

Abstract
Despite prolonged and intensive application, combined antiretroviral therapy cannot eradicate human immunodeficiency virus (HIV)-1 because it is harbored as a latent infection, surviving for long periods of time. Alternative approaches are required to overcome the limitations of current therapy. We have been developing a short interfering RNA (siRNA) gene silencing approach. Certain siRNAs targeting promoter regions of genes induce transcriptional gene silencing. We previously reported substantial transcriptional gene silencing of HIV-1 replication by an siRNA targeting the HIV-1 promoter in vitro. In this study, we show that this siRNA, expressed as a short hairpin RNA (shRNA) (shPromA-JRFL) delivered by lentiviral transduction of human peripheral blood mononuclear cells (PBMCs), which are then used to reconstitute NOJ mice, is able to inhibit HIV-1 replication in vivo, whereas a three-base mismatched variant (shPromA-M2) does not. In shPromA-JRFL–treated mice, HIV-1 RNA in serum is significantly reduced, and the ratio of CD4+/CD8+ T cells is significantly elevated. Expression levels of the antisense RNA strand inversely correlates with HIV-1 RNA in serum. The silenced HIV-1 can be reactivated by T-cell activation in ex vivo cultures. HIV-1 suppression is not due to offtarget effects of shPromA-JRFL. These data provide “proof-of principle” that an shRNA targeting the HIV-1 promoter is able to suppress HIV-1 replication in vivo.
Introduction
Currently available combined antiretroviral therapy has markedly improved both morbidity and mortality associated with human immunodeficincy cirus (HIV)-1 infection, reducing the viral load ((VL) HIV-1 RNA in serum) and rescuing CD4+ T cells from HIV-1 infection.1,2,3,4 However, HIV-1 persists in its proviral form in cellular reservoirs.5,6,7 On cessation of even prolonged combined antiretroviral therapy, rapid viral recrudescence occurs in the overwhelming majority of cases.8,9 Alternative therapeutic approaches are required to overcome these limitations. We have been investigating a transcriptional gene silencing (TGS) approach using short interfering RNAs (siRNAs) gene targeting the promoter region of HIV-1. Unlike siRNA targeting HIV-1 messenger RNA (mRNA), which induces the post-TGS (PTGS) pathway to degrade mRNA in the cytoplasm, we and others have shown that specific siRNAs targeting viral promoter regions can induce TGS within the nucleus.10,11,12,13,14,15,16 TGS has also been demonstrated in an in vivo model targeting the promoter of vascular endothelial growth factor (VEGF-A).17
Although initial reports demonstrated inhibition of HIV-1 replication by siRNA through PTGS,18,19 further in vitro studies revealed a number of modes of resistance: directly, through rapid development of mutations within20,21,22 or near the siRNA targeted region,23 and indirectly, via mutations in regions separate from the RNA interference targets.24 Nonetheless, both direct and indirect modes of resistance compromise the efficacy of combinations of siRNAs targeting multiple HIV mRNA regions.25 Therefore, PTGS approaches appear to have fundamental limitations.
siRNA-induced TGS was originally reported in plants.26,27,28,29 TGS has more recently been observed in certain mammalian cells.11,30,31 TGS has potential advantages over PTGS when silencing of HIV is the objective. The high mutation rate of HIV-1, due to its nonproof reading reverse transcriptase (RT) and high replication rates, allows rapid adaptation to environmental pressures including the development of resistance or escape mutations.32,33 As TGS results in marked reduction of HIV-1 transcription through induction of epigenetic modifications in the HIV-1 promoter,34 production of new viral RNA is limited and the HIV-1 RT enzyme has no substrate on which to act. Therefore, resistance mutations are less likely to develop in a TGS approach.16 However, TGS approaches may have their own pitfalls. Offtarget effects must be carefully excluded as they have been described with siRNAs or antisense RNA designed to induce TGS.35 Sequence-specific offtarget effects are difficult to predict and even slight offsetting of target sequences can make substantial changes to the extent of offtarget effects.36 Furthermore, sequence-nonspecific offtarget effects can be induced by the triggering of interferon (IFN) pathways by double-stranded RNA through endosomal receptors such as Toll-like receptor (TLR)3, TLR7, and TLR8.37,38
We have reported sustained, profound, highly specific viral suppression of viral replication by siRNA- and short heparin RNA (shRNA)–induced TGS of HIV-1 and simian immunodeficiency virus in various in vitro models, through a mechanism that results in chromatin compaction.34,39,40,41,42 Because HIV-1 has identical long terminal repeats (LTRs) at the 5′ and 3′ ends of the integrated virus, any promoter-targeted siRNA can potentially act through PTGS. With our lead candidate, called PromA, we have found that the contribution of PTGS is limited.34 In this study, we used a lentiviral delivery system to express the previously described shRNA targeting the HIV-1 promoter region to transduce human PBMCs. We first assessed shRNA-mediated TGS approach using a PBMC infection model in vitro. We then demonstrated an antiviral effect of this construct on HIV-1 infection in vivo using NOJ mice43 transplanted with the lenivirus-transduced PBMCs.
Results
shRNA targeting the promoter of HIV-1JRFL suppresses viral expression in PBMCs obtained from healthy donors
Our previous in vitro TGS studies were based on PromA targeting the NF-κβ region of the U3 promoter region of HIV-1 (Figure 1a). The humanized NOD/SCID Janus kinase 3 knockout mice model has been developed to use the HIV-1JRFL strain.43 We sequenced the HIV-1JRFL promoter region, which demonstrated that there was a one-base mismatch compared with the original sequence targeted by PromA (Figure 1a). Because induction of TGS is sequence specific, and a two-base mismatched siRNA failed to induce effective TGS in vitro,42 we constructed U6 promoter driven–shRNA expression self-inactivated lentivirus vector plasmids with a GFP expression unit (Figure 1b) specifically targeting this region of HIVJRFL (shPromA-JRFL), as well as shPromA-M2, a three-base mismatched control, and shPromA-Sc (a scrambled control) (Figure 1b). VSV-G envelope pseudotype lentiviruses expressing each of these constructs were used to transduce human PBMCs. A transduction efficiency of 38.4% for shPromA-JRFL, 31.7% for shPromA-M2, and 34.7% for shPromA-Sc was achieved as assessed by EGFP expression 5 days after transduction (Figure 1c). PBMCs transduced with shPromA-JRFL, but not those transduced with control lentivirus, challenged with HIV-1 in vitro, showed significant reduction of HIV-1 gag mRNA (Figure 1d).
Figure 1.

Human immunodeficiency virus (HIV)-1 transcription is inhibited by lenti-shPromA-JRFL in peripheral blood mononuclear cells (PBMCs). (a) Alignment of self-inactivating (SIN) lentivirus vector constructs along with HIV-1JRFL target sequences. A map of HIV-1 5′ long terminal repeat (LTR) region is illustrated: the blue bar indicates the location of NF-κβ binding region; the arrow indicates the HIV-1 transcription start site; red text in the alignment highlights nucleic acids that differ from the HIV-1JRFL sequence; numbers indicate the nucleic acid location relative to the transcription start site; and underlined text indicates NF-κβ binding region in the HIV-1 promoter. (b) Structure of SIN lentivirus vector. SIN vector consists of central polypurine tract (cPPT), U6 promoter (U6 P), short hairpin RNA (shRNA), ubiquitin C promoter (Ubc), and enhanced green fluorescent protein (EGFP). WPREmt, mutant woodchuck promoter response element, and modified LTR, allow integration but not expression of viral genome. We constructed U6 promoter–driven shRNA expression SIN lentivirus vector plasmid with EGFP expression unit targeting shPromA-JRFL, shPromA-M2 (three-base mismatched control), and shPromA-Sc (scramble control). (c) Expression of EGFP after transduction of lenti-shPromA, shM2, shSc into PBMCs. PBMCs prepared from a healthy donor were stimulated with interleukin-2 for 6 hours, followed by transduction of the lentivirus with a multiplicity of infection of 10. Five days later, EGFP expression was determined by flow cytometric analysis. (d) HIV-1 transcription is inhibited in PBMCs transduced by lenti-shPromA-JRFL. 2 × 106 transduced PBMCs were infected with 50 ng HIV-1JRFL, as determined by the reverse transcriptase assay. The cell-associated HIV gag messenger RNA (mRNA) copy number normalized to 1,000 copies of GAPDH is shown along with time after HIV-1JRFL infection. (e) HIV-1 spliced-tat expression is modulated in PBMCs transduced with lenti-shPromA-JRFL. Spliced-tat mRNA copy number normalized to 1,000,000 copies of GAPDH is shown following after HIV-1JRFL infection. (f) The ratio of spliced-tat over unspliced HIV-1 mRNA, measured by HIV-1 gag mRNA, is shown following HIV-1JRFL infection. In panels d, e, and f the mean values and SEM of three independent experiments are plotted.
The detection of gag mRNA reflects transcription of unspliced viral RNA. We also investigated whether the transcription of spliced viral RNA was modulated by shPromA-JRFL by measuring levels of spliced-tat RNA (Figure 1e). As expected, shPromA-JRFL spliced-tat expression was significantly reduced, but with a different kinetic to the suppression of gag RNA. This results in a marked difference in the kinetics of the ratio of spliced (tat): unspliced (gag) RNA in the shPromA-JRFL–treated cultures compared with the control cultures with a peak in the spliced:unspliced ratio at day 7 (Figure 1f). By day 14, the levels of both gag and spliced-tat RNA are similar in each of the cultures, consistent with loss of effect. Sequence of the virus obtained from the culture supernatant of PBMCs transduced with lenti-shPromA-JRFL at day 14 did not show any mutations in U3 region and, in particular, in the shRNA target sequence. Given that only 38.4% of cells in these bulk cultures were transduced, these results suggest that the elevated HIV-1 replication by day 14, as assessed by both spliced and unspliced viral RNA, is likely due to overgrowth of virus from untransduced cells. Having demonstrated the in vitro efficacy of our new construct, we proceeded to in vivo experiments using shPromA-M2 as a control, because this three-base mismatched control is a more rigorous specificity control than the scrambled shPromA sequence.
shPromA-JRFL inhibits HIV-1 replication in a humanized NOJ mouse model
We evaluated the in vivo antiviral effect of shPromA-JRFL in a previously established model of acute HIV-1 infection based on the nonobese diabetic (NOD)/SCID/Janus kinase 3 knockout (NOJ) mice reconstituted with human PBMCs and then infected with HIVJRFL.43 First, we transduced healthy human PBMCs with lentivirus-expressing shPromA-JRFL or shPromA-M2. Transduction efficiency before transplantation was 22% for shPromA-JRFL and 25% for shPromA-M2. Seven days later, mice (n = 8 per group) were transplanted with 1 × 107 (nonselected) lentivirus-transduced PBMCs permouse by intraperitoneal injection and the cells allowed to engraft. Five days later, mice were infected by intraperitoneal inoculation of HIV-1JRFL (Figure 2a). This is a model of rapidly progressive HIV-1 infection with high VLs, massive CD4+ T-cell depletion, and profound immunodeficiency occurring within weeks of infection.43
Figure 2.

Transduction with lentivirus-shPromA-JRFL shows antiviral effects in NOJ mouse. (a) Time line for in vivo NOJ mouse experiment. LV denotes lentivirus. (b) Amount of viral load (VL) (HIV-1 RNA in serum) in mice with lentivirus-transduced peripheral blood mononuclear cells (PBMCs). Blood samples were collected from mice orbit on day 14 after HIV-1JRFL infection. Horizontal bars indicate the medians. *P < 0.05. (c) Effects on the ratio of CD4+/CD8+ cells in mice with lentivirus-transduced PBMCs. Short bars indicate the medians. *P < 0.05, **P < 0.01. (d) Effect on intracellular p24-positive cells. Peritoneal cavity cells and splenocytes recovered on day 14 after HIV-1 inoculation were analyzed by flow cytometry. The percentage of p24-positive cells among CD4+ T cells (gated as mCD45− hCD45+ hCD3+ hCD8−) is shown (n = 8). Horizontal bars indicate the medians. *P < 0.05. NS, not significant; PC, peritoneal cavity; Sp, Spleen.
Mononuclear cells were recovered at sacrifice (day 14 after HIV-1 infection) from the peritoneal cavity and the spleen. VL in serum was detected by RT quantitative real-time PCR (RT-qPCR). VL in the mice transplanted with PBMCs expressing shPromA-JRFL was significantly lower (P = 0.014) than in shPromA-M2 control mice (Figure 2b). CD4+ T cells were reduced relative to CD8+ T cells in shPromA-M2–transplanted mice, whereas the CD4+ to CD8+ T-cell ratio was better preserved in mice transplanted with shPromA-JRFL both in the peritoneal cavity (P = 0.038) and in the spleen (P = 0.002) (Figure 2c). Furthermore, the extent of downregulation of CD4 surface expression is reduced by shPromA-JRFL (Supplementary Figure S1). Thus, shPromA-JRFL appears to protect CD4+ T cells against HIV-1–mediated depletion and downregulation of CD4 surface expression. By contrast, there was no significant difference in CD8+ T-cell numbers between the two groups, indicating successful human PBMC engraftment in all the mice (Supplementary Figure S2). Intracellular staining after gating on human CD3+ CD8− spleen cells demonstrated that the percentage of p24-expressing (p24+) cells was significantly lower in the mice transplanted with shPromA-JRFL–transduced PBMCs (P = 0.014) (Figure 2d).
Expression levels of the antisense strand of shPromA-JRFL inversely correlated with VL
The above data indicate a reduction in viral replication and relative protection from CD4+ T-cell destruction, but there was substantial variability in the extent of these effects among the mice within the PromA-JRFL–treated group. Previous observations have suggested that the antisense strand of double-stranded siRNA is responsible for induction of TGS in mammalian cells.14,39,44 After transduction of the shPromA-JRFL lentivirus, the shRNA expression unit is transcribed from the U6 promoter, by RNA polymerase III, which terminates at a poly(T) motif within this expression unit. The short hairpin loop sequence is then processed by cellular ribonucleases to form mature/processed double-stranded siRNA.45 We, therefore, quantified the antisense strand of the shPromA-JRFL transcript by real-time PCR, as previously described,42 to determine whether the degree of expression and processing of the shRNA constructs impacted the antiviral effects. It was found that the degree of antisense-strand expression had a strong inverse correlation with VL in serum in the mice transplanted with PromA-JRFL–transduced PBMCs (r = 0.83; P = 0.0015), but not in those transplanted with the control shRNA PromA-M2 (Figure 3a). Similarly, there was an inverse correlation between expression of cell-associated HIV-1 gag mRNA in CD4+ T cells from the spleen and expression antisense strand of the shPromA-JRFL (r = 0.84; P = 0.0014; Supplementary Figure S3a) and strong linear correlation between VL and expression of cellular-associated HIV-1 gag mRNA in CD4 cells (r = 0.84; P = 0.0014; Supplementary Figure S3b). Consistent with these observations, there was a strong linear correlation between VL and both the percentage of p24-positive CD3+ CD8− cells (r = 0.98; P < 0.0001; Figure 3b) and the cell-associated viral mRNA from splenocytes (r = 0.84; P = 0.0014) in shPromA-JRFL–expressing mice, indicating that serum VL correlates with cellular expression of HIV-1 Gag protein in CD4+ T cells and gag mRNA in splenocytes. These data all point to the fact that the presence of the processed antisense strand of shPromA-JRFL is a strong correlate of inhibition of viral replication.
Figure 3.

Transcriptional gene silencing (TGS) is induced by lenti-shPromA-JRFL. (a) Inverse correlation of expression level of the antisense strand of shPromA-JRFL and viral load (VL). Relative antisense RNA expression of shPromA-JRFL was detected by the primer-specific reverse transcriptase–polymerase chain reaction. (b) Linear correlation of intracellular p24 expression level and VL. Intercellular p24 staining in CD3+ CD8− cells obtained from peritoneal cavity was analyzed by flow cytometry and was plotted against VL. (c) Transcriptionally suppressed human immunodeficiency virus (HIV)-1 is reactivated by phorbol myristate acetate (PMA). Splenocytes recovered from day 14 after inoculation of HIV-1JRFL were divided into two cultures with or without addition of PMA. After ex vivo culture for 24 hours, cellular-associated messenger RNA (mRNA) was extracted for analysis of HIV gag mRNA. Substantially increased expression of HIV-1 gag mRNA was found after PMA activation in three mice with highly suppressed HIV-1 (Nos. 3, 7, and 8), obtained from shPromA-JRFL treated mice. U1 is a positive control for HIV-1 latently infected cells. The mean values and SEM of three independent experiments are plotted.
Phorbol myristate acetate, a strong stimulating reagent, reactivates silenced transcription of HIV-1 in ex vivo culture
Latent HIV-1 has silenced transcription, which is able to be switched on by strong cellular activating stimuli such as phorbol myristate acetate (PMA).46 The U1 cell line is a latently infected monocytoid cell line that contains the proviral form of HIV-1, with heterochromatin formation in the viral promoter region, the 5′LTR. The silenced provirus can be activated by treatment of cells with PMA with concomitant relaxation and opening of the chromatin structure.47,48,49 Given that we have previously shown that si/shPromA acts by inducing biochemical changes in histone tails resulting in a heterochromatic structure associated with the 5′LTR, we hypothesized that the silenced HIV-1 induced by shPromA-JRFL would be activated by PMA. We conducted ex vivo culture of the splenic CD4+ T cells from all mice in the shPromA-JRFL–treated group, including the three highly suppressed mice (nos. 3, 7, and 8, as indicated in Figure 3a). We found that PMA treatment resulted in elevated levels of gag mRNA in the PBMCs from the three suppressed mice, but not in those where VL was poorly suppressed. Of note, these were the same mice in which the antisense strand of shPromA-JRFL was poorly expressed (Figure 3c). We also found that PMA treatment did not increase the levels of gag mRNA in the PBMCs from the 8 mice treated with shPromA-M2 (Figure 3c). These data are consistent with TGS induced by shPromA-JRFL being responsible for the observed suppression of HIV-1 transcription.
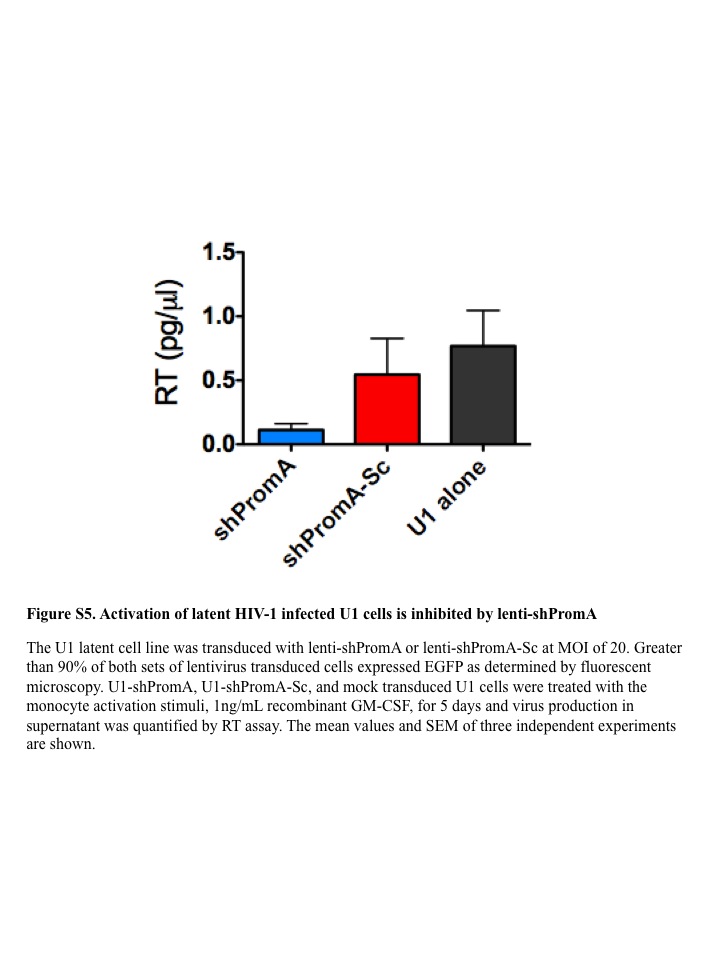
We also confirmed the activation of HIV-1 transcription using an in vitro experimental model. We transduced PM1-CCR5 T cells, with lenti-shPromA-JRFL. We conducted limiting dilution of transduced PM1-CCR5 T cells to isolate strongly positive EGFP clonal populations (Figure 4a,b). After confirming expression of EGFP in more than 99% of cells in this clone, (PromA-JRFL No.3), we infected these cells with two concentrations of HIV-1JRFL (Figure 4c). We also measured the expression level of the antisense strand of the shPromA-JRFL transcript by RT-qPCR in PM1-ccr5 cells in the presence of ongoing active HIV-1JRFL infection. HIV infection did not make a difference to the expression levels of the antisense strand of the shPromA-JRFL transcript (Supplementary Figure S4a,b).42 After confirming that shPromA-JRFL–expressing PM1-CCR5 cells completely suppressed HIV-1 replication, we then assessed whether activation of the suppressed HIV-1 transcription could be induced by various stimuli, including PMA and the histone deacetylase inhibitor, tricostatin A (Figure 4d). Both stimuli resulted in reactivation of viral replication, with tricostatin A having a greater effect than that of PMA. The powerful effect of tricostatin A in viral reactivation in the presence of shPromA-JRFL strongly suggests that this shRNA is causing viral suppression through TGS, because recruitment of histone deacetylase is a classic mark of TGS. It is interesting that not all activation stimuli result in reactivation of Prom-A–suppressed infection. GM-CSF stimulation of untransduced U1 cells results in reactivation of latent virus. However, GM-CSF stimulation of U1 cells lentivirally transduced to express shPromA did not result in increased viral replication (Supplementary Figure S5). These data are concordant with our previous in vitro data, demonstrating that siRNA and shPromA cause suppression of HIV-1 replication through TGS.34,40,41 The data also suggest that TGS mediated through shRNA can be sustained even in the presence of certain cytokines, such as GM-CSF.
Figure 4.
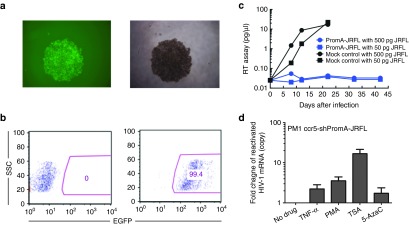
Transcriptional gene silencing (TGS) is induced by lenti-shPromA-JRFL in vitro. (a) PM1-ccr5 cells were transduced with lenti-shPromA with a multiplicity of infection of 10, subjected to limited dilution to isolate EGFP-positive colonies. A fluorescent image of clone PromA-JRFL No. 3. is shown on the left and the corresponding phase contrast image is on the right. (b) Expression of EGFP after expansion of clone PromA-JRFL No.3 as determined by flow cytometric analysis. (c) HIV-1 replication is inhibited in Lenti-shPromA-JRFL–transduced PM1-ccr5 cells. HIV-1 in the culture supernatant was detected by the colorimetric reverse transcriptase (RT) assay. (d) HIV-1 transcription is reactivated from the transcriptionally suppressed PM1-ccr5 cells, transduced with Lenti-shPromA-JRFL. 24 hours after activation with PMA or TSA, cell-associated mRNA was extracted for analysis of HIV gag mRNA. Fold change of reactivated HIV-1 gag mRNA is shown. 5Aza, 5-azacytidine; PMA, phorbol myristate acetate; TNF-α, tumor necrosis factor-α; TSA, trichostatin A. The mean values and SEM of three independent experiments are plotted.
TGS induced by shPromA-JRFL was not associated with offtarget effects
Endosomal innate immune receptors, such as TLR3, TLR7, and TLR8, recognize long single- or double-stranded RNAs, triggering type I IFN and IFN-stimulated gene expression that can result in viral suppression by both nonspecific offtarget effects and undesirable toxicities.37,38 We evaluated the extent of induction of IFN-α gene expression using RT-qPCR on splenocytes from shPromA-JRFL, shPromA-M2, and untreated mice. We used polyI:C (polyinosinepolycytosine)-treated PBMCs as a positive control.50,51 There was no difference in IFN-α expression levels (Figure 5a). Furthermore, by RT-qPCR there was no difference in expression of the IFN-α response genes, OSA1, ISG20, and IFIT1 between groups of mice (Figure 5b), which is consistent with a lack of induction of IFN.
Figure 5.
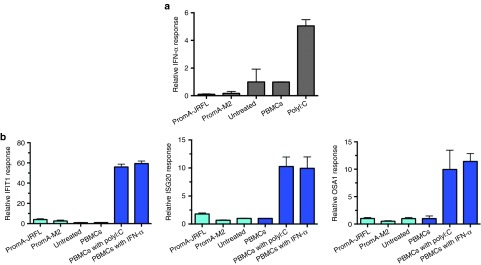
No significant offtarget effects are induced by lenti-shPromA-JRFL. (a) Effect of lentiviral transduction of peripheral blood mononuclear cells (PBMCs) on interferon-α (IFN-α) expression. Splenocytes were prepared from mice transplanted with lentivirus-transduced PBMCs. Cell-associated messenger RNA (mRNA) was extracted for analysis of IFN-α by reverse transcriptase–polymerase chain reaction (RT-PCR). PolyI:C–treated PBMCs were used a positive control, indicated in dark blue. (b) Effect of lentiviral transduction of PBMCs on IFN response genes. Cell-associated mRNA was extracted for analysis of three IFN-α response genes (OSA1, ISG20, and IFITM1) by RT-PCR. PolyI:C or IFN-α–treated PBMCs were used as positive controls, these are indicated in dark blue. The mean values and SEM of three independent experiments are plotted.
To exclude offtarget effects mediated through the targeting by shPromA-JRFL of other NF-κβ binding motifs of host genes as distinct from the NF-κβ motif in the HIV-1 LTR, a PCR-based assay was used to assess the expression levels of 86 NF-κβ-driven host genes, including IFN-α, β, and γ. The shPromA-JRFL was not associated with altered expression of NF-κβ driven host genes, including the IFN genes (Supplementary Figure S6a,b). These data are concordant to our previous analyses of offtarget effects induced by siRNA and shRNA forms of PromA in HeLa and MOLT-4 cell and strongly suggest that the observed HIV-1 suppression is not the result of offtarget effects induced by shPromA-JRFL.42
Discussion
Our previous in vitro data based on HeLa or T-cell lines suggested that TGS of HIV-1 can be induced by promoter targeted si/shRNAs through the induction of epigenetic modifications to form heterochromatin structures, which resemble the biochemical modifications of the HIV-1 promoter in latently infected cell lines.12,13 In this report, we extend our si/shRNA-mediated TGS approach into an in vivo NOJ humanized mouse model.43 Although there are several reports of PTGS mediated by siRNA using in vivo humanized mouse models,50,51,52,53,54 this report demonstrates that HIV-1 gene silencing based on the TGS pathway is possible in vivo. Our NOJ model is a model of acute rapidly progressive HIV infection in which massive infection occurs: hCD4/CD8 cell ratio significantly decreases, and high VL is achieved within 14 days of intraperitoneal inoculation of HIV-1JRFL.43 Despite this highly activated, destructive, and rapidly progressive infection with high levels of viral transcription, we were still able to successfully demonstrate a degree of viral suppression using an shRNA that induces transcriptional silencing.
We demonstrated substantial antiviral effects that resulted in significant alterations in a number of surrogate markers of disease progression in the shPromA-JRFL–treated group, including reduced serum VL, reduced percentage of HIV Gag p24 protein–positive CD3+ CD8− T cells, and an improved ratio of CD4+/CD8+ T cells. Of note, the extent of each of these effects correlated with the extent of expression of the processed shRNA antisense strand. These data also suggest that in mice that showed adequate expression of the antisense strand of shPromA-JRFL in CD4+ T cells, there was an observable HIV-1 antiviral effect, which we conclude is occurring through TGS. Because the CD4+ T cells were relatively protected from active HIV-1 infection through shPromA-JRFL–mediated TGS, we could see significant reduction of VL in serum in shPromA-JRFL mice. The data from the in vitro PBMC experiments show that shPromA-JRFL has marked effects on production of both spliced and unspliced viral mRNA. Reduction in the production of spliced-tat is likely to be important in the effective silencing of latently infected cells, as Tat, through its interaction with the TAR region of the 5′LTR allows efficient upregulation of transcription of long unspliced HIV-1 mRNA.55,56,57 Furthermore, the ex vivo reactivation of HIV-1 infection by PMA stimulation is consistent with our previously reported observations that siPromA and shPromA constructs result in viral suppression by TGS40,41 and with other models of HIV-1 latency.47,48,49,58 In addition, the ex vivo reversal of viral suppression by TNF-α and, in particular, by the histone deacetylase inhibitor, tricostatin A, is consistent with suppression being induced by TGS. We confirmed that shPromA-JRFL did not induce any significant offtarget effects, determined by expression of type I and II IFNs, IFN response genes and NF-κβ–regulated host genes. We also demonstrated that not all activating stimuli reverse this process, for example, the GM-CSF–induced activation of latent HIV-1 in shPromA–transduced U1 cell was inhibited.
This acute human PBMC-NOJ mouse model has been used to demonstrate proof of principle of potential in vivo efficacy of shPromA delivered by a retroviral vector, focusing on the relative protection of human CD4+ T cells against HIV-1 infection. To further this approach, we are investigating the use of newborn NOJ mouse engrafted via intrahepatic injection of human cord blood–derived CD34+ cells transduced with retroviral constructs expressing shPromA and appropriate controls.59 This will enable us to evaluate the effect of this approach on engraftment and hematopoietic cell differentiation and reconstitution, and subsequently on HIV-1 infection. An advantage of the CD34+ NOJ model is that the cell number required in this model is 100-fold lower (5 × 104 CD34+ cells per mouse) than that of required in the current NOJ mouse model reconstituted by human PBMCs (1 × 107 PBMCs per mouse). The titer of our current lentiviruses is ~2 × 108 infectious viral particle per milliliter. Therefore, a higher multiplicity of infection can be achieved to obtain a greater transduction rate, which will potentially provide greater efficacy.
Using the CD34+ cell–reconstituted NOJ model, we wish to explore a scenario closer to that which we envisage these constructs will be used in human HIV-1 treatment, primarily on cessation of antiretroviral drugs in controlled chronic infection to determine whether lentivirally delivered shPromA constructs can stabilize the viral reservoir on withdrawal of antiretroviral therapy. If the latent viral reservoir could be maintained as effectively silenced by shPromA treatment, these constructs would represent a substantial step forward on the road toward a functional cure, by providing an alternative to the currently proposed eradication strategies than using various viral transcription activating agents, such as histone deacetylase and demethylases.49,60,61 Rather than activating virus and abolishing infected cells, we propose that constructs such as shPromA could be used to lock HIV-1 into latency maintaining transcriptionally inactive virus even in patients ceasing conventional antiretroviral therapy, thus achieving a prolonged remission or functional cure of HIV-1 infection.
Materials and methods
Production of lentivirus. The construction of lentiviral vector lenti-shPromA-JRFL, lenti-shPromA-M2, and lenti-Sc were previously described.41 An outline of the construction of self-inactivated lentivirus vector plasmid with GFP expression unit is illustrated in Figure 1b. Vesicular stomatitis virus-G (VSV-G) pseudotyped lentiviral vectors were prepared by transduction of plasmid DNA into 293T cells using HilyMax (Dojindo Molecular Technologies, Osaka, Japan), a lipofectamine-based transfection reagent. The resulting virus was concentrated from supernatant as previously described62,63, and stocks were titrated on 293T cells based on EGFP expression.
PBMC transduction with lentivirus. Peripheral blood was collected from healthy volunteers after informed consent was obtained, according to the institutional guidelines approved by the Faculty of Life Sciences and Pharmaceutical Sciences, Kumamoto University, Kumamoto, Japan. Healthy donor PBMCs were prepared by standard density gradient centrifugation using Ficoll-Hypaque (VWR, Murarrie, Australia). Cells were cultured in RPMI-1640 medium supplemented with penicillin (100 U/ml), streptomycin (100 µg/ml), 20% fetal calf serum (R20) in the presence of 20 units/ml of interleukin-2 (Roche Diagnostic, Castle, Hill, Australia) for 7 hours, followed by overnight transduction with either lenti-shPromA-JRFL, lenti-shPromA-Sc, or lenti-shPromA-M2 using a multiplicity of infection of 1.5–2.0. Cells were then cultured in R20 for a further 7 days before transplantation.
Transplantation of human PBMCs into NOJ mice and HIV-1 infection of mice. Human PBMC-transplanted NOJ (hu-PBMC-NOJ) mice were generated as described previously.43 Briefly, NOJ mice were irradiated (1.0 Gy), and bulk lenti-shPromA-JRFL– or lenti-shPromA-M2–transduced PBMCs (1 × 107) were resuspended in phosphate-buffered saline (PBS) (0.1 ml) and infused intraperitoneally into each mouse. Seven days after PBMC implantation, a dose of 200 ng of HIV-1JRFL, which was determined by HIV-1 p24 antigen ELISA (ZeproMetrix), suspended in 0.1 ml of PBS, was inoculated intraperitoneally into each mouse. On day 14 after HIV-1JRFL infection, mice were killed and blood samples were collected from the mouse orbit, and peritoneal cavity and spleen cells were harvested and resuspended in PBS (see Figure 2a). All animal experiments were performed according to the guidelines of the Kumamoto University Graduate School of Medical Science.
RT-PCR analysis and RT assay. Cellular RNA was extracted using High Pure RNA Tissue Kit (Roche Diagnostic), followed by the RT-PCR analysis as described previously.39,42 Detection of spliced-tat was conducted using the same RT-PCR conditions with the primer set: Tat-F: ATG GAG CCA GTA GAT CCT AGA CTA and Tat-B: ATT CCT TCG GGC CTG TCG using RT-PCR (SensiFAST Probe one-step RT-PCR: Bioline). Both HIV-1 gag mRNA and spliced-tat mRNA levels were normalized against GAPDH. Colorimetric RT activity (RT assay) in culture supernatants was determined as previously described.64
Flow analysis of CD4+ CD8+ T cells and internal p24 staining. Lymphocyte subsets from human mononuclear cells obtained from the transplanted mice were characterized by flow cytometric analysis as described previously.43 Briefly, cells were treated with red cell lysing buffer (155 mM NH4Cl, 10 mM KHCO3, and 0.1 mM EDTA) to lyse erythrocytes, and single-cell suspensions were prepared in staining medium (PBS with 2% fetal bovine serum and 0.05% sodium azide) and stained with monoclonal antibodies: allophycocyanin (APC)-Cy7–conjugated antimouse CD45 (BD Pharmingen, Kobe, Japan), APC-conjugated anti-hCD4 (Dako, Tokyo, Japan), phycoerythrin-Cy7–conjugated anti-hCD3 (e-Bioscience, Tokyo, Japan), Pacific Blue–conjugated anti-hCD8 (BioLegend, Tokyo, Japan), and Pacific Orange–conjugated antihuman CD45 (anti-hCD45) (Invitrogen, Tokyo, Japan). After 30 minutes, cells were washed twice and fixed in PBS with 1% paraformaldehyde for 20 minutes and permeabilized in PBS with 0.1% saponin. After a 10-minute incubation, cells were stained with phycoerythrin-conjugated anti-HIV-1 p24 monoclonal antibody (Beckman Coulter, Tokyo, Japan) for 30 minutes. All washes and staining procedures were conducted at 4 °C. Following staining, the cells were analyzed on an LSR II flow cytometer (BD Bioscience). Data were analyzed with FlowJo software (Tree Star, Tokyo, Japan).
Statistical analysis. RT-PCR analysis and RT assay values are given as mean and SEM. Ratio of CD4+/CD8+ cells and VL were tested for significance using a nonparametric Mann–Whitney U test. A P value <0.05 was considered statistically significant. All analyses were performed using GraphPad Prism Version 5.0a (Graphpad Software, San Diego, CA).
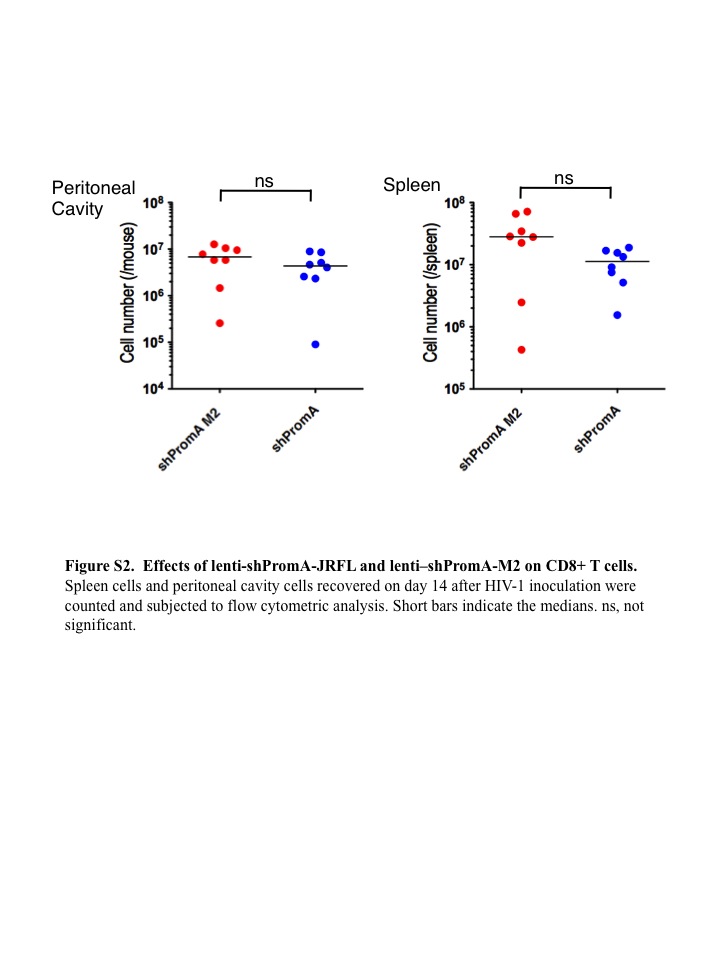
SUPPLEMENTARY MATERIAL Figure S1. Effects of lenti-shPromA-JRFL and lenti-shPromA-M2 on CD4− T cells. Figure S2. Effects of lenti-shPromA-JRFL and lenti-shPromA-M2 on CD8+ T cells. Figure S3. HIV-1 gag HIV mRNA level is inhibited through TGS induced by lenti-shPromA-JRFL. Figure S4. The antisense strand expression level is not altered with HIV-1JRFL infection. Figure S5. Activation of latent HIV-1–infected U1 cells is inhibited by lenti-shPromA. Figure S6. No significant difference in comparison of 86 NF-κβ driven genes in PBMCs trasduced with lenti-shPromA.
Acknowledgments
The authors thank the Australian Government Department of Health and Ageing (RM07292) and National Health and Medical Research Council (455350 to A.K. and 630571 for K.S. and C.H.). Funding for open-access charge was provide by St. Vincent's Centre for Applied Medical Research.
Supplementary material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- Ho DD, Neumann AU, Perelson AS, Chen W, Leonard JM, Markowitz M. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature. 1995;373:123–126. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- Mellors JW, Rinaldo CR, Gupta P, White RM, Todd JA, Kingsley LA. Prognosis in HIV-1 infection predicted by the quantity of virus in plasma. Science. 1996;272:1167–1170. doi: 10.1126/science.272.5265.1167. [DOI] [PubMed] [Google Scholar]
- Perelson AS, Essunger P, Cao Y, Vesanen M, Hurley A, Saksela K, et al. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature. 1997;387:188–191. doi: 10.1038/387188a0. [DOI] [PubMed] [Google Scholar]
- Palella FJ, Delaney KM, Moorman AC, Loveless MO, Fuhrer J, Satten GA, et al. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV Outpatient Study Investigators. N Engl J Med. 1998;338:853–860. doi: 10.1056/NEJM199803263381301. [DOI] [PubMed] [Google Scholar]
- Chun TW, Finzi D, Margolick J, Chadwick K, Schwartz D, Siliciano RF. In vivo fate of HIV-1-infected T cells: quantitative analysis of the transition to stable latency. Nat Med. 1995;1:1284–1290. doi: 10.1038/nm1295-1284. [DOI] [PubMed] [Google Scholar]
- Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science. 1997;278:1295–1300. doi: 10.1126/science.278.5341.1295. [DOI] [PubMed] [Google Scholar]
- Siliciano RF, Greene WC. HIV latency. Cold Spring Harb Perspect Med. 2011;1:a007096. doi: 10.1101/cshperspect.a007096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong JK, Hezareh M, Günthard HF, Havlir DV, Ignacio CC, Spina CA, et al. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science. 1997;278:1291–1295. doi: 10.1126/science.278.5341.1291. [DOI] [PubMed] [Google Scholar]
- Chun TW, Justement JS, Moir S, Hallahan CW, Maenza J, Mullins JI, et al. Decay of the HIV reservoir in patients receiving antiretroviral therapy for extended periods: implications for eradication of virus. J Infect Dis. 2007;195:1762–1764. doi: 10.1086/518250. [DOI] [PubMed] [Google Scholar]
- Turner AM, De La Cruz J, Morris KV. Mobilization-competent Lentiviral Vector-mediated Sustained Transcriptional Modulation of HIV-1 Expression. Mol Ther. 2009;17:360–368. doi: 10.1038/mt.2008.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malecová B, Morris KV. Transcriptional gene silencing through epigenetic changes mediated by non-coding RNAs. Curr Opin Mol Ther. 2010;12:214–222. [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Kelleher AD. Transcriptional regulation by promoter targeted RNAs. Curr Top Med Chem. 2009;9:1079–1087. doi: 10.2174/156802609789630875. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Kelleher AD. Lessons from viral latency in T cells: manipulating HIV-1 transcription by siRNA. HIV Therapy. 2010;4:199–213. [Google Scholar]
- Ahlenstiel CL, Lim HG, Cooper DA, Ishida T, Kelleher AD, Suzuki K. Direct evidence of nuclear Argonaute distribution during transcriptional silencing links the actin cytoskeleton to nuclear RNAi machinery in human cells. Nucleic Acids Res. 2012;40:1579–1595. doi: 10.1093/nar/gkr891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowling S, Stapleton K, Turner AM, Uhlmann E, Lehmann T, Vollmer J, et al. Chemically Modified Oligonucleotides Modulate an Epigenetically Varied and Transient Form of Transcription Silencing of HIV-1 in Human Cells. Mol Ther Nucleic Acids. 2012;1:e16. doi: 10.1038/mtna.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner AM, Ackley AM, Matrone MA, Morris KV. Characterization of an HIV-targeted transcriptional gene-silencing RNA in primary cells. Hum Gene Ther. 2012;23:473–483. doi: 10.1089/hum.2011.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turunen MP, Lehtola T, Heinonen SE, Assefa GS, Korpisalo P, Girnary R, et al. Efficient regulation of VEGF expression by promoter-targeted lentiviral shRNAs based on epigenetic mechanism: a novel example of epigenetherapy. Circ Res. 2009;105:604–609. doi: 10.1161/CIRCRESAHA.109.200774. [DOI] [PubMed] [Google Scholar]
- Novina CD, Murray MF, Dykxhoorn DM, Beresford PJ, Riess J, Lee SK, et al. siRNA-directed inhibition of HIV-1 infection. Nat Med. 2002;8:681–686. doi: 10.1038/nm725. [DOI] [PubMed] [Google Scholar]
- Jacque JM, Triques K, Stevenson M. Modulation of HIV-1 replication by RNA interference. Nature. 2002;418:435–438. doi: 10.1038/nature00896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boden D, Pusch O, Lee F, Tucker L, Ramratnam B. Human immunodeficiency virus type 1 escape from RNA interference. J Virol. 2003;77:11531–11535. doi: 10.1128/JVI.77.21.11531-11535.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das AT, Brummelkamp TR, Westerhout EM, Vink M, Madiredjo M, Bernards R, et al. Human immunodeficiency virus type 1 escapes from RNA interference-mediated inhibition. J Virol. 2004;78:2601–2605. doi: 10.1128/JVI.78.5.2601-2605.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Eije KJ, ter Brake O, Berkhout B. Human immunodeficiency virus type 1 escape is restricted when conserved genome sequences are targeted by RNA interference. J Virol. 2008;82:2895–2903. doi: 10.1128/JVI.02035-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerhout EM, Ooms M, Vink M, Das AT, Berkhout B. HIV-1 can escape from RNA interference by evolving an alternative structure in its RNA genome. Nucleic Acids Res. 2005;33:796–804. doi: 10.1093/nar/gki220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah PS, Pham NP, Schaffer DV. HIV develops indirect cross-resistance to combinatorial RNAi targeting two distinct and spatially distant sites. Mol Ther. 2012;20:840–848. doi: 10.1038/mt.2012.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ter Brake O, Konstantinova P, Ceylan M, Berkhout B. Silencing of HIV-1 with RNA interference: a multiple shRNA approach. Mol Ther. 2006;14:883–892. doi: 10.1016/j.ymthe.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Wassenegger M, Heimes S, Riedel L, Sänger HL. RNA-directed de novo methylation of genomic sequences in plants. Cell. 1994;76:567–576. doi: 10.1016/0092-8674(94)90119-8. [DOI] [PubMed] [Google Scholar]
- Wassenegger M. RNA-directed DNA methylation. Plant Mol Biol. 2000;43:203–220. doi: 10.1023/a:1006479327881. [DOI] [PubMed] [Google Scholar]
- Mette MF, Matzke AJ, Matzke MA. Resistance of RNA-mediated TGS to HC-Pro, a viral suppressor of PTGS, suggests alternative pathways for dsRNA processing. Curr Biol. 2001;11:1119–1123. doi: 10.1016/s0960-9822(01)00315-3. [DOI] [PubMed] [Google Scholar]
- Jones L, Ratcliff F, Baulcombe DC. RNA-directed transcriptional gene silencing in plants can be inherited independently of the RNA trigger and requires Met1 for maintenance. Curr Biol. 2001;11:747–757. doi: 10.1016/s0960-9822(01)00226-3. [DOI] [PubMed] [Google Scholar]
- Sibley CR, Seow Y, Wood MJ. Novel RNA-based strategies for therapeutic gene silencing. Mol Ther. 2010;18:466–476. doi: 10.1038/mt.2009.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts TC, Andaloussi SE, Morris KV, McClorey G, Wood MJ. Small RNA-Mediated Epigenetic Myostatin Silencing. Mol Ther Nucleic Acids. 2012;1:e23. doi: 10.1038/mtna.2012.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller V, Larder BA. Mutational patterns in the HIV genome and cross-resistance following nucleoside and nucleotide analogue drug exposure. Antivir Ther (Lond) 2001;6 suppl. 3:25–44. [PubMed] [Google Scholar]
- Klenerman P, Wu Y, Phillips R. HIV: current opinion in escapology. Curr Opin Microbiol. 2002;5:408–413. doi: 10.1016/s1369-5274(02)00339-9. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Juelich T, Lim H, Ishida T, Watanebe T, Cooper DA, et al. Closed chromatin architecture is induced by an RNA duplex targeting the HIV-1 promoter region. J Biol Chem. 2008;283:23353–23363. doi: 10.1074/jbc.M709651200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg MS, Barichievy S, Schaffer L, Han J, Morris KV. An RNA targeted to the HIV-1 LTR promoter modulates indiscriminate off-target gene activation. Nucleic Acids Res. 2007;35:7303–7312. doi: 10.1093/nar/gkm847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson AL, Bartz SR, Schelter J, Kobayashi SV, Burchard J, Mao M, et al. Expression profiling reveals off-target gene regulation by RNAi. Nat Biotechnol. 2003;21:635–637. doi: 10.1038/nbt831. [DOI] [PubMed] [Google Scholar]
- Hornung V, Guenthner-Biller M, Bourquin C, Ablasser A, Schlee M, Uematsu S, et al. Sequence-specific potent induction of IFN-alpha by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat Med. 2005;11:263–270. doi: 10.1038/nm1191. [DOI] [PubMed] [Google Scholar]
- Judge AD, Sood V, Shaw JR, Fang D, McClintock K, MacLachlan I. Sequence-dependent stimulation of the mammalian innate immune response by synthetic siRNA. Nat Biotechnol. 2005;23:457–462. doi: 10.1038/nbt1081. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Shijuuku T, Fukamachi T, Zaunders J, Guillemin G, Cooper D, et al. Prolonged transcriptional silencing and CpG methylation induced by siRNAs targeted to the HIV-1 promoter region. J RNAi Gene Silencing. 2005;1:66–78. [PMC free article] [PubMed] [Google Scholar]
- Lim HG, Suzuki K, Cooper DA, Kelleher AD. Promoter-targeted siRNAs induce gene silencing of simian immunodeficiency virus (SIV) infection in vitro. Mol Ther. 2008;16:565–570. doi: 10.1038/sj.mt.6300380. [DOI] [PubMed] [Google Scholar]
- Yamagishi M, Ishida T, Miyake A, Cooper DA, Kelleher AD, Suzuki K, et al. Retroviral delivery of promoter-targeted shRNA induces long-term silencing of HIV-1 transcription. Microbes Infect. 2009;11:500–508. doi: 10.1016/j.micinf.2009.02.003. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Ishida T, Yamagishi M, Ahlenstiel C, Swaminathan S, Marks K, et al. Transcriptional gene silencing of HIV-1 through promoter targeted RNA is highly specific. RNA Biol. 2011;8:1035–1046. doi: 10.4161/rna.8.6.16264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori S, Ide K, Nakata H, Harada H, Suzu S, Ashida N, et al. Potent activity of a nucleoside reverse transcriptase inhibitor, 4'-ethynyl-2-fluoro-2'-deoxyadenosine, against human immunodeficiency virus type 1 infection in a model using human peripheral blood mononuclear cell-transplanted NOD/SCID Janus kinase 3 knockout mice. Antimicrob Agents Chemother. 2009;53:3887–3893. doi: 10.1128/AAC.00270-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg MS, Villeneuve LM, Ehsani A, Amarzguioui M, Aagaard L, Chen ZX, et al. The antisense strand of small interfering RNAs directs histone methylation and transcriptional gene silencing in human cells. RNA. 2006;12:256–262. doi: 10.1261/rna.2235106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- Colin L, Van Lint C. Molecular control of HIV-1 postintegration latency: implications for the development of new therapeutic strategies. Retrovirology. 2009;6:111. doi: 10.1186/1742-4690-6-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folks TM, Justement J, Kinter A, Dinarello CA, Fauci AS. Cytokine-induced expression of HIV-1 in a chronically infected promonocyte cell line. Science. 1987;238:800–802. doi: 10.1126/science.3313729. [DOI] [PubMed] [Google Scholar]
- Della Chiara G, Crotti A, Liboi E, Giacca M, Poli G, Lusic M. Negative regulation of HIV-1 transcription by a heterodimeric NF-κB1/p50 and C-terminally truncated STAT5 complex. J Mol Biol. 2011;410:933–943. doi: 10.1016/j.jmb.2011.03.044. [DOI] [PubMed] [Google Scholar]
- Matalon S, Rasmussen TA, Dinarello CA. Histone deacetylase inhibitors for purging HIV-1 from the latent reservoir. Mol Med. 2011;17:466–472. doi: 10.2119/molmed.2011.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Neff CP, Liu X, Zhang J, Li H, Smith DD, et al. Systemic administration of combinatorial dsiRNAs via nanoparticles efficiently suppresses HIV-1 infection in humanized mice. Mol Ther. 2011;19:2228–2238. doi: 10.1038/mt.2011.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neff CP, Zhou J, Remling L, Kuruvilla J, Zhang J, Li H, et al. An aptamer-siRNA chimera suppresses HIV-1 viral loads and protects from helper CD4(+) T cell decline in humanized mice. Sci Transl Med. 2011;3:66ra6. doi: 10.1126/scitranslmed.3001581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ter Brake O, Legrand N, von Eije KJ, Centlivre M, Spits H, Weijer K, et al. Evaluation of safety and efficacy of RNAi against HIV-1 in the human immune system (Rag-2(-/-)gammac(-/-)) mouse model. Gene Ther. 2009;16:148–153. doi: 10.1038/gt.2008.124. [DOI] [PubMed] [Google Scholar]
- Shimizu S, Hong P, Arumugam B, Pokomo L, Boyer J, Koizumi N, et al. A highly efficient short hairpin RNA potently down-regulates CCR5 expression in systemic lymphoid organs in the hu-BLT mouse model. Blood. 2010;115:1534–1544. doi: 10.1182/blood-2009-04-215855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centlivre M, Legrand N, Klamer S, Liu YP, Eije KJ, Bohne M, et al. Preclinical In Vivo Evaluation of the Safety of a Multi-shRNA-based Gene Therapy Against HIV-1. Mol Ther Nucleic Acids. 2013;2:e120. doi: 10.1038/mtna.2013.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed EO, Martin MA. Fields Virology. Lippincott Williams & Wilkins: New York; 2001. HIVs and their replication. [Google Scholar]
- Jeang KT, Xiao H, Rich EA. Multifaceted activities of the HIV-1 transactivator of transcription, Tat. J Biol Chem. 1999;274:28837–28840. doi: 10.1074/jbc.274.41.28837. [DOI] [PubMed] [Google Scholar]
- Greene WC, Peterlin BM. Charting HIV's remarkable voyage through the cell: basic science as a passport to future therapy. Nat Med. 2002;8:673–680. doi: 10.1038/nm0702-673. [DOI] [PubMed] [Google Scholar]
- Kalebic T, Kinter A, Poli G, Anderson ME, Meister A, Fauci AS. Suppression of human immunodeficiency virus expression in chronically infected monocytic cells by glutathione, glutathione ester, and N-acetylcysteine. Proc Natl Acad Sci USA. 1991;88:986–990. doi: 10.1073/pnas.88.3.986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada S, Harada H, Ito T, Saito T, Suzu S. Early development of human hematopoietic and acquired immune systems in new born NOD/Scid/Jak3null mice intrahepatic engrafted with cord blood-derived CD34 + cells. Int J Hematol. 2008;88:476–482. doi: 10.1007/s12185-008-0215-z. [DOI] [PubMed] [Google Scholar]
- Fernandez G, Zeichner SL. Cell line-dependent variability in HIV activation employing DNMT inhibitors. Virol J. 2010;7:266. doi: 10.1186/1743-422X-7-266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wightman F, Ellenberg P, Churchill M, Lewin SR. HDAC inhibitors in HIV. Immunol Cell Biol. 2012;90:47–54. doi: 10.1038/icb.2011.95. [DOI] [PubMed] [Google Scholar]
- Miyoshi H, Takahashi M, Gage FH, Verma IM. Stable and efficient gene transfer into the retina using an HIV-based lentiviral vector. Proc Natl Acad Sci USA. 1997;94:10319–10323. doi: 10.1073/pnas.94.19.10319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Xia HQ, Cleghorn G, Gobe G, West M, Wei MQ. A highly efficient and consistent method for harvesting large volumes of high-titre lentiviral vectors. Gene Ther. 2001;8:1745–1751. doi: 10.1038/sj.gt.3301587. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Craddock BP, Okamoto N, Kano T, Steigbigel RT. Poly A-linked colorimetric microtiter plate assay for HIV reverse transcriptase. J Virol Methods. 1993;44:189–198. doi: 10.1016/0166-0934(93)90054-u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.