

Background: Glyoxalases are a varied group of enzymes that detoxify methylglyoxal by converting it to d-lactate.
Results: The Candida albicans glyoxalase Glx3 is important for yeast growth, especially in glycerol.
Conclusion: Many yeasts contain a novel group of glyoxalases that are not redundant with previously characterized enzymes.
Significance: This is the first demonstration of physiologically relevant glutathione-independent glyoxalases in fungi.
Keywords: Candida albicans, DJ-1, Glycolysis, Structural Biology, X-ray Crystallography, Hsp31, Glyoxalase, Methylglyoxal
Abstract
Methylglyoxal is a cytotoxic reactive carbonyl compound produced by central metabolism. Dedicated glyoxalases convert methylglyoxal to d-lactate using multiple catalytic strategies. In this study, the DJ-1 superfamily member ORF 19.251/GLX3 from Candida albicans is shown to possess glyoxalase activity, making this the first demonstrated glutathione-independent glyoxalase in fungi. The crystal structure of Glx3p indicates that the protein is a monomer containing the catalytic triad Cys136-His137-Glu168. Purified Glx3p has an in vitro methylglyoxalase activity (Km = 5.5 mm and kcat = 7.8 s−1) that is significantly greater than that of more distantly related members of the DJ-1 superfamily. A close Glx3p homolog from Saccharomyces cerevisiae (YDR533C/Hsp31) also has glyoxalase activity, suggesting that fungal members of the Hsp31 clade of the DJ-1 superfamily are all probable glutathione-independent glyoxalases. A homozygous glx3 null mutant in C. albicans strain SC5314 displays greater sensitivity to millimolar levels of exogenous methylglyoxal, elevated levels of intracellular methylglyoxal, and carbon source-dependent growth defects, especially when grown on glycerol. These phenotypic defects are complemented by restoration of the wild-type GLX3 locus. The growth defect of Glx3-deficient cells in glycerol is also partially complemented by added inorganic phosphate, which is not observed for wild-type or glucose-grown cells. Therefore, C. albicans Glx3 and its fungal homologs are physiologically relevant glutathione-independent glyoxalases that are not redundant with the previously characterized glutathione-dependent GLO1/GLO2 system. In addition to its role in detoxifying glyoxals, Glx3 and its close homologs may have other important roles in stress response.
Introduction
Reactive carbonyl species are produced in many organisms as a consequence of central metabolism. Among these, methylglyoxal (MG3; H3C–CO–CHO) has attracted considerable interest dating from the early 20th century for its role as a branch point metabolite for alternative catabolic pathways. MG is an α-ketoaldehyde metabolite produced principally by the spontaneous dephosphorylation of triose phosphates and, in some prokaryotes, by the enzyme-catalyzed dephosphorylation of dihydroxyacetone phosphate (DHAP) (1). A small fraction of MG is also generated by catabolism of acetone and threonine (2, 3) and as a rare elimination side reaction of triose-phosphate isomerase (4). In the model yeast Saccharomyces cerevisiae, ∼0.3% of all glycolytic carbon flux is converted to MG, making it a potentially abundant metabolite under growth conditions that favor high rates of glycolysis (5).
MG is cytotoxic at elevated concentrations because, as a carbonyl electrophile, it can damage proteins, nucleic acids, and lipids by modification of arginine, lysine, cysteine, adenine, and guanine to form various advanced glycation end products (6, 7). Consequently, many organisms produce glyoxalases that detoxify MG by converting it to d-lactate. These enzymes were initially discovered in rabbit and dog tissues and first reported 100 years ago (8). Curiously for a toxic metabolite, MG is produced enzymatically from DHAP by a dedicated methylglyoxal synthase in certain prokaryotes. In combination with the glyoxalases, methylglyoxal synthase constitutes the initial portion of the methylglyoxal bypass, which converts DHAP to MG, then to d-lactate and, ultimately, to pyruvate (Fig. 1) (9). The methylglyoxal bypass has been proposed to serve an important catabolic role in phosphate-limited conditions, as it permits the oxidation of DHAP to pyruvate without having carbon flux through the more phosphate-demanding Embden-Meyerhof-Parnas (EMP) glycolytic pathway (10).
FIGURE 1.

A flowchart of central carbon metabolism emphasizing the EMP pathway and two known glyoxalase systems. The glutathione-independent glyoxalase Glx3 is shown in the shaded box. HTA is the hemithioacetal adduct that spontaneously forms between MG and reduced glutathione, and SDLGSH is the (RS)-lactoylglutathione product of glyoxalase I enzyme. The catalytic actions of glyoxalases are indicated with dashed lines.
In contrast to prokaryotes, eukaryotes do not possess a clearly identified methylglyoxal synthase, and therefore MG production from triose phosphates is presumed to be predominantly nonenzymatic in these organisms (11). Enzymes such as semicarbazide-sensitive amine oxidase can produce some MG, although the magnitude of their contribution to total MG pools in eukaryotes is not clear (12). However, the absence of a known methylglyoxal synthase does not necessarily mean that MG serves no metabolic purpose in eukaryotes. Accumulation of triose phosphates is inhibitory to glycolysis and can be partially relieved by their spontaneous, nonenzymatic dephosphorylation to produce MG. Subsequent detoxification of MG by glyoxalases could then facilitate a minor alternative pathway to generate pyruvate and acetyl-CoA under nonoptimal growth conditions when triose phosphates would otherwise accumulate to high levels. In contrast, under optimal growth conditions, carbon flux is directed to the conventional EMP pathway for pyruvate production.
Glyoxalases are required for the detoxification of MG and may serve a metabolic role in producing d-lactate for further catabolism. The best characterized glyoxalase is the glutathione-dependent two enzyme system (glyoxylases I and II) that detoxifies MG by converting it to d-lactate and regenerating free glutathione. In this system, glutathione and MG are converted to R-(S)-lactoylglutathione by the metalloenzyme glyoxylase I (GLO1 in the yeasts) (13). The lactoylglutathione product is then hydrolyzed by a second enzyme, glyoxylase II (GLO2), to give d-lactate and glutathione (14). In addition to the glutathione-dependent system, a less studied glutathione-independent glyoxalase (glyoxalase III) in Escherichia coli can catalyze the conversion of MG to d-lactate without any cofactor (Fig. 1). Although this activity was known to exist in lysates of E. coli for many years (15), it was only recently ascribed to Hsp31 (HchA), a previously characterized chaperone of the DJ-1 superfamily (16). Consistent with its dual role in the heat and carbonyl stress responses, transcription of E. coli Hsp31/glyoxalase III is rpoS-dependent and thus highest in stationary phase (17, 18). An important lingering question is whether the activity of glutathione-independent glyoxalases is physiologically relevant, as it is unclear why this activity would be needed in addition to the established glutathione-dependent system for detoxifying glyoxals. Notably, both the glutathione-dependent and -independent systems are present in many organisms, including the model yeasts S. cerevisiae and Candida albicans, suggesting that they may not be functionally redundant.
Investigating the physiological role of putative eukaryotic glutathione-independent glyoxalases is aided by the abundance of organisms that contain homologs from the Hsp31 clade of the DJ-1 superfamily. S. cerevisiae is a natural choice for such a study; however, S. cerevisiae contains four Hsp31 homologs (YDR533C, YMR322C, YOR391C, and YPL280W), thus presenting the challenge of generating knock-outs targeting multiple and very similar genes and then determining the degree of functional redundancy among them (19, 20). Additionally, S. cerevisiae switches from respiratory to fermentative metabolism when presented with fermentable carbon sources such as glucose in aerobic growth conditions (the Crabtree effect) and can survive in fermentative growth conditions with nonfunctional mitochondria (i.e. a “petite positive” yeast). Both of these metabolic features act as serious confounding factors to the study of endogenous MG toxicity as a function of carbon source, as the entire metabolic program of the organism shifts in a carbon source-dependent manner (21). In contrast, the yeast C. albicans has only one Hsp31 homolog (ORF 19.251), which simplifies the generation of knock-out strains and the characterization of their phenotypes, thereby making C. albicans a simpler model for studying the functions of these proteins. Unlike S. cerevisiae, C. albicans respires in aerobic growth conditions regardless of carbon source (i.e. it is a Crabtree negative yeast) and therefore must retain functional mitochondria, making it a “petite-negative” yeast as well (21). These metabolic features can be used to alter the level of endogenously produced MG in C. albicans by growing the cells in carbon sources whose catabolism produces different levels of triose phosphates, the direct precursors to MG. Because C. albicans is a Crabtree negative yeast, it will not switch between respiratory and fermentative metabolic programs as a consequence of changing carbon sources, thus eliminating a complication of using Crabtree positive yeasts such as S. cerevisiae in this study.
In this study, the C. albicans YDR533C/Hsp31 homolog ORF 19.251 is shown to possess glutathione-independent glyoxalase III activity. As a consequence, we propose that this gene be named GLX3. Unlike the homologous proteins from S. cerevisiae and E. coli, the crystal structure of CaGlx3 indicates that it is a monomer. As expected, the protein possesses a Cys-His-Glu catalytic triad found in other members of the Hsp31 clade. A combination of site-directed mutagenesis and enzyme kinetics indicates that the active site cysteine residue is critical for glyoxalase activity and that a nearby histidine residue is also important. The in vivo relevance of glyoxalase III activity was established by generating a glx3 knock-out mutant of C. albicans and testing complementation with the wild-type allele. The homozygous glx3 knockout was significantly more sensitive to both exogenously administered MG and to MG produced endogenously by glucose or glycerol catabolism, especially when grown in glycerol. A rationale for the existence and physiological significance of glutathione-independent glyoxalases in fungi is discussed.
EXPERIMENTAL PROCEDURES
Yeast Strains, Media, and Growth Conditions
C. albicans wild-type clinical isolate SC5314 was the generous gift of Dr. Alexander Johnson, University of California at San Francisco. C. albicans strains were grown and maintained in yeast extract/peptone/dextrose (YPD) medium (10 g of yeast extract, 5 g of peptone, and 20 g of glucose per liter) at 30 °C. Growth curves were measured in YPD that had been inoculated with cells washed three times in 50 mm potassium phosphate, pH 6.5, at a density of 1 × 107 cells/ml. Cells were grown aerobically in 250-ml Erlenmeyer flasks containing 50 ml of media that were incubated at 30 °C with 225 rpm rotary agitation. For determination of the role of Glx3p in C. albicans metabolism, multiple media were used as follows: YPD medium supplemented with 0–40 mm methylglyoxal, YPG (yeast extract/peptone medium containing 2% v/v of glycerol), and YP (yeast extract/peptone medium containing neither glucose nor glycerol). All experiments were done in triplicate and means ± S.D. were calculated.
Cloning and Purification of Glx3
The gene encoding Glx3 (ORF19.251) was PCR-amplified from C. albicans SC5314 genomic DNA using primers (Table 1) that introduced 5′-NdeI and 3′-XhoI restriction sites. The PCR product was purified, digested with NdeI and XhoI, and cloned between the corresponding restriction sites of the bacterial protein expression vector pET15b (Novagen). The C136S and H137F point mutations were introduced using site-directed mutagenesis, and all constructs were verified using DNA sequencing (Operon). E. coli strain BL21 (DE3) (Novagen) was transformed with GLX3-pET15b and grown at 37 °C in terrific broth medium supplemented with 100 μg/ml ampicillin and with shaking at 270 rpm to an OD600 ∼2.0. Expression of Glx3 was induced by the addition of isopropyl β-d-1-thiogalactopyranoside (Sigma) to a final concentration of 1 mm for 18 h at 25 °C. Two hours before harvesting the cells, chloramphenicol was added to the culture to a final concentration of 100 μg/ml to arrest protein synthesis and enhance the solubility of the expressed protein (22). The cells were harvested by centrifugation at 4 °C and then resuspended in ice-cold lysis buffer (50 mm HEPES, pH 7.5, 300 mm NaCl, and 10 mm imidazole, 1 mm DTT) at a ratio of 5 ml of buffer per g of wet cell mass. Cells were lysed by the addition of lysozyme to a final concentration of 1 mg/ml and incubation for 30 min, followed by sonication. The lysate was clarified by centrifugation at 12,000 × g to remove cellular debris.
TABLE 1.
Primers used in this study
| Sequence | Name | Reference | Purpose |
|---|---|---|---|
| 5′-TCC GAG GGG GGT ACC TCC TTC-3′ | KpnI | This study | Glx3 knockout |
| 5′-TTG AAA CAC TCG AGG AAG GGG-3′ | XhoI | This study | Glx3 knockout |
| 5′-GGG CAT ACA AAA GCC GCG GAT TTA-3′ | SacI | This study | Glx3 knockout |
| 5′-AAC AAA ATT AAT TGA GCT CTC TTG-3′ | SacII | This study | Glx3 knockout |
| 5′-TAT TGT TGA TGA ATC GAA GAA AAC-3′ | Upstream check | This study | Southern blot |
| 5′-TGT TTA GTA GAT CTT TCT AGA TCT-3′ | Downstream check | This study | Southern blot |
| 5′-TAA AAA TAT CAT ATG GTG AAA GTT TTA CTC-3′ | NdeI | This study | Glx3 insertion in pET15b |
| 5′-TAA TTT CTC GAG TTA ATT ACA TTC AAA AGG-3′ | XhoI | This study | Glx3 insertion in pET15b |
| 5′-CTT TCT GCT GTT AGT CAT GGT CCT GCC AT-3′ | Forward C136S | This study | Glx3 (C136S) point mutation |
| 5′-ATG GCA GGA CCA TAA ACA GCA GAA AC-3′ | Reverse C136S | This study | Glx3 (C136S) point mutation |
| 5′-GTT TGT TTT GGT CCT GCC ATT TTT-3′ | Forward H137F | This study | Glx3 (H137F) point mutation |
| 5′-AAA AAT GGC AGG ACC AAA ACA AAC-3′ | Reverse H137F | This study | Glx3 (H137F) point mutation |
Cleared lysate was mixed with His-Select Ni2+ metal affinity resin (Sigma) for 30 min with gentle stirring at 4 °C. The resin was transferred to a glass column and washed extensively with wash buffer (25 mm HEPES, pH 7.5, 300 mm NaCl, 20 mm imidazole, 2 mm DTT) until no protein was detected in the eluate by Bradford's reagent (Bio-Rad). N-terminal hexahistidine-tagged Glx3 was eluted from the resin with ice-cold elution buffer (25 mm HEPES, pH 7.5, 300 mm NaCl, 200 mm imidazole, 2 mm DTT) and then incubated with bovine thrombin (MP Biomedicals) at a ratio of 1.5 units of thrombin/mg of Glx3 for 3 h at room temperature, followed by overnight dialysis against storage buffer (25 mm HEPES, pH 7.5, 100 mm KCl, and 2 mm DTT). After digestion, any protein retaining the histidine tag was separated from the tag-cleaved protein by a second passage over His-select resin that had been equilibrated in storage buffer. The flowthrough fractions, containing purified tag-cleaved protein Glx3, were then passed over benzamidine-Sepharose (Amersham Biosciences) resin to remove thrombin. Unexpectedly, a considerable amount of Glx3 binds to benzamidine-Sepharose under these conditions. The purified protein was concentrated using a stirred cell concentrator with a nominal molecular mass cutoff of 10 kDa to 20 mg/ml, as determined using UV-visible spectrophotometry with a calculated extinction coefficient at 280 nm (ϵ280) of 21,555 m−1 cm−1 for Glx3 (ExPASy). The purified protein ran as a single band on Coomassie Blue-stained SDS-PAGE and was aliquoted into 50–100-μl volumes, flash-frozen in liquid nitrogen, and stored at −80 °C until needed.
Crystal Structure Determination of Glx3
Glx3 (20 mg/ml in storage buffer) was crystallized using sitting drop vapor equilibration. Initial crystallization conditions were identified using a 96-well plate and commercially available sparse matrix crystallization screens. Conditions delivering crystals were further optimized by fine screening using sitting drop vapor equilibration of drops containing 2 μl of Glx3 with 2 μl of reservoir solution. Obelisk-shaped crystals of wild-type Glx3 measuring ∼20 × 20 × 100 μm grew from 100 mm sodium acetate, pH 4.1, 120 mm ammonium acetate, 27% PEG4000, and 3% ethylene glycol at 4 °C after 1–3 days. The crystals were cryoprotected by serial transfer through reservoir solution supplemented with increasing concentrations of ethylene glycol to a final concentration of 20% v/v. Crystals were removed with nylon loops and cryocooled by rapid immersion into liquid nitrogen.
X-ray diffraction data were collected from a single crystal maintained at 100 K at SSRL beamline 9-2 using 13.79 keV incident x-rays and a MarMosaic 325 charge coupled device detector. The crystal was exposed to x-rays during 0.5° oscillations, and 300 images were collected and processed. The data were indexed, integrated, and scaled using HKL2000 (23), and final data statistics are shown in Table 2.
TABLE 2.
Crystallographic data collection and refinement statistics
| Data collection | |
| X-ray source | SSRL 9-2 |
| X-ray wavelength | 0.90 Å |
| Space group | P42212 |
| Cell dimensions | |
| a = b, c | 92.23 Å, 59.37 Å |
| α = β = γ | 90° |
| Molecules in asymmetric units | 1 |
| Wilson B factor | 21 Å2 |
| Resolutiona | 41 to 1.6 Å |
| No. of reflections | 34,216 |
| Completeness | 100.0% (100.0%) |
| Redundancy | 12.1 (12.2) |
| Rmergeb | 0.05 (1.00) |
| 〈I〉/〈σ(I)〉 | 51.1 (2.2) |
| Refinement | |
| Resolution | 41 to 1.6 Å |
| Rworkc; Rfreed; Ralle | 15.8%; 18.5%; 15.9% |
| No. of protein atoms | 1843 |
| No. of solvent atoms | 279 |
| No. of heteroatoms | 8 |
| Root mean square deviations | |
| Bond lengths | 0.006 Å |
| Bond angles | 1.08° |
| Ramachandran plot favored; allowed; forbidden | 97%; 100%; 0% |
a Values in parentheses are for highest resolution shell (1.63 to 1.60 Å).
b Rmerge = ΣhklΣi|Ihkli − 〈Ihkl〉|/ΣhklΣiIhkli, where i is the ith observation of a reflection with indices h,k,l, and angle brackets indicate the average over all i observations.
c Rwork = Σhkl|Fhklo − Fhklc|/Σ Fhklo, where Fhklc is the calculated structure factor amplitude with index h,k,l, and Fhklo is the observed structure factor amplitude with index h,k,l.
d Rfree is calculated as Rwork, where the Fhklo are taken from a test set comprising 5% of the data (1730 reflections) that were excluded from the refinement.
e Rall is calculated as Rwork, where the Fhklo includes all measured data (including the Rfree test set).
Phases and an initial model for Glx3 were obtained using maximum likelihood molecular replacement in PHASER (24), part of the CCP4 suite of programs (25). A homology model based on the coordinates of one monomer (chain A) of YDR533C (PDB code 1RW7) (20) was generated by SwissModel (26) and used for the rotation and translation searches. A clear top solution was obtained, which was then subjected to stereochemically restrained refinement with riding hydrogen atoms against an amplitude-based maximum likelihood target in REFMAC5 (27). All measured data were used for the refinement, and a bulk solvent correction was applied. The translation-libration-screw model was used for refinement of anisotropic atomic displacement parameters based on a rigid body displacement model that treated the entire protein as a single rigid group (28, 29). Ordered solvent placement and manual adjustments to the model were performed in COOT (30), and the final model was validated using the validation tools in COOT (30) and MOLPROBITY (31). All structural figures were made with UCSF Chimera (32).
Glutathione-independent Glyoxalase Assay
Glyoxalase activity was assayed in vitro using a previously described fixed time point assay that follows the enzymatic depletion of the substrate methylglyoxal (33). Methylglyoxal (Sigma, 40% solution) is unstable and was thus stored at 4 °C until immediately before use. The reaction was initiated by adding enzyme in reaction buffer (100 mm HEPES, pH 7.5, 50 mm KCl, 2 mm DTT) to the specified initial concentration of methylglyoxal in reaction buffer, followed by incubation at 30 °C. The final enzyme concentration was 10 μm. The methylglyoxal remaining at each time point was determined by reaction with 2,4-dinitrophenylhydrazine (DNPH) to generate the purple chromophore methylglyoxal-bis-2, 4-dinitrophenylhydrazone after alkali treatment. As this was a substrate depletion assay, a relatively high concentration of enzyme (10 μm) was used to observe clear differences in remaining substrate concentration over the first 1–2 min of reaction. In all cases, however, substrate was saturating at >10-fold excess, justifying the steady-state assumption.
The assay was performed by removing a 50-μl sample of the reaction at fixed time points after enzyme addition (0, 15, 30, 45, 60, and 75 s) and rapidly mixing with 0.9 ml of distilled water. To this solution, 0.33 ml of a freshly prepared stock of DNPH reagent (0.2% DNPH dissolved in 2 n HCl) was immediately added and incubated at 30 °C for 15 min. This highly acidic solution stops the enzymatic reaction, whose rate was already greatly diminished by the initial 19-fold dilution into water. The characteristic purple color of the hydrazone was developed by addition of 1.67 ml of 3.8 m NaOH and incubation for 5–10 min at room temperature followed by measurement of absorbance at 550 nm in a SpectraMax Plus spectrophotometer (Molecular Devices). All measurements were made in triplicate and multiple preparations of Glx3 were assayed to verify consistent levels of activity. The measured absorbance values at 550 nm were converted to concentration of methylglyoxal using interpolation onto the best-fit line for a methylglyoxal-DNPH calibration curve. The rate of spontaneous loss of MG in solution, determined as above but without added enzyme, was measured and subtracted from all enzymatic rates. Steady-state enzymatic parameters were obtained by fitting the Michaelis-Menten model to the data using Prism (GraphPad).
The glyoxalase assay based on methylglyoxal depletion was complemented by an assay based on the formation of d-lactic acid. As above, 50-μl samples were taken from buffered 10 μm enzyme with varying initial concentrations of methylglyoxal at 30-s increments for the first 150 s of the reaction. These samples were heat-inactivated at 80 °C for 30 s, cooled, supplemented with 10 mm NAD+, and 1 unit of d-lactate dehydrogenase (Sigma catalog no. L-3888 from Lactobacillus leichmanii), and incubated for 30 min at 37 °C. d-Lactate dehydrogenase will oxidize any d-lactate that was produced by glyoxalase activity, producing pyruvate and NADH. The absorbance of NADH was measured at 340 nm and converted to a molar concentration using an extinction coefficient at 340 nm of 6220 m−1 cm−1.
Specific Activity Measurements
Initial rates of methylglyoxal depletion or d-lactate production were measured as described above with 10 μm enzyme acting on 6 mm substrate at both 30 °C (methylglyoxal consumption) and at 37 °C (d-lactate production). These initial rates were divided by the amount of enzyme in the reaction to calculate the reported specific activities, and all measurements were made in triplicate. Means ± S.D. for triplicate measurements are reported in Table 3.
TABLE 3.
Comparative glyoxalase-specific activities of selected DJ-1 superfamily proteins
| Human DJ-1 | Sz. pombe DJ-1 | S. cerevisiae YDR533C | C. albicans Glx3 | C. albicans C136S Glx3 | C. albicans H137F Glx3 | |
|---|---|---|---|---|---|---|
| Methylglyoxal consumption (μmol/min/mg enzyme) | 1.0 ± 0.3a | 2.4 ± 0.2 | 6.4 ± 0.1 | 11.5 ± 0.1 | 0.2 ± 0.1b | 1.7 ± 0.1 |
| d-Lactate production (μmol/min/mg enzyme) | NDc | ND | 10.5 ± 1.0 | 7.0 ± 1.0 | ND | ND |
a Values shown are the average of triplicate experiments ± S.D. using purified proteins as expressed in E. coli. The methylglyoxal assay was performed at 30 °C and the d-lactate assay at 37 °C.
b This value is near the noise level of the measurement and is consistent with no significant activity.
c Data not detected with an estimated detection limit of ∼0.2 μmol/min/mg enzyme for NADH production.
Generation of the glx3 Null Mutant
The upstream fragment of glx3 was amplified by PCR using primers introducing a distal restriction site for KpnI and a proximal site for XhoI (Table 1). A downstream fragment of the gene was amplified with primers that introduced a proximal SacII and a distal SacI restriction site (Table 1). The fragments were cloned into the pSFS2A vector at positions up- and downstream of a SAT1 flipper cassette, creating the construct pSFS2A-GLX3-flank. This SAT1 flipper cassette contains a nourseothricin marker (caSAT1) and an adapted FLP gene (CaFLP) encoding a site-specific recombinase under the control of C. albicans MAL2 promoter (34). A linear fragment containing the disruption cassette was created by digesting the pSFS2A-GLX3-flank construct with KpnI and SacI. The fragment was purified by agarose gel electrophoresis followed by gel extraction of the excised target band (Qiagen). The fragment was introduced by electroporation into C. albicans according to the procedure described in Reuss et al. (34). Briefly, 1 μg of linear DNA was mixed with 40 μl of electrocompetent SC5314 cells and subjected to electroporation (0.2-cm cuvette, 1.5 kV, 400 ohms). The cells were incubated with a 1:1 ratio of 1 m sorbitol/YPD at 30 °C for 2 h with shaking at 225 rpm followed by spreading onto a YPD plate containing 200 μg/ml nourseothricin, which was incubated at 30 °C overnight. The following day, the resistant colonies were picked and streaked again onto a YPD plate containing 200 μg/ml nourseothricin. Resistant cells were analyzed for the first allelic replacement by PCR and Southern blotting (primers, see Table 1). After confirming that one of the GLX3 alleles had been replaced with the caSAT1 cassette (glx3-a1), the cells were inoculated into YPM liquid medium (10 g of yeast extract, 5 g of peptone, and 20 g of maltose per liter) without selective pressure to allow FLP-mediated excision of the cassette as described by Reuss et al. (34). After 6 h of incubation at 30 °C, serially diluted cells were spread on a YPD plate containing 25 μg/ml nourseothricin to assay for restoration of antibiotic sensitivity resulting from FLP-mediated excision of CaSAT1 (glx3-a2). These cells were used for the second allelic deletion following the same procedure.
Measurement of Intracellular Methylglyoxal Concentration
Cellular MG concentrations were measured using a modification of the procedure described by Scheijen and Schalkwijk (35) and McLellan et al. (36). Late log phase cells were harvested from liquid culture by centrifugation and immediately frozen at −80 °C. Cell pellets were resuspended in buffer containing 0.3 m sucrose, 10 mm histidine, and 0.23 mm phenylmethylsulfonyl fluoride, pH 7.4 (2 μl of buffer per mg of pellet). Cell suspensions were then sonicated three times for 5 s (setting 4, Fisher M100 sonicator) within 15-s intervals. Thereafter, the sonicated samples were added to equal volumes of 5.0 m perchloric acid and placed on ice for 10 min. Samples were then centrifuged for 10 min at 3000 rpm (Eppendorf 5417R, EL 129 FA 45-24-11 rotor). Supernatants (50 μl) were then mixed with 150 μl of 0.1 m o-phenylenediamine in 1.6 m perchloric acid and allowed to react for 16 h at room temperature in the dark. After this reaction, mixtures were extracted three times with 2.0 ml of chloroform, and pooled extracts from each sample were dried over anhydrous sodium sulfate. Chloroform was then evaporated from each sample using a gentle stream of air, and samples were resuspended in 500 μl of methanol. High performance liquid chromatography (SCL-Shimadzu 10A) employing a Kinetex 5-μm C18 100R column, a mobile phase of methanol/water/trifluoroacetic acid (52:48:0.1) at a flow rate of 1 ml/min, and a detector wavelength of 312 nm were then used to determine the amount of 2-methylquinoxaline, the product formed following the reaction of MG and o-phenylenediamine. For these experiments 2-methylquinoxaline concentrations ranging from 1 to 100 μm were used to generate the standard curve for absolute concentration determination.
RESULTS
Crystal Structure of C. albicans Glx3p
The crystal structure of Glx3p was determined at 1.6 Å resolution using molecular replacement with a homology model based on S. cerevisiae YDR533C (PDB code 1RW7) (20). Glx3 contains the standard DJ-1 superfamily flavodoxin-like core fold with several noncontiguous secondary structural elements (helices B, H, and I and strands 2 and 3) that form a “cap” region that has been previously called the P domain in other homologs of Hsp31 (Fig. 2A) (20, 37). The structure is very similar to that of YDR533C (core Cα root mean squared deviation = 0.81 Å), as expected based on their 62% sequence identity.
FIGURE 2.

C. albicans Glx3 is a monomeric member of the Hsp31 clade of the DJ-1 superfamily. A, ribbon diagram of Glx3 is shown with β-strands numbered and in red and α-helices lettered in blue. The N and C termini are labeled. Active site residues are shown in bond representation. B shows a close-up view lattice contract that represents the largest intermolecular interface between Glx3 proteins in the crystal. The two proteins are colored pale blue and gold, with primes on residues from the other chain. 2mFo − DFc electron density is shown contoured at 1σ and colored blue. The carboxylic acid-rich contact region between two proteins and the close 2.44-Å hydrogen bond between Glu13 and Glu48 requires low pH to be stable and would not form under physiological conditions. Therefore, there are no intermolecular contacts of probable physiological relevance in the Glx3 crystal.
All other structurally characterized members of the Hsp31 clade, including E. coli Hsp31 (PDB code 1N57) (37) and the S. cerevisiae proteins YDR533C (PDB code 1RW7) (20) and Y0R391C (PDB code 3MII) (38), are homodimers. Although all previously characterized Hsp31-like proteins are dimeric, E. coli Hsp31 contains a 45-amino acid N-terminal extension that results in a formation of a different dimer than observed for the yeast proteins YDR533C or YOR391C, which lack this extension (20, 37–41). This N-terminal 45-amino acid region has been shown to be important for chaperone activity of the E. coli Hsp31 (42, 43), and its absence in YDR533C indicates a potential functional difference between these proteins. The structure of C. albicans Glx3 also lacks this 45-amino acid N-terminal extension but, unlike YDR533C and YOR391C, is a monomer in the crystal. The best candidate for a potential dimer interface buries 606 Å2 of surface area per monomer as calculated by PISA (44). However, this interface features a cluster of acidic residues (Glu13, Glu48, and Asp117) in which Glu13 makes a short hydrogen bond (2.44 Å) with Glu48 from the other monomer, requiring at least one of the side chains to be protonated as a carboxylic acid (Fig. 2B). Crystals of Glx3 could only be obtained at pH values below 4.1, suggesting that the protonation of these carboxylic acids is essential to crystallize Glx3 in this lattice. Because this lattice contact involves multiple carboxylate side chains that would likely be negatively charged at physiological pH, this intermolecular interface is likely not physiologically relevant.
The active site for C. albicans Glx3 contains the triad residues Cys136, His137, and Glu168, as well as nearby residues Glu28 and His106 (Figs. 2A and 3A). These residues are conserved in Hsp31 proteins from yeast and bacteria, and the Glx3 active site aligns well with that of the E. coli enzyme (Fig. 3B). The catalytic Cys136 sits at the bottom of a narrow active site pocket that can accommodate only small molecule substrates (Fig. 3C), consistent with a role in detoxifying low molecular mass glyoxals. As observed in many other DJ-1 superfamily crystal structures (20, 45–49), the active site cysteine residue is surrounded by unusual electron density indicative of probable partial oxidation of the reactive thiol(ate). Two oxygen atoms are modeled at 2.25 and 2.06 Å from the Sγ atom of Cys136 (Fig. 3A). These distances are shorter than the van der Waals contact distances expected for these atoms (∼3.3 Å) but too long to be fully formed covalent bonds, similar to a previous observation in a crystal structure of human DJ-1 (50). Although the cysteine residue of human DJ-1 and its close homologs are easily oxidized to cysteine sulfinic acid (51), the electron density for Cys136 in Glx3 is not completely consistent with this covalent modification. Therefore, these two oxygen atoms were conservatively modeled as water molecules in the crystal structure. We speculate that photoreduction and subsequent redox chemistry caused by synchrotron radiation may be responsible for this modification, as noted previously in Pseudomonas fluorescens isocyanide hydratase (46). In total, the structural features of the Glx3 active site are consistent with a hydrolase employing a Cys-His-Glu catalytic triad with a reactive cysteine that attacks small electrophilic substrates.
FIGURE 3.
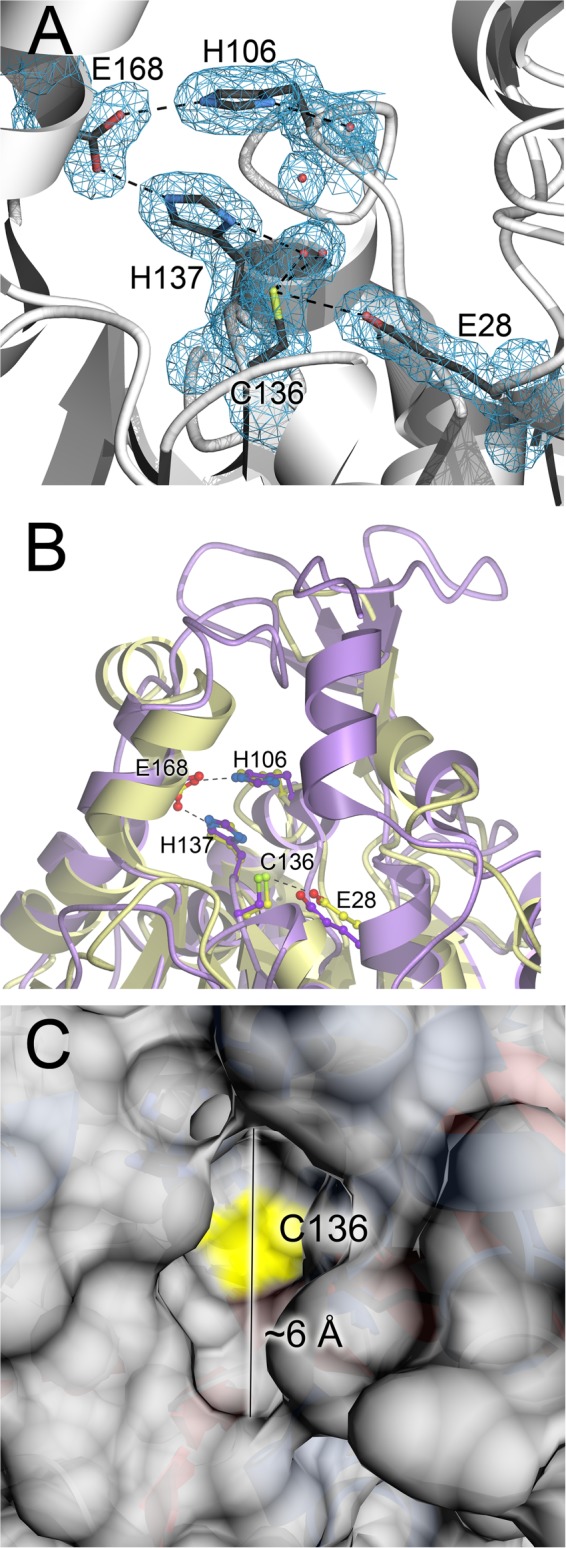
Glx3 contains a Glu-His-Cys catalytic triad at the bottom of a narrow active site cleft. A, catalytic triad and surrounding residues of the Glx3 active site are shown, with 2mFo − DFc electron density contoured at 1σ and shown in blue. Hydrogen bonds are indicated with dashed lines, and ordered water molecules are represented as red spheres. The Glu168-His137-Cys136 triad is surrounded by the other highly conserved residues His106 and Glu28, and these residues may serve as general acids/bases. B, comparison of CaGlx3 (gold) and E. coli Hsp31 (purple) shows that the EcHsp31 active site is somewhat more occluded by an additional α-helix. This may be responsible for the lower reported kcat value of EcHsp31 compared with CaGlx3. C, surface of CaGlx3 is shown with the location of the catalytically essential Cys136 indicated in yellow. The narrow 6-Å opening of this active site is selective for small substrates such as MG.
Glx3 Has Glutathione-independent Glyoxalase Activity in Vitro
The in vitro glyoxalase activity of C. albicans Glx3 was assayed using a fixed time point assay that follows the depletion of the substrate methylglyoxal as detected by formation of the purple-colored methylglyoxal-bis-2,4-dinitrophenylhydrazone adduct (see “Experimental Procedures”). Purified C. albicans Glx3 possessed robust glyoxalase activity in vitro (Table 3), and this activity was confirmed by a complementary assay based on the appearance of d-lactate (Table 3). Glutathione was not required for this activity, consistent with previous studies of E. coli Hsp31 (16). The kinetic constants for C. albicans Glx3 (Km = 5.5 mm, kcat = 7.8 s−1 (see Fig. 4)) are comparable with those reported for E. coli Hsp31 (Km = 1.43 mm, kcat = 2.7 s−1) (16). A notable structural difference between the fungal and prokaryotic enzymes is the more exposed active site in C. albicans Glx3 that is partially occluded by an α-helix in E. coli Hsp31 (Fig. 3B). As expected, mutation of the proposed catalytic nucleophile Cys136 to serine resulted in an inactive enzyme (Table 3 and Fig. 4). Mutation of His137 to phenylalanine in the catalytic triad gave ∼15% of wild-type activity (Table 3), suggesting that His137 also plays an important role during catalysis. It may serve as a general acid/base for one or more of the proton shuffling steps that are required to convert MG to lactate without a change in redox state. A proposed mechanism for glyoxalase III enzymatic activity is presented under “Discussion.”
FIGURE 4.

Glx3 is a glutathione-independent methylglyoxalase. The kinetics of wild-type Glx3 (filled circles) and the C136S mutant (open circles) are shown in hyperbolic representation, and the best fit Michaelis-Menten model is indicated with a solid line. Each rate was measured in triplicate, and mean values are plotted with standard deviations shown as error bars. At each concentration, the background uncatalyzed rate of MG degradation was measured and subtracted from the rate measured with enzyme present. The inactive C136S mutant demonstrates that the proposed catalytic nucleophile Cys136 is essential for catalysis.
Methylglyoxalase Activity Is Common to Yeast Members of the Hsp31 Clade
Three recombinant yeast enzymes exhibited significant glyoxalase activity (Table 3), whereas under our assay conditions, we could detect only very weak glyoxalase activity for recombinant human DJ-1 (Table 3). The S. cerevisiae protein YDR533C had a comparable specific activity for both MG consumption and d-lactate production to C. albicans Glx3 (Table 3), reflecting the aforementioned structural similarities of these two members of the Hsp31 clade of the DJ-1 superfamily. Interestingly, significant consumption of MG was also measured for Sz. pombe DJ-1 (SPAC22E12.03c), which is a close homolog of human DJ-1 and not a member of the Hsp31 clade (52, 53). However, unlike C. albicans Glx3 and S. cerevisiae YDR533C, no d-lactate production was detected for the human or Schizosaccharomyces pombe enzymes, suggesting either that another product is generated or that the d-lactate assay was not sensitive enough to detect the small quantity of product formed. The physiological relevance of the glyoxalase activities of the human and Sz. pombe proteins are therefore unclear. The Hsp31-like yeast proteins CaGlx3 and ScHsp31/YDR533C, in contrast, have robust glyoxalase activities and produce the expected d-lactate product, suggesting conservation of function for this clade of the DJ-1 superfamily.
Glx3 Confers Protection against Both Exogenous and Endogenously Generated Methylglyoxal
C. albicans is a diploid organism, and consequently the biological significance of Glx3 must be assessed via a double knock-out mutant. The double knock-out (glx3-Δ1/glx3-Δ1) and reconstituted strains were created by standard procedures (34), confirmed by Southern blots (Fig. 5), and then tested for their respective sensitivities to exogenous MG in yeast extract/peptone + 2% glucose (YPD) medium under aerobic culture conditions. The glx3-Δ1/glx3-Δ1 mutant was more sensitive to 20 and 40 mm exogenous MG than was its wild-type parent (Fig. 6). In contrast, 10 mm MG had no effect on the growth of any strain (Fig. 6). This experiment establishes that Glx3 can detoxify MG in vivo; however, we note that the high levels of exogenous MG used here are of uncertain physiological relevance.
FIGURE 5.

Southern blot analysis of the GLX3 gene in C. albicans. Genomic DNA from parental (1st lane), reconstituted (2nd lane), and the glx3-Δ1/glx3-Δ1 null mutant (3rd lane) was digested with XbaI, blotted, and probed with a 1623-bp fragment covering the GLX3 gene. β-Actin was used as the loading control.
FIGURE 6.

Glx3 is protective against exogenously added methylglyoxal. Cultures were grown aerobically in yeast extract/peptone + 2% glucose (YPD) medium at 30 °C with increasing methylglyoxal concentrations (noted on figure). The measured optical density is plotted on semi-log axes for C. albicans SC5314 (A) and the glx3-Δ1/glx3-Δ1 homozygous deletion mutant (B). Values shown are the average of triplicate experiments ± S.D.
Endogenous MG is generated as an unavoidable consequence of central carbon metabolism and was modulated by altering the carbon source provided to cultured C. albicans. Of the commonly used carbon sources in media, glycerol has been shown to produce the highest levels of triose phosphates, the immediate precursors to MG (54). The metabolic defects caused by Glx3 deficiency are evident when cell yields obtained by growth in yeast extract/peptone (YP), yeast extract/peptone/glucose (YPD), and yeast extract/peptone/glycerol (YPG) media are compared (Fig. 7). Note that following microbiological convention, the OD600 values of all growth curves in Figs. 6–8 are plotted on logarithmic y axes. Both the wild-type and Glx3-reconstituted (glx3-Δ1/glx3-Δ1/GLX3) C. albicans had equivalent cell yields (OD600 ∼10) on YPD and YPG but achieved only OD600 = 3.5 on YP alone. Thus, cells that expressed Glx3 used glucose and glycerol equivalently, resulting in an ∼2.5-fold increased yield compared with YP alone. In contrast, the glx3-Δ1/glx3-Δ1 mutant had markedly reduced OD600 values of ∼7 and 2.5 on YPD and YPG media, respectively (Fig. 7). Cell yield in YP medium was not strongly affected by the glx3 knockout. The 4-fold reduction in cell yield of the glx3 null mutant grown in YPG is quite large and indicates that the knock-out strain gains no metabolic benefit from the 2% glycerol present in the medium and may even be somewhat inhibited by it. The strong growth defect of the glx3 null mutant in YPG is likely connected to elevated endogenous MG levels under these growth conditions, because glycerol elevates the levels of triose phosphates in the cell and has been shown to substantially increase the production of MG in S. cerevisiae (54). Therefore, Glx3 plays an important role in central carbon metabolism and cellular viability in C. albicans, particularly when glycerol is the principal carbon source.
FIGURE 7.

Glx3 protects against endogenous MG generated by basal catabolism. Cell yields for parental (filled circles), glx3 null mutants (open circles), and reconstituted (filled squares) C. albicans were measured by optical density in multiple media and plotted on semi-log axes as a function of time. A shows that all cell types grow comparably well on yeast extract/peptone (YP) medium. B, glx3 null cells (Δglx3; open circles) show a marked growth defect in YPD (YP + 2% glucose) medium that is complemented by restoration of the wild-type GLX3 locus (Δglx3/GLX3; filled squares). C, glx3 null cells (Δglx3; open circles) show a more pronounced growth defect in YPG (YP + 2% glycerol) medium that is complemented by restoration of the wild-type GLX3 locus (Δglx3/GLX3). YPG medium is expected to produce the highest levels of endogenous MG. Values shown are the average of triplicate experiments ± S.D.
FIGURE 8.

Inorganic phosphate increases cell yields for the glx3 null mutant in glycerol. In all panels, cell yields for parental cells in the absence (filled circles) or presence (filled squares) of 10 mm phosphate are shown, as are glx3 null mutants in the absence (open circles) or presence (open squares) of 10 mm supplemental phosphate. C. albicans were measured by optical density in multiple media and plotted on semi-log axes as a function of time. A shows that added phosphate has little effect on either wild-type or mutant grown in yeast extract/peptone (YP) medium. B, growth defect of the glx3 null cells (Δglx3; open circles) in YPD (YP + 2% glucose) medium is only slightly improved by adding additional phosphate, and there is no effect on the wild-type cells. C shows that the strong growth defect of glx3 null cells (Δglx3; open circles) in YPG (YP + 2% glycerol) medium is significantly complemented by supplementation with inorganic phosphate. There is no effect of added phosphate on the yield of the wild-type cells in any condition. Values shown are the average of triplicate experiments ± S.D.
Because Glx3 has glyoxalase activity in vitro, we tested the hypothesis that the observed growth defects of the knockout are due to elevated levels of endogenously produced MG. The levels of intracellular MG in late log phase cells were directly measured using the chemical capture of MG by o-phenylenediamine to form the UV-absorbing compound 2-methylquinoxaline that was monitored by HPLC (see “Experimental Procedures”). This quantitative method is highly selective for the target α-oxoaldehydes and is more efficient that previous approaches, which are important advantages when measurements are being made in the cellular milieu (35). Greater than 95% of the MG in solution reacted with o-phenylenediamine, and chloroform extraction recovered >96% of 2-methylquinoxaline from solution. The results are shown in Table 4. As expected, wild-type cells produced more endogenous MG in media containing glycerol (YPG) than in media containing glucose (YPD). Cells grown in media lacking either carbon source (YP) have an unexpectedly high level of MG, which is comparable with that of cells grown in YPD medium. The importance of Glx3 for detoxification of endogenously produced MG is shown by the 3–5-fold increase in levels of intracellular MG in glx3-Δ1/glx3-Δ1 cells grown in YPD or YPG media compared with wild-type cells grown in the same media. Re-introduction of the GLX3 gene into the knock-out strain restored intracellular MG concentrations to levels that are slightly below those of wild-type cells grown in either YPD or YPG media, confirming that elevated MG in the knock-out strain is due to the absence of Glx3. In contrast, MG levels in glx3-Δ1/glx3-Δ1 cells grown in YP are elevated and not reduced to base-line levels by restoration of GLX3. This is a topic for future investigation. These experiments allow for the simultaneous measurement of cellular glyoxal, a closely related α-oxoaldehyde. Glyoxal levels were unaffected by the presence or absence of Glx3 in all media, indicating that the enzyme is selective for MG as a substrate in vivo.
TABLE 4.
Levels of intracellular methylglyoxal in C. albicans
The experiment was repeated in technical duplicate with replicate values differing by less than 5% for all measurements.
a YP, yeast extract/peptone; YPD, yeast extract/peptone/dextrose; YPG, yeast extract/peptone/glycerol.
b All units are in nanomoles of methylglyoxal/g wet cell weight.
Phosphate Supplementation Partially Complements the Growth Defect of Glx3 Null Cells in Glycerol
Inorganic phosphate (orthophosphate) plays a key role in the metabolism of triose phosphate sugars, and phosphate-rich trioses accumulate when yeasts are grown on glycerol as a carbon source (54). Adding 10 mm of supplemental potassium phosphate to the growth medium had no effect on the cell yields for wild-type C. albicans under any condition (Fig. 8, A–C) and only a slightly ameliorative effect for the glx3-Δ1/glx3-Δ1 mutant in YPD (Fig. 8B), but it doubled the cell yield for the glx3 null mutant in YPG (Fig. 8C). Because this additional inorganic phosphate has no effect on the yields of the wild-type cells, we conclude that YP medium is not limited in total phosphate. However, we found that the free orthophosphate concentration in this preparation of YP medium is only ∼0.7 mm using the ammonium molybdate/ascorbate assay (55), indicating that free phosphate levels are low. The selectively positive effect that addition of inorganic phosphate has on the glx3-Δ1/glx3-Δ1 mutant in glycerol suggests that supplemental orthophosphate decreases MG levels in the cell, possibly by favoring more rapid catabolism of the triose phosphates that generate MG.
DISCUSSION
Functionally diverse members of the DJ-1 superfamily are found throughout the evolutionary tree (52, 56), and this diversity allows us to take a comparative approach by examining the enzymatic and physiological functions of these proteins in the lower eukaryotes. The proteins of interest in this study belong to the Hsp31 clade of the DJ-1 superfamily, which is well represented in bacteria and lower eukaryotes but largely absent in plants and animals (52, 56). Among the fungi, C. albicans, a well studied human pathogen (57), has only one Hsp31 homolog (ORF 19.251), whereas the model yeast S. cerevisiae has four as follows: YDR533C (also called Hsp31) along with the three additional proteins YPL280W (Hsp32), YOR391C (Hsp33), and YMR322C (SNO4). The latter three proteins are 69% identical at the amino acid level to YDR533C and ∼99% identical to each other (20). In S. cerevisiae, Hsp31 has been knocked out, and the mutant is viable but with increased sensitivity to some oxidative stressors (19). E. coli Hsp31, a chaperone and the charter member of this clade, is 23% identical to YDR533C from S. cerevisiae and 22% identical to ORF 19.251 from C. albicans. Because E. coli Hsp31 was recently shown to have a glutathione-independent glyoxalase activity (16), we sought to determine whether this enzymatic activity was conserved and physiologically relevant in the eukaryotic members of the Hsp31 clade of the DJ-1 superfamily. The results of this work indicate that fungal Hsp31-like proteins are robust glyoxalases with physiologically relevant roles in detoxifying endogenously produced MG, particularly when glycerol is the principal carbon source.
Mechanism of the Glutathione-independent Glyoxalases
The Hsp31-like glutathione-independent glyoxalases possess a Glu-His-Cys catalytic triad, where the cysteine is highly conserved among nearly all DJ-1 superfamily members and is catalytically essential in diverse enzymes in the superfamily (58). From a mechanistic standpoint, the conserved cysteine in the glutathione-independent glyoxalase substitutes for the thiol of glutathione in glyoxalase I, producing a glyoxal-derived thiohemiacetal in Glx3 that obviates the need for glutathione and accounts for the absolute requirement for this conserved cysteine for glyoxalase III activity (16). Moreover, Glx3 enzymes use no cofactor to convert methylglyoxal to lactate, and the catalyzed reaction does not involve a redox component. These constraints are factored into a proposed general mechanism (Fig. 9) that is based on the currently accepted mechanism for glyoxalase I and a prior proposal for E. coli Hsp31 (16). In this mechanism, the conserved cysteine residue (Cys136 in Glx3) acts as the catalytic nucleophile, and a general base abstracts a proton from the first tetrahedral thiohemiacetal intermediate to form an enediolate intermediate. This enediolate intermediate was first proposed in the reaction mechanism of glyoxalase I (lactoylglutathione lyase) (59, 60) and facilitates the critical C1-C2 proton shift that is, in effect, a keto-enol tautomerization. Subedi et al. (16) proposed an enediolate intermediate for the glutathione-independent E. coli glyoxalase III, and more recently, Kwon et al. (61) have proposed an essentially identical mechanism for the more distantly related DJ-1 superfamily glutathione-independent glyoxalases from Arabidopsis thaliana. Therefore, we suggest that this may be a general mechanism for glutathione-independent glyoxalases, even those with significantly different structures. Unresolved aspects of the mechanism, however, are the identities of the general bases/acids involved. Previous work (16) and our structural and mutagenesis data suggest that the conserved residues Glu28, His106, and His137 are viable general acid/base candidates that deserve further scrutiny.
FIGURE 9.

Proposed enzymatic mechanism for Glx3. Electron flow is indicated with arrows, and the mechanism proceeds through the numbered steps. The proposed general acid (HA) and general base (B) are not yet identified, although His137 and Glu28 are reasonable candidates.
Close Homologs of Human DJ-1 Are Weaker Glyoxalases of Unclear Physiological Significance
A recent report indicates that human DJ-1 and its close homologs, Caenorhabditis elegans DJR-1.1 and DJR-1.2, also possess glyoxalase activity (62). This result is somewhat surprising, because these proteins are members of a distinct clade of the DJ-1 superfamily and possess active sites and dimeric structures that are substantially different from the Hsp31-like proteins. In particular, there is no histidine residue in human DJ-1, DJR-1.1, or DJR-1.2 that is structurally equivalent to the important His137 residue in CaGlx3. As expected from these major structural differences, we detect only weak methylglyoxalase activity for human DJ-1, although we find somewhat higher activity for its Sz. pombe homolog SPAC22E12.03c. We could not detect production of the expected product d-lactate by either protein. This could be because these enzymes generate l-lactate or some other product or that our assay is insufficiently sensitive to detect the small amounts of d-lactate produced by these less active DJ-1 homologs.
There are two methodological differences that may account for the discrepancies between our observations and previously reported results. First, we used only MG as a substrate, whereas Lee et al. (62) also used glyoxal, which appears to be a superior substrate for some of the DJ-1 proteins. Second, we performed our assays at 30 and 37 °C, which are standard physiological temperatures for C. albicans, whereas Lee et al. (62) performed their in vitro assays at 45 °C. We note, however, that Lee et al. (62) were also able to detect glutathione-independent glyoxalase activity in mouse and worm extracts at 22 and 37 °C but not in extracts prepared from DJ-1 knockouts (62). Although the specific reason for these differences is unclear, our results indicate that Hsp31-like proteins are considerably more active methylglyoxalases than human DJ-1. This conclusion is consistent with a recent report on the glyoxalase activities of several A. thaliana members of the DJ-1 superfamily, which found that the closest homologs of human DJ-1 in that organism (AtDJ-1A and AtDJ-1B) have only modest glyoxalase activity (61). Therefore, it remains to be established if the glyoxalase activities of human DJ-1 and its close homologs are physiologically relevant. In contrast, the glyoxalase activities of the Hsp31-like members of the DJ-1 superfamily are of clear physiological significance.
Functions of Hsp31-like Glx3 Proteins in Yeasts
A persistent question about MG metabolism is what, if any, purpose does this compound serve in the cell? In bacteria, one answer to this question is the MG bypass, which directs carbon flux from DHAP to pyruvate through MG and d-lactate intermediates and thus avoids the phosphate-intensive synthesis of 1,3-bisphosphoglycerate under phosphate-limiting conditions. In many eukaryotes, MG production from triose phosphates is primarily nonenzymatic, and thus no dedicated MG bypass exists. Therefore, eukaryotic glyoxalases may serve simply to detoxify MG that is unintentionally produced by metabolism. However, these glyoxalases may also serve a role in eukaryotes as a metabolic “spillway” for triose phosphate pools that accumulate under nonoptimal growth conditions, including when using glycerol as a primary carbon source. This MG spillway serves two ends. First, it can produce pyruvate from triose phosphates in a GAPDH-independent pathway that includes d-lactate dehydrogenase. Second, it ensures flux through glycolysis by preventing the inhibitory accumulation of large pools of triose phosphates. The accumulation of triose phosphates is a particular risk in inorganic phosphate-limited conditions where GAPDH activity (which requires inorganic phosphate as a substrate) may be compromised. Because this alternative pathway also sacrifices all of the ATP-synthesizing steps of glycolysis, it can only provide an energetic benefit for yeast growing aerobically, as MG-derived pyruvate can only feed into the TCA cycle and produce NADH to drive oxidative phosphorylation in the presence of oxygen. Therefore, in order for cells to use the EMP pathway, they must have evolved a way to address two related issues: avoiding the large scale accumulation of inhibitory triose phosphates and detoxifying the low levels of MG that arise even if the carbon flux and phosphate availability are sufficient to keep the triose phosphate levels low. These evolutionary constraints are both satisfied by glyoxalases such as Glx3, possibly accounting for its wide distribution and high degree of conservation in lower eukaryotes.
In this study, we have shown that the glutathione-independent glyoxalase Glx3 is an important component of central metabolism in C. albicans, especially when glycerol is the primary carbon source. C. albicans is particularly well suited to the study of endogenously produced MG because it does not switch between respiratory and fermentative growth in aerobic atmosphere as a function of carbon source (i.e. it is a Crabtree negative yeast). C. albicans also contains the glutathione-dependent Glo1/Glo2 glyoxalase system, and therefore we conclude that the glutathione-independent Glx3 enzyme studied here is functionally distinct and provides a growth advantage even when the Glo1/Glo2 system is functional. This is somewhat surprising, given that Glx3 is a significantly less efficient enzyme (kcat/Km = 1.4 × 103 m−1 s−1) than is Glo1 (kcat/Km = 1.2 × 107 m−1 s−1) (63). However, unlike Glx3, Glo1 also requires reduced glutathione as a co-substrate and has complex kinetics as a result (64). Therefore, the in vivo rates of MG detoxification by Glo1 and Glx3 may be less divergent than suggested by their differing catalytic efficiencies. Furthermore, Glx3 activity would be particularly valuable to the cell in conditions where the Glo1/Glo2 system is less effective due to diminished levels of reduced glutathione, such as chronic oxidative stress, stationary phase, or sulfur limitation. Importantly, the physiological relevance of Glx3 activity was directly demonstrated by the 3–5-fold elevation of intracellular MG levels in glx3 knock-out cells grown in YPD and YPG media compared with wild-type cells, confirming that Glx3 detoxifies MG in vivo.
As glycerol catabolism produces high levels of triose phosphates (54), our observation that growth of the glx3 null mutant is strongly impaired when glycerol is the primary carbon source is consistent with a spillway hypothesis for MG and Glx3 activity. This hypothesis was confirmed by the marked elevation in the intracellular MG level measured in glx3 knock-out cells grown in YPG media compared with those grown in YPD. These MG levels, which are higher than any other measured level in our experiments, are responsible for the strong growth defect in YPG media because both phenomena can be complemented by restoration of the wild-type GLX3 gene. Additionally, our observation that supplemental inorganic phosphate partially complements the growth defect of the glx3 null mutant only in glycerol medium suggests that accumulated triose phosphates generated by glycerol catabolism create a metabolic bottleneck that phosphate supplementation helps mitigate. A reduction in the steady-state triose phosphate pools would decrease the level of endogenously produced MG and increase viability for the Glx3 null cells, consistent with our observations. Finally, a role for Glx3 in both MG detoxification and as an auxiliary part of glycolysis is also consistent with the observation that Glx3 is up-regulated 6-fold when C. albicans is grown under hypoxic conditions. Synnott et al. (65) compared C. albicans cultures grown in YPD with 21% oxygen versus 1% oxygen and found increased expression for genes associated with glycolysis and ergosterol synthesis. At that time (65), CaGlx3 (ORF 19.251) was listed under “miscellaneous.” Recognition that ORF 19.251 has glyoxalase activity means that it too fits nicely under the hypoxia-regulated glycolysis umbrella.
Several proteins in the DJ-1 superfamily are noted for having multiple physiological functions, and it is possible that the glx3 null mutant will exhibit multiple phenotypes upon further study. The S. cerevisiae protein YDR533C, an exemplar of the eukaryotic members of this clade, suggests that multiple functions are plausible for many of the yeast Hsp31-like proteins. The transcription of YDR533C is up-regulated 10–30-fold during entry into stationary phase by hydrogen peroxide or diamide stress, by heat stress, and by growth in the presence of the proline analog azetidine-2-carboxylic acid, indicating that it plays an important role in oxidative and protein unfolding stress responses (20). Consistent with this hypothesis, a recent study showed that YDR533C mitigates oxidative stress in S. cerevisiae (19). Therefore, our future studies will examine the expression of GLX3 and Glx3p in wild-type C. albicans under stress conditions in conjunction with the stress responses of the glx3 null mutant.
Acknowledgments
We thank Drs. Audrey Atkins (University of Nebraska-Lincoln), Wayne Riekhof (University of Nebraska-Lincoln), and Todd Holyoak (University of Waterloo) for useful discussions. Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource, a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the United States Department of Energy Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the Department of Energy Office of Biological and Environmental Research and by National Institutes of Health Grant P41GM103393 from NIGMS.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 GM092999 (to M. A. W.). This work was also supported by Ann L. Kelsall and the Farnesol and Candida albicans Research Fund, University of Nebraska Foundation (to K. W. N.).
The atomic coordinates and structure factors (code 4LRU) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- MG
- methylglyoxal
- DHAP
- dihydroxyacetone phosphate
- DNPH
- 2,4-dinitrophenylhydrazine
- EMP
- Embden-Meyerhof-Parnas
- PDB
- Protein Data Bank
- YP
- yeast extract-peptone broth
- YPD
- yeast extract-peptone-dextrose broth
- YPG
- yeast extract-peptone-glycerol broth.
REFERENCES
- 1. Meyerhof O., Lohmann K. (1934) On the enzymatic equilibrium reaction between hexose diphosphate and dihydroxyacetone diphosphate. Biochem. Z. 271, 89–110 [Google Scholar]
- 2. Casazza J. P., Felver M. E., Veech R. L. (1984) The metabolism of acetone in Rat. J. Biol. Chem. 259, 231–236 [PubMed] [Google Scholar]
- 3. Green M. L., Lewis J. B. (1968) The oxidation of aminoacetone by a species of Arthrobacter. Biochem. J. 106, 267–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Richard J. P. (1991) Kinetic parameters for the elimination reaction catalyzed by triosephosphate isomerase and an estimation of the reaction's physiological significance. Biochemistry 30, 4581–4585 [DOI] [PubMed] [Google Scholar]
- 5. Martins A. M., Cordeiro C. A., Ponces Freire A. M. (2001) In situ analysis of methylglyoxal metabolism in Saccharomyces cerevisiae. FEBS Lett. 499, 41–44 [DOI] [PubMed] [Google Scholar]
- 6. Bucala R., Cerami A. (1992) Advanced glycosylation: chemistry, biology, and implications for diabetes and aging. Adv. Pharmacol. 23, 1–34 [DOI] [PubMed] [Google Scholar]
- 7. Ahmed M. U., Thorpe S. R., Baynes J. W. (1986) Identification of Nϵ-carboxymethyllysine as a degradation product of fructoselysine in glycated protein. J. Biol. Chem. 261, 4889–4894 [PubMed] [Google Scholar]
- 8. Dakin H. D., Dudley H. W. (1913) On glyoxalase. J. Biol. Chem. 14, 423–431 [Google Scholar]
- 9. Cooper R. A., Anderson A. (1970) The formation and catabolism of methylglyoxal during glycolysis in Escherichia coli. FEBS Lett. 11, 273–276 [DOI] [PubMed] [Google Scholar]
- 10. Ferguson G. P., Tötemeyer S., MacLean M. J., Booth I. R. (1998) Methylglyoxal production in bacteria: suicide or survival? Arch. Microbiol. 170, 209–218 [DOI] [PubMed] [Google Scholar]
- 11. Phillips S. A., Thornalley P. J. (1993) The formation of methylglyoxal from triose phosphates–Investigation using a specific assay for methylglyoxal. Eur. J. Biochem. 212, 101–105 [DOI] [PubMed] [Google Scholar]
- 12. Lyles G. A., Chalmers J. (1992) The metabolism of aminoacetone to methylglyoxal by semicarbazide-sensitive amine oxidase in human umbilical artery. Biochem. Pharmacol. 43, 1409–1414 [DOI] [PubMed] [Google Scholar]
- 13. Thornalley P. J. (2003) Glyoxalase I–Structure, function and a critical role in the enzymatic defence against glycation. Biochem. Soc. Trans. 31, 1343–1348 [DOI] [PubMed] [Google Scholar]
- 14. Vander Jagt D. L. (1993) Glyoxalase II: Molecular characteristics, kinetics and mechanism. Biochem. Soc. Trans. 21, 522–527 [DOI] [PubMed] [Google Scholar]
- 15. Misra K., Banerjee A. B., Ray S., Ray M. (1995) Glyoxalase III from Escherichia coli–A single novel enzyme for the conversion of methylglyoxal into d-lactate without reduced glutathione. Biochem. J. 305, 999–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Subedi K. P., Choi D., Kim I., Min B., Park C. (2011) Hsp31 of Escherichia coli K-12 is glyoxalase III. Mol. Microbiol. 81, 926–936 [DOI] [PubMed] [Google Scholar]
- 17. Mujacic M., Baneyx F. (2007) Chaperone Hsp31 contributes to acid resistance in stationary-phase Escherichia coli. Appl. Environ. Microbiol. 73, 1014–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sastry M. S., Korotkov K., Brodsky Y., Baneyx F. (2002) Hsp31, the Escherichia coli yedU gene product, is a molecular chaperone whose activity is inhibited by ATP at high temperatures. J. Biol. Chem. 277, 46026–46034 [DOI] [PubMed] [Google Scholar]
- 19. Skoneczna A., Miciałkiewicz A., Skoneczny M. (2007) Saccharomyces cerevisiae Hsp31p, a stress response protein conferring protection against reactive oxygen species. Free Radic. Biol. Med. 42, 1409–1420 [DOI] [PubMed] [Google Scholar]
- 20. Wilson M. A., St Amour C. V., Collins J. L., Ringe D., Petsko G. A. (2004) The 1.8 Å resolution crystal structure of YDR533Cp from Saccharomyces cerevisiae: a member of the DJ-1/ThiJ/PfpI superfamily. Proc. Natl. Acad. Sci. U.S.A. 101, 1531–1536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen X. J., Clark-Walker G. D. (2000) The petite mutation in yeasts: 50 years on. Int. Rev. Cytol. 194, 197–238 [DOI] [PubMed] [Google Scholar]
- 22. Carrió M. M., Villaverde A. (2001) Protein aggregation as bacterial inclusion bodies is reversible. FEBS Lett. 489, 29–33 [DOI] [PubMed] [Google Scholar]
- 23. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 24. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Collaborative Computational Project No. 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 26. Arnold K., Bordoli L., Kopp J., Schwede T. (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201 [DOI] [PubMed] [Google Scholar]
- 27. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 28. Schomaker V., Trueblood K. N. (1968) On the rigid-body motion of molecules in crystals. Acta Crystallogr. B24, 63–76 [Google Scholar]
- 29. Winn M. D., Isupov M. N., Murshudov G. N. (2001) Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. D Biol. Crystallogr. 57, 122–133 [DOI] [PubMed] [Google Scholar]
- 30. Emsley P., Cowtan K. (2004) Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 31. Davis I. W., Leaver-Fay A., Chen V. B., Block J. N., Kapral G. J., Wang X., Murray L. W., Arendall W. B., 3rd, Snoeyink J., Richardson J. S., Richardson D. C. (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF Chimera–A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 33. Cooper R. A. (1975) Methylglyoxal synthase. Methods Enzymol. 41, 502–508 [DOI] [PubMed] [Google Scholar]
- 34. Reuss O., Vik A., Kolter R., Morschhäuser J. (2004) The SAT1 flipper, an optimized tool for gene disruption in Candida albicans. Gene 341, 119–127 [DOI] [PubMed] [Google Scholar]
- 35. Scheijen J. L., Schalkwijk C. G. (2013) Quantification of glyoxal, methylglyoxal and 3-deoxyglucosone in blood and plasma by ultraperformance liquid chromatography tandem mass spectrometry: evaluation of blood specimen. Clin. Chem. Lab. Med. 10.1515/cclm-2012-0878 [DOI] [PubMed] [Google Scholar]
- 36. McLellan A. C., Phillips S. A., Thornalley P. J. (1992) The assay of methylglyoxal in biological systems by derivatization with 1,2-diamino-4,5-dimethoxybenzene. Anal. Biochem. 206, 17–23 [DOI] [PubMed] [Google Scholar]
- 37. Quigley P. M., Korotkov K., Baneyx F., Hol W. G. (2003) The 1.6 Å crystal structure of the class of chaperones represented by Escherichia coli Hsp31 reveals a putative catalytic triad. Proc. Natl. Acad. Sci. U.S.A. 100, 3137–3142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Guo P. C., Zhou Y. Y., Ma X. X., Li W. F. (2010) Structure of Hsp33/YOR391Cp from the yeast Saccharomyces cerevisiae. Acta Crystallogr. F Struct. Biol. Cryst. Commun. 66, 1557–1561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Graille M., Quevillon-Cheruel S., Leulliot N., Zhou C. Z., Li de la Sierra Gallay I., Jacquamet L., Ferrer J. L., Liger D., Poupon A., Janin J., van Tilbeurgh H. (2004) Crystal structure of the YDR533c S. cerevisiae protein, a class II member of the Hsp31 family. Structure 12, 839–847 [DOI] [PubMed] [Google Scholar]
- 40. Lee S. J., Kim S. J., Kim I. K., Ko J., Jeong C. S., Kim G. H., Park C., Kang S. O., Suh P. G., Lee H. S., Cha S. S. (2003) Crystal structures of human DJ-1 and Escherichia coli Hsp31, which share an evolutionarily conserved domain. J. Biol. Chem. 278, 44552–44559 [DOI] [PubMed] [Google Scholar]
- 41. Zhao Y., Liu D., Kaluarachchi W. D., Bellamy H. D., White M. A., Fox R. O. (2003) The crystal structure of Escherichia coli heat shock protein YedU reveals three potential catalytic active sites. Protein Sci. 12, 2303–2311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sastry M. S., Zhou W., Baneyx F. (2009) Integrity of N and C termini is important for E. coli Hsp31 chaperone activity. Protein Sci. 18, 1439–1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sastry M. S., Quigley P. M., Hol W. G., Baneyx F. (2004) The linker-loop region of Escherichia coli chaperone Hsp31 functions as a gate that modulates high affinity substrate binding at elevated temperatures. Proc. Natl. Acad. Sci. U.S.A. 101, 8587–8592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 45. Fioravanti E., Durá M. A., Lascoux D., Micossi E., Franzetti B., McSweeney S. (2008) Structure of the stress response protein DR1199 from Deinococcus radiodurans: a member of the DJ-1 superfamily. Biochemistry 47, 11581–11589 [DOI] [PubMed] [Google Scholar]
- 46. Lakshminarasimhan M., Madzelan P., Nan R., Milkovic N. M., Wilson M. A. (2010) Evolution of new enzymatic function by structural modulation of cysteine reactivity in Pseudomonas fluorescens isocyanide hydratase. J. Biol. Chem. 285, 29651–29661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Premkumar L., Dobaczewska M. K., Riedl S. J. (2011) Identification of an artificial peptide motif that binds and stabilizes reduced human DJ-1. J. Struct. Biol. 176, 414–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wilson M. A., Collins J. L., Hod Y., Ringe D., Petsko G. A. (2003) The 1.1 Å resolution crystal structure of DJ-1, the protein mutated in autosomal recessive early onset Parkinson's disease. Proc. Natl. Acad. Sci. U.S.A. 100, 9256–9261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wilson M. A., Ringe D., Petsko G. A. (2005) The atomic resolution crystal structure of the YajL (ThiJ) protein from Escherichia coli: A close prokaryotic homologue of the Parkinsonism-associated protein DJ-1. J. Mol. Biol. 353, 678–691 [DOI] [PubMed] [Google Scholar]
- 50. Witt A. C., Lakshminarasimhan M., Remington B. C., Hasim S., Pozharski E., Wilson M. A. (2008) Cysteine pKa depression by a protonated glutamic acid in human DJ-1. Biochemistry 47, 7430–7440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wilson M. A. (2011) The role of cysteine oxidation in DJ-1 function and dysfunction. Antioxid. Redox Signal. 15, 111–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lucas J. I., Marín I. (2007) A new evolutionary paradigm for the Parkinson disease gene DJ-1. Mol. Biol. Evol. 24, 551–561 [DOI] [PubMed] [Google Scholar]
- 53. Madzelan P., Labunska T., Wilson M. A. (2012) Influence of peptide dipoles and hydrogen bonds on reactive cysteine pKa values in fission yeast DJ-1. FEBS J. 279, 4111–4120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Penninckx M. J., Jaspers C. J., Legrain M. J. (1983) The glutathione-dependent glyoxalase pathway in the yeast Saccharomyces cerevisiae. J. Biol. Chem. 258, 6030–6036 [PubMed] [Google Scholar]
- 55. Chen P. S., Toribara T. Y., Warner H. (1956) Microdetermination of phosphorus. Anal. Chem. 28, 1756–1758 [Google Scholar]
- 56. Bandyopadhyay S., Cookson M. R. (2004) Evolutionary and functional relationships within the DJ1 superfamily. BMC Evol. Biol. 2004. February 19;4:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nickerson K. W., Atkin A. L., Hornby J. M. (2006) Quorum sensing in dimorphic fungi: farnesol and beyond. Appl. Environ. Microbiol. 72, 3805–3813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wei Y., Ringe D., Wilson M. A., Ondrechen M. J. (2007) Identification of functional subclasses in the DJ-1 superfamily proteins. PLoS Comput. Biol. 3, e10. [DOI] [PubMed] [Google Scholar]
- 59. Hall S. S., Doweyko A. M., Jordan F. (1976) Glyoxalase I enzyme studies. 2. Nuclear magnetic resonance evidence for an enediol-proton transfer mechanism. J. Am. Chem. Soc. 98, 7460–7461 [DOI] [PubMed] [Google Scholar]
- 60. Hall S. S., Doweyko A. M., Jordan F. (1978) Glyoxalase I enzyme studies. 4. General base catalyzed enediol proton transfer rearrangement of methyl- and phenylglyoxalglutathionylhemithiol acetal to S-lactoyl-glutathione and S-mandeloylglutathione followed by hydrolysis. A model for glyoxalase enzyme system. J. Am. Chem. Soc. 100, 5934–5939 [Google Scholar]
- 61. Kwon K., Choi D., Hyun J. K., Jung H. S., Baek K., Park C. (2013) Novel glyoxalases from Arabidopsis thaliana. FEBS J. 280, 3328–3339 [DOI] [PubMed] [Google Scholar]
- 62. Lee J. Y., Song J., Kwon K., Jang S., Kim C., Baek K., Kim J., Park C. (2012) Human DJ-1 and its homologs are novel glyoxalases. Hum. Mol. Genet. 21, 3215–3225 [DOI] [PubMed] [Google Scholar]
- 63. Clugston S. L., Barnard J. F., Kinach R., Miedema D., Ruman R., Daub E., Honek J. F. (1998) Overproduction and characterization of a dimeric non-zinc glyoxalase I from Escherichia coli: evidence for optimal activation by nickel ions. Biochemistry 37, 8754–8763 [DOI] [PubMed] [Google Scholar]
- 64. Lages N. F., Cordeiro C., Sousa Silva M., Ponces Freire A., Ferreira A. E. (2012) Optimization of time-course experiments for kinetic model discrimination. PLoS One 7, e32749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Synnott J. M., Guida A., Mulhern-Haughey S., Higgins D. G., Butler G. (2010) Regulation of the hypoxic response in Candida albicans. Eukaryot. Cell 9, 1734–1746 [DOI] [PMC free article] [PubMed] [Google Scholar]