

Background: The function of the clathrin adaptor AP-2 in the regulation of GPCR coupling to G protein signaling is not known.
Results: AP-2 controls GPCR signaling by modulating receptor surface expression and, unexpectedly, through RGS protein recruitment to G proteins.
Conclusion: AP-2 has diverse functions in the regulation of GPCR signaling.
Significance: AP-2 provides a new mode of GPCR signal regulation.
Keywords: Adaptor Proteins, G Protein-coupled Receptors (GPCR), G Proteins, Intracellular Trafficking, Thrombin
Abstract
The G protein-coupled protease-activated receptor 1 (PAR1) is irreversibly proteolytically activated by thrombin. Hence, the precise regulation of PAR1 signaling is important for proper cellular responses. In addition to desensitization, internalization and lysosomal sorting of activated PAR1 are critical for the termination of signaling. Unlike most G protein-coupled receptors, PAR1 internalization is mediated by the clathrin adaptor protein complex 2 (AP-2) and epsin-1, rather than β-arrestins. However, the function of AP-2 and epsin-1 in the regulation of PAR1 signaling is not known. Here, we report that AP-2, and not epsin-1, regulates activated PAR1-stimulated phosphoinositide hydrolysis via two different mechanisms that involve, in part, a subset of R4 subfamily of “regulator of G protein signaling” (RGS) proteins. A significantly greater increase in activated PAR1 signaling was observed in cells depleted of AP-2 using siRNA or in cells expressing a PAR1 420AKKAA424 mutant with defective AP-2 binding. This effect was attributed to AP-2 modulation of PAR1 surface expression and efficiency of G protein coupling. We further found that ectopic expression of R4 subfamily members RGS2, RGS3, RGS4, and RGS5 reduced activated PAR1 wild-type signaling, whereas signaling by the PAR1 AKKAA mutant was minimally affected. Intriguingly, siRNA-mediated depletion analysis revealed a function for RGS5 in the regulation of signaling by the PAR1 wild type but not the AKKAA mutant. Moreover, activation of the PAR1 wild type, and not the AKKAA mutant, induced Gαq association with RGS3 via an AP-2-dependent mechanism. Thus, AP-2 regulates activated PAR1 signaling by altering receptor surface expression and through recruitment of RGS proteins.
Introduction
The coagulant protease thrombin is generated in response to vascular injury and in thrombotic disease where it promotes hemostasis, thrombosis, and inflammatory responses. Thrombin drives fibrin deposition and mediates cellular responses through a family of protease-activated G protein-coupled receptors (GPCRs)2 (1). Protease-activated receptor 1 (PAR1) is the family prototype and the predominant mediator of thrombin signaling in most cell types. Thrombin cleaves the N terminus of PAR1, unmasking a new N-terminal domain that functions as a peptide ligand by binding to the receptor, inducing a conformational change that facilitates coupling to heterotrimeric G proteins (2). Because of the proteolytic mechanism of activation and generation of a tethered ligand that cannot diffuse away, signaling by PAR1 is tightly regulated. Similar to other GPCRs, activated PAR1 signaling is rapidly desensitized by phosphorylation and β-arrestin binding, which uncouples the receptor from heterotrimeric G protein signaling (3, 4). Activated PAR1 is then internalized from the cell surface, sorted directly to lysosomes, and degraded, which prevents continued signaling by previously activated receptors that return to the cell surface with their tethered ligands intact (5). In metastatic breast carcinoma, activated PAR1 is internalized and recycled back to the cell surface rather than sorted to lysosomes (6). Consequently, activated PAR1 signals persistently and promotes breast carcinoma invasion and tumor growth in vivo (7). These findings indicate that internalization and lysosomal sorting of PAR1 are important for regulating the magnitude and duration of G protein signaling.
In contrast to many classic GPCRs, PAR1 internalization occurs through clathrin-coated pits independent of β-arrestins (4). Several other GPCRs have also been shown to internalize independently of β-arrestins (8). We showed previously that the clathrin adaptor protein complex 2 (AP-2) and epsin-1 are essential for agonist-induced PAR1 internalization (9, 10). The clathrin adaptor AP-2 is a heterotetrameric complex comprised of α, β2, μ2, and σ2 adaptin subunits and has critical functions in the assembly and recruitment of cargo to clathrin-coated pits. The μ2-adaptin subunit of AP-2 binds directly to tyrosine-based “YXXØ” motifs (where Y is a tyrosine, X is any amino acid, and Ø is a bulky hydrophobic residue) (11). Using a bioinformatic approach, we discovered the presence of tyrosine-based motifs within the cytoplasmic (C)-tail domain of PAR1 and ∼30 other mammalian GPCRs (12). The μ2-adaptin subunit of AP-2 binds directly to a PAR1 tyrosine-based motif (420YKKLL424) localized within the distal C-tail region and is required for constitutive internalization and cellular resensitization (13). In addition, agonist-promoted internalization of PAR1 is dually regulated by AP-2 and epsin-1 through phosphorylation- and ubiquitination-dependent mechanisms (10). However, it is not known if AP-2 or epsin-1 regulates activated PAR1 coupling to G protein signaling. In fact, the function of the endocytic machinery in signal regulation of a GPCR that does not require β-arrestins for internalization has not been examined previously.
The regulation of GPCR signaling is mediated through various mechanisms that occur at the level of the receptor and signaling effectors. The family of “regulator of G protein signaling” (RGS) proteins function as GTPase-accelerating proteins for heterotrimeric G proteins, which effectively enhance GTP hydrolysis by the Gα-subunit to shut off G protein signaling. The conventional family of RGS proteins includes 22 members that share a central function in regulation of the Gi and Gq families (14). The R4 family is the largest family of RGS proteins, with many individual members exhibiting overlapping functions in regulation of Gα subunits in vitro and in distinct cell types (15, 16). Individual R4 subfamily members have been shown to specifically regulate different GPCR signaling pathways (17). However, the mechanisms that govern RGS protein activity and specificity toward particular GPCRs represent a major gap in our knowledge. We previously employed an RNA interference screen targeting all conventional RGS proteins in HEK293 cells to define RGS proteins that act specifically at PAR1 (18). Surprisingly, depletion of RGS8 expression resulted in an attenuation of PAR1 signaling that was attributed to decreased receptor surface expression (18). However, the mechanism responsible for RGS8 effects on PAR1 surface expression have yet to be determined. It also remains unclear whether RGS8 or other RGS proteins function similarly in other cell types to control PAR1 signaling.
We hypothesize that the cellular signaling activity of PAR1 is regulated by multiple mechanisms. The first involves desensitization mediated by PAR1 phosphorylation and β-arrestin binding. The second mechanism is mediated by the endocytic machinery. However, unlike most GPCRs, internalization of PAR1 is regulated by AP-2 and epsin-1, rather than by β-arrestins. AP-2 binds directly to PAR1 via a C-tail tyrosine-based motif (420YKKAA424) (19). In this study, we examined the regulation of PAR1 signaling by AP-2 and epsin-1. We further explored the possibility that AP-2 regulates PAR1 signaling through an involvement of RGS proteins. Our findings suggest that AP-2 functions as a critical regulator of PAR1 signaling activity both by modulating receptor surface expression and through recruitment of a subset of the R4 family of RGS proteins. These findings reveal a novel role for AP-2 in the regulation of RGS protein recruitment to G proteins for certain GPCRs.
EXPERIMENTAL PROCEDURES
Reagents and Antibodies
Human α-thrombin was purchased from Enzyme Research Laboratories (South Bend, IN). The PAR1 peptide agonist Ser-Phe-Leu-Leu-Arg-Asn (SFLLRN) was synthesized as the carboxyl amide and purified by reverse phase high-pressure liquid chromatography at the Tufts University Core Facility (Boston, MA). Carbachol and UTP were purchased from Sigma-Aldrich (St. Louis, MO). Rabbit polyclonal anti-FLAG antibody was purchased from Rockland Immunochemicals (Gilbertsville, PA). Mouse monoclonal M2 anti-FLAG antibody and anti-β-actin were purchased from Sigma-Aldrich. The anti-PAR1 C5433 rabbit polyclonal antibody has been described previously (13). Rabbit polyclonal anti-Gαq/11 (C-19) antibody and anti-epsin-1 (H-130) antibody were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). HRP-conjugated secondary goat anti-mouse and anti-rabbit antibodies were obtained from Bio-Rad. Monoclonal anti-HA antibody conjugated to HRP was from Roche. The anti-AP50 (μ2) monoclonal antibody was obtained from BD Biosciences.
cDNAs and Cell Lines
A cDNA encoding human PAR1 WT and C-tail AKKAA mutant containing an N-terminal FLAG epitope cloned into the pBJ vector have been described previously (13, 20). An HA epitope-tagged Gαq placed within an internal loop was provided by Dr. Philip Wedegaertner (Thomas Jefferson University, Philadelphia, PA). HA-tagged RGS2, RGS4-C2S, and RGS5-C2S have been described previously (21). HA-tagged RGS3 was purchased from the Missouri S&T cDNA Resource Center (Rolla, MO).
HeLa cells expressing the FLAG-tagged PAR1 WT or AKKAA mutant were generated and maintained as described previously (20). Human umbilical vein endothelial-derived EA.hy926 cells were maintained as described (22). COS7 cells were grown in DMEM containing 10% (v/v) fetal bovine serum, 100 units/ml penicillin, and 0.1 mg/ml streptomycin.
siRNA
HeLa cells stably expressing FLAG-tagged PAR1 WT or AKKAA mutant were plated at 0.5–1 × 105 cells/well in fibronectin-coated 24-well plates and grown overnight at 37 °C. HeLa cells were transiently transfected with 50 nm of nonspecific, epsin-1, μ2, or RGS4 siRNAs or 100 nm RGS2-, 100 nm RGS5-, or 200 nm RGS3-specific siRNA using Lipofectamine 2000 or Oligofectamine according to the instructions of the manufacturer (Invitrogen). Endothelial cells were plated at 3.5 × 105 cells/well in fibronectin-coated 12-well plates and grown overnight at 37 °C. Endothelial cells were transiently transfected with 50 nm nonspecific or μ2-specific siRNA using Oligofectamine. The nonspecific siRNA 5′-CTACGTCCAGGAGCGCACC-3′, μ2 siRNA 5′-GTGGATGCCTTTCGGGTCA-3′, and epsin-1 siRNA 5′-GGAAGACGCCGGAGTCATT-3′ have been described previously (13). The RGS2-specific siRNA 5′-AACGTGGTGTCTCACTCTGAA-3′, RGS3-specific siRNA 5′-CAGACGGATAGACATACGGAA-3′, RGS4-specific siRNA 5′-AACATGCTAGAGCCTACAATA-3′, and RGS5-specific siRNA 5′-AACGAGAGCAATGACTATTTA-3′ were purchased from Qiagen (Valencia, CA).
Immunoblotting
Protein concentrations were determined from total cell lysates using BCA (Thermo Fisher Scientific, Rockford, IL). Equivalent amounts of cell lysates were resolved by SDS-PAGE, transferred to PVDF membranes, and immunoblotted with the appropriate antibodies.
Phosphoinositide (PI) Hydrolysis
HeLa cells stably expressing FLAG-tagged the PAR1 WT or AKKAA mutant were plated at 1 × 105 cells/well in fibronectin-coated 24-well plates and grown overnight at 37 °C. After transfection, cells were labeled with 1μCi/ml myo-[3H]inositol (American Radiolabeled Chemicals, St. Louis, MO) overnight, treated with or without agonist in the presence of 20 mm lithium chloride (LiCl) for various times at 37 °C, and then accumulated [3H]inositol phosphates (IPs) were measured from 3 wells/time point of an independent experiment as described previously (4). To determine the desensitization rates, cells were labeled as described above. Cells were then stimulated with 10 nm thrombin for 10 min at 37 °C, LiCl was added as indicated, and the accumulation of [3H]IPs were measured as described previously (23).
Immunoprecipitation
COS7 cells were plated at 1.5 × 106 cells/60-mm dish and grown overnight at 37 °C. Cells were transfected with HA-tagged Gαq and FLAG-tagged PAR1 WT or AKKAA mutant for 48 h, serum-starved for 1 h, and then incubated with or without agonist at 37 °C. Cells were washed with ice-cold PBS and lysed in Triton X-100 lysis buffer (50 mm Tris-HCl (pH 7.4), 100 mm NaCl, 5 mm EDTA, 50 mm NaF, 10 mm sodium pyrophosphate, and 1% Triton X-100) supplemented with 100 μg/ml PMSF, 1 μg/ml leupeptin, 2 μg/ml aprotinin, 1 μg/ml soybean trypsin inhibitor, 1 μg/ml pepstatin, and 10 μg/ml benzamidine. Cell lysates were cleared by centrifugation, protein concentrations were determined, and equivalent amounts of cell lysates were immunoprecipitated with anti-FLAG antibody overnight at 4 °C. Immunoprecipitates were washed with lysis buffer, and proteins were eluted in 2× Laemmli buffer (62.5 mm Tris-HCl, (pH 6.8), 10% glycerol, 5% SDS, 0.01% bromphenol blue). Immunoprecipitates were resolved by SDS-PAGE, transferred to PVDF membranes, and immunoblotted with anti-Gαq/11 antibody or anti-FLAG antibody to detect PAR1 expression. Cell lysates were immunoblotted with anti-Gαq/11 and anti-β-actin antibodies as controls. Immunoblot analyses were developed using ECL, exposed to film, and quantified by densitometry using ImageJ.
To examine RGS and Gα protein association, HeLa cells expressing FLAG-tagged PAR1 WT or AKKAA mutant were plated at 1.5 × 106 cells/60-mm dish and grown overnight at 37 °C. Cells were transfected with HA-tagged RGS3 for 48 h or with 50 nm siRNA targeting the μ2-adaptin subunit in combination with HA-tagged RGS3 for 72 h. Cells were serum-starved for 1 h and incubated with or without agonist at 37 °C. Cells were lysed in Triton lysis buffer, and equivalent amounts of cell lysates were immunoprecipitated with anti-Gαq/11 antibody. Immunoprecipitates were processed, and membranes were probed with anti-HA antibody to detect RGS3 associated with Gαq/11, developed with ECL, and quantitated by densitometry using ImageJ.
Reverse Transcription Polymerase Chain Reaction
The first-strand cDNA was generated from total mRNA extracted from either HeLa or endothelial cells using SuperScript® II reverse transcriptase (RT), an engineered version of Moloney murine leukemia virus reverse transcriptase (M-MLVRT) and oligo(dT)12–18 following the instructions of the manufacturer (Invitrogen). The RT enzyme was omitted from the cDNA synthesis reaction in the negative (-) control samples. The first-strand cDNA was amplified by PCR using primers specific for all conventional RGS mRNA transcripts. The RGS primer sequences and predicted PCR amplicon sizes have been described previously (24). The PCR amplification products were resolved by 1.8% (w/v) agarose gel electrophoresis and visualized by ethidium bromide staining.
Cell Surface ELISA
HeLa cells stably expressing FLAG-tagged PAR1 WT or AKKAA mutant were plated at 1 × 105 cells/well in fibronectin-coated 24-well plates and grown overnight at 37 °C. After transfection, cells were placed on ice for 10 min and washed once with ice-cold PBS. Cells were fixed in 4% paraformaldehyde for 5 min on ice and washed twice with PBS. Cells were incubated with anti-FLAG antibody or anti-PAR1 antibody for 60 min at room temperature. Cells were washed twice with medium and incubated with HRP-conjugated secondary antibody for 60 min at room temperature. Cells were washed three times with PBS before incubating with 1-step 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid)-diammonium salt) solution (Thermo Fisher Scientific, Rockford, IL). The absorbance of an aliquot was read at 405 nm using a Molecular Devices SpectraMax Plus microplate reader (Sunnyvale, CA).
Data Analysis
Data were analyzed using GraphPad Prism 4.0 and JMP statistical software. Statistical analysis was determined by performing Student's t test, one-way ANOVA, Dunnett's multiple test, Tukey's post hoc honest significant difference (HSD) test, or two-way ANOVA and Bonferroni post-test.
RESULTS
AP-2 Regulates Activated PAR1 Signaling
In recent work, we showed that activated PAR1 internalization is mediated by AP-2 and epsin-1, not β-arrestins (4, 10), suggesting that they may regulate receptor signaling. However, precisely how AP-2 or epsin-1 function in the regulation of PAR1 signaling is not known. Previous studies indicate that activated PAR1 stimulates Gαq-mediated phospholipase C-induced PI hydrolysis in numerous cell types (25, 26). Thus, to examine the function of AP-2 and epsin-1 in PAR1 signaling, HeLa cells stably expressing PAR1 WT were transfected with nonspecific, μ2-, and/or epsin-1-specific siRNAs, labeled with myo-[3H]inositol, incubated with or without thrombin for various times, and then the accumulation of [3H]IPs was measured as described (4). Activated PAR1 signaling rapidly declined and reached near steady state at 2.5 min in cells transfected with nonspecific or epsin-1-specific siRNA (Fig. 1A), indicating that epsin-1 is not required for the regulation of PAR1 signaling. In contrast, activation of PAR1 in cells lacking AP-2 resulted in a marked increase in PI hydrolysis (Fig. 1A), which was comparable with cells depleted of both AP-2 and epsin-1. The increase in PAR1 signaling observed in AP-2 deficient cells also correlated with an increase in PAR1 expression at the cell surface (Fig. 1A). Unlike PAR1, activation of the Gαq-linked muscarinic receptors with carbachol in AP-2-depleted HeLa cells failed to affect the signaling response compared with nonspecific siRNA-transfected control cells (Fig. 1B). To confirm these findings, we examined the effects of AP-2 depletion on endogenous PAR1 signaling in a cultured human endothelial EA.hy926 cell line. A 60-min incubation with thrombin caused a significantly greater accumulation of [3H]IPs in endothelial cells deficient in AP-2 expression compared with nonspecific siRNA-transfected control cells (Fig. 1C), whereas signaling by UTP (an endogenous agonist for the purinergic P2Y2 or P2Y4 GPCRs) resulted in comparable signaling responses regardless of AP-2 expression (D). Elevated expression of endogenous PAR1 at the cell surface was also observed in AP-2-depleted cells (Fig. 1C). Thus, increased expression of PAR1 at the cell surface may contribute, in part, to greater signaling observed in AP-2-deficient cells.
FIGURE 1.

The activation kinetics of PAR1 signaling are regulated by AP-2. A and B, HeLa cells expressing the PAR1 WT transfected with nonspecific (ns), epsin-1-, μ2-adaptin-, or epsin-1/μ2-adaptin-specific siRNAs labeled with myo-[3H]inositol were incubated with 10 nm thrombin for varying times (A) or 500 μm carbachol (CARB) for 60 min (B) at 37 °C, and [3H]IPs formed were measured. The data (mean ± S.D., n = 3) are representative of three independent experiments. The differences observed in thrombin-stimulated signaling were significant as determined by two-way ANOVA and Bonferroni post-tests (**, p < 0.01; ***, p < 0.001). The differences in PAR1 surface expression (mean ± S.D., n = 3) were significant as determined by one-way ANOVA and Dunnett's multiple comparison tests (**, p < 0.01). Cell lysates were immunoblotted (IB) with the indicated antibodies. Ctrl, control. C and D, endothelial EA.hy926 cells expressing endogenous PAR1 transfected with nonspecific or μ2-adaptin-specific siRNAs labeled with myo-[3H]inositol were incubated with 10 nm thrombin (α-Th) (C) or 100 μm UTP (D) for 60 min at 37 °C, and [3H]IPs formed were measured. The data (mean ± S.D., n = 3) are representative of three independent experiments. The differences observed in thrombin-stimulated signaling were significant as determined by Student's t test (**, p < 0.01). The differences in PAR1 surface expression (mean ± S.D., n = 3) were determined by Student's t test (***, p < 0.001). Cell lysates were immunoblotted with the indicated antibodies.
Activated PAR1 Signaling Efficiency, but Not Desensitization, Is Regulated by AP-2
We next examined the signaling properties of PAR1 in cells lacking AP-2 expression. The concentration effect curves for thrombin at PAR1 expressed in control or AP-2-deficient cells was determined by incubating cells labeled with myo-[3H]inositol with varying concentrations of thrombin for 5 min at 37 °C. The accumulation of [3H]IPs was then measured. The effective thrombin concentration to stimulate a half-maximal response was markedly decreased in AP-2-deficient cells compared with control cells (Fig. 2A). In addition, activation of PAR1 in cells lacking AP-2 expression resulted in an enhanced maximal signaling response compared with siRNA-transfected control cells (Fig. 2A). We also examined whether the efficacy or potency of carbachol-stimulated muscarinic receptor signaling was affected in AP-2-deficient cells. Using PAR1-expressing cells, the concentration-effect curves for carbachol stimulation of muscarinic receptor signaling were determined in control and AP-2-deficient cells. The EC50 values for carbachol-stimulated IP accumulation at 10 min were comparable under both conditions (Fig. 2B). The maximal signaling response induced by saturating carbachol concentrations were also similar (Fig. 2B). These results suggest that AP-2 depletion does not globally affect GPCR signaling activity. We also examined whether the differences in PAR1 signaling were related to desensitization. To assess PAR1 desensitization in control and AP-2-depleted cells, we measured the extent of PAR1 signaling activity remaining after various times of thrombin incubation and found that the apparent rates of desensitization were not significantly different (Fig. 2C). Together, these findings suggest that AP-2 specifically modulates activated PAR1 signaling activity independently of receptor desensitization.
FIGURE 2.
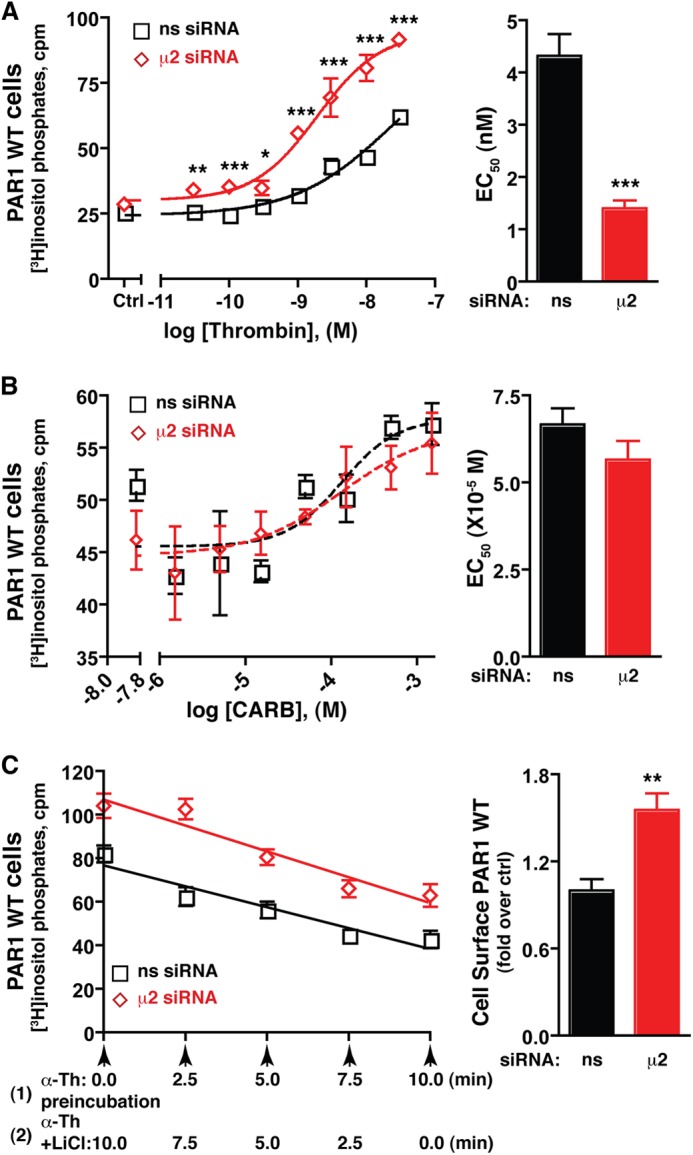
Activated PAR1 signaling efficiency, and not desensitization, is regulated by AP-2. HeLa cells expressing PAR1 WT were labeled with myo-[3H]inositol incubated with varying concentrations of thrombin for 5 min (A) or carbachol (CARB) or 10 min (B) at 37 °C. The concentration effect curve (mean ± S.D., n = 3) shown is a representative experiment. The EC50 values (mean ± S.E.) from multiple independent experiments are shown in the bar graph. The differences in thrombin-stimulated signaling were significant as determined by two-way ANOVA and Bonferroni post-tests (*, p < 0.05; **, p < 0.01; ***, p < 0.001). The differences in EC50 values were determined by Student's t test (***, p < 0.001). ns, not significant; Ctrl, control; M, molar. C, PAR1 WT-expressing HeLa cells labeled with myo-[3H]inositol were incubated with 10 nm thrombin for 10 min at 37 °C (1), and LiCl was added after various times of thrombin preincubation (2). [3H]IPs formed were then measured. The data (mean ± S.D., n = 3) are representative of three independent experiments. The differences in PAR1 surface expression (mean ± S.D., n = 3) were significant as determined by Student's t test (**, p < 0.01). The rates of desensitization (slope of the line) between nonspecific (-3.85 ± 0.43, n = 3) and μ2-adaptin-specific siRNA-treated cells (-4.76 ± 0.43, n = 3) were not significant as determined by Student's t test (p = 0.157). α-Th, thrombin.
A PAR1 AKKAA Mutant Exhibits Enhanced G Protein Coupling Efficiency
To determine whether AP-2 regulates PAR1 signaling via binding to the C-tail tyrosine motif, we utilized a PAR1 AKKAA tyrosine motif mutant that exhibits defective basal AP-2 binding in vitro (13). We examined whether AP-2 retained the capacity to regulate PAR1 AKKAA mutant signaling. HeLa cells stably expressing the PAR1 AKKAA mutant displayed a marked increase in PI hydrolysis following activation with saturating concentrations of thrombin in control siRNA-transfected cells (Fig. 3A). Activated PAR1 AKKAA mutant signaling was similarly enhanced in AP-2-deficient cells (Fig. 3A), suggesting that an intact tyrosine motif is important for AP-2-negative regulation of PAR1 signaling. To further examine differences in PAR1 WT versus AKKAA mutant signaling, thrombin concentration effect curves were performed. HeLa cells expressing the PAR1 WT or AKKAA mutant labeled with myo-[3H]inositol were incubated with varying concentrations of thrombin for 10 min at 37 °C, and then the accumulation of [3H]IPs was examined. The effective concentration of thrombin to stimulate a half-maximal response was modestly but significantly greater for the PAR1 WT compared with the AKKAA mutant (Fig. 3B), suggesting that the activated PAR1 AKKAA mutant couples more efficiently to G protein signaling. To further examine this effect, the relationship between the amount of PAR1 WT and AKKAA mutant expressed at the cell surface to the extent of PI hydrolysis was determined. As the level of PAR1 WT expression at the cell surface was increased (by transfection of cDNA), a greater accumulation of [3H]IPs was detected following stimulation with either saturating or subsaturating concentrations of thrombin (Fig. 3C). Thrombin activation of the PAR1 AKKAA mutant resulted in a similar increase in thrombin-induced PI hydrolysis compared with the wild-type receptor expressed at comparable levels (Fig. 3C). Thus, the amount of PAR1 present on the cell surface contributes to the extent of thrombin-stimulated PI hydrolysis, suggesting that the greater capacity of PAR1 AKKAA mutant to stimulate G protein signaling (Fig. 3A) is attributed, at least in part, to increased surface expression (Fig. 1).
FIGURE 3.
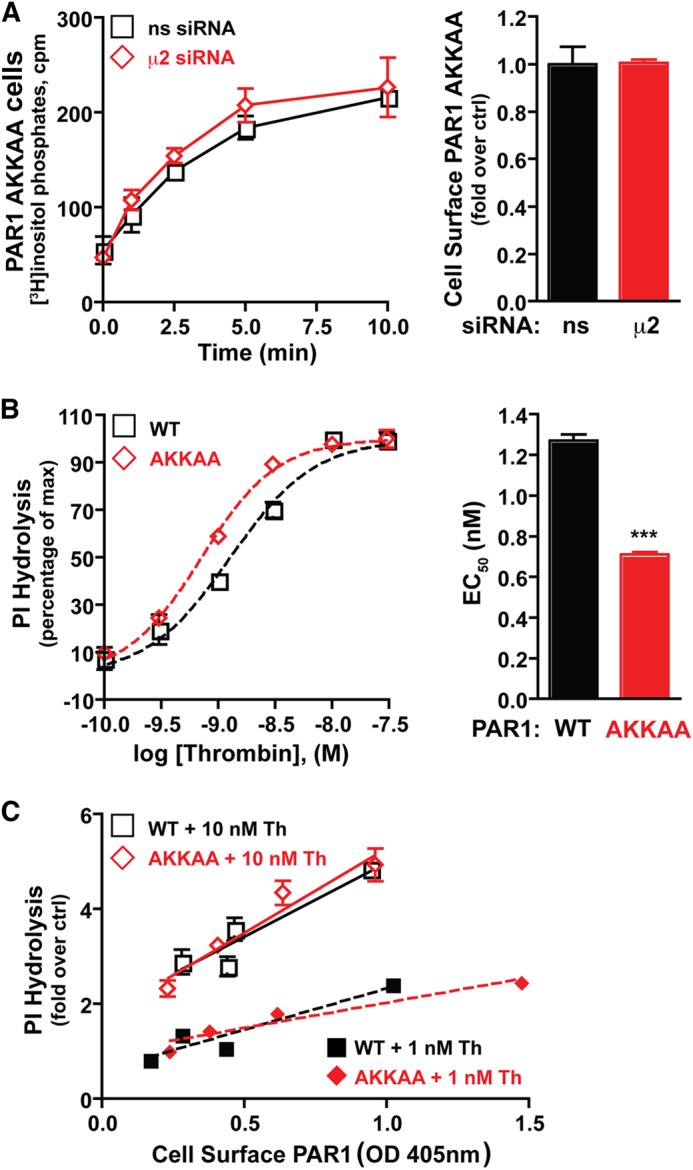
The activated PAR1 AKKAA mutant with defective AP-2 binding exhibits enhanced G protein coupling efficiency. A, HeLa cells expressing the PAR1 AKKAA mutant transfected with nonspecific (ns) or μ2-adaptin-specific siRNAs labeled with myo-[3H]inositol were incubated with 10 nm thrombin for varying times at 37 °C, and [3H]IPs formed were measured. The data (mean ± S.D., n = 3) are representative of three independent experiments. PAR1 surface expression (mean ± S.D., n = 3) was determined by ELISA. B, HeLa cells expressing the PAR1 WT or AKKKAA mutant labeled with myo-[3H]inositol were incubated with varying concentrations of thrombin for 5 min at 37 °C. The concentration effect curve data (mean ± S.D., n = 3) and EC50 values are representative of at least three independent experiments. The difference in EC50 was significant (***, p < 0.001) as determined by Student's t test. M, molar. C, HeLa cells transiently expressing varying amounts of PAR1 WT or PAR1 AKKAA mutant were labeled with myo-[3H]inositol, incubated with 10 nm or 1 nm thrombin for 10 min at 37 °C, and [3H]IPs formed were measured. The data (mean ± S.D., n = 3) are representative of three independent experiments. Surface expression of PAR1 (mean ± S.D., n = 3) was determined by ELISA in the same experiment.
The PAR1 Wild Type and AKKAA Mutant Associate with Gq Similarly
To determine whether differences in PI signaling exhibited by the PAR1 WT and AKKAA mutant were because of Gq association, we examined receptor interaction with Gαq by coimmunoprecipitation using COS7 cells. COS7 cells were utilized because they exhibit high transfection efficiency and because transfected PAR1 stimulates PI hydrolysis following incubation with thrombin (23). Cells were transiently cotransfected with the FLAG-tagged PAR1 WT or AKKAA mutant together with or without Gαq containing an internal HA epitope tag. Cell lysates were immunoprecipitated with anti-FLAG antibodies, and the amount of Gαq associated with the receptor was detected by immunoblotting. Both the PAR1 WT and AKKAA mutant were expressed at comparable amounts as detected by immunoblotting (Fig. 4, A and B). The PAR1 WT and AKKAA mutant interaction with Gαq occurred basally, and the amount of Gαq coimmunoprecipitated was similar with both the wild-type and mutant receptor (Fig. 4A). Moreover, stimulation of either the PAR1 WT or AKKAA mutant with peptide agonist failed to affect the extent of receptor association with Gαq (Fig. 4B). These findings suggest that PAR1 association of Gαq is not likely to contribute to differences observed between wild-type and mutant receptor signaling.
FIGURE 4.

The PAR1 wild type and AKKAA mutant associate with Gαq. COS-7 cells were transiently transfected with FLAG-tagged PAR1 WT or AKKAA mutant together with HA-tagged Gαq. Cells were then either left untreated (A) or treated with 100 μm SFLLRN (B) for 5 min at 37 °C, lysed, and then equivalent amounts of lysates were immunoprecipitated (IP) with anti-FLAG antibody. HA-Gαq associated with PAR1 WT or AKKAA mutant was detected by immunoblot (IB) analysis with an anti-Gαq antibody. The membrane was stripped and reprobed with anti-FLAG antibody to detect PAR1. Total cell lysates were probed for anti-Gαq protein or anti-actin antibodies as a control. The amount of immunoprecipitated Gαq associated with PAR1 WT or AKKAA mutant was quantitated and expressed as a fraction of PAR1 WT (A) or untreated control cells (B).
Signaling by the PAR1 Wild Type and AKKAA Mutant Is Differentially Regulated by RGS Proteins
The regulation of GPCR signaling occurs through multiple mechanisms and at both the receptor and G protein effector levels. The “regulator of G protein signaling” (RGS) proteins accelerate the hydrolysis of GTP by the Gα subunit and, thereby, function as negative regulators of G protein signaling (14). To determine whether PAR1 WT and AKKAA mutant signaling is differentially regulated by RGS proteins, the expression of RGS proteins in the HeLa and human EA.hy926 endothelial cell lines were first profiled using RT-PCR and primers that detect all conventional RGS mRNA transcripts, as described previously (18). Only a subset of transcripts for conventional RGS proteins were detected in HeLa and endothelial cells (Fig. 5). These included RGS2, RGS3, RGS4, and RGS5 of the R4 family of RGS proteins, which are known to effectively regulate Gq subtype Gα subunits (27). The RZ family members RGS19 and RGS20 and the R12 family member RGS10 were also detected in HeLa and endothelial cells (Fig. 5) but are not critical regulators of Gq signaling (28).
FIGURE 5.

Detection of RGS gene transcripts in HeLa and endothelial cells. A and B, RT-PCR was performed on mRNA extracted from either HeLa cells (A) or endothelial cells (B). RGS transcript-specific primers were used in RT-PCR amplification with (+) or without (-) RT. C, the data represent the RGS proteins identified in HeLa and endothelial cells.
To assess the activity of the R4 family members on PAR1 signaling, HeLa cells were cotransfected with PAR1 and HA-tagged RGS proteins of the R4 family. RGS2, RGS4, and RGS5 were tagged with an HA epitope at the C terminus, whereas RGS3 contained three tandem HA epitopes at the N terminus (Fig. 6A). RGS4 and RGS5 also contained cysteine-to-serine mutations at position 2 to increase protein stability and expression in HeLa cells (21) (Fig. 6A). Transfected cells were lysed for immunoblot analysis (Fig. 6A) or labeled with myo-[3H]inositol, stimulated with thrombin for 60 min at 37 °C, and then the accumulation of [3H]IPs were measured. To control for variances in PAR1 expression, thrombin-stimulated PI hydrolysis was plotted against the amount of PAR1 expressed at the cell surface in the different transfection conditions. In cells expressing wild-type PAR1 alone, the extent of thrombin-induced PI hydrolysis increased with the amount of wild-type PAR1 expressed at the cell surface (Fig. 6B), whereas coexpression of varying amounts of RGS2, RGS3, RGS4, and RGS5 proteins significantly reduced thrombin-stimulated PI hydrolysis (Fig. 6B). Untransfected cells not expressing PAR1 or RGS proteins were stimulated with thrombin in parallel and showed a minimal increase in PI hydrolysis (Fig. 6, B and C). In contrast, only coexpression of RGS3 was observed to cause a modest but significant decrease in PAR1 AKKAA mutant signaling, although its overall effect was reduced substantially compared with its effect at wild-type PAR1 (Fig. 6, B and C). These findings suggest that RGS2, RGS3, RGS4, and RGS5 function as negative regulators of activated PAR1-induced PI signaling, whereas the PAR1 AKKAA mutant with defective AP-2 interaction is less sensitive to RGS protein regulation of agonist-stimulated G protein signaling.
FIGURE 6.

Activated PAR1 signaling is negatively regulated by the R4 family of RGS proteins. A, domain structure of the HA-tagged R4 family of RGS proteins expressed in HeLa and endothelial cells. HeLa cells transiently expressing FLAG-tagged PAR1 WT and AKKAA mutant together with increasing amounts of HA-tagged RGS proteins, pcDNA3 vector, or untransfected (UT) control were either lysed or processed as described in B and C. Cell lysates were immunoblotted (IB) with anti-HA antibody to detect RGS protein expression or with anti-actin antibodies as a control. B and C, cells labeled with myo-[3H]inositol were incubated with 10 nm thrombin for 60 min at 37 °C, and [3H]IPs formed were measured. The data (mean ± S.D., n = 3) are representative of three independent experiments. The amount of PAR1 expressed on the cell surface (mean ± S.D., n = 3) for each transfected condition was determined by ELISA. The results are plotted as the fraction of [3H]IPs formed relative to the maximal response versus the amount of PAR1 expressed on the cell surface. The difference between the PI signaling normalized to receptor surface expression (mean ± S.D., n = 3) observed with PAR1 WT expressed alone in cells compared with cells coexpressing the various RGS proteins was significant as determined by single ANOVA and Tukey's HSD post hoc test (**, p < 0.01). However, only coexpression of RGS3 with the PAR1 AKKAA mutant caused a significant decrease in PI signaling compared with PAR1 AKKAA expressed alone as determined by single ANOVA and Tukey's HSD post hoc test (**, p < 0.01).
To confirm RGS protein function on PAR1 signaling, we examined whether depletion of individual endogenous R4 family members by siRNA affects PAR1 signaling. We employed transfection conditions optimized for effective siRNA-mediated depletion of individual epitope-tagged RGS proteins expressed in HeLa cells (Fig. 7A) and then measured the accumulation of [3H]IPs following activation of the PAR1 wild type or AKKAA mutant with thrombin for 60 min. To control for variances in signaling because of changes in PAR1 expression, the data were normalized to the amount of receptor detected on the cell surface in the same experiment. Only siRNA-mediated depletion of RGS5 caused a significant increase in activated PAR1 wild type signaling (Fig. 7B). In contrast to wild-type PAR1, neither siRNA-induced loss of RGS5 or other RGS proteins had a significant effect on PAR1 AKKAA mutant signaling (Fig. 7C), suggesting that AP-2 binding is required for RGS5-dependent regulation of PAR1 signaling. These findings suggest that, at least in HeLa cells, RGS5 has a particularly important role that is not redundant with other R4 family members. However, we cannot exclude the possibility that a residual amount of endogenous RGS protein remains in siRNA-transfected cells and contributes to signal termination in some instances. It is also likely that other R4 family members have redundant functions or important functions in the regulation of PAR1 signaling in other cell types.
FIGURE 7.

Effect of siRNA-mediated RGS protein depletion on activated PAR1 signaling. A, HeLa cells were transiently transfected with HA-tagged RGS2, RGS3, RGS4, or RGS5 together with the corresponding isoform-specific siRNA and lysed. Cell lysates were immunoblotted (IB) with anti-HA antibody to detect RGS protein or an anti-actin antibody as a control. ns, nonspecific. B and C, HeLa cells stably expressing FLAG-tagged PAR1 WT (B) or AKKAA mutant (C) were transiently transfected with 50 nm nonspecific, 100 nm RGS2-, 200 nm RGS3-, 50 nm RGS4-, or 100 nm RGS5-specific siRNA. Cells were then labeled with myo-[3H]inositol, incubated with 10 nm thrombin for 60 min at 37 °C, and then the amounts of [3H]IPs formed were measured. The data (mean ± S.D., n = 9) are normalized to the amount of PAR1 detected on the cell surface. The differences in thrombin-stimulated signaling in nonspecific versus RGS5 siRNA-transfected cells was significant as determined by one-way ANOVA and Dunnett's multiple comparison tests (**, p < 0.01).
Coimmunoprecipitation experiments were then performed to determine whether RGS proteins form a complex with endogenous Gαq following activation of PAR1. The expression of RGS5 is low in HeLa cells, and because robust expression of RGS proteins is necessary for the coimmunoprecipitation analysis with Gαq, we used the 3× HA-tagged RGS3 protein (∼100 kDa), which was more easily detectable in post-precipitation immunoblotting as compared with the other RGS proteins. HeLa cells stably expressing the PAR1 WT or AKKAA mutant were transiently transfected with HA-tagged RGS3, stimulated with or without thrombin for 5 min, lysed, and immunoprecipitated with anti-Gαq antibody. The presence of RGS3 was detected by immunoblotting. Activation of wild-type PAR1 with thrombin enhanced the association of Gαq and RGS3 (Fig. 8A). In contrast, thrombin stimulation of the PAR1 AKKAA mutant failed to increase RGS3 protein association with Gαq (Fig. 8A). These findings suggest that AP-2 may function in PAR1-induced interaction of Gαq with RGS3. To assess the function of AP-2 in agonist-induced RGS3 protein association with Gαq we used siRNA to deplete cells of AP-2 expression. In control cells transfected with nonspecific siRNA, the activation of PAR1 resulted in a marked association of Gαq and RGS3 (Fig. 8B). However, in AP-2-deficient cells, the activation of PAR1 failed to cause Gαq association with RGS3 (Fig. 8B). Taken together, these findings suggest that AP-2 may function as an adaptor to facilitate RGS protein recruitment to Gαq protein.
FIGURE 8.
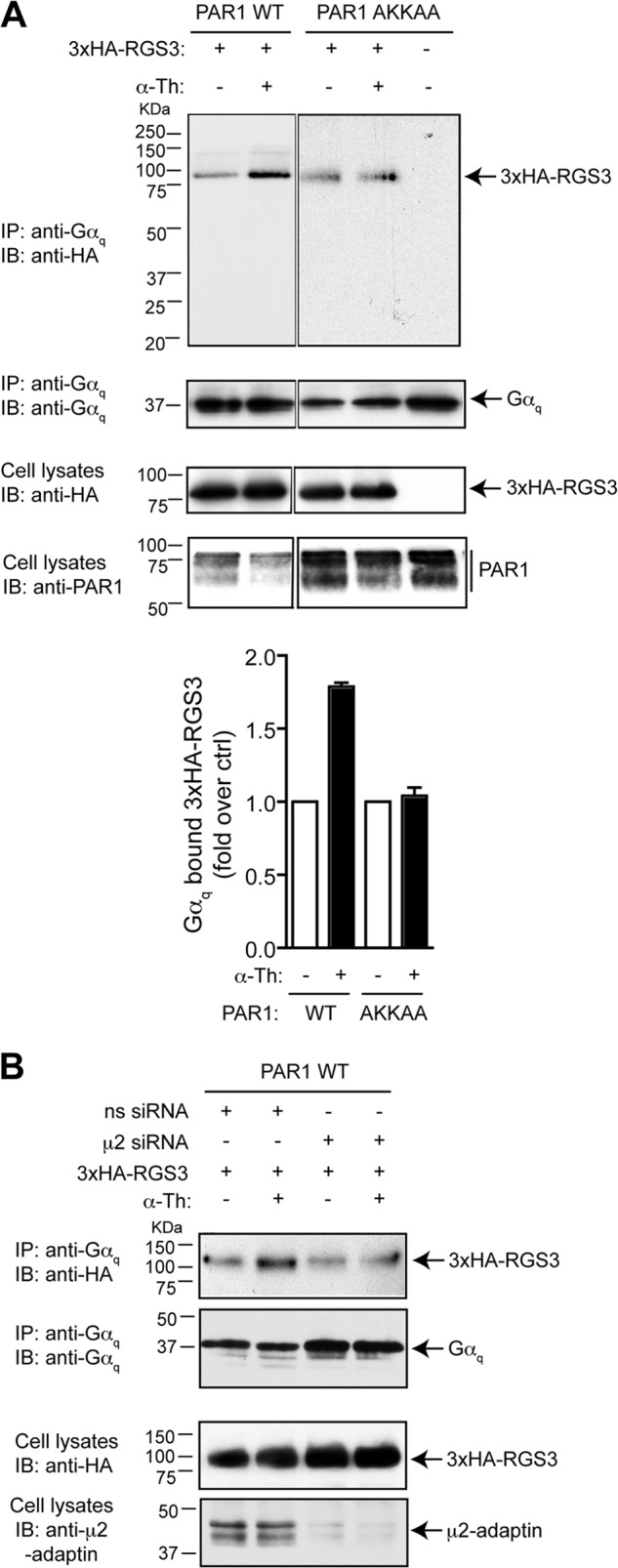
AP-2 mediates activated PAR1-stimulated Gαq-RGS protein complex formation. A, HeLa cells stably expressing FLAG-tagged PAR1 WT or AKKAA mutant were transiently transfected with HA-tagged RGS3 (3×HA-RGS3) and incubated with 10 nm thrombin (α-Th) for 5 min at 37 °C. Equivalent amounts of cells lysates were immunoprecipitated (IP) with anti-Gαq protein antibody, and coassociated RGS3 was detected with anti-HA antibody. Cell lysates were probed with anti-HA, anti-actin, and anti-PAR1 antibody as controls. IB, immunoblot. B, HeLa cells stably expressing FLAG- tagged PAR1 WT were transfected with nonspecific (ns) or μ2-adaptin-specific siRNA together with HA-tagged RGS3. Cells were then incubated with 10 nm thrombin for 5 min at 37 °C, and immunoprecipitated as described above. Cell lysates were immunoblotted with anti-μ2-adaptin antibody to evaluate the efficiency of AP-2 depletion. The data are representative of three independent experiments.
DISCUSSION
GPCR signaling is precisely regulated through various mechanisms mediated by β-arrestins, which function to uncouple the receptor from G protein signaling and promote receptor internalization (29). However, not all GPCRs utilize β-arrestins for desensitization or internalization. We showed previously that internalization of activated PAR1 occurs independently of β-arrestins and requires the clathrin adaptor proteins AP-2 and epsin-1 (4, 10). However, it remained unclear whether AP-2 or epsin-1 regulates PAR1 signaling. Here, we now report that AP-2, and not epsin-1, regulates PAR1 signaling through two distinct mechanisms. Depletion of AP-2 by siRNA or expression of a PAR1 mutant defective in AP-2 binding resulted in enhanced signaling, which correlated with elevated PAR1 expression at the cell surface. Thus, AP-2 regulates PAR1 signaling in part by modulating the amount of receptor expressed at the cell surface. Our data also revealed that certain isoforms of the R4 subfamily of RGS proteins control PAR1 signaling through an AP-2-dependent mechanism that involves agonist-stimulated recruitment of RGS proteins to Gαq protein. These findings suggest a second mechanism by which AP-2 regulates activated PAR1 signaling, namely through the formation of an RGS-G protein complex that efficiently shuts off signaling.
AP-2 is a stable complex, composed of four distinct subunits, that binds to phosphatidylinositol-4,5-bisphosphate and has established functions in clathrin-coated pit assembly and cargo recruitment (30). However, the function of AP-2 in regulating signaling responses is less clear. Previous studies showed that the μ2-adaptin subunit of AP-2 interacts with the type I 4-phosphate 5-kinase core domain to affect phosphatidylinositol-4,5-bisphosphate synthesis (31). Moreover, the binding of endocytic cargo proteins to the μ2-adaptin subunit results in the potent stimulation of type I 4-phosphate 5-kinase activity (31), suggesting that AP-2 can modulate clathrin-mediated endocytosis by controlling phosphatidylinositol-4,5-bisphosphate production. In more recent work, AP-2 has been shown to be an essential structural component of a Wnt-induced “signalosome” at the cell surface (32). The Wnt3A ligand binds to low-density lipoprotein-related proteins 5 and 6 (LRP5/6) and Frizzled seven transmembrane receptors, which stimulates phosphatidylinositol-4,5-bisphosphate production and LRP6 signalosome formation via the recruitment of AP-2 and clathrin. The μ2-adaptin subunit of AP-2 binds directly to a tyrosine-based motif within LRP6, promoting its aggregation and consequent signaling (32). These studies demonstrate that AP-2 has the capacity to bind directly to and modulate effector signaling activity.
In addition to its established function as an endocytic clathrin adaptor protein, AP-2 appears to regulate post-endocytic trafficking. AP-2 has been shown to associate with the N-formyl peptide receptor, a GPCR, on perinuclear endosomes via β-arrestins to facilitate recycling (33). Intriguingly, however, depletion of endogenous AP-2 resulted in the initiation of apoptosis induced by multiple GPCR-specific ligands. These findings suggest that AP-2 has a critical role in GPCR recycling that appears to be linked to cell survival. Recently, AP-2 and phosphatidylinositol clathrin assembly lymphoid-myeloid have been shown to regulate the cellular level of the Alzheimer amyloid precursor protein cleaved C-terminal fragment via the autophagy pathway (34). These findings indicate that AP-2/phosphatidylinositol clathrin assembly lymphoid-myeloid function as autophagic adaptors that recognize and recruit cargo from the endocytic pathway to the LC-3-mediated degradation pathway. Given the emerging evidence that AP-2 has diverse regulatory functions in signaling and trafficking, we hypothesized that AP-2 might regulate activated PAR1-induced G protein signaling.
In this study, we found that AP-2 has a role in controlling PAR1 signaling by modulating receptor expression at the cell surface and through recruitment of RGS proteins. We showed previously that constitutive internalization of PAR1 is mediated by the μ2-adaptin subunit of AP-2 that binds directly to a C-tail distal tyrosine-based motif. In HeLa cells and endothelial cells, siRNA-mediated depletion of AP-2 caused a significant increase in PAR1 expression at the cell surface and enhanced thrombin-stimulated signaling. A PAR1 AKKAA mutant defective in AP-2 binding exhibited similar effects, suggesting that the amount of PAR1 present on the cell surface is related to the extent of signaling. In invasive breast carcinoma cells, increased expression of PAR1 has been correlated with invasion and metastasis (6, 35). We discovered that PAR1 trafficking is severely altered in metastatic breast carcinoma but not in non-metastatic or normal breast epithelial cells. Consequently, PAR1 is not sorted to lysosomes but, rather, internalized and recycled back to the cell surface and results in sustained signaling and enhanced cellular invasion. Thus, maintaining an appropriate amount of PAR1 at the cell surface is critical for proper signaling and appropriate cellular responses.
In addition to modulating PAR1 surface expression, AP-2 also appears to regulate the recruitment of RGS proteins to facilitate termination of PAR1 signaling. The R4 subfamily of RGS proteins functions mainly as GTPase-accelerating proteins for Gαq/11 and Gαi/o proteins (14). We found that a subset of R4 subfamily of RGS proteins, including RGS2, RGS3, RGS4, and RGS5, are expressed in HeLa and endothelial cells. In addition, ectopic expression of RGS2, RGS3, RGS4, and RGS5 markedly attenuated PAR1 signaling, whereas the PAR1 AKKAA mutant defective in AP-2 binding was unaffected. Intriguingly, only siRNA-mediated depletion of RGS5 caused a significant increase in thrombin-activated PAR1 signaling, whereas loss of RGS2, RGS3, and RGS4 expression had no effect. However, signaling by the PAR1 AKKAA mutant was not affected by the loss of RGS5 expression, suggesting that an intact AP-2 binding site is important for regulation by the RGS5 protein. These findings further indicate that RGS5 has a unique function in PAR1 signaling in HeLa cells on the basis of the siRNA-mediated depletion approach. Interestingly, a prior siRNA screen of RGS protein specificity at PAR1 signaling examined in HEK293 cells revealed an important role for RGS2 and RGS8 but not for any other R4 family members (18). RGS8 expression was not detected in HeLa or endothelial cells, but RGS2 expression was confirmed (Fig. 5). Previous studies have clearly established a role for RGS2 in regulation of thrombin signaling in vitro and in vivo (36), suggesting that the failure of RGS2 depletion to affect thrombin signaling in HeLa cells may be due to cell type-specific responses. Indeed, in platelets, thrombin signaling (mediated predominantly by PAR1) occurs through regulation of RGS18 phosphorylation, which modulates its interaction with 14-3-3, spinophilin, and SHP-1 to control G protein signaling (37, 38). However, RGS18 expression was not detected in HeLa and endothelial cells in this study, suggesting that RGS proteins are expressed and regulated uniquely in distinct cell types.
In summary, our work illustrates an important function for the clathrin adaptor AP-2 in the regulation of PAR1 signaling. AP-2 controls the level of PAR1 present at the cell surface, which correlates with the magnitude of signaling, at least for Gαq-stimulated PI hydrolysis. In addition, AP-2 modulates RGS protein recruitment to Gq protein in response to PAR1 activation and, thereby, provides an additional mode by which GPCR signaling can be regulated. Future studies will be important to determine whether other GPCRs that associate with AP-2, such as the N-formyl peptide receptor or thromboxane A2 receptor TPβ, are regulated similarly (33, 39). The precise mechanism by which AP-2 affects RGS protein recruitment to G proteins remains unclear and is an important future pursuit.
Acknowledgments
We thank members of the Trejo laboratory for comments and suggestions.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 GM090689 (to J. T.).
- GPCR
- G protein-coupled receptor
- C-tail
- cytoplasmic tail
- RGS
- regulator of G protein signaling
- PI
- phosphoinositide
- IP
- inositol phosphate(s)
- ANOVA
- analysis of variance.
REFERENCES
- 1. Coughlin S. R. (2005) Protease-activated receptors in hemostasis, thrombosis and vascular biology. J. Thromb. Haemost. 3, 1800–1814 [DOI] [PubMed] [Google Scholar]
- 2. Vu T. K., Hung D. T., Wheaton V. I., Coughlin S. R. (1991) Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell 64, 1057–1068 [DOI] [PubMed] [Google Scholar]
- 3. Ishii K., Chen J., Ishii M., Koch W. J., Freedman N. J., Lefkowitz R. J., Coughlin S. R. (1994) Inhibition of thrombin receptor signaling by a G protein-coupled receptor kinase. Functional specificity among G protein-coupled receptor kinases. J. Biol. Chem. 269, 1125–1130 [PubMed] [Google Scholar]
- 4. Paing M. M., Stutts A. B., Kohout T. A., Lefkowitz R. J., Trejo J. (2002) β-arrestins regulate protease-activated receptor-1 desensitization but not internalization or down-regulation. J. Biol. Chem. 277, 1292–1300 [DOI] [PubMed] [Google Scholar]
- 5. Trejo J., Hammes S. R., Coughlin S. R. (1998) Termination of signaling by protease-activated receptor-1 is linked to lysosomal sorting. Proc. Natl. Acad. Sci. U.S.A. 95, 13698–13702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Booden M. A., Eckert L. B., Der C. J., Trejo J. (2004) Persistent signaling by dysregulated thrombin receptor trafficking promotes breast carcinoma cell invasion. Mol. Cell Biol. 24, 1990–1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Arora P., Cuevas B. D., Russo A., Johnson G. L., Trejo J. (2008) Persistent transactivation of EGFR and ErbB2/HER2 by protease-activated receptor-1 promotes breast carcinoma cell invasion. Oncogene 27, 4434–4445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wolfe B. L., Trejo J. (2007) Clathrin-dependent mechanisms of G protein-coupled receptor endocytosis. Traffic 8, 462–470 [DOI] [PubMed] [Google Scholar]
- 9. Wolfe B. L., Marchese A., Trejo J. (2007) Ubiquitination differentially regulates clathrin-dependent internalization of protease-activated receptor-1. J. Cell Biol. 177, 905–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen B., Dores M. R., Grimsey N., Canto I., Barker B. L., Trejo J. (2011) Adaptor protein complex-2 (AP-2) and epsin-1 mediate protease-activated receptor-1 internalization via phosphorylation- and ubiquitination-dependent sorting signals. J. Biol. Chem. 286, 40760–40770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bonifacino J. S., Traub L. M. (2003) Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu. Rev. Biochem. 72, 395–447 [DOI] [PubMed] [Google Scholar]
- 12. Marchese A., Paing M. M., Temple B. R. S., Trejo J. (2008) G protein-coupled receptor sorting to endosomes and lysosomes. Annu. Rev. Pharmcol. Toxicol. 48, 601–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Paing M. M., Johnston C. A., Siderovski D. P., Trejo J. (2006) Clathrin adaptor AP2 regulates thrombin receptor constitutive internalization and endothelial cell resensitization. Mol. Cell Biol. 28, 3231–3242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kimple A. J., Bosch D. E., Giguère P. M., Siderovski D. P. (2011) Regulators of G-protein signaling and their Gα substrates. Promises and challenges in their use as drug discovery targets. Pharmacol. Rev. 63, 728–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hains M. D., Siderovski D. P., Harden T. K. (2004) Application of RGS bos proteins to evaluate G-protein selectivity in receptor-promoted signaling. Methods Enzymol. 389, 71–88 [DOI] [PubMed] [Google Scholar]
- 16. De Vries L., Zheng B., Fischer T., Elenko E., Farquhar M. G. (2000) The regulator of G protein signaling family. Annu. Rev. Pharmacol. Toxicol. 40, 235–271 [DOI] [PubMed] [Google Scholar]
- 17. Wang Q., Liu M., Mullah B., Siderovski D. P., Neubig R. R. (2002) Receptor-selective effects of endogenous RGS3 and RGS5 to regulate mitogen-activated protein kinase activation in rat vascular smooth muscle cells. J. Biol. Chem. 277, 24949–24958 [DOI] [PubMed] [Google Scholar]
- 18. Laroche G., Giguère P. M., Roth B. L., Trejo J., Siderovski D. P. (2010) RNA interference screen for RGS protein specificity at muscarinic and protease-activated receptors reveals bidirectional modulation of signaling. Am. J. Physiol. Cell Physiol. 299, C654–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Paing M. M., Temple B. R., Trejo J. (2004) A tyrosine-based sorting signal regulates intracellular trafficking of protease-activated receptor-1. Multiple regulatory mechanisms for agonist-induced G protein-coupled receptor internalization. J. Biol. Chem. 279, 21938–21947 [DOI] [PubMed] [Google Scholar]
- 20. Trejo J., Coughlin S. R. (1999) The cytoplasmic tails of protease-activated receptor-1 and substance P receptor specify sorting to lysosomes versus recycling. J. Biol. Chem. 274, 2216–2224 [DOI] [PubMed] [Google Scholar]
- 21. Bodenstein J., Sunahara R. K., Neubig R. R. (2007) N-terminal residues control proteasomal degradation of RGS2, RGS4, and RGS5 in human embryonic kidney 293 cells. Mol. Pharm. 71, 1040–1050 [DOI] [PubMed] [Google Scholar]
- 22. Russo A., Soh U. J., Paing M. M., Arora P., Trejo J. (2009) Caveolae are required for protease-selective signaling by protease-activated receptor-1. Proc. Natl. Acad. Sci. U.S.A. 106, 6393–6397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen C. H., Paing M. M., Trejo J. (2004) Termination of protease-activated receptor-1 signaling by β-arrestins is independent of receptor phosphorylation. J. Biol. Chem. 279, 10020–10031 [DOI] [PubMed] [Google Scholar]
- 24. Lin H., Trejo J. (2013) Transactivation of the PAR1-PAR2 heterodimer by thrombin elicits β-arrestin endosomal signaling. J. Biol. Chem. 288, 11203–11215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Offermanns S., Toombs C. F., Hu Y. H., Simon M. I. (1997) Defective platelet activation in G α (q) -deficient mice. Nature 389, 183–186 [DOI] [PubMed] [Google Scholar]
- 26. Benka M. L., Lee M., Wang G. R., Buckman S., Burlacu A., Cole L., DePina A., Dias P., Granger A., Grant B. (1995) The thrombin receptor in human platelets is coupled to a GTP binding protein of the G α q family. FEBS Lett. 363, 49–52 [DOI] [PubMed] [Google Scholar]
- 27. Neubig R. R., Siderovski D. P. (2002) Regulators of G-protein signalling as new central nervous system drug targets. Nat. Rev. Drug Discov. 1, 187–197 [DOI] [PubMed] [Google Scholar]
- 28. Soundararajan M., Willard F. S., Kimple A. J., Turnbull A. P., Ball L. J., Schoch G. A., Gileadi C., Fedorov O. Y., Dowler E. F., Higman V. A., Hutsell S. Q., Sundström M., Doyle D. A., Siderovski D. P. (2008) Structural diversity in the RGS domain and its interaction with heterotrimeric G protein α-subunits. Proc. Natl. Acad. Sci. U.S.A. 105, 6457–6462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Reiter E., Lefkowitz R. J. (2006) GRKs and β-arrestins. Roles in receptor silencing, trafficking and signaling. Trends Endocrinol. Metab. 17, 159–165 [DOI] [PubMed] [Google Scholar]
- 30. Traub L. M. (2009) Tickets to ride. Selecting cargo for clathrin-regulated internalization. Nat. Rev. Mol. Cell Biol. 10, 583–596 [DOI] [PubMed] [Google Scholar]
- 31. Krauss M., Kukhtina V., Pechstein A., Haucke V. (2006) Stimulation of phosphatidylinositol kinase type I-mediated phosphatidylinositol (4,5)-bisphosphate synthesis by AP-2μ-cargo complexes. Proc. Natl. Acad. Sci. U.S.A. 103, 11934–11939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kim I., Pan W., Jones S. A., Zhang Y., Zhuang X., Wu D. (2013) Clathrin and AP2 are required for PtdIns(4,5)P2-mediated formation of LRP6 signalosomes. J. Cell Biol. 200, 419–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wagener B. M., Marjon N. A., Revankar C. M., Prossnitz E. R. (2009) Adaptor protein-2 interaction with arrestin regulates GPCR recycling and apoptosis. Traffic 10, 1286–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tian Y., Chang J. C., Fan E. Y., Flajolet M., Greengard P. (2013) Adaptor complex AP2/PICALM, through interaction with LC3, targets Alzheimer's APP-CTF for terminal degradation via autophagy. Proc. Natl. Acad. Sci. U.S.A. 110, 17071–17076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Boire A., Covic L., Agarwal A., Jacques S., Sherifi S., Kuliopulos A. (2005) PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell 120, 303–313 [DOI] [PubMed] [Google Scholar]
- 36. Tang K. M., Wang G. R., Lu P., Karas R. H., Aronovitz M., Heximer S. P., Kaltenbronn K. M., Blumer K. J., Siderovski D. P., Zhu Y., Mendelsohn M. E. (2003) Regulator of G-protein signaling-2 mediates vascular smooth muscle relaxation and blood pressure. Nat. Med. 9, 1506–1512 [DOI] [PubMed] [Google Scholar]
- 37. Ma P., Cierniewska A., Signarvic R., Cieslak M., Kong H., Sinnamon A. J., Neubig R. R., Newman D. K., Stalker T. J., Brass L. F. (2012) A newly identified complex of spinophilin and the tyrosine phosphatase, SHP-1, modulates platelet activation by regulating G protein-dependent signaling. Blood 119, 1935–1945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gegenbauer K., Elia G., Blanco-Fernandez A., Smolenski A. (2012) Regulator of G-protein signaling 18 integrates activating and inhibitory signaling in platelets. Blood 119, 3799–3807 [DOI] [PubMed] [Google Scholar]
- 39. Parent J. L., Labrecque P., Orsini M. J., Benovic J. L. (1999) Internalization of the TXA2 receptor α and β isoforms. J. Biol. Chem. 274, 8941–8948 [DOI] [PubMed] [Google Scholar]