

Abstract
Plasma calcium concentration is maintained within a narrow range (8.5-10.5 mg/dL) by the coordinated action of parathyroid hormone (PTH), 1,25(OH)2D3, calcitonin, and ionized calcium (iCa2+) itself. The kidney plays a key role in this process by the fine regulation of calcium excretion. More than 95% of filtered calcium is reabsorbed along the renal tubules. In the proximal tubules, 60% of filtered calcium is reabsorbed by passive mechanisms. In the thick ascending limb, 15% of calcium is reabsorbed by paracellular diffusion through paracellin-1 (claudin-16). The calcium sensing receptor (CaSR) in the basolateral membrane of the thick ascending limb senses the change in iCa2+ and inhibits calcium reabsorption independent to PTH and 1,25(OH)2D3. The fine regulation of calcium excretion occurs in the distal convoluted tubules and connecting tubules despite the fact that only 10-15% of filtered calcium is reabsorbed there. Transient receptor potential vanilloid 5 (TRPV5) and 6 (TRPV6) in the apical membrane act as the main portal of entry, calbindin-D28K delivers Ca2+ in the cytoplasm, and then Na2+/Ca2+ exchanger (NCX1) and plasma membrane Ca2+-ATPase in the basolateral membrane serve as an exit. In the cortical collecting duct, TRPV6 is expressed, but the role might be negligible. In addition to PTH and 1,25(OH)2D3, acid-base disturbance, diuretics, and estrogen affect on these calcium channels. Recently, klotho and fibroblast growth factor 23 (FGF23) are suggested as new players in the calcium metabolism. Klotho is exclusively expressed in the kidney and co-localized with TRPV5, NCX1, and calbindin-D28K. Klotho increases calcium reabsorption through trafficking of TRPV5 to the plasma membrane, and also converts FGF receptor to the specific FGF23 receptor. FGF23:klotho complex bound to FGF receptor inhibits 1α-hydroxylase of vitamin D, and contributes to calcium reabsorption and phosphate excretion in the kidney.
Keywords: kidney, TRPV cation channels, klotho, fibroblast growth factor 23
Introduction
The maintenance of calcium homeostasis is very important because calcium is the main component of bony skeleton and serves as the intracellular and extracellular messenger in numerous essential cellular events such as neuronal network, immune response, muscle contraction, and hormone secretion. Total body calcium in the adult human is about 1-2 kg and 99% of total calcium exists in bone. Even though only less than 1% of body calcium is in the extracellular space, maintaining the extracellular calcium concentration within a narrow range (8.5-10.5 mg/dL) is very important for calcium homeostasis. Approximately 40% of plasma calcium is protein-bound and 10% of calcium is in a complex with anions like phosphate, citrate, and sulfate etc. Only half of plasma calcium is in its free form (ionized form, iCa2+) and physiologically important1). The ionized calcium is tightly regulated by hormones like parathyroid hormone (PTH), 1,25-dihydroxyvitamin D3 (1,25(OH)2D3), calcitonin, and calcium itself. The kidney, intestine, and bone are the main target organs of these regulators, and the kidney plays a key role in the fine regulation of calcium excretion1).
This review will focus on how the kidney works for calcium homeostasis on a molecular basis and discuss new players in the regulation of calcium excretion which were identified recently.
Overview of renal Ca2+ handling
About 50% of plasma calcium (ionized and complexed form; ultrafilterable fraction, excluding the protein bound form) is freely filtered through the renal glomerulus, and 99% of the filtered calcium is actually reabsorbed along renal tubules (Table 1). The excreted calcium in the final urine is about 200 mg per day in an adult person with an average diet. Several factors are involved in the regulation of calcium in renal tubules. PTH and activated vitamin D enhance calcium reabsorption in the thick ascending limb (TAL), distal convoluted tubule (DCT) and/or connecting tubule (CNT), and estrogen promotes calcium absorption in the DCT/CNT1, 2). Acidosis contributes to hypercalciuria by reducing calcium reabsorption in the proximal tubule (PT) and DCT, and alkalosis vice versa3). Diuretics like thiazide and furosemide also alter calcium absorption in the renal tubules; thiazide promotes calcium reabsorption and furosemide inhibits it4, 5). Plasma calcium itself also controls renal calcium absorption through altered PTH secretion as well as via binding to the calcium sensing receptor (CaSR) in the TAL. To facilitate Ca2+ reabsorption along renal tubules; (i) voltage difference between the lumen and blood compartment should be favorable for Ca2+ passage, i.e., a positive voltage in the lumen; (ii) concentration difference should be favorable for Ca2+ passage with a higher Ca2+ concentration in the lumen; (iii) an active transporter should exist if the voltage or concentration difference is not favorable for Ca2+ reabsorption. Each renal tubular segment has a different Ca2+ concentration difference or voltage environment for its unique mechanism for calcium reabsorption.
Table 1.
Renal Ca2+ handling along the tubules
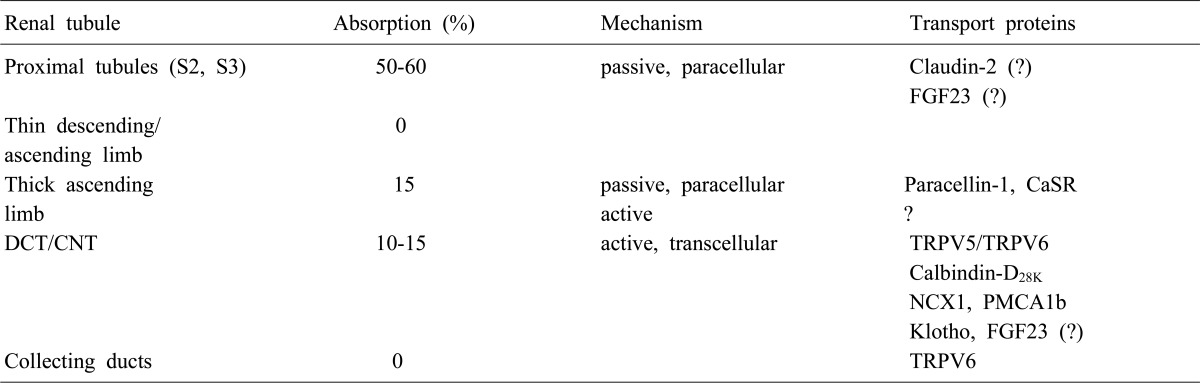
S2, segment 2; S3, segment 3; FGF23, fibroblast growth factor 23; CaSR, calcium sensing receptor; DCT, distal convoluted tubule; CNT, connecting tubule; TRPV, transient receptor potential vallinoid; NCX1, Na2+/Ca2+ exchanger; PMCA1b, plasma membrane Ca2+-ATPase.
Renal Ca2+ handling along the tubules
Fifty to sixty percent of filtered calcium is absorbed in parallel with sodium and water in the PT, suggesting that the passive pathway is the main route of Ca2+ absorption in this segment. Claudin-2 is especially concentrated in the tight junction and also expressed in the basolateral membrane of the PT as the candidate for paracellular Ca2+ channel in the PT6). There is no evidence that Ca2+ reabsorption occurs in the thin descending and ascending limb. In the TAL, 15% of filtered calcium is absorbed, and the passive absorption through paracellular space is known as the main mechanism (Fig. 1). Paracellin-1 (claudin-16) is exclusively expressed in the tight junction of TAL and has been known as the important magnesium channel in the TAL6). Paracellin-1 mutation caused hypercalciuria and nephrocalcinosis in addition to hypomagnesemia2). This finding supports that paracellin-1 is not only the main Mg2+ channel, but also works as the paracellular Ca2+ channel in the TAL. There are some evidences that active transport occurs in the TAL, but no specific channel has yet been identified1). The CaSR is a member of G protein-coupled receptors and suppresses PTH secretion by sensing high plasma Ca2+ level in the parathyroid glands7). In the kidney, the CaSR is most highly expressed in the TAL. Familial hypocalciuric hypercalcemia (FHH) is an autosomal dominant disease due to the mutation of CaSR gene, and is manifested as hypercalcemia, hypophosphatemia, parathyroid hyperplasia, and unusually low renal clearance of calcium. Hypocalciuria, despite of hyperactivity of PTH in FHH, suggests that CaSR plays a direct role in Ca2+ absorption, especially in the TAL independent to PTH action8).
Fig. 1.

Ca2+ absorption in the thick ascending limb of Henle (TAL). (a) The schematic view of Ca2+ reabsorption in the TAL. Paracellin-1 (claudin-16) is located in the tight junction of TAL and serves as the paracellular route for divalent cations. (b) Immunofluorescence image for paracellin-1 (claudin-16) in the mouse TAL cells. Paracellin-1 is co-localized with THP (the marker of TAL) and highly expressed in the tight junction1). BSC1, bumetanide sensitive channel; ROMK, renal outer medullary potassium channel; ClCNKB, chloride channel Kb.
Although only 10-15% of filtered Ca2+ is absorbed in the DCT and CNT, these are the main sites in which the fine regulation of Ca2+ excretion and the major action of PTH and activated vitamin D occur. In the DCT and CNT, the luminal voltage is negative and Ca2+ concentration in the lumen is lower than that of plasma. Thus, active transport mechanism against voltage and concentration gradient should exist in these segments. Several Ca2+ transporting proteins are involved in this active transmembrane transport of Ca2+ in the DCT and CNT. Transcellular Ca2+ reabsorption can occur by three steps; (i) entry of Ca2+ through the calcium channels (TRPV5, TRPV6) in the apical membrane, (ii) binding of Ca2+ with calciumbinding protein (calbindin) and diffusion in the cytoplasm (which enables no significant change in the intracellular i[Ca2+], and (iii) Ca2+ extrusion via an ATP-dependent plasma membrane Ca2+-ATPase (PMCA1b) and an Na2+/Ca2+ exchanger (NCX1) in the basolateral membrane (Fig. 2). In the collecting duct (CD), there is no evidence that Ca2+ reabsorption occurs even though calcium channel (TRPV6) was documented to be expressed in CD cells. Each renal tubule has a unique environment and plays a different role in Ca2+ reabsorption. The coordinated play of different renal tubules could maintain harmony of renal Ca2+ handling.
Fig. 2.

The mechanism of Ca2+ absorption in the renal epithelium. Transcellular Ca2+ reabsorption in the distal convoluted tubule (DCT) and connecting tubule (CNT) occurs by three steps; (i) entry of Ca2+ through the calcium channels [transient receptor potential vanilloid (TRPV) 5, TRPV6] in the apical membrane, (ii) binding of Ca2+ with calcium-binding protein (calbindin) and diffusion in the cytoplasm (without significant change in the intracellular i[Ca2+]), and (iii) Ca2+ extrusion via an ATP-dependent Ca2+-ATPase (PMCA1b) or an Na2+/Ca2+ exchanger (NCX1) in the basolateral membrane.
Renal calcium transport proteins
The important renal calcium transport proteins are exclusively expressed in the DCT and CNT. The characteristics and regulation of calcium transporting proteins will be discussed as follows.
TRPV5 and TRPV6
Transient receptor potential (TRP) channel is a superfamily of ion channels permeable to monovalent and/or divalent cations with six-transmembrane domains. The mammalian TRP family consists of six subfamilies like TRPC (canonical), TRPV (vanilloid), TRPM (melastatin), TRPP (polycystin), TRPML (mucolipin), and TRPA (ankyrin). TRPV is one of them and consists of six members in mammalians; TRPV1 to TRPV6. TRPV5 (previously known as ECaC1) and TRPV6 (ECaC2), both cloned in 1999, have characteristics distinguished from other TRPV channels; (i) constitutively active at low intracellular Ca2+ concentration, and (ii) exclusively selective for Ca2+ (PCa/PNa >100)9). TRPV5 and TRPV6 have the highest sequence homology (~730 amino acids, amino-terminal ankyrin repeats, TM5 and TM6 each forming the pore-region composed with tetramer, on human chromosome 7q34-35) (Fig. 3a). TRPV5 is exclusively expressed in the DCT and CNT in the kidney10) (Fig. 3b). On the contrary, TRPV6 is more ubiquitously distributed, especially in the intestine, and also found from the DCT to the CD in the kidney11) (Fig. 3b). Both TRPV5 and TRPV6 are located in the apical plasma membrane of the tubular epithelium, and serve as the entrance of Ca2+ from the lumen into the cytoplasm. TRPV5 knockout mice exhibited severe hypercalciuria (more than 6 times of wild type mouse) and low bone densities, but without hypocalcemia due to the compensatory elevation of activated vitamin D, clearly demonstrating that TRPV5 plays a crucial role in renal calcium reabsorption12). TRPV6 knockout mice also showed significant hypercalciuria and bone disease13). Even though TRPV5 and TRPV6 knockout mice showed congenital hypercalciuria, the mutation of the proteins has not been found in the human. Until now, TRPV5 is known as the main entry of Ca2+ in renal tubular epithelial cells in the DCT and CNT, and TRPV6 is also known to contribute to renal Ca2+ reabsorption in the distal nephron.
Fig. 3.

Ca2+ transport proteins in the distal convoluted tubule (DCT) and connecting tubule (CNT). (a) The molecular structure of transient receptor potential vanilloid (TRPV) 5 and TRPV6. TRPV 5/6 consists of six transmembrane domains and ankyrin repeat at the N-terminal. The functional pore is composed with TM5 and TM6 of the tetramer2). (b) TRPV5 is exclusively expressed in the apical membrane of the DCT and CNT cells (especially DCT-2). TRPV6 is co-localized with TRPV5, but also observed in the collecting duct (the lower panel). Na+/Cl- cotransporter (NCC) is the DCT and CNT marker10, 11). ECaC, epithelial calcilulm channel 1 (TRPV5); NCX, Na2+/Ca2+ exchanger; CaBP28K, calbindin-D28K; AQP2, aquaporin 2.
Several factors (PTH, 1,25(OH)2D3, calcitonin, estrogen, i[Ca2+], acid-base status, klotho, diuretics, and immunosuppressive drugs, etc) are involved in the regulation of both TRPV5 and TRPV610) (Table 2). Alteration of TRPV5 and TRPV6 by these factors contributes in disturbance of calcium metabolism: dyscalcemia, hypo- and hypercalciuria. 1,25(OH)2D3-depleted rats showed decreased expression of TRPV5 and calbindin-D28K mRNA and protein, and repletion of the hormone restored the expression of them10). The promoter region of TRPV5 gene contains putative vitamin D responsive element (VDRE) surrounded by AP-1 sites, which synergistically enhance the action of 1,25(OH)2D310). 1,25(OH)2D3 also upregulated the renal expression of TRPV611). Therefore, 1,25 (OH)2D3 acts as one of the key regulators of renal Ca2+ handling by alteration of the main renal Ca2+ transport proteins; TRPV5, TRPV6 and calbindin-D28K. Acid-base status also has been known to affect the renal Ca2+ handling. Chronic metabolic acidosis was documented to induce hypercalciuria accompanied with decreased renal expression of TRPV5 and calbindin-D28K, but not in TRPV5 knockout mice, whereas metabolic alkalosis by NaHCO3 administration induced the opposite results3). Nephrocalcinosis and nephrolithiasis observed in the patients of renal tubular acidosis could be explained by the decreased expression of the renal calcium channels by acidosis. Diuretics like furosemide and thiazide have been used for the therapy of hypercalcemia or hypercalciuria. Recently, several studies were performed to document whether these drugs affect renal calcium transport proteins. Furosemide has been used for the treatment of hypercalcemia for many years because it was well known to reduce paracellular Ca2+ transport via reducing lumen-positive voltage in the TAL. Recently, it was documented that furosemide also increased the mRNA levels of TRPV5, TRPV6, and calbindin-D28K in the DCT4). The conflict between hypercalciuria and the increased calcium transporting proteins by furosemide might be explained by the response to the increased distal calcium delivery and compensatory adaptation of the downstream renal segments4). On the contrary, the effect of thiazide on the renal calcium transport proteins is still controversial because conflicting results have been documented. Nijenhuis et al. demonstrated that chronic hydrochlorothiazide treatment significantly reduced urinary Ca2+ excretion which was accompanied by decreased mRNA expression of TRPV5, calbindin-28K, and NCX1 regardless of volume status5). However, the same authors reported later that TRPV5 mRNA was not changed by thiazide14). They also suggested that thiazide-induced hypocalciuria was caused by the enhanced passive Ca2+ transport in the proximal tubule rather than by the action of Ca2+ transport proteins in the DCT using TRPV5 knockout mice14). On the other hand, Lee et al. reported that the effect of thiazide on calcium transport proteins depended upon the volume status; thiazide increased the expression of TRPV5 and calbindin-D28K only when there was no volume contraction15). According to Jang et al., TRPV5 and calbindin-D28K were upregulated by thiazide in hypercalciuric rats16). Therefore, the mechanism of hypocalciuria induced by thiazide needs to be clarified in the future. It has been demonstrated that the channel activation of TRPV5 and TRPV6 require other associated proteins like S100A10 (p11 or annexin 2 light chain)/annexin 2, Rab11b, 80K-H, and NHERF9). In addition, klotho is also known to be involved in the trafficking of TRPV5, which will be discussed in detail later. The coordinated regulation of the abundance, trafficking, and channel activity of TRPV5/6 by these factors can maintain balanced renal Ca2+ excretion.
Table 2.
The regulation of calcium transporting proteins in the DCT and CNT
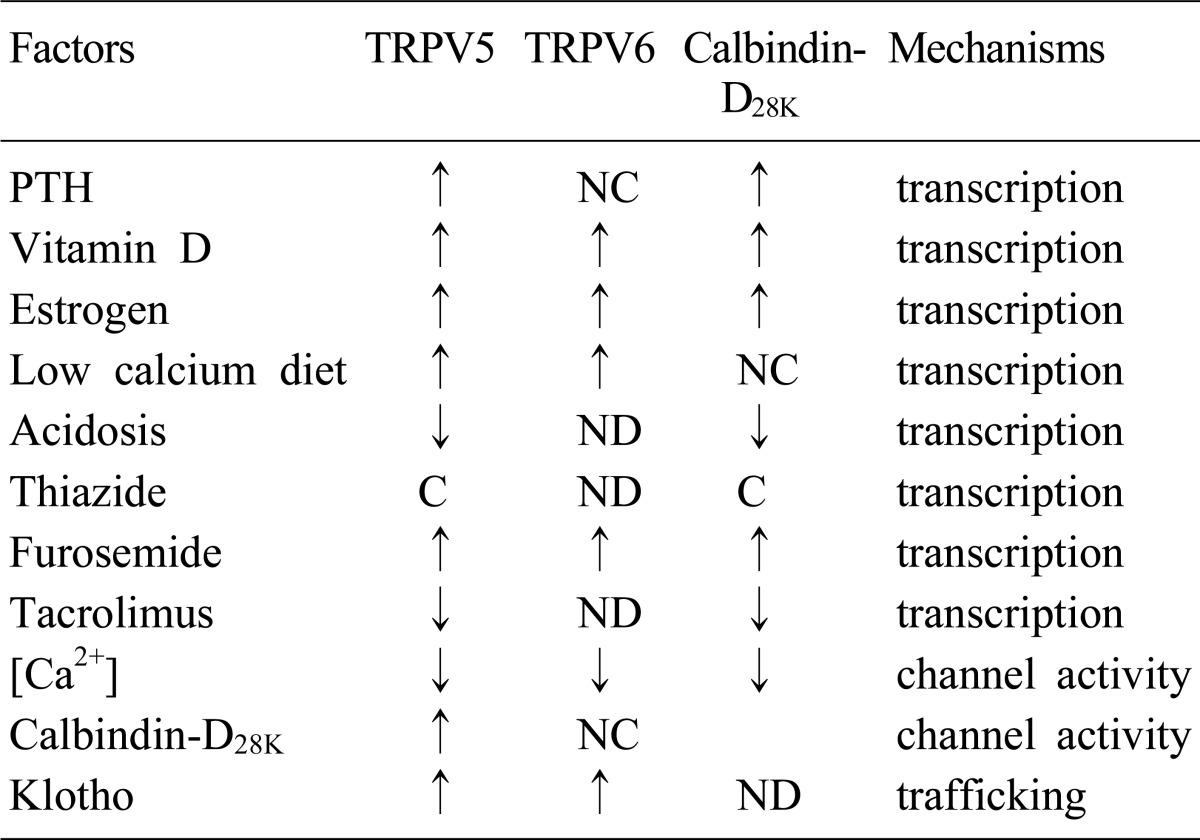
DCT, distal convoluted tubule; CNT, connecting tubule; transient receptor potential vanilloid, TRPV; PTH, parathyroid hormone; ↑, stimulation; ↓, inhibition; NC, no change; ND, not done; C, Controversial.
Calbindin-D28K
Calbindin is a cytosolic calcium-binding protein which is involved in transcellular calcium diffusion and buffers cytosolic Ca2+ to maintain a very low physiologic intracellular Ca2+ concentration. Two isoforms of calbindin exist; calbindin-D28K which is exclusively expressed in the mammalian kidney and calbindin-D9K primarily in the intestine1). Calbindin-D28K is expressed in the cytoplasm of DCT, CNT cells, and principal cells of CD, and colocalized with TRPV5 and NCX1 in the same cells (Fig. 3b). It is regulated by the same factors for TRPV5 such as 1,25(OH)2D3 11), acid-base status3), and furosemide4). Thus, calbindin-D28K is one of the essential components of transepithelial Ca2+ transport, not only as the cytoplasmic delivery vehicle of Ca2+, but also as the dynamic regulatory factor of TRPV5 channel activity17).
NCX1 and PMCA1b
NCX1 and PMCA1b are expressed in the basolateral membrane of the DCT and CNT, and act as the main exit of absorbed Ca2+ to the blood compartment (Fig. 3b). Since the luminal voltage and transcellular Ca2+ concentration difference in the DCT and CNT are not favorable for Ca2+ reabsorption, an active transport mechanism is necessary in these segments. NCX1 is a secondary active transporter generated by the action Na+,K+-ATPase for calcium countertransport1). PMCA1b is a primary active calcium pump and most of its activity is seen in the DCT1). Both NCX1 and PMCA1b are regulated by PTH, calcitonin, and 1,25(OH)2D3 in the distal nephron (Fig. 4).
Fig. 4.

The summary of the distribution of major renal calcium transport proteins. G, glomerulus; PCT, proximal convoluted tubule; PST, proximal straight tubule; S1, segment 1; S2, segment 2; S3, segment 3; DTL, descending thin limb; ATL, ascending thin limb; mTAL, medullary thick ascending limb; cTAL, cortical thick ascending limb; MD, macula densa; DCT, distal convoluted tubule; CNT, connecting tubule; ICD, initial collecting duct; CCD cortical collecting duct; OMCD, outer medullary collecting duct; IMCD, inner medullary collecting duct; TRPV, transient receptor potential vallinoid; PMCA1b, plasma membrane Ca2+-ATPase; NCX1, Na2+/Ca2+ exchanger; NCC, Na+/Cl- cotransporter; AQP2, aquaporin 2.
New players of renal calcium handling
During the past decade, two new proteins; klotho and fibroblast growth factor 23 (FGF23) emerged as the new players of calcium, magnesium, and phosphate metabolism. Klotho and FGF23 are closely linked and play a role in calcium metabolism by their action on parathyroid glands, bone and kidney.
Klotho
Klotho [Clotho] was the youngest of the Moirae of Greek mythology (three sisters who determined the destiny and life of humans; Clotho, Lackesis and Atropos) and the "spinner" who controlled the thread of life. In 1997, the Klotho gene was first discovered by Kuro-o et al. and named it as it is now, because it was originally known as an aging-suppressor gene18). Klotho mutant mice resembled human aging; a short lifespan, infertility, arteriosclerosis, skin atrophy, uncoordinated movement, osteoporosis, emphysema, etc.18). Klotho protein belongs to type 1 membrane protein (single transmembrane domain, 1,014 amino acids, ~130 kDa) and has an extracellular domain with similarity to -glycosidase, β the enzyme involved in the digestion of sugar moieties of substrates. Although klotho is a membrane protein, it is also abundant in the cytoplasm and the cleaved extracellular domain is secreted into blood, cerebrospinal fluid and urine19).
Klotho has been suggested to play an important role in the kidney because it is highly expressed in the kidney and klotho knockout mice showed severe hyperphosphatemia associated with increased 1,25(OH)2D3 levels18). α-Klotho (α-Kl) is highly co-α α expressed with TRPV5, NCX1, and calbindin-D28K in the kidney20) (Fig. 5a), and also abundant in the organs involved in calcium homeostasis such as the parathyroid glands and the choroid plexus. The data strongly indicate that klotho is a regulator of calcium metabolism both in the parathyroid glands and the kidney. The mechanism of calcium regulation by klotho is not clear yet, but there are several suggestions about it. Imura et al. documented that α-Kl made complexes with the α1- and β subunit of Na+,K+-ATPase19). α-KI was required for the rapid recruitment of Na+,K+-ATPase to the cell surface in response to high extracellular Ca2+ concentration19). This activation of Na+,K+-ATPase by α-Kl enhanced calcium reabsorption in the kidney and secretion of PTH in the parathyroid glands19, 21) (Fig. 5b). Chang et al. documented that α-Kl was secreted in the urine and hydrolyzed the extracellular sugar residue on TRPV5 by β-glucuronidase activity20). This hydrolyzed TRPV5 was entrapped in the plasma membranes and then caused increase in Ca2+ reabsorption20) (Fig. 5b). In addition, klotho has interactions with FGF23 and contributes to renal Ca2+ handling. Klotho binds to canonical FGF receptor (FGFR) and converts it into a specific receptor for FGF23, and then α-Kl in combination with FGF23 negatively regulates vitamin D production in the kidney21, 22). Therefore, klotho acts as a new player in renal Ca2+ handling by the regulation of multiple steps of calcium metabolism.
Fig. 5.

Klotho in the kidney. (a) The renal distribution of klotho proteins. Klotho is co-expressed with transient receptor potential vallinoid 5 (TRPV5), Na2+/Ca2+ exchanger (NCX1) and calbindin-D28K in the distal convoluted tubule (DCT) and connecting tubule (CNT) 20). (b) The suggested mechanism of the regulation of calcium homeostasis by klotho. PTH, parathyroid hormone; FGF23, fibroblast growth factor 23.
FGF23
FGF23, a member of the FGF family (type I transmembrane phosphotyrosine kinase receptors), is a 30 kDa secreted protein and inactivated by cleavage into two smaller fragments (N-terminal 18 kDa fragment and C-terminal 12 kDa fragment) by a pro-convertase enzyme, furin (Fig. 6a). It was first cloned as the candidate gene for autosomal dominant hypophosphatemic rickets (ADHR)22). FGF23 is primarily expressed in the osteoblasts and osteocytes. Because Fgf23 knockout mice showed very similar phenotype to Klotho knockout mice including severe hyperphophatemia and osteoporosis, and gain of function mutation of Fgf23 gene was observed in ADHR patients. The main studies about the role of FGF23 in the kidney have focused on phosphate metabolism rather than calcium metabolism23, 24).
Fig. 6.

Fibroblast growth factor 23 (FGF23) in the kidney. (a) The structure of FGF23 proteins. The full length of FGF23 is about 30 kDa, and then inactivated by cleavage to 18 kDa N-terminal fragment and 12 kDa C-terminal fragment24). (b) Suggested mechanism of the FGF23:klotho complex action; Klotho binds to FGF receptor (FGFR) and converts it to a specific receptor for FGF23. Extracellular signalregulated kinase (ERK) 1/2 signaling pathway is activated by binding of the FGF23:klotho complex to FGFR. The FGF23:klotho complex reduces phosphate reabsorption in the proximal tubules via the inhibition of 1α-hydrxylase and NaPi-2a. It might also stimulate parathyroid hormone (PTH) secretion in parathyroid glands. NaPi-2a/c, Type IIa/IIc Na+/Pi contransporter.
For phosphate metabolism, FGF23 acts mainly on the kidney and increases renal phosphate excretion by the inhibition of type IIa Na+/Pi cotransporter expression in the PT and 1α-hydroxylation of vitamin D24, 25). Klotho converts canonical FGFR to specific FGF23 receptor to make it possible for the FGF23:klotho complex to bind to the receptor, and then this complex regulates the activation of vitamin D (Fig. 6b). However, the exact mechanism how FGF23:klotho regulates renal phosphate excretion is still unknown. It is still unknown how the FGF23:klotho complex from the DCT acts in the PT because the main action site of FGF23 in the kidney is the PT, whereas the FGF23:klotho complex is most abundant in the DCT. Both overexpression and deficiency of FGF23 cause several clinical diseases including ADHR and HFTC (hyperphosphatemic familial tumorial calcinosis). Recently, FGF23 was suggested as a potential biomarker for management of phosphate balance in chronic kidney disease (CKD) patients because the circulating FGF23 level was higher in CKD patients than healthy controls and the increased FGF23 level was an independent risk factor for higher mortality among dialysis patients26). FGF23 also plays some roles in the parathyroid glands and other organs like the choroid plexus, pituitary gland, and bone. However, further studies are needed to clarify the roles and the mechanisms.
Conclusion
The kidney has been known as the central organ for calcium homeostasis through fine regulation of renal calcium excretion. For the past decade, there has been big progress in the understanding of the roles of the kidney in calcium homeostasis. The identification of calcium transport proteins and the molecular approach to the regulatory mechanisms achieved a major contribution to this progress. TRPV5, TRPV6, calbindin-D28K, NCX1, and PMCA1b have been identified as the main calcium transport proteins in the distal nephron. PTH, vitamin D, i[Ca2+], CaSR, and other various conditions control renal calcium excretion through the regulation of these transport proteins. Klotho and FGF23 emerged as new players in calcium metabolism in the kidney. Thus, the role of the klotho-FGF23 axis in the regulatory mechanisms of calcium transport needs to be addressed.
References
- 1.Mount DB, Yu AS. Transport of Inorganic Solutes: Sodium, Chloride, Potassium, Magnesium, Calcium, and Phosphate. In: Brenner BM, editor. Brenner & Rector's the kidney. 8th ed. Philadelphia: 2008. pp. 185–192. [Google Scholar]
- 2.Hoenderop JG, Bindels RJ. Epithelial Ca2+ and Mg2+ channels in health and disease. J Am Soc Nephrol. 2005;16:15–26. doi: 10.1681/ASN.2004070523. [DOI] [PubMed] [Google Scholar]
- 3.Nijenhuis T, Renkema KY, Hoenderop JG, Bindels RJ. Acid-base status determines the renal expression of Ca2+ and Mg2+ transport proteins. J Am Soc Nephrol. 2006;17:617–626. doi: 10.1681/ASN.2005070732. [DOI] [PubMed] [Google Scholar]
- 4.Lee CT, Chen HC, Lai LW, Yong KC, Lien YH. Effects of furosemide on renal calcium handling. Am J Physiol Renal Physiol. 2007;293:F1231–F1237. doi: 10.1152/ajprenal.00038.2007. [DOI] [PubMed] [Google Scholar]
- 5.Nijenhuis T, Hoenderop JG, Loffing J, van der Kemp AW, van Os CH, Bindels RJ. Thiazide-induced hypocalciuria is accompanied by a decreased expression of Ca2+ transport proteins in kidney. Kidney Int. 2003;64:555–564. doi: 10.1046/j.1523-1755.2003.00128.x. [DOI] [PubMed] [Google Scholar]
- 6.Kiuchi-Saishin Y, Gotoh S, Furuse M, Takasuga A, Tano Y, Tsukita S. Differential expression patterns of claudins, tight junction membrane proteins, in mouse nephron segments. J Am Soc Nephrol. 2002;13:875–886. doi: 10.1681/ASN.V134875. [DOI] [PubMed] [Google Scholar]
- 7.Ward DT, Riccardi D. Renal physiology of the extracellular calcium-sensing receptor. Pflugers Arch. 2002;445:169–176. doi: 10.1007/s00424-002-0914-x. [DOI] [PubMed] [Google Scholar]
- 8.Attie MF, Gill JR, Jr, Stock JL, Spiegel AM, Downs RW, Jr, Levine MA, et al. Urinary calcium excretion in familial hypocalciuric hypercalcemia. Persistence of relative hypocalciuria after induction of hypoparathyroidism. J Clin Invest. 1983;72:667–676. doi: 10.1172/JCI111016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van de Graaf SF, Hoenderop JG, Bindels RJ. Regulation of TRPV5 and TRPV6 by associated proteins. Am J Physiol Renal Physiol. 2006;290:F1295–F1302. doi: 10.1152/ajprenal.00443.2005. [DOI] [PubMed] [Google Scholar]
- 10.Hoenderop JG, Muller D, Van Der Kemp AW, Hartog A, Suzuki M, Ishibashi K, et al. Calcitriol controls the epithelial calcium channel in kidney. J Am Soc Nephrol. 2001;12:1342–1349. doi: 10.1681/ASN.V1271342. [DOI] [PubMed] [Google Scholar]
- 11.Nijenhuis T, Hoenderop JG, van der Kemp AW, Bindels RJ. Localization and regulation of the epithelial Ca2+ channel TRPV6 in the kidney. J Am Soc Nephrol. 2003;14:2731–2740. doi: 10.1097/01.asn.0000094081.78893.e8. [DOI] [PubMed] [Google Scholar]
- 12.Hoenderop JG, van Leeuwen JP, van der Eerden BC, Kersten FF, van der Kemp AW, Merillat AM, et al. Renal Ca2+ wasting, hyperabsorption, and reduced bone thickness in mice lacking TRPV5. J Clin Invest. 2003;112:1906–1914. doi: 10.1172/JCI19826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoenderop JG, Bindels RJ. Calciotropic and magnesiotropic TRP channels. Physiology (Bethesda) 2008;23:32–40. doi: 10.1152/physiol.00039.2007. [DOI] [PubMed] [Google Scholar]
- 14.Nijenhuis T, Vallon V, van der Kemp AW, Loffing J, Hoenderop JG, Bindels RJ. Enhanced passive Ca2+ reabsorption and reduced Mg2+ channel abundance explains thiazide-induced hypocalciuria and hypomagnesemia. J Clin Invest. 2005;115:1651–1658. doi: 10.1172/JCI24134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee CT, Shang S, Lai LW, Yong KC, Lien YH. Effect of thiazide on renal gene expression of apical calcium channels and calbindins. Am J Physiol Renal Physiol. 2004;287:F1164–F1170. doi: 10.1152/ajprenal.00437.2003. [DOI] [PubMed] [Google Scholar]
- 16.Jang HR, Lee JW, Heo NJ, Lee JH, Oh YK, Na KY, et al. Effects of thiazide on the expression of transient receptor potential vanilloid 5 and calbindin-D28K in a hypercalciuria rat model [Abstract] J Am Soc Nephrol. 2006;17:355A. [Google Scholar]
- 17.Lambers TT, Mahieu F, Oancea E, Hoofd L, de Lange F, Mensenkamp AR, et al. Calbindin-D28K dynamically controls TRPV5-mediated Ca2+ transport. EMBO J. 2006;25:2978–2988. doi: 10.1038/sj.emboj.7601186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390:45–51. doi: 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- 19.Imura A, Tsuji Y, Murata M, Maeda R, Kubota K, Iwano A, et al. alpha-Klotho as a regulator of calcium homeostasis. Science. 2007;316:1615–1618. doi: 10.1126/science.1135901. [DOI] [PubMed] [Google Scholar]
- 20.Chang Q, Hoefs S, van der Kemp AW, Topala CN, Bindels RJ, Hoenderop JG. The beta-glucuronidase klotho hydrolyzes and activates the TRPV5 channel. Science. 2005;310:490–493. doi: 10.1126/science.1114245. [DOI] [PubMed] [Google Scholar]
- 21.Nabeshima Y, Imura H. alpha-Klotho: a regulator that integrates calcium homeostasis. Am J Nephrol. 2008;28:455–464. doi: 10.1159/000112824. [DOI] [PubMed] [Google Scholar]
- 22.Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 2006;444:770–774. doi: 10.1038/nature05315. [DOI] [PubMed] [Google Scholar]
- 23.Yamashita T, Yoshioka M, Itoh N. Identification of a novel fibroblast growth factor, FGF-23, preferentially expressed in the ventrolateral thalamic nucleus of the brain. Biochem Biophys Res Commun. 2000;277:494–498. doi: 10.1006/bbrc.2000.3696. [DOI] [PubMed] [Google Scholar]
- 24.Razzaque MS, Lanske B. The emerging role of the fibroblast growth factor-23-klotho axis in renal regulation of phosphate homeostasis. J Endocrinol. 2007;194:1–10. doi: 10.1677/JOE-07-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu S, Quarles LD. How fibroblast growth factor 23 works. J Am Soc Nephrol. 2007;18:1637–1647. doi: 10.1681/ASN.2007010068. [DOI] [PubMed] [Google Scholar]
- 26.Gutierrez OM, Mannstadt M, Isakova T, Rauh-Hain JA, Tamez H, Shah A, et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med. 2008;359:584–592. doi: 10.1056/NEJMoa0706130. [DOI] [PMC free article] [PubMed] [Google Scholar]