

Background: AR splice variants may play a critical role in prostate cancer.
Results: AR3 modulates expression of tumor-promoting growth factors and promotes epithelial-mesenchymal transition, leading to development of prostatic intraepithelial neoplasia.
Conclusion: AR3 promotes prostate cancer by modulating multiple tumor-associated autocrine/paracrine factors.
Significance: Our findings provide new insights into mechanisms by which AR3 contributes to prostate cancer despite its heterogeneous expression pattern.
Keywords: Androgen Receptor, EMT, Insulin-like Growth Factor (IGF), MicroRNA, Prostate Cancer, mir-29, Splicing Variant
Abstract
Deregulation of androgen receptor (AR) splice variants has been implicated to play a role in prostate cancer development and progression. To understand their functions in prostate, we established a transgenic mouse model (AR3Tg) with targeted expression of the constitutively active and androgen-independent AR splice variant AR3 (a.k.a. AR-V7) in prostate epithelium. We found that overexpression of AR3 modulates expression of a number of tumor-promoting autocrine/paracrine growth factors (including Tgfβ2 and Igf1) and expands prostatic progenitor cell population, leading to development of prostatic intraepithelial neoplasia. In addition, we showed that some epithelial-mesenchymal transition-associated genes are up-regulated in AR3Tg prostates, suggesting that AR3 may antagonize AR activity and halt the differentiation process driven by AR and androgen. This notion is supported by our observations that the number of Ck5+/Ck8+ intermediate cells is increased in AR3Tg prostates after castration, and expression of AR3 transgene in these intermediate cells compromises prostate epithelium regeneration upon androgen replacement. Our results demonstrate that AR3 is a driver of prostate cancer, at least in part, through modulating multiple tumor-promoting autocrine/paracrine factors.
Introduction
Androgen ablation therapy is the first-line treatment for patients with advanced metastatic prostate cancer. Despite an initial favorable response, a majority of patients will eventually develop incurable castration-resistant cancer. Understanding the mechanisms underlying castration resistance is critical for developing more effective treatment of this deadly disease. The androgen receptor (AR)2 is a key transcription factor which is activated by androgens and transduces androgen signaling mainly through regulating expression of genes that are essential for prostate homeostasis (1). It is reported that androgen receptor may directly modulate >200 protein-coding genes as well as a group of microRNAs in a cell type-dependent manner (2–5). In the majority of castration-resistant tumors, AR transcription activity is largely maintained despite androgen deprivation. Multiple underlying mechanisms have been proposed, including deregulation of the AR gene, steroid metabolism enzymes, steroid hormone receptor coactivators, autocrine/paracrine factors, as well as ligand-independent AR splicing variants (6–16). In addition, it has been speculated that androgen ablation therapy may target mainly the bulk of tumor cells expressing AR, but fail to eliminate cancer-initiating cells (or cancer stem cells) which are responsible for initiating and maintaining tumor growth (14, 17).
Prostate stem cells play a critical role in maintaining prostate tissue integrity, and their potential involvement in prostate tumorigenesis has been implicated (14). Several studies have identified and validated the markers such as stem cell antigen-1 (Sca-1), CD49f and CD117 for enriching murine stem/progenitor cells both in vitro and in vivo (18–20). Purified basal cells are able to regenerate prostatic lumens, suggesting that at least a portion of prostatic stem cells reside in basal compartment (21). A single Lin−Sca-1+CD133+CD44+CD117+ cell has been reported to generate functional prostatic ducts in renal grafts (19). A recent study has shown that Lin-Sca-1+CD49fhigh stem cells are increased in response to castration in normal prostate gland and also represent a fraction of tumor-initiating cells in the Pten-null prostate cancer model (22). These data suggest that tumor-initiating cells may share some common features with normal stem cells such as expression of primitive markers and the ability to self-renew. Meanwhile, a second more differentiated luminal Nkx3.1+ progenitor population (castration-resistant Nkx3–1-expressing cells, CARNs) may also exist and play an essential role during mouse prostate regeneration after castration (23). Furthermore, multiple autocrine/paracrine factors, such as EGF, IGF, TGFβ, WNT, NOTCH, and FGF, have been shown to play important roles in regulating proliferation, differentiation, and maintenance of prostatic stem cells (24, 25). Deregulation of these autocrine/paracrine loops is intimately associated with cancer development and progression.
Recently, multiple independent studies showed that a number of AR splicing variants lacking the ligand binding domain are up-regulated in hormone-resistant prostate cancer cells and promote castration-resistant growth in cell culture and xenograft models (10–13). Expression of AR3 (a.k.a. AR-V7), a constitutively active and androgen-independent AR splice variant, predicts biochemical recurrence as well as cancer-specific survival (11, 12, 26). Gene expression profiling revealed that AR3 may have an independent role in regulating a set of target genes in prostate cancer cells. Interestingly, AR3 is expressed primarily in the basal compartment in benign prostatic acini while being virtually undetectable in luminal epithelial cells (11, 27). However, AR3 expression is significantly elevated in luminal epithelial cells in prostatic intraepithelial neoplasia (PIN) lesions, suggesting that aberrant expression of AR3 in luminal epithelial cells may potentially play a causal role in initiation of prostate cancer. Nevertheless, AR3 staining in prostate tumors appears to be quite heterogeneous, suggesting that not all tumor cells express and/or depend on AR3. Therefore, the role of AR3 in prostate cancer initiation and progression remains elusive.
The present study was undertaken to examine the effects of aberrant expression of AR3 in mouse prostate epithelium. We generated the AR3 transgenic (AR3Tg) mouse model and found that overexpression of AR3 can modulate expression of a number of tumor-promoting autocrine/paracrine growth factors including Tgfβ2 and Igf1 and increase prostatic progenitor cell population, leading to development of prostatic intraepithelial neoplasia.
EXPERIMENTAL PROCEDURES
Transgenic Mice
The full-length human AR3 cDNA was cloned into ARR2PB promoter expression cassette and injected into fertilized FVB mouse eggs. The eggs were transplanted into pseudopregnant females. Newborn mice were screened for genomic integration of the human AR3 transgene by PCR of mouse tail DNA and confirmed by RT-PCR, Western blotting, and immunofluorescence. Tail genomic DNA was purified, and PCR was performed as described previously (28). Three independent transgenic lines were established by mating the founder animal with naïve FVB mice. The studies were approved by Institutional Animal Care and Use Committees of University of Maryland.
Western Blot Analysis
Prostate tissues were extracted by using the T-PER protein extraction reagent (Pierce). An equal amount of prostate tissue or total cell lysates was subjected to SDS-PAGE, followed by Western blot analysis using anti-AR(441), anti-actin (C2), anti-AR (N20; Santa Cruz Biotechnology), anti-TGF-β, anti-Smad3, anti-p-Smad3, anti-Smad1, anti-p-Smad1/5 (Cell Signaling), and anti-AR3 (11).
RNA Isolation, Microarrays, Quantitative Real-time Reverse Transcription-PCR, and MicroRNAs
Total RNA including miRNAs was isolated from cells or tissues by using an miRNeasy mini kit (Qiagen) or RNeasy mini kit, and then RNA was treated with RQ1 RNase-free DNase (Promega) according to the manufacturer's instructions. Affymatrix Mouse Gene-1.0st-v1 arrays were used for comparison of gene expression profiles between AR3Tg and WT controls prostates (n = 3). Microarray analysis was performed using BRB-ArrayTools (developed by Richard Simon and BRB-ArrayTools Development Team) to identify differentially expressed genes. Genes were considered statistically significant if their p value was <0.05 and at least 1.2-fold change. The raw data of gene expression profiling have been deposited in GEO repository under the accession GSE52092. The changes of some identified genes were validated by the quantitative real-time PCR. Quantitative RT-PCR was performed using the Fastart SYBR Green Master mix (Roche Applied Science) on a Two Color Real-Time PCR Detection System (Bio-Rad). For the reverse transcription (RT) reaction 1 μg of total RNA was used. Stem-loop RT primer GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACTAACCG was used in the mature miR-29c cDNA synthesis reaction. The primers used for mature miR-29c were 5′-GCGTCTAGCACCATTTGAAAT-3′ and 5′-GTGCAGGGTCCGAGGT-3′; U6, 5′-CTCGCTTCGGCAGCACA-3′ and 5′-AACGCTTCACGAATTTGCGT-3′. The relative abundance of each target transcript was quantified by using the comparative ΔΔCt method with U6 small RNA or 18S as an internal control. The lentiviral vector expressing miR-29 was purchased from GeneCopoeia. The miR-29 inhibitor and its negative control were purchased from Qiagen.
Flow Cytometry
Dissociated murine prostate cells were suspended in DMEM/10% FBS and stained with antibodies for 30 min on ice. The antibodies used were APC-anti Sca-1, FITC-anti CD31, CD45, and Ter119 and PE-anti-CD49f (eBioscience, San Diego, CA). Fluorescence-activated cell sorting (FACS) analyses were performed by using the BD LSR II (BD Biosciences).
Prostate Sphere Assay
Dissociated prostate epithelial cells were prepared from 5–6-week-old adult WT and AR3Tg mice as described previously (20). Prostate sphere assay was performed as described (29). Prostate cells were counted by hemocytometer and cultured in 1:1 Matrigel (BD Biosciences)/prostate epithelial growth medium (Lonza, Walkersville, MD). Ten days after plating, spheres with a diameter >50 μm were counted.
Histological, Immunohistochemical, and Immunofluorescent Analysis
The prostates were microdissected under a dissecting microscope. Tissues were fixed in 10% buffered formalin overnight, then washed and transferred to 70% alcohol. These paraffin-embedded tissues were sectioned (5 μm) and stained with hematoxylin and eosin. Antigen retrieval was performed by incubating slides in Antigen Unmasking Solution (H3300; Vector Laboratories). A Vectastain Elite ABC Kit (Vector Laboratories) was used for immunohistochemical staining according to the protocol recommended by the manufacturer. Rhodamine anti-mouse secondary antibody and Alexa Fluor 488 goat anti-rabbit secondary antibody (Invitrogen) were used for immunofluorescence analysis. To quantify the immunohistochemical staining, the total cells were counted from five independent view fields for each section, and the percentage was presented as the number of cells positively stained with the indicated antibodies/100 total nucleated cells.
Cell Culture, Transfection, and Infection
LNCaP cells were purchased from American Type Culture Collection. Prostate cancer cell lines C4-2 and CWR-R1 were kindly provided by Drs. Tindall and E. Wilson (30), respectively. The cells were transfected with FuGENE 6 (Roche Applied Science) following the manufacturer's instruction. The shRNAs specific for AR3 (shAR3) were described previously (11, 31).
Construction of Reporter Plasmids and Luciferase Reporter Assays
The putative hs-miR-29c target sites from the human IGF1 3′-UTR were amplified by PCR with primer1-F 5′-TACGTCTAGATAGTTGCAACTTTGAGGCCA-3′ and primer1-R 5′-TACGTCTAGACATCTTTGGCTCCAGGCTTC-3′ (first IGF1 3′-UTR site), primer2-F 5′-TACGTCTAGACAAAAAGCCTGTCCACCCTTG-3′ and primer2-R 5′-TACGTCTAGAGTTGAAAGGTGGTGGTGGCT-3′ (second IGF1 3′-UTR site). After digestion with XbaI, the two DNA fragments were, respectively, cloned into the unique XbaI site downstream of firefly luciferase gene in the pGL3-control vector (Promega). Luciferase assays were performed using the dual-luciferase reporter assay system (Promega). 0.2 μg/well of the above pGL3-control vector containing the first or second IGF1 3′-UTR site and the internal control Renilla luciferase phRL-SV40 plasmid and corresponding miR-control or miR-29c mimic were cotransfected into HEK293 cells by using Lipofectamine 2000, and firefly luciferase activity was measured 24 h after transfection. The results were normalized to Renilla luciferase according to the manufacturer's instructions.
RESULTS
To better understand the role of AR3 in prostate, we generated the AR3Tg mouse by using the modified probasin promoter ARR2PB (Fig. 1A). This promoter has been shown to be mainly activated in prostate epithelial cells (32, 33). The presence of the AR3 transgene in genomic DNA from the transgenic founders was confirmed by PCR (Fig. 1B, left). Expression of AR3 transgene in mouse prostates was further examined by RT-PCR, Western blotting, and immunofluorescence staining analyses. As predicted, we could only detect AR3 expression in the prostates of AR3Tg mice, not in their wild-type littermates (Fig. 1, B and C). RT-PCR results also indicated that transgene expression levels in the AR3Tg prostate vary in different lobes: highest in dorsolateral prostate (DLP) and ventral prostate (VP), least in anterior lobes (Fig. 1B, middle). Thus, we focused our study of AR3 function in DLP and VP. As the ARR2PB promoter is activated in mouse prostate as early as 3 weeks of age, we performed gene expression profiling in AR3Tg to identify differentially expressed genes in the AR3Tg at the age of 4 weeks, shortly after the expression of AR3 transgene was turned on. Our microarray analysis revealed that 414 genes were differentially expressed in AR3Tg prostates compared with their wild-type controls. Expression of some well established AR target genes such as kallikrein family proteins (Klk1 and Klk1b27) and androgen-binding proteins (Abpb and Abpd) were increased 20–100-fold, suggesting that overexpression of AR3 could enhance the expression of some known AR target genes. Interestingly, a modest but significant increase of a set of genes associated with stem/progenitor cells, including Ly6a/Sca-1, aldehyde dehydrogenase (Aldh1a1), Tacstd2/Trop2, and Tgfβ2, was detected in AR3Tg prostates. Meanwhile, expression of some microRNAs, such as mir-29c, was altered. Significant changes in expression of Sca-1, Tgfβ2, and mature miR-29 were validated by quantitative real-time PCR (Fig. 1D).
FIGURE 1.

Generation of the AR3Tg mouse model. A, schematic diagram of the ARR2PB-hAR3 construct. B, detection of the AR3 transgene. Left, genotyping of transgenic model AR3Tg. The transgene was exclusively detected in genomic DNA from the AR3Tg prostate, but not in that of the wild-type control (WT). Laminin was used as an internal control. Middle, AR3 expression was examined by RT-PCR using AR3-specific primers. Right, AR3 protein expression was determined by Western blot analysis using AR (N-20) and AR3 antibody. Actin served as a loading control. C, immunofluorescence staining performed by using AR3 antibody in frozen tissue sections of prostates from WT or AR3Tg. Nucleus was visualized using DAPI staining. D, levels of Sca-1 and Tgfβ2 transcripts in a pooled RNAs (n = 3) from WT or AR3Tg prostates determined by real-time PCR analysis with 18S rRNA as an internal control. The level of mir-29 expression in WT or AR3Tg was determined by real-time PCR using U6 (U6 small nuclear RNA) as an internal control. Error bars, S.D. *, p < 0.05.
Sca-1 is one of the stem cell markers that have been used to enrich murine prostatic stem/progenitor cells (18–20). Based on our microarray data that Sca-1 expression is up-regulated in the AR3Tg prostate, we hypothesized that AR3 may play a role in modulating expansion of prostate progenitor cells. To test this hypothesis, we took several approaches, including FACS analysis and sphere-forming analyses, to study the effects of AR3 on prostate progenitor cells. The Lin-Sca-1+CD49fhigh (LSC) subpopulation has been identified by enrichment of Sca-1+CD49fhigh and depletion of hematopoietic and endothelial lineage (CD45+CD31+Ter119+) in several studies (18–20). Previous studies also showed that the LSC subpopulation contains sufficient progenitor activity (18, 19). As shown in Fig. 2A, the LSC subpopulation was significantly increased in AR3Tg mice compared with age-matched wild-type controls. Trop2 has been used to identify a subpopulation of murine and human prostate basal cells with stem cell characteristics in a previous study (34). In this study, we observed an increase in the Sca-1+Trop2+ cell population in AR3Tg prostate (Fig. 2B). Furthermore, in sphere-forming assays, cells from the AR3Tg prostate formed more spheres than those from the wild-type control, indicating that more LSC cells are present in the AR3Tg prostate (Fig. 2C). P63 is expressed mainly in prostate basal cells and required for prostate development (35). P63-expressing cells possess higher self-renewal activity (29). A previous study showed that Pten deletion leads to expansion of a subset of prostate cancer cells positive for the basal epithelial markers such as p63 (36). Immunohistochemistry analyses revealed a significant increase of p63 positive cells in the AR3Tg prostate (data not shown). Taken together, these results demonstrated that overexpression of AR3 increased prostate progenitor cell population.
FIGURE 2.

Increase of prostate progenitor cell subpopulation in AR3Tg mice. A, detection of Lin-Sca-1+CD49f+ subpopulation in 5-week-old WT and AR3Tg prostates (n = 4) by FACS analysis is described under “Experimental Procedures.” The value in the WT controls was set as 1. Error bars, S.D. *, p < 0.05. B, FACS analysis demonstrates that Sca-1+Trop2+ population in prostates of AR3Tg mice was increased compared with their age and genetic background-matched littermate controls (n = 3). *, p < 0.05. C, prostate sphere assays were carried out as described under “Experimental Procedures.” Left, phase-contrast and H&E staining images show prostate spheres derived from prostate cells from the WT and AR3Tg mice (n = 3). Right, prostate spheres are quantified. The average number of spheres derived from the WT prostates was set as 1. **, p < 0.01.
TGFβ is well known for its role in promoting self-renewal and maintenance of human embryonic stem cells as well as prostate stem cells (37–39). Our microarray analysis and real-time quantitative PCR results showed that TGFβ2 expression was increased in AR3Tg prostates. The level of matured Tgfβ protein was also significantly increased in AR3Tg compared with wild-type controls (Fig. 3A). As a result, the levels of phosphorylation of the downstream effectors of Tgfβ, including Smad1 and Smad3, were elevated in AR3Tg, suggesting that the Tgfβ pathway was activated in AR3Tg prostates. We also examined whether TGFβ2 expression in human prostate cancer cells is regulated by AR3. As shown in Fig. 3B, TGFβ2 expression was decreased in AR3 knocked-down CWR-R1 cells compared with control cells. Consistent with the change of TGFβ2 transcript, the level of TGFβ protein and phosphorylation of Smad3 and Smad1/5 was also reduced when AR3 expression was knocked down (Fig. 3C). These data suggest that AR3 plays a role in regulating TGFβ2 expression both in vitro and in vivo. Accompanied with the activation of TGFβ signaling, expression of a number of epithelial-mesenchymal transition (EMT)-associated genes, such as N-cadherin, vimentin, snail, and twist, were up-regulated in AR3Tg prostates (Fig. 3, D and E), suggesting that overexpression of AR3 may poise the luminal epithelial cells for greater plasticity. Therefore, it is possible that activation of the Tgfβ pathway in prostate may halt epithelial differentiation and lead to accumulation of progenitor cells in AR3Tg prostates.
FIGURE 3.

AR3 modulates TGFβ2 expression in prostate cells and induces expression of EMT associated genes. A, increase of TGFβ signaling in AR3Tg prostates. Lysates from 1-month-old WT or AR3Tg prostates (n = 2) were subjected to Western blot analysis with antibodies as indicated. Actin was used as a loading control. B, effects of AR3 knockdown in prostate cancer cells on TGFβ2 expression. CWR-R1 cells were infected with lentivirus encoding the control shRNA (vector) or AR3 shRNA (shAR3) for 48 h in medium containing 5% charcoal-stripped FBS. Total RNAs were then extracted, and the level of TGFβ2 mRNA was determined by real-time PCR. 18S rRNA served as an internal control. Error bars, S.D. *, p < 0.05. C, effects of AR3 knockdown in prostate cancer cells on TGFβ signaling. CWR-R1 cells were treated as above. Total cell lysates were immunoblotted with antibodies as indicated. Tubulin was used as a loading control. D, expression of N-cadherin, slug1, Lcn2, vimentin, snail, and twist1 determined by quantitative PCR in the AR3Tg prostate compared with WT. E, immunofluorescence staining of N-cadherin and E-cadherin in AR3Tg prostate compared with WT.
Our microarray and real-time PCR data showed that miR-29 expression was decreased in AR3Tg prostate, suggesting that AR3 may negatively regulate miR-29 expression in mouse prostate cells. We examined whether AR3 plays a role in regulating miR-29 expression in human prostate cancer cells by either knockdown or overexpression of AR3. Fig. 4A shows that miR-29 expression was decreased in C4-2 cells overexpressing AR3 but increased in CWR-R1 cells when endogenous AR3 was knocked down. This was consistent with the results that we observed in AR3Tg model. Furthermore, the expression level of miR-29 was down-regulated in human prostate tumor tissues compared with their matched benign tissues (n = 3), whereas the relative level of AR3 was increased in the tumor tissues (Fig. 4B). These data suggested that AR3 could regulate miR-29 expression in both human and mouse prostate cells.
FIGURE 4.

AR3 regulates IGF1 expression through regulating mir-29. A, regulation of miR-29 expression by AR3. C4-2 cells were transfected with AR3 or vector control (left) or infected with the lentivirus encoding AR3 shRNA (shRA3) or the vector control shRNA (right) for 48 h, and total RNAs were isolated. The level of miR-29 was determined by quantitative real-time PCR analyses. Error bars, S.D. *, p < 0.05. B, inverse correlation between the expression levels of mature mir-29 and AR3 in human normal versus prostates cancer samples (n = 3). Total RNA was purified from three pairs of matched human normal (N) and prostate cancer (T) samples. The levels of mature mir-29, AR3, and AR mRNAs were determined by real-time PCR. The value of normal sample was set as 1. C, left, putative miR-29 binding sites in the 3′-UTR of Igf1 gene in mouse and human. Right, HEK293T cells cotransfected with luciferase reporters fused with IGF1 3′-UTRs or their reverse insertion with vector control or miR-29 mimics. Dual luciferase assays were carried out to determine the effects of miR-29 on the LUC reporter activity. The normalized luciferase activity of the vector control was set as 1. A representative experiment is shown in triplicate (n = 3). *, p < 0.05; **, p < 0.01. D, mir-29 negative regulation of IGF-1 expression in vitro. Left, C4-2 cells infected with the lentivirus encoding miR-29 or vector control for 48 h. Right, C4-2 cells transfected with the miR-29 inhibitor or negative control for 48 h. The levels of IGF1 and pAKT in these cells were then examined by Western blot analyses after infection or transfection. GAPDH served as a loading control. E, AR3 regulation of IGF1 expression in vitro. C4-2 cells were infected with the lentivirus encoding the control shRNA (vector) or AR3 shRNA (shAR3) for 48 h in medium containing 5% charcoal-stripped FBS. The levels of IGF1 and pAKT were examined by Western blot analysis. GAPDH served as an internal control. F, IGF1 protein level increased in AR3Tg prostates. Lysates from prostate tissues of 1-month-old WT or AR3Tg mice were subjected to Western blot analysis with antibodies as indicated. Actin was used as a loading control.
We then performed bioinformatic analysis using DIANA-microT v3.0 to search for potential miR-29 target genes (40, 41). In addition to known miR-29 target genes such as DNMT3a and DNMT3b (42), we found that both human and mouse IGF1 transcripts contain at least one putative miR-29 binding site in their 3′-UTR (Fig. 4C). To determine whether human IGF1 could be regulated by miR-29 through direct binding to its 3′-UTR, we inserted the DNA fragments of IGF1 3′-UTR containing each of the predicted miR-29 binding sites downstream of the luciferase reporter gene in the pGL3 vector. Luciferase activity of the reporters containing miR-29 binding sites was significantly decreased in the presence of miR-29, whereas activity of the reporters containing reversed inserts remained largely unchanged (Fig. 4C), indicating that miR-29 can directly regulate IGF1 expression through binding to its 3′-UTR. In addition, we detected a reduction of endogenous IGF1 protein and phosphorylation of its downstream AKT in C4-2 cells when miR-29 was overexpressed (Fig. 4D, left). However, inhibition of endogenous miR-29 expression in C4-2 cells significantly increased the level of IGF1 protein and phosphorylated AKT (Fig. 4D, right). Because AR3 inhibited miR-29 expression as shown earlier, we wondered whether AR3 could regulate IGF1 expression. As shown in Fig. 4E, knocking down AR3 expression in C4-2 cells significantly reduced the level of IGF1 and phosphorylated AKT. Meanwhile, both were elevated in AR3Tg prostate compared with wild-type control (Fig. 4F). Taken together, these results indicate that AR3 can regulate IGF1 expression through modulating miR-29 both in vitro and in vivo.
We have shown that overexpression of AR3 increases prostate progenitor cell population and up-regulates IGF1 expression. Transgenic mice overexpressing human IGF1 developed prostate neoplasia (43). We expected to detect similar pathological changes in our AR3Tg model. At the age of 1 year, approximately 43% of AR3Tg mice (n = 22) displayed pathologic changes resembling human PIN, a precancerous lesion that is limited within prostatic acini without frank stromal invasion. These PIN lesions were composed of atypical proliferating luminal epithelial cells displaying nuclear atypia, and the acini were often surrounded by thickened and reactive stroma (Fig. 5, B, D, and F). 7.4 ± 2.1% of AR3-positive cells in the PIN lesion regions are Ki67-positive. N-cadherin, which is elevated in primary and metastatic tumors of individuals with castration-resistant prostate cancer (44), was also highly expressed in PIN lesions of AR3Tg (Fig. 5J). Up-regulation of N-cadherin expression in LNCaP cells when AR3 was overexpressed (data not shown) suggested that AR3 could promote N-cadherin expression in prostate cells. Interestingly, AR3 expression in these 1-year-old AR3Tg mice was quite heterogeneous, and a high expression level of AR3 protein was found in PIN lesions (Fig. 5H), implying that sustained AR3 expression is associated with PIN and likely required for the development of PIN lesions in this model.
FIGURE 5.
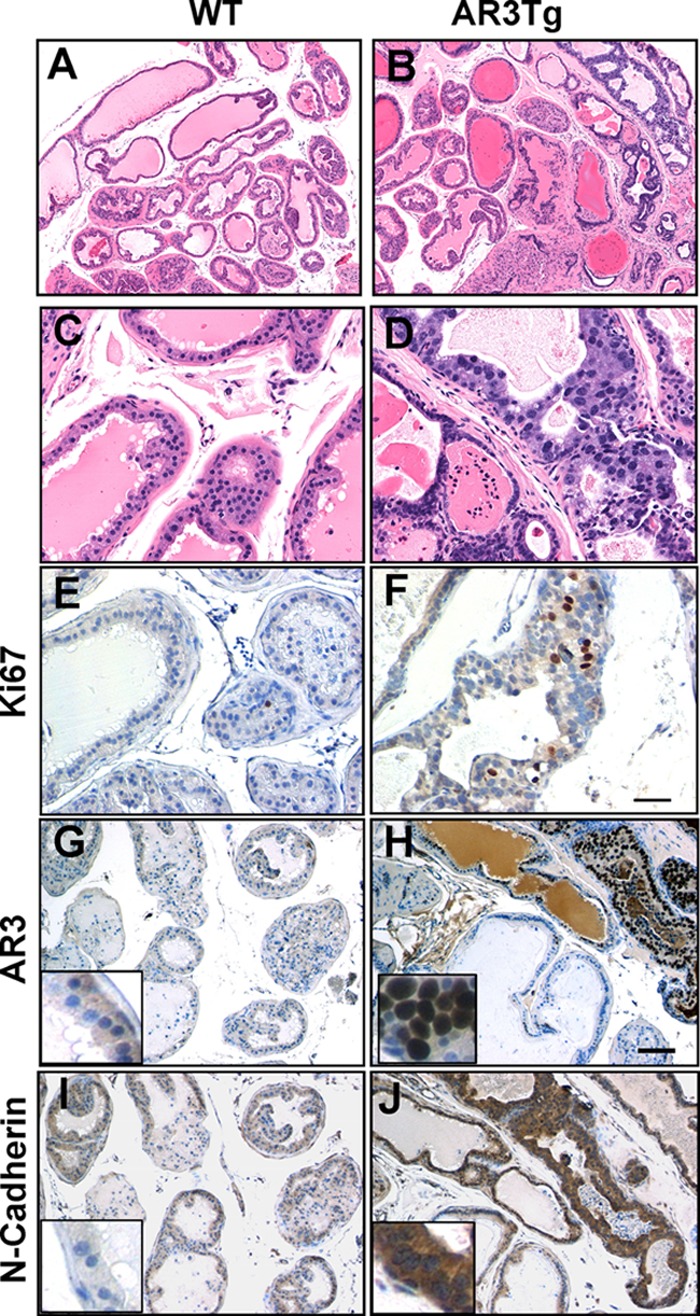
Overexpression of AR3 in mouse prostate epithelium leads to pathologic changes resembling PIN. The dorsolateral lobes of prostates were dissected from the 1-year-old WT or AR3Tg mice. A–D, H&E staining of dorsolateral prostates from WT (A and C) and AR3Tg (B and D). E and F, Ki67 staining showing that proliferation was increased in AR3Tg prostate (F) compared with WT prostate (E). G–J, immunohistochemistry staining showing that AR3 and N-cadherin were highly expressed in PIN lesions in AR3 transgenic mice (H and J) compared with WT mice (G and I).
Our previous study showed that AR3 may play an important role in castration-resistant prostate cancer. To determine whether overexpression of AR3 in prostate epithelial cells is sufficient to confer castration resistance, we castrated 6-week-old AR3Tg male mice along with age-matched wild-type mice. At 2 weeks after castration, prostates from these mice were collected and subjected to H&E staining. There was no apparent morphologic difference between AR3Tg and wild-type prostates (Fig. 6B, top panel). Because the AR3 transgene was driven by the modified probasin promoter, which could be affected by androgen depletion, we also examined whether the transgene was still expressed in AR3Tg prostate after castration. Immunohistochemistry staining revealed that AR expression was low but quite homogeneous and comparable in prostates from both castrated wild-type and transgenic mice (Fig. 6A, a and d). However, AR3 expression was highly heterogeneous in the degenerated acini (Fig. 6Ad), suggesting that the transgene was still expressed in a subset of prostate epithelial cells that are resistant to castration, possibly due to androgen-independent activity of endogenous Ar/Ar isoforms and/or AR3. Interestingly, we found that the majority (approximately 72%) of AR3-positive cells were Ck5+/Ck8+ intermediate cells (Fig. 6Ae), and the number of Ck5+/Ck8+ intermediate cells was increased in castrated AR3Tg prostates compared with WT controls (Fig. 6A, c and f), suggesting that overexpression of AR3 in luminal epithelial cells could lead to an increase of Ck5 expression. This is consistent with our observation that overexpression of AR3, but not AR, in LNCaP cells (which mainly express CK8) increased the number of CK5-positive cells (data not shown). We further investigated whether the increase of Ck5+/Ck8+ intermediate cells could affect prostate regeneration upon readministration of testosterone in castrated AR3Tg mice. H&E analysis revealed a significant difference between regenerated prostates from AR3Tg and WT mice. In contrast to lumens with well organized single-layer epithelial cells in the prostates from WT mice, lumens in regenerated AR3Tg prostates were filled with multiple layers of condensed and disorganized epithelial cells (Fig. 6B, bottom panel), which contained significantly higher number of Ki67-positive cells (data not shown) and Ck5+/Ck8+ intermediate cells (Fig. 6C), suggesting that AR3 may promote expansion of Ck5+/Ck8+ cells or halt terminal differentiation of these intermediate cells in the prostate.
FIGURE 6.

Effects of AR3 overexpression on mouse prostate in response to castration and subsequent regeneration. A, immunohistochemistry staining showing that AR3 expression was heterogeneous (d), whereas AR expression was low but homogeneous in both castrated WT and AR3Tg mice (a and d). The majority of AR3-positive cells are Ck5-positive cells (e). The number of Ck5+/Ck8+ intermediate cells was increased in prostates from castrated AR3Tg mice compared with WT controls (c and f). B, H&E staining of castrated (CS) and regenerated (RE) prostate lobes (ventral prostate (VP)) from WT and AR3 transgenic mice. WT and AR3Tg mice (n = 3) were castrated at 6-weeks of age. After 2 weeks they were injected with hormone (10 μg/g of body weight) for 7 days. C, immunofluorescence staining showing an increase of Ck5+/Ck8+ intermediate cells in regenerated AR3Tg prostates. The number of double positive cells was quantified manually in blind sections by using NIS-Elements image analysis software. Error bars, S.D. *, p < 0.05.
DISCUSSION
Although androgen ablation therapy is one of the most effective treatments of advanced prostate cancer, the role of AR in prostate cancer initiation remains unclear. An early study showed transgenic mice overexpressing wild-type AR under the control of rat probasin promoter develop PIN (45), whereas in another study, enforced expression of AR-WT did not cause significant prostate disease in vivo (46). However, specific knock-out of Ar in mouse prostatic epithelium increased proliferation of epithelial basal cells in the ventral prostate and led to apoptosis of epithelial luminal cells and enlargement of the prostate gland (47). Therefore, the exact role of AR in prostate cancer is still under debate. Our previous study suggests that the AR splice variant AR3 may play a role in prostate cancer initiation and progression (11, 27). In this study, enforced expression of AR3 under the control of the ARR2PB promoter in mice increases prostatic progenitor cell subpopulation and promotes development of lesions resembling PIN. Most importantly, our microarray analysis uncovered a group of genes preferentially regulated by AR3 in mouse prostates, which include a set of genes associated with prostate stem/progenitor cells and microRNAs deregulated in human cancers. We showed that AR3 can up-regulate Tgfβ2 expression in prostate cells, which in turn modulates the maintenance of stem/progenitor cells and prevents or delays the differentiation of luminal epithelial cells. Accompanied with the activation of TGFβ signaling, expression of a number of EMT-associated genes, such as N-cadherin, vimentin, snail and twist, are also up-regulated in AR3Tg prostates, suggesting that overexpression of AR3 in prostate epithelium may promote EMT. Furthermore, we showed that AR3 suppresses mir-29c expression and identified IGF1 as a new target gene of miR-29 in prostate cells. The miR-29 family of microRNAs are deregulated in prostate cancers (48, 49). We also observed that miR-29 expression is reduced in prostate tumors compared with their matched benign tissues, and the relative miR-29 expression in prostate tissues is inversely correlated with the relative expression level of AR3. These data suggest a role of AR3 in regulating miR-29 expression in prostate cells. Although TGFβ was previously shown to induce apoptosis in normal prostate epithelial cells, we did not detect an increase of apoptotic cells in AR3Tg prostates. This is probably due to simultaneous elevated survival pathway controlled by IGF1 and possibly other growth factors. High levels of circulating IGF1 are associated with prostate cancer risk (50–54). Levels of TGFβ are higher in prostate carcinoma cells than either the surrounding stromal cells or their normal prostatic epithelial counterparts (55). TGFβ has been shown to promote EMT and tumor invasion (56). Therefore, AR3 may promote tumor initiation and progression via modulating autocrine/paracrine factors, such as Tgfβ and Igf1. Our study provides new insights into the mechanisms by which AR3 plays a driving role in human prostate cancer despite of its heterogeneous expression pattern in human prostate tumors.
It seems that overexpression of AR3 in mouse prostate could increase the number of Ck5+/Ck8+ intermediate cells, whereas a previous study has shown that basal epithelial AR plays a suppressor role in proliferation of Ck5+/Ck8+ intermediate cells in pes_ARKO-TRAMP mice model (57). These observations suggest that AR3 may antagonize AR activity in regulating proliferation of Ck5+/Ck8+ intermediate cells. It has been reported that the growth-promoting effects of AR splice variants are mediated through the prototype AR in LNCaP cells (58). It is possible that AR activity is required for the function of AR3 and some other splice variants in some cell types, such as luminal epithelial cells. However, in the prostates of castrated AR3Tg mice, the majority of AR3-positive cells are Ck5+/Ck8+ intermediate cells, suggesting that AR3 can be functional to maintain the transgene expression in these intermediate cells in the absence of active AR. Therefore, the requirement of AR for AR3 activity is likely cell context-dependent. This notion is supported by previous studies that 22Rv1 cells are less dependent on AR, and the truncated AR isoforms can drive castration-resistant growth in these cells when AR is knocked down (59, 60). Interestingly, a recent study showed that the anti-androgens targeting the AR ligand binding domain promotes tumor metastasis via increasing TGFβ expression whereas the anti-AR N-terminal drug ASC-J9 inhibits tumor metastasis (61). ASC-J9 has been shown to inhibit AR3 activity in a number of prostate cancer cells (59). It is possible that ASC-J9 attenuates tumor metastasis at least in part through inhibiting AR3 activity whose activity is elevated upon castration, leading to TGFβ production and inhibition of miR-29. Because both TGFβ and miR-29 have been shown to play a critical role in metastasis, future study should be performed to evaluate the role of AR3 in tumor metastasis in compound mouse models in which AR3 transgene expression is independent of androgen. Such study would allow us to fully understand the role of AR3 in promoting metastasis under androgen depletion conditions.
This work was supported, in whole or in part, by National Institutes of Health Grant CA106504 (to Y. Q.). This work was also supported by Department of Defense Grant W81XWH-10-1-0309 (to Y. Q.).
The data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE52092).
- AR
- androgen receptor
- AR3Tg
- AR3 transgenic
- EMT
- epithelial-mesenchymal transition
- LSC
- Lin-Sca-1+CD49fhigh
- miRNA
- microRNA
- PIN
- prostatic intraepithelial neoplasia
- Sca-1
- stem cell antigen-1.
REFERENCES
- 1. Heinlein C. A., Chang C. (2004) Androgen receptor in prostate cancer. Endocr. Rev. 25, 276–308 [DOI] [PubMed] [Google Scholar]
- 2. Bolton E. C., So A. Y., Chaivorapol C., Haqq C. M., Li H., Yamamoto K. R. (2007) Cell- and gene-specific regulation of primary target genes by the androgen receptor. Genes Dev. 21, 2005–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang Q., Li W., Liu X. S., Carroll J. S., Jänne O. A., Keeton E. K., Chinnaiyan A. M., Pienta K. J., Brown M. (2007) A hierarchical network of transcription factors governs androgen receptor-dependent prostate cancer growth. Mol. Cell 27, 380–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Narayanan R., Jiang J., Gusev Y., Jones A., Kearbey J. D., Miller D. D., Schmittgen T. D., Dalton J. T. (2010) MicroRNAs are mediators of androgen action in prostate and muscle. PLoS One 5, e13637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Takayama K., Tsutsumi S., Katayama S., Okayama T., Horie-Inoue K., Ikeda K., Urano T., Kawazu C., Hasegawa A., Ikeo K., Gojyobori T., Ouchi Y., Hayashizaki Y., Aburatani H., Inoue S. (2011) Integration of cap analysis of gene expression and chromatin immunoprecipitation analysis on array reveals genome-wide androgen receptor signaling in prostate cancer cells. Oncogene 30, 619–630 [DOI] [PubMed] [Google Scholar]
- 6. Dai B., Chen H., Guo S., Yang X., Linn D. E., Sun F., Li W., Guo Z., Xu K., Kim O., Kong X., Melamed J., Qiu S., Qiu Y. (2010) Compensatory up-regulation of tyrosine kinase Etk/BMX in response to androgen deprivation promotes castration-resistant growth of prostate cancer cells. Cancer Res. 70, 5587–5596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Guo Z., Dai B., Jiang T., Xu K., Xie Y., Kim O., Nesheiwat I., Kong X., Melamed J., Handratta V. D., Njar V. C., Brodie A. M., Yu L. R., Veenstra T. D., Chen H., Qiu Y. (2006) Regulation of androgen receptor activity by tyrosine phosphorylation. Cancer Cell 10, 309–319 [DOI] [PubMed] [Google Scholar]
- 8. Mahajan N. P., Liu Y., Majumder S., Warren M. R., Parker C. E., Mohler J. L., Earp H. S., Whang Y. E. (2007) Activated Cdc42-associated kinase Ack1 promotes prostate cancer progression via androgen receptor tyrosine phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 104, 8438–8443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. DaSilva J., Gioeli D., Weber M. J., Parsons S. J. (2009) The neuroendocrine-derived peptide parathyroid hormone-related protein promotes prostate cancer cell growth by stabilizing the androgen receptor. Cancer Res. 69, 7402–7411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dehm S. M., Schmidt L. J., Heemers H. V., Vessella R. L., Tindall D. J. (2008) Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 68, 5469–5477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Guo Z., Yang X., Sun F., Jiang R., Linn D. E., Chen H., Kong X., Melamed J., Tepper C. G., Kung H. J., Brodie A. M., Edwards J., Qiu Y. (2009) A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 69, 2305–2313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hu R., Dunn T. A., Wei S., Isharwal S., Veltri R. W., Humphreys E., Han M., Partin A. W., Vessella R. L., Isaacs W. B., Bova G. S., Luo J. (2009) Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 69, 16–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sun S., Sprenger C. C., Vessella R. L., Haugk K., Soriano K., Mostaghel E. A., Page S. T., Coleman I. M., Nguyen H. M., Sun H., Nelson P. S., Plymate S. R. (2010) Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J. Clin. Invest. 120, 2715–2730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shen M. M., Abate-Shen C. (2010) Molecular genetics of prostate cancer: new prospects for old challenges. Genes Dev. 24, 1967–2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Scher H. I., Buchanan G., Gerald W., Butler L. M., Tilley W. D. (2004) Targeting the androgen receptor: improving outcomes for castration-resistant prostate cancer. Endocr. Relat. Cancer 11, 459–476 [DOI] [PubMed] [Google Scholar]
- 16. Scher H. I., Sawyers C. L. (2005) Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J. Clin. Oncol. 23, 8253–8261 [DOI] [PubMed] [Google Scholar]
- 17. Niu Y., Chang T. M., Yeh S., Ma W. L., Wang Y. Z., Chang C. (2010) Differential androgen receptor signals in different cells explain why androgen-deprivation therapy of prostate cancer fails. Oncogene 29, 3593–3604 [DOI] [PubMed] [Google Scholar]
- 18. Lawson D. A., Xin L., Lukacs R. U., Cheng D., Witte O. N. (2007) Isolation and functional characterization of murine prostate stem cells. Proc. Natl. Acad. Sci. U.S.A. 104, 181–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Leong K. G., Wang B. E., Johnson L., Gao W. Q. (2008) Generation of a prostate from a single adult stem cell. Nature 456, 804–808 [DOI] [PubMed] [Google Scholar]
- 20. Xin L., Lawson D. A., Witte O. N. (2005) The Sca-1 cell surface marker enriches for a prostate-regenerating cell subpopulation that can initiate prostate tumorigenesis. Proc. Natl. Acad. Sci. U.S.A. 102, 6942–6947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Goldstein A. S., Huang J., Guo C., Garraway I. P., Witte O. N. (2010) Identification of a cell of origin for human prostate cancer. Science 329, 568–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mulholland D. J., Xin L., Morim A., Lawson D., Witte O., Wu H. (2009) Lin-Sca-1+CD49fhigh stem/progenitors are tumor-initiating cells in the Pten-null prostate cancer model. Cancer Res. 69, 8555–8562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang X., Kruithof-de Julio M., Economides K. D., Walker D., Yu H., Halili M. V., Hu Y. P., Price S. M., Abate-Shen C., Shen M. M. (2009) A luminal epithelial stem cell that is a cell of origin for prostate cancer. Nature 461, 495–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hayward S. W., Haughney P. C., Rosen M. A., Greulich K. M., Weier H. U., Dahiya R., Cunha G. R. (1998) Interactions between adult human prostatic epithelium and rat urogenital sinus mesenchyme in a tissue recombination model. Differentiation 63, 131–140 [DOI] [PubMed] [Google Scholar]
- 25. Thomson A. A., Cunha G. R. (1999) Prostatic growth and development are regulated by FGF10. Development 126, 3693–3701 [DOI] [PubMed] [Google Scholar]
- 26. Hörnberg E., Ylitalo E. B., Crnalic S., Antti H., Stattin P., Widmark A., Bergh A., Wikström P. (2011) Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration resistance and short survival. PLoS One 6, e19059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Guo Z., Qiu Y. (2011) A new trick of an old molecule: androgen receptor splice variants taking the stage. Int. J. Biol. Sci. 7, 815–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dai B., Kim O., Xie Y., Guo Z., Xu K., Wang B., Kong X., Melamed J., Chen H., Bieberich C. J., Borowsky A. D., Kung H. J., Wei G., Ostrowski M. C., Brodie A., Qiu Y. (2006) Tyrosine kinase Etk/BMX is up-regulated in human prostate cancer and its overexpression induces prostate intraepithelial neoplasia in mouse. Cancer Res. 66, 8058–8064 [DOI] [PubMed] [Google Scholar]
- 29. Xin L., Lukacs R. U., Lawson D. A., Cheng D., Witte O. N. (2007) Self-renewal and multilineage differentiation in vitro from murine prostate stem cells. Stem Cells 25, 2760–2769 [DOI] [PubMed] [Google Scholar]
- 30. Gregory C. W., Johnson R. T., Jr., Mohler J. L., French F. S., Wilson E. M. (2001) Androgen receptor stabilization in recurrent prostate cancer is associated with hypersensitivity to low androgen. Cancer Res. 61, 2892–2898 [PubMed] [Google Scholar]
- 31. Guo S., Sun F., Guo Z., Li W., Alfano A., Chen H., Magyar C. E., Huang J., Chai T. C., Qiu S., Qiu Y. (2011) Tyrosine kinase ETK/BMX is up-regulated in bladder cancer and predicts poor prognosis in patients with cystectomy. PLoS One 6, e17778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wu X., Wu J., Huang J., Powell W. C., Zhang J., Matusik R. J., Sangiorgi F. O., Maxson R. E., Sucov H. M., Roy-Burman P. (2001) Generation of a prostate epithelial cell-specific Cre transgenic mouse model for tissue-specific gene ablation. Mech. Dev. 101, 61–69 [DOI] [PubMed] [Google Scholar]
- 33. Zhang J., Thomas T. Z., Kasper S., Matusik R. J. (2000) A small composite probasin promoter confers high levels of prostate-specific gene expression through regulation by androgens and glucocorticoids in vitro and in vivo. Endocrinology 141, 4698–4710 [DOI] [PubMed] [Google Scholar]
- 34. Goldstein A. S., Lawson D. A., Cheng D., Sun W., Garraway I. P., Witte O. N. (2008) Trop2 identifies a subpopulation of murine and human prostate basal cells with stem cell characteristics. Proc. Natl. Acad. Sci. U.S.A. 105, 20882–20887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Signoretti S., Waltregny D., Dilks J., Isaac B., Lin D., Garraway L., Yang A., Montironi R., McKeon F., Loda M. (2000) p63 is a prostate basal cell marker and is required for prostate development. Am. J. Pathol. 157, 1769–1775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang S., Garcia A. J., Wu M., Lawson D. A., Witte O. N., Wu H. (2006) Pten deletion leads to the expansion of a prostatic stem/progenitor cell subpopulation and tumor initiation. Proc. Natl. Acad. Sci. U.S.A. 103, 1480–1485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. James D., Levine A. J., Besser D., Hemmati-Brivanlou A. (2005) TGFβ/activin/nodal signaling is necessary for the maintenance of pluripotency in human embryonic stem cells. Development 132, 1273–1282 [DOI] [PubMed] [Google Scholar]
- 38. Salm S. N., Burger P. E., Coetzee S., Goto K., Moscatelli D., Wilson E. L. (2005) TGF-β maintains dormancy of prostatic stem cells in the proximal region of ducts. J. Cell Biol. 170, 81–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Blum R., Gupta R., Burger P. E., Ontiveros C. S., Salm S. N., Xiong X., Kamb A., Wesche H., Marshall L., Cutler G., Wang X., Zavadil J., Moscatelli D., Wilson E. L. (2009) Molecular signatures of prostate stem cells reveal novel signaling pathways and provide insights into prostate cancer. PLoS One 4, e5722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Maragkakis M., Alexiou P., Papadopoulos G. L., Reczko M., Dalamagas T., Giannopoulos G., Goumas G., Koukis E., Kourtis K., Simossis V. A., Sethupathy P., Vergoulis T., Koziris N., Sellis T., Tsanakas P., Hatzigeorgiou A. G. (2009) Accurate microRNA target prediction correlates with protein repression levels. BMC Bioinformatics 10, 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Maragkakis M., Reczko M., Simossis V. A., Alexiou P., Papadopoulos G. L., Dalamagas T., Giannopoulos G., Goumas G., Koukis E., Kourtis K., Vergoulis T., Koziris N., Sellis T., Tsanakas P., Hatzigeorgiou A. G. (2009) DIANA-microT web server: elucidating microRNA functions through target prediction. Nucleic Acids Res. 37, W273–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fabbri M., Garzon R., Cimmino A., Liu Z., Zanesi N., Callegari E., Liu S., Alder H., Costinean S., Fernandez-Cymering C., Volinia S., Guler G., Morrison C. D., Chan K. K., Marcucci G., Calin G. A., Huebner K., Croce C. M. (2007) MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc. Natl. Acad. Sci. U.S.A. 104, 15805–15810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. DiGiovanni J., Kiguchi K., Frijhoff A., Wilker E., Bol D. K., Beltrán L., Moats S., Ramirez A., Jorcano J., Conti C. (2000) Deregulated expression of insulin-like growth factor 1 in prostate epithelium leads to neoplasia in transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 97, 3455–3460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tanaka H., Kono E., Tran C. P., Miyazaki H., Yamashiro J., Shimomura T., Fazli L., Wada R., Huang J., Vessella R. L., An J., Horvath S., Gleave M., Rettig M. B., Wainberg Z. A., Reiter R. E. (2010) Monoclonal antibody targeting of N-cadherin inhibits prostate cancer growth, metastasis and castration resistance. Nat. Med. 16, 1414–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Stanbrough M., Leav I., Kwan P. W., Bubley G. J., Balk S. P. (2001) Prostatic intraepithelial neoplasia in mice expressing an androgen receptor transgene in prostate epithelium. Proc. Natl. Acad. Sci. U.S.A. 98, 10823–10828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Han G., Buchanan G., Ittmann M., Harris J. M., Yu X., Demayo F. J., Tilley W., Greenberg N. M. (2005) Mutation of the androgen receptor causes oncogenic transformation of the prostate. Proc. Natl. Acad. Sci. U.S.A. 102, 1151–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wu C. T., Altuwaijri S., Ricke W. A., Huang S. P., Yeh S., Zhang C., Niu Y., Tsai M. Y., Chang C. (2007) Increased prostate cell proliferation and loss of cell differentiation in mice lacking prostate epithelial androgen receptor. Proc. Natl. Acad. Sci. U.S.A. 104, 12679–12684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Porkka K. P., Pfeiffer M. J., Waltering K. K., Vessella R. L., Tammela T. L., Visakorpi T. (2007) MicroRNA expression profiling in prostate cancer. Cancer Res. 67, 6130–6135 [DOI] [PubMed] [Google Scholar]
- 49. Ozen M., Creighton C. J., Ozdemir M., Ittmann M. (2008) Widespread deregulation of microRNA expression in human prostate cancer. Oncogene 27, 1788–1793 [DOI] [PubMed] [Google Scholar]
- 50. Mantzoros C. S., Tzonou A., Signorello L. B., Stampfer M., Trichopoulos D., Adami H. O. (1997) Insulin-like growth factor 1 in relation to prostate cancer and benign prostatic hyperplasia. Br. J. Cancer 76, 1115–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wolk A., Mantzoros C. S., Andersson S. O., Bergström R., Signorello L. B., Lagiou P., Adami H. O., Trichopoulos D. (1998) Insulin-like growth factor 1 and prostate cancer risk: a population-based, case-control study. J. Natl. Cancer Inst. 90, 911–915 [DOI] [PubMed] [Google Scholar]
- 52. Chan J. M., Stampfer M. J., Giovannucci E., Gann P. H., Ma J., Wilkinson P., Hennekens C. H., Pollak M. (1998) Plasma insulin-like growth factor-I and prostate cancer risk: a prospective study. Science 279, 563–566 [DOI] [PubMed] [Google Scholar]
- 53. Harman S. M., Metter E. J., Blackman M. R., Landis P. K., Carter H. B. (2000) Serum levels of insulin-like growth factor I (IGF-I), IGF-II, IGF-binding protein-3, and prostate-specific antigen as predictors of clinical prostate cancer. J. Clin. Endocrinol. Metab. 85, 4258–4265 [DOI] [PubMed] [Google Scholar]
- 54. Chokkalingam A. P., Pollak M., Fillmore C. M., Gao Y. T., Stanczyk F. Z., Deng J., Sesterhenn I. A., Mostofi F. K., Fears T. R., Madigan M. P., Ziegler R. G., Fraumeni J. F., Jr., Hsing A. W. (2001) Insulin-like growth factors and prostate cancer: a population-based case-control study in China. Cancer Epidemiol. Biomarkers Prev. 10, 421–427 [PubMed] [Google Scholar]
- 55. Perry K. T., Anthony C. T., Steiner M. S. (1997) Immunohistochemical localization of TGFβ1, TGFβ2, and TGFβ3 in normal and malignant human prostate. Prostate 33, 133–140 [DOI] [PubMed] [Google Scholar]
- 56. Ao M., Williams K., Bhowmick N. A., Hayward S. W. (2006) Transforming growth factor-β promotes invasion in tumorigenic but not in nontumorigenic human prostatic epithelial cells. Cancer Res. 66, 8007–8016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Niu Y., Altuwaijri S., Lai K. P., Wu C. T., Ricke W. A., Messing E. M., Yao J., Yeh S., Chang C. (2008) Androgen receptor is a tumor suppressor and proliferator in prostate cancer. Proc. Natl. Acad. Sci. U.S.A. 105, 12182–12187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Watson P. A., Chen Y. F., Balbas M. D., Wongvipat J., Socci N. D., Viale A., Kim K., Sawyers C. L. (2010) Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc. Natl. Acad. Sci. U.S.A. 107, 16759–16765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yamashita S., Lai K. P., Chuang K. L., Xu D., Miyamoto H., Tochigi T., Pang S. T., Li L., Arai Y., Kung H. J., Yeh S., Chang C. (2012) ASC-J9 suppresses castration-resistant prostate cancer growth through degradation of full-length and splice variant androgen receptors. Neoplasia 14, 74–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tsai H. C., Boucher D. L., Martinez A., Tepper C. G., Kung H. J. (2012) Modeling truncated AR expression in a natural androgen-responsive environment and identification of RHOB as a direct transcriptional target. PLoS One 7, e49887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lin T. H., Lee S. O., Niu Y., Xu D., Liang L., Li L., Yeh S. D., Fujimoto N., Yeh S., Chang C. (2013) Differential androgen deprivation therapies with anti-androgens of Casodex or MDV3100 vs. anti-androgen receptor of ASC-J9 lead to promote vs. suppress prostate cancer metastasis. J. Biol. Chem. 288, 19359–19369 [DOI] [PMC free article] [PubMed] [Google Scholar]