

Background: Aryl hydrocarbon receptor (AhR) is a protein regulating differentiation and function of immune cells.
Results: NF-κB activates transcription of AhR and enhances activity of AhR-regulated genes.
Conclusion: Activation of NF-κB involves RelA-mediated expression of AhR.
Significance: Inflammatory stimuli and cytokines that regulate NF-κB induce AhR expression during activation and differentiation of immune cells.
Keywords: Aryl Hydrocarbon Receptor, Dendritic Cells, Gene Regulation, Inflammation, NF-Kappa B (NF-KB)
Abstract
The aryl hydrocarbon receptor (AhR) is involved in the regulation of immune responses, T-cell differentiation, and immunity. Here, we show that inflammatory stimuli such as LPS induce the expression of AhR in human dendritic cells (DC) associated with an AhR-dependent increase of CYP1A1 (cytochrome P4501A1). In vivo data confirmed the elevated expression of AhR by LPS and the LPS-enhanced 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-mediated induction of CYP1A1 in thymus of B6 mice. Inhibition of nuclear factor-κB (NF-κB) repressed both normal and LPS-enhanced, TCDD-inducible, AhR-dependent gene expression and canonical pathway control of RelA-regulated AhR-responsive gene expression. LPS-mediated induction of AhR was NF-κB-dependent, as shown in mouse embryonic fibroblasts (MEFs) derived from Rel null mice. AhR expression and TCDD-mediated induction of CYP1A1 was significantly reduced in RelA-deficient MEF compared with wild type MEF cells and ectopic expression of RelA restored the expression of AhR and induction of CYP1A1 in MEF RelA null cells. Promoter analysis of the human AhR gene identified three putative NF-κB-binding elements upstream of the transcription start site. Mutation analysis of the AhR promoter identified one NF-κB site as responsible for mediating the induction of AhR expression by LPS and electrophoretic shift assays demonstrated that this NF-κB motif is recognized by the RelA/p50 heterodimer. Our results show for the first time that NF-κB RelA is a critical component regulating the expression of AhR and the induction of AhR-dependent gene expression in immune cells illustrating the interaction of AhR and NF-κB signaling.
Introduction
The AhR2 is a member of basic helix-loop-helix-PAS (Per-ARNT-Sim) transcription factors, including Per (period), ARNT (AhR nuclear translocator), and Sim (single-minded), regulating hypoxia, circadian rhythm, and cellular processes such as differentiation and apoptosis. Similar to its murine ortholog, the human AhR promoter bears multiple transcription initiation sites that are clustered in a GC-rich region and contains neither TATA nor CCAAT boxes (1–3). The GC-rich region includes four consensus sequences for Sp-1 binding sites, which seem to be necessary for basal expression of the AhR-promoter construct.
There is increasing evidence showing that the AhR plays an important role in regulating immune responses and that exposure to AhR-activating toxicants contributes to the development of immune disorders (4). The AhR affects the expression of immunoregulatory genes and the function as well as differentiation of inflammatory DC (5–8). Furthermore, recent reports show that the AhR plays a critical role in T-cell differentiation and immunity of the gut (9–12), but the molecular mechanisms that control its activity in immune cells during inflammation have remained unclear. Immunohistochemical analysis of embryonic tissues showed that AhR expression is developmentally regulated (13). Stimulation of resting T cells with mitogens resulted in a marked increase of AhR expression (14, 15), whereas TGF-β inhibits or increases the expression of AhR and genes of the AhR gene battery in a cell-specific manner (16, 17).
In this study, we elucidated the molecular mechanisms responsible for regulating AhR expression during inflammatory responses. We demonstrate for the first time that LPS markedly induces AhR expression through activation of RelA and binding of RelA/p50 to an NF-κB binding site identified in the human AhR gene promoter.
EXPERIMENTAL PROCEDURES
Reagents and Antibodies
Dimethyl sulfoxide (DMSO), phorbol 12-myristate 13-acetate, and LPS were obtained from Sigma. [γ-32P]ATP (6000 Ci/mmol) was purchased from ICN Biochemicals, Inc. (Costa Mesa, CA). NF-κB inhibitors (pyrrolidinedithiocarbamate (PDTC), (E)-capsaicin (CAPS), and caffeic acid phenethyl ester (CAPE) were purchased from Calbiochem. TCDD (>99% purity) was originally obtained from Dow Chemical Co. (Midland, MI). Other molecular biological reagents were purchased from Qiagen (Valencia, CA) and Roche Clinical Laboratories (Indianapolis, IN). Poly dI·dC, polyclonal RelA, ARNT (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), NF-κB member p50 (Active Motif, Carlsbad, CA) and AhR (Novus Biologicals, Littleton, CO; Abnova, Walnut, CA) were used for Western blot analyses and Supershift in EMSA.
Animals and Treatment
Male C57BL/6J wild type (WT) mice, 8 weeks old, purchased from The Jackson Laboratory (West Sacramento, CA). Male mice were housed (four per cage) in a selective pathogen-free facility and humidity- and temperature-controlled room. Ahr null mice (Ahr−/−) were a kind gift of Christopher Bradfield (University of Wisconsin). The animals were maintained on a 12:12 h light/dark cycle and had free access to water and food according to the guidelines set by the University of California Davis. Mice were allowed to adapt to the facility for 1 week. TCDD and LPS were administered via a single intraperitoneal injection. TCDD was prepared from a stock solution and diluted in corn oil. A stock solution of LPS was prepared in water. Control animals received an equal amount of corn oil (5 ml/kg) or water alone. C57BL/6J and Ahr−/− mice received a single dose of 15 μg/kg TCDD for 24 h. After 18 h of TCDD treatment, mice were injected with a dose of 250 μg/kg LPS. Four animals from each group were killed, and organs were excised, weighed, and, if not otherwise stated, quickly frozen in liquid nitrogen and stored at −80 °C for analysis.
Cloning of the Human AhR Promoter and Site-directed Mutagenesis
The promoter region of human AhR was generated by PCR amplification of genomic DNA isolated from the human colon carcinoma cell line LS180 as described previously (17). The luciferase expression plasmid under control of the human AhR promoter was termed pGL3-hAhRP, including 5640 nt upstream of the putative start site (17). To produce deletion constructs of pGL3-hAhRP, the clone was digested with KpnI and several endonucleases (EcoRV, EcoRI, NdeI, SauI, ApaI) that possess single recognition sequences within the full-length construct and are termed AhRΔ(−2510), AhRΔ(−1980), AhRΔ(−1892), AhRΔ(−881), and AhRΔ(−120), respectively (17). DNA promoter analysis of the human AhR gene was performed using the TFSEARCH program (19) and identified three putative NF-κB binding sites. Mutation of these three NF-κB sequences in the human AhR gene promoter (at −2757 bp 5′-GGGGAATTTC-3′, at −1452 bp 5′-GGGAATTTGC-3′, and at −399 bp 5′-GGAAACTCCT-3′) was carried out by site-directed mutagenesis (Stratagene, La Jolla, CA) using the following primers synthesized by Integrated DNA Technologies, Inc. (Coralville, IA): NF-κB M1 mutant, 5′-TGGGGAGGAAGGTACCTTTCATGCAGACTG-3′; NF-κB M2 mutant, 5′-TACACTGTCTTCTTTGGTACCTTGCTCCATCTTTTTCCTT-3′; and NF-κB M3 mutant, 5′-AAAAGGTCAAGGTACCTCCTAGCCTTCAAG-3′. Insertion of the mutated bases (underlined) was confirmed by sequencing. Expression vectors for p50 and RelA were kindly provided by W. Greene (J. David Gladstone Institute, San Francisco, CA), and the RelB expression plasmid was kindly provided by U. Siebenlist (National Institutes of Health, Bethesda, MD).
Cell Culture, Transfection Experiments, and Luciferase Assay
Primary MEFs were isolated from 14-day post coitus wild type, Rel null B6 mice, and Ahr−/− B6 mouse embryos as described (20–22). Embryos were surgically removed and separated from maternal tissues and the yolk sack. The bodies were minced finely and then incubated in a solution of trypsin and EDTA (0.05% trypsin; 1 mm EDTA) at 37 °C for 30 min. The supernatant was centrifuged for 5 min at 1000 × g. The resulting pellet was resuspended in culture medium, and cells were plated in 100-mm culture dishes. Primary MEFs were maintained in DMEM:F12 (Invitrogen). Cell culture medium contained 10% fetal bovine serum and 100 units of penicillin and 100 μg/ml streptomycin. Human monocyte-derived DC were generated from CD14+ monocytes freshly isolated from healthy individuals as described previously (23). Cells were maintained in RPMI 1640 medium containing 50 μm 2-mercaptoethanol, 10 mm HEPES, 10% endotoxin-free fecal calf serum, recombinant human GM-CSF, and IL-4 (1000 units/ml each) for 7 days. The human monocytic cell line U937 was obtained from ATCC and maintained in RPMI 1640 medium. To generate DC, U937 cells were cultured for 2 days in growth medium supplemented with GM-CSF (20 ng/ml) and IL-4 (40 ng/ml) as described previously (8). Transfection of plasmid DNA or siRNA into U937-derived DC was performed via Nucleofector technology as described (24). Briefly, 106 U937-derived DC were resuspended in 100 μl of Nucleofector Solution V (Amaxa GmbH, Köln, Germany) and nucleofected with 1.0 μg of plasmid DNA or siRNA using program V-001, which is preprogrammed into the Nucleofector device (Amaxa GmbH). siRNA to target human RelA and a negative control siRNA were synthesized by Qiagen. For transient transfection of MEF, cells were plated in 24-well plates (1 × 105 cells per well) and transfected using jetPEI (PolyTransfection; Qbiogene, Irvine, CA), according to the manufacturer's instructions. Briefly, 0.3 μg of the RelA construct was suspended in 25 μl of 150 mm sterile NaCl solution. Also, 0.3 μl of jetPEI solution was suspended in 25 μl of 150 mm sterile NaCl solution. The jetPEI/NaCl solution was then added to the DNA/NaCl solution and incubated at room temperature for 30 min. The medium in the wells was changed to fresh medium, and 50 μl of the DNA/jetPEI was added to each well. The transfection was allowed to proceed for 24 h, and cells were treated with 10 nm TCDD or 0.1% Me2SO (control) for 6 h. To control the transfection efficiency, cells were cotransfected with 0.1 μg per well β-galactosidase reporter construct. Luciferase activities were measured with the luciferase reporter assay system (Promega Corp., Madison, WI) using a luminometer (Berthold Lumat LB 9501/16; Pittsburgh, PA). Relative light units were normalized to β-galactosidase activity and to protein concentration, using Bradford dye assay (Bio-Rad).
EMSA
Nuclear extracts were isolated from U937 cells as described previously (24). In brief, 5 × 106 cells were treated with LPS or TCDD for 90 min and harvested in Dulbecco's PBS containing 1 mm PMSF and 0.05 μg/μl of aprotinin. After centrifugation, the cell pellets were gently resuspended in 1 ml of hypotonic buffer (20 mm HEPES, 20 mm NaF, 1 mm Na3VO4, 1 mm Na4P2O7, 1 mm EDTA, 1 mm EGTA, 0.5 mm PMSF, 0.13 μm okadaic acid, 1 mm dithiothreitol, pH 7.9, and 1 μg/ml each leupeptin, aprotinin, and pepstatin). The cells were allowed to swell on ice for 15 min and then homogenized by 25 strokes of a Dounce homogenizer. After centrifugation for 1 min at 16,000 × g, nuclear pellets were resuspended in 300 μl ice-cold high-salt buffer (hypotonic buffer with 420 mm NaCl, and 20% glycerol). The samples were passed through a 21-gauge needle and stirred for 30 min at 4 °C. The nuclear lysates were microcentrifuged at 16,000 × g for 20 min, aliquoted, and stored at −80 °C. DNA-protein binding reactions were carried out in a total volume of 15 μl containing 10 μg of nuclear protein, 60,000 cpm of double-stranded DRE consensus oligonucleotide (5′-GCCCCGGAGTTGCGTGAGAAGAGCCTGG-3′), AhR-NF-κB1 oligonucleotide (5′-TGGGGAGGAAGGGGAATTTCATGCAGACTG-3′), AhR-NF-κB2 oligonucleotide (5′-TACACTGTCTTCTTTGGGAATTTGCTCCATCTTTTTCCTT-3′), or AhR-NF-κB3 (5′-AAAAGGTCAAGGAAACTCCTAGCCTTCAAG-3′), 25 mm Tris buffer (pH 7.5), 50 mm NaCl, 1 mm EDTA, 0.5 mm dithiothreitol, 5% glycerol, and 1 μg of poly(dI·dC). The samples were incubated at room temperature for 20 min. Competition experiments were performed in the presence of a 100-fold molar excess of unlabeled DNA fragments. Protein-DNA complexes were resolved on a 4% nondenaturating polyacrylamide gel and visualized by exposure of the dried gels to x-ray films. Protein-DNA complexes were quantified using a ChemiImagerTM 4400 (Alpha Innotech Corp., San Leandro, CA).
Quantitative Real-time RT-PCR
Total RNA was isolated from cells using a Quick-RNA Mini prep isolation kit (Zymo Research, Irvine, CA), and cDNA synthesis was done as described previously (24). Quantitative detection of β-actin and differentially expressed genes was performed with a LightCycler LC480 Instrument (Roche Diagnostics) using the Fast SYBR Green Master Mix (Invitrogen) according to the manufacturer's instructions. The primers for each gene were designed on the basis of the respective cDNA or mRNA sequences using OLIGO primer analysis software provided by Steve Rozen and the Whitehead Institute/Massachusetts Institute of Technology Center for Genome Research so that the targets were 100–200 bp in length (Table 1). PCR amplification was carried out in a total volume of 20 μl containing 2 μl of cDNA, 10 μl of 2× Fast SYBR Green Master Mix, and 0.2 μm of each primer. The PCR cycling conditions were 95 °C for 30 s followed by 40 cycles of 94 °C for 3 s and 60 °C for 30 s. Detection of the fluorescent product was performed at the end of the 72 °C extension period. Negative controls were concomitantly run to confirm that the samples were not cross-contaminated. A sample with DNase- and RNase-free water instead of RNA was concomitantly examined for each of the reaction units described above. To confirm the amplification specificity, the PCR products were subjected to melting curve analysis.
TABLE 1.
Primers used to amplify mRNAs via quantitative real-time PCR based on published GenBank sequences for mouse and human
| Gene | Forward primer (5′–3′) | Reverse primer (5′–3′) |
|---|---|---|
| Human β-actin | GGACTTCGAGCAAGAGATGG | AGCACTGTGTTGGCGTACAG |
| Human Ahr | TCAACAGCAACAGTCCTTGG | TCCAATTTTCAAACATGCCA |
| Human ARNT | AACCTCACTTCGTGGTGGTC | CAATGTTGTGTCGGGAGATG |
| Human CYP1A1 | TAGACACTGATCTGGCTGCAG | GGGAAGGCTCCATCAGCATC |
| Mouse β-actin | AGCCATGTACGTAGCCATCC | CTCTCAGCTGTGGTGGTGAA |
| Mouse AhR | ACCAGAACTGTGAGGGTTGG | TCTGAGGTGCCTGAACTCCT |
| Mouse ARNT | TGCCTCATCTGGTACTGCTG | GAACATGCTGCTCACTGGAA |
| Mouse CYP1A1 | GGCCACTTTGACCCTTACAA | CAGGTAACGGAGGACAGGAA |
Western Blot Analysis
To analyze the level of AhR and CYP1A1 protein in human DC, whole cell protein (25 μg) was separated on a 10% SDS-polyacrylamide gel and blotted onto a polyvinylidine difluoride membrane (Immuno-Blot; Bio-Rad). The antigen-antibody complexes were visualized using the chemoluminescence substrate SuperSignal West Pico (Pierce) as recommended by the manufacturer.
Statistical Analysis
All experiments were repeated a minimum of three times, and data are expressed as mean ± S.D. Differences were considered significant at p < 0.05. A comparison of two groups was made with an unpaired, two-tailed Student's t test. A comparison of multiple groups was made with analysis of variance followed by a Dunnett's or Tukey's test.
RESULTS
LPS-induced Expression of AhR and CYP1A1 in Human DC and Thymus of B6 Mice
Treatment of human DC derived from whole blood with LPS for 24 h significantly induced the expression of AhR mRNA of ∼3-fold compared with control (Fig. 1A). TCDD had no significant effect on AhR mRNA expression in human DC. LPS also increased the background expression of CYP1A1 by ∼2.5-fold and further elevated TCDD-induced expression of CYP1A1 to 85-fold compared with a 25-fold increase of CYP1A1 by TCDD in the absence of LPS. Furthermore, while LPS induced the expression of AhR and CYP1A1 mRNA in the thymus of B6 wt mice (Fig. 1B), it failed to do so in the thymus of Ahr−/− mice (Fig. 1C). Similar to human DC, treatment with LPS significantly increased the TCDD-induced expression of CYP1A1 in the thymus of B6 mice (Fig. 1B). To monitor the expression level of AhR mRNA during inflammation, U937-derived DC were activated with LPS for 1 to 24 h. Maximal LPS-dependent induction of AhR (∼4.5-fold above control) was found after 12 h of LPS treatment in human U937 DC. The increase in AhR by LPS occurred concomitantly with a 2-fold increase of CYP1A1 mRNA at 6 and 12 h and reached a maximum of a 3-fold increase of CYP1A1 mRNA at 24 h (Fig. 1D). The expression of ARNT did not significantly change after treatment with TCDD or LPS in DC or thymus. Western blot analysis confirmed that the LPS-induced mRNA expression of AhR and CYP1A1 also occurred at the protein level, showing increased level of AhR and CYP1A1 at 6 and 24 h in whole cell lysates of human DC (Fig. 1E). EMSA with nuclear extracts from U937-derived DC revealed that the LPS-induced expression of AhR is associated with a small increase in protein-DRE complex formation in LPS-treated cells compared with control cells (Fig. 1F, lanes 1 and 3). Similar to the enhanced CYP1A1 mRNA expression in TCDD-treated DC and thymus of TCDD-treated B6 mice, LPS incubation led to an enhanced TCDD-induced protein-DNA complex formation (Fig. 1F, lane 4). EMSA supershift analysis revealed that the enhanced protein-DNA complex observed in the presence of TCDD and LPS contained both AhR and ARNT (Fig. 1G, lanes 5 and 6).
FIGURE 1.

Increased expression of AhR and CYP1A1 by LPS in human DC and thymus of B6 mice. A, LPS induces AhR and CYP1A1 in human DC. Human monocyte-derived DC were generated as described under “Experimental Procedures.” DC were treated with 10 nm TCDD for 7 days or 0.05 μg/ml LPS for 24 h. TCDD-treated DC were co-treated with LPS for 24 h. B6 wild type (B) and Ahr−/− (C) mice were treated with 15 μg/kg TCDD for 24 h or with 0.5 mg/kg LPS for 6 h. For co-treatment, mice were treated with 15 μg/kg TCDD for 18 h and then treated with 0.5 mg/kg LPS for 6 h. D, time course study of AhR, ARNT, and CYP1A1 mRNA induction in U937-derived DC. Cells were treated for 1 to 48 h with 0.1 μg/ml LPS or 1 μl/ml water (Control). E, Western blot analysis of AhR, ARNT, and CYP1A1 protein level in human DC. 25 μg of whole cell protein was loaded per lane. AhR and CYP1A1 protein levels were quantitated and normalized to actin. Values represent the mean ± S.D. of three independent experiments. An asterisk indicates significantly different from control cells (p < 0.05). Double asterisks indicate significantly higher than only TCDD-treated cells (p < 0.05). F, LPS-induced AhR binding to a DRE consensus element of the Cyp1a1 promoter. Nuclear extracts from untreated control (lane 1) and LPS-treated (lanes 3 and 5) U937-derived DC were used for EMSA. Cells were treated with 1 nm TCDD for 1 h or with 0.1 μg/ml LPS for 6 h. A possible enhancement of TCDD-induced AhR-binding activity as shown in lane 2 was tested by treatment of cells with LPS (0.1 μg/ml) for 6 h followed by treatment with TCDD (1 nm) for 1 h (lane 4). EMSA was performed using double-stranded, 32P-labeled oligonucleotide containing the DRE binding sequence of the rat Cyp1a1 promoter. To confirm specificity, a 100-fold excess of unlabeled DRE oligonucleotide (lane 5) was added. G, the specific binding of AhR and ARNT was identified by EMSA supershift analyses using AhR- and ARNT-specific antibodies (lanes 5 and 6). For EMSA, one representative experiment of three independently performed experiments is shown. Ab., antibody; Comp., competition; Ctrl, control; Treat., treatment.
AhR Expression Is Regulated by NF-κB RelA
To determine whether activation of AhR by LPS is NF-κB-dependent, U937-derived DC were pretreated for 30 min with the NF-κB inhibitor PDTC or water as vehicle. LPS had no significant effect on the expression of AhR or CYP1A1 when cells were preincubated with PDTC (Fig. 2A), indicating a requirement for NF-κB to stimulate AhR gene expression in LPS-treated cells. Our results examining DRE luciferase reporter (containing the DRE1 sequence of the rat Cyp1a1 gene promoter region −1029 to −997) activity in U937-derived DC show that LPS enhances the TCDD-induced DRE-dependent luciferase activity (Fig. 2B). We further examined what role NF-κB might play in the LPS enhancement effect as well as in AhR-dependent gene expression. The optimal treatment conditions for NF-κB inhibition were determined to be 200 μm PDTC, 50 μm CAPS, and 5 μg/ml CAPE for 30 min (data not shown) before treatment with 10 nm TCDD. Not only did each NF-κB inhibitor block the LPS enhancement effect, they also reduced the magnitude of induction of luciferase by TCDD alone. These results further support a role for NF-κB in normal AhR-dependent signal transduction as well as in the LPS enhancement effect (Fig. 2B). These results confirm that the repressive effect of NF-κB inhibitors on AhR-dependent gene expression is DRE-dependent. Furthermore, gene silencing of RelA decreased the expression of AhR and suppressed the TCDD-induced and LPS-enhanced expression of CYP1A1 in U937-derived DC (Fig. 2C). The protein level of RelA was significantly reduced by gene silencing using RelA-specific siRNA (Fig. 2D).
FIGURE 2.

NF-κB RelA dependent expression of AhR. A, U937-derived DC were treated with 0.1 μg/ml LPS, 1 nm TCDD, and co-treated with TCDD and LPS for 6 h in absence or presence of 200 μm PDTC. PDTC or water was added 30 min prior to the addition of DMSO as vehicle, TCDD, or LPS. AhR, ARNT, and CYP1A1 mRNA levels were analyzed using real-time PCR. B, effect of NF-κB inhibitors on TCDD-induced and LPS-enhanced DRE luciferase reporter activity. U937-derived DC were incubated with carrier solvent alone (1 μl/ml), 0.1 μg/ml LPS, 1 nm TCDD, 200 μm PDTC, 5 μg/ml CAPE, 50 μm CAPS, or the indicated combination. TCDD was dissolved in DMSO, LPS, and PDTC in water, and CAPE and CAPS were dissolved in ethanol. Cells were treated with LPS for 16 h and with TCDD for 4 h. PDTC, CAPE, and CAPS were added 30 min prior to LPS and TCDD. Luciferase activity was determined as described under “Experimental Procedures.” Values are expressed as relative luminescence units/mg protein and represent the mean ± S.D. of triplicate determinations. C, siRNA-mediated RelA gene ablation was performed in U937-derived DC. After transient transfection for 48 h, cells were treated with 1 nm TCDD, 0.1 μg/ml LPS, or 0.1% DMSO (Ctrl; control) for 6 h. mRNA levels of AhR and CYP1A1 were determined using real-time PCR. Values for AhR and CYP1A1 mRNA expression are normalized to the expression of β-actin. Values are the mean ± S.D. of three independent experiments. Single asterisk indicates significantly different from control (p < 0.05); double asterisks indicate significantly higher than only TCDD-treated cells (p < 0.05); triple asterisks indicate significantly lower than only TCDD- or LPS-treated cells (p < 0.05). D, RelA protein ablation was confirmed by Western blot analysis of U937-derived DC transfected with RelA siRNA (siRelA) or scrambled siRNA (siCtrl) for 48 h. 25 μg of whole cell protein was loaded per lane.
To test which NF-κB subunit mediates the induction of AhR by LPS, we utilized MEFs deficient in RelA and RelB controlling the canonical and non-canonical NF-κB signaling pathway. MEF cells derived from WT, AhR-, RelA-, and RelB-deficient mice were treated with LPS and TCDD for 6 h. As shown in DC, LPS up-regulated the expression of AhR (2.2-fold) and CYP1A1 (4.5-fold) in WT MEFs as well as in RelB−/− MEFs (Fig. 3A). In contrast, LPS did not significantly increase the expression of AhR or CYP1A1 in RelA−/− or Ahr−/− MEFs. Furthermore, the constitutive expression of AhR was drastically reduced by ∼90% and the TCDD-induced expression of CYP1A1 was suppressed by 60% in RelA−/− MEF compared with WT MEF (Fig. 3, A and B), which was also confirmed on protein level in Western blot analysis (Fig. 3, E and F). The expression of ARNT did not significantly change after treatment with TCDD or LPS in MEFs (Fig. 3C). Transient transfection of RelA−/− MEF with a RelA expression plasmid for 24 h increased the basal expression of AhR (Fig. 3D). Furthermore, the LPS-dependent induction of AhR expression and enhanced CYP1A1 expression by TCDD was restored in MEF RelA−/− cells after transient transfection with a RelA expression vector (pRelA) for 24 h and incubation with TCDD or LPS for 6 h (Fig. 3D).
FIGURE 3.
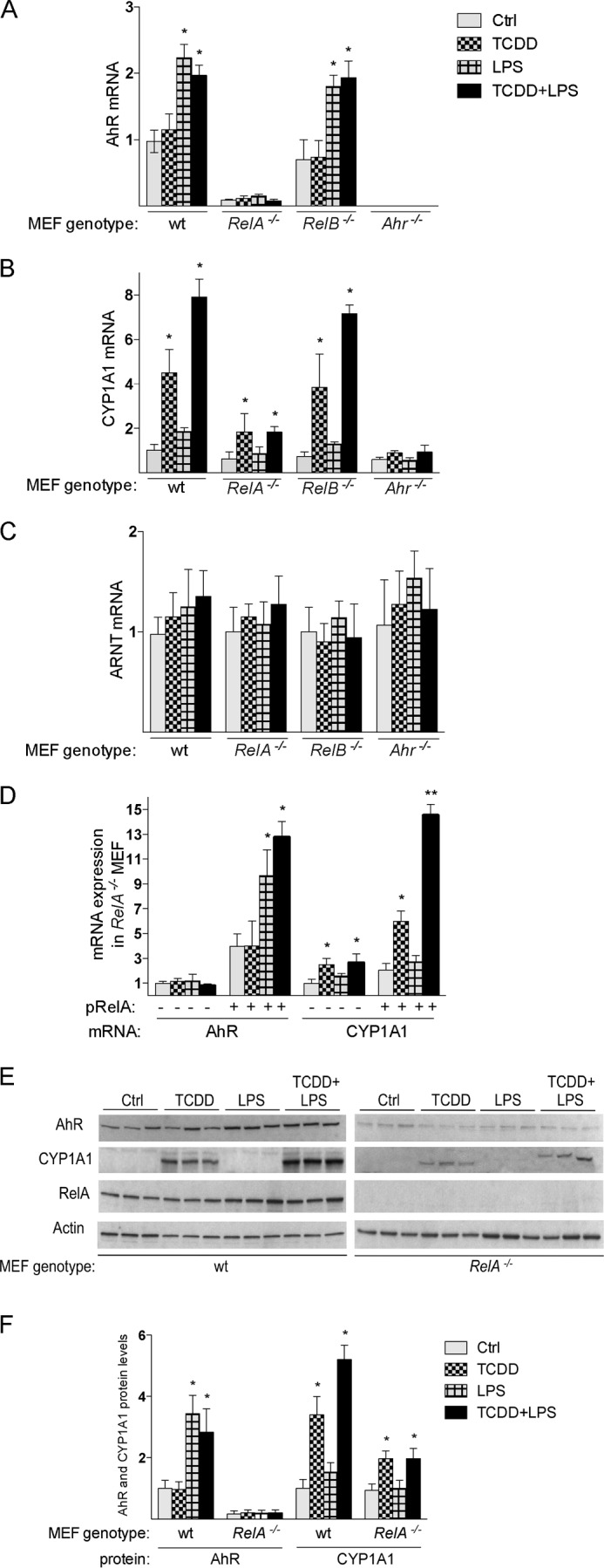
Expression of AhR and CYP1A1 in MEF cells from wt and Rel null mice. A, expression of AhR mRNA; B, CYP1A1 mRNA; C, ARNT mRNA in MEF cells were analyzed using real-time PCR. MEF cells derived from wild type (wt), RelA-deficient mice (RelA−/−), RelB-deficient mice (RelB−/−), and AhR-deficient mice (Ahr−/−) were treated with 1 nm TCDD, 0.1 μg/ml LPS, or 0.1% DMSO (Ctrl; control) for 6 h. D, induction of AhR and CYP1A1 is restored in MEF RelA−/− cells after transient transfection with a RelA. Cells were transiently transfected with a control or RelA expression plasmid (pRelA) for 24 h and treated with 1 nm TCDD, 0.1 μg/ml LPS, or 0.1% DMSO (Ctrl) for 6 h. E, Western blot analysis of AhR, CYP1A1, and RelA protein levels in WT and RelA−/− MEF. 25 μg of whole cell protein was loaded per lane. F, AhR and CYP1A1 protein levels were quantitated and normalized to actin. Values represent the mean ± S.D. of three independent experiments. A single asterisk indicates significantly different from control cells (p < 0.05). Values for AhR, ARNT, and CYP1A1 mRNA expression are normalized to the expression of β-actin. Values are the mean ± S.D. of three independent experiments. A single asterisk indicates significantly different from control cells (p < 0.05).
Identification of NF-κB Binding Sites on the AhR Promoter
DNA promoter analysis of the 5′-upstream regulatory region of the human AhR gene using the TFSEARCH program (19) revealed three putative NF-κB binding sites, AhR-NF-κB1 at −2757 bp, AhR-NF-κB2 at −1452 bp, and AhR-NF-κB3 at −399 bp within the 5′-upstream regulatory region of the AhR gene (Fig. 4A). To examine the role that these NF-κB binding sites may play in the regulation of AhR gene expression, transfection experiments were carried out with deletion reporter constructs of the AhR promoter. Our data revealed that only the full length promoter containing 5640 nt upstream of the putative start site of the AhR promoter is sufficient to induce promoter activity by LPS in human U937 DC (Fig. 4B). The reporter activity of the deletion constructs AhRΔ(−2510), AhRΔ(−881), or AhRΔ(−120) was not significantly increased by LPS, indicating the importance of the distal upstream AhR-NF-κB1 site in regulating this response. Promoter studies with mutations of each of the three AhR-NF-κB sites revealed that only mutation of the AhR-NF-κB1 site eliminated the LPS-induced AhR promoter response (Fig. 4C). A mutation in the AhR-NF-κB2 site reduced the LPS-mediated AhR activity by ∼20%, but the effect was not statistically significant. Mutation of the AhR-NF-κB3 at −399 bp had no significant effect on LPS-induced AhR gene promoter activity.
FIGURE 4.
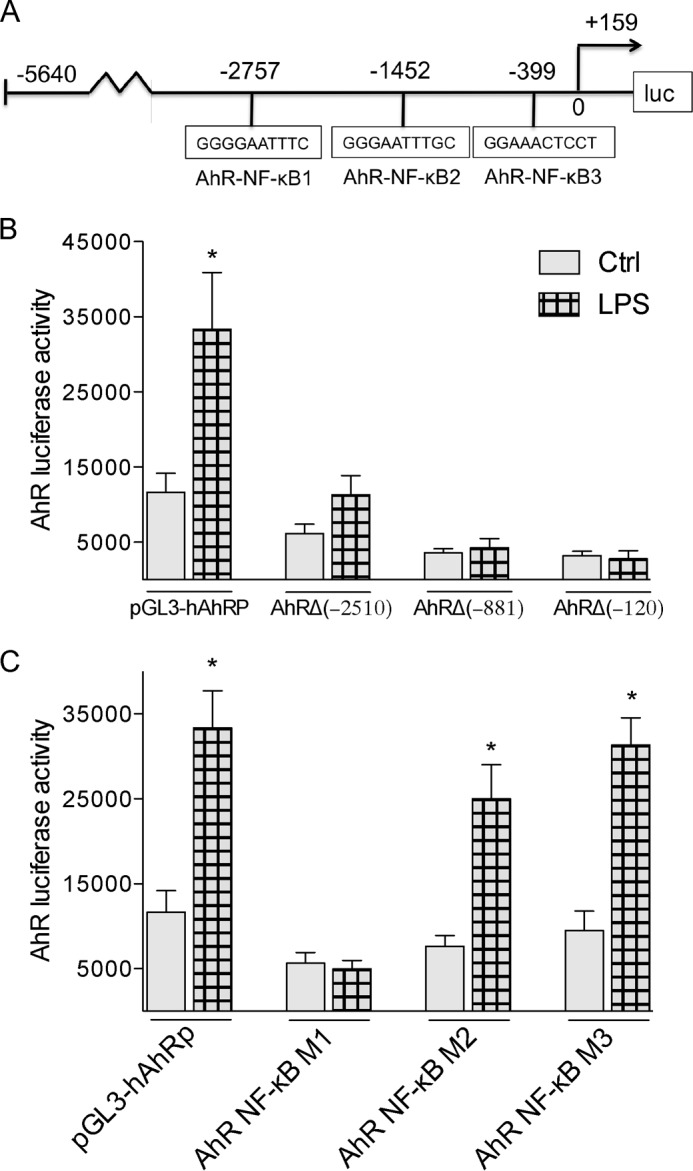
LPS-specific effects on deletion and mutation constructs of the human AhR promoter. A, schematic illustration of the full-length promoter construct of the human AhR gene containing 5640 bp upstream of the transcriptional start site (indicated by an arrow) cloned into a luciferase (luc) reporter vector. Positions of the three putative NF-κB recognition sites are presented. B, effect of LPS on AhR deletion constructs. U937-derived DC were transiently transfected with pGL3-hAhRP, and the deletion constructs AhRΔ(−2510), AhRΔ(−881), or AhRΔ(−120). C, effect of LPS on AhR promoter constructs mutated in NF-κB-binding sites. U937-derived DC were transiently transfected with equimolar amounts of the designated deletion or mutation constructs and treated with 100 ng/ml LPS for 6 h. Mean + S.D. of three independent experiments are given. A single asterisk indicates significantly different from control (p < 0.05).
Enhanced Binding of RelA to an NF-κB Binding Element of the Human AhR Promoter
Because RelA is a subunit of the NF-κB family that binds to NF-κB consensus sequences, we examined the binding of RelA to each of the three putative NF-κB elements from the human AhR gene promoter. Using nuclear extracts of U937 DC results from EMSA show that LPS stimulates NF-κB binding to DNA containing the AhR-NF-κB1 site (Fig. 5A, lane 10). NF-κB DNA binding to the AhR-NF-κB3 element was slightly reduced by LPS (lane 2), whereas binding to the AhR-NF-κB2 site was unaffected by LPS treatment (lane 6). Supershift analyses with RelA-specific antibodies revealed that RelA binds to the AhR-NF-κB1 element present in the upper complex of the classical LPS-activated NF-κB complex (Fig. 5B, lane 3), which also contains the NF-κB subunit p50 (Fig. 5B, lane 4). EMSA using AhR-NF-κB2 and AhR-NF-κB3 oligonucleotides did not reveal any binding of RelA (Fig. 5A, lanes 3 and 7).
FIGURE 5.

LPS induces nuclear protein binding to a NF-κB-binding element of the AhR promoter. Nuclear extracts from untreated control (lanes 1, 5, and 9) and LPS-treated (lanes 2, 6, and 10) U937-derived DC were used for EMSA. A, EMSA was performed using double-stranded, 32P-labeled oligonucleotides containing the AhR-NF-κB1, AhR-NF-κB2, or AhR-NF-κB3 binding sequence of the human AhR promoter. A possible binding of RelA was identified by supershift analyses using RelA-specific antibodies (lanes 3, 7, and 11). B, EMSA was performed using double-stranded, 32P-labeled oligonucleotides containing the AhR-NF-κB1 site. The bands corresponding to the specific LPS-induced RelA and p50 NF-κB subunits are indicated by arrows (lanes 3 and 4). To confirm specificity, a 100-fold excess of unlabeled AhR-NF-κB oligonucleotides from the AhR promoter was added (lane 5). One representative experiment of three independently performed experiments is shown. Ab., antibody; Comp., competition; Ctrl, control; Treat., treatment.
Expression of AhR in Inflammatory Disease
Here, we tested whether the expression of AhR would change under the conditions of an inflammatory disease, which is associated with an increased NF-κB activity. First, we investigated AhR expression in peripheral blood mononuclear cells (PBMCs) from rheumatoid arthritis (RA) patients compared with PBMCs from healthy patients. PBMCs were isolated as described previously (25). Blood samples from patients with RA and control subjects were obtained by venipuncture after informed consent. The clinical diagnosis was verified using published criteria (26). The protocol was approved by the Institutional Review Board of the University of California at Davis. We found that AhR mRNA levels were significantly elevated (∼2-fold) in PBMCs from RA patients compared with PBMCs from healthy patients (Fig. 6A). We next measured AhR gene expression during allergic airway inflammation in a murine model. To induce allergic airway inflammation, BALB/c mice were exposed to ovalbumin aerosol as described elsewhere (27). The level of AhR mRNA was 2.4-fold increased in the lungs of BALB/c mice after ovalbumin-induced lung inflammation compared with control mice exposed to filtered air (Fig. 6B).
FIGURE 6.
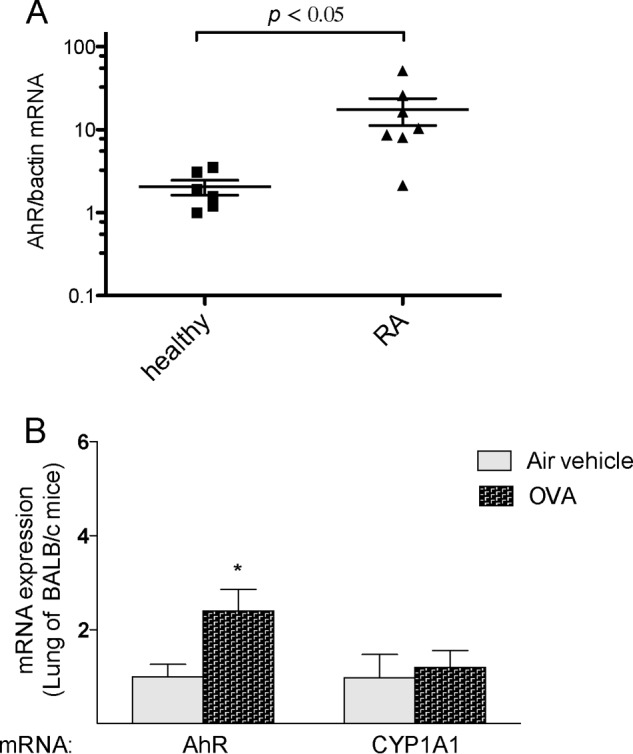
Elevated expression of AhR in inflammatory disease. A, AhR expression in PBMCs from healthy and RA patients. Total RNA was harvested from PBMCs from six healthy and seven RA patients matched by gender and age. B, expression of AhR in lungs of mice after ovalbumin (OVA)-induced allergic lung inflammation and airway hyperreactivity. BALB/c mice (five mice per group) were sensitized by intraperitoneal (intraperitoneal) injection of ovalbumin and alum solution for 4 weeks and then exposed to ovalbumin aerosol six times over a period of 12 days as described elsewhere (26). Age-matched control animals were injected intraperitoneal with ovalbumin + alum adjuvant (sensitized) and were exposed only to filtered air. Experiments were performed in triplicate for each of the samples. AhR and CYP1A1 mRNA levels were analyzed by real-time PCR. A single asterisk indicates significantly different from control (p < 0.05).
DISCUSSION
The results of the current study provide new insight into the mechanism of the regulation of the human AhR gene through NF-κB signaling enhancing the expression of AhR-regulated genes by inflammatory stimuli. LPS activated NF-κB signaling increased the expression of AhR and CYP1A1 in vitro as well as in vivo. Results from EMSA and transfection assays indicate that the NF-κB-mediated increase of AhR involves elevated AhR activity. The clear increase of TCDD-induced expression of CYP1A1 by LPS in human DC as well as in thymus of B6 mice suggest that an enhanced expression of AhR during inflammation would lead to an increased sensitivity toward AhR ligands as suggested in previous reports (28–30). The reduction in both TCDD- and LPS-enhanced expression of AhR and CYP1A1 through inhibition of NF-κB indicate the importance of NF-κB in regulating the expression of AhR- and AhR-dependent genes. This mechanism is strongly supported by engineered fibroblasts from RelA null mice showing a clear-cut reduction in AhR expression and a suppressed TCDD-mediated induction of CYP1A1 compared with WT MEF.
Recently, we showed that the absence of the AhR impairs the LPS-mediated induction of inflammatory marker genes and CYP1A1 (31). In contrast to the current findings in thymus, LPS reduced the basal expression of CYP1A1 in liver, which has been shown previously in mouse hepatoma cells (32). The LPS-mediated decrease of CYP1A1 was enhanced and accelerated in liver of Ahr−/− mice compared with B6 WT mice (31), demonstrating that the suppression of CYP1A1 by LPS is mediated via an AhR-independent mechanism, whereas the induction of CYP1A1 by LPS depends on the presence of a functional AhR as shown in MEF and thymus of B6 WT and Ahr−/− mice. Interestingly, irradiation with ultraviolet B also induces the expression of CYP1A1 in human epidermal keratinocytes (33) and the human hepatoma cell line HepG2 in an AhR-dependent manner (34). Together these data show that the NF-κB-mediated suppression of CYP1A1 in liver and the increased expression of CYP1A1 in DC and thymus are regulated through different mechanisms.
In the present study, we identified three putative NF-κB sites on the promoter sequence of the human AhR gene. In reporter gene assays through generation of different deletion constructs and mutational analysis, we were able to show that only one NF-κB binding site at −2757 bp (AhR-NF-κB1) of the AhR promoter has functional activity as indicated by LPS responsiveness. To verify the functional importance of the identified NF-κB site, we confirmed the binding of RelA and p50 heterodimer in supershift analysis. These findings clearly show the regulation of AhR gene expression through activation of TLR4 and binding of the NF-κB RelA/p50 complex providing a new cross-talk between NF-κB and AhR signaling. Both transcription factors are also involved in the regulation of cellular processes such as apoptosis and immune functions (4, 35). Recent reports including own data demonstrated the interaction between the two signaling pathways affecting the regulation of inflammatory responsive genes (36, 37). Particularly in cells of the innate immune system such as macrophages, DC, natural killer cells, and lymphoid tissue inducer-like cells, the interaction of both factors seem to result in significant effects on gene regulation and cellular responses (7, 8, 24, 36). Furthermore, non-stimulated peripheral human B cells either lack or express low levels of AhR. However, activation of B cells with CpG or CD40 ligand activated the expression of AhR mRNA and protein (38), which was associated with increased sensitivity of B cells to AhR ligands. On the other hand activation of AhR by TCDD may lead to profound impairment of humoral immune responses in B cells (39). Crawford et al. (15) showed that phorbol 12-myristate 13-acetate-activated splenocytes exhibit a rapid and robust increase in steady state AhR expression associated with enhanced AhR binding activity and phorbol 12-myristate 13-acetate-induced expression of CYP1A1, which supports results from the present study showing an NF-κB-mediated stimulation of DRE-reporter activity. However, the phorbol 12-myristate 13-acetate-mediated increase of CYP1A1 may also dependent upon functional PKC as shown previously (40–42).
Activation of differentiating monocytes or macrophages has been shown to be associated with increased levels of AhR (43, 36). The AhR is also differentially expressed in various T-cell subsets. The expression and activation of the AhR has been recently shown to be a critical event in T-cell differentiation. For instance, the AhR is usually not expressed in naïve T-cells, Th1, or Th2 cells; however, Treg, Tr1, and especially Th17 cells express a high level of AhR (9, 10, 44). Because NF-κB is activated downstream of the IL-17 pathway, these data support the control of AhR expression by NF-κB signaling, leading to an increased expression of functional AhR in T-cells.
Th17 cells are key effector T cells in human autoimmune diseases, including RA, and recent reports suggest a critical role of AhR not only in Th17 cell development but also in the context of RA pathogenesis (45). Results of the current study indicate that AhR mRNA is modestly induced in two disease models such as RA and allergic airway inflammation, but the experiments do not necessarily define a role for NF-κB in these responses. These data are in line with a report demonstrating elevated AhR levels in RA synovial tissue compared with osteoarthritis tissue (46). Interestingly, AhR antagonists have been shown to ameliorate inflammation associated with RA and the deficiency or antagonism of AhR may cause a lower inflammatory response mediated by IL-1β (47).
In summary, these data support the important role of the AhR in differentiating immune cells and that activation of NF-κB involves RelA-dependent AhR expression and enhanced activity of AhR-regulated genes. First, these findings may explain altered metabolism of xenobiotics and drugs by inflammatory stimuli and cytokines that regulate NF-κB; second, the data may provide a mechanism causing activated immune cells to be more sensitive to immune modulation by AhR ligands than are resting cells.
Acknowledgments
We thank Sarah Kado and Jaeeun Baek for technical assistance. We thank Christopher Bradfield for kindly providing AhR null mice and W. Greene and Ulrich Siebenlist for providing RelA, p50, and RelB expression plasmids. We also thank Bruce Hammock and Jun Yang for kindly providing lung tissue from BALB/c mice treated with ovalbumin to induce allergic lung inflammation.
This work was supported, in whole or in part, by NIEHS, National Institutes of Health Grants R01 ES019898-02 (to C. F. A. V.) and R01 ES007685 (to M. S. D.).
- AhR
- aryl hydrocarbon receptor
- ARNT
- AhR nuclear translocator
- CYP1A1
- cytochrome P4501A1
- DC
- dendritic cell(s)
- DRE
- dioxin-responsive element
- MEF
- mouse embryonic fibroblast
- RA
- rheumatoid arthritis
- TCDD
- 2,3,7,8-tetrachlorodibenzo-p-dioxin
- DMSO
- dimethyl sulfoxide
- PDTC
- pyrrolidinedithiocarbamate
- CAPE
- caffeic acid phenethyl ester
- PBMC
- peripheral blood mononuclear cell.
REFERENCES
- 1. Schmidt J. V., Carver L. A., Bradfield C. A. (1993) Molecular characterization of the murine Ahr gene. Organization, promoter analysis, and chromosomal assignment. J. Biol. Chem. 268, 22203–22209 [PubMed] [Google Scholar]
- 2. Fitzgerald C. T., Nebert D. W., Puga A. (1998) Regulation of mouse Ah receptor (Ahr) gene basal expression by members of the Sp family of transcription factors. DNA Cell. Biol. 17, 811–822 [DOI] [PubMed] [Google Scholar]
- 3. FitzGerald C. T., Fernandez-Salguero P., Gonzalez F. J., Nebert D. W., Puga A. (1996) Differential regulation of mouse Ah receptor gene expression in cell lines of different tissue origins. Arch. Biochem. Biophys. 333, 170–178 [DOI] [PubMed] [Google Scholar]
- 4. Marshall N. B., Kerkvliet N. I. (2010) Dioxin and immune regulation: emerging role of aryl hydrocarbon receptor in the generation of regulatory T cells. Ann. N. Y. Acad. Sci. 1183, 25–3720146706 [Google Scholar]
- 5. Ruby C. E., Leid M., Kerkvliet N. I. (2002) 2,3,7,8-Tetrachlorodibenzo-p-dioxin suppresses tumor necrosis factor-α and anti-CD40-induced activation of NF-κB/Rel in dendritic cells: p50 homodimer activation is not affected. Mol. Pharmacol. 62, 722–728 [DOI] [PubMed] [Google Scholar]
- 6. Jin G. B., Moore A. J., Head J. L., Neumiller J. J., Lawrence B. P. (2010) Aryl hydrocarbon receptor activation reduces dendritic cell function during influenza virus infection. Toxicol. Sci. 116, 514–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bankoti J., Rase B., Simones T., Shepherd D. M. (2010) Functional and phenotypic effects of AhR activation in inflammatory dendritic cells. Toxicol. Appl. Pharmacol. 246, 18–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vogel C. F., Goth S. R., Dong B., Pessah I. N., Matsumura F. (2008) Aryl hydrocarbon receptor signaling mediates expression of indoleamine 2,3-dioxygenase. Biochem. Biophys. Res. Commun. 375, 331–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Quintana F. J., Basso A. S., Iglesias A. H., Korn T., Farez M. F., Bettelli E., Caccamo M., Oukka M., Weiner H. L. (2008) Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 453, 65–71 [DOI] [PubMed] [Google Scholar]
- 10. Veldhoen M., Hirota K., Westendorf A. M., Buer J., Dumoutier L., Renauld J. C., Stockinger B. (2008) The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 453, 106–109 [DOI] [PubMed] [Google Scholar]
- 11. Kiss E. A., Vonarbourg C., Kopfmann S., Hobeika E., Finke D., Esser C., Diefenbach A. (2011) Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science 334, 1561–1565 [DOI] [PubMed] [Google Scholar]
- 12. Lawrence B. P., Sherr D. H. (2012) You AhR what you eat? Nat. Immunol. 13, 117–119 [DOI] [PubMed] [Google Scholar]
- 13. Abbott B. D., Probst M. R. (1995) Developmental expression of two members of a new class of transcription factors: II. Expression of aryl hydrocarbon receptor nuclear translocator in the C57BL/6N mouse embryo. Dev. Dyn. 204, 144–155 [DOI] [PubMed] [Google Scholar]
- 14. Lawrence B. P., Leid M., Kerkvliet N. I. (1996) Distribution and behavior of the Ah receptor in murine T lymphocytes. Toxicol. Appl. Pharmacol. 138, 275–284 [DOI] [PubMed] [Google Scholar]
- 15. Crawford R. B., Holsapple M. P., Kaminski N. E. (1997) Leukocyte activation induces aryl hydrocarbon receptor up-regulation, DNA binding, and increased Cyp1a1 expression in the absence of exogenous ligand. Mol. Pharmacol. 52, 921–927 [DOI] [PubMed] [Google Scholar]
- 16. Vogel C., Döhr O., Abel J. (1994) Transforming growth factor-beta 1 inhibits TCDD-induced cytochrome P450IA1 expression in human lung cancer A549 cells. Arch. Toxicol. 68, 303–307 [DOI] [PubMed] [Google Scholar]
- 17. Wolff S., Harper P. A., Wong J. M., Mostert V., Wang Y., Abel J. (2001) Cell-specific regulation of human aryl hydrocarbon receptor expression by transforming growth factor-β(1). Mol. Pharmacol. 59, 716–724 [DOI] [PubMed] [Google Scholar]
- 18.Deleted in proof
- 19. Heinemeyer T., Wingender E., Reuter I., Hermjakob H., Kel A. E., Kel O. V., Ignatieva E. V., Ananko E. A., Podkolodnaya O. A., Kolpakov F. A., Podkolodny N. L., Kolchanov N. A. (1998) Databases on Transcriptional Regulation: TRANSFAC, TRRD, and COMPEL. Nucleic Acids Res. 26, 362–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Reznikoff C. A., Brankow D. W., Heidelberger C. (1973) Establishment and characterization of a cloned line of C3H mouse embryo cells sensitive to postconfluence inhibition of division. Cancer Res. 33, 3231–3238 [PubMed] [Google Scholar]
- 21. Vogel C. F., Matsumura F. (2003) Interaction of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) with induced adipocyte differentiation in mouse embryonic fibroblasts (MEFs) involves tyrosine kinase c-Src. Biochem. Pharmacol. 66, 1231–1244 [DOI] [PubMed] [Google Scholar]
- 22. Shih V. F., Davis-Turak J., Macal M., Huang J. Q., Ponomarenko J., Kearns J. D., Yu T., Fagerlund R., Asagiri M., Zuniga E. I., Hoffmann A. (2012) Control of RelB during dendritic cell activation integrates canonical and noncanonical NF-κB pathways. Nat. Immunol. 13, 1162–1170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chang W. L., Baumgarth N., Yu D., Barry P. A. (2004) Human cytomegalovirus-encoded interleukin-10 homolog inhibits maturation of dendritic cells and alters their functionality. J. Virol. 78, 8720–8731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vogel C. F., Sciullo E., Li W., Wong P., Lazennec G., Matsumura F. (2007) RelB, a new partner of aryl hydrocarbon receptor-mediated transcription. Mol. Endocrinol. 21, 2941–2955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tsuda M., Ambrosini Y. M., Zhang W., Yang G. X., Ando Y., Rong G., Tsuneyama K., Sumida K., Shimoda S., Bowlus C. L., Leung P. S., He X. S., Coppel R. L., Ansari A. A., Lian Z. X., Gershwin M. E. (2011) Fine phenotypic and functional characterization of effector cluster of differentiation 8 positive T cells in human patients with primary biliary cirrhosis. Hepatology 54, 1293–1302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Aletaha D., Neogi T., Silman A.J., Funovits J., Felson D. T., Bingham C. O., 3rd, Birnbaum N. S., Burmester G. R., Bykerk V. P., Cohen M. D., Combe B., Costenbader K. H., Dougados M., Emery P., Ferraccioli G., Hazes J. M., Hobbs K., Huizinga T. W., Kavanaugh A., Kay J., Kvien T. K., Laing T., Mease P., Ménard H. A., Moreland L. W., Naden R. L., Pincus T., Smolen J. S., Stanislawska-Biernat E., Symmons D., Tak P. P., Upchurch K. S., Vencovský J., Wolfe F., Hawker G. (2010) Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 62, 2569–2581 [DOI] [PubMed] [Google Scholar]
- 27. Kenyon N. J., Bratt J. M., Linderholm A. L., Last M. S., Last J. A. (2008) Arginases I and II in lungs of ovalbumin-sensitized mice exposed to ovalbumin: sources and consequences. Toxicol. Appl. Pharmacol. 230, 269–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Morris D. L., Karras J. G., Holsapple M. P. (1993) Direct effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on responses to lipopolysaccharide (LPS) by isolated murine B-cells. Immunopharmacology 26, 105–112 [DOI] [PubMed] [Google Scholar]
- 29. Tucker A. N., Vore S. J., Luster M. I. (1986) Suppression of B cell differentiation by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Mol. Pharmacol. 29, 372–377 [PubMed] [Google Scholar]
- 30. Luster M. I., Germolec D. R., Clark G., Wiegand G., Rosenthal G. J. (1988) Selective effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin and corticosteroid on in vitro lymphocyte maturation. J. Immunol. 140, 928–935 [PubMed] [Google Scholar]
- 31. Wu D., Li W., Lok P., Matsumura F., Vogel C. F. (2011) AhR deficiency impairs expression of LPS-induced inflammatory genes in mice. Biochem. Biophys. Res. Commun. 410, 358–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tian Y., Ke S., Denison M. S., Rabson A. B., Gallo M. A. (1999) Ah receptor and NF-κB interactions, a potential mechanism for dioxin toxicity. J. Biol. Chem. 274, 510–515 [DOI] [PubMed] [Google Scholar]
- 33. Kostyuk V. A., Potapovich A. I., Lulli D., Stancato A., De Luca C., Pastore S., Korkina L. (2013) Modulation of human keratinocyte responses to solar UV by plant polyphenols as a basis for chemoprevention of non-melanoma skin cancers. Curr. Med. Chem. 20, 869–879 [PubMed] [Google Scholar]
- 34. Luecke S., Wincent E., Backlund M., Rannug U., Rannug A. (2010) Cytochrome P450 1A1 gene regulation by UVB involves crosstalk between the aryl hydrocarbon receptor and nuclear factor κB. Chem. Biol. Interact. 184, 466–473 [DOI] [PubMed] [Google Scholar]
- 35. Baeuerle P. A., Henkel T. (1994) Function and activation of NF-kappa B in the immune system. Annu. Rev. Immunol. 12, 141–179 [DOI] [PubMed] [Google Scholar]
- 36. Kimura A., Naka T., Nakahama T., Chinen I., Masuda K., Nohara K., Fujii-Kuriyama Y., Kishimoto T. (2009) Aryl hydrocarbon receptor in combination with Stat1 regulates LPS-induced inflammatory responses. J. Exp. Med. 206, 2027–2035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vogel C. F., Matsumura F. (2009) A new cross-talk between the aryl hydrocarbon receptor and RelB, a member of the NF-κB family. Biochem. Pharmacol. 77, 734–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Allan L. L., Sherr D. H. (2005) Constitutive activation and environmental chemical induction of the aryl hydrocarbon receptor/transcription factor in activated human B lymphocytes. Mol. Pharmacol. 67, 1740–1750 [DOI] [PubMed] [Google Scholar]
- 39. Sulentic C. E., Kaminski N. E. (2011) The long winding road toward understanding the molecular mechanisms for B-cell suppression by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. Sci. 120, S171–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Carrier F., Owens R. A., Nebert D. W., Puga A. (1992) Dioxin-dependent activation of murine Cyp1a-1 gene transcription requires protein kinase C-dependent phosphorylation. Mol. Cell. Biol. 12, 1856–1863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen Y. H., Tukey R. H. (1996) Protein kinase C modulates regulation of the CYP1A1 gene by the aryl hydrocarbon receptor. J. Biol. Chem. 271, 26261–26266 [DOI] [PubMed] [Google Scholar]
- 42. Long W. P., Pray-Grant M., Tsai J. C., Perdew G. H. (1998) Protein kinase C activity is required for aryl hydrocarbon receptor pathway-mediated signal transduction. Mol. Pharmacol. 53, 691–700 [DOI] [PubMed] [Google Scholar]
- 43. Hayashi S., Okabe-Kado J., Honma Y., Kawajiri K. (1995) Expression of Ah receptor (TCDD receptor) during human monocytic differentiation. Carcinogenesis 16, 1403–1409 [DOI] [PubMed] [Google Scholar]
- 44. Cua D. J., Tato C. M. (2010) Innate IL-17-producing cells: the sentinels of the immune system. Nat. Rev. Immunol. 10, 479–489 [DOI] [PubMed] [Google Scholar]
- 45. Nakahama T., Kimura A., Nguyen N. T., Chinen I., Hanieh H., Nohara K., Fujii-Kuriyama Y., Kishimoto T. (2011) Aryl hydrocarbon receptor deficiency in T cells suppresses the development of collagen-induced arthritis. Proc. Natl. Acad. Sci. U.S.A. 108, 14222–14227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kobayashi S., Okamoto H., Iwamoto T., Toyama Y., Tomatsu T., Yamanaka H., Momohara S. (2008) A role for the aryl hydrocarbon receptor and the dioxin TCDD in rheumatoid arthritis. Rheumatology 47, 1317–1322 [DOI] [PubMed] [Google Scholar]
- 47. Lahoti T. S., John K., Hughes J. M., Kusnadi A., Murray I. A., Krishnegowda G., Amin S., Perdew G. H. (2013) Aryl hydrocarbon receptor antagonism mitigates cytokine-mediated inflammatory signalling in primary human fibroblast-like synoviocytes. Ann. Rheum. Dis. 72, 1708–1716 [DOI] [PMC free article] [PubMed] [Google Scholar]