

Background: The angiotensin-1-converting enzyme (ACE) is a zinc protease unique in its chloride ion dependence.
Results: Key substrate interactions with the catalytic site affect the structure and thermodynamics of ACE, which modulate chloride ion-dependent hydrolysis.
Conclusion: Substrate composition influences chloride affinity in ACE.
Significance: The interdependence of ACE substrate composition, structure, and thermodynamics influence its chloride ion activation in biology.
Keywords: Biophysics, Crystallography, Hypertension, Protein Structure, Thermodynamics, Angiotensin-1-converting Enzyme, Chloride Ion Activation, Enzymology
Abstract
Somatic angiotensin-converting enzyme (sACE), a key regulator of blood pressure and electrolyte fluid homeostasis, cleaves the vasoactive angiotensin-I, bradykinin, and a number of other physiologically relevant peptides. sACE consists of two homologous and catalytically active N- and C-domains, which display marked differences in substrate specificities and chloride activation. A series of single substitution mutants were generated and evaluated under varying chloride concentrations using isothermal titration calorimetry. The x-ray crystal structures of the mutants provided details on the chloride-dependent interactions with ACE. Chloride binding in the chloride 1 pocket of C-domain ACE was found to affect positioning of residues from the active site. Analysis of the chloride 2 pocket R522Q and R522K mutations revealed the key interactions with the catalytic site that are stabilized via chloride coordination of Arg522. Substrate interactions in the S2 subsite were shown to affect chloride affinity in the chloride 2 pocket. The Glu403-Lys118 salt bridge in C-domain ACE was shown to stabilize the hinge-bending region and reduce chloride affinity by constraining the chloride 2 pocket. This work demonstrated that substrate composition to the C-terminal side of the scissile bond as well as interactions of larger substrates in the S2 subsite moderate chloride affinity in the chloride 2 pocket of the ACE C-domain, providing a rationale for the substrate-selective nature of chloride dependence in ACE and how this varies between the N- and C-domains.
Introduction
Angiotensin I-converting enzyme (ACE5; EC 3.4.15.1) is a zinc metallopeptidase that functions as a dipeptidyl carboxypeptidase, catalyzing the hydrolysis of a wide range of functional peptides. Its main role is within the renin-angiotensin system, which regulates blood pressure and renal homeostasis and thus is an important drug target for the treatment of hypertension and other cardiovascular and renal diseases (1–3). The systemic renin-angiotensin system relies on a series of proteolytic events involving circulating renin/prorenin and somatic ACE (sACE), present on the external surface of endothelial cells, to process angiotensinogen into angiotensin II (AngII), a hypertensive and mitogenic octapeptide. sACE catalyzes the last step in the production of AngII by cleaving the C-terminal dipeptide of AngI (4). The main form of mammalian sACE consists of two very similar domains (N- and C-domains) each possessing a functional catalytic site (1, 2). There is also a smaller testicular form of ACE (tACE), which is transcribed from the same gene and identical to the C-domain of ACE. The mammalian ACE gene arose from a gene duplication event during the course of vertebrate evolution (5).
The active site for both domains is contained within the large central groove, with the signature HEXXH zinc-binding motif residing on the α15 helix (6, 7). Access to this active site is severely limited, with a small pore in the N-terminal chamber or an occluded slot in the C-terminal chamber providing the only theoretical entry and suggests that a degree of flexibility in the domain movements is required for substrate access (7, 8). The catalytic mechanism has been suggested based on comparison with work done on the structurally analogous zinc metalloprotease, thermolysin, and is proposed to occur via a general base mechanism, whereby a nucleophilic water molecule or hydroxide ion attacks the carbonyl carbon of the scissile bond (9, 10).
Two chloride ions are found buried in the structure of tACE and separated by a distance of 20.3 Å (6). The first chloride (Cl1), 20.7 Å away from the zinc, is bound by four ligands, namely Arg186, Trp485, Arg489, and water, and is encapsulated by a hydrophobic shell of four tryptophans (6). The N-domain crystal structures show no chloride bound in this pocket, with the only difference in residue composition in the pocket being His164, which corresponds to Arg186 in tACE (7). The second chloride ion (Cl2), which is observed in both the C- and N-domain structures, is located much closer to the active site zinc at 10.4 Å and is coordinated by Arg522, Tyr224 (Arg500 and Tyr202 in the N-domain), and a water molecule (6).
ACE activity is dependent on chloride ions, with the degree of dependence being substrate-specific (11). ACE is situated mainly on the surface of endothelial tissue, an environment where blood plasma chloride levels rarely fluctuate from the 100 mm average, sufficient for ACE to be considered fully active in angiotensin I hydrolysis (11). However, ACE has been found in other non-vascular tissues, including kidney, brain, bone marrow, pancreas, and adipose tissues (12), where the chloride levels can vary substantially, and also intracellularly (13), where the chloride concentration is ∼5 mm (14). These varying concentrations would have an impact on ACE catalytic efficiency.
Variance in chloride activity is also seen in inhibitor binding. The difference in affinities for the ACE inhibitors trandolaprilat, enalaprilat, and lisinopril between the N- and C-domains is greater at high chloride concentration (300 mm), whereas for captopril, the difference is larger at low chloride concentration (20 mm) (15). The variability in chloride-mediated enhancement of inhibitor affinity may have potential applications in design of inhibitors targeting tissue-specific ACE activity. Indeed, a full delineation of the exact structural and mechanistic aspects of ACE chloride dependence for each domain could be important for the development of domain-selective inhibitors.
In this study, key single point mutations were generated to investigate the mechanism of chloride activation and substrate specificity. Activity of these mutants was characterized using an isothermal titration calorimetry (ITC)-based assay providing in depth information on the enzyme thermodynamic and kinetic parameters under varying chloride concentrations. Furthermore, the high resolution x-ray crystal structures of these mutants were determined, revealing the key molecular interactions involved in chloride binding. Together, these data identified the close relationship between the nature of the substrate and chloride activation through the chloride 2 pocket and the S2 subsite. The role for the chloride 1 pocket remains elusive and could be mainly structural. A key salt bridge specific to C-domain ACE between Glu403 and Lys118 was shown to stabilize the hinge-bending region and reduce chloride affinity by constraining the chloride 2 pocket. This work provides a rationale for the substrate-selective nature of chloride dependence in ACE and how this varies between the N- and C-domains.
EXPERIMENTAL PROCEDURES
Chemicals
All chemicals were purchased from Merck and Sigma-Aldrich. Benzyloxycarbonyl-l-Phe-l-His-Leu (Z-FHL) was purchased from Bachem AG (Bubendorf, Switzerland).
Mutations
A fully soluble, minimally N-glycosylated version of human tACE, tACEΔ36-g13sol (16) (referred to as the C-domain hereafter), truncated after Ser625 and lacking the 36 O-glycosylated N-terminal residues (17), was cloned into the BamHI and NotI restriction sites of pcDNA3.1(+) (Invitrogen) in order to facilitate expression in mammalian cells. A series of C-domain mutants were generated (Table 1 and Fig. 1) using a cassette-based subcloning strategy, as described previously (16), whereby PCR-based site-directed mutagenesis was used to introduce single amino acid mutations into fragments of the C-domain, contained within pGEM11Zf(+) (Promega), which were sequenced bidirectionally to verify the mutation and then subcloned into the C-domain. Human soluble N-domain Asp629 (18), containing residues 1–629, was previously cloned into EcoRI and XbaI restriction sites of pcDNA 3.1(+)(Invitrogen) for expression.
TABLE 1.
Site-directed mutagenesis of C-domain ACE
Shown is the location of the mutations with respect to potential role, the name of the mutation, and residue in the C-domain that was mutated along with its numerical position, identity of the amino acid if it is being converted to its corresponding N-domain residue, and the identity of the residue if that mutation is completely novel (i.e. same in both domains).
| Location | Mutant name | C-domain | N-domain | Non-domain mutation |
|---|---|---|---|---|
| Chloride 1 pocket | R186H | Arg186 | His | |
| S2 pocket (chloride channel) | E403R | Glu403 | Arg | |
| Chloride 2 pocket | D465T | Asp465 | Thr | |
| R522K | Arg522 | Lys | ||
| R522Q | Arg522 | Gln |
FIGURE 1.

Overall structure of C-domain ACE mutants. A, location of mutated residues Glu403, Asp465, and Arg522 (magenta) in C-domain ACE (cyan; Protein Data Bank code 1O8A (6)), with chloride and zinc ions shown as spheres (green and gray, respectively). B, superposition of C-domain ACE (cyan; Protein Data Bank code 1O8A (6)) with the mutant structures E403R (light pink), D465T (pink), R522K (violet), and R522Q (purple).
Enzymes
N- and C-domain human ACE proteins were generated by expression in cultured mammalian CHO cells (human ACE) and purified to homogeneity as described previously (16, 19).
X-ray Crystallography
Crystals were obtained with 1 μl of the C-domain mutant sample (5–10 mg/ml in 50 mm HEPES, pH 7.5, 0.1 mm PMSF) mixed with an equal volume of reservoir solution (100 mm MIB buffer, pH 4.0, 10 μm zinc sulfate, 5% glycerol, and 15% PEG 3350) and suspended above the well as a hanging drop. Diffraction quality crystals of C-domain mutants appeared after ∼3–5 days. The R522K·captopril complex was obtained by co-crystallization with a 1 mm concentration of the ligand.
X-ray diffraction data for C-domain mutants were collected on PX station IO2, IO4, and IO4-1 at the Diamond Light Source (Oxon, UK). 25% PEG 3350 was added to the drop as a cryoprotectant to keep the crystal at constant temperature (100 K) under the liquid nitrogen jet during data collection. For each mutant, 100–150 images were collected by using a PILATUS-2M, -6M (Dectris, Switzerland) or a Quantum-315 CCD (Area Detector Systems Corp., Poway, CA) detector. Raw data images were processed and scaled with XDS (20), MOSFLM (21), or XIA2 and SCALA using the CCP4 suite (22). Initial phases for structure solution were obtained using the molecular replacement routines of the PHASER program (23). The atomic coordinates of native C-domain (Protein Data Bank code 1O8A (6)) were used as a search model for structure determination. The resultant models were refined using REFMAC5 (24). Five percent of reflections were separated as an Rfree set and used for cross-validation (25). Manual adjustments of the model were carried out using COOT (26). Water molecules were added at positions where Fo − Fc electron density peaks exceeded 3σ, and potential hydrogen bonds could be made. Validation was conducted with the aid of the program MOLPROBITY (27). Crystallographic data statistics are summarized in Table 2. All figures were drawn with PyMOL (Schrödinger, LLC, New York) and rendered with POV-ray.
TABLE 2.
Crystallographic statistics
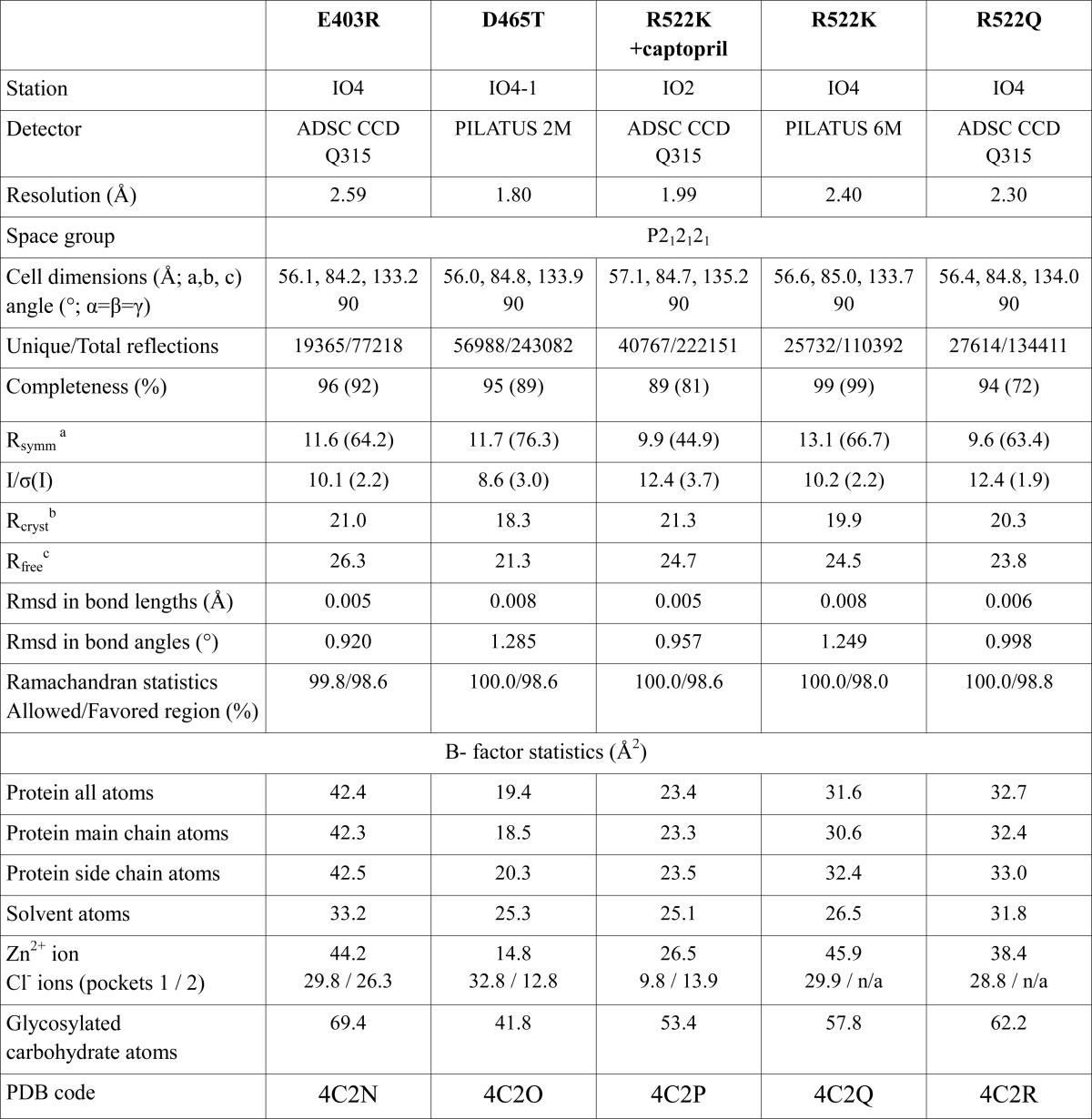
a Rsymm = ΣhΣi[|Ii(h) − 〈I(h)〉|/ΣhΣi Ii(h)], where Ii is the ith measurement, and 〈I(h)〉 is the weighted mean of all the measurements of I(h).
b Rcryst = Σh|Fo − Fc|/ΣhFo, where Fo and Fc are observed and calculated structure factor amplitudes of reflection h, respectively.
c Rfree is equal to Rcryst for a randomly selected 5% subset of reflections.
Chloride Titration Assays Using Hippuryl-l-His-Leu (HHL) and Z-FHL
Chloride titrations were performed using HHL and Z-FHL solutions containing varying amounts of NaCl (0–1 m) prepared using Milli-Q distilled H2O (chloride concentration <0.06 mm). All substrates used were HPLC-purified (>98% purity), so any chloride or chloride salts would elute with the breakthrough peak; hence, the effects on chloride concentrations are negligible. Assays were performed using a protocol adapted from that described previously (28), with the primary difference being use of a different buffer (50 mm TAPSO (pH 7.5)). Milliunits of ACE activity, with 1 unit defined as 1 nmol of HL produced/min/ml (min−1·ml−1) at 37 °C in assay buffer, were calculated, divided by the total number of μg in the reaction volume, and expressed as specific activity (milliunits·μg−1). Substrate concentrations used were ≥5 times Km, and the total percentage hydrolysis in each reaction was kept below 20% in order to approximate first order rates (kcat/Km). The measure of chloride binding (Kd(app)) was calculated from activation plots of specific activity versus the logarithm of chloride concentration, with the resultant curves analyzed via non-linear regression using a sigmoidal dose-response curve with the formula,
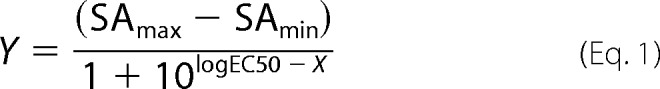 |
where SAmin is the specific activity in the absence of NaCl, SAmax is the maximum specific activity upon titration with chloride, X is the logarithm of chloride concentration, Y is the response (specific activity), and EC50 is the X value when the response is halfway between SAmax and SAmin.
Isothermal Titration Calorimetry Sample Preparation
Enzyme samples of high concentration (>10 μm) were dialyzed extensively against 3× 1 liter of reaction buffer, which consisted of 50 mm TAPSO buffer (pH 7.5), 10 μm ZnSO4, and either 0, 20, or 300 mm NaCl.
Substrate/inhibitor was prepared by either dissolution directly into dialysate, to the desired concentration (for HHL, Z-FHL, and lisinopril), or by equilibrating against a 1-ml G10 Sephadex desalting column (for angiotensin I). Angiotensin I (Asp-Arg-Val-Tyr-Ile-His-Pro-Phe-His-Leu) was dissolved in dialysate at high concentration then passed through a 1-ml G10 column (equilibrated with dialysate in order to reduce TFA concentrations) under gravity. 200-μl fractions were collected, and absorbance was measured at 275 nm. These were pooled, and the final concentration was determined spectrophotometrically via absorbance at 275 nm, using the empirically calculated extinction coefficient of 1280 m−1·cm−1.
Isothermal Titration Calorimetry Binding Assays
Assays were performed using an iTC200 microcalorimeter and according to the guidelines in the iTC200 microcalorimeter user manual provided by MicroCal. Buffer conditions for both enzyme and lisinopril consisted of 50 mm TAPSO buffer (pH 7.5), 10 μm ZnSO4, and either 0, 20, or 300 mm NaCl, prepared using Milli-Q distilled H2O (chloride concentration <0.06 mm). The reaction cell was maintained at 20 °C, with the lisinopril concentration in the syringe (800–1400 μm) at 10 times the enzyme concentration in the cell (8–12 μm) to account for lisinopril dilution (∼6-fold) and ensure a complete 1:1 binding curve. Binding assays were performed with every preparation of enzyme used in the kinetic assays; this served as an active site titration to determine active enzyme concentration in order to improve the accuracy of the kcat (equal to Vmax/[E]) values obtained in those experiments. The Kd(app), ΔG, ΔH, and ΔS values for lisinopril binding were calculated using Origin 7 with the iTC200 MicroCal Software Addon.
Isothermal Titration Calorimetry Kinetic Assays
All ITC experiments were performed using an iTC200 microcalorimeter (MicroCal LLC), with raw data either extracted for custom calculations using Microsoft Excel or analyzed using Origin 7 software with a proprietary MicroCal analysis module. Assays were performed at 37 °C using an adapted version of the progress curve method described by Stockbridge and Wolfenden (29), where the variation comprised the use of substrate concentrations of 3–5 times Km. The assay setup for the titration calorimeter involves a single injection of substrate into enzyme or enzyme into substrate and the continuous monitoring of thermal power as substrate is catalyzed to completion. A means of converting this thermal power into a reaction curve of product formed over time has been described previously (29). Given that heat released or absorbed by the enzyme reaction is directly proportional to the amount of substrate hydrolyzed, the amount of product formed over time can be calculated, and a progress curve can be generated. Integration of the total area under the curve represents the total heat turned over in catalyzing all of the substrate. Integrating the area between substrate injection (t = 0) and each time point (t) and then dividing by the total integrated area gives a percentage of the product formed at time t. Multiplying this fraction at each time point by the total substrate concentration at t = 0 (S0) relates it to molar concentration. The formula describing this relationship is as follows,
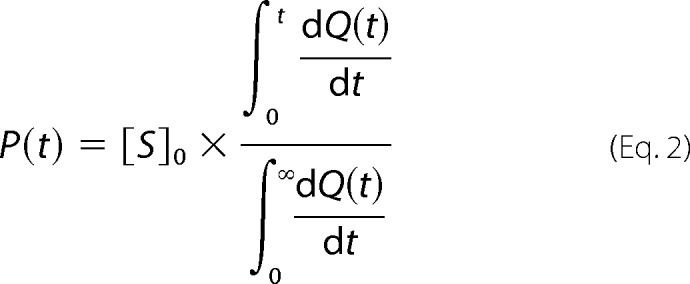 |
with the resultant progress curve being a plot of P(t) (product formed) versus t (time).
Progress curves were generated and analyzed according to functions described by Golicnik (30). The temporal closed form solution of the Michaelis-Menten equation used is given by Equation 3,
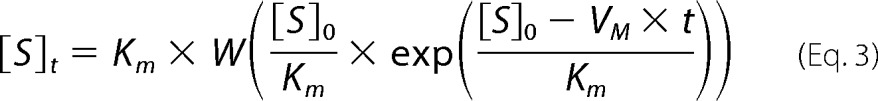 |
where W is the Lambert W(x) function (31). Golicnik (30) evaluated a number of approximations of W and found one version that produced an acceptably low amount of systemic error when applied to Equation 3 in evaluating progress curves,
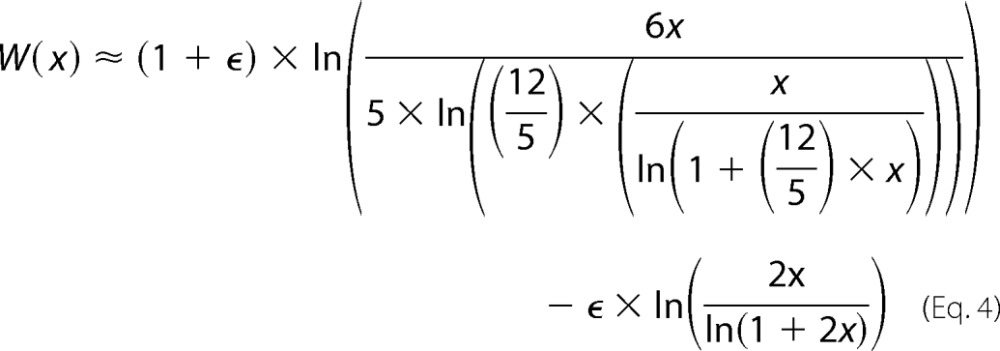 |
where ϵ = 0.4586887. Progress curves were generated in Microsoft Excel using Equation 2 and analyzed using the combined Equations 3 and 4, which were written into Graphpad Prism 5 software according to instructions provided by Golicnik (30). Assays were performed in triplicate as a minimum, and all kinetic constants (Km, kcat, and kcat/Km) are reported as the mean of the three separate determinations along with the S.E. value.
The thermodynamic parameters (ΔG, ΔH, and ΔS) for the hydrolysis of substrates (HHL, Z-FHL, and angiotensin I) were determined using the kinetic data (kcat/Km) and the raw data from each of those assays. The Gibbs free energy (ΔG) was calculated by the formula ΔG = −RT ln(kcat/Km), with R being the gas constant (1.9858 cal·K−1·mol−1) and T being assay temperature (310 K). The apparent enthalpy (ΔHapp) was determined for each assay via integration of the area under the curve using the following formula.
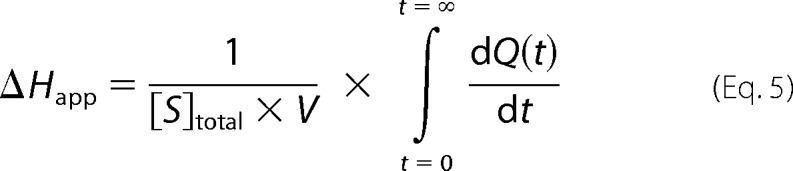 |
To exclude the contribution of buffer to enthalpy, the intrinsic enthalpy (ΔHint) was calculated using the relationship, ΔHapp = ΔHint + nH × ΔHion (ΔHion for TAPSO buffer = 9.378 kcal/mol; nH = −0.37 for all substrates). Incorporation of ΔG and ΔHint into ΔG = ΔHint − TΔS allows calculation of −TΔS, which is the entropy as a function of temperature. These values were calculated separately for each assay, which was done in triplicate as a minimum, with values shown as mean and S.E.
RESULTS AND DISCUSSION
Crystal Structures of Human C-domain sACE Mutants
Human C-domain mutants E403R, D465T, R522K, and R522Q were crystallized and data were collected at 2.59, 1.80, 1.99, and 2.30 Å resolution, respectively (Fig. 1 and Table 2). An unambiguous difference density map confirmed the mutations at the expected sites (Fig. 2). None of the mutations created caused any major conformational change in the overall architecture of the protein, with the root mean square deviation over an all-atom superposition being ≤0.4 Å for each of the mutants when compared with the C-domain (Fig. 1B). The consequences appeared to be localized around the mutated sites, which were designed to investigate the role of these specific residues in the chloride-dependent enzymatic activity. Mutations R522K (ligand-free) and R522Q showed the loss of the chloride ion in proximity to the active site (Fig. 3).
FIGURE 2.

Electron density map of mutated residues in C-domain ACE. A weighted difference map was calculated with REFMAC5 and is displayed at 1σ level for E403R (A), D465T (B), R522K + captopril (C), R522K (D), and R522Q (E).
FIGURE 3.

Structural changes from mutations in C-domain ACE. Comparison of local structural changes from mutations in C-domain ACE. Shown are native (cyan) and mutant (magenta) structures where E403R (A), D465T (B), R522K + captopril (C), R522K (D), and R522Q (E) are represented. Chloride and zinc ions are shown as spheres (green and gray, respectively), and water molecules are shown in red. Possible hydrogen bonds are shown as dashed lines.
The E403R mutation is in a highly accessible solvent area, and thus the longer side chain presented lower stability. Furthermore, whereas Glu403 made a salt bridge with Lys118, mutation to Arg403 resulted in local disorder with a higher B factor seen for Lys118. The side chain of Lys118 was refined in this position as in the C-domain (Fig. 3A) but is expected to be very mobile, as illustrated by the lack of electron density (Fig. 2A and Table 3). E403R also presented less contact with the surrounding residues by the loss of several hydrogen bonds with Ala400, Leu401, and Arg402. No water molecule was seen interacting with the mutated residue despite its solvent accessibility, and overall, no conformational change of the main carbon chain was visible.
TABLE 3.
Side chain hydrogen interactions in mutated residues
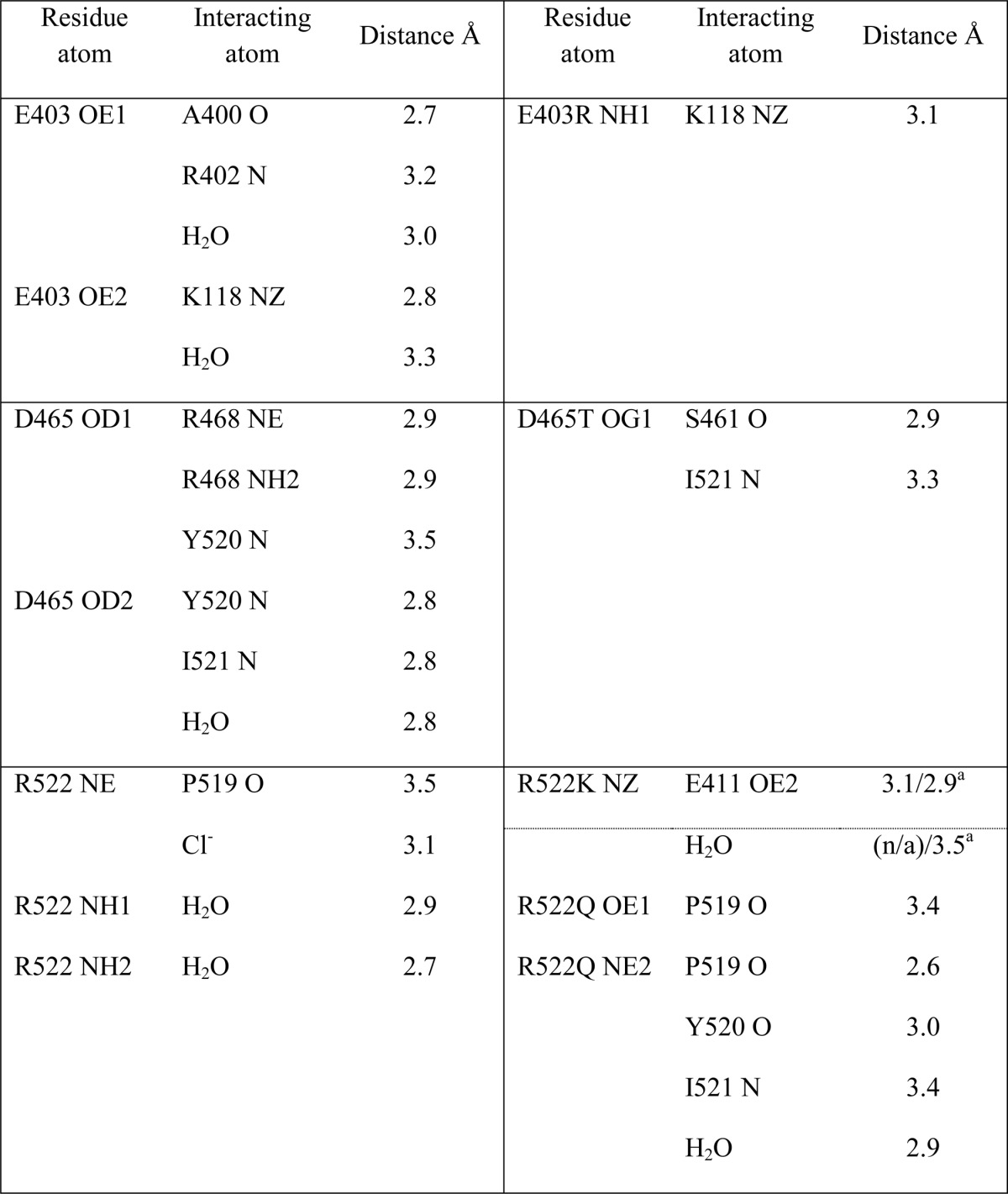
a R522K structure with and without captopril, respectively.
A high resolution data set was collected for the D465T mutant. The native Asp465 side chain is stabilized by a salt bridge with Arg468 as well as hydrogen bonds with the main chain of Tyr520 and Ile521 (Fig. 3B and Table 3). Although it did not disrupt the local secondary α-helical structure, mutation to Thr465 resulted in reorientation of the side chain with a 60° rotation that allows it to interact with Ser461 and Ile521, thus losing the interaction with Arg468. Interestingly, this also led to a change in rotamer conformation of Trp469, which went from a water-mediated interaction with Asp465 to a hydrogen bond with Arg221. This created a new pocket that permitted the presence of a malonate molecule (a component of the crystallization buffer; data not shown). The malonic acid is coordinating with the same Arg221 on one side and mimicking the interactions previously made by Asp465 on the other side, including the salt bridge with Arg468 and hydrogen bonds with Tyr520 and Ile521. Noticeably, loop Ser434–Ser439 was visible for the first time in a C-domain crystal structure (Fig. 1B) with clear electron density observed for this flexible loop region.
The chloride ion is directly coordinated by Arg522 in the native C-domain, along with Tyr224 and a water-mediated interaction to Trp220 (Fig. 3C). The R522K mutant was crystallized with and without captopril (Fig. 2, C and D, respectively). Whereas the Cl− ion is not visible in the ligand-free structure, the ion was clearly visible in the bound complex. There is no evidence from the electron density map of the R522K·captopril structure that the chloride is linked to Lys522 (Fig. 2C) with the amino group located more than 4 Å away from a water in the ion's hydration shell. The chloride ion is coordinated by the hydroxyl group of Tyr224 and the surrounding water molecules linked to Trp220, Trp469, and Asp465. Importantly, Lys522 now forms a salt bridge with Glu411, which is part of the zinc coordination motif and thus involved in the enzyme catalytic activity. Lys522 is also within the distance of a cation-π interaction with Tyr523, thus further directing the side chain away from the Cl− ion. It should be added that captopril is present at the active site in the same position as that of the wild-type C-domain·captopril complex structure (32). Although its presence did not disturb the catalytic pocket, the ligand interacts with Tyr523 through a hydrogen bond and π stacking of its proline group with the phenol of Tyr523. In the ligand-free structure, the chloride ion is replaced by a water molecule within coordinating distance of Tyr224 (Fig. 3D). The position of the residues within the chloride pocket remains unchanged, and Lys522 is therefore making interactions similar to those described for the ligand-bound structure. An additional water-mediated contact was seen with Glu411, reinforcing the link with the catalytic pocket.
Mutation R522Q also resulted in the loss of the chloride ion (Fig. 2E), with Gln522 making strong hydrogen bonds with the backbone of residues Pro519, Tyr520, and Ile521 (Fig. 3E and Table 3) and a weak cation-π interaction with Tyr523. In place of the Cl− is now a water molecule stabilized by Tyr224 and a network of other waters themselves linked to Asp465 and Trp469. Furthermore, a sulfate ion was present in the active site, making direct coordination with the zinc ion and surrounding residues of the S1 subsite, including Tyr523.
Effect of Chloride on Energetic Contributions in ACE Binding and Catalysis
Mutant C-domain constructs were characterized using the substrates HHL, Z-FHL, and AngI in the absence and presence of 20 mm NaCl. Kinetic constants and thermodynamic parameters are displayed in Tables 4 and 5, respectively.
TABLE 4.
Kinetic parameters for the cleavage of HHL, Z-FHL, and angiotensin I by ACE constructs
Shown is cleavage of HHL, Z-FHL, and angiotensin I by the C-domain, the N-domain, and a number of C-domain mutant constructs. Constants were determined using an ITC assay at 0 and 20 mm NaCl (including 300 mm for the C-domain with HHL) in 50 mm TAPSO (pH 7.5) and 10 μm ZnSO4. Values are shown as the mean of three separate determinations of Km, kcat, and kcat/Km along with S.E.
| Construct | Substrate | [NaCl] | Km | kcat | kcat/Km |
|---|---|---|---|---|---|
| mm | mm | s−1 | mm−1 s−1 | ||
| N-domain | HHL | 0 | 3.910 ± 0.060 | 14.15 ± 0.53 | 3.63 ± 0.14 |
| 20 | 0.720 ± 0.020 | 38.84 ± 0.13 | 53.97 ± 0.15 | ||
| Z-FHL | 0 | 1.360 ± 0.010 | 73.38 ± 0.62 | 54.10 ± 0.47 | |
| 20 | 0.520 ± 0.010 | 509.50 ± 15.61 | 991.25 ± 31.06 | ||
| Angiotensin I | 0 | 0.520 ± 0.010 | 4.04 ± 0.06 | 36.23 ± 0.47 | |
| 20 | 0.077 ± 0.006 | 24.98 ± 1.59 | 325.40 ± 20.54 | ||
| C-domain | HHL | 0 | 1.960 ± 0.020 | 3.26 ± 0.46 | 1.66 ± 0.24 |
| 20 | 1.650 ± 0.030 | 127.00 ± 1.78 | 77.15 ± 1.03 | ||
| 300 | 0.779 ± 0.008 | 606.00 ± 21.86 | 777.50 ± 28.11 | ||
| Z-FHL | 0 | 0.580 ± 0.010 | 80.25 ± 1.20 | 140.00 ± 2.13 | |
| 20 | 0.120 ± 0.010 | 282.00 ± 11.16 | 2372.50 ± 93.67 | ||
| Angiotensin I | 0 | 0.049 ± 0.001 | 1.70 ± 0.11 | 35.28 ± 2.11 | |
| 20 | 0.036 ± 0.001 | 7.38 ± 0.05 | 204.67 ± 1.21 | ||
| R186H | HHL | 0 | 2.740 ± 0.030 | 2.09 ± 0.08 | 0.77 ± 0.03 |
| 20 | 2.060 ± 0.020 | 122.00 ± 1.16 | 59.27 ± 0.62 | ||
| Z-FHL | 0 | 1.000 ± 0.040 | 76.24 ± 2.94 | 76.44 ± 2.94 | |
| 20 | 0.150 ± 0.010 | 467.34 ± 36.45 | 3193.34 ± 247.68 | ||
| Angiotensin I | 0 | 0.085 ± 0.003 | 1.52 ± 0.03 | 17.93 ± 0.29 | |
| 20 | 0.095 ± 0.003 | 7.83 ± 0.19 | 82.65 ± 2.02 | ||
| E403R | HHL | 0 | 4.360 ± 0.050 | 20.48 ± 0.43 | 4.70 ± 0.10 |
| 20 | 1.540 ± 0.040 | 459.00 ± 24.12 | 298.67 ± 15.90 | ||
| Z-FHL | 0 | 0.530 ± 0.010 | 159.34 ± 2.91 | 304.34 ± 5.21 | |
| 20 | 0.540 ± 0.010 | 929.34 ± 14.77 | 1736.67 ± 28.49 | ||
| Angiotensin I | 0 | 0.069 ± 0.003 | 3.96 ± 0.28 | 57.46 ± 4.07 | |
| 20 | 0.061 ± 0.004 | 9.08 ± 0.41 | 123.00 ± 5.42 | ||
| R522Q | HHL | 0 | 2.820 ± 0.020 | 6.18 ± 0.17 | 2.20 ± 0.06 |
| 20 | 1.680 ± 0.010 | 4.80 ± 0.18 | 2.87 ± 0.11 | ||
| Z-FHL | 0 | 0.110 ± 0.010 | 92.24 ± 2.16 | 860.34 ± 20.01 | |
| 20 | 0.200 ± 0.010 | 124.67 ± 9.36 | 653.34 ± 47.73 | ||
| Angiotensin I | 0 | 0.092 ± 0.003 | 12.98 ± 0.29 | 141.75 ± 3.20 | |
| 20 | 0.077 ± 0.003 | 13.75 ± 0.84 | 179.50 ± 11.14 | ||
| R522K | HHL | 0 | 3.720 ± 0.020 | 5.31 ± 0.19 | 1.43 ± 0.06 |
| 20 | 2.250 ± 0.020 | 2.03 ± 0.05 | 0.91 ± 0.02 | ||
| Z-FHL | 0 | 0.570 ± 0.010 | 126.00 ± 1.00 | 223.25 ± 1.94 | |
| 20 | 0.880 ± 0.020 | 193.50 ± 5.13 | 220.75 ± 5.97 | ||
| Angiotensin I | 0 | 0.130 ± 0.002 | 0.83 ± 0.03 | 6.37 ± 0.21 | |
| 20 | 0.119 ± 0.002 | 3.79 ± 0.07 | 32.05 ± 0.53 |
TABLE 5.
Thermodynamic parameters for the cleavage of HHL, Z-FHL and angiotensin I by ACE constructs
Shown are thermodynamic parameters associated with the cleavage of HHL, Z-FHL, and angiotensin I by the C-domain, N-domain, and a number of C-domain mutant constructs. Values were determined at 0 and 20 mm NaCl (including 300 mm for the C-domain with HHL). Values are shown as the mean of three separate determinations of ΔG, ΔH, and −TΔS along with S.E.
| Construct | Substrate | [NaCl] | ΔHint | ΔG | −TΔS |
|---|---|---|---|---|---|
| mm | kcal mol−1 | kcal mol−1 | kcal K−1 mol−1 | ||
| N-domain | HHL | 0 | 2.48 ± 0.03 | −5.04 ± 0.02 | −7.52 ± 0.05 |
| 20 | 2.59 ± 0.01 | −6.93 ± 0.00 | −9.52 ± 0.01 | ||
| Z-FHL | 0 | 2.25 ± 0.01 | −6.71 ± 0.01 | −8.96 ± 0.01 | |
| 20 | 2.80 ± 0.06 | −8.50 ± 0.02 | −11.29 ± 0.08 | ||
| Angiotensin I | 0 | 2.80 ± 0.06 | −6.46 ± 0.01 | −11.29 ± 0.08 | |
| 20 | 2.71 ± 0.10 | −7.81 ± 0.05 | −10.52 ± 0.10 | ||
| C-domain | HHL | 0 | 2.37 ± 0.07 | −4.19 ± 0.01 | −6.56 ± 0.07 |
| 20 | 2.73 ± 0.03 | −6.93 ± 0.01 | −9.66 ± 0.03 | ||
| 300 | 2.60 ± 0.01 | −8.33 ± 0.01 | −10.93 ± 0.01 | ||
| Z-FHL | 0 | 2.27 ± 0.03 | −7.30 ± 0.01 | −9.57 ± 0.04 | |
| 20 | 2.70 ± 0.04 | −9.06 ± 0.02 | −11.80 ± 0.03 | ||
| Angiotensin I | 0 | 2.16 ± 0.07 | −6.44 ± 0.04 | −8.60 ± 0.10 | |
| 20 | 2.19 ± 0.04 | −7.53 ± 0.00 | −9.72 ± 0.04 | ||
| R186H | HHL | 0 | 2.25 ± 0.01 | −3.75 ± 0.01 | −6.00 ± 0.01 |
| 20 | 2.72 ± 0.02 | −6.77 ± 0.01 | −9.49 ± 0.02 | ||
| Z-FHL | 0 | 2.53 ± 0.06 | −6.92 ± 0.02 | −9.45 ± 0.08 | |
| 20 | 2.74 ± 0.08 | −9.22 ± 0.05 | −11.95 ± 0.07 | ||
| Angiotensin I | 0 | 2.27 ± 0.04 | −6.03 ± 0.01 | −8.30 ± 0.03 | |
| 20 | 2.19 ± 0.01 | −6.97 ± 0.02 | −9.16 ± 0.02 | ||
| E403R | HHL | 0 | 2.62 ± 0.01 | −5.21 ± 0.01 | −7.82 ± 0.02 |
| 20 | 2.93 ± 0.05 | −7.76 ± 0.03 | −10.69 ± 0.04 | ||
| Z-FHL | 0 | 2.41 ± 0.03 | −7.77 ± 0.01 | −10.18 ± 0.04 | |
| 20 | 2.83 ± 0.10 | −8.85 ± 0.01 | −11.67 ± 0.11 | ||
| Angiotensin I | 0 | 2.63 ± 0.05 | −6.73 ± 0.05 | −9.35 ± 0.10 | |
| 20 | 2.75 ± 0.11 | −7.21 ± 0.03 | −9.97 ± 0.10 | ||
| R522Q | HHL | 0 | 2.48 ± 0.03 | −4.46 ± 0.02 | −6.94 ± 0.05 |
| 20 | 2.65 ± 0.16 | −4.90 ± 0.02 | −7.54 ± 0.18 | ||
| Z-FHL | 0 | 2.38 ± 0.05 | −8.41 ± 0.02 | −10.79 ± 0.06 | |
| 20 | 2.51 ± 0.06 | −8.24 ± 0.05 | −10.75 ± 0.01 | ||
| Angiotensin I | 0 | 2.28 ± 0.01 | −7.30 ± 0.01 | −9.59 ± 0.02 | |
| 20 | 2.36 ± 0.11 | −7.45 ± 0.04 | −9.80 ± 0.15 | ||
| R522K | HHL | 0 | 2.29 ± 0.05 | −4.47 ± 0.02 | −6.76 ± 0.07 |
| 20 | 2.24 ± 0.04 | −4.46 ± 0.01 | −6.69 ± 0.04 | ||
| Z-FHL | 0 | 2.22 ± 0.02 | −7.58 ± 0.01 | −9.81 ± 0.03 | |
| 20 | 2.78 ± 0.04 | −7.58 ± 0.02 | −10.36 ± 0.05 | ||
| Angiotensin I | 0 | 2.21 ± 0.04 | −5.39 ± 0.02 | −7.24 ± 0.02 | |
| 20 | 1.85 ± 0.00 | −6.39 ± 0.01 | −8.60 ± 0.04 |
In evaluating the effect of chloride on the thermodynamic profile of the C- and N-domains, the thermodynamic parameters were determined in the presence and absence of chloride (Fig. 4, A and B). Overall, the enthalpy (ΔH) is positive and very similar between the N- and C-domains as well as between substrates for both domains, indicating that the enthalpic contributions are very similar between the domains. What is also evident for all substrates with both domains is the larger contribution to ΔG by entropy (−TΔS) than enthalpy (ΔH), showing that binding is most likely driven by hydrophobic interactions, with desolvation of substrate and structural changes being possible contributions.
FIGURE 4.

Effect of chloride concentration on thermodynamic parameters associated with ACE hydrolysis. Shown is the thermodynamic signature for the hydrolysis of HHL, Z-FHL, and angiotensin I at 0 and 20 mm NaCl by the C-domain (A) and N-domain (B). The values for the C-domain with HHL at 300 mm NaCl are included because this is the concentration of maximal activity and chloride saturation for this substrate and domain (whereas 20 mm is the maximum for the other values). C, the ΔΔG, ΔΔH, and −TΔΔS values for the C-domain and N-domain represent the difference in ΔG, ΔH, and −TΔS between 0 and 20 mm (between 0 and 300 mm for HHL with the C-domain). D, relative Kd(app) values (left y axis) and -fold increase in kcat/Km from 0 mm to maximal activity (right y axis) for C- and N-domains with HHL, Z-FHL, and AngI. Error bars, S.E.
There was a significant decrease in ΔG, associated with reduced catalysis, in the absence of chloride for C- and N-domains using AngI, HHL, and Z-FHL (Fig. 4, A and B). For all three substrates, there were negligible changes in the ΔH values, suggesting that increased activity in the presence of chloride is entropically driven. The fairly consistent ΔH, with and without chloride, between substrates suggests that the main interactions that form part of the catalytic mechanism are preserved, which is consistent with the observation that catalysis does occur in the absence of chloride.
In order to better evaluate the chloride-dependant shifts in thermodynamic data, the changes in ΔG, ΔH, and −TΔS (ΔΔG, ΔΔH, and −TΔΔS) between 0 mm NaCl and the concentration of maximal activity (300 mm for HHL, 20 mm NaCl for Z-FHL and AngI) were calculated for both domains (Fig. 4C). For the C-domain, the greatest change in ΔG is observed with HHL, with reduced change seen for Z-FHL and even less for AngI. These changes in free energy are almost completely entropically-driven, as evidenced by the −TΔS values showing an almost identical trend. There is also almost no change in the enthalpy for AngI, with minor increases for HHL (0.24 kcal/mol) and Z-FHL approaching 0.5 kcal/mol (0.48 kcal/mol). The cleavage of a peptide bond would presumably lead to a lowering of enthalpy by 2–5 kcal/mol. However, it can be assumed that the conformational changes suggested by the negative −TΔΔS values would result in the formation of sufficient interactions, presumably stabilizing in nature, to counterbalance the effect of peptide bond cleavage and result in an overall slightly positive ΔΔH. The entropy-linked increase in enthalpy is probably the result of hydrophobic forces, either via desolvation of hydrophobic groups upon binding of substrate or via the formation of new bonds with a structural change. By contrast, the corresponding ΔΔG, ΔΔH, and −TΔΔS values for the N-domain present far less variation between the different substrates. The ΔΔG and −TΔΔS for HHL are considerably less negative (∼2-fold), whereas, in comparison, those for Z-FHL are relatively unchanged, with the AngI values being slightly more negative than those for the C-domain. The larger entropic shift for AngI hydrolysis with the N-domain is driven by a ∼0.5 kcal/mol increase in the enthalpy, which could be a loss of specific interactions due to variability of interactions with AngI between the two domains.
The largest variation in energetic contributions between domains is seen with HHL, suggesting that the difference in substrate composition, and hence interactions, compared with Z-FHL and AngI could be responsible for the observed shift. To highlight this variation, the Kd(app) values were compared with the -fold increase in kcat/Km between 0 mm NaCl and maximal activity for HHL, Z-FHL, and AngI for both domains (Fig. 4D). The Kd(app) and -fold increase in activity for the C- and N-domains are considerably higher for HHL. This indicates that the chloride dependence mechanisms of the C- and N-domains are most pronounced with the shorter HHL substrate and modulated by the increased length of the Z-FHL and AngI substrates.
Chloride 1 Pocket
The chloride 1 pocket was proposed to be involved in C-terminal stabilization of substrates (10, 33), and amino acid mutations in this pocket abrogated chloride activation with AngI (34). To further investigate the role of the chloride 1 pocket and how it might affect interactions in the S1′ and S2′ subsites, the R186H mutation was evaluated. In this mutation, the chloride-coordinating Arg in the C-domain was converted to a His, the corresponding residue in the N-domain and the only difference in the respective chloride 1 pockets between the two domains. The initial chloride titration results indicated that R186H produced no significant change in chloride binding for both HHL and Z-FHL. A comparison of the relative levels of enzymatic activity for R186H with the C-domain and for HHL, Z-FHL, and AngI at 0 and 20 mm NaCl is shown in Fig. 5, A and B. At 0 mm NaCl, all substrates show an approximate halving of activity relative to the C-domain (Fig. 5A), which can be attributed to increased Km values (Table 4). In the absence of free chloride, this indicates that the chloride 1 pocket probably affects substrate binding. At 20 mm chloride, the AngI kcat/Km for R186H relative to the C-domain is 40.4% (Fig. 5B), which compares favorably with results previously reported for a R186Q mutant (34). These variations in kcat/Km for AngI are due to the Km for R186H (0.095 mm) being almost 3-fold higher than for the C-domain (0.036 mm), with very little change in kcat (7.83 and 7.38 s−1, respectively). This is in contrast to Z-FHL, where the change is due to an almost 2-fold increase in kcat from the C-domain (282.0 s−1) to R186H (509.5 s−1), with a concomitant minor increase in Km (0.120 and 0.150 mm, respectively). HHL hydrolysis showed a similar pattern to AngI, with an increased Km from the C-domain to R186H, yet a change in kcat with Z-FHL is observed, suggesting that interactions in the chloride 1 pocket play a role in mediating both substrate binding and catalysis.
FIGURE 5.

Kinetic and thermodynamic comparisons for R186H. The kcat/Km for the C-domain (black bars) and R186H (white bars) taken as the percentage of the kcat/Km for C-domain at 0 mm (A) and 20 mm (B) NaCl. C, the ΔΔG, ΔΔH, and −TΔΔS values for C-domain and R186H represent the difference in ΔG, ΔH, and −TΔS between 0 and 20 mm NaCl.
Supporting this, a large change in binding affinity with lisinopril for R186H relative to the C-domain is also observed, with the Kd for R186H (132 ± 6.2 nm) being ∼3-fold higher than for the C-domain (45.5 ± 4.24 nm) at 0 mm NaCl, as well as ∼3-fold lower at 20 mm NaCl (4.42 ± 0.5 and 13.5 ± 2.06, respectively) (Table 6). This further indicates that chloride is modulating the conformation of the prime binding sites via the chloride 1 pocket in the C-domain. When comparing the enthalpic change for the C-domain with the R186H mutant, there is some difference (10–50%) upon chloride binding (ΔΔH) for the substrates yet not more than 11% change in the overall ΔH values (Fig. 5C). This suggests that there is no significant variation in interactions and that the effect is probably due to minor structural changes. The lack of a distinct effect on activity-associated chloride binding in the chloride 1 pocket of the R186H mutant supports the theory that the chloride 2 pocket is the main mechanistic effector of chloride dependence and that the chloride 1 pocket exerts its influence primarily via affinity modulation in the prime binding site. This also serves as a key differentiator in chloride-mediated substrate/inhibitor affinity between the domains.
TABLE 6.
Determination of lisinopril binding for all constructs
The Kd values (nm) for lisinopril binding at 0 and 20 mm for the C-domain, the N-domain and a number of C-domain mutant constructs. Constants were determined using an ITC assay at 0 and 20 mm NaCl in 50 mm TAPSO (pH 7.5) and 10 μm ZnSO4. The S.D. value was calculated using the non-linear regression fitting function provided.
| Construct | [NaCl] | Kd |
|---|---|---|
| mm | nm | |
| N-domain | 0 | 47.80 ± 4.94 |
| 20 | 4.61 ± 0.63 | |
| C-domain | 0 | 45.50 ± 4.24 |
| 20 | 13.50 ± 2.06 | |
| R186H | 0 | 132.00 ± 6.72 |
| 20 | 4.42 ± 0.50 | |
| E403R | 0 | 2.67 ± 0.31 |
| 20 | 6.76 ± 0.65 | |
| R522Q | 0 | 22.00 ± 3.31 |
| 20 | 15.90 ± 2.09 | |
| R522K | 0 | 35.30 ± 2.85 |
| 20 | 26.30 ± 1.86 |
In the absence of a crystal structure of the R186H mutant, it is difficult to make any prediction as to what may be occurring and whether chloride would be present in the pocket. In the absence of chloride, the histidine of R186H could potentially form stacking interactions with Trp182 as (N-domain) His164 does with Trp160 and with Arg489 showing similar dual coordination of the Trp279 carbonyl. However, this would not explain why R186Q (34) abrogated chloride dependence.
Chloride 2 Pocket
Although the nature of the prime binding dipeptide and interactions within the chloride 1 pocket may partially explain the varying levels of overall activity between domains, it does not adequately describe how these interactions affect binding of chloride in the chloride 2 pocket, which has been shown to be mechanistically important (35).
In evaluating the crystal structures of the C-domain and the D465T mutant, extensive hydrogen bonding between the Asp465 carboxylate and primary amides of both Tyr520 and Ile521 is seen (Fig. 3B). Tyr520, Ile521, Arg522, and Tyr523 are the first four residues on the C-terminal end of the α23 helix (Tyr520–Ala541), with Pro519 representing the end of the helix and the beginning of a highly variable loop. However, this region of the α23 helix is within subdomain II of both domains, a region predicted to have little major structural variation relative to the proposed hinge region that is part of subdomain I (8). The C-terminal end of α23 is the main point of contact with α20, supporting a structural role for Asp465. Furthermore, in the C-domain, Asp465 hydrogen-bonds with Arg468, which interacts strongly with the variable loop via hydrogen bonds with backbone primary carbonyls of His513 and Val518. In the N-domain, Asp443 (which corresponds to C-domain Asp465) does not interact with Arg446 (Arg468 in the C-domain), but Arg446 does hydrogen-bond with Thr496, which corresponds to Val518 in the C-domain. Both Val518, in the S1 subpocket, and His513, which interacts with Tyr523 (10), are important residues for peptide binding that reside on this variable loop (36). Given that Asp465 is one of very few contacts between helices α23 and α20, its role is most likely in stabilization of the C-terminal end of helix α23.
The chloride titration data determined for HHL and Z-FHL using the D465T mutant (Table 7) support this assertion. The maximum specific activity in the presence of chloride (SAmax) and the activity at 0 mm NaCl as a percentage of maximal activity (%SAmax) for D465T with both HHL and Z-FHL were lower than those of the C-domain. An increase in Kd(app) for Z-FHL and a decrease for HHL with D465T was also observed. The location of Asp465 on the side of the chloride 2 pocket opposite from the proposed chloride channel area precludes any direct interaction with the chloride ion and involvement in the chloride channel mechanism. These results may be explained by the fact that Asp465 stabilizes the C-terminal end of helix α23 and the variable loop via hydrogen bonding with the primary amides of Tyr520 and Ile521. The Thr in this position is capable of fewer potential hydrogen bonds (Fig. 3B and Table 3), resulting in more structural movement that affects the C-terminal carboxylate coordinating Tyr520 (lowering of SAmax). Its effect on %SAmax and Kd(app) is probably due to structural destabilization, illustrated by the rearrangement of Trp469 (Fig. 3B), which suggests that structural rotation or movement around the C-terminal end of helix α23, which contains Arg522 and Tyr523, moderates not only activity but chloride affinity.
TABLE 7.
Effect of NaCl on the hydrolysis of HHL and Z-FHL synthetic peptides by ACE constructs
All values were obtained from chloride-titrated activity assays. Units for SAmin and SAmax are milliunits·μg−1, and Kd(app) values are in mm. Data shown represent the average of two independent determinations, each in triplicate. The approximation symbol indicates that accurate estimates of Kd(app) and SAmax could not be obtained because the chloride concentration range was insufficient for saturation to occur. SAmax for these values is activity at 500 and 50 mm NaCl for HHL and Z-FHL, respectively.
| Construct | HHL |
Z-FHL |
||||||
|---|---|---|---|---|---|---|---|---|
| Kd(app) | SAmin | SAmax | SAmin % of SAmax | Kd(app) | SAmin | SAmax | SAmin % of SAmax | |
| mm | milliunits/μg | milliunits/μg | % | mm | milliunits/μg | milliunits/μg | % | |
| N-domain | 0.2 | 10.5 | 24.2 | 43.5 | 0.8 | 36.5 | 206.7 | 17.6 |
| C-domain | 46.9 | 2.1 | 83.2 | 2.5 | 1.8 | 93.5 | 339.3 | 27.5 |
| R186H | 76.9 | 3.7 | 113.3 | 3.2 | 2.1 | 130.3 | 467.2 | 27.8 |
| E403R | 2.7 | 6.3 | 99.8 | 6.3 | 0.3 | 214.5 | 398.5 | 53.8 |
| D465T | 12.7 | 0.4 | 46.6 | 0.8 | 2.8 | 2.1 | 110.7 | 1.8 |
| R522K | ∼339.7 | 4.1 | ∼10.0 | 40.5 | ∼104.6 | 97.2 | ∼193.2 | 50.3 |
| R522Q | ∼417.8 | 4.6 | ∼11.2 | 40.8 | ∼231.9 | 161.9 | ∼218.0 | 74.3 |
Thus, the interaction between Arg522 and Tyr523 is likely to influence chloride activation. The crystal structure of native C-domain showed the possible cation-π bond between these two residues, whereas the structures of the two 522 mutants demonstrated little structural change apart from the loss of chloride coordination.
Given these potential interactions, the kinetic data obtained for R522K and R522Q mutants relative to C-domain can be assessed. Comparing the Kd(app) values for HHL and Z-FHL (Fig. 6A), the largest increases relative to the C-domain are seen for HHL with R522K (7-fold) and R522Q (9-fold). A similar trend is seen with the Kd(app) values for Z-FHL and AngI (despite the lack of R552Q Kd(app)) although not to the same degree as for HHL.
FIGURE 6.

Trends in chloride binding and activity for R522Q and R522K. Shown is a graphical representation of chloride binding and kinetic values obtained for C-domain (black bars), R522K (gray bars), and R522Q (white bars) with HHL, Z-FHL, and AngI as substrates. A, the Kd(app) values (mm) for chloride binding. *, AngI values were estimated from the report of Liu et al. (35). The kcat/Km value was taken as the percentage of the kcat/Km for the C-domain at 0 mm (B) and 20 mm (C) NaCl. D, change in thermodynamic parameters for R522Q and R522K. The ΔΔG, ΔΔH, and −TΔΔS values for C-domain, R522Q, and R522K with HHL, Z-FHL, and AngI are shown and represent the difference in ΔG, ΔH, and −TΔS between 0 and 20 mm NaCl.
The replacement of Arg522 with a lysine or a glutamine causes a decrease in chloride affinity, in agreement with the structural data. Furthermore, R522K and R522Q follow the same Kd(app) trend as the C-domain (AngI < Z-FHL < HHL) although proportionally higher than the C-domain for each. This indicates that substrate interactions are somehow moderating access and binding of the chloride ion. This is confirmed by the presence of chloride in the pocket of R522K only when a ligand is bound at the active site (and captopril is a peptide mimetic). Substrate binding would cause local restructuring to interact with the catalytic site and thereby also close access to the chloride pocket. The sulfhydral group of captopril coordinates the zinc ion, thereby causing a loss of the hydrogen bond from Tyr523 seen in the tACE·lisinopril complex (32) and the transition state intermediate of the substrate. This could allow for more favorable interaction with the chloride ion.
Other interactions in the chloride 2 pocket are the salt bridge between Lys522 and Glu411 as well as the cation-π interaction of charged 522 residues with Tyr523. As would be expected with disruption of a key active site residue, the maximal kcat/Km values for R522K and R522Q are lower than that for the C-domain, with R522K lower than R522Q (Table 3). To better gauge the relative effects, the kcat/Km values for R522K and R522Q at 0 and 20 mm NaCl are represented as a percentage of the kcat/Km values for the C-domain (Fig. 6, B and C). At 0 mm NaCl, R522Q shows a marked increase in activity over both C-domain and R522K with Z-FHL and AngI (Fig. 6B), suggesting that additional interactions for the 522 position beyond chloride coordination are important. At 20 mm NaCl, there is an increase in activity for both R522K and R522Q with larger substrates that interact more tightly with the S1 and S2 active site pockets (Fig. 6C). Although R522K shows only a marginal increase over HHL (AngI (15.6%) > Z-FHL (9.3%) > HHL (0.1%)) relative to C-domain at 20 mm NaCl, there is a marked increase for R522Q (AngI (87.7%) > Z-FHL (27.5%) > HHL (0.36%)) to the point where R522Q activity is almost as high as C-domain activity with AngI. This would indicate that the bonding pattern is different between R522K and R522Q and that interactions of longer substrates have an effect on either the chloride 2 pocket or a key catalytic mechanism. To further probe this trend, the contributions of Km and kcat to the catalytic efficiency of R522Q and R522K are evaluated. For HHL, the mechanistic lack of activation is demonstrated by the low variation in Km but marked differences in the kcat, where values are similar at 0 mm chloride and only C-domain showing an increase at 20 mm. Interestingly, R522Q shows a 5-fold lower Km for Z-FHL at 0 mm NaCl than C-domain, yet this trend is reversed at 20 mm, with the Km for R522Q being ∼2-fold higher than that for the C-domain. The data show that Km for R522K and R522Q with Z-FHL is higher at 20 mm than at 0 mm, a reversal of the general trend for the C-domain with all substrates. Both mutants show less than 2-fold increases in kcat from 0 to 20 mm NaCl, versus the greater than 3-fold increase observed for the C-domain. Thus, the large decrease in activation for R522Q and R522K with Z-FHL is influenced by the increased Km as well as the reduced kcat associated with the presence of chloride, in contrast to HHL, which is predominantly influenced by kcat. Interestingly, a similar lack of variance is seen in Km values for R522Q and R522K with AngI and greater variation in kcat. The increases in Km observed for all substrates with R522K and, to a lesser degree, R522Q indicate that coordination by chloride in this pocket affects substrate affinity as well as catalysis, suggesting some structural movement around helix α23. The most noticeable effect of the R522Q mutation is the greater kcat values that increase with substrate length and are even higher than C-domain for AngI. Taking into account the structural data, it is likely that the lack of chloride coordination allows for more freedom of movement at the S1 recognition site, particularly for Tyr523, thus resulting in more efficient catalysis. It is therefore unlikely that the cation-π interaction is favored over the hydrogen bonds between Lys522 and Glu411 and between Gln522 and α23, respectively.
The changes in thermodynamic profiles in the presence of chloride for R522Q and R522K were evaluated (Fig. 6D). For R522Q, a reduced endothermic change in enthalpy is seen for HHL (0.165 kcal/mol) and ZHFL (0.105 kcal/mol) relative to the C-domain (0.36 and 0.48 kcal/mol, respectively), which would indicate the loss of an interaction(s) with the binding site. That the ΔΔH values do not exceed the minimum hydrogen bonding energy (1 kcal/mol) might be due to differing hydration effects associated with the different residue counteracting the enthalpic change.
For R522K, there is a different pattern with the enthalpy changes, with HHL (−0.07 kcal/mol) showing an exothermic shift relative to the C-domain (0.36 kcal/mol), AngI (0.35 kcal/mol) showing an endothermic shift relative to the C-domain (0.02 kcal/mol), and Z-FHL relatively unchanged. As with R522Q, an exothermic shift would suggest a bond breaking for HHL, which would be expected with the lack of direct chloride coordination. The lack of variation in enthalpy with Z-FHL for R522K is interesting, considering that there is no ΔΔG and the kcat/Km is lower than C-domain and R522Q at both 0 and 20 mm NaCl. Given the assertions that Lys neither coordinates chloride nor cation-π-bonds with Tyr523, the interactions of Z-FHL in the S1 and S2 pockets may be compensating via minor structural shifts. The endothermic shift in enthalpy observed with AngI for R522K is accompanied by a relatively large shift in free energy (−0.99 kcal/mol) similar in magnitude to that in the C-domain (−1.09 kcal/mol), which actually results in a larger −TΔΔS value due to the more endothermic enthalpy shift. There is a 5-fold increase in kcat/Km from 0 to 20 mm NaCl, resulting in the observed ΔΔG, although these values are considerably lower than in the C-domain. The modeling of AngI bound to C-domain (Fig. 7) based on the structure of the bound (cleaved) peptide complex structure (36) showed the possible interaction between Arg522 and the substrate through a number of water-mediated hydrogen bonds. Although it is unlikely that specific side chains are involved, the peptide backbone (positions P1 and P3) is within the distance (5.5 Å) of the Arg522 hydration shell. These important interactions may account for the chloride-mediated ΔΔG as R522K and the catalytic site overall (through bonding with Glu411 and α17) lose the stability provided by the chloride ion.
FIGURE 7.
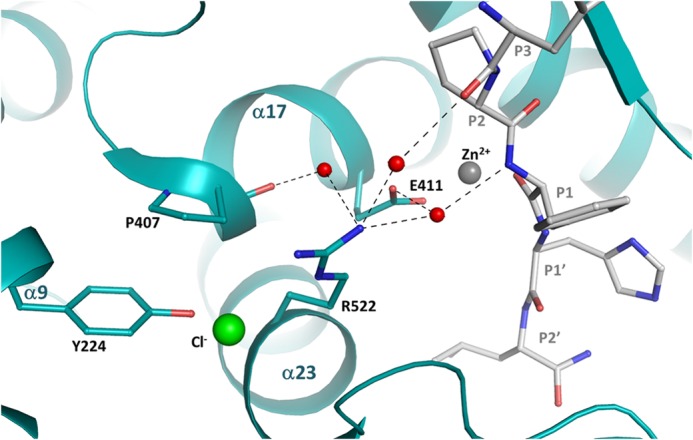
Model of angiotensin I binding to C-domain ACE. The structure of the C-domain ACE·Ang complex (Protein Data Bank code 4APH (36)) was used to model Angiotensin I before catalysis. The AngI peptide is represented in gray, chloride is shown in green, zinc ion is shown in gray, and water molecules are shown in red. The potential hydrogen bonds are shown in dashed lines. Residue 522 is presented in a stick representation.
Chloride coordination appears necessary to stabilize Arg522 toward the catalytic channel, thereby promoting catalysis of substrate. Substrate interactions within the S1/S2 pockets affect positioning of this complex and provide a rationale whereby substrate binding modulates chloride affinity and hence its own catalysis.
Chloride Channel
The E403R mutation was generated to investigate a proposed chloride channel, where Glu403 may act as an ionic gate mediating access of chloride to the chloride 2 pocket (37). For the E403R mutation, the Glu was converted to an Arg, corresponding to Arg381 in the N-domain (Table 1). The E403R mutation significantly reduced Kd(app) for chloride binding with both HHL and Z-FHL relative to the C-domain (Table 6). This effect was reversible in the N-domain when Arg381 was converted to a Glu, where the Kd(app) values showed 29- and 15-fold increases for HHL (4.7 mm) and Z-FHL (11.4 m) over the N-domain (0.2 and 0.8 mm, respectively). Furthermore, substrate interactions in the S2 subpocket where Glu403 resides might disrupt any ion gating, as evidenced by the large discrepancy in Kd(app) seen between HHL and Z-FHL for the C-domain, which was largely abolished with E403R.
Interestingly, Kd(app) varies with different substrates, suggesting that chloride coordination is affected, but changes in overall activity and degrees of activation are also seen, thus highlighting further functions for Glu403. The E403R kcat/Km values are shown relative to the C-domain at 0 mm NaCl (Fig. 8A) and at maximal activity (Fig. 8B) for HHL, Z-FHL, and AngI. The kcat/Km values for E403R with all substrates at maximal activity are generally lower relative to the C-domain, whereas at 0 mm NaCl, they are all higher. This indicates that Glu403 affects activity as well as potentially mediating access to the chloride 2 pocket. Glu403 makes a salt bridge with Lys118 in the native structure (Fig. 9). Mutation of the Lys118-corresponding residues, Lys154 in rabbit testicular ACE (38) and Lys694 of rabbit lung ACE (39), drastically reduced chloride sensitivity, thus highlighting its critical role. Intriguingly, the Glu403-Lys118 salt bridge is disrupted in the complex structure of ACE bound to the inhibitory peptide BPPb (36), where Glu403 was seen to make a salt bridge with the P2 residue (Lys) of the peptide (Fig. 9). Thus, Glu403 certainly plays a role in substrate binding through the S2 subsite. The E403R mutation resulted in a loss of the strong interaction with Lys118, although the residues are within hydrogen bonding distance (Fig. 3A). Lys118 proved less stable (as indicated by the weak electron density for its side chain; Fig. 2A). Thus, E403R and the N-domain lack this stabilizing salt bridge and show lower Kd(app) values for chloride binding (Table 7), suggesting that the chloride pocket is in a conformation that can more readily coordinate chloride.
FIGURE 8.

Trends in chloride binding and activity for E403R. Shown is a graphical representation of kcat/Km kinetic values obtained for the C-domain (black bars) and E403R (white bars) with HHL, Z-FHL, and AngI as substrates. The kcat/Km value was taken as the percentage of the kcat/Km for the C-domain at 0 mm (A) and 20 mm (B) NaCl. C, change in thermodynamic parameters for E403R. The ΔΔG, ΔΔH, and −TΔΔS values for the C-domain and E403R are shown and represent the difference in ΔG, ΔH, and −TΔS between 0 and 20 mm NaCl (0 and 300 mm for HHL with C-domain).
FIGURE 9.
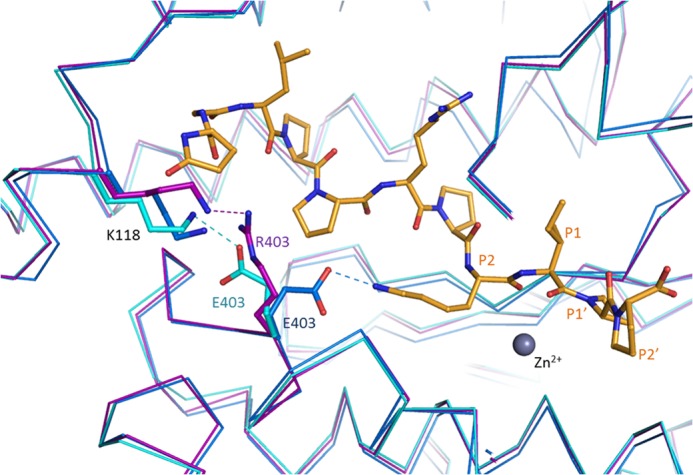
Comparison of C-domain ACE bound to peptide inhibitor BPPb and the E403R mutant structures. The structures of the C-domain ACE (cyan; Protein Data Bank code 4APJ (36)) and C-domain ACE·BPPb complex (blue; Protein Data Bank code 4APJ (36)) were superposed onto the C-domain E403R structure (magenta). The BPPb peptide is represented in yellow, and the zinc ion is shown in gray (not present in the BPPb complex structure). Interactions with position 403 are shown as dashed lines (colors correspond to the respective structures).
Further evidence of the stabilizing role of the Glu403-Lys118 interaction is seen in the change in thermodynamic parameters for E403R relative to the C-domain (Fig. 8C). The ΔΔG values are reduced for all substrates relative to the C-domain, which is due to the observed increase in kcat/Km at 0 mm NaCl and decrease at 20 mm NaCl (Fig. 8, A and B). That the pattern and magnitude of the enthalpies (ΔΔH) for the substrates correlates well with the C-domain indicates that there should be no major structural rearrangement. The lack of significant enthalpy changes do not discount minor structural differences, which are evidenced by less favorable changes in entropy (−TΔΔS). These minor structural effects would help explain the Km and kcat data for E403R relative to C-domain (Table 4). E403R shows increased kcat values at maximal activity for Z-FHL and AngI (929.34 and 9.08 s−1) relative to C-domain (282.00 and 7.38 s−1), which is countered by higher Km values in the presence of chloride for E403R (0.530 and 0.061 mm) over the C-domain (0.120 and 0.036 mm). These result in lower maximal kcat/Km values with both substrates for E403R but serve to confirm that some structural variation is present. The HHL substrate does not appear to interact strongly within the S2 region, and it is likely that its affinity is predominantly affected by S1′/S2′ interactions.
These results provide evidence for a stabilizing role for Glu403 in the C-domain via its interaction with Lys118 within the hinge-bending region rather than a functional ion-gated channel. The salt bridge serves to reduce the affinity of chloride within the chloride 2 pocket of C-domain as well as affect the structural architecture of the S2 pocket. Substrate interactions within this pocket, exemplified by Z-FHL and to a greater extent AngI, appear to disrupt the Glu403-Lys118 interaction and allow for improved chloride binding. The absence of interaction in the N-domain means that the chloride 2 pocket would have a higher affinity for chloride, even in the absence of any substrate interaction within the S2 pocket. The size of the substrate and, more importantly, how far it extends into the non-prime subsites has been shown to be important where substrate interactions in the S2 pocket modulate chloride affinity directly in the C-domain of ACE. These interactions are likely to be propagated via structural perturbations around the active site rather than any specific interaction, which is consistent with the high degree of variability in ACE substrate identity.
Substrate-mediated Chloride Dependence
The interactions and mechanisms within the two chloride pockets have been characterized using structural, mutagenic, and kinetic data to highlight the close relationship between the chloride dependence and substrate composition. Arg522 interacts indirectly with Glu411 and Tyr523, thus affecting the catalytic site by stabilizing the enzyme-substrate transition state intermediate. Importantly, the affinity of chloride coordinating Arg522 is moderated by structural constraint via the Glu403-Lys118 salt bridge in the C-domain of ACE, which is a point of major difference with the N-domain. Arg522 is part of α23, which includes other important residues for recognition of the substrate C-terminal carboxylate along with Lys511 situated on the 310 H6 helix (36). An adjacent variable loop completes the S1 subsite and includes His513, probably involved in stabilizing the transition state (40); thus, minor structural movement in this area can have a large effect on catalysis and chloride binding and is influenced by substrate interactions.
The effect of substrate composition on activity can be inferred from the kinetic and structural data. The tripeptide HHL shows a highly chloride-dependent profile exemplified by a high Kd(app). The P1′ and P2′ residue interactions within their respective pockets have an unfavorable influence on chloride affinity, presumably via interaction or non-interaction with Tyr523 and Tyr520 (Fig. 10). Longer peptides (Z-FHL and AngI), although sharing the same P′ composition as HHL, have been shown not to reduce chloride binding. The results with both Z-FHL and AngI show a much tighter Kd(app) for chloride binding; thus, they are likely to moderate chloride affinity via interaction with the S2 pocket. This may be mediated by disruption of the Glu403-Lys118 salt bridge. It is possible that the greater length of AngI has a significant structural effect on the chloride pocket, which may also account for the relatively lower kcat values.
FIGURE 10.
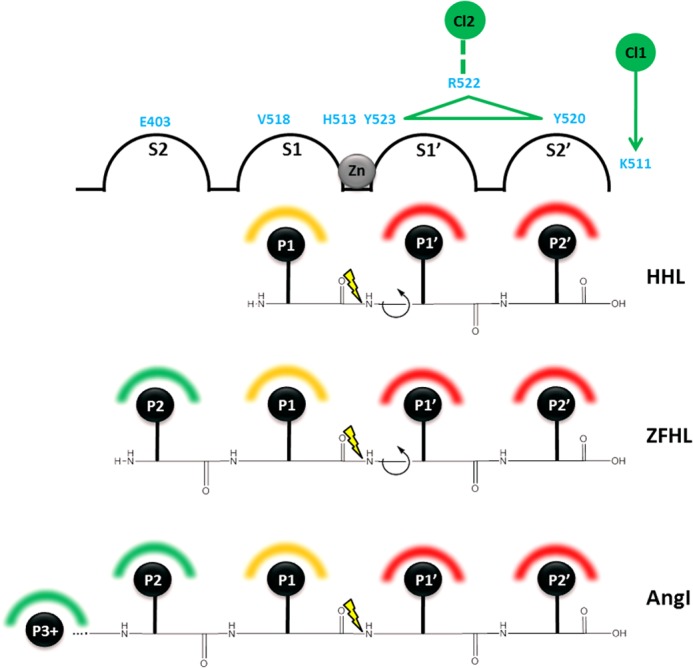
Schematic representation of substrate binding modes in C-domain ACE. Binding of substrate to the active site is shown using an adapted Schechter and Berger diagram (42), with active site subpockets (S2, S1, S1′, and S2′) shown as linked half-spheres and peptides shown as labeled black spheres (P2, P1, P1′, and P2′, with P3+ indicating residues P3–P10 for AngI). Positions of C-domain ACE residues (light blue) are shown above the relevant subsites, with helix α23 containing Arg522, Tyr523, and Tyr520 shown as a green triangle. Chloride ions (numbered green spheres) are shown, with chloride coordination to Arg522 indicated by a dashed green line and the influence of chloride on Lys511 shown by a green arrow. The active site zinc (gray sphere) indicates the position of the catalytic mechanism between S1 and S1′, with yellow lightning showing the position of the scissile bond for each peptide. Interactions that increase chloride affinity (green-shaded semicircles), those that do not increase chloride affinity (red-shaded semicircles), and interactions that may affect chloride affinity (orange-shaded semicircles) are indicated above each peptide residue.
The role of the chloride 1 pocket in C-domain ACE is less clear; however, we show that it does have some effect on chloride dependence. The chloride within this pocket may be present in a structural role, and there may not be any significant dynamic movement in this pocket. Alternatively, Moiseeva et al. (41) suggested that chloride binding in this pocket may have an inhibitory role at high chloride concentrations. The latter possibility agrees with our proposal, where binding of chloride could affect C-terminal carboxylate coordination within the S2′ pocket and thereby moderate activity via those interactions; however, this requires further investigation.
CONCLUSION
Key amino acid mutations in the C-domain of ACE showed marked effects on chloride dependence when evaluated using kinetic and thermodynamic data determined for the hydrolysis of HHL, Z-FHL, and AngI under various chloride concentrations. The interpretation was complemented with the molecular details provided by the determination of the crystal structures for four of the mutants. A model of interactions in the chloride 1 pocket was developed to explain how chloride binding might modulate Lys511 coordination and potentially S2′ pocket conformation in the C-domain. Interactions between Arg522 and the zinc-coordinating Glu411 as well as with the transition state-stabilizing residue Tyr523 are described, the relevance of which was supported by interpretation of the kinetic and thermodynamic effects of the R522Q and R522K mutants. This also suggested that chloride binding was most likely affected by subtle structural effects that could be modulated by differing substrate interactions. Interpretation of the E403R mutant kinetics and thermodynamics allowed refinement of this concept and demonstrated that Glu403 in the C-domain of ACE plays a role in reducing chloride affinity in the chloride 2 pocket via a salt bridge with Lys118, an interaction that is not present in the N-domain and which probably represents the primary differentiation in chloride dependence between the domains. Finally, the different ways in which substrate interactions can modulate chloride affinity via the S′ or S2 pockets are described. This study has provided a framework to describe chloride dependence of the ACE N- and C-catalytic sites and will contribute considerably to substrate selectivity studies as well as the development of domain-selective inhibitors for the improved treatment of hypertension (C-selective ACEi) and fibrosis (N-selective ACEi).
Acknowledgments
We thank the scientists at stations I02, I04, and I04-1 at the Diamond Light Source (Didcot, Oxon, UK) for support during x-ray diffraction data collection.
This work was supported by Medical Research Council Project Grant G1001685 and Wellcome Trust Equipment Grant 088464 (to K. R. A.). This work was also supported by the University of Cape Town and the South African National Research Foundation (to E. D. S. and C. J. Y.).
- ACE
- angiotensin-I-converting enzyme
- sACE
- somatic angiotensin-converting enzyme
- tACE
- testicular ACE
- AngI and AngII
- angiotensin I and II, respectively
- ITC
- isothermal titration calorimetry
- HHL
- hippuryl-l-His-Leu
- Z
- benzyloxycarbonyl
- SA
- specific activity
- BppB
- bradykinin potentiating peptide B
- TAPSO
- 3-[[1,3-dihydroxy-2-(hydroxymethyl)propan-2-yl]amino]-2-hydroxypropane-1-sulfonic acid.
REFERENCES
- 1. Acharya K. R., Sturrock E. D., Riordan J. F., Ehlers M. R. (2003) ACE revisited. A new target for structure-based drug design. Nat. Rev. Drug Discov. 2, 891–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Watermeyer J. M., Kroger W. L., Sturrock E. D., Ehlers M. R. (2009) Angiotensin-converting enzyme–New insights into structure, biological significance and prospects for domain-selective inhibitors. Curr. Enzyme Inhib. 5, 134–147 [Google Scholar]
- 3. Anthony C. S., Masuyer G., Sturrock E. D., Acharya K. R. (2012) Structure based drug design of angiotensin-I converting enzyme inhibitors. Curr. Med. Chem. 19, 845–855 [DOI] [PubMed] [Google Scholar]
- 4. Corvol P., Eyries M., Soubrier F. (2004) Handbook of Proteolytic Enzymes, (Barrett A. J., Rawlings N. D., Woessner J. F., eds) pp. 332–346, Elsevier Academic Press, San Diego [Google Scholar]
- 5. Hubert C., Houot A. M., Corvol P., Soubrier F. (1991) Structure of the angiotensin I-converting enzyme gene. Two alternate promoters correspond to evolutionary steps of a duplicated gene. J. Biol. Chem. 266, 15377–15383 [PubMed] [Google Scholar]
- 6. Natesh R., Schwager S. L., Sturrock E. D., Acharya K. R. (2003) Crystal structure of the human angiotensin-converting enzyme-lisinopril complex. Nature 421, 551–554 [DOI] [PubMed] [Google Scholar]
- 7. Corradi H. R., Schwager S. L., Nchinda A. T., Sturrock E. D., Acharya K. R. (2006) Crystal structure of the N domain of human angiotensin I-converting enzyme provides a structural basis for domain specific inhibitor design. J. Mol. Biol. 357, 964–974 [DOI] [PubMed] [Google Scholar]
- 8. Watermeyer J. M., Sewell B. T., Schwager S. L., Natesh R., Corradi H. R., Acharya K. R., Sturrock E. D. (2006) Structure of testis ACE glycosylation mutants and evidence for conserved domain movement. Biochemistry 45, 12654–12663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Matthews B. W. (1988) Structural basis of the action of thermolysin and related zinc peptidases. Acc. Chem. Res. 21, 333–340 [Google Scholar]
- 10. Sturrock E. D., Natesh R., van Rooyen J. M., Acharya K. R. (2004) Structure of angiotensin I-converting enzyme. Cell Mol. Life Sci. 61, 2677–2686 [DOI] [PubMed] [Google Scholar]
- 11. Shapiro R., Holmquist B., Riordan J. F. (1983) Anion activation of angiotensin converting enzyme. Dependence on nature of substrate. Biochemistry 22, 3850–3857 [DOI] [PubMed] [Google Scholar]
- 12. Paul M., Poyan Mehr A., Kreutz R. (2006) Physiology of local renin-angiotensin systems. Physiol. Rev. 86, 747–803 [DOI] [PubMed] [Google Scholar]
- 13. Abadir P. M., Foster D. B., Crow M., Cooke C. A., Rucker J. J., Jain A., Smith B. J., Burks T. N., Cohn R. D., Fedarko N. S., Carey R. M., O'Rourke B., Walston J. D. (2011) Identification and characterization of a functional mitochondrial angiotensin system. Proc. Natl. Acad. Sci. U.S.A. 108, 14849–14854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lodish H. F., Berk A., Zipursky L., Matsudaira P., Baltimore D., Darnell J. (1999) Molecular Cell Biology, 4th Ed., Scientific American Books, New York [Google Scholar]
- 15. Wei L., Clauser E., Alhenc-Gelas F., Corvol P. (1992) The two homologous domains of human angiotensin I-converting enzyme interact differently with competitive inhibitors. J. Biol. Chem. 267, 13398–13405 [PubMed] [Google Scholar]
- 16. Gordon K., Redelinghuys P., Schwager S. L., Ehlers M. R., Papageorgiou A. C., Natesh R., Acharya K. R., Sturrock E. D. (2003) Deglycosylation, processing and crystallization of human testis angiotensin-converting enzyme. Biochem. J. 371, 437–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yu X. C., Sturrock E. D., Wu Z., Biemann K., Ehlers M. R., Riordan J. F. (1997) Identification of N-linked glycosylation sites in human testis angiotensin-converting enzyme and expression of an active deglycosylated form. J. Biol. Chem. 272, 3511–3519 [DOI] [PubMed] [Google Scholar]
- 18. Balyasnikova I. V., Metzger R., Franke F. E., Danilov S. M. (2003) Monoclonal antibodies to denatured human ACE (CD 143), broad species specificity, reactivity on paraffin sections, and detection of subtle conformational changes in the C-terminal domain of ACE. Tissue Antigens 61, 49–62 [DOI] [PubMed] [Google Scholar]
- 19. Anthony C. S., Corradi H. R., Schwager S. L., Redelinghuys P., Georgiadis D., Dive V., Acharya K. R., Sturrock E. D. (2010) The N domain of human angiotensin-I converting enzyme. The role of N-glycosylation and the crystal structure in complex with an N-domain-specific phosphinic inhibitor, RXP407. J. Biol. Chem. 285, 35685–35693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Leslie A. G. W., Powell H. R. (2007) Processing diffraction data with Mosflm. in Evolving Methods for Macromolecular Crystallography (Read R. J., Sussman J. L., eds) pp. 41–51, Springer, Dordrecht, The Netherlands [Google Scholar]
- 22. Collaborative Computing Project, Number 4 (1994) The CCP4 suite. Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 23. McCoy A. J., Grosse-Kuntsleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 25. Brünger A. T. (1992) Free R-value. A novel statistical quantity for assessing the accuracy of crystal structures. Nature 355, 472–475 [DOI] [PubMed] [Google Scholar]
- 26. Emsley P., Cowtan K. (2004) Coot. Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 27. Davis I. W., Leaver-Fay A., Chen V. B., Block J. N., Kapral G. J., Wang X., Murray L. W., Arendall W. B., 3rd, Snoeyink J., Richardson J. S., Richardson D. C. (2007) MolProbity. All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Friedland J., Silverstein E. (1976) A sensitive fluorimetric assay for serum angiotensin-converting enzyme. Am. J. Clin. Pathol. 66, 416–424 [DOI] [PubMed] [Google Scholar]
- 29. Stockbridge R. B., Wolfenden R. (2009) The intrinsic reactivity of ATP and the catalytic proficiencies of kinases acting on glucose, N-acetylgalactosamine, and homoserine. A thermodynamic analysis. J. Biol. Chem. 284, 22747–22757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Golicnik M. (2010) Explicit reformulations of time-dependent solution for a Michaelis-Menten enzyme reaction model. Anal. Biochem. 406, 94–96 [DOI] [PubMed] [Google Scholar]
- 31. Corless R. M., Gonnet G. H., Hare D. E. G., Jeffrey D. J., Knuth D. E. (1996) On the Lambert W function. Adv. Comput. Math. 5, 329–359 [Google Scholar]
- 32. Natesh R., Schwager S. L., Evans H. R., Sturrock E. D., Acharya K. R. (2004) Structural details on the binding of hypertensive drugs captopril and enalaprilat to human testicular angiotensin I-converting enzyme. Biochemistry 43, 8718–8724 [DOI] [PubMed] [Google Scholar]
- 33. Guy J. L., Jackson R. M., Acharya K. R., Sturrock E. D., Hooper N. M., Turner A. J. (2003) Angiotensin-converting enzyme-2 (ACE2). Comparative modeling of the active site, specificity requirements, and chloride dependence. Biochemistry 42, 13185–13192 [DOI] [PubMed] [Google Scholar]
- 34. Rushworth C. A., Guy J. L., Turner A. J. (2008) Residues affecting the chloride regulation and substrate selectivity of the angiotensin-converting enzymes (ACE and ACE2) identified by site-directed mutagenesis. FEBS J. 275, 6033–6042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu X., Fernandez M., Wouters M. A., Heyberger S., Husain A. (2001) Arg1098 is critical for the chloride dependence of human angiotensin I-converting enzyme C-domain catalytic activity. J. Biol. Chem. 276, 33518–33525 [DOI] [PubMed] [Google Scholar]
- 36. Masuyer G., Schwager S. L., Sturrock E. D., Isaac R. E., Acharya K. R. (2012) Molecular recognition and regulation of human angiotensin-I converting enzyme (ACE) activity by natural inhibitory peptides. Sci. Rep. 2, 717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tzakos A. G., Galanis A. S., Spyroulias G. A., Cordopatis P., Manessi-Zoupa E., Gerothanassis I. P. (2003) Structure-function discrimination of the N- and C-catalytic domains of human angiotensin-converting enzyme. Implications for Cl− activation and peptide hydrolysis mechanisms. Protein Eng. 16, 993–1003 [DOI] [PubMed] [Google Scholar]
- 38. Sen I., Kasturi S., Abdul Jabbar M., Sen G. C. (1993) Mutations in two specific residues of testicular angiotensin-converting enzyme change its catalytic properties. J. Biol. Chem. 268, 25748–25754 [PubMed] [Google Scholar]
- 39. Chen Y. N., Riordan J. F. (1990) Identification of essential tyrosine and lysine residues in angiotensin converting enzyme. Evidence for a single active site. Biochemistry 29, 10493–10498 [DOI] [PubMed] [Google Scholar]
- 40. Fernandez M., Liu X., Wouters M. A., Heyberger S., Husain A. (2001) Angiotensin I-converting enzyme transition state stabilization by His1089. Evidence for a catalytic mechanism distinct from other gluzincin metalloproteinases. J. Biol. Chem. 276, 4998–5004 [DOI] [PubMed] [Google Scholar]
- 41. Moiseeva N. A., Binevski P. V., Baskin I. I., Palyulin V. A., Kost O. A. (2005) Role of two chloride-binding sites in functioning of testicular angiotensin-converting enzyme. Biochemistry 70, 1167–1172 [DOI] [PubMed] [Google Scholar]
- 42. Schechter I., Berger A. (1967) On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 27, 157–162 [DOI] [PubMed] [Google Scholar]