

TO THE EDITOR: In lymphoplasmacytic lymphoma/Waldenström's macroglobulinemia (LPL/WM), the immunophenotype of the neoplastic population generally does not show positivity for CD5, CD23, or CD10 monoclonal antibodies, with only 5-20% showing positivity. However, this immunophenotypic particularity does not exclude a diagnosis of Waldenström's macroglobulinemia; moreover, CD5 and/or CD23 monoclonal antibody positivity can characterize a subset of patients with high IgM values; these patients are at risk for hyperviscosity-related complications.
WM was first described by Jan Waldenström in 1944 [1] and is classified as an indolent B-cell lymphoplasmacytic lymphoma (LPL) or LPL/WM [2]. It is a lymphoproliferative neoplasm characterized by the presence of a serum IgM monoclonal component associated with bone marrow lymphocytic infiltration. According to the Workshop on WM held in Athens in 2002 [2], the diagnosis (Table 1) is principally based on the following two essential findings: 1) the quantitative presence of any serum IgM monoclonal component and 2) bone marrow infiltration by lymphoid cells with a lymphoplasmacytic aspect.
Table 1.
Diagnostic criteria for Wladenström's macroglobulinemia and IgM monoclonal gammopathy of undetermined significance.
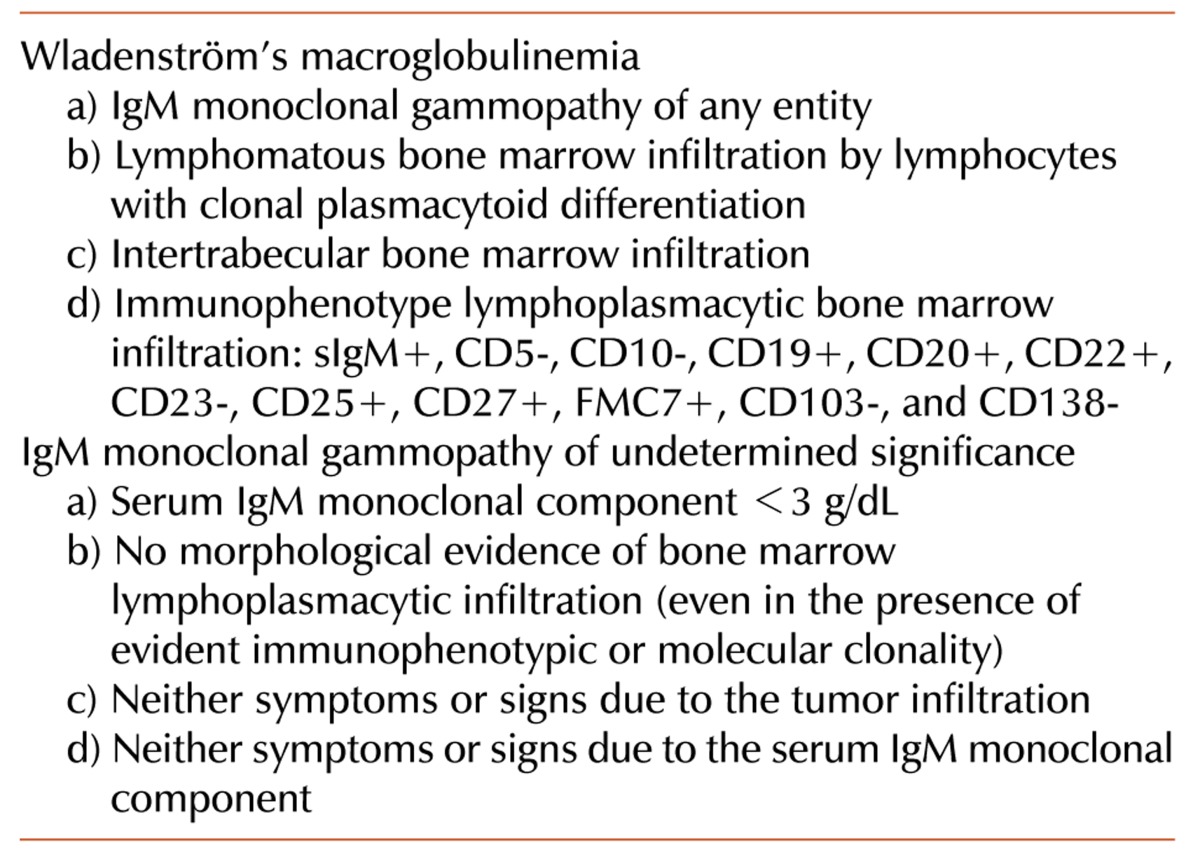
In WM patients, the serum IgM monoclonal component concentration varies widely. However, while finding a monoclonal IgM component is a prerequisite for the LPL/WM diagnosis, it is not a specific feature of WM. In fact, an IgM monoclonal component can be found in other lymphoproliferative disorders, such as chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL), splenic marginal zone lymphoma (SMZL), diffuse large cell lymphoma (DLCL), mantle cell lymphoma (MCL), myeloma and IgM monoclonal gammopathy of undetermined significance (IgM-MGUS)(Table 1).
The lymphomatous nature of bone marrow infiltration must be confirmed using flow cytometry and immunohistochemistry to define the immunophenotype. Bone marrow infiltration, usually intertrabecular, is represented by small lymphocytes with evidence of clonal plasmacytoid differentiation. Mast cells are often present, supporting growth of the lymphoplasmacytic population [3]. WM does not show cytogenetic chromosome marker alterations, with most patients displaying a normal karyotype. However, various numerical and structural abnormalities have been described, of which the most frequent is deletion of the long arm of chromosome 6q [4]. Recently, a MYD88 L265P mutation has been observed in more than 90% of WM patients. This has an important diagnostic impact and will improve understanding of the WM pathophysiological mechanism and is likely to improve therapeutic outcomes [5].
We report the case of a 74-year-old male who had a clinical history of uric acid arthropathy, prostatic hypertrophy, and diabetes mellitus type II. In 2003, laboratory tests showed the presence of the serum IgM-κ monoclonal component (1.5 g/dL), increased β2-microglobulin level (β2-M, 2.84 mg/dL), increased ESR (62 mm/h), decreased IgA (22 mg/dL) and IgG levels (387 mg/dL), and significantly increased IgM level (up to 3,190 mg/dL). Blood cell count, differential leukocyte counts, and LDH enzyme level were normal.
The bone marrow aspirate (Fig. 1) and immunohistochemistry of the bone marrow biopsy suggested a lymphoproliferative disease with immune secretion, that is, LPL/WM with clinical features suggesting a slow progression. Immunologically, the B-lymphoid population showed positivity for CD5 (Fig. 2) and CD23 (Fig. 3) monoclonal antibodies, in addition to positivity for the classic surface IgM, CD19, CD20, and CD22 monoclonal antibodies, using both flow cytometry and immunohistochemistry; CD10 monoclonal antibody by flow cytometry showed negativity.
Fig. 1.
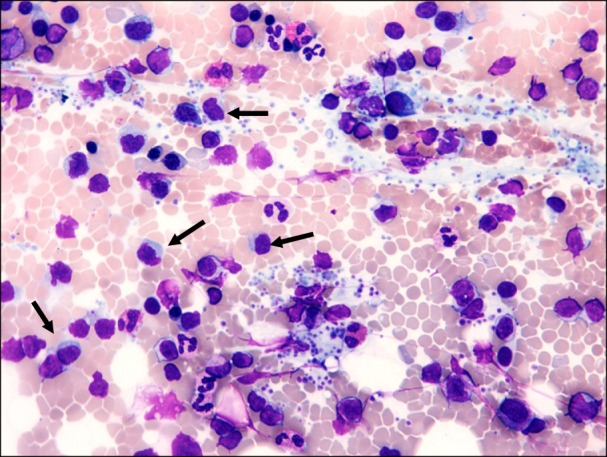
Bone marrow aspirate. Lymphocytes with lymphoplasmacytoid appearance (arrows).
Fig. 2.
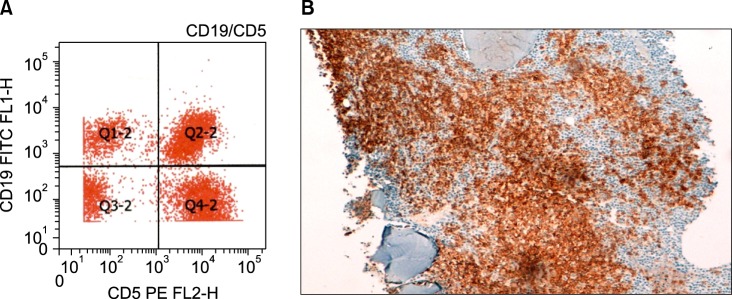
(A) Bone marrow flow cytometry finding of CD5+ lymphoplasmacytic lymphoma/WM. (B) Immunohistochemistry of bone marrow biopsy showing CD5+.
Fig. 3.
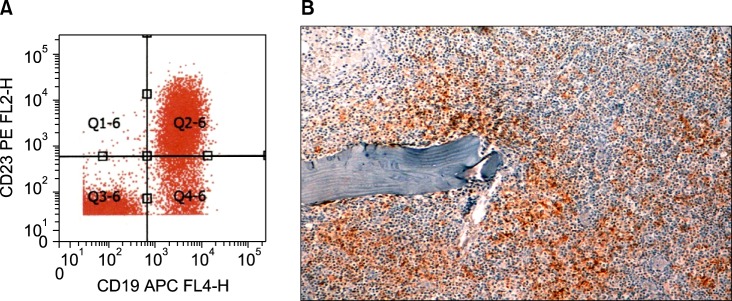
(A) Bone marrow flow cytometry finding of CD23+ lymphoplasmacytic lymphoma/WM. (B) Immunohistochemistry of bone marrow biopsy showing CD23+.
In 2004, laboratory tests showed values of serum IgM-κ monoclonal component <2.0 g/dL. The increase in IgM level (>3,000 mg/dL) and decrease in IgG and IgA levels persisted, while the blood cell and differential leukocyte counts were normal. In 2005, the patient had leukocytosis and lymphocytosis. In 2004 and 2005, bone marrow aspirate and biopsy showed disease stability. In 2006, serum IgM-κ increased as high as 4.0 g/dL; thus, therapy with an alkylating agent (chlorambucil at 4-6 mg/day) was started. The therapy decreased the serum IgM-κ to 1.3 g/dL with the disappearance of splenomegaly. Subsequent checks showed stability of serum IgM-κ with values not higher than 2.0 g/dL, while the increased β2-M and IgM and decreased IgG and IgA persisted; the cell blood and differential leukocyte counts remained normal.
In 2012, serum IgM-κ, started to increase again (2.6 g/dL), as well as β2-M (5.6 mg/dL); moreover, the patient was anemic (Hb level, 109 g/L), and leukocytosis (white blood cell count, 16.0×109/L) and lymphocytosis (lymphocytes, 75%) were present. In 2013, the patient was hospitalized for weight loss, anemia, lymphocytosis, and an increase in IgM and CRP (5.8 mg/dL). In the years following the diagnosis, the neoplastic B-lymphocytes showed persistent positivity for CD5 and CD23 and negativity for CD10.
LPL is a diagnosis of exclusion. The immunological arrangement has an important role in the differential diagnosis of B-cell lymphoproliferative diseases. In WM, the clonal neoplastic proliferation is represented by memory B cells from postgerminal centers after somatic hypermutation. The differential diagnoses were CLL/SLL, MCL, MZL, and plasma cell myeloma. Furthermore, there is a diagnostic overlap between LPL/WM, IgM MGUS, and smoldering WM.
In LPL/WM, the immunological framework is essential to distinguish it from other B-lymphoproliferative diseases with bone marrow involvement and an accompanying IgM monoclonal component. A neoplastic B-lymphoid population expressing CD5 and CD23 positivity simultaneously can suggest a diagnosis of CLL, while positivity for CD5 with simultaneous negativity for CD23 may suggest MCL. A condition of follicular non-Hodgkin lymphoma generally is characteristic for CD10 positivity [6].
In LPL/WM, the typical immunophenotype of the lymphoid population is represented by the following positive panel of monoclonal antibodies: CD19, CD20, CD22, FMC7, BCL2, CD38, CD79a, and monotypic light chains surface [7]. Usually, the immunophenotype of sIgM+, CD5-, CD10-, CD19+, CD20+, CD22+, and CD23-, in association with non-paratrabecular bone marrow infiltration, is diagnostic of WM [7, 8]. In WM, the infiltration pattern and the degree of plasmacytic differentiation can be varied. Given the neoplastic population variability, the positivity of associated antigens PAX5 and CD79a may indicate the correct diagnosis [9].
The present case is reported for immunological peculiarity of the lymphoid population, with CD5 and CD23 positivity and CD10 negativity to monoclonal antibodies. In LPL/WM, monoclonal antibodies CD5, CD10, and CD23 are generally negative. Only 5-20% of cases are positive, while single positivity for CD5 or CD23 is seen in approximately 50% of cases [10, 11]. CD5 and CD23 positivity of the neoplastic lymphoid population is not always easily detectable or acknowledged as definitively positive. The difficulty can be related to data interpretation skills, mainly with flow cytometry [8]. Indeed, low positivity levels observed in some laboratories may not be recognized, while in other laboratories, they may be interpreted as unequivocally positive [12].
Clinical case reports have shown that in LPL/WM, CD5 and CD23 positivity in a lymphoid population can show high IgM values (≥3,000 mg/dL) [11]. This can have important clinical and biological implications because CD23 positivity identifies patients with a great disease burden and at risk of developing important hyperviscosity-related complications. It is also appropriate to test the lymphoid population for CD52 positivity that may be present in many LPL/WM cases. This has an important therapeutic implication for possible therapy with monoclonal anti-CD52 antibody (alemtuzumab) that may have a clinical benefit in a substantial number of patients [13].
This case shows that LPL can display a heterogeneous B-lymphoid neoplastic population, which may show CD5, CD23, and CD10 positivity or CD10 negativity; in these cases, diagnosis of WM is not excluded [11]. Moreover, the presence of CD5+ and/or CD23+ can help to define a subgroup of patients with a high IgM value that may help trigger of possible paraprotein-related complications.
ACKNOWLEDGMENTS
We are grateful to Mr. Giorgio Donelli for his technical contribution in flow cytometry.
Footnotes
No potential conflicts of interest relevant to this article were reported.
References
- 1.Waldenstrom J. Incipient myelomatosis or "essential" hyperglobulinemia with fibrinogenopenia: a new syndrome? Acta Med Scand. 1944;117:216–247. [Google Scholar]
- 2.Owen RG, Treon SP, Al-Katib A, et al. Clinicopathological definition of Waldenstrom's macroglobulinemia: consensus panel recommendations from the Second International Workshop on Waldenstrom's Macroglobulinemia. Semin Oncol. 2003;30:110–115. doi: 10.1053/sonc.2003.50082. [DOI] [PubMed] [Google Scholar]
- 3.Tournilhac O, Santos DD, Xu L, et al. Mast cells in Waldenstrom's macroglobulinemia support lymphoplasmacytic cell growth through CD154/CD40 signaling. Ann Oncol. 2006;17:1275–1282. doi: 10.1093/annonc/mdl109. [DOI] [PubMed] [Google Scholar]
- 4.Mansoor A, Medeiros LJ, Weber DM, et al. Cytogenetic findings in lymphoplasmacytic lymphoma/Waldenström macroglobulinemia. Chromosomal abnormalities are associated with the polymorphous subtype and an aggressive clinical course. Am J Clin Pathol. 2001;116:543–549. doi: 10.1309/6U88-357U-UKJ5-YPT3. [DOI] [PubMed] [Google Scholar]
- 5.Treon SP, Hunter ZR. A new era for Waldenstrom macroglobulinemia: MYD88 L265P. Blood. 2013;121:4434–4436. doi: 10.1182/blood-2013-04-494849. [DOI] [PubMed] [Google Scholar]
- 6.Pangalis GA, Kyrtsonis MC, Kontopidou FN, et al. Differential diagnosis of Waldenstrom's macroglobulinemia from other low-grade B-cell lymphoproliferative disorders. Semin Oncol. 2003;30:201–205. doi: 10.1053/sonc.2003.50046. [DOI] [PubMed] [Google Scholar]
- 7.Naderi N, Yang DT. Lymphoplasmacytic lymphoma and Waldenstrom macroglobulinemia. Arch Pathol Lab Med. 2013;137:580–585. doi: 10.5858/arpa.2012-0034-RS. [DOI] [PubMed] [Google Scholar]
- 8.Konoplev S, Medeiros LJ, Bueso-Ramos CE, Jorgensen JL, Lin P. Immunophenotypic profile of lymphoplasmacytic lymphoma/Waldenstrom macroglobulinemia. Am J Clin Pathol. 2005;124:414–420. doi: 10.1309/3G1X-DX0D-VHBN-VKB4. [DOI] [PubMed] [Google Scholar]
- 9.Owen RG, Barrans SL, Richards SJ, et al. Waldenstrom macroglobulinemia. Development of diagnostic criteria and identification of prognostic factors. Am J Clin Pathol. 2001;116:420–428. doi: 10.1309/4LCN-JMPG-5U71-UWQB. [DOI] [PubMed] [Google Scholar]
- 10.Morice WG, Chen D, Kurtin PJ, Hanson CA, McPhail ED. Novel immunophenotypic features of marrow lymphoplasmacytic lymphoma and correlation with Waldenstrom's macroglobulinemia. Mod Pathol. 2009;22:807–816. doi: 10.1038/modpathol.2009.34. [DOI] [PubMed] [Google Scholar]
- 11.Hunter ZR, Branagan AR, Manning R, et al. CD5, CD10, and CD23 expression in Waldenstrom's macroglobulinemia. Clin Lymphoma. 2005;5:246–249. doi: 10.3816/clm.2005.n.008. [DOI] [PubMed] [Google Scholar]
- 12.San Miguel JF, Vidriales MB, Ocio E, et al. Immunophenotypic analysis of Waldenstrom's macroglobulinemia. Semin Oncol. 2003;30:187–195. doi: 10.1053/sonc.2003.50074. [DOI] [PubMed] [Google Scholar]
- 13.Owen RG, Hillmen P, Rawstron AC. CD52 expression in Waldenstrom's macroglobulinemia: implications for alemtuzumab therapy and response assessment. Clin Lymphoma. 2005;5:278–281. doi: 10.3816/clm.2005.n.016. [DOI] [PubMed] [Google Scholar]