

Background: The phospholipid cardiolipin undergoes acyl chain remodeling after biosynthesis, which has been hypothesized to optimize mitochondrial function.
Results: Δcld1 yeast, containing unremodeled cardiolipin, have no mitochondrial morphology or oxidative phosphorylation defects.
Conclusion: Cardiolipin remodeling is not required for optimal mitochondrial bioenergetic function in yeast.
Significance: Cardiolipin remodeling may be important for presently unknown mitochondrial processes and/or have unappreciated physiological functions.
Keywords: Bioenergetics/Electron Transfer Complex, Cardiolipin, Membrane Biogenesis, Membrane Lipids, Mitochondria, Mitochondrial Diseases, Phospholipid, Yeast
Abstract
After biosynthesis, an evolutionarily conserved acyl chain remodeling process generates a final highly homogeneous and yet tissue-specific molecular form of the mitochondrial lipid cardiolipin. Hence, cardiolipin molecules in different organisms, and even different tissues within the same organism, contain a distinct collection of attached acyl chains. This observation is the basis for the widely accepted paradigm that the acyl chain composition of cardiolipin is matched to the unique mitochondrial demands of a tissue. For this hypothesis to be correct, cardiolipin molecules with different acyl chain compositions should have distinct functional capacities, and cardiolipin that has been remodeled should promote cardiolipin-dependent mitochondrial processes better than its unremodeled form. However, functional disparities between different molecular forms of cardiolipin have never been established. Here, we interrogate this simple but crucial prediction utilizing the best available model to do so, Saccharomyces cerevisiae. Specifically, we compare the ability of unremodeled and remodeled cardiolipin, which differ markedly in their acyl chain composition, to support mitochondrial activities known to require cardiolipin. Surprisingly, defined changes in the acyl chain composition of cardiolipin do not alter either mitochondrial morphology or oxidative phosphorylation. Importantly, preventing cardiolipin remodeling initiation in yeast lacking TAZ1, an ortholog of the causative gene in Barth syndrome, ameliorates mitochondrial dysfunction. Thus, our data do not support the prevailing hypothesis that unremodeled cardiolipin is functionally distinct from remodeled cardiolipin, at least for the functions examined, suggesting alternative physiological roles for this conserved pathway.
Introduction
Cardiolipin (CL),4 a mitochondrial phospholipid with two phosphate headgroups and four acyl chains, is required for the optimal function of numerous mitochondrial processes, including oxidative phosphorylation (OXPHOS) (1–13), protein import (14–17), establishment of cristae morphology (18, 19), fission and fusion (20–22), and apoptosis (23, 24). Despite limited acyl chain specificity of the CL biosynthetic enzymes (25, 26), the acyl chain composition of CL within an organism or cell type displays a remarkable degree of homogeneity (27). This is achieved via acyl chain remodeling that is initiated by a lipase(s), generating monolyso-CL (MLCL, CL lacking one acyl chain), and completed by a transacylase or an acyltransferase that reacylates MLCL (28).
Mutations in the MLCL transacylase tafazzin cause Barth syndrome, resulting in cardiac and skeletal myopathy, cyclic neutropenia, and respiratory chain defects (29–31). In Barth syndrome patients and models of Barth syndrome, CL levels are decreased, MLCL accumulates, and the remaining CL contains an altered acyl chain composition (32–37), although which lipid alteration either individually or in combination leads to mitochondrial dysfunction has not been thoroughly investigated.
Although the pathophysiological importance of defective CL remodeling is firmly established, the physiological importance of this pathway represents a largely unaddressed issue that is preventing a comprehensive molecular understanding of how this evolutionarily conserved and clinically relevant process promotes mitochondrial function. The most widely accepted hypothesis is based on the highly intriguing observation that the final homogeneous molecular form of CL varies between organisms or even between tissues within the same organism (27, 38). As such, it is postulated that CL molecules with different acyl chain compositions are functionally distinct and that the molecular form of CL specifically fits the demands of its host cell (38–41). However, as of yet, this provocative hypothesis has not been directly tested in any model organism.
At present, yeast are not merely the best but also the only model available capable of directly comparing the functionality of distinct molecular forms of CL in otherwise isogenic cells. Three CL remodeling pathways have been identified in higher eukaryotes, although their relative contribution to establishing the final molecular form of CL is unclear (28, 42). In contrast, yeast only undergo tafazzin-mediated CL remodeling. Recently, the phospholipase that initiates CL remodeling was identified in yeast as Cld1p (43). Cld1p has no homolog in higher eukaryotes; however, its function, the removal of an acyl chain from CL to form MLCL, is conserved. A similar phenomenon is seen with the phosphatidylglycerophosphate phosphatase in CL biosynthesis; the yeast (Gep4p) and mammalian (PTPMT1) enzymes are phylogenetically unrelated despite catalyzing the same reaction (44, 45). A calcium-independent phospholipase A2 has been identified as a CL phospholipase in flies (iPLA2-VIA) and mammals (iPLA2-γ) (46–48). However, murine iPLA2-γ is not the lipase that provides the substrate MLCL utilized by tafazzin (49). Consequently, the exact role of these lipases in CL remodeling is unclear. Therefore, in higher eukaryotes not only are there multiple potential CL remodeling pathways but, additionally, a complete inventory of all of the involved players has not been established. As such, it is currently not possible to compare the functionality of distinct molecular forms of CL in metazoans.
In contrast, Cld1p localizes exclusively to mitochondria and is the only lipase that initiates tafazzin-mediated CL remodeling in yeast (43, 50). Here, we utilized Δcld1 yeast to determine whether cardiolipin molecules with different acyl chain compositions, in this case unremodeled versus remodeled cardiolipin, have distinct functional capacities, a central prediction of the prevailing hypothesis. Unexpectedly, unremodeled CL functioned as well as remodeled CL in maintaining mitochondrial morphology and promoting OXPHOS. Furthermore, mutating CLD1, and thus preventing the initiation of CL remodeling, was able to suppress the defects of Δtaz1 yeast. Thus, we conclude that in yeast, unremodeled CL can support known CL-dependent mitochondrial functions as well as remodeled CL.
EXPERIMENTAL PROCEDURES
Yeast Strains and Growth Conditions
All yeast strains used in this study were isogenic to GA74-1A (MATa, his3-11,15, leu2, ura3, trp1, ade8 [rho+, mit+]), except where indicated, and have been described previously (12, 50–52), except for Δcrd1Δcld1, which was generated by replacing the entire open reading frame of CLD1 with HIS3MX6 and CRD1 with TRP1 (53). Strains derived from W303 (MATa, ade2-1, ura3-1,15, his3-11, trp1-1, can1-100 [rho+, mit+]) were generated by replacing CRD1 with TRP1, CLD1 with HIS3MX6, and/or TAZ1 with URA3MX. Strains derived from PTY144 (MATα, leu2-3,112, ura3-52, trp1-Δ1, lys2, his3::hisg [rho+, LYS2]) (54) were generated by replacing CRD1, CLD1, or TAZ1 with HIS3MX6, except for Δcld1Δtaz1, in which CLD1 was replaced with HIS3MX6 and TAZ1 with URA3MX. Yeast were grown in rich lactate media (1% yeast extract, 2% tryptone, 0.05% dextrose, 2% lactic acid, 3.4 mm CaCl2·2H2O, 8.5 mm NaCl, 2.95 mm MgCl2·6H2O, 7.35 mm KH2PO4, 18.7 mm NH4Cl, pH 5.5), except for growth analysis where overnight YPD (1% yeast extract, 2% peptone, 2% dextrose) cultures were spotted on synthetic media (0.17% yeast nitrogen base, 0.5% ammonium sulfate, 0.2% complete amino acid mix with either 2% dextrose or 3% glycerol, 1% ethanol). Genetic knock-outs constructed for this study were generated by replacing the entire open reading frame of the gene using PCR-mediated gene replacement (53).
Multidimensional Mass Spectrometry-based Shotgun Lipidomic Analysis of Mitochondrial Lipids
A modified Bligh and Dyer procedure was used to extract lipids from each yeast mitochondrial preparation. Each lipid extract was reconstituted with a volume of 200 μl/mg mitochondrial protein in chloroform/methanol (1:1; v/v). Internal standards for quantification of individual molecular species of lipid classes were added prior to lipid extraction (55). Shotgun lipidomics analyses were performed with a QqQ mass spectrometer (Thermo Fisher Scientific TSQ Vantage, San Jose, CA) equipped with an automated nanospray device (Triversa Nanomate, Advion Biosciences, Ithaca, NY) and operated with Xcalibur software as described previously (56). Identification and quantification of lipid molecular species were performed using an automated software program as described previously (57).
Electron Microscopy
Cells were harvested and fixed in 3% glutaraldehyde contained in 0.1 m sodium cacodylate, pH 7.4, 5 mm CaCl2, 5 mm MgCl2, and 2.5% (w/v) sucrose for 1 h at room temperature with gentle agitation, spheroplasted, embedded in 2% ultra low temperature agarose (prepared in water), cooled, and subsequently cut into small pieces (∼1 mm3). The cells were then post-fixed in 1% OsO4, 1% potassium ferrocyanide contained in 0.1 m sodium cacodylate, 5 mm CaCl2, pH 7.4, for 30 min at room temperature. The blocks were washed thoroughly four times with double distilled H2O, 10 min total, transferred to 1% thiocarbohydrazide at room temperature for 3 min, washed in double distilled H2O (four times, 1 min each), and transferred to 1% OsO4, 1% potassium ferrocyanide in 0.1 m sodium cacodylate, pH 7.4, for an additional 3 min at room temperature. The cells were washed four times with double distilled H2O (15 min total), stained en bloc in Kellenberger's uranyl acetate for 2 h to overnight, dehydrated through a graded series of ethanol, and subsequently embedded in Spurr resin. Sections were cut on a Reichert Ultracut T ultramicrotome, post-stained with uranyl acetate and lead citrate, and observed on an FEI Tecnai 12 transmission electron microscope at 100 kV. Images were recorded with a Soft Imaging System Megaview III digital camera, and figures were assembled in Adobe Photoshop with only linear adjustments in contrast and brightness.
Assessment of Δψm
The lipophilic cationic dye tetramethylrhodamine methyl ester (TMRM, Molecular Probes), which accumulates in mitochondria in accordance with a Nernstian distribution, was used in quench mode. 2-ml samples of mitochondria (0.1 mg of mitochondrial protein/ml) in measurement buffer (MB: 20 mm Tris-HCl, pH 7.2, 20 mm KCl, 3 mm MgCl2, 4 mm KH2PO4, and 250 mm sucrose) containing 50 nm TMRM (from DMSO stocks, final DMSO concentration 1.0% (v/v)) were added to stirred cuvettes. TMRM emission (λex 547 nm; λem 570 nm; slits at 4 nm) was measured over a time course that included the successive addition of the following: (i) respiratory substrate (2 mm NADH) at 100 s; (ii) 45 μm ADP, pH 7.5, at 300 and 700 s, and (iii) 2.5 μm valinomycin at 1000 s to completely dissipate the potential. The relative measure of Δψm was based on the difference in fluorescence intensity (ΔF) prior to respiratory substrate addition and after establishment of the maximal Δψm. The time dependence of return to state 4 respiration following the initiation of a phosphorylation cycle was calculated graphically (Kaleidagraph) as the time between ADP addition and stable re-establishment of the maximal Δψm. Carboxyatractyloside and oligomycin, each at final concentration of 10 μm, were incubated with mitochondria at 4 °C for 5 min either separately or together. After incubation, TMRM emission was measured over a time course as described above.
Complex III and Complex IV Activity Measurements
Complex III and IV activities were measured as described (58) with a few modifications. To measure complex III activity, 1–25 μg of mitochondria solubilized in 0.5% (w/v) n-dodecyl β-d-maltoside were added to reaction buffer (50 mm KPi, 2 mm EDTA, pH 7.4) with 0.008% (w/v) horse heart cytochrome c and 1 mm KCN. The reaction was started by adding 100 μm decylubiquinol, and the reduction of cytochrome c followed at 550 nm. Complex IV activity was measured by adding mitochondrial extracts to reaction buffer with 0.008% (w/v) ferrocytochrome c and following cytochrome c oxidation at 550 nm.
Antibodies
Most antibodies used in this study were generated in our laboratory or in the J. Schatz (University of Basel, Basel, Switzerland) or C. Koehler (UCLA) laboratories and have been described previously (18, 36, 50, 59–63). Other antibodies used were mouse anti-Aac2p clone 6H8 (64) and horseradish peroxidase (Thermo Fisher Scientific) or fluorescent (Pierce)-conjugated secondary antibodies.
Miscellaneous
Isolation of mitochondria, preparation of yeast cell extracts, blue native-PAGE, mitochondrial respiration, phospholipid analysis, and immunoblotting were performed as described previously (12, 18, 52). Statistical comparisons were performed by one-way analysis of variance compared with wild type using SigmaPlot 11 software (Systat software, San Jose, CA). All graphs show the mean ± S.E.
RESULTS
CLD1 Functions Upstream of TAZ1 in CL Remodeling
The initial characterization of CLD1 revealed that Δcld1 and Δcld1Δtaz1 yeast contained identical mitochondrial phospholipid profiles (43), indicating that CLD1 is epistatic to TAZ1 (the yeast homolog of tafazzin) in the same pathway. In contrast, growth on respiratory media, where ethanol and glycerol are the only available carbon sources thus requiring ATP generated by OXPHOS, suggested that CLD1 functions in a pathway parallel to, or distinct from, TAZ1 (43).
In an attempt to resolve this, we analyzed CL biosynthesis and remodeling mutants in multiple independent yeast strains. As expected, in mitochondria isolated from yeast lacking cardiolipin synthase (Δcrd1), CL was absent and its precursor phosphatidylglycerol accumulated (Fig. 1). Importantly, the same phospholipid profile was seen in the double mutant, Δcrd1Δcld1, indicating that CLD1 functions after CRD1, in one pathway. The Δcld1 mutant displayed a lipid profile similar to that of wild type, whereas Δtaz1 resulted in a reduction of CL and an accumulation of MLCL. The double mutant Δcld1Δtaz1 phenocopied Δcld1 and wild type. Thus, in agreement with previous studies (43, 50), analysis of mitochondrial phospholipids indicates that Cld1p functions upstream of Taz1p and downstream of Crd1p in the CL remodeling pathway.
FIGURE 1.

CLD1 is epistatic to TAZ1 in the CL remodeling pathway by mitochondrial phospholipid analysis. A, mitochondrial phospholipids from the indicated strains derived from GA74-1A were labeled with 32Pi and separated by TLC. B, quantification of A. n = 5–6. ***, p < 0.001. Mitochondrial phospholipids from the indicated strains derived from C PTY144 and D W303 were analyzed as in A. Whole cell extracts from the indicated strains derived from E PTY144 and F W303 were immunoblotted. * indicates a nonspecific cross-reaction of the Cld1p antisera. PA, phosphatidic acid; PE, phosphatidylethanolamine; PG, phosphatidylglycerol; PS, phosphatidylserine; PI, phosphatidylinositol; PC, phosphatidylcholine.
The placement of CLD1 between CRD1 and TAZ1 was also analyzed by growth on respiratory media (Fig. 2). Δcrd1 yeast displayed a growth defect, and both Δcrd1Δcld1 and Δcrd1Δtaz1 mimicked this phenotype, confirming that CRD1 is upstream of both CLD1 and TAZ1. Δtaz1 yeast also displayed a growth defect on respiratory media but only at elevated temperature. Consistent with Beranek et al. (43), Δcld1 yeast grew as well as wild type, but in contrast to their results, our analysis showed that Δcld1Δtaz1 did not exhibit a growth defect and phenocopied Δcld1. To rule out strain-specific differences, this epistasis analysis was confirmed in two additional genetic backgrounds (Figs. 1 and 2). Therefore, Cld1p functions upstream of Taz1p in a single biochemical pathway.
FIGURE 2.
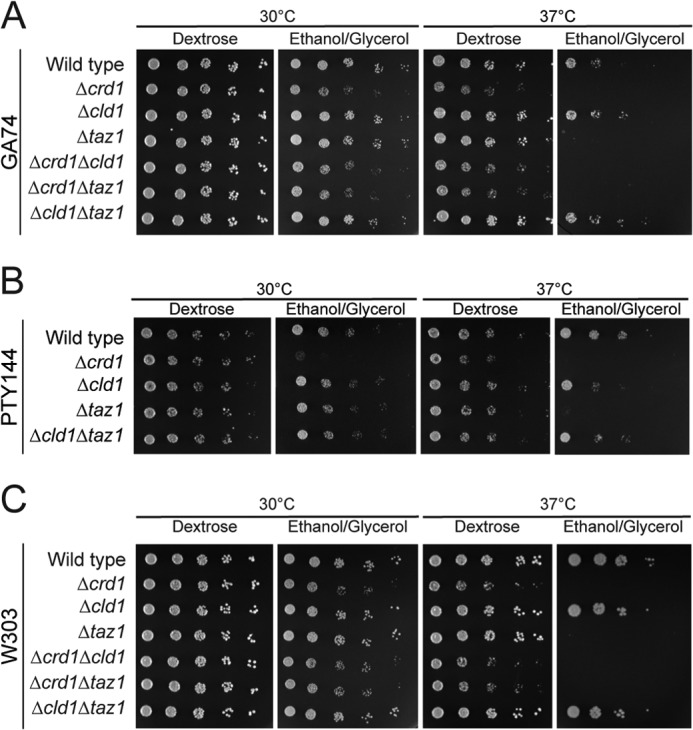
CLD1 is epistatic to TAZ1 in the CL remodeling pathway by respiratory growth analysis. 1:4 serial dilutions of the indicated strains derived from GA74-1A (A), PTY144 (B), or W303 (C) were spotted on the indicated media and incubated at 30 or 37 °C.
Δcld1 Mitochondria Contain Unremodeled CL
CL from Δcld1 yeast was previously shown by GC/MS to contain more C16:0 fatty acyl chains than wild type at the expense of C18:1 and C16:1 (43). This is consistent with the presence of unremodeled CL in Δcld1 mitochondria and prompted us to analyze more comprehensively the acyl chain composition of CL in remodeling mutants by shotgun lipidomics (Fig. 3A and supplemental Table S1). The acyl chain composition of CL from Δcld1 or Δtaz1 mitochondria was clearly altered compared with wild type, and the CL species that accumulated in Δcld1 were similar to that of Δcld1Δtaz1, consistent with CLD1 functioning upstream of TAZ1.
FIGURE 3.

Δcld1 contains unremodeled CL. A, acyl chain composition of CL was determined by multidimensional mass spectrometric array analysis. n = 3. B, CL was categorized by the number of saturated acyl chains and expressed as a % of the total CL. C, quantification of the total amount of CL per mg of protein. D, CL was categorized by the number of saturated acyl chains and expressed as the amount of CL per mg of protein. E, acyl chain composition of MLCL was determined by multidimensional mass spectrometric array analysis. n = 3. Statistical analysis is provided in supplemental Table S1.
Unremodeled CL in yeast is characterized in part by saturated acyl chains of increased heterogeneity. To quantify this, the molecular species of CL from each strain was categorized by the number of saturated acyl chains and expressed as the percent of total CL (Fig. 3B). CL from wild type mitochondria contained mostly unsaturated fatty acyl chains; only 8% contained one saturated acyl chain although the remaining 92% of CL contained no saturated acyl chains. In contrast, only 20% of CL from Δcld1 contained no unsaturated acyl chains, whereas 51 and 28% of CL contained one or two saturated acyl chains, respectively.
Interestingly, the presence of CL with no saturated acyl chains in both Δcld1 and Δtaz1 mitochondria suggests that either an alternative (albeit minor) CL remodeling pathway exists or instead that a subpopulation of newly synthesized CL already contains four unsaturated acyl chains. Although the relative amounts of mature (e.g. a remodeled-like acyl chain composition) CL vary between Δcld1 and Δtaz1 mitochondria, the absolute amounts are similar (Fig. 3, C and D), implying that Cld1p is able to specifically deacylate unremodeled CL. This is further supported by the molecular species of MLCL present in Δtaz1; although 28% of CL in Δcld1 contains two saturated acyl chains, none of the molecular forms of MLCL in Δtaz1 contained two saturated acyl chains (Fig. 3E and supplemental Table S1), suggesting that Cld1p preferentially removes saturated acyl chains from CL.
Thus, we have provided the most extensive analysis to date of the CL acyl chain composition in CL remodeling mutants. CL molecules from Δcld1 yeast contain more saturated acyl chains than wild type, consistent with unremodeled CL. Accordingly, Δcld1 is a genetic tool to determine whether CL molecules with different acyl chain compositions are functionally distinct in a strain that is otherwise isogenic to wild type yeast.
CL Remodeling Is Not Required to Maintain Mitochondrial Morphology
Altered mitochondrial morphology has been observed in Δcrd1 and Δtaz1 yeast (19, 52). To determine what role, if any, CL remodeling plays in the establishment and/or maintenance of mitochondrial morphology, CL remodeling mutants were analyzed by EM (Fig. 4).
FIGURE 4.

Mitochondrial morphology is not affected by unremodeled CL. Mitochondria from the indicated strains derived from GA74 (A–E) and W303 (F–J) yeast were analyzed by transmission electron microscope. A and F, representative micrographs from the indicated strains. m, mitochondria; n, the nucleus. Bars, 0.5 μm. B and G, quantification of cristae length per mitochondrion. C and H, quantification of outer membrane length per mitochondrion. D and I, quantification of aberrant mitochondria for each strain, defined as the appearance of exaggerated cristae >0.5 μm in length. The number of mitochondria analyzed is indicated for each strain. E and J, examples of mitochondria with exaggerated cristae. Bars, 0.5 μm. *, p < 0.05; **, p ≤ 0.01; ***, p ≤ 0.001.
No overt morphological differences were observed between mitochondria in wild type and Δcld1 yeast (Fig. 4A). Surprisingly, the morphology of mitochondria in Δcrd1 and Δtaz1 also appeared unaffected, although measurement of mitochondrial membranes indicated that both mutants contained longer cristae membranes than wild type or Δcld1 (Fig. 4, B and C). Additionally, no difference in the number of aberrant mitochondria, which display exaggerated cristae, was observed between wild type and any CL remodeling mutant (Fig. 4, D and E).
To confirm these results, mitochondria from CL remodeling mutants derived from the W303 genetic background were also analyzed (Fig. 4, F–J). Although no remarkable morphological defects were observed, mitochondria from Δcrd1 and Δcld1 (but not Δtaz1) yeast displayed longer cristae membranes than wild type, and all of the mutants displayed longer outer membranes than wild type (Fig. 4, G and H). Additionally, in the W303 background, no aberrant mitochondria were observed in wild type (Fig. 4, I and J), unlike the mutants that contained a small fraction of aberrant mitochondria.
The mitochondrial morphology in Δcrd1, Δcld1, and Δtaz1 yeast remained largely unperturbed, although subtle differences were noted. Importantly, studies that previously reported abnormal mitochondrial morphology in Δcrd1 and Δtaz1 yeast never reported the penetrance of the observed defects (19, 52). Furthermore, our results indicate that the genetic background contributes to mitochondrial morphology. Thus, we conclude that there is not a general morphological phenotype in mitochondria lacking either remodeled CL or CL entirely.
CL Remodeling Is Not Required for Optimal OXPHOS Function
CL is required for the optimal function of respiratory complexes (1–4, 6–8), as well as for the stability of respiratory supercomplexes (9–12, 28). Respiratory supercomplexes, which in yeast consist of two copies of complex III and either one (III2IV) or two (III2IV2) copies of complex IV, increase the efficiency of electron flux through the electron transport chain via substrate channeling (65, 66). Thus, we used Δcld1 to determine the ability of unremodeled CL to support OXPHOS.
In Δcrd1 (and Δcrd1Δcld1) mitochondria, respiratory supercomplexes were destabilized due to the absence of CL, as seen by the decreased abundance of the III2IV2 supercomplex and the resultant increase in the III2IV supercomplex, as well as the liberated complex III dimer and free complex IV (Fig. 5A). Additionally, the ADP/ATP carrier (Aac2p) did not assemble into higher molecular weight complexes, including with respiratory complexes, in the absence of CL (12). In Δtaz1, respiratory supercomplex stability was not affected, although the association of Aac2p with the supercomplex was diminished. In Δcld1, however, the stability of respiratory supercomplexes, including those containing Aac2p, was preserved, indicating that the acyl chain composition of CL does not affect respiratory supercomplex stability.
FIGURE 5.

OXPHOS function is not affected by unremodeled CL. A, mitochondria were solubilized with digitonin, separated by blue native-PAGE (top panels), and immunoblotted for Cyt1p (complex III), Cox2p (complex IV), Atp2p (complex V), and Aac2p. Bottom panels are immunoblots following SDS-PAGE, which serve as loading controls. B and C, respiration measured in the presence of 2 mm NADH. n = 6–9. B, phosphate/oxygen ratios. C, respiratory control ratios. D, representative TMRM time traces of mitochondria isolated from the indicated yeast strains following the addition of 2 mm NADH (to establish the Δψm) and two sequential additions of 45 μm ADP to induce phosphorylation cycles, manifest as transient depolarizations. E, average times required for the re-establishment of maximal (state 4) Δψm following ADP addition for the yeast strains indicated. *, p < 0.05; ***, p < 0.001.
We further investigated the role of the molecular form of CL in OXPHOS by measuring the rate of O2 consumption in isolated mitochondria. The ratio of ADP consumed per oxygen reduced (phosphate/oxygen ratio, a measure of OXPHOS efficiency) in Δcld1 mitochondria was indistinguishable from wild type, but it was decreased in Δcrd1, Δcrd1Δcld1, and Δtaz1 mitochondria (Fig. 5B). Likewise, no change in the respiratory control ratio (a measure of OXPHOS coupling) was observed in Δcld1 compared with wild type (Fig. 5C). Notably, the OXPHOS defects observed in Δtaz1 mitochondria were suppressed after the additional deletion of CLD1.
As an alternative method to measure OXPHOS function, we tracked the membrane potential (Δψm) of isolated mitochondria using the potentiometric fluorescent probe TMRM (Fig. 5D). Δψm was established via NADH addition, and state 3 respiration was induced by adding ADP, which caused a transient depolarization due to the utilization of the proton gradient to drive ADP/ATP transport by Aac2p and ATP production by complex V. After the ADP was consumed, the inner membrane repolarized and state 4 respiration resumed. Δcld1 and Δcld1Δtaz1 mitochondria were able to repolarize at rates identical to wild type, whereas Δcrd1, Δcrd1Δcld1, and Δtaz1 mitochondria repolarized more slowly (Fig. 5E). Taken together, these results indicate that OXPHOS coupling is not dependent on the acyl chain composition of CL that is generated by tafazzin-mediated remodeling.
Interestingly, the individual state 3 and state 4 respiration rates in Δcrd1 and Δtaz1 mitochondria were higher than in wild type (Fig. 6A), which is consistent with some reports but different from others (1, 2, 12, 14, 67). This observation potentially could be due to a lower steady-state Δψm for mitochondria from these strains which, due to respiratory control, would result in higher respiration rates (e.g. easier to pump protons against a lower electrochemical potential); but relative Δψm measurements in Δcrd1 and Δcrd1Δcld1 mitochondria were not significantly different from wild type (Fig. 6B). However, TMRM time traces revealed that immediately after establishing the Δψm, Δcrd1, Δcrd1Δcld1, and Δtaz1 mitochondria began to depolarize, whereas those from the other strains maintained a high Δψm (Fig. 5D). This depolarization may originate from breaches in the inner membrane permeability barrier that, although not large enough to resolve on measurements of relative steady-state Δψm (Fig. 6B), could be resolved on individual traces (Fig. 5D). To identify the source of the putative proton leak, we tested the effects of inhibiting two key OXPHOS components, complex V using oligomycin and Aac2p using carboxyatractyloside (Fig. 6C), both of which create regulated aqueous conduits in the membrane and require CL for assembly (12, 13). The lack of transient depolarization after ADP addition confirmed the efficacy of both oligomycin and carboxyatractyloside. Interestingly, both inhibitors curtailed the immediate depolarization in Δcrd1, Δcrd1Δcld1, and Δtaz1 mitochondria, suggesting that in these strains the time-dependent decrease in Δψm was mediated by proton leak through complex V and Aac2p.
FIGURE 6.

Mitochondrial proton leak is increased in the absence of CL. A, respiration of isolated mitochondria measured in the presence of NADH (state 4) or NADH and ADP (state 3). ***, p < 0.001. B, relative membrane potentials from five independent experiments were plotted (circles). The mean ± S.E. are displayed as black bars. C, membrane potential of the indicated strains measured in the presence of oligomycin (left), carboxyatractyloside (middle), or oligomycin + carboxyatractyloside (right).
When respiration was analyzed under uncoupled conditions to measure maximum electron transport capacity, a small but significant decrease in mitochondria lacking CRD1 was measured, but no decrease was seen in Δtaz1 or Δcld1 (Fig. 7A). Measurement of individual complex III and complex IV activities revealed that the defect was specific to complex III (Fig. 7, B and C), consistent with CL participating in its catalytic mechanism (68). Furthermore, the steady-state abundance of respiratory complex subunits (as well as other mitochondrial proteins) was not affected in any mutant (Fig. 7D). Thus, unremodeled and remodeled CL, which differ significantly in their acyl chain composition, have the same capacity to promote the expression, assembly, and activity of the OXPHOS system.
FIGURE 7.

Individual components of OXPHOS are not affected by unremodeled CL. A, uncoupled respiration measured in the presence of 2 mm NADH and 10 μm CCCP. B, complex III; C, complex IV activity, measured in n-dodecyl β-d-maltoside mitochondrial extracts. **, p < 0.01. D, mitochondrial proteins from the indicated strains were separated by SDS-PAGE and immunoblotted. * indicates a nonspecific cross-reaction of the Cld1p antisera.
DISCUSSION
Despite the pervasiveness of the hypothesis that CL remodeling establishes a molecular form of CL that is optimized to support mitochondrial function, direct evidence for this proposition is lacking. Using Δcld1 yeast, which cannot initiate CL remodeling, we have provided the most comprehensive comparison to date of the intrinsic functional capacity of distinct molecular forms of CL, remodeled versus unremodeled CL, in otherwise isogenic cells. Our data indicate that in yeast unremodeled and remodeled CL are equally able to maintain mitochondrial morphology and promote OXPHOS and are thus at variance with the prevailing model that CL remodeling is critical for mitochondrial function. Still, it is possible that the acyl chain composition in mammals plays a larger role in controlling OXPHOS function than in yeast and that this capacity has been a relatively recent addition to the functionality of this remodeling pathway. Consistent with this possibility is that cardiolipin remodeling attributed to acyl-CoA:lysocardiolipin acyltransferase-1, which localizes to the endoplasmic reticulum, is associated with mitochondrial dysfunction (69–71). Alternatively, mitochondrial processes other than OXPHOS that are presently not known and thus not interrogated in this study, may be dependent on a specific CL acyl chain composition.
Our results, however, suggest that CL remodeling evolved to achieve other biological outcomes instead of simply establishing a tissue-specific molecular form of CL. The ability to remodel CL acyl chains may be more important than the establishment of a specific molecular form. CL is susceptible to oxidative damage due to its tight association with respiratory complexes, the major sites of reactive oxygen species production in a cell (72). Thus, CL remodeling may be used as a repair mechanism that removes and replaces damaged acyl chains, restoring OXPHOS capacity (50, 73). Indeed, increased oxidative damage is observed in Δtaz1 yeast and Barth syndrome lymphoblasts (74, 75). Why then, do most tissues/organisms contain only a few molecular forms of CL? Tafazzin has no acyl chain specificity (76). Thus, the acyl chain composition of remodeled CL may instead reflect the acyl chain composition of the surrounding lipids in the microenvironment containing tafazzin (77). Additionally, when compared with Δcld1, the acyl chain composition of CL in Δtaz1 suggests that saturated acyl chains are the preferred substrate of Cld1p. As such, the specificity of the lipase may also contribute to the final molecular form of CL in a given tissue/cell (78).
These results have important implications regarding the pathological causes of Barth syndrome. Great emphasis has been placed on the altered CL acyl chain composition, but Barth syndrome patients (and models) also exhibit decreased levels of CL with concurrent increases in MLCL (32–37). Our data suggest that the absolute levels of lipids (either decreased CL or increased MLCL) and/or the absence of an active remodeling pathway may exert a larger role in contributing to the disease state than simple changes in the final acyl chain composition. These conclusions have therapeutic implications. For instance, if alterations in the levels of CL and MLCL are the major drivers of mitochondrial dysfunction, then therapies promoting the accumulation of CL and/or depletion of MLCL may alleviate the symptoms of Barth syndrome. An obvious target to inhibit is the lipase that initiates CL remodeling, as we have shown that the Δcld1Δtaz1 yeast strain phenocopies wild type. Interestingly, Barth syndrome patient lymphoblasts treated with the iPLA2 inhibitor bromoenol lactone partially corrects the MLCL/CL ratio, as does knocking out iPLA2 in TAZ−/− flies (47). However, this strategy is currently hampered in patients because the relevant lipase(s) that functions upstream of tafazzin has not been molecularly identified (49). Thus, further investigation into the basic biology of CL remodeling is required before plausible treatments can be realized.
Acknowledgments
We thank Drs. Jeff Schatz and Carla Koehler for antibodies and Dr. Peter Thorsness for the PTY144 strain.
This work was supported, in whole or in part, by National Institutes of Health Grants R00HL089185 and 1R01HL108882 (to S. M. C.) and Grant SRR022588A from NCRR (to J. M. M.). This work was also supported by National Science Foundation Grant MCB-1024908 (to N. N. A.) and intramural institutional research funds (to X. H.).

This article contains supplemental Table 1.
- CL
- cardiolipin
- Δψm
- mitochondrial membrane potential
- MLCL
- monolysocardiolipin
- OXPHOS
- oxidative phosphorylation
- TMRM
- tetramethylrhodamine methyl ester
- iPLA2
- calcium-independent phospholipase A2.
REFERENCES
- 1. Koshkin V., Greenberg M. L. (2000) Oxidative phosphorylation in cardiolipin-lacking yeast mitochondria. Biochem. J. 347, 687–691 [PMC free article] [PubMed] [Google Scholar]
- 2. Koshkin V., Greenberg M. L. (2002) Cardiolipin prevents rate-dependent uncoupling and provides osmotic stability in yeast mitochondria. Biochem. J. 364, 317–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fry M., Green D. E. (1980) Cardiolipin requirement by cytochrome oxidase and the catalytic role of phospholipid. Biochem. Biophys. Res. Commun. 93, 1238–1246 [DOI] [PubMed] [Google Scholar]
- 4. Fry M., Green D. E. (1981) Cardiolipin requirement for electron transfer in complex I and III of the mitochondrial respiratory chain. J. Biol. Chem. 256, 1874–1880 [PubMed] [Google Scholar]
- 5. Eble K. S., Coleman W. B., Hantgan R. R., Cunningham C. C. (1990) Tightly associated cardiolipin in the bovine heart mitochondrial ATP synthase as analyzed by 31P nuclear magnetic resonance spectroscopy. J. Biol. Chem. 265, 19434–19440 [PubMed] [Google Scholar]
- 6. Gomez B., Jr., Robinson N. C. (1999) Phospholipase digestion of bound cardiolipin reversibly inactivates bovine cytochrome bc1. Biochemistry 38, 9031–9038 [DOI] [PubMed] [Google Scholar]
- 7. Sedlák E., Robinson N. C. (1999) Phospholipase A2 digestion of cardiolipin bound to bovine cytochrome c oxidase alters both activity and quaternary structure. Biochemistry 38, 14966–14972 [DOI] [PubMed] [Google Scholar]
- 8. Schwall C. T., Greenwood V. L., Alder N. N. (2012) The stability and activity of respiratory complex II is cardiolipin-dependent. Biochim. Biophys. Acta 1817, 1588–1596 [DOI] [PubMed] [Google Scholar]
- 9. Zhang M., Mileykovskaya E., Dowhan W. (2002) Gluing the respiratory chain together. Cardiolipin is required for supercomplex formation in the inner mitochondrial membrane. J. Biol. Chem. 277, 43553–43556 [DOI] [PubMed] [Google Scholar]
- 10. Zhang M., Mileykovskaya E., Dowhan W. (2005) Cardiolipin is essential for organization of complexes III and IV into a supercomplex in intact yeast mitochondria. J. Biol. Chem. 280, 29403–29408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pfeiffer K., Gohil V., Stuart R. A., Hunte C., Brandt U., Greenberg M. L., Schägger H. (2003) Cardiolipin stabilizes respiratory chain supercomplexes. J. Biol. Chem. 278, 52873–52880 [DOI] [PubMed] [Google Scholar]
- 12. Claypool S. M., Oktay Y., Boontheung P., Loo J. A., Koehler C. M. (2008) Cardiolipin defines the interactome of the major ADP/ATP carrier protein of the mitochondrial inner membrane. J. Cell Biol. 182, 937–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Acehan D., Malhotra A., Xu Y., Ren M., Stokes D. L., Schlame M. (2011) Cardiolipin affects the supramolecular organization of ATP synthase in mitochondria. Biophys. J. 100, 2184–2192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jiang F., Ryan M. T., Schlame M., Zhao M., Gu Z., Klingenberg M., Pfanner N., Greenberg M. L. (2000) Absence of cardiolipin in the crd1 null mutant results in decreased mitochondrial membrane potential and reduced mitochondrial function. J. Biol. Chem. 275, 22387–22394 [DOI] [PubMed] [Google Scholar]
- 15. van der Laan M., Meinecke M., Dudek J., Hutu D. P., Lind M., Perschil I., Guiard B., Wagner R., Pfanner N., Rehling P. (2007) Motor-free mitochondrial presequence translocase drives membrane integration of preproteins. Nat. Cell Biol. 9, 1152–1159 [DOI] [PubMed] [Google Scholar]
- 16. Gebert N., Joshi A. S., Kutik S., Becker T., McKenzie M., Guan X. L., Mooga V. P., Stroud D. A., Kulkarni G., Wenk M. R., Rehling P., Meisinger C., Ryan M. T., Wiedemann N., Greenberg M. L., Pfanner N. (2009) Mitochondrial cardiolipin involved in outer-membrane protein biogenesis: implications for Barth syndrome. Curr. Biol. 19, 2133–2139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Marom M., Safonov R., Amram S., Avneon Y., Nachliel E., Gutman M., Zohary K., Azem A., Tsfadia Y. (2009) Interaction of the Tim44 C-terminal domain with negatively charged phospholipids. Biochemistry 48, 11185–11195 [DOI] [PubMed] [Google Scholar]
- 18. Claypool S. M., McCaffery J. M., Koehler C. M. (2006) Mitochondrial mislocalization and altered assembly of a cluster of Barth syndrome mutant tafazzins. J. Cell Biol. 174, 379–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mileykovskaya E., Dowhan W. (2009) Cardiolipin membrane domains in prokaryotes and eukaryotes. Biochim. Biophys. Acta 1788, 2084–2091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. DeVay R. M., Dominguez-Ramirez L., Lackner L. L., Hoppins S., Stahlberg H., Nunnari J. (2009) Coassembly of Mgm1 isoforms requires cardiolipin and mediates mitochondrial inner membrane fusion. J. Cell Biol. 186, 793–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ban T., Heymann J. A., Song Z., Hinshaw J. E., Chan D. C. (2010) OPA1 disease alleles causing dominant optic atrophy have defects in cardiolipin-stimulated GTP hydrolysis and membrane tubulation. Hum. Mol. Genet. 19, 2113–2122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Montessuit S., Somasekharan S. P., Terrones O., Lucken-Ardjomande S., Herzig S., Schwarzenbacher R., Manstein D. J., Bossy-Wetzel E., Basañez G., Meda P., Martinou J. C. (2010) Membrane remodeling induced by the dynamin-related protein Drp1 stimulates Bax oligomerization. Cell 142, 889–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ostrander D. B., Sparagna G. C., Amoscato A. A., McMillin J. B., Dowhan W. (2001) Decreased cardiolipin synthesis corresponds with cytochrome c release in palmitate-induced cardiomyocyte apoptosis. J. Biol. Chem. 276, 38061–38067 [DOI] [PubMed] [Google Scholar]
- 24. Gonzalvez F., Schug Z. T., Houtkooper R. H., MacKenzie E. D., Brooks D. G., Wanders R. J., Petit P. X., Vaz F. M., Gottlieb E. (2008) Cardiolipin provides an essential activating platform for caspase-8 on mitochondria. J. Cell Biol. 183, 681–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Houtkooper R. H., Akbari H., van Lenthe H., Kulik W., Wanders R. J., Frentzen M., Vaz F. M. (2006) Identification and characterization of human cardiolipin synthase. FEBS Lett. 580, 3059–3064 [DOI] [PubMed] [Google Scholar]
- 26. de Kroon A. I., Rijken P. J., De Smet C. H. (2013) Checks and balances in membrane phospholipid class and acyl chain homeostasis, the yeast perspective. Prog. Lipid Res. 52, 374–394 [DOI] [PubMed] [Google Scholar]
- 27. Schlame M., Ren M., Xu Y., Greenberg M. L., Haller I. (2005) Molecular symmetry in mitochondrial cardiolipins. Chem. Phys. Lipids 138, 38–49 [DOI] [PubMed] [Google Scholar]
- 28. Claypool S. M., Koehler C. M. (2012) The complexity of cardiolipin in health and disease. Trends Biochem. Sci. 37, 32–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Barth P. G., Scholte H. R., Berden J. A., Van der Klei-Van Moorsel J. M., Luyt-Houwen I. E., Van 't Veer-Korthof E. T., Van der Harten J. J., Sobotka-Plojhar M. A. (1983) An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J. Neurol. Sci. 62, 327–355 [DOI] [PubMed] [Google Scholar]
- 30. Bione S., D'Adamo P., Maestrini E., Gedeon A. K., Bolhuis P. A., Toniolo D. (1996) A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat. Genet. 12, 385–389 [DOI] [PubMed] [Google Scholar]
- 31. Schlame M., Ren M. (2006) Barth syndrome, a human disorder of cardiolipin metabolism. FEBS Lett. 580, 5450–5455 [DOI] [PubMed] [Google Scholar]
- 32. Xu Y., Condell M., Plesken H., Edelman-Novemsky I., Ma J., Ren M., Schlame M. (2006) A Drosophila model of Barth syndrome. Proc. Natl. Acad. Sci. U.S.A. 103, 11584–11588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gu Z., Valianpour F., Chen S., Vaz F. M., Hakkaart G. A., Wanders R. J., Greenberg M. L. (2004) Aberrant cardiolipin metabolism in the yeast taz1 mutant: a model for Barth syndrome. Mol. Microbiol. 51, 149–158 [DOI] [PubMed] [Google Scholar]
- 34. Valianpour F., Mitsakos V., Schlemmer D., Towbin J. A., Taylor J. M., Ekert P. G., Thorburn D. R., Munnich A., Wanders R. J., Barth P. G., Vaz F. M. (2005) Monolysocardiolipins accumulate in Barth syndrome but do not lead to enhanced apoptosis. J. Lipid Res. 46, 1182–1195 [DOI] [PubMed] [Google Scholar]
- 35. Schlame M., Kelley R. I., Feigenbaum A., Towbin J. A., Heerdt P. M., Schieble T., Wanders R. J., DiMauro S., Blanck T. J. (2003) Phospholipid abnormalities in children with Barth syndrome. J. Am. Coll. Cardiol. 42, 1994–1999 [DOI] [PubMed] [Google Scholar]
- 36. Whited K., Baile M. G., Currier P., Claypool S. M. (2013) Seven functional classes of Barth syndrome mutation. Hum. Mol. Genet. 22, 483–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Soustek M. S., Falk D. J., Mah C. S., Toth M. J., Schlame M., Lewin A. S., Byrne B. J. (2011) Characterization of a transgenic short hairpin RNA-induced murine model of Tafazzin deficiency. Hum. Gene. Ther. 22, 865–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cheng H., Mancuso D. J., Jiang X., Guan S., Yang J., Yang K., Sun G., Gross R. W., Han X. (2008) Shotgun lipidomics reveals the temporally dependent, highly diversified cardiolipin profile in the mammalian brain: temporally coordinated postnatal diversification of cardiolipin molecular species with neuronal remodeling. Biochemistry 47, 5869–5880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Houtkooper R. H., Turkenburg M., Poll-The B. T., Karall D., Pérez-Cerdá C., Morrone A., Malvagia S., Wanders R. J., Kulik W., Vaz F. M. (2009) The enigmatic role of tafazzin in cardiolipin metabolism. Biochim. Biophys. Acta 1788, 2003–2014 [DOI] [PubMed] [Google Scholar]
- 40. Kiebish M. A., Yang K., Sims H. F., Jenkins C. M., Liu X., Mancuso D. J., Zhao Z., Guan S., Abendschein D. R., Han X., Gross R. W. (2012) Myocardial regulation of lipidomic flux by cardiolipin synthase: setting the beat for bioenergetic efficiency. J. Biol. Chem. 287, 25086–25097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schlame M., Towbin J. A., Heerdt P. M., Jehle R., DiMauro S., Blanck T. J. (2002) Deficiency of tetralinoleoyl-cardiolipin in Barth syndrome. Ann. Neurol. 51, 634–637 [DOI] [PubMed] [Google Scholar]
- 42. Schlame M. (2013) Cardiolipin remodeling and the function of tafazzin. Biochim. Biophys. Acta 1831, 582–588 [DOI] [PubMed] [Google Scholar]
- 43. Beranek A., Rechberger G., Knauer H., Wolinski H., Kohlwein S. D., Leber R. (2009) Identification of a cardiolipin-specific phospholipase encoded by the gene CLD1 (YGR110W) in yeast. J. Biol. Chem. 284, 11572–11578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Osman C., Haag M., Wieland F. T., Brügger B., Langer T. (2010) A mitochondrial phosphatase required for cardiolipin biosynthesis: the PGP phosphatase Gep4. EMBO J. 29, 1976–1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang J., Guan Z., Murphy A. N., Wiley S. E., Perkins G. A., Worby C. A., Engel J. L., Heacock P., Nguyen O. K., Wang J. H., Raetz C. R., Dowhan W., Dixon J. E. (2011) Mitochondrial phosphatase PTPMT1 is essential for cardiolipin biosynthesis. Cell Metab. 13, 690–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mancuso D. J., Sims H. F., Han X., Jenkins C. M., Guan S. P., Yang K., Moon S. H., Pietka T., Abumrad N. A., Schlesinger P. H., Gross R. W. (2007) Genetic ablation of calcium-independent phospholipase A2γ leads to alterations in mitochondrial lipid metabolism and function resulting in a deficient mitochondrial bioenergetic phenotype. J. Biol. Chem. 282, 34611–34622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Malhotra A., Edelman-Novemsky I., Xu Y., Plesken H., Ma J., Schlame M., Ren M. (2009) Role of calcium-independent phospholipase A2 in the pathogenesis of Barth syndrome. Proc. Natl. Acad. Sci. U.S.A. 106, 2337–2341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schlame M., Blais S., Edelman-Novemsky I., Xu Y., Montecillo F., Phoon C. K., Ren M., Neubert T. A. (2012) Comparison of cardiolipins from Drosophila strains with mutations in putative remodeling enzymes. Chem. Phys. Lipids 165, 512–519 [DOI] [PubMed] [Google Scholar]
- 49. Kiebish M. A., Yang K., Liu X., Mancuso D. J., Guan S., Zhao Z., Sims H. F., Cerqua R., Cade W. T., Han X., Gross R. W. (2013) Dysfunctional cardiac mitochondrial bioenergetic, lipidomic, and signaling in a murine model of Barth syndrome. J. Lipid Res. 54, 1312–1325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Baile M. G., Whited K., Claypool S. M. (2013) Deacylation on the matrix side of the mitochondrial inner membrane regulates cardiolipin remodeling. Mol. Biol. Cell 24, 2008–2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jarosch E., Tuller G., Daum G., Waldherr M., Voskova A., Schweyen R. J. (1996) Mrs5p, an essential protein of the mitochondrial intermembrane space, affects protein import into yeast mitochondria. J. Biol. Chem. 271, 17219–17225 [DOI] [PubMed] [Google Scholar]
- 52. Claypool S. M., Boontheung P., McCaffery J. M., Loo J. A., Koehler C. M. (2008) The cardiolipin transacylase, tafazzin, associates with two distinct respiratory components providing insight into Barth syndrome. Mol. Biol. Cell 19, 5143–5155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wach A., Brachat A., Pöhlmann R., Philippsen P. (1994) New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast 10, 1793–1808 [DOI] [PubMed] [Google Scholar]
- 54. Thorsness M. K., White K. H., Thorsness P. E. (2002) Migration of mtDNA into the nucleus. Methods Mol. Biol. 197, 177–186 [DOI] [PubMed] [Google Scholar]
- 55. Christie W., Han X. (2010) Lipid Analysis: Isolation, Separation, Identification and Lipidomic Analysis, 4th Ed., pp. 55–66, The Oily Press, Bridgewater, UK [Google Scholar]
- 56. Han X., Yang K., Gross R. W. (2008) Microfluidics-based electrospray ionization enhances the intrasource separation of lipid classes and extends identification of individual molecular species through multi-dimensional mass spectrometry: development of an automated high-throughput platform for shotgun lipidomics. Rapid Commun. Mass Spectrom. 22, 2115–2124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yang K., Cheng H., Gross R. W., Han X. (2009) Automated lipid identification and quantification by multidimensional mass spectrometry-based shotgun lipidomics. Anal. Chem. 81, 4356–4368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tzagoloff A., Akai A., Needleman R. B. (1975) Assembly of the mitochondrial membrane system: isolation of nuclear and cytoplasmic mutants of Saccharomyces cerevisiae with specific defects in mitochondrial functions. J. Bacteriol. 122, 826–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Daum G., Böhni P. C., Schatz G. (1982) Import of proteins into mitochondria. cytochrome b2 and cytochrome c peroxidase are located in the intermembrane space of yeast mitochondria. J. Biol. Chem. 257, 13028–13033 [PubMed] [Google Scholar]
- 60. Maccecchini M. L., Rudin Y., Blobel G., Schatz G. (1979) Import of proteins into mitochondria: precursor forms of the extramitochondrially made F1-ATPase subunits in yeast. Proc. Natl. Acad. Sci. U.S.A. 76, 343–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ohashi A., Gibson J., Gregor I., Schatz G. (1982) Import of proteins into mitochondria. The precursor of cytochrome c1 is processed in two steps, one of them heme-dependent. J. Biol. Chem. 257, 13042–13047 [PubMed] [Google Scholar]
- 62. Poyton R. O., Schatz G. (1975) Cytochrome c oxidase from bakers' yeast. IV. Immunological evidence for the participation of a mitochondrially synthesized subunit in enzymatic activity. J. Biol. Chem. 250, 762–766 [PubMed] [Google Scholar]
- 63. Riezman H., Hay R., Gasser S., Daum G., Schneider G., Witte C., Schatz G. (1983) The outer membrane of yeast mitochondria: isolation of outside-out sealed vesicles. EMBO J. 2, 1105–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Panneels V., Schüssler U., Costagliola S., Sinning I. (2003) Choline head groups stabilize the matrix loop regions of the ATP/ADP carrier ScAAC2. Biochem. Biophys. Res. Commun. 300, 65–74 [DOI] [PubMed] [Google Scholar]
- 65. Cruciat C. M., Brunner S., Baumann F., Neupert W., Stuart R. A. (2000) The cytochrome bc1 and cytochrome c oxidase complexes associate to form a single supracomplex in yeast mitochondria. J. Biol. Chem. 275, 18093–18098 [DOI] [PubMed] [Google Scholar]
- 66. Lapuente-Brun E., Moreno-Loshuertos R., Acín-Pérez R., Latorre-Pellicer A., Colás C., Balsa E., Perales-Clemente E., Quirós P. M., Calvo E., Rodríguez-Hernández M. A., Navas P., Cruz R., Carracedo Á., López-Otín C., Pérez-Martos A., Fernández-Silva P., Fernández-Vizarra E., Enríquez J. A. (2013) Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science 340, 1567–1570 [DOI] [PubMed] [Google Scholar]
- 67. Ma L., Vaz F. M., Gu Z., Wanders R. J., Greenberg M. L. (2004) The human TAZ gene complements mitochondrial dysfunction in the yeast taz1Δ mutant. Implications for Barth syndrome. J. Biol. Chem. 279, 44394–44399 [DOI] [PubMed] [Google Scholar]
- 68. Wenz T., Hielscher R., Hellwig P., Schägger H., Richers S., Hunte C. (2009) Role of phospholipids in respiratory cytochrome bc1 complex catalysis and supercomplex formation. Biochim. Biophys. Acta 1787, 609–616 [DOI] [PubMed] [Google Scholar]
- 69. Cao J., Liu Y., Lockwood J., Burn P., Shi Y. (2004) A novel cardiolipin-remodeling pathway revealed by a gene encoding an endoplasmic reticulum-associated acyl-CoA:lysocardiolipin acyltransferase (ALCAT1) in mouse. J. Biol. Chem. 279, 31727–31734 [DOI] [PubMed] [Google Scholar]
- 70. Cao J., Shen W., Chang Z., Shi Y. (2009) ALCAT1 is a polyglycerophospholipid acyltransferase potently regulated by adenine nucleotide and thyroid status. Am. J. Physiol. Endocrinol. Metab. 296, E647–E653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Li J., Romestaing C., Han X., Li Y., Hao X., Wu Y., Sun C., Liu X., Jefferson L. S., Xiong J., Lanoue K. F., Chang Z., Lynch C. J., Wang H., Shi Y. (2010) Cardiolipin remodeling by ALCAT1 links oxidative stress and mitochondrial dysfunction to obesity. Cell Metab. 12, 154–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kim J., Minkler P. E., Salomon R. G., Anderson V. E., Hoppel C. L. (2011) Cardiolipin: characterization of distinct oxidized molecular species. J. Lipid Res. 52, 125–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Musatov A. (2006) Contribution of peroxidized cardiolipin to inactivation of bovine heart cytochrome c oxidase. Free Radic. Biol. Med. 41, 238–246 [DOI] [PubMed] [Google Scholar]
- 74. Gonzalvez F., D'Aurelio M., Boutant M., Moustapha A., Puech J. P., Landes T., Arnauné-Pelloquin L., Vial G., Taleux N., Slomianny C., Wanders R. J., Houtkooper R. H., Bellenguer P., Møller I. M., Gottlieb E., Vaz F. M., Manfredi G., Petit P. X. (2013) Barth syndrome: Cellular compensation of mitochondrial dysfunction and apoptosis inhibition due to changes in cardiolipin remodeling linked to tafazzin gene mutation. Biochim. Biophys. Acta 1832, 1194–1206 [DOI] [PubMed] [Google Scholar]
- 75. Chen S., He Q., Greenberg M. L. (2008) Loss of tafazzin in yeast leads to increased oxidative stress during respiratory growth. Mol. Microbiol. 68, 1061–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Xu Y., Malhotra A., Ren M., Schlame M. (2006) The enzymatic function of tafazzin. J. Biol. Chem. 281, 39217–39224 [DOI] [PubMed] [Google Scholar]
- 77. Schlame M., Acehan D., Berno B., Xu Y., Valvo S., Ren M., Stokes D. L., Epand R. M. (2012) The physical state of lipid substrates provides transacylation specificity for tafazzin. Nat. Chem. Biol. 8, 862–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Zhang L., Bell R. J., Kiebish M. A., Seyfried T. N., Han X., Gross R. W., Chuang J. H. (2011) A mathematical model for the determination of steady-state cardiolipin remodeling mechanisms using lipidomic data. PLoS One 6, e21170. [DOI] [PMC free article] [PubMed] [Google Scholar]