

Background: Diacylglycerol kinases (DGKs) synthesize phosphatidic acid (PA), and PA can activate growth-regulatory mTOR signaling.
Results: The ζ isoform of DGK is necessary for a mechanically induced increase in PA-mTOR signaling, and overexpression of DGKζ induces skeletal muscle hypertrophy.
Conclusion: PA synthesized by DGKζ regulates the mechanical activation of mTOR signaling and hypertrophy.
Significance: DGKζ is a potential target for treating muscle atrophy/wasting.
Keywords: Mechanotransduction, mTOR, Phosphatidic Acid, Phospholipase D, Skeletal Muscle, Diacylglycerol Kinase ζ, Hypertrophy
Abstract
The activation of mTOR signaling is essential for mechanically induced changes in skeletal muscle mass, and previous studies have suggested that mechanical stimuli activate mTOR (mammalian target of rapamycin) signaling through a phospholipase D (PLD)-dependent increase in the concentration of phosphatidic acid (PA). Consistent with this conclusion, we obtained evidence which further suggests that mechanical stimuli utilize PA as a direct upstream activator of mTOR signaling. Unexpectedly though, we found that the activation of PLD is not necessary for the mechanically induced increases in PA or mTOR signaling. Motivated by this observation, we performed experiments that were aimed at identifying the enzyme(s) that promotes the increase in PA. These experiments revealed that mechanical stimulation increases the concentration of diacylglycerol (DAG) and the activity of DAG kinases (DGKs) in membranous structures. Furthermore, using knock-out mice, we determined that the ζ isoform of DGK (DGKζ) is necessary for the mechanically induced increase in PA. We also determined that DGKζ significantly contributes to the mechanical activation of mTOR signaling, and this is likely driven by an enhanced binding of PA to mTOR. Last, we found that the overexpression of DGKζ is sufficient to induce muscle fiber hypertrophy through an mTOR-dependent mechanism, and this event requires DGKζ kinase activity (i.e. the synthesis of PA). Combined, these results indicate that DGKζ, but not PLD, plays an important role in mechanically induced increases in PA and mTOR signaling. Furthermore, this study suggests that DGKζ could be a fundamental component of the mechanism(s) through which mechanical stimuli regulate skeletal muscle mass.
Introduction
Mechanical stimuli play a critical role in the regulation of skeletal muscle mass, and the maintenance of muscle mass contributes significantly to health and issues associated with quality of life (1, 2). However, the mechanism(s) via which mechanical stimuli are converted into the molecular events that regulate muscle mass remain poorly defined. Nevertheless, advancements in our understanding are being made, and it is becoming increasingly evident that a protein kinase called the mammalian (or mechanistic) target of rapamycin (mTOR)2 is a key determinant in the mechanical regulation of muscle mass. For example, many studies have demonstrated that mechanical stimulation leads to the activation of mTOR signaling, and its inhibition with the drug rapamycin prevents mechanically induced increases in muscle mass (3–5). Furthermore, it has been shown that the activation of mTOR signaling is sufficient to induce an increase in muscle fiber size (i.e. hypertrophy) (6). These observations are important because they provide the cornerstone (i.e. mTOR signaling) for investigations that are aimed at developing pharmacological treatments that can mimic the effects of mechanical stimuli on muscle mass. However, to develop such treatments, we will first need to understand how mechanical stimuli regulate mTOR signaling.
Based on previous studies, it has been concluded that mechanical stimuli activate mTOR signaling through a unique mechanism that does not require typical candidates such as PI3K, protein kinase B, ERK, exogenous nutrients, or intracellular calcium (7–11). However, the identity of the unique mechanism remains unknown (4). Nonetheless, an emerging body of evidence suggests that phosphatidic acid (PA) may play a key role in this process. For instance, various forms of mechanical stimuli can promote an increase in the intracellular concentration of PA (8, 10, 12). Furthermore, just like mechanical stimulation, PA can activate mTOR signaling via a PI3K- and ERK-independent mechanism (8, 11). It has also been shown that PA can bind to the FKBP12-rapamycin binding (FRB) domain of mTOR, and it can directly activate mTOR kinase activity in vitro (11, 13, 14). Based on these points, it has been proposed that a mechanically induced increase in PA could lead to an enhanced binding of PA to mTOR and in turn facilitate the activation of mTOR signaling and ultimately muscle growth.
The potential role of PA in the mechanical activation of mTOR signaling was first exposed in 2006 when it was shown that: (i) mechanical stretch induces a transient increase in the activity of phospholipase D (PLD), (ii) the use of 1-butanol to inhibit the synthesis of PA by PLD completely prevents the mechanically induced increase in PA and mTOR signaling, and (iii) mTOR signaling in mechanically stimulated muscles exhibits resistance to the inhibitory effects of rapamycin (10). With regard to this last point, it is important to consider that rapamycin forms a complex with the FKBP12 protein, and like PA, this complex can bind to the FRB domain in mTOR (an event that is responsible for the inhibitory effects of rapamycin on mTOR signaling) (15, 16). Furthermore, it has been suggested that PA and the rapamycin-FKBP12 complex can compete with each other for binding to the FRB domain, and due to this competition, an enhanced binding of PA to the FRB domain would be expected to induce resistance to the inhibitory effects of rapamycin (17, 18). For these reasons it has been concluded that the rapamycin resistance that is observed in mechanically stimulated muscles indicates that mechanical stimulation promotes an enhanced binding of PA to mTOR.
A potential role of PA in the mechanical activation of mTOR signaling was further explored in a subsequent study that employed a bout of eccentric contractions as the source of mechanical stimulation (8). Again, it was determined that 1-butanol prevents the mechanical activation of mTOR signaling. Thus, the results from this study provided further support for the conclusion that a PLD-dependent increase in PA is necessary for the mechanical activation of mTOR signaling. However, more recent studies have shown that many of the biological effects of 1-butanol cannot be attributed to the inhibition of the synthesis of PA by PLD (19–21). Moreover, the time course of the mechanically induced increase in PLD activity does not adequately correspond with the increase in PA, and hence, it has been argued that a mechanically induced increase in PLD activity may not fully explain how mechanical stimuli promote an increase in PA (4). In other words the potential role of PLD and/or PA in the mechanical activation of mTOR signaling remains a subject of debate. Therefore, the initial aim of the current study was to re-evaluate the role that PLD plays in controlling the mechanically induced changes in PA and mTOR signaling. Surprisingly, our results demonstrated that changes in PLD activity are not required for these events. Instead, we obtained evidence which indicates that the ζ isoform of diacylglycerol kinase (DGKζ), an enzyme that synthesizes PA via the phosphorylation of diacylglycerol (DAG), is largely responsible for the mechanically induced increase in PA and significantly contributes to the mechanical activation of mTOR signaling. Furthermore, we determined that the overexpression of DGKζ is sufficient to induce hypertrophy, and this effect occurs through a rapamycin-sensitive mechanism that requires DGKζ kinase activity (i.e. the synthesis of PA). Combined, these results highlight a novel role for DGKζ, but not PLD, in the mechanical activation of PA-mTOR signaling and reveal new insights into the potential mechanism(s) through which mechanical stimuli ultimately regulates muscle mass.
EXPERIMENTAL PROCEDURES
Materials
PA (1,2-dioctanoyl-sn-glycero-3-phosphate (C8 PA)), phosphatidylcholine (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (PC)), DAG (1–2-dioleoyl-sn-glycerol), and phosphatidylbutanol (1,2-dioleoyl-sn-glycero-3-phosphobutanol (PtdBut)) were purchased from Avanti Lipids (Alabaster, AL). Phosphatidylserine (1,2-dipalmitoyl-sn-glycero-3-phosphoserine (PS)) were purchased from Echelon Biosciences (Salt Lake City, UT). [3H]Myristic acid and [γ-32P]ATP were purchased from Perkin Elmer (Waltham, MA). Rapamycin was purchased from LC laboratories (Woburn, MA). 5-Fluoro-2-indolyl des-chlorohalopemide (FIPI) was purchased from Sigma. 12-O-Tetradecanoylphorbol-13-acetate (TPA), and R59949 was purchased from Enzo Life Sciences (Farmingdale, NY). These drugs were all dissolved in DMSO to make stock solutions.
Rabbit anti-ribosomal S6 kinase (p70S6k), anti-phospho-ERK1/2 (Thr-202/Tyr-204), anti-ERK1/2, anti-regulatory-associated protein of mTOR (Raptor), anti-mTOR, anti-GFP, and anti-lactate dehydrogenase (LDH) antibodies were purchased from Cell Signaling (Danvers, MA). Rabbit anti-phospho-p70S6k (Thr-389), goat anti-DGKζ, and FITC-conjugated goat anti-rat IgG antibodies were purchased from Santa Cruz Biotechnologies (Santa Cruz, CA). Mouse anti-Na+/K+-ATPase antibody was purchased from Developmental Studies Hybridoma Bank (Iowa City, IA). Rat anti-HA antibody was purchased from Roche Applied Science. Rabbit anti-laminin antibody was purchased from Sigma. HRP-conjugated anti-rabbit IgG antibody was purchased from Vector Laboratories (Burlingame, CA). HRP-conjugated anti-mouse IgG, HRP-conjugated anti-rat IgG, and DyLight 594-conjugated anti-rabbit IgG antibodies were purchased from Jackson ImmunoResearch Laboratories Inc. (West Grove, PA).
Plasmid Constructs and Purification
pEGFP-C3 (GFP) was purchased from Clontech (Mountain View, CA). pRK5-myc-p70S6k-glutathione transferase (GST-p70S6k) was kindly provided by Dr. Karyn Esser (University of Kentucky, Lexington, KY). YFP-tagged Ras homologue enriched in brain (YFP-Rheb) was obtained from Dr. Kun-Liang Guan (University of California San Diego, La Jolla, CA). pHA3-DGKζ with three tandem N-terminal HA epitope tags (HA-WT-DGKζ) was a generous gift from Dr. Matthew Topham (University of Utah, Salt Lake City, UT). pHA3-kinase dead DGKζ (HA-KD-DGKζ) was generated from the pHA3-DGKζ via a guanine-to-adenosine change (22) at nucleotide 1756 of DGKζ (NCBI Reference Sequence NM_001105540.1) using a QuikChange Lightning Mutagenesis kit (Agilent Technologies, Inc., Santa Clara, CA). All plasmid DNA was grown in DH5α Escherichia coli, purified with an Endofree plasmid kit (Qiagen, Valencia, CA), and resuspended in sterile PBS.
Animals
Wild-type FVB/N and C57BL/6 mice were purchased from Charles River Laboratories (Wilmington, MA), Taconic (Hudson, NY), or The Jackson Laboratory (Bar Harbor, MA). DGKζ−/− C57BL/6 mice have been described previously (23) and were kindly provided by Dr. Xiao-Ping Zhong (Duke University, Durham, NC). All animals were housed in a room maintained at 25 °C with a 12-h:12-h light:dark cycle and received food and water ad libitum. 8–10-Week-old animals were used for all experiments, and they were anesthetized with an intraperitoneal injection of ketamine (100 mg/kg) and xylazine (10 mg/kg) before all surgical procedures. After muscle collection, mice were sacrificed by cervical dislocation under anesthesia. For all non-histochemical analyses, the muscles were immediately frozen in liquid nitrogen at the end of the experimental procedures. All methods were approved by the Institutional Animal Care and Use Committee of the University of Wisconsin-Madison under protocol # V01324.
Ex Vivo Mechanical Stimulation
Mouse extensor digitorum longus (EDL) muscles were mechanically stimulated in an ex vivo organ culture system that consisted of a refined myograph apparatus (Kent Scientific, Torrington, CT) and an organ culture bath as previously described (7). Briefly, the proximal and distal tendons of EDL muscles were connected to a micromanipulator and the lever arm of a force transducer, respectively. Then the length of the muscle was adjusted until a passive tension of 13.5 mN was obtained (note: preliminary studies demonstrated that the optimal length (Lo) of EDL muscles was obtained at 13.5 mN). The muscles were then subjected to intermittent 15% passive stretch as a source of mechanical stimulation or held static at Lo as a control condition. The bath incubation media consisted of Krebs-Henseleit buffer (120 mm NaCl, 4.8 mm KCl, 25 mm NaHCO3, 2.5 mm CaCl2, 1.2 mm KH2PO4, 2 mm MgSO4, and 5 mm HEPES) supplemented with 1× minimum essential medium amino acid mixture (Invitrogen) and 25 mm glucose. The medium was maintained at 37 °C with continuous 95% O2 and 5% CO2 gassing, and it was exchanged with fresh medium at 30 min intervals.
Skeletal Muscle Transfection and Rapamycin Injections
Mouse tibialis anterior (TA) muscles were transfected by electroporation as previously described (6). In brief, a small incision was made through the skin covering the TA muscle. A total of 30–50 μg of plasmid DNA (GFP, HA-WT-DGKζ, or HA-KD-DGKζ) solution was then injected into the proximal (6 μl) and distal (6 μl) ends of the muscle belly with a 27-gauge needle. After the injections, 2 stainless steel pin electrodes (1-cm gap) connected to an ECM 830 electroporation unit (BTX/Harvard Apparatus, Holliston, MA) were laid on top of the proximal and distal myotendinous junctions. Then, eight 20-ms square-wave electric pulses at a frequency of 1 Hz were delivered onto the muscle with a field strength of 160 V/cm. After the electroporation procedure, incisions were closed with Vetbond surgical glue, and the animals were allowed to recover for 3 or 7 days. In some experiments vehicle or rapamycin solution was administered into the animals via intraperitoneal injections immediately after the electroporation procedure. Rapamycin solution was prepared by dissolving the appropriate volume of the stock solution (in DMSO) that was needed to inject mice with 1.5 or 4.5 mg/kg of body weight in 200 μl of PBS. For the vehicle control solution, an equivalent amount of DMSO was dissolved in 200 μl of PBS. These injections were repeated every 24 h.
Cell Culture and Transfection
C2C12 myoblasts were cultured in growth media consisting of high glucose DMEM (HyClone, Logan, UT) supplemented with antibiotics and antimycotics (100 μg/ml streptomycin, 100 units/ml penicillin, and 0.25 μg/ml amphotericin) and 10% fetal bovine serum (Invitrogen). For mTOR kinase activity assay, C2C12 myoblasts stably expressing FLAG-tagged mTOR (FLAG-mTOR) were obtained from Dr. Jie Chen (University of Illinois, Urbana, IL) and cultured in the growth medium that was further supplemented with 0.2 mg/ml G418 (HyClone). For stimulation experiments, the growing myoblasts were plated on 6-well dishes at 40–60% confluence. During this time, if needed, cells were co-transfected with 2 μg of GFP, HA-WT-DGKζ, or HA-KD-DGKζ and 0.2 μg of GST-p70S6k constructs by using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. The following day the cells were serum-starved in the absence of any antibiotics and antimycotics before being subjected to experimental treatments. Cell cultures were maintained at 37 °C in a humidified atmosphere of 95% air and 5% CO2.
Fractionation
Frozen muscles were homogenized with a Polytron homogenizer in ice-cold fractionation buffer (20 mm Tris (pH 7.5), 250 mm sucrose, 1 mm EDTA, 1 mm EGTA, 1 mm DTT, 50 mm NaF, 1 mm Na3VO4, 1 mm PMSF, and 20 μg/ml each leupeptin, pepstatin, aprotinin, and soybean trypsin inhibitor), and the homogenates were precleared by centrifugation at 1000 × g (4 °C) for 10 min. The precleared supernatants (whole lysate) were further centrifuged at 100,000 × g (4 °C) for 1 h to separate the soluble supernatants (cytosolic fraction) and insoluble pellets (crude membrane fraction) as previously described (24). The membrane pellets were washed once and resuspended in the ice-cold fractionation buffer for DGK activity assay or Laemmli buffer for Western blot analysis. The protein concentration in each whole lysate sample was determined with the DC protein assay kit (Bio-Rad) and used to control the loading amounts of cytosolic and/or membrane protein in the DGK activity assay and Western blot analysis.
In Vitro DGK Activity Assay
DGK activity was measured by the standard octyl glucoside (OG)/PS-mixed micelle assay as previously described (25). In brief, OG/PS-mixed micelles were prepared by resuspending dried DAG and PS in 5× OG buffer (275 mm OG, 1 mm diethylenetriaminepentaacetic acid) with vortexing and sonication. Then the reaction was initiated by adding 10 μl of each diluted sample into 90 μl of reaction mixture (20 μl of mixed micelles added to 70 μl of reaction buffer), which yielded a final concentration of (55 mm OG, 0.25 mm DAG (0.87 mol% in micelles), 1 mm PS, 50 mm imidazole (pH 6.6), 50 mm NaCl, 12.5 mm MgCl2, 1 mm EGTA, 0.2 mm diethylenetriaminepentaacetic acid, 1 mm DTT, 500 μm ATP, and 1 μCi [γ-32P]ATP). After a 30-min incubation at 25 °C, the reaction was terminated by the addition of 0.45 ml chloroform-methanol 1:2 (v/v) and 0.15 ml of 1% perchloric acid. After vortexing, 0.15 ml of chloroform and 0.15 ml of 1% perchloric acid were added to the mixture and vortexed again. The mixture was centrifuged at 2000 × g for 5 min, and the lower organic phase was washed twice with 1% perchloric acid. [γ-32P]PA in the organic phase was separated on LK5D silica gel plates by TLC with a solvent system consisting of ethyl acetate-isooctane-acetic acid-water 13:2:3:10 (v/v) as previously described (10). The amount of [γ-32P]PA was then visualized and quantified with a Storm PhosphorImager (GE Healthcare).
In Vitro mTOR Kinase Activity Assay
Growing C2C12 myoblasts stably expressing FLAG-mTOR were plated and then maintained in G418-free growth media. Upon confluence, the cells were serum-starved overnight and then collected in either ice-cold CHAPS lysis buffer (40 mm HEPES (pH 7.4), 2 mm EDTA, 0.3% CHAPS, 10 mm sodium pyrophosphate, 10 mm β-glycerophosphate, and 1 tablet of EDTA-free protease inhibitors (Roche Applied Science) per 25 ml) or Triton X-100 lysis buffer (40 mm Tris (pH 7.5), 1 mm EDTA, 5 mm EGTA, 0.5% Triton X-100, 25 mm β-glycerol phosphate, 25 mm NaF, 1 mm Na3VO4, 10 μg/ml leupeptin, and 1 mm PMSF). Fresh cell lysates were centrifuged at 2500 × g for 5 min, and 100–150 μg of protein from the supernatant was immunoprecipitated for the FLAG tag by incubating with 10 μl of EZview red ANTI-FLAG M2-agarose affinity gel beads (Sigma) at 4 °C for 2 h. After the incubation, the beads were pelleted by centrifugation at 500 × g for 30 s and washed with fresh ice-cold wash buffer (40 mm HEPES (pH 7.4), 150 mm NaCl, 2 mm EDTA, 0.3% CHAPS, 10 mm sodium pyrophosphate, 10 mm β-glycerophosphate, and 1 tablet of EDTA-free protease inhibitors per 25 ml) 3 times and kinase wash buffer (25 mm HEPES (pH 7.4) and 20 mm KCl) 2 times.
For lipid stimulations, C8 PA vesicles (50% C8 PA and 50% PC) or PC vesicles (100% PC) were prepared by drying lipids under nitrogen gas and dissolving in vesicle buffer (150 mm NaCl and 10 mm Tris (pH 8.0)) with 5 min of sonication at a concentration of 6 mm. The lipid vesicles were then diluted to 150 μm in 1.5× reaction buffer (see below), and 10 μl of this solution was incubated with the immunoprecipitates at 30 °C for 15 min. The mTOR kinase reaction toward purified GST-p70S6k (11) was initiated by adding 5 μl of 750 μm ATP into the immunoprecipitates, which yields a final mTOR kinase assay buffer that contains (25 mm HEPES (pH 7.4), 50 mm KCl, 10 mm MgCl2, 250 μm ATP, 50 ng of GST-p70S6k, and 100 μm C8 PA or PC vesicles). After 20 min of incubation, the reaction was terminated by the addition of 25 μl of 2× Laemmli buffer. Samples were boiled for 5 min and subjected to Western blot analysis as described below.
Western Blot Analysis
Frozen muscles were homogenized in the ice-cold Triton X-100 lysis buffer and the whole homogenates were used for analysis. Myoblasts were lysed in the ice-cold Triton X-100 buffer and centrifuged at 500 g (4 °C) for 5 min, and the supernatant was used for further analyses. The protein concentration in each sample was determined with the DC protein assay kit.
Equal amounts of protein from each sample were dissolved in 2× Laemmli buffer before being subjected to SDS-PAGE. After the electrophoretic separation, proteins were transferred to a PVDF membrane, blocked with 5% milk in TBST (TBS with 1% Tween 20) for 1 h, and probed with the indicated primary antibodies overnight at 4 °C. The membranes were then washed for 30 min in TBST and incubated with the appropriate HRP-conjugated secondary antibodies for 45 min at room temperature. After 30 min of washing in TBST, the blots were incubated in regular ECL (Pierce) or ECL Prime (GE healthcare) for 5 min at room temperature. Images of the blots were either captured on film or with a Chemi410 camera mounted to a UVP Autochemi system (UVP, Upland, CA). Once the appropriate image was captured, the membranes were stained with Coomassie Blue to verify equal loading throughout all lanes. Densitometric measurements of each blot were carried out using ImageJ (National Institutes of Health).
Immunohistochemical Analysis
Transfected muscles were subjected to immunohistochemistry as previously described (6). Briefly, collected muscles were incubated in PBS containing 4% paraformaldehyde with gentle rocking at 4 °C for 30 min. The muscles were then submerged in optimal cutting temperature compound (Tissue-Tek; Sakura, Torrance, CA) at resting length and frozen in liquid nitrogen-chilled isopentane. Cross-sections (10 μm in thickness) from the mid-belly of the muscle were obtained with a cryostat and fixed in −20 °C acetone for 10 min. Sections were warmed to room temperature for 5 min and then rehydrated with cool steam vapors followed by a 15-min incubation in PBS. Note that in experiments that did not include GFP-transfected muscles, the above paraformaldehyde and steam vapor treatments were omitted as in Goodman et al. (26). The rehydrated sections were then incubated in solution A (PBS containing 0.5% BSA and 0.5% Triton X-100) for 20 min and probed with the indicated primary antibodies dissolved in solution A for 45 min at room temperature. Sections were washed with PBS and then incubated with the appropriate fluorophore-conjugated secondary antibodies dissolved in solution A for 1 h at room temperature. Finally, the sections were washed with PBS, and images of the different fluorophores were captured with a DS-QiMc camera on an 80i epifluorescence microscope (both from Nikon, Tokyo, Japan). The monochrome images were merged with NIS-Elements D image analysis software (Nikon), and the average cross-sectional area (CSA) of 35–120 randomly selected fibers per sample was measured by tracing the periphery of individual fibers along the laminin stain. All analyses were performed by investigators that were unaware of the sample identification.
Analysis of PLD Activity and Lipid Concentrations
PLD activity and PA, and DAG concentrations in skeletal muscles ex vivo were measured with modifications of previously described methods (10). Briefly, EDL muscles were prelabeled in the organ culture system with media containing [3H]myristic acid (2.5 μCi/ml) for 2 h and then subjected to experimental treatments. For the measurement of PLD activity, the PLD-mediated transphosphatidylation reaction was initiated by incubating the muscles with fresh medium containing 0.5% 1-butanol during the final 15 min of the treatments. The muscles were homogenized with a Polytron in chloroform-methanol 2:1 (v/v), and total lipids were extracted according to Folch et al. (56). The extracted lipids were combined with 10 μg of PtdBut, PA, or DAG standards and then subjected to TLC as described in the DGK activity assay section. The solvent system consisted of ethyl acetate-isooctane-acetic acid-water 13:2:3:10 (v/v) for separation of PtdBut and PA or hexane-diethyl ether-acetic acid 50:50:1 (v/v) for separation of DAG. The standard PtdBut, PA, or DAG spots containing the 3H-labeled PtdBut, PA, or DAG, respectively, was visualized by iodine staining and scraped off the TLC plate to count the amount of radioactivity by liquid scintillation spectrometry. An aliquot of the extracted lipids was also subjected to the liquid scintillation spectrometry to count radioactivity in the total lipids. Final calculations for PLD activity, PA, and DAG were made by dividing the amount of radioactivity in the Ptdbut, PA, or DAG spot by the amount of radioactivity in the total lipid extract.
For analysis of PLD activity and PA in cells, myoblasts were prelabeled in serum-free medium containing [3H]myristic acid (2.5 μCi/ml) overnight and then washed twice with fresh serum-free medium. After an additional 2-h incubation in serum-free medium, the cells were subjected to experimental treatments in the presence (for PLD activity) or absence (for PA) of 0.3% 1-butanol during the final 15 min of the treatments. The cells were collected in chloroform-methanol 2:1 (v/v) and then processed as described above.
Statistical Analysis
All values are expressed as the means (±S.E. in graphs). Statistical significance was determined by using the student's t test (2-tailed, unpaired) or analysis of variance (one-way or two-way) followed by Student-Newman-Keuls post hoc analysis. Differences between groups were considered significant when p ≤ 0.05. All statistical analyses were performed on SigmaStat software (San Jose, CA).
RESULTS
Evidence That PA Can Function as a Direct Upstream Activator of mTOR Signaling in Response to Mechanical Stimulation
In this study we employed the same ex vivo model of mechanical stimulation that was previously used to expose the potential role of PLD and PA in the mechanical activation of mTOR signaling (10). With this model, it has been shown that mechanical stimulation induces an increase in PA and rapamycin-sensitive mTOR signaling (10, 11); however, whether the increase in PA is driven through a rapamycin-sensitive mechanism has not been addressed. This is an important question because recent studies have revealed that rapamycin-sensitive mTOR can phosphorylate and inhibit the activity of the phosphatidic acid phosphatase called Lipin 1 (27, 28). Thus, it remained possible that the mechanically induced increase in PA could be driven through a pathway that lies downstream, rather than upstream, to the activation of rapamycin-sensitive mTOR. Therefore, to test this we subjected muscles to mechanical stimulation in the presence or absence of rapamycin and then examined the muscles for changes in mTOR signaling and PA. As expected, rapamycin eliminated the mechanical activation of rapamycin-sensitive mTOR signaling as revealed by changes in the phosphorylation of p70S6k on the Thr-389 residue (29) (Fig. 1A). However, rapamycin did not affect the mechanically induced increase in PA (Fig. 1B). Based on these results, it can be concluded that the mechanically induced increase in PA is driven through a pathway that lies upstream and/or parallel to the activation of the rapamycin-sensitive elements of mTOR signaling.
FIGURE 1.

Evidence that PA can function as a direct upstream activator of mTOR signaling in response to mechanical stimulation. A and B, mouse EDL muscles were held at Lo in an ex vivo organ culture system and treated as follows. A, preincubated with 150 nm rapamycin (RAP +) or the vehicle (RAP −, DMSO) for 30 min and then subjected to 90 min of a stretch (Stretch +) or control condition (Stretch −) followed by Western blot analysis for phosphorylated (P) and total (T) p70. B, prelabeled with [3H]myristic acid for 2 h. During the final 30 min of the prelabeling, the muscles were incubated with rapamycin or the vehicle as in A and then subjected to 90 min of the stretch or control conditions. The concentration of 3H-labeled PA was measured and expressed as a percentage of values obtained in the vehicle control samples. C, C2C12 myoblasts stably expressing FLAG-mTOR were serum-starved overnight and collected in either CHAPS or Triton X-100 lysis buffer. The lysates were subjected to immunoprecipitation (IP) for the FLAG epitope, and then the immunoprecipitates were incubated for 15 min with 150 μm PA vesicles (PA +) or 150 μm PC vesicles as a control condition (PA −). The kinase activity of mTOR was then assayed with GST-p70 as a substrate. The resulting samples were subjected to Western blot analysis for the indicated proteins, and the phosphorylated:total ratios of GST-p70 were divided by the amount of mTOR in each reaction. These values were then expressed as a ratio of the values obtained in the PC-treated samples collected in CHAPS lysis buffer. The values were obtained from four independent experiments. All values are presented as the mean (±S.E. in graphs, n = 3–11 per group). *, significantly different from the drug (B)- or lysis buffer (C)-matched control group. †, significantly different from the stimulation-matched CHAPS group; p ≤ 0.05.
mTOR is typically found in multiprotein complexes such as the mTOR complex 1 (mTORC1) and the mTOR complex 2 (mTORC2). mTORC1 is defined by the presence of a protein called Raptor, whereas mTORC2 contains a protein called the rapamycin-insensitive companion of mTOR (Rictor) but not Raptor. Previous studies have suggested that signaling by mTORC1, but not mTORC2, is responsible for controlling the rapamycin-sensitive phosphorylation of substrates such as p70S6k(Thr-389) (30). However, more recent studies have shown that mTOR, in the absence of Raptor, can also regulate p70S6k(Thr-389) phosphorylation through a rapamycin-sensitive mechanism (16). In other words it appears that there is an uncharacterized rapamycin-sensitive pool of mTOR that does not involve the classic mTORC1. Furthermore, it was recently proposed that mechanical stimuli might specifically regulate this uncharacterized pool of mTOR rather than mTORC1 (11). Based on this possibility, we wanted to determine if PA could activate mTOR signaling in the absence of Raptor. To test this, we collected C2C12 myoblasts that stably express FLAG-mTOR in CHAPS lysis buffer to preserve the mTOR/Raptor interaction or Triton X-100 lysis buffer to dissociate the mTOR/Raptor interaction. mTOR from the different lysis conditions was then immunoprecipitated and incubated with lipid vesicles composed of 50% C8 PA and 50% PC or 100% PC as a control condition. The immunoprecipitates were then subjected to an in vitro mTOR kinase activity assay with p70S6k as a substrate. In this experiment we confirmed an enhanced mTOR kinase activity in the absence of Raptor as previously demonstrated (Fig. 1C) (31). Importantly, the results also demonstrated that PA can induce an increase in mTOR kinase activity and that Raptor is not required for this effect (Fig. 1C). Combined, the results presented in Fig. 1 establish that a mechanically induced increase in PA could function as a direct upstream activator of mTOR signaling.
Changes in PLD Activity Are Not Required for a Mechanically Induced Increase in PA or mTOR Signaling
To reexamine the role of PLD in the mechanical regulation of PA and mTOR signaling, we first performed measurements of PLD activity in muscles that were prelabeled with [3H]myristic acid, which preferentially labels the PLD substrate PC (32). In contrast to the previous study that used [3H]arachidonic acid for labeling (10), our results indicated that mechanical stimulation does not alter the activity of PLD toward [3H]myristic acid-labeled substrates (Fig. 2A). However, mechanical stimulation was still able to induce an increase in the concentration of [3H]myristic acid-labeled PA (Fig. 2E). This observation suggested that changes in PLD activity are not required for a mechanically induced increase in PA.
FIGURE 2.

Changes in PLD activity are not required for a mechanically induced increase in PA or mTOR signaling. A, mouse EDL muscles were held at Lo in an ex vivo organ culture system, prelabeled with [3H]myristic acid for 2 h, and then subjected to 15 or 90 min of a stretch (Stretch +) or control condition (Stretch −). PLD activity was measured throughout a 15-min period and expressed as a percentage of the values obtained in the time-matched control samples. B and C, C2C12 myoblasts were prelabeled in serum-free medium containing [3H]myristic acid overnight. After washing with fresh medium, the cells were preincubated with 1, 10, or 100 nm FIPI or the vehicle (FIPI 0, DMSO) for 30 min and then stimulated with 100 nm TPA (TPA +) or the vehicle as a control condition (TPA −, DMSO) for 15 min in the presence (B) or absence (C) of 0.3% 1-butanol. The cells were collected, and PLD activity (B) or the concentration of 3H-labeled PA (C) was measured and expressed as a percentage of the values obtained in the vehicle control samples. The values were obtained from five independent experiments. D--F, mouse EDL muscles were held at Lo in an ex vivo organ culture system and treated as follows. D and E, prelabeled with [3H]myristic acid for 2 h. During the final 30 min of prelabeling, the muscles were incubated with 100 nm FIPI (FIPI +) or the vehicle (FIPI−, DMSO). The muscles were then subjected to 15 or 90 min of stimulation with 1 μm TPA (TPA +) or the vehicle as a control condition (TPA −, DMSO) (D) or subjected to 15 or 90 min of the stretch or control conditions (E). PLD activity (D) or the concentration of 3H-labeled PA (E) was measured and expressed as a percentage of values obtained in the time-matched vehicle control samples. F, preincubated with FIPI or the vehicle as described above and then subjected to 15 or 90 min of the stretch or control conditions followed by Western blot analysis for phosphorylated (P) and total (T) p70. The phosphorylated:total ratios of p70 were expressed relative to the values obtained in the time-matched vehicle control samples. All values are presented as the mean (±S.E. in graphs, n = 3–8 per group). *, significantly different from the time- and drug-matched control group. †, significantly different from the time- and stimulation-matched vehicle group; p ≤ 0.05.
To further determine if changes in PLD activity are required for a mechanically induced increase in PA, we employed FIPI, which is known to be a more potent and specific inhibitor of PLD than 1-butanol (19). With this compound, we first performed a dose-response analysis in C2C12 myoblasts to identify the minimal concentration of FIPI that was needed to fully inhibit an agonist-induced increase in PLD activity. When TPA was used as a PLD agonist, it was determined that 100 nm FIPI could completely prevent the TPA-induced increase in both PLD activity and PA (Fig. 2, B and C). We also determined that 100 nm FIPI could completely block the TPA-induced increase in PLD activity when muscles were incubated in the ex vivo organ culture system (Fig. 2D).
Having established that 100 nm FIPI could block agonist-induced changes in PLD activity, we next set out to determine if FIPI would impair the ability of mechanical stimuli to induce an increase in PA and mTOR signaling. As shown in Fig. 2E, the increase in PA induced by both 15 and 90 min of mechanical stimulation was not impaired when muscles were treated with FIPI. Furthermore, FIPI did not alter the ability of mechanical stimuli to induce an increase in mTOR signaling (Fig. 2F). When combined, these results demonstrate that mechanical stimuli induce an increase in PA and mTOR signaling via a PLD-independent mechanism.
Mechanical Stimulation Increases DAG and Membrane DGK Activity
Our finding that changes in PLD activity are not required for a mechanically induced increase in PA inspired us to search for another enzyme(s) that could contribute to this event. During this search, we realized that several forms of mechanical stimuli, including contractions and stretch, have been reported to induce a rapid increase in the intracellular concentration of DAG (12, 33). Based on these reports, we envisioned the potential for a mechanism in which mechanical stimulation induces an accumulation of DAG, and this in turn would provide an enhanced level of substrate for the DGKs. Because the DGKs can synthesize PA via the phosphorylation of DAG, the net result of the DAG accumulation would be an enhanced synthesis of PA. To begin testing this possibility, we first wanted to determine if our model of mechanical stimulation also promotes an increase in DAG. As shown in Fig. 3A, DAG, just like PA, was significantly elevated after both 15 and 90 min of mechanical stimulation. Next, we asked if the elevated levels of DAG are associated with an increase in DGK activity. Interestingly, we found that mechanical stimulation induced a biphasic increase in membranous DGK activity (Fig. 3, B and C). However, the activity of DGK in whole muscle lysates was not increased by mechanical stimulation (Fig. 3D). Thus, it appears that mechanical stimulation causes a pool of DGK to translocate to membranous structures and/or allosterically activates membrane-associated DGK.
FIGURE 3.

Mechanical stimulation increases DAG and membrane DGK activity. Mouse EDL muscles were held at Lo in an ex vivo organ culture system and treated as follows. A, prelabeled with [3H]myristic acid for 2 h and then subjected to 15 or 90 min of a stretch (Stretch +) or control condition (Stretch −). The concentration of 3H-labeled DAG was measured and expressed as a percentage of values obtained in the control samples. B, subjected to 15 min of the stretch or control conditions and then separated into cytosolic (Cyt) and membrane (Memb) fractions. The different fractions were then subjected to Western blot analysis for Na+/K+-ATPase and LDH. C and D, subjected to 5–90 min of the stretch or control conditions, and then DGK activity (32P-PA) in the membrane fraction (C) and whole lysates (D) was measured and expressed as a percentage of the values obtained in the time-matched control samples. All values are presented as the mean ± S.E. (n = 3–4 per group). *, Significantly different from the time-matched control group; p ≤ 0.05.
R59949 Does Not Inhibit the Mechanically Induced Increase in PA or the Activation of mTOR Signaling
The aforementioned findings prompted us to further explore the potential role of the DGKs in the mechanical regulation of PA and mTOR signaling. To begin this analysis, we first took advantage of the DGK inhibitor R59949. Before describing our results, it is important to mention that the DGK family of enzymes is composed of at least 10 different isoforms, and currently available inhibitors of DGK (R59949 and its weaker analog R59902) preferentially inhibit only the α, β, γ, and θ isoforms of DGK (34, 35). Consistent with these points, we found that R59949 (100 μm) induces a partial, but significant, inhibition of basal DGK activity (Fig. 4A). However, R59949 did not inhibit the mechanically induced increase in PA or mTOR signaling (Fig. 4, B and C). Based on these results, we have concluded that the α, β, γ, and θ isoforms of DGK are not necessary for a mechanically induced increase in PA and mTOR signaling.
FIGURE 4.
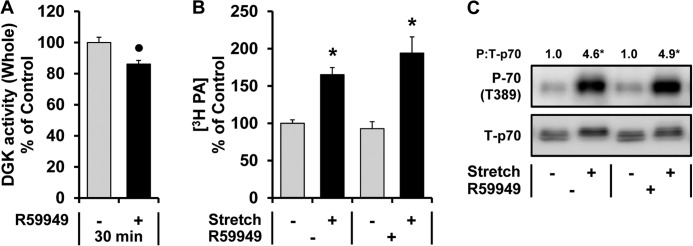
R59949 does not inhibit the mechanically induced increase in PA or the activation of mTOR signaling. Mouse EDL muscles were held at Lo in an ex vivo organ culture system and treated as follows. A, incubated with 100 μm R59949 (R59949 +) or the vehicle (R59949 −, DMSO) for 30 min. DGK activity from the whole lysates was measured and expressed as a percentage of values obtained in the vehicle control samples. B, prelabeled with [3H]myristic acid for 2 h. During the final 30 min of the prelabeling, the muscles were incubated with R59949 or the vehicle as described in A. The muscles were then subjected to 90 min of the stretch (Stretch +) or control conditions (Stretch −), and the concentration of 3H-labeled PA was measured and expressed as a percentage of values obtained in the vehicle control samples. C, preincubated with R59949 or the vehicle as described in A and then subjected to 90 min of the stretch or control conditions followed by Western blot analysis for phosphorylated (P) and total (T) p70. The phosphorylated:total ratios of p70 were expressed relative to the values obtained in the vehicle control samples. All values are presented as the mean (±S.E. in graphs, n = 3–8 per group). ●, significantly different from the vehicle group. *, significantly different from the drug-matched control group; p ≤ 0.05.
The Mechanically Induced Increase in PA and mTOR Signaling Is Impaired in Muscles from DGKζ −/− Mice
Previous studies have shown that the δ, ϵ, and ζ isoforms of DGK are highly expressed in skeletal muscle (36). Of these isoforms, we became particularly interested in DGKζ because this isoform is resistant to the inhibitory effects of R59949, and in HEK 293 cells it has been reported that the synthesis of PA by DGKζ contributes to the serum-induced activation of mTOR signaling (34, 37). Moreover, we were able to further validate this latter point in C2C12 myoblasts by demonstrating that the overexpression of DGKζ enhances the serum-induced activation of mTOR signaling, and this effect requires the kinase activity of DGKζ (i.e. the synthesis of PA) (Fig. 5). To the best of our knowledge, DGKζ is the only isoform of DGK that has been shown to exert a regulatory effect on mTOR signaling. Therefore, we set out to perform a series of experiments that were specifically aimed at addressing the role of DGKζ in the mechanical regulation of PA and mTOR signaling. To accomplish this, we employed muscles from homozygous DGKζ knock-out (DGKζ−/−) mice. As originally described (23), DGKζ−/− mice were viable, fertile, and had a normal appearance. Furthermore, our initial analysis of the muscles from DGKζ−/− mice did not reveal any overt abnormality in phenotypes such as muscle weight to body weight ratio, muscle fiber type distributions, and histological appearance (data not shown).
FIGURE 5.

Overexpression of DGKζ enhances the serum-induced activation of mTOR signaling in a kinase activity-dependent manner. C2C12 myoblasts were co-transfected with plasmid DNA encoding GFP, wild type DGKζ (HA-WT-DGKζ), or kinase dead DGKζ (HA-KD-DGKζ) and GST-p70. The following day the cells were serum-starved overnight and then stimulated with 20% fetal bovine serum (Serum +) for 30 min. The cells were collected and then subjected to Western blot analysis for the indicated proteins. The phospho (P):total (T) ratios of GST-p70 were then expressed as a percentage of the values obtained in the GFP non-stimulated (control, Serum −) samples. Values are presented as the mean ± S.E. and were obtained from five independent experiments (n = 5–9 per group). *, significantly different from the plasmid-matched control group. †, significantly different from the stimulation-matched GFP group; p ≤ 0.05.
Previous studies have suggested that the loss of DGKs can result in an accumulation of DAG and a concomitant elevation in the PKC-ERK signaling pathway (25, 37, 38). Consistent with these reports, we found that EDL muscles from DGKζ−/− mice exhibit elevated basal levels of DAG (140% of control, p < 0.0001) and ERK phosphorylation (Fig. 6A). Because signaling through ERK has been widely implicated in the activation of mTOR signaling, we were not surprised to find that the DGKζ−/− muscles also exhibit elevated basal levels of mTOR signaling under both in vivo and ex vivo conditions (Fig. 6, A and C). Combined, these results indicate that muscles from DGKζ−/− mice display the expected molecular phenotypes.
FIGURE 6.

The mechanically induced increase in PA and the activation of mTOR signaling is impaired in muscles from DGKζ−/− mice. A, EDL muscles from WT and DGKζ−/− mice were collected and subjected to Western blot analysis for the indicated proteins. B--E, EDL muscles from WT and DGKζ−/− mice were held at Lo in an ex vivo organ culture system and treated as follows. B, prelabeled with [3H]myristic acid for 2 h and then subjected to 90 min of the stretch (Stretch +) or control conditions (Stretch −). The concentration of 3H-labeled PA was measured and expressed as a percentage of values obtained in WT control samples. C and D, subjected to 30 or 90 min of the stretch or control conditions followed by Western blot analysis for phosphorylated (P) and total (T) p70. The phosphorylated:total ratios of p70 were expressed relative to the values obtained in the time-matched WT control samples (C) or expressed as a percentage of the time- and genotype-matched control samples (D). E, preincubated with 5, 10, or 20 nm rapamycin (RAP) or the vehicle (RAP 0, DMSO) for 30 min and then subjected to 90 min of stretch followed by Western blot analysis for phosphorylated (P-) and total (T-) p70. The phosphorylated:total ratios of p70 were expressed as a percentage of values obtained in the genotype-matched vehicle samples. All values are presented as the mean (±S.E. in graphs, n = 3–9 per group). *, significantly different from the time- and genotype-matched control group. †, significantly different from the time- and stimulation-matched WT group. ●, significantly different from the genotype-matched vehicle group. #, significantly different from the drug-matched WT group; p ≤ 0.05.
To address the role of DGKζ in the mechanical regulation of PA and mTOR signaling, EDL muscles from WT and DGKζ−/− mice were subjected to mechanical stimulation and examined for changes in PA and mTOR signaling. Intriguingly, we found that the mechanically induced increase in PA was almost completely abolished in the muscles from DGKζ−/− mice (Fig. 6B). Furthermore, the mechanical activation of mTOR signaling was also significantly impaired in the DGKζ−/− muscles (Fig. 6, C and D). Finally, when mechanically stimulated muscles from WT and DGKζ−/− mice were treated with increasing doses of rapamycin (0–20 nm), it was determined that mTOR signaling in the DGKζ−/− muscles was significantly more sensitive to the inhibitory effects of rapamycin (Fig. 6E). As mentioned in the introduction, mTOR signaling in mechanically stimulated muscles exhibits resistance to the inhibitory effects of rapamycin, and this is most likely driven by an enhanced binding of PA to the FRB domain in mTOR. Thus, the results from these experiments not only demonstrate that DGKζ is largely responsible for the mechanically induced increase in PA but also suggest that the DGKζ-dependent increase in PA leads to an enhanced binding of PA to mTOR and, in-turn, the activation of mTOR signaling.
Overexpression of DGKζ Induces Hypertrophy through a Kinase-dependent and Rapamycin-sensitive Mechanism
Previous studies have shown that signaling through mTOR is necessary for a mechanically induced hypertrophic response and that the activation of mTOR signaling is sufficient to induce hypertrophy (3, 6). Thus, we wanted to know if an enhanced synthesis of PA by DGKζ could also promote a hypertrophic response. To address this question, we transfected TA muscles with plasmid DNA encoding WT-DGKζ, a kinase dead mutant of DGKζ (KD-DGKζ), or GFP as a control condition. As expected, the overexpression of WT-DGKζ, but not KD-DGKζ, led to an enhanced synthesis of PA as assessed by an in vitro DGK activity assay (Fig. 7C). Furthermore, the examination of CSA revealed that the WT-DGKζ-transfected fibers were 33% larger than non-transfected fibers of the same muscles, whereas the CSA of fibers transfected with KD-DGKζ or GFP was not altered (Fig. 7, A and B). Based on these results, it can be concluded that the overexpression of DGKζ is sufficient to induce hypertrophy, and this effect requires the kinase activity of DGKζ (i.e. the synthesis of PA).
FIGURE 7.
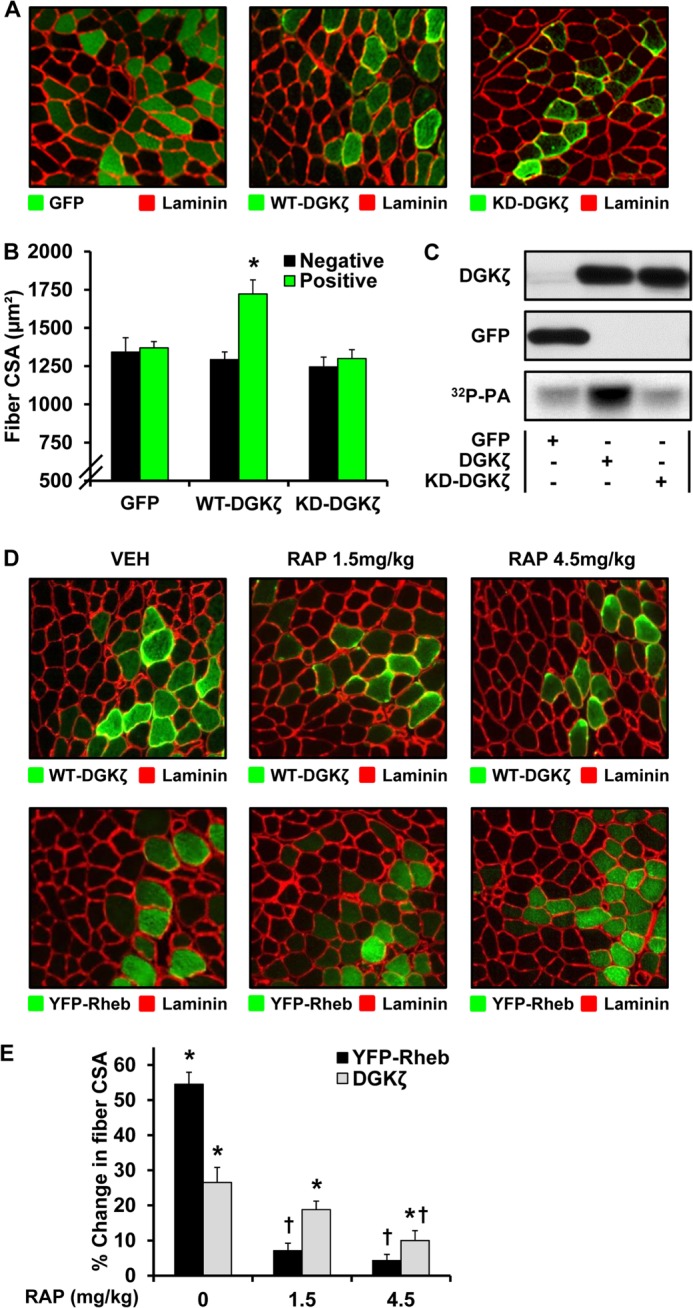
Overexpression of DGKζ induces hypertrophy through a kinase-dependent and rapamycin-sensitive mechanism. A–C, mouse TA muscles were transfected with plasmid DNA encoding GFP, HA-WT-DGKζ, or HA-KD-DGKζ. A, the muscles were collected at 7 days post transfection, and cross-sections from the mid-belly of the muscles were subjected to immunohistochemistry for GFP and laminin or for HA and laminin. B, CSA of the transfected fibers (Positive) and non-transfected fibers (Negative) within each of the GFP-, HA-WT-DGKζ-, and HA-KD-DGKζ-transfected muscles. C, muscles were collected at 3 days post transfection, and whole lysates were subjected to a DGK activity assay (32P-PA) or Western blot analysis for the indicated proteins. D and E, mouse TA muscles were transfected with plasmid DNA encoding HA-WT-DGKζ or YFP-Rheb. Immediately after transfection the mice were subjected to a regime of daily 1.5 or 4.5 mg/kg rapamycin (RAP) or vehicle (RAP 0, DMSO) injections. D, the muscles were collected at 7 days post transfection, and cross-sections from the mid-belly of the muscles were subjected to immunohistochemistry for HA and laminin or for YFP and laminin. E, the CSA of the transfected fibers and non-transfected fibers within each of the HA-WT-DGKζ- and YFP-Rheb-transfected muscles was determined, and then the percent difference between the averaged CSA of the transfected fibers and non-transfected fibers of each muscle was calculated. All values are presented as the mean ± S.E. (n = 3–8 muscles per group, 35–120 fibers per muscle). *, significantly different from the drug- and plasmid-matched non-transfected fibers. †, significantly different from the plasmid-matched vehicle group; p ≤ 0.05.
Next, we wanted to determine if DGKζ induces hypertrophy through an mTOR-dependent mechanism. To this end, mouse TA muscles were transfected with WT-DGKζ or Rheb as a positive control. Immediately after transfection, the mice were subjected to daily injections of rapamycin or the solvent vehicle as a control condition. As shown in Fig. 7, D and E, a daily dose of 1.5 mg/kg rapamycin prevented the robust hypertrophic response that is induced by the overexpression of Rheb. However, the same dose of rapamycin did not significantly inhibit the hypertrophic effect of WT-DGKζ (Fig. 7, D and E). This result was not entirely surprising because the fibers transfected with WT-DGKζ would presumably have higher levels of PA which in turn would be expected to confer resistance to the inhibitory effects of rapamycin. Therefore, we performed an additional experiment in which the mice were injected with a higher dose of rapamycin (4.5 mg/kg/day). In this case the results demonstrated that the DGKζ-induced hypertrophic response was significantly inhibited (Fig. 7, D and E). Thus, it appears that the DGKζ-induced hypertrophic response is driven at least in part through an mTOR-dependent mechanism. Furthermore, just like the mechanical activation of mTOR signaling, the DGKζ-induced hypertrophic response is partially resistant to the inhibitory effects of rapamycin, which suggests that it is mediated by an enhanced binding of PA to mTOR.
DISCUSSION
How mechanical stimuli are converted into the molecular events that regulate skeletal muscle mass has been one of the long-standing questions in the field of muscle biology. Accordingly, several hypotheses have been proposed to explain this phenomenon, and one that has attracted increasing interest predicts that a mechanically induced increase in PA leads to an enhanced binding of PA to mTOR and in turn promotes the activation of mTOR signaling and ultimately muscle growth (4). In this study we have obtained several novel lines of evidence that support this hypothesis by identifying that (i) mechanical stimulation induces an increase in PA via a DGKζ-dependent mechanism, (ii) loss of DGKζ impairs mechanically induced increases in both mTOR signaling and a sign of enhanced PA-mTOR binding, and (iii) overexpression of DGKζ promotes fiber hypertrophy through a mTOR-dependent mechanism that requires the ability of DGKζ to synthesize PA.
In the early 2000s, a potential role for PLD and PA in the regulation of mTOR signaling was unveiled by studies that used primary alcohols (e.g. 1-butanol) to inhibit the PLD-catalyzed synthesis of PA. For example, these studies concluded that the PLD-dependent synthesis of PA is necessary for the serum- and phenylephrine-induced activation of mTOR signaling (18, 39). Based on these studies, it was subsequently hypothesized that the mechanical activation of mTOR signaling might also be mediated by the PLD-dependent synthesis of PA. Indeed, this appeared to be confirmed when it was demonstrated that 1-butanol can prevent mechanically induced increases in both PA and mTOR signaling (10). However, the involvement of PLD in these events became uncertain when it was shown that a newly developed PLD-specific inhibitor (FIPI) did not affect many of the biological processes that are inhibited by 1-butanol (19, 20). Moreover, a recent study has also shown that several ethanol (a two-carbon primary alcohol)-sensitive events were not observed when more reliable techniques, such as a genetic deletion of PLD, were employed (21). Consistent with the concerns raised by these studies, we found that, although FIPI could block agonist-induced changes in PLD activity, it did not prevent the mechanically induced increase in PA or mTOR signaling. Therefore, we have concluded that the previously reported effects of 1-butanol on mechanically induced changes in PA and mTOR signaling were most likely due to off-target/PLD-independent events.
Despite the wide use of primary alcohols, there have been many studies that did not rely solely on primary alcohols to modulate PLD activity, and these studies still demonstrate that PLD can play an important role for the regulation of mTOR signaling. For example, using shRNA-mediated knockdown of PLD1, it has been shown that an increase in PLD1 activity is required for the activation of mTOR signaling that occurs during myoblast differentiation and in response to amino acid stimulation (40, 41). A more recent study has also found that modulations of PLD1 expression are sufficient to induce changes in both mTOR signaling and the size of L6 myotubes (42). Interestingly, this study also determined that, like DGKζ, overexpression of PLD1 is sufficient to induce hypertrophy in vivo. Although the link between the regulation of PLD and muscle mass under physiologically relevant conditions is still not known, these results provide fundamental support for the conclusion that an elevation in PA can induce the activation of mTOR signaling and ultimately hypertrophy of skeletal muscle.
Our finding that DGKζ contributes to the mechanical activation of PA-mTOR signaling is novel; however, the mechanism through which mechanical stimuli use DGKζ to promote an increase in PA and mTOR signaling remains unknown. Nevertheless, recent progress in mTOR biology may have revealed some important clues. Specifically, it has been demonstrated that mTOR can colocalize with late endosomal/lysosomal structures (LELs) where its direct activators, such as Rheb, reside (24, 41, 43, 44). Furthermore, it has been shown that forced targeting of mTOR to the LEL is sufficient to activate mTOR signaling, indicating that the LEL is a key integration site for the regulation of mTOR signaling (43, 45). More recently, it was also shown that the LEL is highly enriched with PA and that mechanical stimulation leads to an increase in the colocalization of mTOR with the LEL (24, 46). This latter observation is of particular interest because it suggests that mechanical stimulation could induce an enhanced binding of PA to mTOR at least in part by increasing the association of mTOR with the PA-enriched LEL.
Based on the information mentioned above, it is tempting to suggest that DGKζ could regulate PA-mTOR signaling by directly controlling the level of PA at the LEL. In support of this possibility, it has been reported that DGKζ is able to physically interact with β-arrestins and sorting nexin 27 (SNX27), both of which are found on early endosomal structures (47, 48). Because early endosomes can transform into LEL, the recruitment of DGKζ into early endosomes by β-arrestins and/or SNX27 might ultimately contribute to an enrichment of PA at the newly formed LEL. Interestingly, it has been shown that mechanical stretching of bladder tissues results in a very rapid and robust increase in the endocytosis of the apical membrane, which is then delivered to LEL for degradation (49). Because endocytosis transports β-arrestins and its associated proteins (e.g. DGKζ) to early endosomes, it is possible that mechanical stimulation induces an enhanced targeting of DGKζ to early endosomes, and ultimately the LEL, by promoting endocytosis (48). Consistent with this possibility, we have recently shown that mechanical stimulation induces an increase in the number of LEL (45). Furthermore, in the current study we found that mechanical stimulation promotes an increase in membranous DGK activity, which might reflect an enhanced localization of DGKζ with the early endosomes/LEL. If correct, such an event would be expected to promote an increase in PA at the LEL and, in-turn, an enhanced binding of PA to mTOR and ultimately the activation of mTOR signaling. Clearly, this hypothesis is quite speculative, and the exact mechanism(s) via which DGKζ contributes to the mechanical activation of PA-mTOR signaling remains to be defined. Nonetheless, examining the potential role of the LEL in this process appears to be an area that is worthy of further investigation.
Another novel finding in this study is that overexpression of DGKζ induces fiber hypertrophy via a mechanism that requires its kinase activity. The requirement of kinase activity is noteworthy because it suggests that the DGKζ-induced hypertrophy was mediated by an increase in the synthesis of PA. As shown in previous studies, an enhanced synthesis of PA can trigger mTOR-dependent (rapamycin-sensitive) cellular responses, but these responses tend to be resistant to the inhibitory effects of rapamycin (10, 50). For instance, PLD activity/PA is frequently elevated in human breast cancer cells, and the growth rate of these cells becomes partially resistant to rapamycin in a manner that is correlated with the higher levels of PLD activity (50). Therefore, our observation that overexpression of DGKζ induces hypertrophy through an mTOR-dependent (rapamycin-sensitive), but partially rapamycin-resistant mechanism, further suggests that the hypertrophic effect of DGKζ is exerted through an increase in PA that in turn would lead to an enhanced binding of PA to mTOR and the subsequent activation of mTOR-dependent growth regulatory events.
Previous studies have demonstrated that signaling through mTOR is necessary for a mechanically induced hypertrophic response and that the activation of mTOR signaling is sufficient to induce hypertrophy (3, 5, 6). Thus, the results of this study strongly suggest that DGKζ could also play a role in the molecular process through which mechanical stimuli regulate skeletal muscle mass. Consistent with this possibility, a recent clinical study found that subjects who displayed the greatest degree of myofiber hypertrophy in response to heavy resistance exercise also had the highest pre-training levels of skeletal muscle DGKζ transcript expression (51). Additional evidence implicating DGKζ in mechanically induced hypertrophy is that DGKζ can regulate several stages of skeletal myogenesis including myoblast differentiation and fusion, a process that has been widely implicated in the regulation of mechanically induced hypertrophy (52–54). Because the process of myogenesis is also largely controlled by mTOR signaling, it is possible that DGKζ could regulate mechanically induced hypertrophy via the control of mTOR-dependent myogenesis (55). Combined, several pieces of evidence suggest that DGKζ could be a key component of the mechanism(s) through which mechanical stimuli induce hypertrophy, and addressing this possibility will be an essential topic for future studies.
In summary, the results from this study indicate that DGKζ contributes to the mechanical activation of PA-mTOR signaling and induces hypertrophy via an mTOR-dependent mechanism that requires the ability of DGKζ to synthesize PA. In the future it will be important to determine how DGKζ contributes to the mechanical activation of PA-mTOR signaling and to define the role of DGKζ in mechanically induced hypertrophy in vivo. The resulting knowledge will further advance our understanding of the mechanism(s) through which mechanical stimuli regulate skeletal muscle mass and may ultimately lead to the development of therapies that can prevent the loss of muscle mass during conditions such as aging, diseases, and inactivity.
Acknowledgments
We thank Dr. Karyn Esser (University of Kentucky, Lexington, KY), Dr. Kun-Liang Guan (University of California San Diego, La Jolla, CA), and Dr. Matthew Topham (University of Utah, Salt Lake City, UT) for providing the GST-p70S6k, YFP-Rheb, and HA-WT-DGKζ plasmids, respectively. Special thanks are extended to Dr. Jie Chen (University of Illinois, Urbana, IL) for providing the C2C12 myoblasts stably expressing FLAG-mTOR. We also thank Dr. Xiao-Ping Zhong (Duke University, Durham, NC) for providing DGKζ−/− C57BL/6 mice.
This work was supported, in whole or in part, by National Institutes of Health Grant AR057347 (to T. A. H.).
- mTOR
- mammalian (or mechanistic) target of rapamycin
- mTORC1
- mammalian target of rapamycin complex 1
- mTORC2
- mammalian target of rapamycin complex 2
- CSA
- cross-sectional area
- DAG
- diacylglycerol
- DGK
- diacylglycerol kinase
- EDL
- extensor digitorum longus
- FIPI
- 5-fluoro-2-indolyl des-chlorohalopemide
- FRB
- FKBP12-rapamycin binding
- LEL
- late endosomal/lysosomal structures
- OG
- octyl glucoside
- p70S6k
- ribosomal S6 kinase
- PA
- phosphatidic acid
- C8 PA
- 1,2-dioctanoyl-sn-glycero-3-phosphate
- PC
- phosphatidylcholine
- PLD
- phospholipase D
- PS
- phosphatidylserine
- PtdBut
- phosphatidylbutanol
- Raptor
- regulatory associated protein of mTOR
- Rheb
- Ras homologue enriched in brain
- TA
- tibialis anterior
- TPA
- 12-O-tetradecanoylphorbol-13-acetate
- Lo
- optimal length.
REFERENCES
- 1. Hurley B. F., Hanson E. D., Sheaff A. K. (2011) Strength training as a countermeasure to aging muscle and chronic disease. Sports Med. 41, 289–306 [DOI] [PubMed] [Google Scholar]
- 2. Goldberg A. L., Etlinger J. D., Goldspink D. F., Jablecki C. (1975) Mechanism of work-induced hypertrophy of skeletal muscle. Med. Sci. Sports 7, 185–198 [PubMed] [Google Scholar]
- 3. Goodman C. A., Frey J. W., Mabrey D. M., Jacobs B. L., Lincoln H. C., You J. S., Hornberger T. A. (2011) The role of skeletal muscle mTOR in the regulation of mechanical load-induced growth. J. Physiol. 589, 5485–5501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hornberger T. A. (2011) Mechanotransduction and the regulation of mTORC1 signaling in skeletal muscle. Int. J. Biochem. Cell Biol. 43, 1267–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Avilés Mendoza G. J., Seidel N. E., Otsu M., Anderson S. M., Simon-Stoos K., Herrera A., Hoogstraten-Miller S., Malech H. L., Candotti F., Puck J. M., Bodine D. M. (2001) Comparison of five retrovirus vectors containing the human IL-2 receptor γ chain gene for their ability to restore T and B lymphocytes in the X-linked severe combined immunodeficiency mouse model. Mol. Ther. 3, 565–573 [DOI] [PubMed] [Google Scholar]
- 6. Goodman C. A., Miu M. H., Frey J. W., Mabrey D. M., Lincoln H. C., Ge Y., Chen J., Hornberger T. A. (2010) A phosphatidylinositol 3-kinase/protein kinase B-independent activation of mammalian target of rapamycin signaling is sufficient to induce skeletal muscle hypertrophy. Mol. Biol. Cell 21, 3258–3268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hornberger T. A., Stuppard R., Conley K. E., Fedele M. J., Fiorotto M. L., Chin E. R., Esser K. A. (2004) Mechanical stimuli regulate rapamycin-sensitive signalling by a phosphoinositide 3-kinase-, protein kinase B-, and growth factor-independent mechanism. Biochem. J. 380, 795–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. O'Neil T. K., Duffy L. R., Frey J. W., Hornberger T. A. (2009) The role of phosphoinositide 3-kinase and phosphatidic acid in the regulation of mammalian target of rapamycin following eccentric contractions. J. Physiol. 587, 3691–3701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hornberger T. A., Chien S. (2006) Mechanical stimuli and nutrients regulate rapamycin-sensitive signaling through distinct mechanisms in skeletal muscle. J. Cell Biochem. 97, 1207–1216 [DOI] [PubMed] [Google Scholar]
- 10. Hornberger T. A., Chu W. K., Mak Y. W., Hsiung J. W., Huang S. A., Chien S. (2006) The role of phospholipase D and phosphatidic acid in the mechanical activation of mTOR signaling in skeletal muscle. Proc. Natl. Acad. Sci. U.S.A. 103, 4741–4746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. You J. S., Frey J. W., Hornberger T. A. (2012) Mechanical stimulation induces mTOR signaling via an ERK-independent mechanism. Implications for a direct activation of mTOR by phosphatidic acid. PloS ONE 7, e47258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cleland P. J., Appleby G. J., Rattigan S., Clark M. G. (1989) Exercise-induced translocation of protein kinase C and production of diacylglycerol and phosphatidic acid in rat skeletal muscle in vivo. Relationship to changes in glucose transport. J. Biol. Chem. 264, 17704–17711 [PubMed] [Google Scholar]
- 13. Yoon M. S., Sun Y., Arauz E., Jiang Y., Chen J. (2011) Phosphatidic acid activates mammalian target of rapamycin complex 1 (mTORC1) kinase by displacing FK506 binding protein 38 (FKBP38) and exerting an allosteric effect. J. Biol. Chem. 286, 29568–29574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Veverka V., Crabbe T., Bird I., Lennie G., Muskett F. W., Taylor R. J., Carr M. D. (2008) Structural characterization of the interaction of mTOR with phosphatidic acid and a novel class of inhibitor. Compelling evidence for a central role of the FRB domain in small molecule-mediated regulation of mTOR. Oncogene 27, 585–595 [DOI] [PubMed] [Google Scholar]
- 15. Chen J., Zheng X. F., Brown E. J., Schreiber S. L. (1995) Identification of an 11-kDa FKBP12-rapamycin-binding domain within the 289-kDa FKBP12-rapamycin-associated protein and characterization of a critical serine residue. Proc. Natl. Acad. Sci. U.S.A. 92, 4947–4951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yip C. K., Murata K., Walz T., Sabatini D. M., Kang S. A. (2010) Structure of the human mTOR complex I and its implications for rapamycin inhibition. Mol. Cell 38, 768–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Toschi A., Lee E., Xu L., Garcia A., Gadir N., Foster D. A. (2009) Regulation of mTORC1 and mTORC2 complex assembly by phosphatidic acid. Competition with rapamycin. Mol. Cell. Biol. 29, 1411–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fang Y., Vilella-Bach M., Bachmann R., Flanigan A., Chen J. (2001) Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science 294, 1942–1945 [DOI] [PubMed] [Google Scholar]
- 19. Su W., Yeku O., Olepu S., Genna A., Park J. S., Ren H., Du G., Gelb M. H., Morris A. J., Frohman M. A. (2009) 5-Fluoro-2-indolyl des-chlorohalopemide (FIPI), a phospholipase D pharmacological inhibitor that alters cell spreading and inhibits chemotaxis. Mol. Pharmacol. 75, 437–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yanase Y., Carvou N., Frohman M. A., Cockcroft S. (2010) Reversible bleb formation in mast cells stimulated with antigen is Ca2+/calmodulin-dependent and bleb size is regulated by ARF6. Biochem. J. 425, 179–193 [DOI] [PubMed] [Google Scholar]
- 21. Sato T., Hongu T., Sakamoto M., Funakoshi Y., Kanaho Y. (2013) Molecular mechanisms of N-formyl-methionyl-leucyl-phenylalanine-induced superoxide generation and degranulation in mouse neutrophils. Phospholipase D is dispensable. Mol. Cell. Biol. 33, 136–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Topham M. K., Bunting M., Zimmerman G. A., McIntyre T. M., Blackshear P. J., Prescott S. M. (1998) Protein kinase C regulates the nuclear localization of diacylglycerol kinase-ζ. Nature 394, 697–700 [DOI] [PubMed] [Google Scholar]
- 23. Zhong X. P., Hainey E. A., Olenchock B. A., Jordan M. S., Maltzman J. S., Nichols K. E., Shen H., Koretzky G. A. (2003) Enhanced T cell responses due to diacylglycerol kinase ξ deficiency. Nat. Immunol. 4, 882–890 [DOI] [PubMed] [Google Scholar]
- 24. Jacobs B. L., You J. S., Frey J. W., Goodman C. A., Gundermann D. M., Hornberger T. A. (2013) Eccentric contractions increase the phosphorylation of tuberous sclerosis complex-2 (TSC2) and alter the targeting of TSC2 and the mechanistic target of rapamycin to the lysosome. J. Physiol. 591, 4611–4620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chibalin A. V., Leng Y., Vieira E., Krook A., Björnholm M., Long Y. C., Kotova O., Zhong Z., Sakane F., Steiler T., Nylén C., Wang J., Laakso M., Topham M. K., Gilbert M., Wallberg-Henriksson H., Zierath J. R. (2008) Down-regulation of diacylglycerol kinase δ contributes to hyperglycemia-induced insulin resistance. Cell 132, 375–386 [DOI] [PubMed] [Google Scholar]
- 26. Goodman C. A., McNally R. M., Hoffmann F. M., Hornberger T. A. (2013) Smad3 induces atrogin-1, inhibits mTOR and protein synthesis, and promotes muscle atrophy in vivo. Mol. Endocrinol. 27, 1946–1957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Eaton J. M., Mullins G. R., Brindley D. N., Harris T. E. (2013) Phosphorylation of lipin 1 and charge on the phosphatidic acid head group control its phosphatidic acid phosphatase activity and membrane association. J. Biol. Chem. 288, 9933–9945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Harris T. E., Huffman T. A., Chi A., Shabanowitz J., Hunt D. F., Kumar A., Lawrence J. C., Jr. (2007) Insulin controls subcellular localization and multisite phosphorylation of the phosphatidic acid phosphatase, lipin 1. J. Biol. Chem. 282, 277–286 [DOI] [PubMed] [Google Scholar]
- 29. Magnuson B., Ekim B., Fingar D. C. (2012) Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. Biochem. J. 441, 1–21 [DOI] [PubMed] [Google Scholar]
- 30. Zoncu R., Efeyan A., Sabatini D. M. (2011) mTOR. From growth signal integration to cancer, diabetes, and ageing. Nat. Rev. Mol. Cell Biol. 12, 21–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kim D. H., Sarbassov D. D., Ali S. M., King J. E., Latek R. R., Erdjument-Bromage H., Tempst P., Sabatini D. M. (2002) mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110, 163–175 [DOI] [PubMed] [Google Scholar]
- 32. el Bawab S., Macovschi O., Thevenon C., Goncalves A., Némoz G., Lagarde M., Prigent A. F. (1996) Contribution of phosphoinositides and phosphatidylcholines to the production of phosphatidic acid upon concanavalin A stimulation of rat thymocytes. J. Lipid Res. 37, 2098–2108 [PubMed] [Google Scholar]
- 33. Sadoshima J., Izumo S. (1993) Mechanical stretch rapidly activates multiple signal transduction pathways in cardiac myocytes. Potential involvement of an autocrine/paracrine mechanism. EMBO J. 12, 1681–1692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jiang Y., Sakane F., Kanoh H., Walsh J. P. (2000) Selectivity of the diacylglycerol kinase inhibitor 3-[2-(4-[bis-(4-fluorophenyl)methylene]-1-piperidinyl)ethyl]-2, 3-dihydro-2-thioxo-4(1H)quinazolinone (R59949) among diacylglycerol kinase subtypes. Biochem. Pharmacol. 59, 763–772 [DOI] [PubMed] [Google Scholar]
- 35. Baldanzi G., Alchera E., Imarisio C., Gaggianesi M., Dal Ponte C., Nitti M., Domenicotti C., van Blitterswijk W. J., Albano E., Graziani A., Carini R. (2010) Negative regulation of diacylglycerol kinase theta mediates adenosine-dependent hepatocyte preconditioning. Cell Death Differ. 17, 1059–1068 [DOI] [PubMed] [Google Scholar]
- 36. van Blitterswijk W. J., Houssa B. (2000) Properties and functions of diacylglycerol kinases. Cell. Signal. 12, 595–605 [DOI] [PubMed] [Google Scholar]
- 37. Avila-Flores A., Santos T., Rincón E., Mérida I. (2005) Modulation of the mammalian target of rapamycin pathway by diacylglycerol kinase-produced phosphatidic acid. J. Biol. Chem. 280, 10091–10099 [DOI] [PubMed] [Google Scholar]
- 38. Gorentla B. K., Wan C. K., Zhong X. P. (2011) Negative regulation of mTOR activation by diacylglycerol kinases. Blood 117, 4022–4031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ballou L. M., Jiang Y. P., Du G., Frohman M. A., Lin R. Z. (2003) Ca2+- and phospholipase D-dependent and -independent pathways activate mTOR signaling. FEBS Lett. 550, 51–56 [DOI] [PubMed] [Google Scholar]
- 40. Yoon M. S., Chen J. (2008) PLD regulates myoblast differentiation through the mTOR-IGF2 pathway. J. Cell Sci. 121, 282–289 [DOI] [PubMed] [Google Scholar]
- 41. Yoon M. S., Du G., Backer J. M., Frohman M. A., Chen J. (2011) Class III PI-3-kinase activates phospholipase D in an amino acid-sensing mTORC1 pathway. J. Cell Biol. 195, 435–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jaafar R., De Larichaudy J., Chanon S., Euthine V., Durand C., Naro F., Bertolino P., Vidal H., Lefai E., Némoz G. (2013) Phospholipase D regulates the size of skeletal muscle cells through the activation of mTOR signaling. Cell Commun. Signal 11, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sancak Y., Bar-Peled L., Zoncu R., Markhard A. L., Nada S., Sabatini D. M. (2010) Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141, 290–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Saito K., Araki Y., Kontani K., Nishina H., Katada T. (2005) Novel role of the small GTPase Rheb. Its implication in endocytic pathway independent of the activation of mammalian target of rapamycin. J. Biochem. 137, 423–430 [DOI] [PubMed] [Google Scholar]
- 45. Jacobs B. L., Goodman C. A., Hornberger T. A. (2013) The mechanical activation of mTOR signaling. An emerging role for late endosome/lysosomal targeting. J. Muscle Res. Cell Motil. 10.1007/s10974-013-9367-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhao K., Zhou H., Zhao X., Wolff D. W., Tu Y., Liu H., Wei T., Yang F. (2012) Phosphatidic acid mediates the targeting of tBid to induce lysosomal membrane permeabilization and apoptosis. J. Lipid Res. 53, 2102–2114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rincón E., Santos T., Avila-Flores A., Albar J. P., Lalioti V., Lei C., Hong W., Mérida I. (2007) Proteomics identification of sorting nexin 27 as a diacylglycerol kinase ζ-associated protein. New diacylglycerol kinase roles in endocytic recycling. Mol. Cell Proteomics 6, 1073–1087 [DOI] [PubMed] [Google Scholar]
- 48. Nelson C. D., Perry S. J., Regier D. S., Prescott S. M., Topham M. K., Lefkowitz R. J. (2007) Targeting of diacylglycerol degradation to M1 muscarinic receptors by β-arrestins. Science 315, 663–666 [DOI] [PubMed] [Google Scholar]
- 49. Truschel S. T., Wang E., Ruiz W. G., Leung S. M., Rojas R., Lavelle J., Zeidel M., Stoffer D., Apodaca G. (2002) Stretch-regulated exocytosis/endocytosis in bladder umbrella cells. Mol. Biol. Cell 13, 830–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chen Y., Zheng Y., Foster D. A. (2003) Phospholipase D confers rapamycin resistance in human breast cancer cells. Oncogene 22, 3937–3942 [DOI] [PubMed] [Google Scholar]
- 51. Thalacker-Mercer A., Stec M., Cui X., Cross J., Windham S., Bamman M. (2013) Cluster analysis reveals differential transcript profiles associated with resistance training-induced human skeletal muscle hypertrophy. Physiol. Genomics 45, 499–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Evangelisti C., Riccio M., Faenza I., Zini N., Hozumi Y., Goto K., Cocco L., Martelli A. M. (2006) Subnuclear localization and differentiation-dependent increased expression of DGK-ζ in C2C12 mouse myoblasts. J. Cell Physiol. 209, 370–378 [DOI] [PubMed] [Google Scholar]
- 53. Abramovici H., Gee S. H. (2007) Morphological changes and spatial regulation of diacylglycerol kinase-ζ, syntrophins, and Rac1 during myoblast fusion. Cell Motil. Cytoskeleton 64, 549–567 [DOI] [PubMed] [Google Scholar]
- 54. Guerci A., Lahoute C., Hébrard S., Collard L., Graindorge D., Favier M., Cagnard N., Batonnet-Pichon S., Précigout G., Garcia L., Tuil D., Daegelen D., Sotiropoulos A. (2012) Srf-dependent paracrine signals produced by myofibers control satellite cell-mediated skeletal muscle hypertrophy. Cell Metab. 15, 25–37 [DOI] [PubMed] [Google Scholar]
- 55. Sun Y., Ge Y., Drnevich J., Zhao Y., Band M., Chen J. (2010) Mammalian target of rapamycin regulates miRNA-1 and follistatin in skeletal myogenesis. J. Cell Biol. 189, 1157–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Folch J., Lees M., Sloane Stanley G. H. (1957) A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 226, 497–509 [PubMed] [Google Scholar]