

Background: The Na+/Cl−-dependent serotonin transporter contains two putative Na+-binding sites (Na1 and Na2).
Results: Mutations at Na1, but not Na2, allow Ca2+ to replace Na+ and alter the conducting properties of the transporter.
Conclusion: Ca2+ binds at Na1, but in the Na1 mutant, it does not appear to be transported.
Significance: This work uncovers distinct roles of the Na+-binding sites for serotonin transporter function.
Keywords: Electrophysiology, Homology Modeling, Monoamine Transporters, Serotonin Transporters, Transporters, SLC6, Ion Dependence, Structure-Function
Abstract
Neurotransmitter transporters of the SLC6 family of proteins, including the human serotonin transporter (hSERT), utilize Na+, Cl−, and K+ gradients to induce conformational changes necessary for substrate translocation. Dysregulation of ion movement through monoamine transporters has been shown to impact neuronal firing potentials and could play a role in pathophysiologies, such as depression and anxiety. Despite multiple crystal structures of prokaryotic and eukaryotic SLC transporters indicating the location of both (or one) conserved Na+-binding sites (termed Na1 and Na2), much remains uncertain in regard to the movements and contributions of these cation-binding sites in the transport process. In this study, we utilize the unique properties of a mutation of hSERT at a single, highly conserved asparagine on TM1 (Asn-101) to provide several lines of evidence demonstrating mechanistically distinct roles for Na1 and Na2. Mutations at Asn-101 alter the cation dependence of the transporter, allowing Ca2+ (but not other cations) to functionally replace Na+ for driving transport and promoting 5-hydroxytryptamine (5-HT)-dependent conformational changes. Furthermore, in two-electrode voltage clamp studies in Xenopus oocytes, both Ca2+ and Na+ illicit 5-HT-induced currents in the Asn-101 mutants and reveal that, although Ca2+ promotes substrate-induced current, it does not appear to be the charge carrier during 5-HT transport. These findings, in addition to functional evaluation of Na1 and Na2 site mutants, reveal separate roles for Na1 and Na2 and provide insight into initiation of the translocation process as well as a mechanism whereby the reported SERT stoichiometry can be obtained despite the presence of two putative Na+-binding sites.
Introduction
Secondary active solute transporters are a critical feature of biological systems that utilize chemiosmotic gradients to support energetically unfavorable concentrative movement of substrates across the plasma membrane. Evidence suggests that substrate translocation occurs via an alternating access mechanism in which ion and substrate binding facilitate conformational changes in the transporter allowing a central binding domain to be alternatively open to either side of the membrane (1–3). Recently, a number of solute transporters from different families have been crystallized, providing critical details about the structural features of these carriers (4–12). Despite these transporters possessing little to no sequence homology, all share a 5 + 5 arrangement of transmembrane helices termed the FIRL (“five-transmembrane helix-inverted topology repeat, LeuT-like”) or “LeuT-fold” (13), where the second bundle of five helices have inverted symmetry in relation to the first group of five helices, supporting the idea of a conserved functional mechanism (3, 14, 15). Importantly, a collection of crystal structures have been solved representing the “open-to-out”, “occluded,” and “open-to-in” conformations of the carriers, suggesting mechanisms that may support substrate translocation (4, 16–19). Unfortunately, these static poses are limited in their ability to explain how ion binding is mechanistically and energetically coupled to the conformationally mediated transport of substrate.
In this study, we focus on the human serotonin transporter (hSERT),3 a member of the SLC6 family that plays a major regulatory role in synaptic transmission by clearing 5-HT from the synaptic cleft, thereby terminating 5-HT receptor activation (20–25). hSERT has importance in human health, being the target of a number of therapeutic and illicit drugs, including tricyclic antidepressants, selective serotonin reuptake inhibitors, cocaine, and 3,4-methylenedioxymethamphetamine (“ecstasy”) (25–28). Although SERT shares only ∼21% overall sequence identity with the SLC6 bacterial amino acid transporter LeuT, the residues in the core of the two proteins, where substrate and ions bind, approach ∼60% identity, suggesting that the two Na+-binding sites in LeuT, termed Na1 and Na2 (4), are conserved in hSERT (29). Evidence for two Na+-binding sites in monoamine transporters was recently bolstered by the solving of the eukaryotic Na+/Cl−-dependent dopamine transporter from Drosophila melanogaster (dDAT) with sodium-binding sites comparable with those in LeuT (12). Crystal structures of dDAT and a LeuT Cl−-dependent mutant (E290S) (30) have greatly advanced our understanding of the Cl−-binding site in these proteins and support previous biochemical studies (31–33).
Interestingly, 5-HT uptake analysis indicates that during coupled transport, only one Na+ is translocated per cycle (34), suggesting that the Na1 and Na2 sites probably have distinct but as yet unknown roles. Recent computational analysis of the Na2 binding site in proteins with the LeuT-fold predicted that transition to an inward facing conformation destabilizes Na+ coordination at Na2, resulting in Na+ release followed by substrate dissociation (19, 35–40). Were this true in hSERT, which appears to translocate a single Na+ per transport cycle, the cation at Na1 would not be mobile. This surmise is consistent with data from crystal structure and molecular dynamic analysis of the bacterial galactose transporter, vSGLT, because this transporter possesses the homologous Na2 site but lacks the Na1 site (5, 41). Despite these implications, however, the distinct role of the Na1 and Na2 in hSERT remains elusive. To understand the roles that each bound Na+ performs in hSERT, we used site-directed mutagenesis in combination with biochemical and electrophysiological analyses to characterize how alterations at either of the Na+ coordination sites affect ion dependence and selectivity as well as ion and 5-HT transport. Using a mutation that alters Na1 coordination yet retains 5-HT transport, we uncovered distinct roles for the Na1 and Na2 coordination sites as well as molecular interactions that appear to be important in the 5-HT transport mechanism.
EXPERIMENTAL PROCEDURES
Site-directed Mutagenesis
Mutagenesis of hSERT cDNA in pcDNA 3.1 was accomplished using the Change-IT multiple mutation site-directed mutagenesis kit (Affymetrix, Cleveland, OH). Mutations were verified by DNA sequencing via Northwoods DNA, Inc. (Bemidji, MN).
5-HT Uptake Analysis
All transport studies of the mutants were conducted using either HEK-293 cells transfected with Trans-IT LTI (Mirus Inc.) in Opti-MEM medium as described previously (42) or stably expressing HEK-293 cells under G418 (800 μg/ml) selection. Cell lines were plated on 24-well poly-d-lysine-coated culture plates at a density of 50,000 cells/well and maintained at 37 °C, with 5% CO2 and under high humidity. Prior to uptake, plates were washed using the appropriate buffer as follows. Standard complete buffer contained 120 mm NaCl, 5.4 mm KCl, 1.2 mm CaCl2, 10 mm glucose, 7.5 mm HEPES, pH 7.4. Cation-only replacement buffers were prepared by replacing NaCl with a specific cation, giving XCl, where X represents Li+, K+, Ca2+, Mg2+, Ba2+, NH4+, N-methyl-d-glucamine (NMDG+), or choline+ (120 mm for single valence cations or 60 mm for divalent cations). When Ca2+ was completely removed from the buffer, 10 mm mannitol was added to balance the osmolality. A small amount of cell detachment was detected when assays were conducted in buffer lacking Ca2+, suggesting that could be responsible for lower uptake activity under “Na+-only” conditions; however, cell viability studies show that the cell wash off is minimal. Furthermore, a similar decrease in activity was observed in the Na+-only buffer supplemented with 50 μm Ca2+, which eliminated detachment during uptake, indicating that any cell loss had little to no impact on the decreased activity observed in the Na+-only buffers. Buffers were adjusted to pH 7.4 using KOH or NMDG. Anion replacement buffers were made similarly, with all buffer salts having Cl− replaced by Y (120 mm NaY, 5.4 mm KY, 1.2 mm CaY, where Y represents acetate, gluconate, or sulfate). Assays measured transport of 50 nm [3H]5-HT (5-hydroxy[3H]tryptamine-trifluoroacetate, 28.5 Ci/mmol, PerkinElmer Life Sciences) as described previously (42). Assays were conducted for 10 min in order to stay within the linear range of uptake, with the exception of 5-HT saturation analysis (15 min) and 5-HT equilibrium analysis (2–90 min). Saturation assays were performed as described, except [3H]5-HT was diluted 50-fold with non-radiolabeled 5-HT to achieve the highest concentration of 50 μm. Transport assays were terminated by washing with cold assay buffer. Cells were dissolved in Microscint 20 (PerkinElmer Life Sciences) scintillation fluid, and counts/min were determined using a TopCountNXT (PerkinElmer Life Sciences). Basal activity from non-transfected (or parental) cells was subtracted from experimental wells to obtain specific activity. Km, Vmax, and EC50 values were calculated by fitting non-linear curves to the data as a function of 5-HT concentration (GraphPad Software Prism 5). All experiments were conducted in triplicate and were independently replicated a minimum of three times.
Protein Expression Analysis
Transporter surface expression was determined in stably expressing HEK-293 cells plated on poly-d-lysine-coated 24-well plates at a density of 100,000 cells/well. 24–48 h after plating, cell surface proteins were biotinylated, quantitated, and analyzed using Western blotting as described previously (42).
Cysteine Accessibility Analysis
HEK-293 hSERT cells were plated at 50,000 cells/well and transfected as described above. Plates were washed with a cation-specific assay buffer, followed by treatment using the same buffer supplemented with 1 mm (2-trimethylammonium)ethyl)methanethiosulfonate bromide (MTSET) (Toronto Research Chemicals Inc.), 1 mm MTSET + 20 μm 5-HT, or vehicle. In wells receiving 5-HT and MTSET, 5-HT was added 2 min prior to MTSET. Plates were incubated at room temperature for 10 min. To terminate the reaction, MTSET was removed via multiple washes with MKRH+G (120 mm NaCl, 4.7 mm KCl, 2.2 mm CaCl2, 1.2 mm MgSO4, 1.2 mm KH2PO4, 10 mm Glucose, 10 mm HEPES, pH 7.4). The remaining transport activity was determined using 50 nm [3H]5-HT as described previously.
Two-electrode Voltage Clamp Analysis
cRNA Preparation
Plasmids encoding hSERT were linearized and transcribed into RNA with a T7 RNA polymerase kit mMessage mMachine (Ambion). A total of 5 ng of cRNA was microinjected into each oocyte. Electrophysiological recordings were performed 3–9 days following injection.
Oocyte Preparation
Xenopus laevis frogs (Nasco, Fort Atkinson, WI) were anesthetized with ethyl 3-aminobenzoate methanesulfonate (FLUKA A5040) (2 mg/ml in H2O). The frog was decapitated, and the ovarian lobes were removed and transferred to sterile Ca2+-free OR2 solution (82.5 mm NaCl, 2.5 mm KCl, 2 mm MgCl2, 10 mm HEPES, pH adjusted to 7.4 with NaOH). The lobes were manually dissected to produce groups of 5–10 oocytes and incubated in OR2, containing 1 mg/ml collagenase from Clostridium histolyticum (Sigma). Incubation for 45–60 min at 18 °C was sufficient to digest and remove the follicular layer. Oocytes were then selected and transferred to a Ringer solution (100 mm NaCl, 2 mm KCl, 1.8 mm CaCl2, 1 mm MgCl2, 5 mm Hepes, pH adjusted to 7.6 with NaOH). Oocytes were kept at 18 °C for a minimum of 2 h prior to injection. Injected oocytes were kept for 6–9 days at 18 °C in a Ringer solution containing 2.5 mm Na+ pyruvate, 100 μg/ml penicillin, 100 μg/ml streptomycin. Solutions were changed daily.
Electrophysiological Recordings in X. laevis Oocytes
A CA-1B high performance oocyte clamp (Dagan Corp.) was employed for the measurements. The recorded signal was digitized with a Digidata 13222A system (Axon Instruments). pCLAMP 9.2 (Axon Instruments) was used for data acquisition. Borosilicate glass capillaries were pulled to a final resistance of 0.4–1.2 megaohms and filled with 3 m KCl. Oocytes were impaled, and the membrane potential was clamped to a holding potential of −60 mV. For continuous superfusion with Na+ solution (120 mm NaCl, 2 mm KCl, 1 mm BaCl2, 1 mm MgCl2, 10 mm HEPES, pH adjusted to 7.4 with NaOH) a gravity-driven superfusion system (Warner Instruments, Eight Channel Perfusion Valve Control System (VC-8)) was used. For the Ca2+ solutions, NaCl was replaced by CaCl2. The osmolarity of all solutions was kept the same. For measurements of the substrate-independent leak current, we used 10 μm paroxetine. The leak was defined by subtraction of the respective currents (INa+ − Iparoxetine). Recordings were started after a stable current base line had been established. The current was sampled with 100 Hz and low pass-filtered with 20 Hz.
Whole Cell Patch Clamp
For patch clamp recordings, HEK293 cells stably expressing hSERT N101A were seeded at low density for 24 h before measuring currents. To measure substrate-induced hSERT currents, cells were voltage-clamped using the whole cell patch clamp technique. Briefly, glass pipettes were filled with a solution consisting of 133 mm potassium gluconate, 5.9 mm NaCl, 1 mm CaCl2, 0.7 mm MgCl2, 10 mm EGTA, and 10 mm HEPES adjusted to pH 7.2 with KOH. For some experiments, the internal Cl− concentration had to be increased. In these instances, the pipette solution consisted of 133 mm KCl, 5.9 mm NaCl, 1 mm CaCl2, 0.7 mm MgCl2, 10 mm EGTA, and 10 mm HEPES adjusted to pH 7.2 with KOH. The cells were continuously superfused with external solution: 140 mm NaCl, 3 mm KCl, 2.5 mm CaCl2, 2 mm MgCl2, 20 mm glucose, and 10 mm HEPES adjusted to pH 7.4 with NaOH. In those experiments where external Na+ was replaced by Ca2+, we used the following solution: 15 mm CaCl2, 150 mm choline chloride, 1 mm MgCl2, and 10 mm HEPES, adjusted to pH 7.2 with KOH.
Currents were recorded at room temperature (20–24 °C) using an Axopatch 200B amplifier and pClamp version 10.2 software (MDS Analytical Technologies). Cells were voltage-clamped to potentials between −100 and −10 mV, and 10 μm 5-HT was applied for 5 s once every 60 s. Current traces were filtered at 1 kHz and digitized at 2 kHz using a Digidata 1320A (MDS Analytical Technologies). The liquid junction potentials were calculated, and measurements were compensated accordingly. Drugs were applied using a DAD-12 device (Adams & List, Westbury, NY), which permits complete solution exchange around the cells within 100 ms (43). Current amplitudes in response to 5-HT application were quantified using Clampfit version 10.2 software. Passive holding currents were subtracted, and the traces were filtered using a 100-Hz digital Gaussian low pass filter.
Molecular Dynamic (MD) Simulations of hSERT·5-HT·Ion Complexes
MD simulations were conducted using hSERT and N101A mutant homology-modeled structures complexed with 5-HT and ions. Homology models of hSERT were based on the “open to out” conformation of the leucine transporter (Protein Data Bank code 3F3A) and built using Prime (Prime, version 3.1, Schrödinger, LLC, New York). The alignment obtained in the structure prediction module of Prime was manually edited to match the comprehensive alignment by Beuming et al. (23, 44). The loops were modeled and refined in Prime using an ab initio loop prediction method. Side chain optimization and minimization was conducted on loop candidates, and the models were validated for quality based on Ramachandran plots (45) by PROCHECK validation (46) using the ADIT site. hSERT structural models were prepared for docking in the Protein Preparation Wizard (47) (Schrödinger Suite 2012 Protein Preparation Wizard, Epik version 2.3, Schrödinger, LLC, New York; Impact version 5.8, Schrödinger, LLC, New York; Prime version 3.1, Schrödinger, LLC, New York) using default options. Protonated 5-HT conformers generated by LigPrep (LigPrep, version 2.5, Schrödinger, LLC, New York) were docked into hSERT homology models containing various combinations of Na+, Ca2+, and Cl− utilizing the induced fit docking protocol (48). The best scoring 5-HT-docked hSERT and N101A complexes were placed in the center (along the z axis, coinciding with the normal of the 1-palmitoyl-2-oleoyl-phosphatidylcholine bilayer) of the pre-equilibrated 1-palmitoyl-2-oleoyl-phosphatidylcholine lipid bilayer using the VMD visualization package (49). The dimensions of the simulation box were 20 × 20 × 14 Å, containing 1 protein, 1 ligand, and ∼50,340 TIP3P water molecules and 222 1-palmitoyl-2-oleoyl-phosphatidylcholine molecules. 186 Na+ ions and 191 Cl− counterions were added to obtain an electroneutral system with a salt concentration of 150 mm. All calculations were carried out with GROMACS version 4.5.4 (50), using a CHARMM27 force field under periodic boundary conditions. The topologies and parameters files for the ligand were generated by the SwissParam tool (51) based on the Merck molecular force fields that are compatible with CHARMM and GROMACS. All simulations were performed in the NPT ensemble with velocity scaling (V-rescale) thermostat and Parrinello-Rahman barostat. For the CHARMM force field operating in GROMACS, electrostatics were calculated using particle mesh Ewald with appropriate cut-offs: rlist = 1.3, rcoulomb = 1.3, rvdw = 1.2, vdwtype = switch, rvdw_switch = 0.8. Fourier spacing of 0.12 nm and a particle mesh Ewald order of 4 were employed. V-rescale thermostat with a coupling constant of 0.1 ps was used to separately couple protein, lipid, and solvent, including water and ions. The pressure was coupled using the Parrinello-Rahman algorithm at 1 bar with a coupling constant ρ = 1 ps and a uniform compressibility of 4.5 × 10−5 bar−1. The coordinates were saved every 100 ps with an integration time step of 2 fs. The LINCS (linear constraint solver) algorithm was used to restrain all bond lengths (52). The steepest descent algorithm in GROMACS was used to minimize the energy of the 5-HT-docked and ion-incorporated WT and N101A mutant hSERT structures in 1-palmitoyl-2-oleoyl-phosphatidylcholine bilayer followed by the equilibration phase. At the temperature of 303 K, the initial velocities were generated following a Maxwellian distribution. The system was equilibrated for 1 ns at a temperature of 303 K by fixing the position of the docked complex by applying position restraints of 1000 kJ mol−1 nm−2 on each heavy atom, whereas lipids and water were allowed to move normally. After initial equilibration for 1 ns, the production runs were performed for 12 ns. The pressure was maintained at 1 atmosphere using semi-isotropic pressure coupling to a Parrinello-Rahman barostat with a coupling constant of 5 ps. Conformations resulting from the production phase of each simulation were stored at intervals of 100 ps and analyzed. PyMOL (53) was used to generate the molecular graphic diagrams.
RESULTS
Asn-101 Mutation Specifically Modifies Cation Dependence, Allowing Ca2+ to Functionally Replace Na+ for 5-HT Transport
Previously, we demonstrated that mutation of Asn-101 to Ala or Cys conferred Cl−-independent transport to hSERT (54) while maintaining little to no loss in transport activity. Sequence analysis between hSERT and LeuT shows that the hSERT Asn-101 residue corresponds to Asn-27 in LeuT and Asn-49 in dDAT, residues that directly coordinate Na+ at the Na1 site. Direct coordination of Asn-101 with Na+ at Na1 is further supported by the fact that lengthening the side chain by one carbon through a N101Q mutation yields a non-functional transporter, whereas smaller side chains are tolerated (54). We examined the impact that mutations at Asn-101 have on Na+ coupling to 5-HT transport in hSERT. First, we looked for alterations in cation selectivity through measurement of [3H]5-HT uptake in cells stably expressing the hSERT, hSERT N101A, or hSERT N101C mutant transporters in simple buffers containing only a single cation, where the cation was either Na+, Ca2+, Li+, Ba2+, NH4+, choline+, NMDG+, K+, or Mg2+ (Fig. 1). Appreciable 5-HT transport was observed with hSERT in the presence of Na+-only buffer, which was 85% ± 6.3% of the uptake observed with complete buffer (see “Experimental Procedures”). Under the same conditions, the N101A and N101C mutants yielded uptake levels of 69 ± 2.8 and 63 ± 2.3%, respectively (54). The inability of K+, Li+, Mg2+, Ba2+, NH4+, choline+, and NMDG+ to functionally replace Na+ in the Asn-101 mutants for 5-HT uptake revealed that cation selectivity for 5-HT uptake is relatively intact. In fact, replacement of Na+ with Ba2+ (hSERT), Li+ (Asn-101 mutants), and K+ actually inhibited 5-HT uptake to levels below those observed with NMDG+ (Fig. 1). Remarkably, whereas native hSERT exhibited only 5.2% ± 0.6% activity when Ca2+ replaced Na+, Ca2+ could fully substitute for Na+ in the N101A mutant, exhibiting the same degree of uptake observed in the Na+-only buffer. Likewise, Ca2+ could substitute for Na+ in the N101C mutant, albeit with less efficacy (64.2 ± 2.9%), suggesting that Ca2+ coordination may be suboptimal in the N101C mutant compared with N101A. Uptake of 5-HT under Ca2+-only conditions maintained the Cl− independence observed in Na+-containing buffers (Fig. 2).
FIGURE 1.

5-HT uptake by Asn-101 mutants under cation substitution reveals Ca2+ can functionally replace Na+ in 5-HT transport. Transport activity was measured in HEK cells stably expressing hSERT, N101A, or N101C. Initial transport rates (10 min) were obtained in complete buffer or in the presence of a cation-only buffer, as indicated on the x axis. Activity is given as the percentage of uptake in complete buffer. A small amount of K+ (5.4 mm) was also added to each buffer because transport activity was lost upon complete removal of K+. Notably, K+ alone is unable to support 5-HT uptake. The Ca2+ effect on uptake by the Asn-101 mutants was specific because 5-HT transport was not observed in Ca2+-only buffers using the parental cell line lacking hSERT. Bars, mean and S.E. (error bars) of at least three independent experiments, each of which was performed in quadruplicate. A two-way analysis of variance was performed on sample sets with a Bonferroni post hoc test; ***, p < 0.001.
FIGURE 2.

5-HT uptake in Asn-101 mutants under cation and/or anion replacement. Transport activity was measured in HEK cells stably expressing hSERT, N101A, or N101C. Initial transport rates (10 min) were obtained in complete buffer or in the presence of a buffer in which NaCl was replaced with 60 mm CaCl2, 120 mm sodium acetate (NaCH3CO2), 60 mm calcium acetate (Ca(CH3CO2)2), or 60 mm calcium gluconate (Ca(HOCH2(CHOH)4COOH)2). Activity was normalized to activity in complete buffer. Bars, mean and S.E. (error bars) of three separate experiments, each performed in quadruplicate. Data were analyzed using a two-way analysis of variance and Bonferroni post hoc test.
Na+, but Not Ca2+, Imparts Conformational Changes in Native hSERT, whereas Both Na+ and Ca2+ Can Promote 5-HT-induced Conformational Changes in the N101A Mutant
The ability of Ca2+ to support 5-HT translocation by N101A and N101C mutants suggests that Ca2+ binding to SERT induces conformational changes similar to those obtained with Na+. Inactivation of SERT-mediated 5-HT transport by cysteine-directed methanothiosulfanate (MTS) reagents (42, 55, 56) can be used as an indicator of conformational changes in hSERT (54, 57–59). We used the membrane-impermeant (MTS) reagent MTSET to examine altered accessibility of residue Cys-109 in the presence of Na+, Ca2+, and the large monovalent cation, NMDG+, as well as the anions Cl− and acetate−. Cys-109 is positioned on the extracellular end of TM1, a domain thought to undergo significant conformational change during substrate translocation (19). The extent of inactivation of hSERT by MTS adduction at Cys-109 can be modulated by both cation and anion interactions with the transporter (54, 55). In the presence of NaCl, hSERT was relatively insensitive to 1 mm MTSET, and co-incubation with 20 μm 5-HT had no detectable effect (Fig. 3A). Substitution of Na+ with either Ca2+ or NMDG+ resulted in a significant increase in sensitivity of hSERT to MTSET, and similarly, co-incubation with 20 μm 5-HT had no effect on sensitivity. In contrast, the same conditions tested in the N101A background revealed that Cys-109 is highly sensitive to MTSET treatment, giving an 83.5 ± 1.5% loss of 5-HT transport in Na+ buffer (Fig. 3B). Importantly, a C109A/N101A mutant is insensitive to MTSET inactivation (54). Unlike in the hSERT WT, co-incubation with 5-HT in the presence of Na+ or Ca2+ significantly protected the N101A mutant from inactivation, yielding only a 55 ± 2.7 and 74 ± 2.6% loss of activity, respectively. Co-incubation of 5-HT with NMDG+ did not afford the same protection from MTSET (Fig. 3B). This shows that, in the N101A mutant, Na+ and Ca2+ are both capable of promoting conditions that, together with 5-HT, result in conformational changes in SERT. This effect was not observed with the larger NMDG+ ion, which due to its bulk is unlikely to access the cation-binding site in hSERT (60). The data also reveal that both Na+ and Cl− are necessary for protection from MTS inactivation at Cys-109 in hSERT, because removal of either ion results in a significant increase in sensitivity (Fig. 3, C and E). However, Ca2+ cannot afford protection in WT hSERT, because the absence or presence of Ca2+ does not alter the sensitivity of Cys-109 (Fig. 3, D and F). In the N101A mutant, increased protection from MTS inactivation was not dependent on Cl− but did require the presence of a cation (Na+ or Ca2+) and 5-HT (Fig. 3, C–F), suggesting that conformational changes normally mediated by binding of only Na+ and Cl− require a cation and 5-HT in the N101A mutant.
FIGURE 3.

Ca2+ and Na+ alter Cys-109 accessibility to MTSET, suggesting that these cations may promote similar conformational changes in the N101A mutant. HEK-293 cells expressing hSERT or hSERT N101A were pretreated with 1 mm MTSET in the absence (gray bars) or presence (black bars) of 5-HT (20 μm) and in the presence of Na+, Ca2+, or NMDG+. Following treatment, 5-HT uptake assays were performed in complete buffer to quantitate activity. The percentage of remaining activity is plotted and is defined as the amount of 5-HT uptake of MTSET-treated cells as a percentage of untreated cells. Bars, mean and S.E. (error bars) of at least three independent experiments, each of which was performed in quadruplicate. A and B, results of MTSET treatment on WT hSERT (A) and hSERT N101A (B) in each cation buffer. C and D, data from A and B replotted with respect to genetic background and the presence or absence of Na+ (C) or Ca2+ (D). E and F highlight the effect of Cl− replacement under full Na+ (E) or full Ca2+ (F) buffer conditions. A two-tailed t test was performed on sample sets as indicated with brackets. ***, p < 0.001; *, p < 0.05; NS, not significant.
hSERT Asn-101 Mutants Display a Loss of Potency for Na+ to Drive 5-HT Transport
Given that LeuT residue Asn-27 and dDAT residue Asn-49 are homologous to hSERT Asn-101 and they directly coordinate Na+ at the Na1 site (4, 12), it is reasonable that mutations at Asn-101 would directly impact Na+ binding in hSERT. To evaluate this possibility, [3H]5-HT uptake was measured using Na+ dose-response assays, where Na+ concentrations ranged from 0 to 120 mm, and NMDG+ or Ca2+ was used to compensate for the reductions in Na+. When NMDG+ was replaced by Na+, the dose-response curves for N101A and N101C shifted rightward compared with WT, yielding increased EC50Na+ values of 10.2 ± 3.0 and 18.3 ± 4.7 mm, respectively, compared with 3.8 ± 1.2 mm in hSERT (Fig. 4A). The loss of Asn-101 coordination of Na+, due to its mutation to Ala or Cys, is consistent with the observed decrease of Na+ potency, as well as the reduced ability of the Asn-101 mutants to efficiently couple the Na+ chemiosmotic gradient to 5-HT transport (Fig. 4D) (54). A similar rank-order increase in the EC50Na+ values for the Asn-101 mutants is observed upon substitution of Na+ with Ca2+ (28 ± 3.7 and 47 ± 6.1 mm for N101A and N101C, respectively, compared with 16 ± 1.8 mm for hSERT (Fig. 4B)). Ca2+ has not previously been reported to modulate 5-HT transport in SERT; however, we found that at low Na+ concentrations, Ca2+ was able to enhance 5-HT uptake (Fig. 4B) in hSERT, whereas NMDG+ did not (Fig. 4A). In contrast, the presence of moderate to low levels of Ca2+ negatively affected 5-HT uptake as Na+ levels increased, as evidenced by a shallower slope and increase in EC50Na+ values compared with NMDG+ replacement. Taken together, these data indicate that Ca2+ may act in a competitive manner to modulate the efficacy of Na+ to support 5-HT transport in native hSERT.
FIGURE 4.

Cation dependence and concentrative uptake studies. Cation dependence analyses reveal that Asn-101 mutants display altered Na+ and Ca2+ efficacies in driving 5-HT transport. Initial transport rates (10 min) of [3H]5-HT (50 nm) were obtained for HEK cells stably expressing hSERT (●), N101A (□), or N101C (♢) over a range of Na+ concentrations (0–120 mm) replaced with NMDG+ (A) or Ca2+ (B). Uptake was also measured (C) using Ca2+ concentrations (0–60 mm) replaced by NMDG+. Transport activity is expressed relative to that at maximal Na+ concentration. EC50 values were determined by fitting data to a sigmoidal dose-response curve (variable slope). Steady-state uptake kinetics reveal a loss of concentrative capacity in Asn-101 mutants. [3H]5-HT uptake by HEK cells stably expressing hSERT (●), N101A (□), or N101C (♢) in assay buffer containing 120 mm Na+ (D) or 60 mm Ca2+ (E) was monitored over 120 min. Data were background-subtracted using data from parental HEK cells. Data were converted to attamoles of 5-HT accumulated/cell unit time. The data were fit to a Michaelis-Menten nonlinear regression equation using Prism 5. Values represent the mean and S.E. (error bars) of a minimum of three independent experiments, each of which was performed using triplicate wells.
Finally, equimolar replacement of NMDG+ with Ca2+ yielded a dose-dependent increase in 5-HT transport that reached levels that were ∼50% (N101A) and ∼35% (N101C) of transport observed with 120 mm Na+. In contrast, the WT displayed minimal uptake (∼5%) even at the highest Ca2+ concentration tested.
Mutation at Asn-101 Diminishes the Ability of the Transporter to Concentrate 5-HT
Because the N101A and N101C mutants display diminished capacity to couple transport to the chemiosmotic gradients of Na+ and Cl− (54), we examined concentrative 5-HT uptake under both Ca2+ and Na+ replacement conditions (Fig. 4, D and E). As expected, concentrative transport of 5-HT by hSERT was all but eliminated (95% reduction) in Ca2+ buffer (Fig. 4E). As previously demonstrated (54), in Na+-only buffer, concentrative uptake of 5-HT was significantly reduced in the N101A and N101C mutants compared with hSERT (Fig. 4D). In Ca2+ buffer, concentrative uptake by the N101A and N101C mutants was reduced 51 and 32%, respectively, compared with equilibrium levels achieved with Na+, suggesting that the hSERT-Ca2+ interaction is less efficient than Na+ at coupling the ion gradient to 5-HT transport. Previously, we reported that the N101A and N101C mutants exhibit increased substrate efflux (54), which could account for part of the dramatic decrease in concentrative uptake. Differences were also observed for the time necessary to reach concentrative equilibrium. In Na+, the t½ for hSERT was 62 ± 10 min compared with 16 ± 3.7 and 12 ± 3.1 min for N101A and N101C, respectively. The time for the mutants to reach steady state in Ca2+ is more rapid than in Na+, with N101A reaching t½ at 5.4 ± 1.1 min and N101C at 7.3 ± 2.1 min, whereas in WT, the t½ is attenuated to 103 ± 59 min. To account for the fast saturation of the mutants, uptake assays were only 10 min in length.
Ca2+ Decreases the Apparent Affinity of 5-HT in both hSERT and the Asn-101 Mutants
Kinetic transport analysis in Na+ buffer yielded indistinguishable Km values for hSERT (1.2 ± 0.4 μm), N101A (1.6 ± 0.5 μm), and N101C (1.2 ± 0.3 μm). As stated above, 5-HT uptake by hSERT under Ca2+-only conditions retains ∼5% activity, which is sufficient to measure transport kinetics. Interestingly, Km values for 5-HT transport increased in all three transporters by ∼10-fold (hSERT, 14 ± 2.6 μm; N101A, 18 ± 3.1 μm; N101C, 11 ± 1.7 μm). This equivalent increase in Km for 5-HT in hSERT and the Asn-101 mutants suggests that Ca2+ binds hSERT and the Asn-101 mutants in a similar manner. When taken together with the fact that the Na1 site is thought to directly coordinate 5-HT, these data suggests that Ca2+ binds at Na1.
Mutations at Na2 Site Fail to Alter Cation Selectivity
Although the amino acid substitutions at Asn-101 would directly implicate the Na1 site as the target for Ca2+ activity in the mutants, studies have suggested that an extensive ion network connects the Na1, Na2, Cl−, and 5-HT binding sites (31, 32, 38, 54). Therefore, it is possible that alterations in amino acids and their coordination at Na1 could structurally influence ionic coupling at Na2, permitting Ca2+ to bind at the Na2 site and activate transport. To investigate Ca2+ binding to Na2, amino acid substitutions were introduced at residues Asp-437 and Ser-438 in the Na2 site to disrupt their side chain carboxyl- and hydroxyl-mediated coordination with Na+. The other amino acids comprising the Na2 site (Gly-94, Val-97, and Leu-434) were not mutated, because they coordinate the Na+ ion via backbone carbonyls. Functional characterization of the mutants with [3H]5-HT uptake assays revealed that the conservative D437E mutant is non-functional, whereas the other substitutions showed 16–93% activity (Table 1). Activity of Ser-438 mutants ranged from 27 to 94%, with the highest activity exhibited by the conservative S438T mutant. None of the substitutions at Asp-437 or Ser-438 conferred Ca2+-mediated 5-HT transport (Table 1).
TABLE 1.
Determination of surface expression, transport activity and ion dependency for the Na1 and Na2 site mutants
Surface expression was calculated by performing densitometry of SERT bands from Western blots of biotinylated surface proteins as described previously (42). Transport activity was measured in HEK cells expressing the hSERT or indicated mutants with assays conducted in complete buffer. Activity of mutants in 120 mm NaCl is expressed as a percentage of WT activity. Activity under Ca2+ and Cl− replacement conditions is expressed as percentage uptake observed of the same construct in normal Na+ and Cl− buffer. Each value represents the mean and S.E. of triplicate wells from independent experiments repeated at least three times. NA, not applicable.
| Surface expression | Activity |
|||
|---|---|---|---|---|
| 120 mm Na+ | 60 mm Ca2+ | 0 mm Cl− | ||
| % | % | % | % | |
| hSERT | 100 ± 11 | 100 ± 4.7 | 3.23 ± 0.9 | 3.51 ± 0.4 |
| N101A | 77.7 ± 8.9 | 77.0 ± 4.2 | 68.9 ± 3.7 | 99.2 ± 8.5 |
| N101C | 62.4 ± 7.2 | 69.2 ± 5.3 | 42.5 ± 1.5 | 97.4 ± 5.5 |
| D437A | 115 ± 11 | 66.5 ± 7.8 | 6.53 ± 1.5 | 8.48 ± 2.0 |
| D437C | 109 ± 9.2 | 93.4 ± 5.4 | 6.91 ± 1.3 | 6.08 ± 0.8 |
| D437E | NA | NA | NA | NA |
| D437T | 85.7 ± 4.0 | 83.3 ± 5.6 | 5.64 ± 1.5 | 11.1 ± 1.6 |
| D437V | 78.7 ± 13 | 15.9 ± 4.3 | 5.93 ± 3.1 | 25.0 ± 6.1 |
| S438A | 101 ± 15 | 62.0 ± 6.5 | 6.08 ± 0.9 | 35.9 ± 3.4 |
| S438C | 105 ± 7.4 | 77.1 ± 4.1 | 1.97 ± 0.5 | 7.01 ± 1.4 |
| S438T | 131 ± 11 | 93.5 ± 5.6 | 4.18 ± 0.8 | 2.31 ± 0.3 |
| S438V | 48.0 ± 2.4 | 26.7 ± 5.1 | 5.13 ± 2.4 | 4.20 ± 1.3 |
Simultaneous mutation of residues Asn-101 and Ser-438 was not well tolerated, resulting in a marked reduction in transport (1–12% remaining activity) (Fig. 5). However, introduction of N101A or N101C into either the S438T or S438A background restored Ca2+-supported translocation, which is consistent with Ca2+ interaction at Na1. Notably, Ca2+ was superior to Na+ in supporting uptake in transporters containing the N101A/S438A, N101A/S438T, or N101C/S438A mutations. Conversely, the N101C/S438T mutant, which showed the most uptake activity of the double mutants, had greater levels of transport in Na+ buffer compared with Ca2+, but uptake in Ca2+ was still ∼6-fold greater than in NMDG buffer. These data further support the idea that Ca2+-mediated influence on transport occurs through interaction at Na1 rather than Na2.
FIGURE 5.

Introduction of the Asn-101 mutations into the Na2 mutant backgrounds restored Ca2+-dependent uptake activity. Transport activity was measured in HEK cells transiently expressing hSERT or a Asn-101/Ser-438 double mutant. Initial transport rates (10 min) were obtained in a Na+-containing buffer (120 mm) or in a buffer in which Na+ was completely replaced by Ca2+ (60 mm) or NMDG+ (120 mm). Bars, mean and S.E. (error bars) for three separate experiments, each performed in quadruplicate. ***, p < 0.001.
The Na1 Site Does Not Contribute to the Substrate-induced or Leak Currents in hSERT
SERT exhibits not only Na+-dependent Na+ and 5-HT flux but also 5-HT-gated suprastoichiometric current (60). Because our data support binding of Ca2+ at the Na1 site in hSERT, we surmised that analysis of the substrate-induced or leak currents in the Asn-101 mutants under Ca2+ replacement conditions would provide insight into the contribution of the Na1-bound cation to the conductive states of hSERT. Therefore, we utilized two-electrode voltage clamping studies to examine currents generated by 5-HT transport via the N101A mutant in the presence of various concentrations of Na+ and Ca2+. Buffers containing different concentrations of Na+, Ca2+, or both cations were perfused into the system and allowed to equilibrate, after which 5-HT was added to induce transport-associated current. In the presence of only Na+, an inward current of ∼−30 nA is generated by the movement of a large amount of Na+ ions through the WT transporter. When Na+ is partially replaced by increasingly larger amounts of Ca2+, the inward current displayed by hSERT decreases in a dose-dependent manner (Fig. 6A), whereas in the N101A mutant, the current persists; however, its magnitude is slightly decreased (Fig. 6B). The large transient current observed upon the first addition of Ca2+ is not dependent on SERT and probably results from activation of endogenous channels because the same transient can be seen in uninjected oocytes upon the addition of Ca2+ (Fig. 6B, inset). Therefore, after the initial addition of Ca2+ to the buffer, readings were taken only after a stable base line was reached.
FIGURE 6.

Whole cell clamp electrophysiology studies. Replacement of Na+ with Ca2+ is shown. Representative current traces of WT (A) and N101A (B), respectively, recorded from X. laevis oocytes using the two-electrode voltage clamp technique are shown. The cells were clamped to −60 mV, and currents were induced with 10 μm 5-HT (black bars). The Ca2+ concentration was successively increased, and the Na+ concentration was simultaneously decreased as indicated. Upon wash in 10 mm Ca2+, a transient inward current was observed. The inset between A and B depicts bars representative of substrate-induced current from oocytes expressing N101A in the presence of Na+ (left bar) or Ba2+ (right bar). C, the current-voltage dependence of 5-HT-induced currents of the N101A mutant assessed in a solution containing 80 mm Ca2+ is plotted (n = 5). For the Ca2+ solutions, NaCl was replaced by CaCl2. Shown are the I/V curve for the substrate-induced current and the I/V curve for the substrate independent leak in Na+. The leak was defined by the current remaining after subtraction of the current upon application of 10 μm paroxetine. D, current amplitudes elicited by 10 μm 5-HT in WT (open circle) and N101A (closed circle) are plotted as a function of increasing Ca2+ concentrations (each data point is the average of six experiments). The remaining fraction of normalized current in WT and N101A at 80 mm Ca2+ was 0.12 ± 0.35 and 0.30 ± 0.30, respectively. E, a representative current trace recorded from HEK293 cells expressing hSERT N101A in the presence of physiological ion gradients (see “Experimental Procedures”). At −90 mV, a 5-s application of 10 μm 5-HT provoked an inwardly directed current. F, current responses to 10 μm 5-HT were measured at successively positive voltages. The currents were normalized to the maximum amplitude at −90 mV and plotted as a function of the respective holding potential. G, a typical trace of a current through hSERT N101A is shown in the absence of external Na+ but in the presence of 15 mm external Ca2+. At −90 mV, the current induced by 10 μm 5-HT was inwardly directed. H, a reversal potential at around −60 mV, when the inner Cl− concentration was low (5 mm) (○) or around 0 mV when the inner Cl− concentration was high (140 mm) (□). Bars, mean and S.E. (error bars).
Previous analysis of the Asn-101 mutants in Na+ buffer revealed a reversal potential of ∼+70 mV (54), indicating that Na+ was the conducting ion. However, when we fully replaced Na+ with Ca2+ in the N101A mutant, the reversal potential dropped to ∼−30 mV, suggesting that Ca2+ does not carry the current when substituted for Na+, because we would expect a more positive reversal potential if Ca2+ were now carrying the current (Fig. 6C). Remarkably, this indicates that, unlike Na+, Ca2+ does not seem to permeate through the transporter, which may also explain why current levels decreased when Na+ was replaced by Ca2+ (Fig. 6B). Under full cation replacement with 80 mm Ca2+, there is a prominent leak current, as defined by current block with 10 μm paroxetine. This leak current also reverses at −30 mV (Fig. 6C). A similar reversal potential for the substrate-induced current and the leak current is expected if both currents are carried by the same conformational intermediate, as recently suggested by Schicker et al. (61). The fact that the reversal potential for I5-HT and Ileak occurs at ∼-30 mV suggests that Cl− is the permeating ion. Unfortunately, we were unable to determine the I/V relationship for WT hSERT when Na+ was replaced with 80 mm Ca2+ because the currents were <2 nA at −60 mV.
Because Ca2+ does not appear to be transported and carry current, we wanted to see if the addition of Ca2+ could inhibit Na+-mediated currents by examining transport-mediated currents at increasing concentrations of Ca2+ (Fig. 6D). We found that hSERT exhibited a linear, dose-dependent decrease in current as Ca2+ concentration increased (and Na+ concentration decreased). In contrast, N101A displayed a hyperbolic curve, indicating that Ca2+ reduces the amount of Na+-generated current.
Finally, although Ba2+ can often functionally replace Ca2+ in channels (62), Ba2+ was unable to support substrate-induced currents in the N101A mutant (data not shown). This is consistent with the failure of Ba2+ to promote transport in radiolabeled 5-HT uptake studies (Fig. 1), revealing that Ca2+ binding is selective and is a prerequisite for current generation.
Whole Cell Clamp of the N101A Mutant in HEK293 Cells Suggests That Cl− Is the Main Charge Carrier When External Na+ Is Substituted by Ca2+
In order to further test if Cl− (and not Ca2+) is the main charge carrier by the N101A mutant, we performed whole cell patch clamp analysis on HEK293 cells stably expressing hSERT N101A. This technique allowed us to control the internal ion composition, thus facilitating the interpretation of the reversal potential. When cells were recorded in the presence of an external solution containing 152 mm Na+, application of 10 μm 5-HT for 5 s provoked an inwardly directed current at a holding potential of −90 mV (Fig. 6E). Current-voltage analysis in the presence of full external Na+ was performed (Fig. 6F); however, excessive noise occurred at voltages positive to −10 mV (probably due to endogenous channels), precluding analysis in that voltage range. When Na+ was removed from the external solution and replaced with 15 mm Ca2+ and 150 mm choline, 10 μm 5-HT was still able to induce current (Fig. 6G). In HEK293 cells, unlike oocytes, full replacement of Na+ by Ca2+ (80 mm) resulted in unstable electrical recordings. For this reason, we employed a solution containing Ca2+ and choline that has been used in studies of voltage-gated Ca2+ channels (63). Importantly, choline could not support 5-HT uptake in WT SERT or the N101A or N101C mutants (Fig. 1); therefore, the data represent the contribution of extracellular Ca2+. In this buffer, application of 5-HT led to currents that were absent in cells expressing WT hSERT (data not shown). Current-voltage relationships were recorded with 15 mm external Ca2+ and two internal Cl− concentrations. The current reversed at −55 mV when the internal Cl− concentration was 9.3 mm compared with a reversal potential of ∼−5 mV when internal Cl− was at 143.3 mm (Fig. 6H). These observed shifts in reversal potential in response to internal Cl− concentration suggest that the current is mediated by Cl− in the N101A mutant when Ca2+ is the supporting cation.
hSERT Asn-101 Mutants Appear to Function as both Active and Passive Transporters
Comparison of 5-HT saturation uptake in hSERT with buffers containing either Na+ or NMDG+ reveals that, when Na+ is completely replaced by NMDG+, increasing the amount of extracellular 5-HT only marginally elevates substrate uptake in a dose-dependent manner (Fig. 7). In contrast, both Asn-101 mutants demonstrate enhanced substrate uptake in response to increasing extracellular 5-HT, suggesting that the inside/outside gradient of 5-HT is better able to drive transport in the mutants. Furthermore, the similar levels of uptake by the Asn-101 mutants in the presence or absence of Na+ suggest that the mutant transporters do not couple efficiently to the Na+ gradient and may act more as passive-facilitative transporters when extracellular 5-HT levels are high.
FIGURE 7.

Asn-101 mutants exhibit increased in 5-HT dose-dependent levels of substrate transport in the absence of Na+. HEK cells stably expressing the hSERT, N101A, or N101C were evaluated for dose-dependent 5-HT uptake after 15 min in the presence (solid black bars) or absence (solid gray bars) of Na+. Data were converted to fmol of 5-HT transported/cell/min. Bars, mean and S.E. (error bars) of a data set representing at least three independent experiments, each of which was performed in triplicate. Numbers above gray bars indicate the -fold decrease in activity when comparing the corresponding experimental condition in Na+ buffer. A two-way analysis of variance performed on sample sets with a Bonferroni post hoc test revealed that all mutant -fold decreases in activity were significantly different from the same experimental paradigm in WT hSERT (p < 0.001).
Molecular Dynamics Simulations Suggest a Mechanism for Ca2+ Gain-of-function Phenotype
The dramatic impact the Asn-101 mutation has on ion- and substrate-coupled translocation suggested that mechanisms important to the translocation process could be revealed through comparison of hSERT and the N101A mutant in molecular dynamic simulations. To carry out these studies, 5-HT and ion-docked comparative models of hSERT and N101A were subjected to 12 ns of MD simulations in a lipid bilayer system to investigate alterations in ion coordination (Table 2 and supplemental Fig. S1). Simulations of hSERT Na1Na+, Na2Na+, and ClCl− with 5-HT bound revealed that coordination of Na+ at Na1 was optimal, with a coordination number of 6 (CN:6) (64) (Table 2) and was identical to that shown for LeuT (4). Likewise, coordination of Na+ at Na2 was similar to that in LeuT and dDAT except that the Oγ Ser-438 (Ser-335 in LeuT, Ser-421 in dDAT) interaction with Na+ (expected by homology to LeuT) was varied based on the system (see below). Cl− coordination was similar to that reported by Forrest et al. (33) with the addition of Gln-332 Nϵ (33, 65). dDAT residue Gln-316, which is homologous to Gln-332, coordinates Cl− in the crystal structure (12). The Cl− ion was omitted from MD simulations with the N101A mutant, because these transporters exhibit Cl− independence (54). As indicated by the reduced potency for Na+ to support 5-HT uptake in the Asn-101 mutants, the simulations reveal that coordination of Na+ at Na1 in N101A is decreased to CN:5, due to the loss of the Asn-101 Oδ interaction.
TABLE 2.
Molecular dynamics simulation occupancies
Occupancies for atomic coordinations were calculated over 12 ns of simulation and are scaled from 0 to 1 with 1 being 100% occupancy within the designated distance cut-offs (Na+, 3.5 Å; Ca2+, 3.5 Å; Cl−, 4.0 Å). Occupancies that were not equal to 1 are notated in subscript parentheses.
| WT | |
| Na1 | Ala-96 O,Asp-98 Oδ, Asn-101 Oδ, Ser-336 O Oγ, Asn-368 Oδ; n = 6 |
| Na2 | Gly-94 O,Val-97 O, Leu-434 O, Asp-437 Oδ1 Oδ2; n = 5 |
| Cl | Tyr-121 HH, Gln-332 Nϵ, Ser-336 Hγ, Asn-368 Hδ, Ser-372 Hγ; n = 5 |
| WT, without Cl | |
| Na1 | Ala-96 O,Asp-98 Oδ1 Oδ2 (0.8), Asn-101 Oδ, Ser-336 O(0.5); n = 5 |
| Na2 | Gly-94 O,Val-97 O, Leu-434 O, Asp-437 Oδ1 Oδ2; n = 5 |
| WT, with Ca, Na, and Cl | |
| Ca1 | Ala-96 O,Asp-98 Oδ1 Oδ2, Asn-101 Oδ, Ser-336 O; n = 5 |
| Na2 | Gly-94 O,Val-97 O, Leu-434 O(0.9), Asp-437 Oδ1 Oδ2; n = 5 |
| Cl | Ca2+, Asn-101 Oδ, Ser-336 Hγ(0.9); Leu-337 N(0.5); n = 4 |
| WT, without Cl, without 5-HT | |
| Na1 | Ala-96 O, Asp-98 Oδ1 Oδ2, Asn-101 Oδ, Ser-336 O(0.945), n = 5 |
| Na2 | Gly-94 O,Val-97 O, Leu-434 O(0.7), Asp-437 Oδ1 Oδ2; Ser-438 Oγ; n = 5.7 |
| WT, without 5-HT | |
| Na1 | Ala-96 O, Asp-98 Oδ1 Oδ2 , Asn-101 Oδ(0.9), Ser-336 O(0.9), n = 4.8 |
| Na2 | Gly-94 O ,Val-97 O, Leu-434 O(0.8), Asp-437 Oδ1 Oδ2 ; Ser-438 Oγ(0.9); n = 5.6 |
| Cl | Tyr-121 HH, Gln-332 Nϵ , Ser-336 Hγ, Ser-372 Hγ; Ser-369 Hγ(0.2); n = 4.1 |
| N101A, without Cl | |
| Na1 | Ala-96 O, Asp-98 Oδ1 Oδ2 (0.2), Ser-336 O Oγ, Asn-368 Oδ; n = 5 |
| Na2 | Gly-94 O,Val-97 O, Leu-434 O(0.5), Asp-437 Oδ1 Oδ2; n = 4.5 |
| N101A, with Ca, without Cl | |
| Ca1 | Asp-98 Oδ1 Oδ2 (0.9), Ser-336 O Oγ, Asn-368 Oδ; n = 5 |
| Na2 | Gly-94 O,Val-97 O, Leu-434 O, Asp-437 Oδ1 Oδ2; n = 5 |
| N101A, with Ca, without Na, without Cl | |
| Ca1 | Ala-96 O(0.6), Asp-98 Oδ1 Oδ2, Ser-336 O(0.6) Oγ(0.7), Asn-368 Oδ; n = 6 |
Notably, we observed a correlation between transporter function and the coordination state of the carboxyl side chain of residue Asp-98 in Na1. 5-HT lacks the carboxyl group found on amino acid substrates, such as leucine, and this Asp at position 98, which is strictly conserved among monoamine transporters, is believed to serve as the functional correlate to the leucine carboxyl group in support of Na+ and 5-HT binding. In the inhibitor bound dDAT structure, this conserved Asp (Asp-46) indirectly coordinates Na+ at Na1 through a water molecule (12). However, it is possible that direct Na+-Asp− interaction may occur in substrate-bound dDAT. In the absence of 5-HT, the side chain of Asp-98 in WT SERT exhibits bidentate coordination (O∂1, O∂2) with the Na+ at Na1 (Fig. 8A). However, in 5-HT-bound models, Asp-98 coordination to Na1 is monodendate, with one O∂ from the carbonyl participating in coordination of the (+)-charged amine of 5-HT (Fig. 8B). Simulations involving an empty Cl site revealed that Asp-98 retains bidentate coordination with Na1 and, importantly, lacks coordination with 5-HT (Fig. 8C). This apparent Cl−-dependent coordination of Asp-98 to 5-HT is consistent with previously published findings, where Cl− binding to hSERT decreased the Km of 5-HT ∼4-fold (54, 66, 67). In contrast, simulations with the N101A mutant with Na+ reveal that Asp-98 (O∂) can coordinate the amine of 5-HT even in the absence of Cl−. This loss of Cl−-dependent coordination of Asp-98 in the N101A background could explain why, under Cl−-free conditions, the N101A mutant exhibits a Km for 5-HT that is comparable with values obtained with the Cl−-bound WT hSERT (54).
FIGURE 8.

Proposed model for the coupling of Na1, Na2, and Cl sites to substrate transport in WT hSERT. Core regions of TM1 and TM6 (gray) and TM7 and TM8 (white) of hSERT are displayed for clarity. Highlighted residues Asp-98, Asn-368, and Ser-438 are shown as sticks. 5-HT is depicted in a ball and stick representation. Na+, Ca2+, and Cl− ions are represented as blue, magenta, and green spheres, respectively. Red dashed lines indicate coordination. The panels demonstrate the following. 1) Asp-98 coordination determines hSERT transport competency. Asp-98 coordinates both Na1 and 5-HT when hSERT is transport-competent (B), whereas Asp-98 exhibits bidentate coordination to Na1 when simulations lack 5-HT (A) or Cl− (C) as well as when Ca2+ is placed at Na1 (D). 2) Bound 5-HT alters Ser-438 coordination of Na2. In simulations lacking 5-HT, Ser-438 interacts with Na2 (A), whereas simulations with 5-HT bound reveal S438 does not coordinate Na+ at Na2 (B–D). 3) Asn-368 does not interact with Na1 in transport-incompetent hSERT (A, C, and D).
Analysis of Ca2+ at Na1 revealed bidentate coordination between Ca2+ and the side chain of Asp-98 under all simulation conditions, which precludes Asp-98–5-HT coordination (Fig. 8D). This loss of 5-HT stabilization is consistent with our finding that under Ca2+-only conditions, WT hSERT as well as the Asn-101 mutants exhibit a 10-fold increase in Km for 5-HT compared with Na+-containing buffers.
Evaluation of the transport-competent and -incompetent complexes with the non-functional transporters represented by hSERT without Cl− and hSERT with Ca2+ in the Na1 site and Cl− in the Cl site revealed reorganization of residues Ser-336 and Asn-368 in the Na1 site. These amino acids have been reported to be critical for Na+ and Cl− coordination (31–33, 38). In the non-functional transporters, the side chain of residue Ser-336 is oriented with Hγ pointing away from the reported Cl−-binding site as per Forrest et al. (31) (Fig. 9A). In contrast, all of the transport competent structures have the Hγ directed toward the Cl−-binding site (Fig. 9B). Likewise, Asn-368, which normally coordinates the Na+ at Na1, as shown in the transport-competent structures, is flipped away from the Na1 site in the non-functional models, losing contact with both the Na1 and Cl−-binding sites. These data suggest that the availability of Ser-336 and Asn-368 to participate in the Na1 and Cl coordination sites plays a critical role for transport function and provide a mechanistic explanation for 1) the inability of Ca2+ to substitute for Na+ in the WT transporter, 2) the loss of function of WT transporter in the absence of Cl−, and 3) the gain of function by the N101A mutant to utilize Ca2+ or Na+ without Cl−.
FIGURE 9.
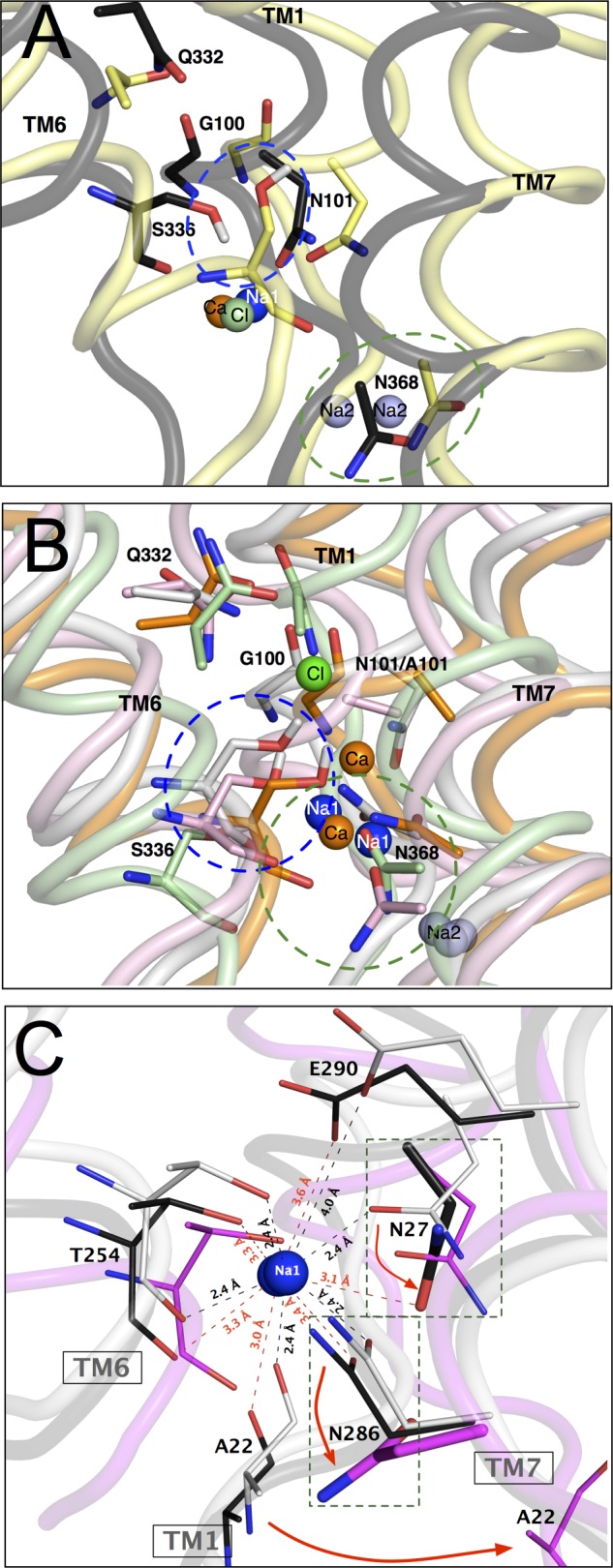
MD simulations of hSERT comparative models and LeuT crystal structures support a mechanistic role for Na1 site coordinating residues. Shown are molecular dynamic simulations of hSERT comparative models under ionic conditions that yield functionally competent or incompetent transporters. A and B, 5-HT-docked comparative hSERT models were simulated in a lipid bilayer system for 12 ns using GROMACS. Models were simulated with various combinations of ions representative of our biochemical analyses. The constructs were named using the convention where hSERT Na1Na+, Na2Na+, and ClCl− represent hSERT with Na+ at Na1, Na+ at Na2, and Cl− at Cl, respectively. Empty ion binding sites are not listed. The peptide backbones are shown as tubes colored as follows: hSERT Na1-Na2-Cl (white); hSERT Na1-Na2 (yellow); hSERT Ca1-Na2-Cl (black); N101A Na1-Na2 (orange); N101A Ca1-Na2 (pink); and N101A Ca1 (green). The structures were aligned using Cαs in PyMOL (53), and for clarity, only TM1, -6, and -7 are shown. Na1Na+, Na2Na+, Cl−, and Ca2+ are represented as blue, purple, green, and orange spheres, respectively. Panels are representative trajectories from simulations under transport-incompetent (A) and -competent (B) conditions. Residues of interest are depicted as sticks. The blue and green dashed ovals highlight the orientation of the Ser-336 and Asn-368 side chains, respectively. C, comparison of Na1 site from LeuT from “outward open,” “outward occluded,” and “inward open” crystal structures reveals significant conformational changes by residues homologous to Ala-96, Asn-101, and Asn-368 in hSERT. LeuT transporter crystal structures representing “outward open” (white), “inward open occluded” (black), and “inward open” (magenta) conformations. Na+ is shown as blue spheres. Residues coordinating Na+ are shown in lines and sticks. Dashed line boxes depict the reorientation of Asn-27 (black sticks) and Asn-286 (magenta sticks) in “outward occluded” (black; S355A/T354V mutant) conformation lacking sodium ion at the Na2 site and “inward open” conformation with both Na+ sites unoccupied.
Finally, we evaluated ion coordination at Na2 in our MD simulations, in light of our biochemical data, to look for possible clues to the functional role of Na2 in transport. In simulations lacking 5-HT, we found that the TM8 residue Ser-438 coordinates the Na+ ion at Na2 (Fig. 8A). However, in 5-HT-bound simulations, the Ser-438 side chain reorients away and no longer participates in coordination of the Na2 ion, suggesting that 5-HT binding may contribute to destabilization of the Na2 site (Fig. 8, B–D). Further analysis of the simulated transporter systems revealed that transporters lacking coordination between Ser-438 and Na+ at Na2 were functionally competent in biochemical analysis, suggesting that loss of Ser-438/Na2 coordination may be important for progression through the transport process (Table 2 and Fig. 8, A and B). However, two simulated systems did not follow this rule. The WT hSERT, 5-HT, Na1Na+, and Na2Na+, which lacks Cl− (Fig. 8C), and the WT hSERT, 5-HT, Na1Ca2+, Na2Na+, and Cl−, which has Ca2+ bound to Na1 (Fig. 8D), are both functionally inactive yet lack the Ser-438/Na2 interaction. This discrepancy can be explained if the loss of the Ser-438/Na2 interaction disturbs the hydrogen bond network linking Na2 to the Cl and Na1 sites, thereby disrupting their function as molecular checkpoints and preventing translocation by the WT transporter unless the appropriate ions and substrate are bound. In contrast, although the N101A transporter also exhibits Na2 destabilization due to loss of Ser-438 interaction, substrate translocation can proceed relatively unchecked because Na2 is uncoupled from the Na1 and Cl sites (as evidenced by functionality in the absence of Cl− or when Ca2+ replaces Na+).
DISCUSSION
This study presents biochemical, electrophysiological, and computational analyses of mutations in a conserved Asn residue (Asn-101) in TM1 of hSERT that is part of the proposed Na1 binding site. The findings of this work reveal that, in addition to the previously reported Cl− independence afforded by mutation of Asn-101 (54), the N101A and N101C mutants can utilize Ca2+, in addition to Na+, to support 5-HT uptake. The unique properties afforded by these substitutions at Asn-101 have allowed us to uncover distinct roles for the Na1 and Na2 sites in the 5-HT transport process.
Conformational studies based on TM1 sensitivity to MTS reagents support the Na+-like behavior of Ca2+ in the Asn-101 mutants, because both Na+ and Ca2+, but not the large cation NMDG+, impart conformational changes to the N101A mutant transporter. Also, Ca2+ alters the dose dependence of Na+ for 5-HT uptake and was able to support small but detectable 5-HT-induced currents in native hSERT. This suggests that Ca2+ can bind to WT hSERT but leads to a primarily non-productive coordination state, one that can be overcome in the Asn-101 mutant background.
The functional interplay between the Na1, Na2, and Cl binding sites (36, 38, 39) presented a challenge to delineate where Ca2+ binds to promote transport in the Asn-101 mutants. However, we uncovered several lines of evidence indicating that Ca2+ binds at Na1. First, mutation at Asn-101 resulted in Ca2+-dependent transport, whereas mutations at Na2 site residues failed to yield Ca2+-dependent uptake. (Interestingly, two of the Na2 mutants displayed significant Cl− independence (Table 1)). Second, introduction of N101A or N101C mutations into the Na2 mutant backgrounds yielded Ca2+-mediated transport. Third, the Km of 5-HT for hSERT, as well as the Asn-101 mutants, increases 10-fold in Ca2+-only buffers. This decrease in apparent affinity of 5-HT is consistent with our MD analyses showing that Ca2+ binding at Na1 results in bidentate coordination between Ca2+ and the conserved Asp (Asp-98) in TM1. The nature of this interaction precludes the ability of Asp-98 to stabilize 5-HT binding, by preventing the Asp-98 side chain from interacting with the (+)-charged amine of 5-HT in agreement with the increased 5-HT Km.
Remarkably, although our data suggest that Ca2+ binds to Na1, reversal potentials from I/V analyses suggest that Cl−, and not Ca2+, is the major carrier of current in the N101A mutant. Furthermore, Ca2+ does not appear to permeate during transport in the Asn-101 mutant. This finding, combined with our evidence that Ca2+ binds at Na1, supports distinct roles for Na1 and Na2 in the translocation process. This assertion is strengthened by sequence comparison analysis and crystal structures (25), which reveal that Na2, in contrast to Na1, is absolutely conserved among LeuT-like transporters and therefore probably serves to couple substrate translocation to the Na+ gradient. This essential role of Na2 is also supported by inward facing crystal structures (19) and molecular dynamic studies (41, 68), which indicate that the destabilization of Na2 and release of its coordinated Na+ ion are critical steps for substrate release. Based on this understanding, if Ca2+ were able to bind at Na2 in the Asn-101 mutants, a Ca2+ ion would probably be released into the cytoplasm during 5-HT translocation. However, our electrophysiological data argue against contribution of Ca2+ to the substrate-induced or leak currents in the N101A mutant under full replacement of Na2+ by Ca2+. Collectively, these findings support the binding of Ca2+ to the Na1 site, which allows us to propose that the ion bound at Na1 (i.e. Na+ or Ca2+) is not co-translocated during stoichiometric transport. However, we acknowledge that we may not be able to detect low level Ca2+ flux. Nevertheless, such a mechanism, where only the Na+ at Na2 is co-transported, agrees with the historical 1Nain:15-HTin:1Clin:1Kout stoichiometry (69–71) and indicates that the Na1 site acts as a molecular checkpoint for 5-HT binding, whereas Na2 primarily functions to couple the Na+ gradient to transport.
Schicker et al. (61) showed that substrate currents in SERT are probably carried by a conducting state in equilibrium with the K+-bound inward facing conformation. In the same study, a model was produced in which 5-HT-induced currents were explained by conformational changes that increased the fraction of transporters in the conducting state. In this model, currents induced by 5-HT were assumed to be carried by an intermediate from which 5-HT had already dissociated. In addition, it was suggested that other known activities of SERT, such as the substrate-independent leak current of Na+ or Li+, might also be explained by the same conducting state. In this current study, we found that the leak current and the substrate-induced current through hSERT N101A reversed at the same potential, consistent with the hypothesis that both activities are related to the same conformational state. WT SERT cannot convert to the inward facing conformation with 5-HT when external Na+ is absent. However, in hSERT N101A Ca2+ supports this conversion. Therefore, if we assume that currents through WT and N101A are mechanistically similar, hSERT N101A may serve as a resource to explore the conducting state in a solution devoid of Na+. Additionally, our data support the idea that the conducting state is capable of carrying Cl−, a conjecture that is difficult to test in WT SERT. However, understanding this mechanism may provide insight into DAT function where Cl− has been identified as the primary conducting ion in the transient channel mode (72–74).
The ability of the Asn-101 mutants to utilize Ca2+ to support 5-HT uptake may indicate that Asn-101 mutations alter ion selectivity at Na1. However, we found that Ca2+ can bind to WT SERT and promote low levels of substrate transport and ion currents. Also, although Ca2+ was able to functionally replace Na+ in the Asn-101 mutant, other mono- and divalent cations could not. These findings argue that the Ca2+-mediated transport gained in the Asn-101 mutants does not originate from altered ion selectivity. Rather, they suggest that the Na1 site has a molecular restriction that prevents Ca2+ from permitting transporter activation. The Asn-101 mutants appear to disrupt this restriction by altering the ionic network connecting the Cl, Na1, and Na2 sites, revealing critical aspects of the molecular interactions between substrate and ions necessary for transporter operation that have, until now, remained enigmatic.
Evaluation of the data from this study, in light of our previous analysis of the Asn-101 residue (54), suggests critical mechanistic roles for a number of hSERT residues in substrate binding and transport. Uptake studies have shown that, in the absence of Cl−, the Km for 5-HT transport is increased significantly, indicating that Cl− binding alters the apparent affinity of 5-HT for hSERT (54, 66, 67). Our MD studies link this positive impact of Cl− on the Km of 5-HT to the residue Asp-98. Under normal Na+ and Cl− conditions, we observed monodentate coordination of the Asp-98 side chain carbonyl with the Na+ at Na1 and the amine of 5-HT. However, removal of Cl− in the simulations resulted in a loss of Asn-368 coordination with Na1, which is replaced by a bidentate interaction of the side chain of Asp-98 with Na1. This eliminates the Asp-98–5-HT interaction, providing a rational explanation for the observed increase in the Km for 5-HT. Likewise, the absence of a cation at the Na1 site does not alter 5-HT binding (66), suggesting that, in the absence of Na+, the carboxyl side chain of Asp-98 is available to stabilize 5-HT binding, although it does not lead to productive transport. Therefore, we propose that upon 5-HT binding, Asp-98 transitions from bidentate to monodentate coordination of Na+ at Na1 and that this change in coordination is critical to initiate transport. This is supported in our MD simulations with Ca2+ bound at Na1, where Asp-98 shows bidentate coordination with Na1 in both WT and Asn-101 mutant backgrounds. This lack of interaction between Asp-98 and 5-HT would lead to decreased stabilization of 5-HT binding. This is backed by uptake studies where Ca2+ replacement of Na+ resulted in a ∼10-fold increase in the Km for 5-HT in both the WT and Asn-101 mutant backgrounds. Therefore, the inability of Asp-98 to interact with 5-HT would prevent the side chain rearrangement in Asp-98 that we speculate is important to initiate transport and provides a rationale for the inability of Ca2+ to functionally replace Na+ in the WT transporter.
Given that our data reveal that Ca2+ can only bind to the Na1 site, this would suggest that the Asn-101 mutant transporters can function despite having an unoccupied Na2 site. This ability of the transporter to function with Na2 in a bound or unbound state suggests at least partial uncoupling of transport to the Na+ gradient, allowing the transporter to function in a “slippage” mode. Support for this idea comes from our MD studies, where we propose that 5-HT binding to SERT results in loss of Ser-438 coordination of Na+ at Na2. Normally, in the absence of Cl− or the presence of Ca2+, our proposed role for the Cl and Na1 sites as molecular checkpoints in WT SERT would keep the transporter from cycling, although Na2 is destabilized by 5-HT binding. However, in the N101A mutant, the regulatory role of the Cl and Na1 sites is uncoupled from Na2, allowing 5-HT binding to destabilize Na2 via S438 and promote transport. This uncoupling could also account for the large Na+ leak currents and increased efflux previously reported for the Asn-101 mutants (54) as well as the observed 5-HT dose-dependent uptake in the absence of Na+ or Ca2+ described in this study. The N101A- and N101C-mediated disruption of the Na1 and Cl sites to act as a molecular switch could explain how 5-HT binding alone to these mutants can promote transport, although Asp-98 has bidentate coordination to Na1 in absence of Cl− or the presence of Ca2+. This uncoupled state could also explain our finding that 5-HT can enhance its own uptake in a dose-dependent manner in Asn-101 mutants, even when Na+ is replaced by NMDG+.
Additional support for uncoupling of substrate to ion binding in the Asn-101 mutants comes from our substituted cysteine accessibility method studies, which reveal differential conformational changes proximal to Cys-109 (extracellular vestibule end of TM1) under various ion and substrate conditions. The findings show that in hSERT, Na+ and Cl−, but not 5-HT, are sufficient and necessary to induce a vestibular conformational change involving TM1, indicating that this movement is coupled to ion occupancy of the Na+- and Cl−-binding sites. In the N101A mutant, however, neither Cl−, Na+, nor Ca2+, alone or in combination, support the vestibular conformational change. Notably, 5-HT is able to promote a vestibular conformational change, suggesting that, whereas cation and anion binding are no longer sufficiently coupled to bring about conformational changes on their own, the uncoupled state can be overcome by 5-HT coordination. Thus, 5-HT binding could act to orient TM1, -3, -6, and -8, allowing transport to proceed, albeit less efficiently. In WT SERT, the absence of Na+ or Cl− in their binding sites would inhibit the ability of 5-HT to engage transport, but in the uncoupled state of the mutant, the ion binding sites no longer tightly regulate initiation of the transport cycle, permitting 5-HT alone to activate transport.
The recent LeuT crystal structures supplement the collection of conformational intermediates such that we now have structures spanning from the “apo open outward” to the “apo open inward” conformations. Comparison of the findings from our study with these LeuT transporter snapshots provides support for our proposed mechanism (4, 16, 19). As LeuT transitions from the outward facing to the inward facing structures, the residues that coordinate Na+ at Na1 (distances around 2.4 Å) move further away from Na+ (to around 3.4 Å), becoming weaker, but notably still coordinate Na+. However, more importantly, there is a large shift in the position of the Ala-22, Asn-27, and Asn-268 side chains in the Na1 site when LeuT transitions to an inward facing conformation (Fig. 9C). These residues correspond to Ala-96, Asn-101, and Asn-368, respectively, in hSERT, which are highly conserved amino acids and may demonstrate a preserved role for these residues in facilitating coupled transport in the SLC6 family. In fact, we found it intriguing that in all of the MD simulations, Asn-368 coordination of the cation bound at Na1 correlated with the functionally competent transporter systems, whereas a lack of Asn-368 coordination was observed in all transporter systems that were non-functional. This is not too surprising, given that in comparative models (31, 54) and by homology to dDAT (12), Asn-368 is proposed to interact with Na1Na+, Cl−, Asn-101, and Ser-336 (which directly coordinates Cl−).
Finally, the ability of the mutation at Asn-101 to affect such a large number of SERT biophysical properties emphasizes the requirement for proper communication between the ion- and substrate-binding sites to provide efficient coupling of transport to the ionic gradients. To that end, mutations such as Asn-101 will offer powerful tools for future studies to gain a better understanding of the molecular features necessary for secondary active transport.
Acknowledgment
We thank Dr. Lucia Carvelli for valuable input.
This work was supported, in whole or in part, by National Institutes of Health K01 Career Development Award DA022378 (to L. K. H.) and by National Institutes of Health, NIGMS, Institutional Development Award (IDeA) P20GM12345. This work was also supported by a National Science Foundation-EPSCoR Faculty Startup Award (to L. K. H.) and Austrian Science Foundation/FWF Grant F3506 (to H. H. S.).

This article contains supplemental Fig. S1.
- hSERT
- human serotonin transporter
- SERT
- serotonin transporter
- dDAT
- dopamine transporter from D. melanogaster
- 5-HT
- 5-hydroxytryptamine
- MTSET
- (2-trimethylammonium)ethyl)methanethiosulfonate bromide
- NMDG+
- N-methyl-d-glucamine
- MTS
- methanothiosulfanate.
REFERENCES
- 1. Jardetzky O. (1966) Simple allosteric model for membrane pumps. Nature 211, 969–970 [DOI] [PubMed] [Google Scholar]
- 2. Forrest L. R., Zhang Y.-W., Jacobs M. T., Gesmonde J., Xie L., Honig B. H., Rudnick G. (2008) Mechanism for alternating access in neurotransmitter transporters. Proc. Natl. Acad. Sci. U.S.A. 105, 10338–10343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Forrest L. R., Rudnick G. (2009) The rocking bundle. A mechanism for ion-coupled solute flux by symmetrical transporters. Physiology 24, 377–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yamashita A., Singh S. K., Kawate T., Jin Y., Gouaux E. (2005) Crystal structure of a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporters. Nature 437, 215–223 [DOI] [PubMed] [Google Scholar]
- 5. Faham S., Watanabe A., Besserer G. M., Cascio D., Specht A., Hirayama B. A., Wright E. M., Abramson J. (2008) The crystal structure of a sodium galactose transporter reveals mechanistic insights into Na+/sugar symport. Science 321, 810–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Weyand S., Shimamura T., Yajima S., Suzuki S., Mirza O., Krusong K., Carpenter E. P., Rutherford N. G., Hadden J. M., O'Reilly J., Ma P., Saidijam M., Patching S. G., Hope R. J., Norbertczak H. T., Roach P. C., Iwata S., Henderson P. J., Cameron A. D. (2008) Structure and molecular mechanism of a nucleobase-cation-symport-1 family transporter. Science 322, 709–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shaffer P. L., Goehring A., Shankaranarayanan A., Gouaux E. (2009) Structure and mechanism of a Na+-independent amino acid transporter. Science 325, 1010–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schulze S., Köster S., Geldmacher U., Terwisscha van Scheltinga A. C., Kühlbrandt W. (2010) Structural basis of Na+-independent and cooperative substrate/product antiport in CaiT. Nature 467, 233–236 [DOI] [PubMed] [Google Scholar]
- 9. Shimamura T., Weyand S., Beckstein O., Rutherford N. G., Hadden J. M., Sharples D., Sansom M. S., Iwata S., Henderson P. J., Cameron A. D. (2010) Molecular basis of alternating access membrane transport by the sodium-hydantoin transporter Mhp1. Science 328, 470–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ressl S., Terwisscha van Scheltinga A. C., Vonrhein C., Ott V., Ziegler C. (2009) Molecular basis of transport and regulation in the Na+/betaine symporter BetP. Nature 458, 47–52 [DOI] [PubMed] [Google Scholar]
- 11. Gao X., Lu F., Zhou L., Dang S., Sun L., Li X., Wang J., Shi Y. (2009) Structure and mechanism of an amino acid antiporter. Science 324, 1565–1568 [DOI] [PubMed] [Google Scholar]
- 12. Penmatsa A., Wang K. H., Gouaux E. (2013) X-ray structure of dopamine transporter elucidates antidepressant mechanism. Nature 503, 85–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Khafizov K., Perez C., Koshy C., Quick M., Fendler K., Ziegler C., Forrest L. R. (2012) Investigation of the sodium-binding sites in the sodium-coupled betaine transporter BetP. Proc. Natl. Acad. Sci. U.S.A. 109, E3035–E3044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Krishnamurthy H., Piscitelli C. L., Gouaux E. (2009) Unlocking the molecular secrets of sodium-coupled transporters. Nature 459, 347–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Abramson J., Wright E. M. (2009) Structure and function of Na+-symporters with inverted repeats. Curr. Opin. Struct. Biol. 19, 425–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Singh S. K., Piscitelli C. L., Yamashita A., Gouaux E. (2008) A competitive inhibitor traps LeuT in an open-to-out conformation. Science 322, 1655–1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhou Z., Zhen J., Karpowich N. K., Goetz R. M., Law C. J., Reith M. E., Wang D.-N. (2007) LeuT-desipramine structure reveals how antidepressants block neurotransmitter reuptake. Science 317, 1390–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Piscitelli C. L., Gouaux E. (2012) Insights into transport mechanism from LeuT engineered to transport tryptophan. EMBO J. 31, 228–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Krishnamurthy H., Gouaux E. (2012) X-ray structures of LeuT in substrate-free outward-open and apo inward-open states. Nature 481, 469–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Blakely R. D., Berson H. E., Fremeau R. T., Jr., Caron M. G., Peek M. M., Prince H. K., Bradley C. C. (1991) Cloning and expression of a functional serotonin transporter from rat brain. Nature 354, 66–70 [DOI] [PubMed] [Google Scholar]
- 21. Hoffman B. J., Mezey E., Brownstein M. J. (1991) Cloning of a serotonin transporter affected by antidepressants. Science 254, 579–580 [DOI] [PubMed] [Google Scholar]
- 22. Reith M. E. A. (2002) Neurotransmitter Transporters: Structure, Function, and Regulation, 2nd Ed., Humana Press, Totowa, NJ [Google Scholar]
- 23. Beuming T., Shi L., Javitch J. A., Weinstein H. (2006) A comprehensive structure-based alignment of prokaryotic and eukaryotic neurotransmitter/Na+ symporters (NSS) aids in the use of the LeuT structure to probe NSS structure and function. Mol. Pharmacol. 70, 1630–1642 [DOI] [PubMed] [Google Scholar]
- 24. Kristensen A. S., Andersen J., Jørgensen T. N., Sørensen L., Eriksen J., Loland C. J., Strømgaard K., Gether U. (2011) SLC6 neurotransmitter transporters. Structure, function, and regulation. Pharmacol. Rev. 63, 585–640 [DOI] [PubMed] [Google Scholar]
- 25. Pramod A. B., Foster J., Carvelli L., Henry L. K. (2013) SLC6 transporters. Structure, function, regulation, disease association and therapeutics. Mol. Aspects Med. 34, 197–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ramamoorthy S., Bauman A. L., Moore K. R., Han H., Yang-Feng T., Chang A. S., Ganapathy V., Blakely R. D. (1993) Antidepressant- and cocaine-sensitive human serotonin transporter. Molecular cloning, expression, and chromosomal localization. Proc. Natl. Acad. Sci. U.S.A. 90, 2542–2546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tatsumi M., Groshan K., Blakely R. D., Richelson E. (1997) Pharmacological profile of antidepressants and related compounds at human monoamine transporters. Eur. J. Pharmacol. 340, 249–258 [DOI] [PubMed] [Google Scholar]
- 28. McCann U. D., Szabo Z., Scheffel U., Dannals R. F., Ricaurte G. A. (1998) Positron emission tomographic evidence of toxic effect of MDMA (“Ecstasy”) on brain serotonin neurons in human beings. Lancet 352, 1433–1437 [DOI] [PubMed] [Google Scholar]
- 29. Barker E. L., Moore K. R., Rakhshan F., Blakely R. D. (1999) Transmembrane domain I contributes to the permeation pathway for serotonin and ions in the serotonin transporter. J. Neurosci. 19, 4705–4717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kantcheva A. K., Quick M., Shi L., Winther A.-M., Stolzenberg S., Weinstein H., Javitch J. A., Nissen P. (2013) Chloride binding site of neurotransmitter sodium symporters. Proc. Natl. Acad. Sci. U.S.A. 110, 8489–8994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Forrest L. R., Tavoulari S., Zhang Y.-W., Rudnick G., Honig B. (2007) Identification of a chloride ion binding site in Na+/Cl−-dependent transporters. Proc. Natl. Acad. Sci. U.S.A. 104, 12761–12766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zomot E., Bendahan A., Quick M., Zhao Y., Javitch J. A., Kanner B. I. (2007) Mechanism of chloride interaction with neurotransmitter:sodium symporters. Nature 449, 726–730 [DOI] [PubMed] [Google Scholar]
- 33. Tavoulari S., Rizwan A. N., Forrest L. R., Rudnick G. (2011) Reconstructing a chloride-binding site in a bacterial neurotransmitter transporter homologue. J. Biol. Chem. 286, 2834–2842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sneddon J. M. (1969) Sodium-dependent accumulation of 5-hydroxytryptamine by rat blood platelets. Br. J. Pharmacol. 37, 680–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shi L., Weinstein H. (2010) Conformational rearrangements to the intracellular open states of the LeuT and ApcT transporters are modulated by common mechanisms. Biophys. J. 99, L103–L105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhao C., Noskov S. Y. (2011) The role of local hydration and hydrogen-bonding dynamics in ion and solute release from ion-coupled secondary transporters. Biochemistry 50, 1848–1856 [DOI] [PubMed] [Google Scholar]
- 37. Zhao C., Stolzenberg S., Gracia L., Weinstein H., Noskov S., Shi L. (2012) Ion-controlled conformational dynamics in the outward-open transition from an occluded state of LeuT. Biophys. J. 103, 878–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zdravkovic I., Zhao C., Lev B., Cuervo J. E., Noskov S. Y. (2012) Atomistic models of ion and solute transport by the sodium-dependent secondary active transporters. Biochim. Biophys. Acta 1818, 337–347 [DOI] [PubMed] [Google Scholar]
- 39. Yu H., Noskov S. Y., Roux B. (2010) Two mechanisms of ion selectivity in protein binding sites. Proc. Natl. Acad. Sci. U.S.A. 107, 20329–20334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Noskov S. Y., Roux B. (2008) Control of ion selectivity in LeuT. Two Na+ binding sites with two different mechanisms. J. Mol. Biol. 377, 804–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Watanabe A., Choe S., Chaptal V., Rosenberg J. M., Wright E. M., Grabe M., Abramson J. (2010) The mechanism of sodium and substrate release from the binding pocket of vSGLT. Nature 468, 988–991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Henry L. K., Adkins E. M., Han Q., Blakely R. D. (2003) Serotonin and cocaine-sensitive inactivation of human serotonin transporters by methanethiosulfonates targeted to transmembrane domain I. J. Biol. Chem. 278, 37052–37063 [DOI] [PubMed] [Google Scholar]
- 43. Boehm S. (1999) ATP stimulates sympathetic transmitter release via presynaptic P2X purinoceptors. J. Neurosci. 19, 737–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kaufmann K. W., Dawson E. S., Henry L. K., Field J. R., Blakely R. D., Meiler J. (2009) Structural determinants of species-selective substrate recognition in human and Drosophila serotonin transporters revealed through computational docking studies. Proteins 74, 630–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ramachandran G. N., Ramakrishnan C., Sasisekharan V. (1963) Stereochemistry of polypeptide chain configurations. J. Mol. Biol. 7, 95–99 [DOI] [PubMed] [Google Scholar]
- 46. Laskowski R., Macarthur M., Moss D., Thornton J. (1993) PROCHECK. A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26, 283–291 [Google Scholar]
- 47. Sastry G. M., Adzhigirey M., Day T., Annabhimoju R., Sherman W. (2013) Protein and ligand preparation. Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 27, 221–234 [DOI] [PubMed] [Google Scholar]
- 48. Sherman W., Day T., Jacobson M. P., Friesner R. A., Farid R. (2006) Novel Procedure for Modeling Ligand/Receptor Induced Fit Effects. J. Med. Chem. 49, 534–553 [DOI] [PubMed] [Google Scholar]
- 49. Humphrey W., Dalke A., Schulten K. (1996) VMD. Visual molecular dynamics. J. Mol. Graph 14, 33–38, 27–28 [DOI] [PubMed] [Google Scholar]
- 50. Hess B., Kutzner C., van der Spoel D., Lindahl E. (2008) GROMACS 4. Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 [DOI] [PubMed] [Google Scholar]
- 51. Zoete V., Cuendet M. A., Grosdidier A., Michielin O. (2011) SwissParam. A fast force field generation tool for small organic molecules. J. Comput. Chem. 32, 2359–2368 [DOI] [PubMed] [Google Scholar]
- 52. Hess B., Bekker H., Berendsen H. J. C., Fraaije J. G. E. M. (1997) LINCS. A linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 [Google Scholar]
- 53. DeLano W. L. (2010) The PyMOL Molecular Graphics System, version 1.3r1, Schrödinger, LLC, New York [Google Scholar]
- 54. Henry L. K., Iwamoto H., Field J. R., Kaufmann K., Dawson E. S., Jacobs M. T., Adams C., Felts B., Zdravkovic I., Armstrong V., Combs S., Solis E., Rudnick G., Noskov S. Y., DeFelice L. J., Meiler J., Blakely R. D. (2011) A conserved asparagine residue in transmembrane segment 1 (TM1) of serotonin transporter dictates chloride-coupled neurotransmitter transport. J. Biol. Chem. 286, 30823–30836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chen J. G., Liu-Chen S., Rudnick G. (1997) External cysteine residues in the serotonin transporter. Biochemistry 36, 1479–1486 [DOI] [PubMed] [Google Scholar]
- 56. Chen J. G., Sachpatzidis A., Rudnick G. (1997) The third transmembrane domain of the serotonin transporter contains residues associated with substrate and cocaine binding. J. Biol. Chem. 272, 28321–28327 [DOI] [PubMed] [Google Scholar]
- 57. Ni Y. G., Chen J. G., Androutsellis-Theotokis A., Huang C. J., Moczydlowski E., Rudnick G. (2001) A lithium-induced conformational change in serotonin transporter alters cocaine binding, ion conductance, and reactivity of Cys-109. J. Biol. Chem. 276, 30942–30947 [DOI] [PubMed] [Google Scholar]
- 58. Androutsellis-Theotokis A., Ghassemi F., Rudnick G. (2001) A conformationally sensitive residue on the cytoplasmic surface of serotonin transporter. J. Biol. Chem. 276, 45933–45938 [DOI] [PubMed] [Google Scholar]
- 59. Androutsellis-Theotokis A., Rudnick G. (2002) Accessibility and conformational coupling in serotonin transporter predicted internal domains. J. Neurosci. 22, 8370–8378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mager S., Min C., Henry D. J., Chavkin C., Hoffman B. J., Davidson N., Lester H. A. (1994) Conducting states of a mammalian serotonin transporter. Neuron 12, 845–859 [DOI] [PubMed] [Google Scholar]
- 61. Schicker K., Uzelac Z., Gesmonde J., Bulling S., Stockner T., Freissmuth M., Boehm S., Rudnick G., Sitte H. H., Sandtner W. (2012) Unifying concept of serotonin transporter-associated currents. J. Biol. Chem. 287, 438–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Partridge L. D., Leach J. K. (1991) Calcium Channels: Their Properties, Functions, Regulation, and Clinical Relevance, pp. 121–123, CRC Press, Boca Raton, FL [Google Scholar]
- 63. Bock G., Gebhart M., Scharinger A., Jangsangthong W., Busquet P., Poggiani C., Sartori S., Mangoni M. E., Sinnegger-Brauns M. J., Herzig S., Striessnig J., Koschak A. (2011) Functional properties of a newly identified C-terminal splice variant of Cav1.3 L-type Ca2+ channels. J. Biol. Chem. 286, 42736–42748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Harding M. M. (2002) Metal-ligand geometry relevant to proteins and in proteins. Sodium and potassium. Acta Crystallogr. D Biol. Crystallogr. 58, 872–874 [DOI] [PubMed] [Google Scholar]
- 65. Ben-Yona A., Bendahan A., Kanner B. I. (2011) A glutamine residue conserved in the neurotransmitter:sodium:symporters is essential for the interaction of chloride with the GABA transporter GAT-1. J. Biol. Chem. 286, 2826–2833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chang A. S., Lam D. M. (1998) Mechanistic analyses of ion dependences in a high-affiinity human serotonin transport system in transfected murine fibroblast cells. J. Physiol. 510, 903–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Nelson P. J., Rudnick G. (1982) The role of chloride ion in platelet serotonin transport. J. Biol. Chem. 257, 6151–6155 [PubMed] [Google Scholar]
- 68. Koldsø H., Noer P., Grouleff J., Autzen H. E., Sinning S., Schiøtt B. (2011) Unbiased simulations reveal the inward-facing conformation of the human serotonin transporter and Na+ ion release. PLoS Comput. Biol. 7, e1002246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Rudnick G., Nelson P. J. (1978) Platelet 5-hydroxytryptamine transport, an electroneutral mechanism coupled to potassium. Biochemistry 17, 4739–4742 [DOI] [PubMed] [Google Scholar]
- 70. Nelson P. J., Rudnick G. (1979) Coupling between platelet 5-hydroxytryptamine and potassium transport. J. Biol. Chem. 254, 10084–10089 [PubMed] [Google Scholar]
- 71. Quick M. (2003) Regulating the conducting states of a mammalian serotonin transporter. Neuron 40, 537–549 [DOI] [PubMed] [Google Scholar]
- 72. Carvelli L., McDonald P. W., Blakely R. D., Defelice L. J. (2004) Dopamine transporters depolarize neurons by a channel mechanism. Proc. Natl. Acad. Sci. U.S.A. 101, 16046–16051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ingram S. L., Prasad B. M., Amara S. G. (2002) Dopamine transporter-mediated conductances increase excitability of midbrain dopamine neurons. Nat. Neurosci. 5, 971–978 [DOI] [PubMed] [Google Scholar]
- 74. Meinild A.-K., Sitte H. H., Gether U. (2004) Zinc potentiates an uncoupled anion conductance associated with the dopamine transporter. J. Biol. Chem. 279, 49671–49679 [DOI] [PubMed] [Google Scholar]