

Background: ALS is a motor neuron disease characterized by the loss of axons and neurons.
Results: NFIL3 protects neurons in cellular and animal models of ALS.
Conclusion: NFIL3 is a neuroprotective molecule intrinsic to neurons.
Significance: NFIL3 is a potential therapeutic target for the treatment of ALS and related neurodegenerative diseases.
Keywords: Amyotrophic Lateral Sclerosis (Lou Gehrig Disease), Clock Genes, Neurodegeneration, Neurodegenerative Diseases, Neuroprotection, NFIL3
Abstract
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease characterized by the loss of motor neurons. Here we show that the basic leucine zipper transcription factor NFIL3 (also called E4BP4) confers neuroprotection in models of ALS. NFIL3 is up-regulated in primary neurons challenged with neurotoxic insults and in a mouse model of ALS. Overexpression of NFIL3 attenuates excitotoxic neuronal damage and protects neurons against neurodegeneration in a cell-based ALS model. Conversely, reduction of NFIL3 exacerbates neuronal demise in adverse conditions. Transgenic neuronal expression of NFIL3 in ALS mice delays disease onset and attenuates motor axon and neuron degeneration. These results suggest that NFIL3 plays a neuroprotective role in neurons and constitutes a potential therapeutic target for neurodegeneration.
Introduction
ALS3 is a progressive, fatal, and untreatable neurodegenerative disease characterized by the death of large motor neurons in the cerebral cortex and spinal cord. Although most ALS cases are sporadic, the familial form of ALS (FALS) with Mendelian inheritance and high penetrance occurs in ∼10% of the cases and is clinically indistinguishable from sporadic ALS (1–3). Mutations in superoxide dismutase 1 (SOD1) identified in ALS 20 years ago account for ∼20% of FALS (4). In recent years, mutations in TARDBP, FUS, PFN1, and C9ORF72 have been discovered to underlie a significant fraction of FALS (1–3). Despite the recent explosion in the genetics of ALS, the pathogenic mechanisms of the disease remain unclear. Studies over the last decades have nevertheless suggested that oxidative stress, excitotoxicity, defective axonal transport, protein misfolding, trophic factor deprivation, and inflammation contribute to motor neuron death through the concerted actions of toxic molecules within motor neurons and factors derived from glial cells (1–3). Although numerous studies aim to attenuate the deleterious neuronal/non-neuronal cell interface, the enhancement of defense mechanisms intrinsic to neurons remains a potential strategy to counteract neuron death in ALS. Thus, uncovering novel neuroprotective molecules within neurons could lead to the development of therapeutic approaches for ALS and related neurodegenerative diseases.
NFIL3 (nuclear factor interleukin 3-regulated, also known as E4BP4) is the mammalian basic leucine zipper transcription factor that was originally identified for its binding activity to the promoters of the adenovirus E4 gene and human IL-3 gene (5, 6). NFIL3 plays important roles in the development and survival of immune cells (7) and in the circadian clock system, particularly in the transcriptional control of clock genes and regulation of circadian output pathways (8–10). Noticeably, NFIL3 is expressed in embryonic rat and chicken motor neurons (11). Ectopic expression of NFIL3 promotes survival of chicken embryonic cultured motor neurons upon deprivation of neurotrophic factors or activation of death receptors. In vivo, overexpression of NFIL3 in the neural tube of chicken embryos reduced the number of dying motor neurons at later stages (11). Taken together, these results suggest a role of NFIL3 in repressing programmed cell death of neurons during development. The roles of NFIL3 in the adult central nervous system neurons remain, however, totally unexplored.
In this study, we found that NFIL3 is up-regulated in primary neurons challenged with various neurotoxic insults relevant to ALS and in a mouse model of ALS. Overexpression of NFIL3 protects neurons against excitotoxicity as well as neurodegeneration in a cell-based ALS model, whereas reduction of NFIL3 exacerbates neurodegeneration. Furthermore, neuronal overexpression of NFIL3 in ALS mice delays the onset of the disease and alleviates motor axon degeneration. These findings reveal that NFIL3 is a neuroprotective molecule intrinsic to the neuronal defense mechanism and represents a novel potential therapeutic target within neurons for the treatment of neurodegeneration.
EXPERIMENTAL PROCEDURES
Plasmids
The full-length open reading frame of mouse Nfil3 was amplified by PCR with Pfu Turbo polymerase (Stratagene) from cDNA derived from the adult mouse brain and was subcloned into the pcDNA3.1 vector (Invitrogen). A pBS/U6 plasmid was provided by Dr. Yang Shi (Harvard Medical School). Plasmids encoding NFIL3 shRNA were generated by inserting the annealed oligonucleotides into the pBS/U6 plasmid (12) digested with ApaI (blunted) and EcoRI. Targeted sequences for shRNA were as follows: NFIL3 shRNA 1, 5′-GGTGTAGTGGGCAAGTCTTC-3′; NFIL3 shRNA 2, 5′-GGAGAAACGGCGGAAAAACA-3′. The pBS/U6 plasmid was used as a control shRNA plasmid. For generating myc epitope-tagged NFIL3, the full-length open reading frame of mouse Nfil3 was subcloned into the pCS2-MT plasmid. A plasmid encoding a silent mutant of Nfil3 was generated by QuikChange mutagenesis using the Nfil3/pCS2-MT plasmid as a template. Plasmids expressing FLAG epitope-tagged human SOD1 and SOD1G93A have been generated previously by Nguyen and colleagues (University of Calgary) (13).
NFIL3 Antibodies
Mouse anti-NFIL3 antibody was prepared from anti-NFIL3 antiserum (9) by purification using protein G-Sepharose (GE Healthcare Life Sciences) and used for immunoblotting of whole cell lysates derived from neuronal cultures. Rabbit anti-NFIL3 antiserum was generated as follows and used for immunohistochemistry, immunocytochemistry, and immunoblotting of nuclear extracts derived from tissues. A peptide corresponding to amino acids 90–250 of mouse NFIL3 was expressed as a GST fusion protein in Escherichia coli, purified by using a glutathione-Sepharose column (GE Healthcare Life Sciences), and immunized to rabbits. Immunization was performed by Medical & Biological Laboratories. The plasmid encoding GST-NFIL3 was a gift from Dr. Toshiya Inaba (Hiroshima University). The specificity of the antiserum was confirmed by depletion of the protein by RNAi and by peptide competition.
Cell Culture and Transfection
Neocortical cells were prepared as described earlier (13, 14). Neocortical cells were plated at a density of 1.0 × 106 cells (for immunoblotting) or 1.0 × 105 cells (for immunostaining) on sterile coverslips precoated with poly-D-lysine and laminin in the wells of 24-well plates and maintained in Neurobasal medium (Invitrogen) supplemented with B27. H2O2 was purchased from Wako Chemicals. Ionomycin was purchased from Calbiochem. Treatments with H2O2, ionomycin, and glutamate were performed at 9–10 days in vitro (div). Neocortical cells were lysed with PBS containing 1.0% Triton X-100, 1.0% deoxycholate, 0.1% sodium dodecyl sulfate, 2 mm EDTA, and protease inhibitor mixture (Roche Molecular Biochemicals). After 30 min on ice, the lysates were centrifuged at 19,000 × g for 10 min at 4 °C, and the extracts were subjected to immunoblotting. Antibodies used were mouse anti-NFIL3 antibody (1:500) and mouse anti-dynein intermediate chain antibody (1:2000, Santa Cruz Biotechnology).
Transfection into neurons was performed using Lipofectamine 2000 (Invitrogen) at 7 div in Neurobasal medium. Transfections were allowed to proceed for 4–5 h.
For evaluation of NFIL3 knockdown by shRNA constructs, HEK293T cells maintained in 10% FBS/DMEM were transiently transfected using Lipofectamine 2000 (Invitrogen). Transfections were allowed to proceed for 4–5 h, and then the cells were cultured in 10% FBS/DMEM for 48 h. The cell extracts were prepared as described above and then subjected to immunoblotting. Antibodies used were mouse anti-NFIL3 antibody (1:500), mouse anti-dynein intermediate chain antibody (1:2000; Santa Cruz Biotechnology), mouse anti-myc epitope antibody (clone 9E10, 1:100, Santa Cruz Biotechnology), and mouse β-actin antibody (clone AC-15, 1:20,000; Sigma-Aldrich).
Transgenic Mice and Analysis of Disease Onset and Survival
Mice expressing human SOD1G93A (G93AGurdl), which were generated from mice harboring human SOD1G93A (B6SJL-TgN[SOD1-G93A]1Gur, hSOD1G93A, The Jackson Laboratory) by backcrossing with the C57BL/6 strain for more than 20 generations (15), were a gift from Drs. Minako Tateno (National Center of Neurology and Psychiatry) and Makoto Urushitani (Shiga University of Medical Science).
To construct the Nfil3 transgene, the full-length open reading frame of mouse Nfil3 was subcloned into the pMX-IRES-GFP plasmid (a gift from Dr. Toshio Kitamura, The University of Tokyo). The Nfil3-IRES-GFP cassette was then isolated, blunt-ended, and inserted into the EcoRV site of the pNN265 (a gift from Dr. Mark Mayford, The Scripps Research Institute) to give rise to pNN265-Nfil3-IRES-GFP. A NotI fragment containing the Nfil3-IRES-GFP, along with the SV40 small-T antigen intron and early polyadenylation sequences, was isolated from the plasmid and inserted into the pMM403 plasmid (a gift from Dr. Mark Mayford, The Scripps Research Institute), which contains the promoter of mouse Ca2+/calmodulin-activated protein kinase IIα gene to obtain the PMM403-Nfil3-IRES-GFP plasmid. The linearized transgene construct, generated from the plasmid by SfiI digestion, was microinjected into fertilized eggs from C57BL/6J mice, and the transgenic offspring was screened by PCR genotyping using tail DNA (PCR product size, 621 bp). The primers used were 5′-GATCCTGCCGATTCGAGCCGCC-3′ and 5′-GCGGGGCTTTCCTGAGTGTGCTC-3′. The Nfil3 hemizygotes obtained (CaMKIIa-E4BP4, accession no. CDB0479T) were crossed with SOD1G93A hemizygotes to obtain double transgenic mice, SOD1G93A transgenic mice, Nfil3 transgenic mice, and non-transgenic wild-type mice. Mice were housed under 12 h light, 12 h dark cycles with food and water available ad libitum.
Disease onset, early stage, and end stage were defined according to previous reports (16, 17). In brief, disease onset was determined as the time when mice reached peak body weight before the denervation-induced muscle atrophy and weight loss. Early disease stage was defined as the time of 10% weight loss of the maximal body weight. End-stage was determined as the time at which a mouse could not right itself within 20 s when placed on its side. All animal experiments were conducted in accordance with guidelines set by The University of Tokyo.
Preparation of Tissue Extracts
Nuclear extracts were prepared from mouse brains and spinal cords as described earlier (9) and subjected to immunoblotting. Antibodies used were rabbit anti-NFIL3 antiserum (1:1,000) and rabbit anti-TFIID antibody (1:1000, Santa Cruz Biotechnology).
For analysis of SOD1 expression, lumbar spinal cords derived from mice at 8–9 weeks of age were lysed with PBS containing 1.0% Triton X-100, 1.0% deoxycholate, 0.1% sodium dodecyl sulfate, 2 mm EDTA, and protease inhibitor mixture (Roche Molecular Biochemicals). After 30 min on ice, the lysates were centrifuged at 19,000 × g for 10 min at 4 °C, and the extracts were subjected to immunoblotting. Antibodies used were rabbit anti-SOD1 antibody (1:1000, Santa Cruz Biotechnology) and mouse anti-dynein intermediate chain antibody (1:2000, Santa Cruz Biotechnology).
Immunocytochemistry and Immunohistochemistry
Cultured cortical neurons were fixed with 4% paraformaldehyde in PBS for 20 min at 37 °C, blocked with 3% BSA/0.2% Triton X-100 in PBS, and incubated with primary antibodies in the blocking solution at 4 °C overnight. Primary antibodies used were rat anti-GFP (clone GF090R, 1:1000, Nakalai Tesque), mouse anti-βIII-tubulin (clone TUJ1, 1:5000, Covance), rabbit anti-microtubule-associated protein 2 (1:1000, Millipore), mouse anti-myc epitope (clone 9E10, 1:100, Santa Cruz Biotechnology), mouse anti-FLAG epitope (clone M2, 1:200, Sigma-Aldrich), and rabbit anti-NFIL3 antiserum (1:10,000) antibodies. Secondary antibodies used were Alexa Fluor 488/Alexa Fluor 568-conjugated anti-IgG antibodies (Invitrogen) and Cy3/Dylight549/Dylight649/Cy5-conjugated anti-IgG antibodies (Jackson ImmunoResearch Laboratories, Inc.). Nuclei were stained with DAPI or TO-PRO-3 iodide (Invitrogen). The coverslips were mounted in a Prolong Gold mounting solution (Invitrogen). Fluorescent images were obtained by using Zeiss LSM5 confocal microscope.
For immunohistochemistry, brains and lumbar spinal cords were isolated and fixed with 4% paraformaldehyde in PBS overnight at 4 °C. Thereafter, thick brain sections (40 μm) and spinal cord sections (60 μm) were made with a vibratome (Leica). For staining of lumbar spinal cord sections with Neurofilament H and glial fibrillary acidic protein (GFAP) antibody, thick sections (20 μm) were made by cryostat (Leica). The sections were incubated with a blocking solution (3% BSA and 0.5% Triton X-100 in PBS) and then incubated with primary antibodies overnight at 4 °C. Nuclei were stained with DAPI or TO-PRO-3 iodide (Invitrogen). Anti-NFIL3 antiserum (1:10,000), goat anti-choline acetyltransferase antibody (1:50, Millipore), mouse Neurofilament H (clone SMI32, 1:5000, Covance), and rabbit anti-GFAP antibodies (1:500, Sigma-Aldrich) were used. The sections were then incubated with Alexa Fluor 488/Alexa Fluor 568/Cy3/Cy5-conjugated secondary antibodies overnight at 4 °C and mounted in Prolong Gold mounting solution (Invitrogen). Images were obtained with Zeiss LSM5 confocal microscope.
For analysis of L5 ventral root axons, L5 roots were dissected and fixed with 4% PFA for 30 min at room temperature. Thick sections (10 μm) were made by cryostat (Leica). The sections were then treated with HistoVT One (Nakalai Tesque) at 70 °C for 15 min, blocked with 3% BSA and 0.5% Triton X-100 in PBS, and incubated with antibodies against myelin basic protein (clone 14, 1:500, Sigma-Aldrich) and βIII-tubulin (clone TUJ1, 1:5000, Covance). Images were obtained with a Zeiss LSM5 confocal microscope, and quantifications of L5 ventral root axons were carried out.
Statistical Analyses
We used two-tailed Student's t tests or Welch's t tests to compare two groups. For disease onset and survival analyses, log-rank tests were used. All data are shown as mean ± S.E. A value of p < 0.05 was considered statistically significant.
RESULTS
Up-regulation of NFIL3 in Neurons Challenged with Neurotoxic Insults
We first examined whether levels of NFIL3 in mouse primary cortical neurons are influenced by various neurotoxic insults, such as excitotoxicity caused by a high concentration of glutamate, the free radical generator hydrogen peroxide that generates oxidative stress, and the calcium ionophore ionomycin (13, 14). Treatment of neurons with 50 μm glutamate rapidly induced NFIL3 protein expression (Fig. 1a). Similarly, hydrogen peroxide (500 μm) and ionomycin (10 μm) caused an increase in NFIL3 protein levels (Fig. 1, b and c). Because excitotoxicity, oxidative stress, and disruption of calcium homeostasis have been shown to contribute to the pathogenesis of ALS (1–3), we next determined the expression of NFIL3 in SOD1G93A mice (G93AGurdl) (15), transgenic mice expressing a mutant form of SOD1 (SOD1G93A) linked to FALS. These mice exhibit progressive motor neuron degeneration in the spinal cord associated with calcium overload and oxidative stress, display progressive weight loss resulting from denervation-induced muscle atrophy, develop paralysis, and, ultimately, die (15–18). In the lumbar spinal cord of mutant SOD1G93A mice at the age of 164–180 days, a stage with slight weight loss and motor neuron degeneration, NFIL3 was slightly up-regulated (Fig. 1d). These results indicate that NFIL3 is up-regulated in primary neurons challenged with insults relevant to the pathogenesis of ALS and in a murine model of the disease.
FIGURE 1.
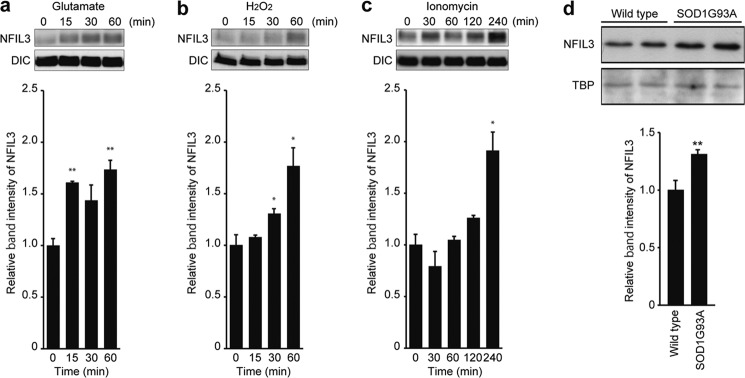
Neurotoxicity induces up-regulation of NFIL3. a–c, primary neurons at 10 div were treated with glutamate (a), H2O2 (b), or ionomycin (c) for the indicated time. The top panels show representative immunoblot analyses of NFIL3 and dynein intermediate chain (DIC, loading control). The density of the NFIL3 band was quantified, and quantifications are presented as mean ± S.E. (n = 3–4). *, p < 0.05; **, p < 0.01 compared with time = 0 by two-tailed t test. d, up-regulation of NFIL3 in the spinal cord of mutant SOD1G93A mice. NFIL3 protein levels in the spinal cord of either SOD1G93A transgenic (SOD1G93A-tg) or wild-type littermate mice at the age of 164–180 days were assessed by Western blot analysis of nuclear extracts. Immunoblot analyses of NFIL3 and TATA-binding protein (TBP, loading control) are shown. The density of the NFIL3 band was quantified, and quantifications are presented as mean ± S.E. n = 4 (two males and two females) for wild-type and 7 (three males and four females) for SOD1G93A-tg. **, p < 0.01 compared with wild-type mice by two-tailed t test.
Neuroprotective role of NFIL3 in Primary Neurons
To understand the significance of NFIL3 up-regulation, we investigated the effect of NFIL3 overexpression in primary cortical neurons treated with a high concentration of glutamate. As reported previously (13, 14), treatment of 50 μm glutamate resulted in extensive neuronal death concomitant with neurite fragmentation and chromatin condensation (13) (Fig. 2, a and b). Overexpression of NFIL3 significantly reduced neuronal demise triggered by glutamate (Fig. 2, a and b). To investigate the effect of NFIL3 knockdown in neurons, we generated DNA-based RNAi plasmids (NFIL3 shRNA#1 and #2) that express shRNA against NFIL3. The NFIL3 shRNA constructs efficiently silenced expression of NFIL3 that was transiently expressed in HEK293T cells (Fig. 2c). In contrast, expression of mutant NFIL3 with two silent mutations within the target sequence of the NFIL3 shRNA#1 was not affected by shRNA#1 (Fig. 2d). Also, the shRNA constructs silenced endogenous NFIL3 in primary neurons (Fig. 2, e and f). Importantly, depletion of NFIL3 with the shRNA constructs exacerbated neuronal degeneration induced by glutamate (Fig. 2, a and b). Furthermore, when Nfil3 mutant cDNA with two silent mutations within the RNAi target sequence was cotransfected, degeneration of stressed NFIL3 shRNA-expressing neurons was prevented almost completely (Fig. 2, a and b).
FIGURE 2.

NFIL3 protects against glutamate-induced neurotoxicity. a, effects of NFIL3 overexpression or NFIL3 depletion on neurons treated with 50 μm glutamate. Primary neurons at 7 div were transfected with NFIL3, NFIL3 shRNA (#1 or #2), and/or mutant NFIL3 (mutNFIL3, myc-tagged NFIL3 containing two silent mutations within the NFIL3 shRNA#1-targeted sequence), as indicated. GFP was also introduced as a transfection marker. Cells at 9 div were treated with either vehicle or 50 μm glutamate for 30 min and immunostained with antibodies against GFP (green), MAP2 (red), Tuj1 (red), and myc tag (blue/white), as indicated. The insets show magnified views of the nucleus of the transfected neurons. Scale bars = 50 μm. b, quantifications of cell death on the basis of the concomitant neurite fragmentation and chromatin condensation in neurons transfected with NFIL3 and/or NFIL3 shRNA after vehicle/glutamate treatment. Data are presented as mean ± S.E. n = 3–4. *, p < 0.05; **, p < 0.01 compared with mock by two-tailed t test. More than 100 cells were analyzed for each condition. Data were obtained from three to four independent experiments. c, evaluation of NFIL3 knockdown by shRNA constructs. HEK293T cells were transiently transfected with plasmids encoding GFP (pEGFP-C1) and either wild-type NFIL3, NFIL3 shRNA#1, and NFIL3 shRNA#2, as indicated. Cell lysates were prepared after 48 h and subjected to Western blot analysis with antibodies against GFP, NFIL3, and dynein intermediate chain (DIC). d, HEK293T cells were transiently transfected with plasmids encoding myc-tagged wild-type NFIL3 (NFIL3-myc), control shRNA, NFIL3 shRNA#1, NFIL3 shRNA#2, and myc-tagged mutant NFIL3 (mutNFIL3-myc, NFIL3 that contains two silent mutations within the NFIL3 shRNA#1-targeted sequence), as indicated. Cell lysates were prepared after 48 h and subjected to Western blot analysis with antibodies against myc and β-actin. The NFIL3 shRNA#1-targeted sequence and mutated sequence (indicated by red) in mutNfil3 are shown. e, primary neurons at 1 div were transfected with GFP and NFIL3 shRNA (#1 or #2). Cells at 3 div were fixed and immunostained with anti-GFP antibody (green), anti-NFIL3 antiserum (red), and anti-βIII tubulin (Tuj1) (blue). GFP-positive and GFP-negative neurons are indicated by green and red arrowheads, respectively. Scale bar = 20 μm. f, The nuclear NFIL3 fluorescence intensity of GFP-positive cells and that of neighboring GFP-negative cells were measured, and the ratio of these intensity values was plotted (mean ± S.E.; n = 21; *, p < 0.05; two-tailed t test).
We also examined the effect of NFIL3 overexpression/knockdown in a cell-based ALS model. Primary cortical neurons expressing SOD1G93A, a mutant form of SOD1 linked to FALS, resulted in extensive neuronal death (13). Overexpression of NFIL3 significantly reduced neuronal demise triggered by SOD1G93A (Fig. 3, a and b), whereas depletion of NFIL3 exacerbated neuronal degeneration (c). Furthermore, coexpression of Nfil3 mutant cDNA with two silent mutations within the RNAi target sequence suppressed degeneration of stressed NFIL3 shRNA-expressing neurons (Fig. 3c). Thus, NFIL3 confers neuroprotection against excitotoxicity and mutant SOD1 toxicity in primary neurons, and this function could be boosted to further enhance neuronal survival in vitro.
FIGURE 3.

NFIL3 protects against mutant SOD1-induced neurotoxicity. a, effects of overexpression of NFIL3 on SOD1G93A -induced toxicity. Primary neurons at 7 div were transfected with either mutant SOD1G93A (FLAG-tagged SOD1G93A), FLAG-tagged wild-type SOD1, or FLAG-tagged SOD1G93A with NFIL3. GFP was also introduced as a transfection marker. The cells at 9 div were immunostained with antibodies against GFP (green), MAP2 (red), and FLAG tag (blue/white). Cell death was scored as in Fig. 2. Scale bars = 50 μm. b, quantifications of cell death in wild-type SOD1-, SOD1G93A -, and NFIL3/SOD1G93A-expressing neurons. Data are presented as mean ± S.E. (n = 3–4; *, p < 0.05; ***, p < 0.001 by two-tailed Welch's t test). Data were obtained from three to four independent experiments. c, effects of NFIL3 depletion on SOD1G93A -induced toxicity. Primary neurons at 7 div were transfected with SOD1G93A and either control shRNA, NFIL3 shRNA (#1 and #2), or NFIL3 shRNA#1 with mutant NFIL3, as indicated. Cells at 9 div were immunostained as in a, and cell death was quantified. Data are presented as mean ± S.E. (n = 3–4; **, p < 0.01; ***, p < 0.001 compared with control shRNA by two-tailed Welch's t test). Data were obtained from three to four independent experiments.
Neuronal Overexpression of NFIL3 in ALS Mice Slows Disease Onset
We next sought to investigate the potential neuroprotective effects of NFIL3 overexpression in adult neurons in vivo. To this end, we generated transgenic mice expressing Nfil3 under control of the CaMKIIα promoter (19). The CaMKIIα promoter is turned on 3 weeks after birth and is strongly expressed in the neocortex, hippocampus, and dorsal/ventral horns of the spinal cord (19, 20). Consistently, Nfil3 transgenic mice overexpressed NFIL3 in the neocortex, hippocampus, and spinal cord, as assessed by immunohistochemistry (Fig. 4a). Further, we detected overexpression of NFIL3 in large spinal cord motor neurons in Nfil3 transgenic mice (Fig. 4b). Almost no NFIL3 immunoreactivity was observed in tissue sections immunostained with NFIL3 antiserum preincubated with its immunogen (Fig. 4a) and in primary neurons depleted of the protein by RNAi (e), thereby confirming the specificity of the antiserum.
FIGURE 4.

Neuronal expression of NFIL3 in Nfil3 transgenic mice. a, NFIL3 expression in the neocortex, hippocampus, and lumbar spinal cord in Nfil3 transgenic (Nfil3-tg) and wild-type littermate mice. The brain/spinal cord sections were immunostained with either rabbit anti-NFIL3 antibody or anti-NFIL3 antibody preincubated with its immunogen (2.5 μg/ml). Nuclei were stained with TO-PRO-3. Scale bars = 200 μm. b, NFIL3 expression in motor neurons in the lumbar spinal cord of Nfil3-tg and wild-type littermate mice. Motor neurons were identified by immunostaining with an antibody against choline acetyl-transferase (ChAT). Scale bar = 20 μm. c, NFIL3 expression was assessed by Western blot analysis of nuclear extracts prepared from the neocortex and lumbar spinal cord of wild-type, Nfil3-tg, mutant SOD1G93A transgenic (SOD1G93A-tg), and SOD1G93A-tg overexpressing Nfil3 (Nfil3/SOD1G93A-tg) mice at 8–20 weeks of age. Representative immunoblot analyses of NFIL3 and TATA-binding protein (TBP, loading control) are shown. d, mutant SOD1G93A expression in the lumbar spinal cord of mutant SOD1G93A mice overexpressing Nfil3 (Nfil3/SOD1G93A-tg) and mutant SOD1G93A transgenic (SOD1G93A-tg) littermate mice at 8–9 weeks of age. Expression of mutant SOD1G93A was assessed by Western blot analysis of whole tissue lysates. Representative immunoblot analyses of SOD1 and dynein intermediate chain (DIC, loading control) are shown. e, the density of the mutant SOD1G93A band was quantified and is presented as mean ± S.E. (n = 3 male mice, p = 0.47 by two-tailed t test). n.s., not significant.
We then crossed Nfil3 transgenic mice with SOD1G93A mice to inspect the effects of neuronal overexpression of NFIL3 on phenotypes in ALS mice. Double transgenic mice for Nfil3/SOD1G93A, single transgenic mice for SOD1G93A, and non-transgenic mice derived from the breeding were used for analyses. In the neocortex and lumbar spinal cord of mice with the Nfil3 transgene (Fig. 4c), NFIL3 levels increased significantly by ∼1.4-fold (versus non-transgenic mice) (spinal cord: 1.4 ± 0.10-fold in Nfil3 transgenic mice, n = 3, p = 0.02; 1.1 ± 0.07-fold in SOD1G93A mice, n = 3, p = 0.4; 1.4 ± 0.04-fold in Nfil3/SOD1G93A mice, n = 3, p = 0.003; neocortex: 1.4 ± 0.07-fold in Nfil3 transgenic mice, n = 3, p = 0.04; 1.1 ± 0.03-fold in SOD1G93A mice, n = 3, p = 0.3; 1.6 ± 0.13-fold in Nfil3/SOD1G93A mice, n = 3, p = 0.04; by two-tailed Student's t test). Further, in Nfil3/SOD1G93A mice, transgenic overexpression of NFIL3 did not alter the expression levels of SOD1G93A in the lumbar spinal cord (Fig. 4, d and e). To determine the effects of NFIL3 overexpression on onset and progression of motor neuron disease in SOD1G93A mice, we performed a simple, objective measure of disease onset defined by initiation of body weight loss that reflects denervation-induced muscle atrophy (15–18). Remarkably, transgenic expression of NFIL3 significantly delayed disease onset (Fig. 5, a and d), yielding a mean delay of 15 days. Also, the early disease phase (defined as the time at which 10% weight loss from the maximal body weight was observed) and end stage (16, 17) were delayed significantly by NFIL3 expression, yielding an average delay of 14 days and 15 days, respectively (Fig. 5, b and c). Of note, there was no significant difference in the duration from onset to early disease and the duration from early disease to end stage (Fig. 5, e and f), suggesting that NFIL3 overexpression has little effect on progression of later stages of ALS. Furthermore, similar rescue phenotypes were observed with the second line of Nfil3 transgenic mice (Fig. 6), thereby confirming the neuroprotective role of NFIL3 in delaying phenotypic disease onset in ALS mice.
FIGURE 5.

Transgenic expression of Nfil3 in neurons in ALS model mice slows disease onset. a–c, ages of disease onset (ages when mice reached peak body weight, p = 0.017 by log-rank test, a), early disease phase (ages of 10% weight loss from maximal weight, p = 0.026 by log-rank test, b), or end-stage disease (p = 0.026 by log-rank test, c) of Nfil3/SOD1G93A-tg (red) and SOD1G93A-tg (blue) mice. Data were obtained from SODG93A-tg (onset and early disease, six males and four females; end-stage, six males and two females) and Nfil3/SOD1G93A-tg mice (onset and early disease, seven males and four females; end-stage, seven males and three females). d–f, mean onset (*, p < 0.05 by two-tailed t test), mean duration of an early disease phase (from onset to 10% weight loss, p = 0.94 by two-tailed t test), and mean duration of a later disease phase (from 10% weight loss to end stage, p = 0.84 by two-tailed t test) for Nfil3/SOD1G93A-tg (red) and SOD1G93A-tg (blue) mice.
FIGURE 6.

Delayed disease onset of Nfil3/SOD1G93A double transgenic mice derived from a second line of Nfil3 transgenic mice. a, NFIL3 expression in the neocortex, hippocampus, and motor neurons in the lumbar spinal cord of a second line of Nfil3 transgenic (Nfil3-tg line#2) and age-matched, wild-type mice. Brain/spinal cord sections were immunostained with rabbit anti-NFIL3 antibody. Nuclei were stained with TO-PRO-3. Motor neurons were identified by immunostaining with an antibody against choline acetyl-transferase (ChAT). Scale bars = 200 μm for neocortex/hippocampus and 20 μm for motor neurons. b, ages at which disease onset (p = 0.019 by log-rank test) of mutant SOD1G93A mice overexpressing Nfil3 (Nfil3/SOD1G93A-tg, red) and mutant SOD1G93A mice (SOD1G93A-tg, blue), both of which were obtained by crossing of Nfil3-tg line#2 mice with mutant SOD1G93A mice. Data were obtained from SOD1G93A-tg (seven males and three females) and Nfil3/SOD1G93A-tg mice (six males and three females). c–e, mean onset (*, p < 0.05 by two-tailed t test), mean duration of an early disease phase (p = 0.43 by two-tailed t test), and mean duration of a later disease phase (p = 0.56 by two-tailed t test) for Nfil3/SOD1G93A-tg (red) and SOD1G93A-tg (blue) mice.
Neuronal NFIL3 Overexpression Attenuates Motor Axon Degeneration in ALS Mice
We then determined whether the delay of the disease-associated phenotypes correlated with an attenuation of neuropathological changes. In SOD1G93A mice, motor axons of the ventral roots gradually degenerate as the disease progresses. We found a significantly larger number of motor axons in L5 ventral roots of Nfil3/SOD1G93A double transgenic mice at 180–190 days of age (Fig. 7, a and b) when compared with age-matched SOD1G93A mice. In addition, a significantly larger number of large spinal cord motor neurons were detected in Nfil3/SOD1G93A double transgenic mice at 220–230 days of age, whereas astrogliosis in the ventral horn of the lumbar spinal cord of SOD1G93A mice was attenuated significantly by NFIL3 overexpression (Fig. 7, c and d). Together, these results indicate that increased neuronal expression of NFIL3 attenuates motor axon and neuron degeneration and delays phenotypic disease onset in ALS mice.
FIGURE 7.

Transgenic expression of Nfil3 in neurons in SOD1G93A mice attenuates degeneration of motor axons. a, L5 ventral root sections derived from 180- to 190-day-old SOD1G93A mice overexpressing Nfil3 (Nfil3/SOD1G93A-tg), SOD1G93A mice (SOD1G93A-tg), and wild-type mice were stained with antibodies against Tuj1 (red/white) and myelin basic protein (MBP, green). Representative images are shown. Images of L5 ventral root sections derived from SOD1G93A mice at the end stage are also shown. The bottom panels are magnified views. Scale bars = 200 μm. b, the number of motor axons was counted and is presented as mean ± S.E. n = 3 (one male and two females) for wild-type, 4 (four males) for SOD1G93A-tg, 3 (three males) for Nfil3/SOD1G93A-tg, and 3 (one male and two females) for SOD1G93A-tg at the end stage. *, p < 0.05 by two-tailed t test. c, lumbar spinal cord sections derived from SOD1G93A-tg, Nfil3/SOD1G93A-tg, and wild-type mice at 220–230 days of age were immunostained with Neurofilament H (SMI32) and GFAP antibodies. d, the number of large motor neurons per section was counted and is presented as mean ± S.E. Data were obtained from three mice (two males and one female). *, p < 0.05; ***, p < 0.001 by two-tailed t test. Mean GFAP fluorescent intensity in the ventral horn per section was quantified and is presented as mean ± S.E. Data were obtained from three mice (two males and one female). *, p < 0.05; **, p < 0.01 by two-tailed t test.
DISCUSSION
In this study, we show a neuroprotective role for NFIL3 in cellular and animal models of ALS. NFIL3 protein levels increased in neurons challenged with various neurotoxic insults relevant to the pathogenesis of ALS and in mutant SOD1 mice. Furthermore, overexpression of NFIL3 exerted neuroprotection in vivo and in vitro, whereas NFIL3-depletion in primary neurons exacerbated neuronal degeneration in adverse conditions. Together, these observations suggest that NFIL3 is up-regulated in neurons in times of adversity to cope with and protect them against neuronal demise. Thus, NFIL3 expression may constitute a cell defense mechanism in neurons. In addition, this study showed that neuronal overexpression of NFIL3 in ALS mice delayed disease onset, alleviated motor axon and neuron degeneration, but had no effect on disease progression. Because disease progression is dictated by neuronal degeneration through non-cell autonomous mutant SOD1 damage in astrocytes and microglia (16, 17), we propose that NFIL3 acts autonomously within motor neurons to counteract neurotoxicity induced by mutant SOD1 in ALS mice.
The detailed mechanisms by which NFIL3 is up-regulated and exerts neuroprotection remain to be established. Nfil3 is trans-activated by elevated intracellular Ca2+ concentration and activation of cAMP response element-binding protein (21, 22). In various neurodegenerative diseases, disruption of Ca2+ homeostasis takes place during the pathogenic process, and various neurotoxic insults, such as Aβ, oxidative stress, and exicitotoxin, induce an increase in intracellular Ca2+ concentration in neurons (23). Considering that NFIL3 was up-regulated in primary neurons challenged with a high concentration of glutamate, hydrogen peroxide, and calcium ionophores, it is possible that excess inflow of Ca2+ may trigger up-regulation of NFIL3 in neurons.
Of note, NFIL3 acts as a regulator of the circadian clock system and has been shown to suppress gene expression of core circadian clock components such as period2 (8–10). Interestingly, PERIOD2, besides its roles in maintaining circadian rhythms, functions as a proapoptotic protein in cancer cells (24, 25). Because inhibition of neuronal apoptosis in ALS model mice results in delayed disease onset (26, 27), neuronal up-regulation of NFIL3 in times of adversity may confer neuroprotection through alterations of circadian clock components (e.g. a decrease in PERIOD2) within neurons that prevent apoptosis.
In summary, our findings are shedding light on the emerging link between circadian clock components and neuroprotection. Modulation of the levels of clock components (e.g. pharmacological targeting of casein kinase Iϵ to alter casein kinase Iϵ-dependent phosphorylation and degradation of NFIL3 and PERIOD2) (9, 28) may affect neuronal survival. The identification of upstream and downstream targets of NFIL3 will likely help to better understand the defense system in neurons, thereby providing new therapeutic strategies to ameliorate neurodegenerative conditions.
Acknowledgments
We thank Drs. Minako Tateno and Makoto Urushitani for SOD1G93A mice and Drs. Toshio Kitamura, Toshiya Inaba, Yang Shi, and Mark Mayford for plasmids.
This work was supported in part by grants-in-aid from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (to N. K., Y. F., and K. S.) and by an operating grant from the Canadian Institutes of Health Research (to M. D. N).
- ALS
- amyotrophic lateral sclerosis
- FALS
- familial amyotrophic lateral sclerosis
- div
- days in vitro
- GFAP
- glial fibrillary acidic protein.
REFERENCES
- 1. Bruijn L. I., Miller T. M., Cleveland D. W. (2004) Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu. Rev. Neurosci. 27, 723–749 [DOI] [PubMed] [Google Scholar]
- 2. Kanning K. C., Kaplan A., Henderson C. E. (2010) Motor neuron diversity in development and disease. Annu. Rev. Neurosci. 33, 409–440 [DOI] [PubMed] [Google Scholar]
- 3. Ferraiuolo L., Kirby J., Grierson A. J., Sendtner M., Shaw P. J. (2011) Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 7, 616–630 [DOI] [PubMed] [Google Scholar]
- 4. Rosen D. R., Siddique T., Patterson D., Figlewicz D. A., Sapp P., Hentati A., Donaldson D., Goto J., O'Regan J. P., Deng H. X. (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62 [DOI] [PubMed] [Google Scholar]
- 5. Cowell I. G., Skinner A., Hurst H. C. (1992) Transcriptional repression by a novel member of the bZIP family of transcription factors. Mol. Cell Biol. 12, 3070–3077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang W., Zhang J., Kornuc M., Kwan K., Frank R., Nimer S. D. (1995) Molecular cloning and characterization of NF-IL3A, a transcriptional activator of the human interleukin-3 promoter. Mol. Cell Biol. 15, 6055–6063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Male V., Nisoli I., Gascoyne D. M., Brady H. J. (2012) E4BP4. An unexpected player in the immune response. Trends Immunol. 33, 98–102 [DOI] [PubMed] [Google Scholar]
- 8. Mitsui S., Yamaguchi S., Matsuo T., Ishida Y., Okamura H. (2001) Antagonistic role of E4BP4 and PAR proteins in the circadian oscillatory mechanism. Genes Dev. 15, 995–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Doi M., Okano T., Yujnovsky I., Sassone-Corsi P., Fukada Y. (2004) Negative control of circadian clock regulator E4BP4 by casein kinase Iϵ-mediated phosphorylation. Curr. Biol. 14, 975–980 [DOI] [PubMed] [Google Scholar]
- 10. Doi M., Nakajima Y., Okano T., Fukada Y. (2001) Light-induced phase-delay of the chicken pineal circadian clock is associated with the induction of cE4bp4, a potential transcriptional repressor of cPer2 gene. Proc. Natl. Acad. Sci. U.S.A. 98, 8089–8094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Junghans D., Chauvet S., Buhler E., Dudley K., Sykes T., Henderson C. E. (2004) The CES-2-related transcription factor E4BP4 is an intrinsic regulator of motoneuron growth and survival. Development 131, 4425–4434 [DOI] [PubMed] [Google Scholar]
- 12. Sui G., Soohoo C., Affar E. B., Gay F., Shi Y., Forrester W. C., Shi Y. (2002) A DNA vector-based RNAi technology to suppress gene expression in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 99, 5515–5520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim D., Nguyen M. D., Dobbin M. M., Fischer A., Sananbenesi F., Rodgers J. T., Delalle I., Baur J. A., Sui G., Armour S. M., Puigserver P., Sinclair D. A., Tsai L. H. (2007) SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer's disease and amyotrophic lateral sclerosis. EMBO J. 26, 3169–3179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee M. S., Kwon Y. T., Li M., Peng J., Friedlander R. M., Tsai L. H. (2000) Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature 405, 360–364 [DOI] [PubMed] [Google Scholar]
- 15. Takeuchi S., Fujiwara N., Ido A., Oono M., Takeuchi Y., Tateno M., Suzuki K., Takahashi R., Tooyama I., Taniguchi N., Julien J. P., Urushitani M. (2010) Induction of protective immunity by vaccination with wild-type apo superoxide dismutase1 in mutant SOD1 transgenic mice. J. Neuropathol. Exp. Neurol. 69, 1044–1056 [DOI] [PubMed] [Google Scholar]
- 16. Boillée S., Yamanaka K., Lobsiger C. S., Copeland N. G., Jenkins N. A., Kassiotis G., Kollias G., Cleveland D. W. (2006) Onset and progression in inherited ALS determined by motor neurons and microglia. Science 312, 1389–1392 [DOI] [PubMed] [Google Scholar]
- 17. Yamanaka K., Chun S. J., Boillee S., Fujimori-Tonou N., Yamashita H., Gutmann D. H., Takahashi R., Misawa H., Cleveland D. W. (2008) Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat. Neurosci. 11, 251–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gurney M. E., Pu H., Chiu A. Y., Dal Canto M. C., Polchow C. Y., Alexander D. D., Caliendo J., Hentati A., Kwon Y. W., Deng H. X. (1994) Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 264, 1772–1775 [DOI] [PubMed] [Google Scholar]
- 19. Mayford M., Bach M. E., Huang Y. Y., Wang L., Hawkins R. D., Kandel E. R. (1996) Control of memory formation through regulated expression of a CaMKII transgene. Science 274, 1678–1683 [DOI] [PubMed] [Google Scholar]
- 20. Krestel H. E., Mayford M., Seeburg P. H., Sprengel R. (2001) A GFP-equipped bidirectional expression module well suited for monitoring tetracycline-regulated gene expression in mouse. Nucleic Acids Res. 29, E39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. MacGillavry H. D., Stam F. J., Sassen M. M., Kegel L., Hendriks W. T., Verhaagen J., Smit A. B., van Kesteren R. E. (2009) NFIL3 and cAMP response element-binding protein form a transcriptional feedforward loop that controls neuronal regeneration-associated gene expression. J. Neurosci. 29, 15542–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nishimura Y., Tanaka T. (2001) Calcium-dependent activation of nuclear factor regulated by interleukin 3/adenovirus E4 promoter-binding protein gene expression by calcineurin/nuclear factor of activated T cells and calcium/calmodulin-dependent protein kinase signaling. J. Biol. Chem. 276, 19921–19928 [DOI] [PubMed] [Google Scholar]
- 23. Bossy-Wetzel E., Schwarzenbacher R., Lipton S. A. (2004) Molecular pathways to neurodegeneration. Nat. Med. 10, S2-S9 [DOI] [PubMed] [Google Scholar]
- 24. Fu L., Pelicano H., Liu J., Huang P., Lee C. (2002) The circadian gene Period2 plays an important role in tumor suppression and DNA damage response in vivo. Cell 111, 41–50 [DOI] [PubMed] [Google Scholar]
- 25. Hua H., Wang Y., Wan C., Liu Y., Zhu B., Yang C., Wang X., Wang Z., Cornelissen-Guillaume G., Halberg F. (2006) Circadian gene mPer2 overexpression induces cancer cell apoptosis. Cancer Sci. 97, 589–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Inoue H., Tsukita K., Iwasato T., Suzuki Y., Tomioka M., Tateno M., Nagao M., Kawata A., Saido T. C., Miura M., Misawa H., Itohara S., Takahashi R. (2003) The crucial role of caspase-9 in the disease progression of a transgenic ALS mouse model. EMBO J. 22, 6665–6674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li M., Ona V. O., Guégan C., Chen M., Jackson-Lewis V., Andrews L. J., Olszewski A. J., Stieg P. E., Lee J. P., Przedborski S., Friedlander R. M. (2000) Functional role of caspase-1 and caspase-3 in an ALS transgenic mouse model. Science 288, 335–339 [DOI] [PubMed] [Google Scholar]
- 28. Eide E. J., Woolf M. F., Kang H., Woolf P., Hurst W., Camacho F., Vielhaber E. L., Giovanni A., Virshup D. M. (2005) Control of mammalian circadian rhythm by CKIϵ-regulated proteasome-mediated PER2 degradation. Mol. Cell Biol. 25, 2795–2807 [DOI] [PMC free article] [PubMed] [Google Scholar]