Scholar]
- 52. Kumar M., Makonchuk D. Y., Li H., Mittal A., Kumar A. (2009) TNF-like weak inducer of apoptosis (TWEAK) activates proinflammatory signaling pathways and gene expression through the activation of TGF-β-activated kinase 1. J. Immunol. 182, 2439–2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hayden M. S., Ghosh S. (2008) Shared principles in NF-κB signaling. Cell 132, 344–362 [DOI] [PubMed] [Google Scholar]
- 54. Hayden M. S., Ghosh S. (2012) NF-κB, the first quater-century: remarkable progress and outstanding questions. Genes Dev. 26, 203–234 [DOI] [PMC free article] [PubMed] [Google Scholar]