

Background: Negative regulation of innate antiviral singling is crucial for preventing cell damage elicited by hyperinflammation.
Results: Gp78 was identified as a negative regulator of innate antiviral signaling.
Conclusion: Gp78 regulates MAVS by both decreasing protein levels and by inhibiting signaling via physical interactions.
Significance: Gp78 regulation of innate antiviral signaling points to a novel and unexpected role for this protein.
Keywords: ERad, Interferon, Mitochondria, Rig-like receptors (RLR), RNA Viruses, Gp78, MAM, MAVS, Antiviral Signaling
Abstract
In a previous study, we identified the E3 ubiquitin ligase Gp78 by RNAi high-throughput screening as a gene whose depletion restricted enterovirus infection. In the current study, we show that Gp78, which localizes to the ER-mitochondria interface, is a regulator of RIG-I-like receptor (RLR) antiviral signaling. We show that depletion of Gp78 results in a robust decrease of vesicular stomatitis virus (VSV) infection and a corresponding enhancement of type I interferon (IFN) signaling. Mechanistically, we show that Gp78 modulates type I IFN induction by altering both the expression and signaling of the mitochondria-localized RLR adaptor mitochondrial antiviral signaling (MAVS). Expression of mutants of Gp78 that abolish its E3 ubiquitin ligase and its participation in ER-associated degradation (ERAD) lost their ability to degrade MAVS, but surprisingly maintained their ability to repress RLR signaling. In contrast, Gp78 lacking its entire C terminus lost both its ability to degrade MAVS and repress RLR signaling. We show that Gp78 interacts with both the N- and C-terminal domains of MAVS via its C-terminal RING domain, and that this interaction is required to abrogate Gp78-mediated attenuation of MAVS signaling. Our data thus implicate two parallel pathways by which Gp78 regulates MAVS signaling; one pathway requires its E3 ubiquitin ligase and ERAD activity to directly degrade MAVS, whereas the other pathway occurs independently of these activities, but requires the Gp78 RING domain and occurs via a direct association between this region and MAVS.
Introduction
Recognition of pathogen-derived nucleic acids is among the most important mechanisms by which a host cell defends against pathogen infection. Upon recognition of these nucleic acids, the transcription of myriad antiviral genes ensues, culminating in a cellular antimicrobial state that equips the cell to resist and/or suppress infection. The cytosolic pattern recognition receptors (PRRs)2 retinoic acid inducible gene-I (RIG-I) and melanoma differentiation-associated gene 5 (MDA5) are largely responsible for initiating the innate immune response to cytosolic dsRNA derived from the replication of viral pathogens (1). The signaling initiated by one or both of these cytosolic sentinels converges on a common mitochondria-localized adaptor molecule, mitochondrial antiviral signaling (MAVS), which in turn leads to nuclear translocation of NF-κB and interferon (IFN) regulatory factor (IRF)-3 for induction of type I interferon (IFN) production (2). MAVS contains an N-terminal caspase recruitment and activation domain (CARD), which is required for both upstream and downstream interactions, as well as a C-terminal mitochondrial localization sequence, which is required for downstream signaling events (2–5).
Because enhanced inflammation can lead to cell damage, mechanisms must exist to tightly regulate antiviral signaling. There are a variety of mechanisms by which regulators specifically modulate MAVS expression and/or signaling. This can be achieved by protein-protein interactions that physically disrupt or enhance upstream or downstream interactions required for propagating MAVS-mediated signaling (6–12). MAVS regulation can also be achieved by post-translational modifications such as ubiquitination that lead to inactivation or proteasomal degradation (12–15). Variations in mitochondrial dynamics have also been reported to play a role in MAVS regulation, such as alterations in mitochondrial fusion/fission (16, 17), membrane potential (17), reactive oxygen species generation (18), and mitochondrial-endoplasmic reticulum contacts (MAMs) (16, 19).
MAMs are defined as sites of close physical contact (∼10–30 nm (20, 21)) between the ER and mitochondria. It is estimated that between 5 and 20% of mitochondria are in direct contact with the ER (22). The MAM is an important cellular domain that regulates a variety of functions involved in cellular homeostasis such as lipid biosynthesis (23, 24), Ca2+ signaling, and cell survival pathways (25–27). Quite interestingly, activated MAVS-containing innate immune synapses form at MAMs, and the population of MAVS at the MAM is targeted by the hepatitis C virus (HCV) NS3/4A protease, underscoring the importance of this compartment in innate antiviral signaling (19). In addition, the mitochondria and MAM have been associated with the induction of inflammasome signaling (28).
The MAM proteome includes Gp78 (29–32), an E3 ubiquitin ligase active in the ER-associated degradation (ERAD) pathway. Gp78 is also a cell surface receptor for the cytokine autocrine motility factor (AMF), the activity of which has been linked with increased cancer metastasis presumably due to its role in cell differentiation, survival and growth (33). Gp78 is responsible for conjugation of ubiquitin to misfolded proteins, which are then directed to the cytosolic proteasome for subsequent degradation (34–36). Gp78-mediated ERAD participation requires its C-terminal RING domain (responsible for ligase activity), Cue domain (responsible for binding of ubiquitin), and E2 (the enzyme responsible for bringing in the ubiquitin) binding site (34–36). The C terminus of gp78 also contains a site of interaction with the AAA ATPase p97 (VCP), which provides the driving force for translocation of the polyubiquitinated substrates to the cytosol for subsequent degradation by the proteasome (37–40). It has also been recently reported that Gp78 induces mitochondrial fragmentation in a ligase-dependent manner, leading to mitophagy upon mitochondrial membrane depolarization (41).
Previously, we conducted RNAi high-throughput screening and identified Gp78 as a gene whose depletion led to a significant reduction in infection of the enteroviruses coxsackievirus B (CVB) and poliovirus (PV) (42). In the current study, we provide a molecular mechanism for these previous findings and show that Gp78 is a novel regulator of RLR signaling. We show that in addition to CVB and PV, depletion of Gp78 results in a robust decrease of vesicular stomatitis virus (VSV) infection. Mechanistically, we show that expression of Gp78 dramatically represses type I interferon (IFN) signaling upstream of IRF3, and that this decrease in signaling corresponds to decreases in MAVS protein levels. Expression of Gp78 mutants defective in E3 ubiquitin ligase activity or ERAD participation lost their ability to decrease MAVS levels, but surprisingly maintained their ability to repress RLR-mediated IFNβ signaling. In contrast, Gp78 lacking its entire C terminus lost both its ability to induce reductions in MAVS expression and repress RLR signaling. These studies point to an unexpected role for the MAM-localized Gp78 E3 ubiquitin ligase in the negative regulation of MAVS signaling. Our data implicate two parallel pathways by which Gp78 regulates MAVS expression and signaling, one pathway requires its E3 ubiquitin ligase and ERAD activity, while the other pathway occurs independently of E3 ubiquitin ligase and ERAD activity, but requires the Gp78 C terminus and occurs via an association between this region and the N- and C-terminal domains of MAVS.
EXPERIMENTAL PROCEDURES
Cells and Viruses
HEK293T cells, human fibrosarcoma HT1080 cells, and human osteosarcoma U2OS cells were cultured in DMEM-H supplemented with 10% FBS and 1× penicillin/streptomycin. Human brain microvascular endothelial cells (HBMEC) were cultured as previously described (42, 43). Experiments were performed with CVB3-RD at 3 particle-forming units (pfu)/cell (expanded as previously described (44)), PV Sabin 2 at 1 pfu/cell (previously described in (45)), recombinant GFP-expressing vesicular stomatitis virus at 2 pfu/cell (VSV, as described in Ref. 44), and Sendai virus at 25 hemagglutination units (HAU)/ml (SeV, Cantell strain purchased from Charles River Laboratories).
Antibodies
Mouse anti-enterovirus VP1 (Ncl-Entero) was obtained from Novocastra Laboratories. Mouse anti-GFP (B-2), mouse anti-V5 (H-9), rabbit and mouse anti-Flag (OctA, D-8 or H-5, respectively), rabbit anti-GAPDH (FL-335), and goat anti-Gp78 (N-18) were obtained from Santa Cruz Biotechnology. Rabbit anti-MAVS was obtained from Bethyl Laboratories. Mouse monoclonal antibody to mitochondria (MTCO2) was obtained from Abcam. Alexa fluor-conjugated secondary antibodies were from Invitrogen. Rat anti-Gp78 IgM (3F3A) was a generous gift from Dr. Ivan Nabi (University of British Columbia, Vancouver, Canada) and was previously described (46).
Plasmids, siRNAs, and Transfections
Unless otherwise specified, all Gp78 constructs were of human origin. PCI-Neo-gp78/JM20 was purchased from Addgene and was previously described (35). For subsequent cloning, Gp78 was amplified by PCR from pCI-Neo-Gp78/JM20 using primers encoding an N-terminal Flag tag, and was then cloned into pcDNA3.1 using BamHI and XbaI sites. C-terminal Gp78 mutants were generated by standard PCR cloning. Primer sequences are available upon request. Flag-tagged mouse Gp78 and the mouse Gp78 RING mutant were provided by Dr. Ivan Nabi and have been previously described (41). EGFP-MAVS, EGFP-MAVS-CT, and -NT, EGFP-RIG-I, V5-IRF3–5D, and EGFP-STING have been described previously (44) (47).
The siRNA targeting Gp78 was purchased from Sigma Aldrich (GGACGAACUCCUCCAGCAAtt). Control (scrambled) siRNAs were purchased from Ambion or Sigma.
Plasmid transfections were performed using X-tremeGENE 9 or HP (Roche) essentially per the manufacturer's protocol. For siRNA transfections, HBMEC or HT1080 were transfected with siRNAs (final concentration 25–75 nm) using DharmaFECT-1 transfection reagent (Thermo-Fisher Scientific) according to the manufacturer's protocol.
Immunoblots
Cells were grown in 24-well plates, and lysates prepared with RIPA buffer (50 mm Tris-HCl, pH 7.4, 1% Nonidet P-40, 0.25% sodium deoxycholate, 150 mm NaCl, 1 mm EDTA, 1 mm phenylmethanesulfonyl fluoride, 1 mg/ml aprotinin, leupeptin, and pepstatin). Lysates were run on 4–20% Tris-HCl gels (Bio-Rad) and transferred to nitrocellulose membranes. Membranes were blocked using 5% nonfat dry milk, probed with the indicated antibodies, and developed using horseradish peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology) and SuperSignal West Pico or Dura chemiluminescent substrates (Pierce Biotechnology). Densitometry was performed using Image J (NIH).
Immunoprecipitations
HEK293T cells transiently transfected with the indicated plasmids were lysed with RIPA buffer (450 mm NaCl, 1 mm EDTA, 50 mm Tris-HCl, pH 7.8, 1% Nonidet P-40, 1 mm phenylmethylsulfonyl fluoride, 0.5 μg/ml leupeptin, and 0.5 μg/ml pepstatin), and insoluble material was cleared by centrifugation. Lysates were incubated with the indicated antibodies for 1–2 h at 4 °C followed by the addition of Sepharose G beads for an additional 1–2 h at 4 °C. After centrifugation, the beads were washed with RIPA buffer a minimum of five times and heated at 95 °C for 10 min in Laemmli sample buffer. Following a brief centrifugation, the supernatant was immunoblotted with the indicated antibodies.
Reporter-Gene Assays
Activation of IFNβ and NF-κB promoters was quantified using dual luciferase reporter-gene assays. Cells were transfected in 96-well plates with p-125 luc (which contains the entire IFNβ promoter upstream of firefly luciferase) or NF-κB reporter (which contains NF-κB responsive promoter elements upstream of firefly luciferase) plasmids together with a control Renilla luciferase plasmid (pRL-null, Promega) and the indicated plasmids. Cells were lysed and prepared for luciferase measurement using the Dual-Luciferase assay kit (Promega) according to the manufacturer's instructions, and luciferase activity was measured using a Synergy 2 luminescence plate reader (Bio-Tek). Data are presented as fold induction over uninfected or vector-transfected controls, and are normalized to Renilla luciferase activity. All experiments were performed in triplicate and conducted a minimum of three times.
RT-qPCR
Total cellular RNA was extracted using TRI reagent (MRC) according to the manufacturer's protocol. RNA samples were treated with RNase-free DNase (Qiagen) prior to cDNA synthesis. Total RNA (1 μg) was reverse transcribed using iScript cDNA synthesis kit (Bio-Rad). RT-qPCR was performed using iQ SYBR Green Supermix (Bio-Rad) in an Applied Biosystems StepOnePlus real-time PCR machine. Gene expression was calculated using the 2-ΔΔCT method and was normalized to actin (48). QuantiTect primers against Gp78 were purchased from Qiagen and actin primer sequences have been described elsewhere (49). Primer sequences are as follows: MAVS (Forward, 5′-GTCACTTCCTGCTGAGA-3′; Reverse, 5′-TGCTCTGAATTCTCTCCT-3′); ISG56 (Forward, 5′-CAACCAAGCAAATGTGAGGA-3′; Reverse, 5′-AGGGGAAGCAAAGAAAATGG-3′).
Immunofluorescence Microscopy
Cells cultured in 8-well chamber slides (LabTek, Nunc) were washed and fixed with either 4% paraformaldehyde or with ice-cold methanol. Cells were then permeabilized with 0.1% Triton X-100 in phosphate-buffered saline (PBS) and incubated with the indicated primary antibodies for 1 h at room temperature (RT). Following washing, cells were incubated with secondary antibodies for 30 min at room temperature, washed, and mounted with Vectashield (Vector Laboratories) containing 4′,6-diamidino-2-phenylindole (DAPI). Images were captured using a FV1000 confocal laser scanning microscope (Olympus), analyzed using Image J (NIH) or FV10-ASW (Olymous) (Bitplane), and contrasted and merged using Photoshop (Adobe).
Statistical Analysis
Data are presented as mean ± S.D. Unpaired, two-tailed t test or one-way analysis of variance (ANOVA) and Bonferroni's correction for multiple comparisons were used to determine statistical significance (*, p < 0.01).
RESULTS
Gp78 Is a Regulator of RNA Virus Infection
We previously conducted high throughput RNAi screens for novel regulators of enterovirus infection in human brain microvascular endothelial cells (HBMEC) (42), and identified Gp78 as a regulator of CVB and PV infection whose depletion led to a robust decrease of infection (42) (Fig. 1A). To expand on these findings, we also determined the effects of Gp78 depletion on CVB infection in the fibrosarcoma cell line HT1080 (a cell type reported to express high levels of Gp78 (50)). Similar to our results in HBMEC, we found that RNAi-mediated Gp78 silencing decreased CVB infection (Fig. 1C). In addition, we found that Gp78 silencing also reduced the infection of the unrelated RNA virus VSV, a member of the rhabdovirus family (Fig. 1, B and C). Efficient reduction of Gp78 expression in the presence of RNAi was achieved in these experiments (Fig. 1D). Taken together, these data show that depletion of Gp78 in both HBMEC and HT1080 cells results in a decrease of CVB, PV, and VSV infection, suggesting a mechanism that is common to two independent families of RNA viruses in disparate cell types.
FIGURE 1.

Gp78 depletion restricts RNA virus replication. A, decreased CVB and PV replication by high-throughput RNAi screening in HBMEC transfected with Gp78 siRNAs (siGp78) compared with an siRNA targeting a gene within the library that had no effect on viral replication (si-No Effect). VP1 staining is shown in green, and DAPI-stained nuclei are shown in blue. White text at bottom left denotes the level of infection (%). B, decreased VSV-GFP replication in HT1080 cells transfected with siGp78 compared with control siRNA (siCON), as assessed by immunofluorescence microscopy at 8 h post-infection. VSV-GFP is shown in green, and DAPI-stained nuclei are shown in blue. C, decreased VSV-GFP and CVB replication in HT1080 cells transfected with siGP78 compared with control siRNA (siCON), as assessed by immunofluorescence microscopy at 8 h (VSV-GFP) or 16 h (CVB) post-infection. D, level of Gp78 expression in HT1080 cells transfected with siGp78 compared with a control siRNA, as assessed by RT-qPCR 48 h post-transfection. All data are representative of at least three independent experiments, and data in C and D are presented as mean ± S.D. (*, p < 0.01).
Gp78 Negatively Regulates Type I IFN Signaling
Because depletion of Gp78 resulted in a decrease of infection by two unrelated families of RNA viruses, we next determined whether Gp78 regulated some aspect of type I IFN signaling. We found that expression of exogenous Gp78 led to a significant decrease in Sendai virus (SeV)-induced signaling to both the IFNβ and NF-κB promoters (Fig. 2, A and B). SeV is specifically recognized by RIG-I (51). In addition, we found that overexpression of Gp78 greatly attenuated the induction of the interferon stimulated gene (ISG)-56 by both cytosolic poly (I:C) or SeV infection (Fig. 2, C and E). In contrast, RNAi-mediated silencing of Gp78 greatly enhanced this induction (Fig. 2, D and E). Of note, the expression levels of ISG56 slightly increased in the absence of Gp78 (by ∼16-fold) even without stimulation by SeV (Fig. 2D), which could point to a steady state regulatory role for Gp78 in type I IFN signaling. Taken together, these data suggest a negative regulatory role for Gp78 in type I IFN.
FIGURE 2.

Gp78 regulates type I interferon signaling. A and B, dual luciferase assays from 293T cells transfected with IFNβ (A) or NF-κB (B) promoted luciferase constructs and the indicated plasmids. Cells were infected with SeV 24 h post-transfection and luciferase activity was measured 16 h post-infection. C and D, levels of ISG56 in untreated 293T (C) or HT1080 (D) cells transfected with the indicated plasmids (C) or siRNAs (D), transfected with 500 ng poly (I:C) (C), or infected with SeV (D) at 48 h post-transfection for 16 h, as assessed by RT-qPCR. E, Gp78 expression from 293T (overexpression) or HT1080 (siRNA) cells transfected with the indicated plasmids (left) or siRNAs (right), as assessed by RT-qPCR. Data are presented as mean ± S.D. and correspond to data shown in C and D. F, level of Gp78 (left) or ISG56 (right) expression in untreated HT1080 cells, or cells transfected with 500 ng poly (I:C), treated with 500 units/ml IFNβ overnight, or infected with SeV for 24 h, as assessed by RT-qPCR. All data are representative of at least three independent experiments and presented as mean ± S.D. (*, p < 0.01).
A common characteristic of many type I IFN mediators is their inducible expression upon treatment with virus infection, purified interferon and/or PRR agonists (2, 28, 52–54). Given the possible role of Gp78 in the regulation of type I IFN signaling, we determined if its expression was inducible under these conditions. We found that expression of Gp78 was not induced by treatment with purified IFNβ or cytosolic poly (I:C) or by infection with SeV (Fig. 2F), a result underscoring the possible steady state regulatory role for Gp78 in type I IFN signaling.
Gp78 Negatively Regulates RLR Signaling
RLR signaling is one of the most important components of the type I IFN response to RNA viruses. Given that depletion of Gp78 restricted the replication of both CVB and VSV, and enhanced type I IFN signaling, we next investigated the role of Gp78 in RLR signaling. We found that expression of exogenous Gp78 greatly decreased signaling to the IFNβ promoter induced by overexpression of RIG-I, MDA5, and MAVS (Fig. 3A). However, exogenously expressed Gp78 had no effect on signaling to the IFNβ promoter induced by overexpression of a constitutively active mutant of IRF-3 (IRF3–5D) (55) (Fig. 3B). These results suggest that Gp78 exerts its regulatory role within the RLR pathway upstream of IRF3 activation.
FIGURE 3.
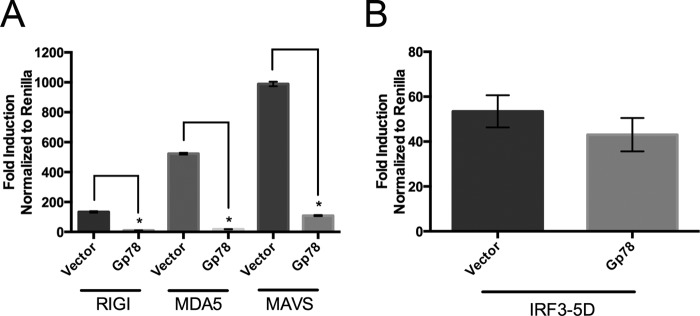
Gp78 regulates RLR signaling. A and B, dual luciferase assays from 293T cells transfected with IFNβ promoted luciferase constructs, RIG-I, MDA5, MAVS (A), or IRF3–5D (B), and the indicated plasmids (vector or Gp78). Luciferase activity was measured 48 h post-transfection. All data are representative of at least three independent experiments and presented as mean ± S.D. (*, p < 0.01).
Gp78 Expression Results in the Post-translational Down-regulation of MAVS
Because silencing of Gp78 restricted the replication of CVB and VSV, which are detected by different RLRs (11, 56, 57), and because Gp78 expression abrogated RLR signaling upstream of IRF3, we next investigated the effect of Gp78 on the expression of various innate immune-associated components. Strikingly, we found that overexpression of Gp78 resulted in a marked decrease in EGFP-MAVS protein levels by immunoblotting (Fig. 4A). In contrast, expression of Gp78 had no effect on the expression of EGFP-RIG-I, V5-IRF3–5D, or the unrelated ER-localized IFN signaling molecule EGFP-STING (Fig. 4A). Importantly, overexpression of Gp78 had no effect on MAVS mRNA levels, suggesting that the Gp78-mediated decrease in MAVS protein levels occurs post-translationally (Fig. 4B).
FIGURE 4.

Gp78 specifically alters MAVS levels. A, immunoblot analysis from 293T cells 48 h post-transfection with EGFP-MAVS, EGFP-RIG-I, V5-IRF3–5D, or EGFP-STING and either vector or Gp78. Antibodies directed against GFP or V5 were used. Immunoblotting for Gp78 (middle panel) is included to demonstrate transfection and GAPDH (bottom panels) is included as a loading control. B, levels of MAVS in 293T cells transfected with vector or Gp78 as assessed by RT-qPCR at 48 h post-transfection. C, immunoblot analysis for endogenous MAVS 48 h post-transfection with increasing amounts of Gp78 (from 0 μg to 2 μg). Immunoblotting for Gp78 (middle panel) is included to demonstrate transfection and GAPDH (bottom panels) is included as a loading control. D, immunofluorescence microscopy for endogenous MAVS in U2OS cells 48 h post-transfection with Flag-Gp78. MAVS is shown in red, and Flag-Gp78 is shown in green. DAPI-stained nuclei are shown in blue. White arrows denote areas of decreased MAVS staining in the presence of Gp78. E, immunoblot analysis from 293T cells 48 h post-transfection with EGFP-MAVS and either vector or Gp78. Antibodies directed against GFP and MTCO2 (an unrelated mitochondrial protein) are included as a measure of specificity, and immunoblotting for Gp78 (middle panel) is included to demonstrate transfection and GAPDH (bottom panels). All data are representative of at least three independent experiments, and data in B are presented as mean ± S.D. (*, p < 0.01).
Because our previous results relied on the overexpression of MAVS, we also determined whether expression of Gp78 reduced levels of endogenous MAVS. Similar to our findings with exogenously expressed MAVS, we found that Gp78 also decreased the levels of endogenous MAVS in a dose-dependent manner (Fig. 4C). In addition, we observed a pronounced loss of endogenous MAVS immunofluorescence in cells transfected with Gp78 (Fig. 4D).
Given that Gp78 has been associated with mitochondrial fragmentation and mitophagy (41), we also determined whether overexpression of Gp78 would lead to the possible degradation of other mitochondria-localized components. We found that expression of Gp78 had no effect on the levels of the constitutive mitochondrial marker MTC02 (Fig. 4E). Taken together, these data show that MAVS protein levels are post-translationally decreased in the presence of Gp78 in a specific manner that does not rely on mitophagy or mitochondrial fragmentation.
Gp78 Colocalizes with MAVS and Specifically Targets the MAVS CARD
Consistent with the work of others (30, 32), we found that exogenously expressed Gp78 partially localized with a marker of mitochondria (Fig. 5A). In addition, we found that endogenous Gp78 colocalized with endogenous MAVS, likely at the ER-mitochondria interface in uninfected cells and in cells infected with SeV (Fig. 5B). MAVS contains a C-terminal domain that mediates its mitochondrial localization and is required for its activity, and an N-terminal CARD-containing region that is required for upstream and downstream interactions (2). To investigate which region of MAVS is required for Gp78-mediated degradation, we cotransfected Flag-Gp78 with either full length MAVS (MAVS-WT), or with deletion mutants of MAVS containing 148 N-terminal amino acids including the CARD (MAVS-NT) or 391 C-terminal amino acids including the transmembrane domain, but lacking the CARD (MAVS-CT) (Fig. 5C). We found that expression of Gp78 induced a reduction in the expression of both MAVS-WT and MAVS-NT, but had no significant effect on the expression of MAVS-CT. These data suggest that the CARD-containing N terminus of MAVS is required for Gp78-mediated decreases in expression.
FIGURE 5.

Gp78 is localized at the mitochondria in close proximity to MAVS, and targets the CARD of MAVS. A, immunofluorescence microscopy of U2OS cells 48 h post-transfection with Flag-Gp78. Mitochondria are shown in red and were stained with MTCO2 antibody and Gp78 is shown in green and was stained with a Flag-specific antibody. B, immunofluorescence microscopy of endogenous MAVS and endogenous Gp78 in U2OS cells in uninfected (Mock) or SeV-infected HT1080 cells. MAVS is shown in red, and Gp78 is shown in green and was stained using the 3F3A antibody specific for Gp78. C, top, schematic of MAVS constructs used. Bottom, immunoblot analysis from 293T cells 48 h post-transfection with EGFP-MAVS, EGFP-MAVS-CT, or EGFP-MAVS-NT with either vector or Gp78. Antibody directed against GFP was used. Immunoblotting for Flag and GAPDH (bottom panel) are included to show expression of Gp78 and as a loading control, respectively. Table at bottom, densitometry (MAVS/GAPDH normalized to vector control) in cells transfected with vector of Gp78. All data are representative of at least three independent experiments.
Gp78-mediated Degradation of MAVS Requires Its E3 Ubiquitin Ligase and ERAD Activity
Several domains within the C terminus of Gp78 are critical for its E3 ubiquitin ligase and ERAD activities (schematic, Fig. 6A, left). These include a RING finger domain, a CUE domain, an E2-binding region (G2BR) (34–36), and a VCP-interacting motif for translocation of the polyubiquitinated substrates to the cytosol for subsequent degradation by the proteasome) (37–40). We next sought to determine the role of the E3 ubiquitin ligase and ERAD functions of Gp78 in its degradation of MAVS using a panel of point and truncation mutants (schematic, Fig. 6A, right). When MAVS was cotransfected with human or mouse Gp78 (hGp78 or mGp78, respectively), a robust decrease in MAVS protein levels was evident by immunoblotting (Fig. 6B, compare lane 1 to lanes 2 and 3). However, MAVS protein levels were unaffected by a point mutant of mGp78 described previously (41) that abolishes its E3 ligase activity (mGp78 RING mut) (Fig. 6B, compare lane 1 to lanes 3 and 4). Furthermore, truncation mutants of hGp78 that have been described previously to inhibit its participation in the ERAD pathway (34, 35) (lacking the VIM (ΔVCP), VIM and G2BR (ΔG2BR), VIM, G2BR and CUE (ΔCUE), or the entire C terminus (ΔC)), lost their ability to decrease MAVS (Fig. 6B, compare lane 1 to lanes 4–8). Importantly, when treated with MG132 (a proteasome inhibitor), MAVS protein levels were partially restored (∼2.5-fold restoration) in the presence of Gp78 (Fig. 6C). Interestingly, consistent with the work of others (53), we found that MAVS protein levels (Fig. 6D), but not RNA levels (not shown), were significantly decreased in response to SeV infection. Together, these data point to a role for the E3 ubiquitin ligase and ERAD activity of Gp78 in MAVS degradation.
FIGURE 6.

The E3 ubiquitin ligase activity of Gp78 and its association with the ERAD pathway is required for Gp78-mediated MAVS degradation. A, left panel, schematic of Gp78 showing important regions for E3 ubiquitin ligase activity. Right panel, schematic of the C terminus of Gp78 illustrating deletion mutants used in this panel. B, immunoblot analysis from 293T cells 48 h post-transfection with EGFP-MAVS and the indicated plasmids. Antibody directed against GFP was used. Immunoblotting for Flag and GAPDH (bottom panel) are included to show expression of wild-type and mutant Gp78 and as a loading control, respectively. Table at bottom, densitometry (MAVS/GAPDH normalized to vector control) in cells transfected with the indicated plasmids. Densitometry was performed, and data are presented as fold change from vector untreated (bottom panel). C, immunoblot analysis from 293T cells 48 h post-transfection with EGFP-MAVS and either vector control or Gp78. MG132 (20 μm) was added 16 h post-transfection. Antibodies directed against GFP and GAPDH were used. Table at bottom, densitometry (MAVS/GAPDH normalized to vector control) in cells transfected with the indicated plasmids and either Mock- or MG132-treated. D, immunoblot analysis of MAVS (top) from 293T cells infected with SeV (50 or 100 HAU/ml) for ∼24 h. Immunoblotting for actin (bottom) is included as a loading control. Table at bottom, densitometry (MAVS/Actin normalized to control (mock infection).
Gp78-mediated Abrogation of MAVS-mediated Signaling Occurs Independently of E3 Ubiquitin Ligase and ERAD Activities
We found that the E3 ubiquitin ligase and ERAD activities of Gp78 were required for its degradation of MAVS. Thus, we next determined whether this activity was also required for Gp78-mediated abrogation of type I IFN signaling. Surprisingly, we found that most of the mutants of Gp78 that ablated its ability to alter MAVS expression (mGp78 RING mut, hGP78-ΔVCP, -ΔG2BR, and -ΔCUE), retained their ability to suppress SeV-induced IFNβ signaling (Fig. 7A). In contrast, only the hGp78 mutant lacking almost the entire C terminus (hGP78-ΔC) lost the ability to attenuate antiviral signaling (Fig. 7A). These data suggested that there are divergent mechanisms by which Gp78 induces MAVS degradation and attenuates MAVS-mediated signaling.
FIGURE 7.

The C terminus of Gp78 interacts with the N- and C-terminal regions of MAVS and is required to ablate MAVS-mediated signaling. A, dual luciferase assays from 293T cells transfected with IFNβ promoted luciferase constructs and the indicated plasmids (a schematic of these constructs is shown in Fig. 6A, right). Cells were mock-infected or infected with SeV 24 h post-transfection and luciferase activity was measured 16 h post-infection. B, immunoblot analysis from 293T cell immunoprecipitates transfected with EGFP-MAVS and the indicated plasmids. Immunoprecipitation was performed with anti-Flag antibody, and immunoblotting was performed with anti-GFP antibody. Input (GFP) is shown at bottom. C, immunofluorescence microscopy for Flag-Gp78 wild-type (left) or ΔC (right) (in green) ∼24 h following transfection in U2OS cells. Mitochondria are shown in red and were detected using anti-MTCO2. DAPI-stained nuclei are shown in blue. D, immunoblot analysis from 293T cell immunoprecipitates transfected with EGFP-MAVS-NT or -CT and either vector, Flag-Gp78 ΔVCP, or Flag-Gp78-ΔC. Immunoprecipitation was performed with anti-Flag antibody, and immunoblotting was performed with anti-GFP antibody. Input (GFP) is shown at bottom. Arrows denote NT and CT fragments, and ns denotes a nonspecific band. Data are representative of at least three independent experiments, and data in A are presented as mean ± S.D. (*, p < 0.01).
The C Terminus of Gp78 Interacts with MAVS and Binds to Both the N- and C-terminal Domains of MAVS
There are several pathways by which cellular components attenuate MAVS-mediated signaling. One of these includes the use of specific protein-protein interactions to inhibit the binding of key upstream and/or downstream innate immune signaling components to MAVS (6–8). Given that Gp78 localizes in close proximity to MAVS (Fig. 5B) and can attenuate MAVS-mediated signaling even in the absence of its degradation (Fig. 7A), we next determined whether Gp78 and MAVS form an interaction by performing coimmunoprecipitation studies. For these studies, we utilized the ΔVCP, ΔCUE, and ΔC mutants of Gp78, but not wild-type Gp78, to avoid experimental difficulties related to the decrease in MAVS levels mediated by full length Gp78. We found that MAVS coimmunoprecipitated with both Gp78-ΔVCP and Gp78-ΔCUE (Fig. 7B). In contrast, MAVS did not coimmunoprecipitate with Gp78-ΔC, despite this modification not significantly altering its localization (Fig. 7, B and C). These data suggest that Gp78 utilizes a domain between amino acids 311 and 455 of its C terminus, most likely its RING region, to interact with MAVS.
Both the N- and C-terminal domains of MAVS play critical roles in its activation by upstream components and propagation of downstream signals. For example, whereas both RIG-I and MDA5 bind to the N-terminal CARD of MAVS (2), tumor necrosis factor (TNF) receptor-associated factor (TRAF)-3 binds to a region within the C terminus of MAVS (58). Because we observed an association between Gp78 and MAVS, and a possible ablation of MAVS-mediated signaling as a result of this interaction, we next determined whether Gp78 interacted with the N- or C-terminal regions of MAVS. We found that Gp78-ΔVCP interacted with both the N- and C-terminal domains of MAVS, whereas we did not detect any association of either of these domains with Gp78-ΔC, as expected (Fig. 7D). These data show that Gp78 utilizes a region within its C terminus to interact with multiple regions of MAVS.
DISCUSSION
Here we report on the regulation of MAVS expression and signaling by the MAM-associated E3 ubiquitin ligase Gp78. We identified Gp78 initially using an unbiased high throughput RNAi screen to identify novel regulators of enterovirus infection (42). In the follow-up studies presented here, we found that RNAi-mediated silencing of Gp78 also restricted VSV infection and correlated with enhancements of type I IFN antiviral signaling. Mechanistically, we found that Gp78 alters RLR signaling by both enhancing the degradation of MAVS via its E3 ubiquitin ligase and ERAD-mediated functions and by specifically interacting with MAVS via a region within its C terminus. Collectively, these data report on the unexpected role of Gp78 in the regulation of MAVS-mediated antiviral signaling and suggest that it specifically functions to attenuate antiviral signaling of MAVS at the MAM by two parallel pathways (reviewed in the schematic shown in Fig. 8).
FIGURE 8.
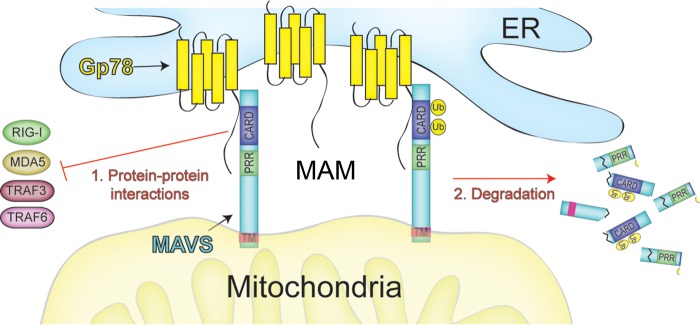
Schematic of the proposed mechanisms of Gp78-mediated regulation of MAVS signaling. Based on the data presented here, we propose a model in which MAM-associated Gp78 regulates MAVS signaling by two mechanisms: (1) protein-protein interactions and (2) ubiquitin-mediated degradation. In (1), Gp78 binding to MAVS might prevent its association with upstream (RIG-I/MDA5) or downstream (TRAF3/TRAF6) components associated with antiviral signaling. In (2), Gp78 utilizes its E3 ubiquitin ligase and ERAD functions to induce the degradation of MAVS. Although not specifically depicted in this schematic, MAVS oligomerization is a critical component of its signaling (60, 69, 70).
Whereas CVB and other enteroviruses are sensed by MDA5, RLR-mediated anti-VSV signaling is specifically mediated by RIG-I (11, 56, 57). Our findings that Gp78 depletion suppressed infection by both viruses supports its specific regulation of an innate-immune associated factor common to both viruses, such as MAVS. Regulation of antiviral signaling at the mitochondrial level is quite strategic given that signals propagated by two independent cytosolic sensors converge on MAVS at the mitochondrial membrane. Therefore, regulators of MAVS such as Gp78 exert a higher level of control than they might if they targeted upstream components of RLR signaling such as RIG-I or MDA5 individually.
The MAM is emerging as a critical platform for MAVS-mediated innate antiviral signaling (19). In light of evidence that the active population of MAVS in virus-infected cells is localized to the MAM (19, 30, 32), negative regulation of MAVS in this compartment is critical to prevent excessive inflammation. Therefore, MAM-localized Gp78 is an ideal candidate to negatively regulate MAVS. Interestingly, we found that type I IFN signaling is enhanced in the absence of Gp78 in uninfected cells. This could indicate that Gp78 plays a housekeeping role in the regulating of MAVS signaling to suppress MAVS-mediated innate immune signaling under basal states, likely as a means of avoiding hyperinflammatory signaling. This notion is supported by our findings that Gp78 expression is not induced by purified IFNβ treatment, transfection of cells with a synthetic ligand of RLR signaling, or SeV infection. Given the lack of a robust induction of Gp78 in response to any RLR ligand, it is possible that other interferon-inducible regulators of MAVS exist within the MAM, and that these would be important for immediate regulation during an acute viral infection. However, in the event of excessive inflammation after a viral infection has been contained, or in the context of excessive inflammation in the absence of a viral threat (i.e. autoimmunity), negative regulation of MAVS at the MAM by a steady-state protein such as Gp78 would become critical for cellular homeostasis.
Ubiquitin-mediated proteasomal degradation of MAVS is a known mechanism for its negative regulation (13–16, 59). Moreover, Gp78 is a well-characterized E3 ubiquitin ligase of the ERAD pathway. Given that Gp78-induced degradation of MAVS was E3 ubiquitin ligase and ERAD dependent, and that the effect was partially rescued with the proteasome inhibitor MG132, we conclude that Gp78-mediated MAVS degradation is achieved at least in part by its Gp78-mediated ubiquitination and proteasomal degradation. Whereas the RING, Cue, and G2BR regions are important for the E3 ubiquitin ligase activity of Gp78 (34, 35), the VIM region is not required for the enzymatic addition of ubiquitin to the substrate and is instead required for translocation of the ubiquitinated substrate out of the ER membrane and to the cytosol for completion of the ERAD pathway (37–40). Importantly, mutant Gp78 lacking this domain did not degrade MAVS, supporting a specific role for ERAD in MAVS degradation. The ERAD pathway is an ER-specific mechanism of protein quality control, and is mainly responsible for destruction of misfolded proteins exiting the ER by marking them with ubiquitin for proteasomal degradation (61). Although the requirement of the VIM region seems to point to a role for the ERAD pathway in Gp78-mediated MAVS degradation, it is important to note that MAVS is a membrane-localized protein in the mitochondria that, like ERAD substrates in the ER, would require translocation into the cytosol for interaction with the proteasome even in the absence of traditional ERAD. In fact, there are specific substrates recognized and negatively regulated by Gp78-mediated ubiquitination and proteasomal degradation in a manner distinct from ERAD's traditional role of nonspecific protein quality control (62, 63), including MAVS.
We show that the CARD-containing N-terminal region of MAVS, but not the C-terminal region of MAVS, is targeted for Gp78-mediated degradation. However, RIG-I, which also contains CARDs that are subject to ubiqutination (64), is not sensitive to Gp78-mediated degradation. These data point to the specificity of the Gp78-mediated degradation of MAVS and suggest that it may not target all CARDs. In addition, as we observed degradation of MAVS-NT, this suggests that the mitochondrial localization of MAVS is not required for its degradation by Gp78.
Surprisingly, we observed a repression of MAVS-mediated signaling by Gp78 mutants incapable of participating in the ERAD pathway. We show that Gp78 binds to both the N- and C-terminal regions of MAVS, and that the region of Gp78 required for this binding is within RING-containing residues 311–455, as binding was lost when these residues were removed. Because RING domains are known to mediate protein-protein interactions (65, 66), it is likely that this region of Gp78 mediates its interaction with MAVS. Protein-protein interaction is a well-defined mechanism of regulating MAVS-mediated signaling. This is often achieved by physically blocking key interactions of MAVS with up or downstream signaling partners (6–10). It is possible that binding of Gp78 to MAVS within its CARD would disrupt the interaction between MAVS and RIG-I/MDA5 that is required for downstream signaling (2–5). However, it is also possible that Gp78 binds to a different region of MAVS, such as the proline-rich region or TRAF interaction motifs (TIMs) required for MAVS interaction with the downstream signaling adaptors TRAF2/3/5/6 (5, 58, 67, 68), effectively disrupting and suppressing signaling. In addition, Gp78 may inhibit some aspect of MAVS oligomerization, which has been shown to play an important role in its signaling (60, 69, 70).
Taken together, our study provides evidence for two possible mechanisms for the regulation of MAVS expression and signaling by Gp78. The first mechanism requires the ERAD activity of Gp78 and likely corresponds enhancements in MAVS ubiquitination and proteosomal degradation while the second occurs independently of ERAD function, but requires Gp78-MAVS interactions (schematic, Fig. 8). Both of these mechanisms likely require physical interaction of Gp78 with MAVS. These results shed light on a novel function of Gp78 in the regulation of MAVS-mediated antiviral signaling. Moreover, our work suggests that other MAM-localized components might also serve to specifically target MAVS as a means to regulate inflammatory signaling within the cell. Defining the specific components of the MAVS regulome specifically within the MAM will undoubtedly provide exciting new insights into the regulation of antiviral signaling.
Acknowledgment
We thank Ivan R. Nabi for generously providing reagents.
This work was supported, in whole or in part, by National Institutes of Health Grant R01-AI081759 (to C. B. C.).
- PRR
- pattern recognition receptor
- RIG
- retinoic acid inducible gene
- MDA
- melanoma differentiation-associated gene
- CARD
- caspase recruitment and activation domain
- MAVS
- mitochondrial antiviral signaling
- MAM
- mitochondrial-endoplasmic reticulum contacts
- ERAD
- endoplasmic reticulum-associated degradation
- CVB
- coxsackievirus B
- PV
- poliovirus
- VSV
- vesicular stomatitis virus.
REFERENCES
- 1. Kawai T., Akira S. (2006) Innate immune recognition of viral infection. Nat. Immunol. 7, 131–137 [DOI] [PubMed] [Google Scholar]
- 2. Seth R. B., Sun L., Ea C. K., Chen Z. J. (2005) Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF3. Cell 122, 669–682 [DOI] [PubMed] [Google Scholar]
- 3. Kawai T., Takahashi K., Sato S., Coban C., Kumar H., Kato H., Ishii K. J., Takeuchi O., Akira S. (2005) IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 6, 981–988 [DOI] [PubMed] [Google Scholar]
- 4. Meylan E., Curran J., Hofmann K., Moradpour D., Binder M., Bartenschlager R., Tschopp J. (2005) Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437, 1167–1172 [DOI] [PubMed] [Google Scholar]
- 5. Xu L. G., Wang Y. Y., Han K. J., Li L. Y., Zhai Z., Shu H. B. (2005) VISA is an adapter protein required for virus-triggered IFN-β signaling. Mol. Cell. 19, 727–740 [DOI] [PubMed] [Google Scholar]
- 6. Moore C. B., Bergstralh D. T., Duncan J. A., Lei Y., Morrison T. E., Zimmermann A. G., Accavitti-Loper M. A., Madden V. J., Sun L., Ye Z., Lich J. D., Heise M. T., Chen Z., Ting J. P. (2008) NLRX1 is a regulator of mitochondrial antiviral immunity. Nature 451, 573–577 [DOI] [PubMed] [Google Scholar]
- 7. Yasukawa K., Oshiumi H., Takeda M., Ishihara N., Yanagi Y., Seya T., Kawabata S., Koshiba T. (2009) Mitofusin 2 inhibits mitochondrial antiviral signaling. Sci. Signal 2, ra47. [DOI] [PubMed] [Google Scholar]
- 8. Xu L., Xiao N., Liu F., Ren H., Gu J. (2009) Inhibition of RIG-I and MDA5-dependent antiviral response by gC1qR at mitochondria. Proc. Natl. Acad. Sci. U.S.A. 106, 1530–1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu X. Y., Wei B., Shi H. X., Shan Y. F., Wang C. (2010) Tom70 mediates activation of interferon regulatory factor 3 on mitochondria. Cell Res. 20, 994–1011 [DOI] [PubMed] [Google Scholar]
- 10. Liu X. Y., Chen W., Wei B., Shan Y. F., Wang C. (2011) IFN-induced TPR protein IFIT3 potentiates antiviral signaling by bridging MAVS and TBK1. J. Immunol. 187, 2559–2568 [DOI] [PubMed] [Google Scholar]
- 11. Wang P., Yang L., Cheng G., Yang G., Xu Z., You F., Sun Q., Lin R., Fikrig E., Sutton R. E. (2013) UBXN1 interferes with Rig-I-like receptor-mediated antiviral immune response by targeting MAVS. Cell Reports 3, 1057–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jacobs J. L., Coyne C. B. (2013) Mechanisms of MAVS Regulation at the Mitochondrial Membrane. J. Mol. Biol. 425, 5009–5019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jia Y., Song T., Wei C., Ni C., Zheng Z., Xu Q., Ma H., Li L., Zhang Y., He X., Xu Y., Shi W., Zhong H. (2009) Negative regulation of MAVS-mediated innate immune response by PSMA7. J. Immunol. 183, 4241–4248 [DOI] [PubMed] [Google Scholar]
- 14. You F., Sun H., Zhou X., Sun W., Liang S., Zhai Z., Jiang Z. (2009) PCBP2 mediates degradation of the adaptor MAVS via the HECT ubiquitin ligase AIP4. Nat. Immunol. 10, 1300–1308 [DOI] [PubMed] [Google Scholar]
- 15. Wang Y., Tong X., Ye X. (2012) Ndfip1 negatively regulates RIG-I-dependent immune signaling by enhancing E3 ligase Smurf1-mediated MAVS degradation. J. Immunol. 189, 5304–5313 [DOI] [PubMed] [Google Scholar]
- 16. Castanier C., Garcin D., Vazquez A., Arnoult D. (2010) Mitochondrial dynamics regulate the RIG-I-like receptor antiviral pathway. EMBO Rep. 11, 133–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Koshiba T., Yasukawa K., Yanagi Y., Kawabata S. (2011) Mitochondrial membrane potential is required for MAVS-mediated antiviral signaling. Sci. Signal 4, ra7. [DOI] [PubMed] [Google Scholar]
- 18. Zhao Y., Sun X., Nie X., Sun L., Tang T. S., Chen D., Sun Q. (2012) COX5B regulates MAVS-mediated antiviral signaling through interaction with ATG5 and repressing ROS production. PLoS Pathog 8, e1003086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Horner S. M., Liu H. M., Park H. S., Briley J., Gale M., Jr. (2011) Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc. Natl. Acad. Sci. U.S.A. 108, 14590–14595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Csordás G., Renken C., Várnai P., Walter L., Weaver D., Buttle K. F., Balla T., Mannella C. A., Hajnóczky G. (2006) Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 174, 915–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Friedman J. R., Lackner L. L., West M., DiBenedetto J. R., Nunnari J., Voeltz G. K. (2011) ER tubules mark sites of mitochondrial division. Science 334, 358–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rizzuto R., Pinton P., Carrington W., Fay F. S., Fogarty K. E., Lifshitz L. M., Tuft R. A., Pozzan T. (1998) Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280, 1763–1766 [DOI] [PubMed] [Google Scholar]
- 23. Stone S. J., Vance J. E. (2000) Phosphatidylserine synthase-1 and -2 are localized to mitochondria-associated membranes. J. Biol. Chem. 275, 34534–34540 [DOI] [PubMed] [Google Scholar]
- 24. Pottekat A., Menon A. K. (2004) Subcellular localization and targeting of N-acetylglucosaminyl phosphatidylinositol de-N-acetylase, the second enzyme in the glycosylphosphatidylinositol biosynthetic pathway. J. Biol. Chem. 279, 15743–15751 [DOI] [PubMed] [Google Scholar]
- 25. Hayashi T., Su T. P. (2007) Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell 131, 596–610 [DOI] [PubMed] [Google Scholar]
- 26. Dennis E. A., Kennedy E. P. (1972) Intracellular sites of lipid synthesis and the biogenesis of mitochondria. J. Lipid Res. 13, 263–267 [PubMed] [Google Scholar]
- 27. Wieckowski M. R., Giorgi C., Lebiedzinska M., Duszynski J., Pinton P. (2009) Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat. Protoc 4, 1582–1590 [DOI] [PubMed] [Google Scholar]
- 28. Zhou R., Yazdi A. S., Menu P., Tschopp J. (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225 [DOI] [PubMed] [Google Scholar]
- 29. Benlimame N., Simard D., Nabi I. R. (1995) Autocrine motility factor receptor is a marker for a distinct membranous tubular organelle. J. Cell Biol. 129, 459–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Goetz J. G., Genty H., St-Pierre P., Dang T., Joshi B., Sauvé R., Vogl W., Nabi I. R. (2007) Reversible interactions between smooth domains of the endoplasmic reticulum and mitochondria are regulated by physiological cytosolic Ca2+ levels. J. Cell Sci. 120, 3553–3564 [DOI] [PubMed] [Google Scholar]
- 31. Wang H. J., Benlimame N., Nabi I. (1997) The AMF-R tubule is a smooth ilimaquinone-sensitive subdomain of the endoplasmic reticulum. J. Cell Sci. 110, 3043–3053 [DOI] [PubMed] [Google Scholar]
- 32. Wang H. J., Guay G., Pogan L., Sauvé R., Nabi I. R. (2000) Calcium regulates the association between mitochondria and a smooth subdomain of the endoplasmic reticulum. J. Cell Biol. 150, 1489–1498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liotta L. A., Mandler R., Murano G., Katz D. A., Gordon R. K., Chiang P. K., Schiffmann E. (1986) Tumor cell autocrine motility factor. Proc. Natl. Acad. Sci. U.S.A. 83, 3302–3306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen B., Mariano J., Tsai Y. C., Chan A. H., Cohen M., Weissman A. M. (2006) The activity of a human endoplasmic reticulum-associated degradation E3, gp78, requires its Cue domain, RING finger, and an E2-binding site. Proc. Natl. Acad. Sci. U.S.A. 103, 341–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fang S., Ferrone M., Yang C., Jensen J. P., Tiwari S., Weissman A. M. (2001) The tumor autocrine motility factor receptor, gp78, is a ubiquitin protein ligase implicated in degradation from the endoplasmic reticulum. Proc. Natl. Acad. Sci. U.S.A. 98, 14422–14427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shimizu K., Tani M., Watanabe H., Nagamachi Y., Niinaka Y., Shiroishi T., Ohwada S., Raz A., Yokota J. (1999) The autocrine motility factor receptor gene encodes a novel type of seven transmembrane protein. FEBS Lett. 456, 295–300 [DOI] [PubMed] [Google Scholar]
- 37. Ballar P., Shen Y., Yang H., Fang S. (2006) The role of a novel p97/valosin-containing protein-interacting motif of gp78 in endoplasmic reticulum-associated degradation. J. Biol. Chem. 281, 35359–35368 [DOI] [PubMed] [Google Scholar]
- 38. Lilley B. N., Ploegh H. L. (2005) Multiprotein complexes that link dislocation, ubiquitination, and extraction of misfolded proteins from the endoplasmic reticulum membrane. Proc. Natl. Acad. Sci. U.S.A. 102, 14296–14301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ye Y., Meyer H. H., Rapoport T. A. (2001) The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 414, 652–656 [DOI] [PubMed] [Google Scholar]
- 40. Zhong X., Shen Y., Ballar P., Apostolou A., Agami R., Fang S. (2004) AAA ATPase p97/valosin-containing protein interacts with gp78, a ubiquitin ligase for endoplasmic reticulum-associated degradation. J. Biol. Chem. 279, 45676–45684 [DOI] [PubMed] [Google Scholar]
- 41. Fu M., St-Pierre P., Shankar J., Wang P. T., Joshi B., Nabi I. R. (2013) Regulation of mitophagy by the Gp78 E3 ubiquitin ligase. Mol. Biol. Cell 24, 1153–1162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Coyne C. B., Bozym R., Morosky S. A., Hanna S. L., Mukherjee A., Tudor M., Kim K. S., Cherry S. (2011) Comparative RNAi screening reveals host factors involved in enterovirus infection of polarized endothelial monolayers. Cell Host Microbe 9, 70–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stins M. F., Badger J., Sik Kim K. (2001) Bacterial invasion and transcytosis in transfected human brain microvascular endothelial cells. Microb. Pathog. 30, 19–28 [DOI] [PubMed] [Google Scholar]
- 44. Bozym R. A., Delorme-Axford E., Harris K., Morosky S., Ikizler M., Dermody T. S., Sarkar S. N., Coyne C. B. (2012) Focal adhesion kinase is a component of antiviral RIG-I-like receptor signaling. Cell Host Microbe 11, 153–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Coyne C. B., Kim K. S., Bergelson J. M. (2007) Poliovirus entry into human brain microvascular cells requires receptor-induced activation of SHP-2. EMBO J. 26, 4016–4028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nabi I. R., Watanabe H., Raz A. (1990) Identification of B16-F1 melanoma autocrine motility-like factor receptor. Cancer Res. 50, 409–414 [PubMed] [Google Scholar]
- 47. Mukherjee A., Morosky S. A., Delorme-Axford E., Dybdahl-Sissoko N., Oberste M. S., Wang T., Coyne C. B. (2011) The coxsackievirus B 3C protease cleaves MAVS and TRIF to attenuate host type I interferon and apoptotic signaling. PLoS Pathog 7, e1001311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 49. Delorme-Axford E., Donker R. B., Mouillet J. F., Chu T., Bayer A., Ouyang Y., Wang T., Stolz D. B., Sarkar S. N., Morelli A. E., Sadovsky Y., Coyne C. B. (2013) Human placental trophoblasts confer viral resistance to recipient cells. Proc. Natl. Acad. Sci. U.S.A. 110, 12048–12053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. St-Pierre P., Dang T., Joshi B., Nabi I. R. (2012) Peripheral endoplasmic reticulum localization of the Gp78 ubiquitin ligase activity. J. Cell Sci. 125, 1727–1737 [DOI] [PubMed] [Google Scholar]
- 51. Li K., Chen Z., Kato N., Gale M., Jr., Lemon S. M. (2005) Distinct poly(I-C) and virus-activated signaling pathways leading to interferon-beta production in hepatocytes. J. Biol. Chem. 280, 16739–16747 [DOI] [PubMed] [Google Scholar]
- 52. Alexia C., Poalas K., Carvalho G., Zemirli N., Dwyer J., Dubois S. M., Hatchi E. M., Cordeiro N., Smith S. S., Castanier C., Le Guelte A., Wan L., Kang Y., Vazquez A., Gavard J., Arnoult D., Bidère N. (2013) The endoplasmic reticulum acts as a platform for ubiquitylated components of nuclear factor κB signaling. Sci. Signal 6, ra79. [DOI] [PubMed] [Google Scholar]
- 53. Castanier C., Zemirli N., Portier A., Garcin D., Bidère N., Vazquez A., Arnoult D. (2012) MAVS ubiquitination by the E3 ligase TRIM25 and degradation by the proteasome is involved in type I interferon production after activation of the antiviral RIG-I-like receptors. BMC Biology 10, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Castanier C., Arnoult D. (2011) Mitochondrial localization of viral proteins as a means to subvert host defense. Biochim. Biophy. Acta 1813, 575–583 [DOI] [PubMed] [Google Scholar]
- 55. Lin R., Heylbroeck C., Pitha P. M., Hiscott J. (1998) Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol. Cell. Biol. 18, 2986–2996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lawlor K. E., Vince J. E. (2013) Ambiguities in NLRP3 inflammasome regulation: Is there a role for mitochondria? Biochim. Biophys. Acta, in press [DOI] [PubMed] [Google Scholar]
- 57. Misawa T., Takahama M., Kozaki T., Lee H., Zou J., Saitoh T., Akira S. (2013) Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat. Immunol. 14, 454–460 [DOI] [PubMed] [Google Scholar]
- 58. Paz S., Vilasco M., Werden S. J., Arguello M., Joseph-Pillai D., Zhao T., Nguyen T. L., Sun Q., Meurs E. F., Lin R., Hiscott J. (2011) A functional C-terminal TRAF3-binding site in MAVS participates in positive and negative regulation of the IFN antiviral response. Cell Res. 21, 895–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Arnoult D., Soares F., Tattoli I., Castanier C., Philpott D. J., Girardin S. E. (2009) An N-terminal addressing sequence targets NLRX1 to the mitochondrial matrix. J. Cell Sci. 122, 3161–3168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hou F., Sun L., Zheng H., Skaug B., Jiang Q. X., Chen Z. J. (2011) MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell 146, 448–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Meusser B., Hirsch C., Jarosch E., Sommer T. (2005) ERAD: the long road to destruction. Nat. Cell Biol. 7, 766–772 [DOI] [PubMed] [Google Scholar]
- 62. Liang J. S., Kim T., Fang S., Yamaguchi J., Weissman A. M., Fisher E. A., Ginsberg H. N. (2003) Overexpression of the tumor autocrine motility factor receptor Gp78, a ubiquitin protein ligase, results in increased ubiquitinylation and decreased secretion of apolipoprotein B100 in HepG2 cells. J. Biol. Chem. 278, 23984–23988 [DOI] [PubMed] [Google Scholar]
- 63. Song B. L., Sever N., DeBose-Boyd R. A. (2005) Gp78, a membrane-anchored ubiquitin ligase, associates with Insig-1 and couples sterol-regulated ubiquitination to degradation of HMG CoA reductase. Mol. Cell 19, 829–840 [DOI] [PubMed] [Google Scholar]
- 64. Jabaut J., Ather J. L., Taracanova A., Poynter M. E., Ckless K. (2013) Mitochondria-targeted drugs enhance Nlrp3 inflammasome-dependent IL-1β secretion in association with alterations in cellular redox and energy status. Free Rad. Biol. Med. 60, 233–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Goulet M. L., Olagnier D., Xu Z., Paz S., Belgnaoui S. M., Lafferty E. I., Janelle V., Arguello M., Paquet M., Ghneim K., Richards S., Smith A., Wilkinson P., Cameron M., Kalinke U., Qureshi S., Lamarre A., Haddad E. K., Sekaly R. P., Peri S., Balachandran S., Lin R., Hiscott J. (2013) Systems analysis of a RIG-I agonist inducing broad spectrum inhibition of virus infectivity. PLoS Pathog. 9, e1003298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Belgnaoui S. M., Paz S., Samuel S., Goulet M. L., Sun Q., Kikkert M., Iwai K., Dikic I., Hiscott J., Lin R. (2012) Linear ubiquitination of NEMO negatively regulates the interferon antiviral response through disruption of the MAVS-TRAF3 complex. Cell Host Microbe 12, 211–222 [DOI] [PubMed] [Google Scholar]
- 67. Nakhaei P., Sun Q., Solis M., Mesplede T., Bonneil E., Paz S., Lin R., Hiscott J. (2012) IκB kinase epsilon-dependent phosphorylation and degradation of X-linked inhibitor of apoptosis sensitizes cells to virus-induced apoptosis. J. Virol. 86, 726–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Belgnaoui S. M., Paz S., Hiscott J. (2011) Orchestrating the interferon antiviral response through the mitochondrial antiviral signaling (MAVS) adapter. Curr. Opin. Immunol. 23, 564–572 [DOI] [PubMed] [Google Scholar]
- 69. Tang E. D., Wang C. Y. (2009) MAVS self-association mediates antiviral innate immune signaling. J. Virol. 83, 3420–3428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Baril M., Racine M. E., Penin F., Lamarre D. (2009) MAVS dimer is a crucial signaling component of innate immunity and the target of hepatitis C virus NS3/4A protease. J. Virol. 83, 1299–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]