

Abstract
Several small molecules and ions, notably carbon monoxide, cyanide, cyanate, and hydrogen sulfide, are potent inhibitors of Ni-containing carbon monoxide dehydrogenases (Ni-CODH) that catalyze very rapid, efficient redox interconversions of CO2 and CO. Protein film electrochemistry, which probes the dependence of steady-state catalytic rate over a wide potential range, reveals how these inhibitors target particular oxidation levels of Ni-CODH relating to intermediates (Cox, Cred1, and Cred2) that have been established for the active site. The following properties are thus established: (1) CO suppresses CO2 reduction (CO is a product inhibitor), but its binding affinity decreases as the potential becomes more negative. (2) Cyanide totally inhibits CO oxidation, but its effect on CO2 reduction is limited to a narrow potential region (between −0.5 and −0.6 V), below which CO2 reduction activity is restored. (3) Cyanate is a strong inhibitor of CO2 reduction but inhibits CO oxidation only within a narrow potential range just above the CO2/CO thermodynamic potential—EPR spectra confirm that cyanate binds selectively to Cred2. (4) Hydrogen sulfide (H2S/HS−) inhibits CO oxidation but not CO2 reduction—the complex on/off characteristics are consistent with it binding at the same oxidation level as Cox and forming a modified version of this inactive state rather than reacting directly with Cred1. The results provide a new perspective on the properties of different catalytic intermediates of Ni-CODH—uniting and clarifying many previous investigations.
INTRODUCTION
In biology, the rapid and direct redox interconversion of CO and CO2 (1), a reaction of enormous technological importance,
| (1) |
is catalyzed by metalloenzymes called carbon monoxide dehydrogenases (CODHs).1,2 The enzymes found in anaerobic organisms contain Ni and are referred to as Ni-CODHs. Important examples are provided by the thermophilic bacterium Carboxydothermus hydrogenoformans (Ch), which can grow chemolithoautotrophically on CO as sole carbon and energy source2,3 and expresses five Ni-CODHs. In the Wood–Ljungdahl pathway for CO2 fixation used by methanogenic and acetogenic bacteria, a Ni-CODH is complexed with acetyl-CoA synthase (ACS), where it provides, internally, the CO necessary for generating acetyl-CoA.4 Using conventional kinetic assays, turnover frequencies as high as 30 000 s−1 at pH 8, 70 °C have been reported5 for CO oxidation by two of these enzymes, CODH ICh and CODH IICh, both of which are monofunctional (i.e., they catalyze only the CO/CO2 interconversion) and have similar spectroscopic properties.
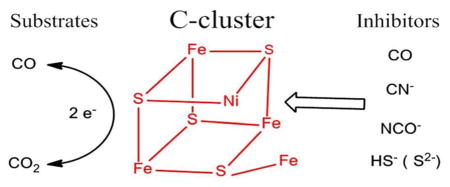
Structural features of Ni-CODHs of relevance to this paper are summarized in Figure 1. Crystal structures of the monofunctional CODHs from Rhodospirillum rubrum (Rr)6 and C. hydrogenoformans7 show that the enzymes are homodimeric. Each subunit contains a buried active site known as the C-cluster which features a [Ni3Fe-4S] cubane-like center with a fourth, “dangling” Fe-atom linked to the [3Fe-4S] fragment via one sulfido ligand. (“Dangling” emphasizes the part-external position of this Fe relative to the main cluster, and we use it in preference to the less-informative terms “ferrous center II” or “unique Fe”.) There has been controversy over whether a fifth sulfido ligand, bridging the dangling Fe and the Ni, is present in active enzyme.1 Electron transfers between the C-cluster and the protein surface are mediated by two [4Fe-4S] clusters: the B-cluster, which has a typical EPR signature of a [4Fe-4S]+ cluster(gav < 2), and the surface-exposed D-cluster that is coordinated equally by both subunits. It is thought that electrons are transferred from ferredoxin to the D-cluster and pass through the B-cluster to the C-cluster, which catalyzes the reduction of CO2 to CO.
Figure 1.

Different structures of the active sites of CODH IICh obtained by X-ray diffraction: (a) −600 mV with CO2, (b) −320 mV, (c) −320 mV with cyanide, and (d) CO-reduced CODH. Two positions are found for the dangling Fe-atom in the crystal structure, labeled Fe1a and Fe1b, respectively. The PDB codes are shown in each case.
This basic model for CO2/CO interconversion has emerged from crystallography, various kinetic and spectroscopic techniques, and theoretical calculations. A particularly significant result was reported by Jeoung and Dobbek,7 who, by incubating crystals of CODH IICh with NaHCO3 under reducing conditions (−0.6 V), revealed an “intermediate” state in which CO2 is bound through the C-atom to the Ni (at 1.96 Å) to complete the square-planar coordination geometry that is quite typical of Ni(II). One of the O-atoms of the bound CO2 is coordinated to the dangling Fe at a distance of 2.05 Å (Figure 1a). This result supported a working mechanistic hypothesis based on simple chemical principles—that the role of the dangling Fe is to abstract (as hydroxide) an O-atom from CO2, via a proton-coupled two-electron reaction, leaving CO bound to Ni: conversely, the reverse reaction involves nucleophilic attack by the Fe-bound hydroxide on the Ni-bound carbonyl C-atom.
The redox transformations have been studied by EPR-monitored titrations.8 The inactive oxidized state Cox is EPR-silent: as studied with the CODH/ACS complex from Moorella thermoacetica (Mt), Cox is reduced by one electron to the active state known as Cred1, which exhibits a characteristic EPR signal with gav ≈ 1.82 (g = 2.01, 1.81, 1.65). The reduction potential for this reaction is approximately −220 mV in CODH/ACSMt8 and −110 mV in CODHRr.9 In CODH/ACSMt, the Cred1 state rapidly reacts with CO (the rate constant at 5 °C is 400 s−1, equating to approximately 1.7 × 104 s−1 at the physiological growth temperature of 55 °C)10 to give CO2 (which is released) and Cred2, which exhibits an EPR signal with gav ≈ 1.86 (g = 1.97, 1.87, 1.75). The reduction potential for the two-electron interconversion between Cred1 and Cred2 in CODH/ACSMt is approximately −520 mV for CODHMt8 and −540 mV for the CODH/ACS from Methanosarcina thermophila11 at pH 7, which is very close to the formal reduction potential for the CO2/CO couple.12 The half-cycle regeneration of Cred1 or Cred2 by long-range electron transfers requires at least transient participation of an EPR-silent intermediate state Cint.13 The electron distribution in the C-cluster in these different oxidation levels is still unresolved. Based on the large 57Fe- and minimal 61Ni-hyperfine values, most of the unpaired spin density is clearly localized on Fe in both the Cred1 and Cred2 states.14 The similar EPR spectra for the C-cluster (gav < 2) further suggest that a [3Fe-4S]− fragment is present in both Cred1 and Cred2; therefore, the two-electron transition might involve Ni(II,0) or participation of a Ni(II) hydride,15 the latter possibility also accounting for the instability of Cint.
Several small molecules such as cyanide (CN−), cyanate (NCO−), and sulfide (introduced as H2S or HS−) are known to be inhibitors of CODH.16–21 Although reactions of these inhibitors with CODH isozymes have been investigated by kinetic methods, spectroscopy, and crystallography, it is still unclear how they selectively target discrete species in the catalytic cycle (e.g., Cox, Cred1, Cred2). Cyanide, an electronic and structural mimic of CO, is a slow-binding competitive inhibitor of CO oxidation,18,20,22,23 and this is consistent with the observation that its binding affects the EPR spectrum of Cred1 but not Cred2.22 It thus follows that NCO−, which structurally and electronically resembles CO2, should bind preferentially to Cred2. The EPR experiments carried out on the CODH/ACSMt complex from Moorella thermoacetica are not easily interpreted in this context, because they showed that NCO− binding causes major changes to the EPR spectrum (gav > 2) under conditions that favor Cred1.16 Less intuitive is any prediction of the state(s) that an exogenous sulfido species should target, and there remains some controversy. An additional S-atom is found in certain crystal structures of Ni-CODHs as a bridging ligand between the Ni-atom and the dangling Fe-atom,17,18 yet reported effects of exogenous sulfide vary between it being an inhibitor19 and an activator.17,18
Protein film electrochemistry (PFE) refers to a suite of techniques that address enzyme molecules directly attached to an electrode surface, allowing catalytic activity to be recorded (as current) as a continuous function of electrode potential and/or time.24 The rates and strengths of binding of an inhibitor are thus addressable as a precise function of potential during steady-state catalysis, and this allows the particular redox level of the enzyme that is being targeted to be identified. Importantly, information on enzyme activity is easily obtained at potentials well below those commonly accessed by chemical mediators and titrants, which is essential when studying an enzyme able to catalyze CO2 reduction, which requires potentials more negative than −0.5 V. The fact that the enzyme is immobilized also allows inhibitors to be added and then removed, a procedure that is impractical in solution kinetics. Many enzymes, including CODH, behave as reversible electrocatalysts and are inspirational for energy technologies such as solar fuel conversion.12,25–27
Here we describe the use of PFE to establish and analyze the strong potential dependence of the inhibition of enzymatic CO oxidation and CO2 reduction by cyanide, cyanate, and sulfide. The experiments focus mainly on one technique, cyclic voltammetry, which provides immediate qualitative information in both time and potential dimensions. The results, obtained for CODH ICh from Carboxydothermus hydrogenoformans, reveal very clearly how each inhibitor targets different states of the enzyme and show how rapidly they bind or leave the active site as the redox level is changed. Selective binding of cyanate to Cred2 is confirmed by EPR spectroscopy. The findings provide a wide, continuous potential landscape for structural and spectroscopic research carried out over three decades, and they explain and unify important aspects of this class of enzyme.
METHODS
Isolation and purification of CODH ICh from Carboxydothermus hydrogenoformans were carried out as previously described.28 All chemicals were of analytical or equivalent grade. The gases, carbon monoxide, carbon dioxide, and argon, were purchased from BOC. Potassium cyanide and potassium thiocyanate were obtained from Fluka, and sodium sulfide and potassium cyanate were purchased from Sigma-Aldrich. All electrochemical experiments were undertaken in a glovebox (Vacuum Atmospheres, O2 < 5 ppm). An all-glass electrochemical cell was used for all experiments. The cell was fitted with inlet and outlet (vented to flue) for gases, a port for injection of solutions, a Luggin side arm that housed the saturated calomel electrode (SCE) reference, a Pt counter electrode, and a housing into which the electrode rotator fitted snuggly and formed a gastight seal. The pyrolytic graphite “edge” (PGE) electrode (diameter 2 mm) was constructed as described previously.12 Before each electrochemical experiment, the PGE electrode was polished using 5 μm sandpaper and sonicated to remove debris. Approximately 2 μL of enzyme solution (16 mg/mL) was discharged from a pipet onto the PGE surface. To improve the stability of the enzyme film on the electrode, polymyxin (Duchefa Biochemie) was added to the coating solution in a 3:1 ratio over enzyme. Instability (we refer to this as film loss) was usually only a problem when conducting experiments over several hours at 25 °C.
All potentials are quoted versus the standard hydrogen electrode (SHE) using the scaling correction E(SHE) = E(SCE) + 241 mV at 25 °C. Voltammetry and chronoamperometry were performed with an Autolab PGSTAT10 or PGSTAT20 electrochemical analyzer. The rotation rate (ω) of the electrode was usually set at 3500 rpm, above which the current was always independent of ω. Unless otherwise stated, experiments were carried out using 0.2 M MES buffer (pH 7.0) as buffer-electrolyte at 25 °C, the high concentration of MES being necessary to minimize changes in pH due to reaction with CO2. Gas mixtures were introduced into the cell headspace without bubbling—the electrode rotation was sufficient to facilitate equilibrium between headspace and solution. The gas flow rate and the composition of gas entering the cell headspace were controlled by mass flow controllers (Sierra).
EPR spectra were recorded using a Bruker ESP300e (with EMS upgrade) spectrometer equipped with an ER 041 X microwave bridge, a B–E 25 magnet, an ER 083 C power source, a 4119HS cavity, and an Oxford Instruments cryostat. As-isolated samples of CODH ICh were buffer-exchanged into 50 mM Tris-HCl, pH 7.6, just before treatments with oxidants or reductants. The Cred1 state of the enzyme was generated by titrating the protein with benzyl viologen. The Cred2 state was generated by purging with CO for 5 min and then adding dithionite to a final concentration of 4 mM, or by simply treating with dithionite alone.
RESULTS
Carbon Monoxide as a Product Inhibitor
Cyclic voltammograms to probe product inhibition by CO during the reduction of 40% CO2 are displayed in Figure 2. The enzyme is adsorbed on the rough PGE surface in such a way that electronic contact is made between the electrode and the active site via the Fe–S clusters that extend to the protein surface. In a cyclic voltammogram the electrode potential is varied in a cyclic manner back and forth between two limits. The potential variation induces a flow of current that depends on the equilibrium potential set by the Nernst equation (i.e., the standard potential adjusted by the logarithmic activities protons and oxidized/reduced forms) and by the corresponding rates of catalytic electron transfer in each direction. Catalytic reduction of CO2 appears as a negative current that begins at electrode potentials more negative than −0.5 V, whereas oxidation appears as a positive current that begins above −0.5 V. The current is directly related to steady-state enzyme activity. Absolute activity is usually difficult to calculate because it requires knowledge of the electroactive coverage of the enzyme, which is usually too low to measure reliably, but the voltammetry gives excellent information on the relative rates of catalysis in either direction. As the potential is varied, inhibitors may become bound or released, thereby altering the current in particular regions of the voltammogram. The kinetics of inhibitor binding and release can be studied by carrying out experiments at different scan rates, or by introducing an inhibitor at a certain fixed potential and monitoring the current as a function of time. The reason for performing cyclic voltammetry as opposed to simply using single linear sweeps of potential is that processes that are not at steady state become visible; importantly, interconversions that are slow compared to the potential scan rate appear as hysteresis, in contrast to the strict overlay in either direction that would occur if all processes conform to steady-state kinetics.
Figure 2.

Inhibition of CODH ICh by CO. Different partial pressures of CO were introduced into the electrochemical cell to measure product inhibition. As the CO concentration increases, the electrocatalytic current for CO2 reduction decreases. The inset enlarges the region in which CO2 reduction occurs. Experimental conditions: 25 °C, 0.2 M MES buffer (pH 7.0), rotation rate 3500 rpm, scan rate 10 mV/s.
In Figure 2, cyclic voltammetry was conducted between limiting potentials of −760 and −160 mV, at different levels of CO, using Ar to maintain the total gas balance. As the CO concentration in the headspace is increased the current due to electrocatalytic CO2 reduction decreases. The attenuation of current is due to product inhibition: as expected for rapid on/off processes, the voltammograms conform to a steady-state condition throughout the cycle with no hysteresis. In addition to the decrease in CO2 reduction current as CO is added, the average potential at which the scans in either direction cross the zero current line becomes more negative. These two observations are connected in an important way because the decrease in zero-current potential—a thermodynamic effect—shows that the driving force has decreased, and this could alone explain the lower CO2 reduction activity. However, even after taking this factor into consideration by adjusting the horizontal position of the voltammogram, the rate of CO2 reduction is clearly retarded if CO is present.
Lineweaver–Burk plots using chronoamperometric data were used to obtain the apparent KI value for CO at two different electrode potentials, −560 and −760 mV, at pH 7 (an experiment at −760 mV is shown in Supporting Information, Figure S1). Data are shown in Table S1. A 7-fold higher inhibition constant is obtained at the more negative potential (KI = 337 vs 46 μM), showing clearly that CO has a much lower affinity for the more reduced state of the active site.
Cyanide—An Inhibitor of CO Oxidation
Cyanide is a well-studied inhibitor of CODH.18,20,23,28–30 According to EPR studies,18,20,21,31 an EPR signal with gav = 1.72 (g = 1.87, 1.78, 1.55) is observed when CN− is added to the Cred1 state of the CODH/ACSMt. Figure 3a, b shows catalytic cyclic voltammograms obtained before and after injection of KCN to a final concentration of 1 mM into the electrochemical cell under 100% CO2. The cycles were initiated at −0.74 V, and the scan rate was 1 mV/s—chosen to reveal hysteresis, reversibility, and changes in these features as the CN− concentration drops due to HCN evaporation. Panel (a) shows the effect of injecting CN− at the beginning of the second cycle (also at −0.74 V): the current due to CO2 reduction does not immediately drop to zero, but does so over the course of several seconds as the potential is scanned in the positive direction. Toward the end of the second cycle, the current increases sharply below −0.6 V, showing that the inhibitor is released rapidly as the enzyme adopts a more reduced steady-state level in the catalytic cycle. Upon commencing the third cycle, inactivation occurs at a slower rate, attributable to evaporation of HCN (pKa = 9.21) from the cell solution; accordingly, this hysteresis was especially evident from a separate experiment (Supporting Information, Figure S2) in which CN− was injected to give a final concentration of just 0.1 mM. Panel (b) shows a similar experiment except that CN− was injected during the latter part of the first cycle, as the potential passes down through −0.6 V. Especially evident now is the slow rate at which CN− inhibits the enzyme (over the 50 s taken to reach −0.65 V) before it dissociates as the potential continues on to −0.74 V. The second cycle continues as expected from panel (a). Further experiments confirmed that CO oxidation activity is totally abolished, regardless of the presence or absence of CO2 (Supporting Information, Figure S3).
Figure 3.

Cyclic voltammograms showing the potential and time dependence of inhibition of CODH ICh by cyanide. Experimental conditions: 25 °C, 0.2 M MES buffer (pH 7.0), scan rate 1 mV/s. (a) After a complete cycle, KCN was injected into the electrochemical cell at −0.74 V to give a final concentration of 1 mM. Subsequent scans reveal the release of CN− as the potential is taken below −0.6 V, and rebinding of CN− on the return oxidative scan. (b) Same as (a) except that CN− is injected (also to a final concentration of 1 mM) at −0.6 V, i.e., near the end of the first cycle while the potential becomes more negative. This experiment clearly shows CN− binding and then dissociating from the enzyme as the potential goes below −0.65 V. (c) Result of fitting the Nernst equation from the red trace of (a): a value of n = 1.3 is obtained.
To establish the stoichiometry and cooperativity of the redox transition between oxidized CN− binding and reduced CN-free states, we studied the cyclic voltammetry in the presence of CN− under conditions designed to minimize hysteresis, i.e., at very slow scan rate (0.4 mV/s) and a high concentration of CN− (10 mM). By plotting log{fractional inhibition} against electrode potential, we obtained an n value between 1.3 and 1.6 in several experiments, where n represents the degree of cooperativity for a reaction involving more than one electron. An example is given in Figure 3c. In the context of these experiments, “cooperativity” means the degree to which a one-electron redox transformation results in spontaneous transfer of the second electron: a fully cooperative reaction would show n = 2.0, meaning that the one-electron intermediate is highly unstable. The significance of an n value between 1.3 and 1.6 is that a one-electron intermediate (in this case Cint) is unstable. In unbiased terms this means that the release of CN− occurs predominantly from a state that is two electrons more reduced than the state to which it binds tightly. This estimation was not trivial. First, the large differences in the rates at which CN− inhibits and is released, and the strong potential dependence of these rates, make it difficult to attain a reversible situation; therefore, a very low scan rate was required. Second, the reactivation potential for CN−-inhibited CODH starts below ~ −620 mV, which limits the range over which a sigmoidal current trace can be defined because the slope due to proton reduction from water at the PGE electrode becomes significant at ~ −1 V. Third, CN− is largely present as HCN, which evaporates from the electrochemical cell at pH 7.0. This evaporation could be minimized by halting the gas flow (CO2) during the experiment.
Cyanate—An Inhibitor of CO2 Reduction and Marginal Inhibitor of CO Oxidation
As for cyanide, we studied cyanate inhibition by cyclic voltammetry, through which both CO2 reduction and CO oxidation are observed in the same experiment, making it possible to deconvolute the effect of inhibitors in both states. As evident from Figure 4a, the current due to CO2 reduction decreases to almost zero when potassium cyanate is injected to a final concentration of 6.67 mM at −0.76 V during the sweep to more positive potential. On the return cycle there is a narrow window, just above −0.6 V, where CO2 reduction is observed before full inhibition is re-established. The dissociation constant of NCO− was estimated to be about 1.9 mM for CO2 reduction at −760 mV (Supporting Information, Figure S4). Figure 4b shows the effect of NCO− on CO oxidation: there is a narrow region of potential where CO oxidation appears inhibited, but above −0.4 V the effect on CO oxidation is almost insignificant (the decrease in limiting current at high potential is attributable to slow film loss). The catalytic voltammogram recorded in the presence of 6.7 mM NCO− resembles that recorded for CO only, except that the sigmoidal wave is shifted by about 50 mV in the positive direction. The clear conclusion from these experiments is that cyanate is a potent inhibitor of CO2 reduction and inhibits CO oxidation only within a narrow window just above the thermodynamic potential for the CO2/CO couple.
Figure 4.

Inhibition of CODH ICh by cyanate. Potassium cyanate was injected (final concentration in the solution is 6.67 mM) into the electrochemical cell under a gas atmosphere of 100% CO2 (Figure 3a) and 50% CO, 50% CO2 (Figure 3b). Experimental conditions: 25 °C, 0.2 M MES buffer (pH 7.0), rotation rate 3500 rpm, scan rate 1 mV/s.
Binding of cyanate to CODH ICh was studied further by EPR spectroscopy, and relevant results are shown in Figure 5. The spectrum of a sample oxidized with benzyl viologen, which results in the state Cred1 characterized by g = 2.01, 1.89, and 1.73, is essentially unchanged in the presence of 1 mM potassium cyanate, and the shape is further unchanged even in the presence of 20 mM cyanate (Figure 5A–C). Using these EPR spectral parameters, i.e., 40 mW at 12 K, the “g = 1.94-type” signals of the B-cluster and the Cred2 signals are unsaturated and the Cred1 features are slightly saturated (Supporting Information, Figure S5, left panel); however, the morphology of the Cred1 spectrum is unchanged relative to a spectrum run at lower powers. These conditions are ideal for studying the benzyl viologen-oxidized sample because the slowly relaxing radical signal, which overlaps with the low-field absorption feature of Cred1, is strongly suppressed (Supporting Information, Figure S5, right panel). When the power is reduced to below 1 mW or the temperature is increased to above 25 K, the strongly saturated signal of the benzyl viologen radical begins to be observed. This narrow S-shaped radical signal becomes predominant at low powers and high temperatures (above 25 K); however, at these temperatures, the spectral features from the fast-relaxing C-cluster are very broad. The g-values for Cred1 are slightly different from those of the CODHs from Rhodospirillum rubrum (CODHRr) (2.03, 1.88, 1.71) and the CODH/ACS from Moorella thermoacetica (CODH/ACSMt) (2.01, 1.81, 1.65)8 and Methanosarcina thermophila (2.02, 1.88, 1.71).11 The CO-treated CODH ICh (Figure 5D) exhibits an EPR spectrum that reflects mainly reduction of the B-cluster to form the Bred state, having g = 2.04, 1.93, and 1.89, with the C-cluster largely persisting in the Cred1 state. It is surprising that CO treatment alone does not convert CODH ICh predominantly to the Cred2 state, as is observed for the CODH/ACSMt32 and CODHRr.33 However, addition of either 1 mM (Figure 5E) or 20 mM (not shown) cyanate results in almost quantitative conversion of Cred1 to the Cred2 state, as shown by the increase in EPR signals with g = 1.96 and 1.77 and loss of the signals from Cred1. Similarly, when cyanate was added to the dithionite-reduced form of CODH ICh, exhibiting the spectra due to Bred and a mixture of Cred1 and Cred2 states (Figure 5F), the intensity of the Cred2 signals increased (Figure 5G). These results demonstrate that cyanate selectively enhances the EPR spectrum of the Cred2 state of CODH ICh, confirming the PFE results.
Figure 5.

EPR spectra of CODH ICh (A) oxidized with benzyl viologen; (B) oxidized with benzyl viologen and in the presence of 1 mM potassium cyanate; (C) oxidized with benzyl viologen and in the presence of 20 mM potassium cyanate; (D) reduced with CO; (E) upon CO reduction, in the presence of 1 mM potassium cyanate; (F) reduced with dithionite; (G) reduced with dithionite and then in the presence of 20 mM potassium cyanate. EPR conditions: modulation amplitude 10 G for all spectra and (temperature, microwave power, frequency) (A) 12 K, 41.2 mW, 9.38 GHz; (B) 12 K, 41.18 mW, 9.38 GHz; (C) 12 K, 41.2 mW, 9.383 GHz; (D) 10 K, 41.22 mW, 9.382 GHz; (E) 12 K, 41.21 mW, 9.379 GHz; (F) 12 K, 41.21 mW, 9.38 GHz; (G) 11 K, 41.21 mW, 9.381 GHz.
Sulfide—An Inhibitor of CO Oxidation That Binds to an Oxidized Inactive State of CODH
Some crystal structures of CODHs have revealed a sulfide ion bridged between the Ni-atom and the dangling Fe-atom, and there has been some controversy as to whether this structural feature is physiologically relevant.7,17–19,34 Experiments to examine the inhibition of CODH by sulfide are shown in Figure 6. No inhibition of catalytic CO2 reduction was observed (Figure 6a), even when a relatively high concentration of buffered sodium sulfide (to a final value of 2 mM) was injected (the loss in current during a cycle is due to natural film instability). This result correlates strongly with the observation that no EPR signal changes are observed when sulfide is added to a sample in the Cred2 state.18,19 In contrast, sulfide inhibits CO oxidation in a complex manner, as shown in Figure 6b. Without addition of sulfide, cyclic voltammograms at low scan rates show slow inactivation as the potential is taken above 0 V, with a partial reactivation observed between −50 and −100 mV on the return scan. The reactivation potential corresponds to values expected for the Cox/Cred1 couple based on several studies of CODH from different species. When 1 mM sodium sulfide (final concentration) is injected at −400 mV (at which potential the CO electrocatalytic oxidation current is small due to the low driving force, but still very evident), there is no immediate drop in current or even the slightest change of shape of the cyclic voltammogram as the potential is increased over the next 0.3 V. This result is consistent with the report from Svetlitchnyi et al.,18 who concluded that sulfide does not inhibit CO oxidation by methyl viologen. Feng and Lindahl, by contrast, found that sulfide partially inhibited CO oxidation activity (limiting values being 30% and 60% for CODHMt and CODHRr, respectively) when using methyl viologen as electron acceptor.
Figure 6.

Potential control of CODH ICh inhibition by sulfide: 1 mM Na2S or 2 mM Na2S (final concentration) was injected at different potentials. The asterisk refers to the reactivation potential. Experimental conditions: 25 °C, 0.2 M MES buffer (pH 7.0), rotation rate 3500 rpm, scan rate 1 mV/s.
However, a very different and more detailed picture emerges when the potential scan is continued in a positive direction, as the current levels off and decreases rapidly above −50 mV. The enzyme remains inactive during much of the return scan to negative potential and then reactivates at potentials below −0.25 V in a sharp transition (which remains sharp when the scan rate is increased to 10 mV/s). Experiments in which the sulfide solution is injected at more positive potentials (Figure 6c, d) show that inhibition occurs immediately, and the degree of inhibition increases as the potential increases, being complete at potentials above 0 V. The potential at which CODH is rapidly inactivated in the presence of sulfide coincides closely with the potential (ca. −50 to −100 mV) at which the enzyme inactivates (albeit very slowly) in the absence of sulfide. Chronoamperometric experiments showed that the reaction with sulfide is much faster than the reaction with CN− at +0.16 V (Supporting Information, Figure S6). Our results therefore show that sulfide does not bind directly (or at least rapidly) to a catalytic state of CODHCh (allowing CO oxidation to occur unaffected over a wide potential range, up to approximately −0.1 V and well above that attainable with oxidized methyl viologen) but enters instead via a more oxidized state, which it stabilizes. Release of the inhibiting sulfide species does not occur until a more reducing potential is applied. This potential (ca. −0.25 V) is more negative than that observed for the normal activation process (−50 to −100 mV) assigned to the reductive one-electron transformation from Cox to Cred1. Continuing the scan down to −560 mV and then scanning back to the region of CO oxidation reveals that only a small amount of further recovery has occurred under highly reducing conditions (compare the currents at −400 mV). The general decrease in catalytic activity that is observed compared to experiments performed without sulfide suggests that sulfide also causes less reversible changes to the enzyme.
DISCUSSION
Several small molecules and ions, carbon monoxide, cyanide, cyanate, and hydrosulfide (sulfide), act as inhibitors of Ni-CODH in a redox-dependent manner, as summarized in Figure 7. In this scheme, red shading indicates the potential region over which the enzyme is inhibited, gray indicates no binding, and green indicates that binding leads to turnover. The dashed arrows indicate reactions that are slow compared to those indicated by full arrows. As a guide, the reduction potentials for CO2/CO as well as values expected on the basis of various EPR titrations for the Cox/Cred1/Cred2 interconversions are also indicated. The results of PFE studies suggest a model for how these inhibitors intercept the catalytic cycle at different stages, as shown in Scheme 1. We assume that all inhibitors target the active site: X-ray crystallography of the enzymes from R. rubrum,6 C. hydrogenoformans,7 and M. thermoacetica35,36 shows that the active site consists of a distorted [Ni–3Fe–4S] cubane coordinated to a unique dangling Fe site (also called ferrous component II (FCII)). Structures of the C. hydrogenoformans7 and M. thermoacetica37 enzymes demonstrate binding of the substrate water molecule to the dangling iron, consistent with spectroscopic studies indicating that water binds to Fe.38 The structure of the CO2-bound C. hydrogenoformans enzyme reveals the carbon bound to Ni and one of the O-atoms bound to the dangling iron.7
Figure 7.

Potential dependence of binding of inhibitors to CODH ICh. Red shading refers to the potential region over which the enzyme is inhibited, gray indicates no binding, and green indicates that binding leads to turnover. The dashed arrows indicate reactions that are slow compared to those indicated by full arrows. The reduction potentials indicated at the top of the chart are guiding estimates based on values reported in the literature for Cred1/Cred28,11 (close to the CO2/CO potential at pH 7) and our own observation from the voltammetry in the case of Cox/Cred1.
Scheme 1. Summary of the Interceptions of the Catalytic Cycle of CODH ICh by Small-Molecule Inhibitorsa.
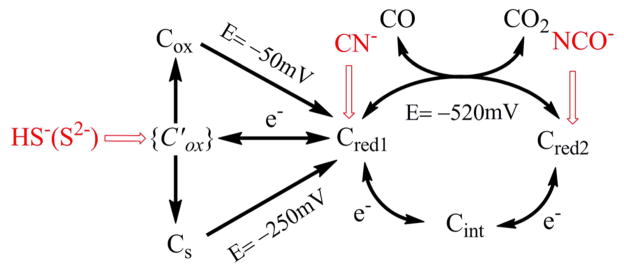
aThe potentials −250 and −50 mV are the values observed for reactivation of enzyme with and without sulfide. The potential −520 mV is the standard potential for the CO2/CO half-cell reaction at pH 7.0.
Our electrochemical results demonstrate that CO inhibits CO2 reduction by binding as a product inhibitor to the Cred1 state of CODH. Product inhibition is further quantified by chronoamperometry, which shows that KI for CO increases as the potential becomes more negative. This binding stabilizes the product complex as Cred1-CO, impeding return to the active Cred2 state for CO2 reduction. In a structure of the CODH from Methanosarcina barkeri (Mb), CO was proposed to bind to Ni with a bent conformation, adjacent to the water–Fe complex.39 Multiple infrared bands at 2078, 2044, 1970, 1959, and 1901 cm−1 were attributed to multiple CO molecules or heterogeneous modes of binding to the C-cluster.40
The electrochemical studies are entirely consistent with previous conclusions that CN− targets the Cred1 state,18,20,22,23 and they show further that it is released at very negative potential (the n-value shows that release occurs from a state that is two electrons more reduced, which must be Cred2), enabling CO2 reduction to occur. The results further highlight the unusually slow rate at which CN− binds, which results in severe hysteresis of the catalytic cyclic voltammetry. All Ni-containing CODHs are subject to potent inhibition by cyanide.18,20–23,41,42 Binding of cyanide results in conversion of the EPR spectrum of Cred1 to a similar spectrum with g = 1.87, 1.78, and 1.55.22 There is significant kinetic and structural evidence that CN− binds to the Ni center of the C-cluster, although some spectroscopic studies suggest that CN− binds to the dangling Fe.33,38 Support for a Ni-CN complex include early studies of the R. rubrum enzyme, which showed that the Ni-deficient enzyme does not bind CN−.20,42 Similarly, crystallographic studies of the C-cluster in the M. thermoacetica CODH/ACSMt indicate that CN− binds to Ni in a bent conformation reminiscent of the Ni-CO complex (above), again with an adjacent water remaining bound to the dangling Fe.37 A conserved isoleucine residue, which is present in both the M. barkeri and M. thermoacetica structures, appears to promote this bent configuration, sterically preventing formation of the expected linear Ni-CO and Ni-CN complexes. On the other hand, a linear Ni-CN complex has been observed in the crystal structure of cyanide-bound CODH IICh from C. hydrogenoformans.43 These two (linear and bent) modes of cyanide binding are consistent with steady-state kinetic studies which demonstrate that CN− acts as a slow inhibitor,20 thus suggesting that cyanide forms a rapid, reversible complex that undergoes a slow rearrangement to form a tight-binding complex.28,43
Based on our electrochemical studies, cyanate (NCO−), an analogue of CO2, binds at the Cred2 level and is released as the potential is raised to favor Cred1 at steady state. As with CN−, the binding of NCO− is slow, on the time scale of several seconds for millimolar concentrations. Cyanate may bind in a bridging mode similar to that of CO2, with carbon bound to Ni and one of the oxygens bound to Fe.7 The EPR results fully support the stabilization of Cred2 provided by cyanate binding (which results in an enhancement of the spectrum); equally, the lack of changes in spectral shape provide indirect evidence that the NCO− ligand binds to the Ni site (which harbors little spin density) within the NiFeS core instead of the [3Fe-4S]− core where most of the spin density is housed. Equally, compared to CN−, NCO− is a much weaker donor and less likely to exert an indirect electronic effect on the [3Fe-4S]− core.
The binding of sulfide to CODH has been a source of much controversy, and studies have indicated that sulfide can act as either an inhibitor19 or an activator.17,18 Initial structures of the C. hydrogenoformans CODH included a sulfide bridge between Ni and the dangling Fe in the C-cluster,17,18 which is not observed in the enzymes from R. rubrum and M. thermoacetica. Subsequent studies of the C. hydrogenoformans enzyme indicated that the sulfide is absent in catalytically competent enzyme species and that its replacement leads to enzyme activation. Our experiments now show that sulfide does not bind (at least rapidly) until a potential above −50 mV is applied, meaning that (at least) one electron must first be removed from the C-cluster. This potential is close to values reported for the Cox/Cred1 redox couple, which is associated with reactivation of oxidatively inactivated CODH, and corresponds also to the small reactivation peak always observed in the absence of sulfide when the electrode potential has been taken to a high value. However, the product formed in the presence of sulfide does not reactivate until a more reducing potential of −0.25 V is applied. Addition of sodium sulfide to the enzyme that had been held at a high potential for a long period (to produce a larger amount of Cox) still showed reactivation at −0.1 V, demonstrating that sulfide does not react, at least rapidly, with Cox itself. Therefore, sulfide (as HS−) rapidly intercepts a state that is also oxidized above Cred1 before the stable form(s) of Cox is established. The explanation for past confusion about sulfide ligation is therefore (at least for CODHCh) that two forms of Cox can coexist, one with an O-atom bridge and the other (Cs in Scheme 1) with a S-atom bridge, the latter state appearing more stable because it requires a more negative potential to release the sulfide ligand. Although we cannot rule out sulfide binding to a more reduced state, it is important to note that if the reactivation process instead corresponded to the reductive removal of sulfide from Cred1 (reports vary from it being a zero or partial inhibitor of CO oxidation by the weak oxidant, methyl viologen17–19), it would not be possible to account for the potential of reactivation that is so clearly observed. Sulfide binding that is specific to Cred1 must stabilize that state relative to Cred2 and thus lower the reduction potential for the Cred1/Cred2 couple; i.e., a much more negative value would be expected for reactivation, as is observed with CN−. The stark contrast to the behavior with respect to the Cred1 inhibitor CN−, observed so clearly using PFE, shows that sulfide acts instead by targeting and stabilizing a higher oxidation level than Cred1. In a previous study of CODH by PFE, we consistently noted two reactivation peaks at high potential,12 although the relative sizes of each varied from one sample to another. This observation could now be explained if that enzyme sample contained a certain fraction of the sulfide-inactivated oxidized form. Interestingly, the structure (Figure 1d) showing a bridging S-atom was prepared by reduction under CO, perhaps reflecting how difficult it is to control the oxidation states of centers during sample preparation for crystallography.
CONCLUSION
In summary, these PFE experiments represent the first investigations of reversible inhibitor binding to a CODH over a wide and continuous range of potential. The results unambiguously reveal the extent of inhibition and rates of binding or release of different inhibitors as the electrode potential is varied; the electrode potential in turn controls the prevailing redox state of the enzyme during steady-state catalysis. Cyanide, which is known to bind to Cred1, ceases to be an inhibitor of CO2 reduction at very negative potentials. Cyanate is established as a selective inhibitor of CO2 reduction with only a marginal effect on CO oxidation; it therefore binds selectively to Cred2, as expected for a molecule that is isoelectronic with CO2. Sulfide, which behaves entirely differently to CN− and does not inhibit CO oxidation under a modest driving force, is established to bind only under more oxidizing conditions. Sulfide thus stabilizes an inactive oxidized state that may be analogous to Cox but with a bound sulfide ligand, its binding and release being poorly reversible. The information thus obtained offers an invaluable guide for interpreting and understanding the complex and diverse observations made through spectroscopic and structural investigations.
Supplementary Material
Acknowledgments
This research was supported by grants from the Biological and Biotechnological Sciences Research Council, Grants BB/H003878-1 and BB/I022309-1 (to F.A.A.), and from the National Insitutes of Health, GM39451 (to S.W.R.).
Footnotes
Notes
The authors declare no competing financial interest.
Results showing CO product inhibition at −760 mV; effect of cyanide on CO2 reduction, using a lower concentration of cyanide; cyanide inhibition of the CO oxidation activity of CODHICh observed at two different potentials; inhibition of the CO2 reduction activity of CODHICh by cyanate; EPR spectra of CODHICh at different powers and temperatures; comparisons of the rates of inhibition by cyanide and sulfide; Km values for CO2 and inhibition constants for CO product inhibition at two different potentials. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Kung Y, Drennan CL. Curr Opin Chem Biol. 2011;15:276. doi: 10.1016/j.cbpa.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ragsdale SW. Crit Rev Biochem Mol Biol. 2004;39:165. doi: 10.1080/10409230490496577. [DOI] [PubMed] [Google Scholar]
- 3.Svetlichny VA, Sokolova TG, Gerhardt M, Ringpfeil M, Kostrikina NA, Zavarzin GA. System Appl Microbiol. 1991;14:254. doi: 10.1007/BF02539140. [DOI] [PubMed] [Google Scholar]
- 4.Ragsdale SW, Pierce E. Biochim Biophys Acta–Proteins Proteom. 2008;1784:1873. doi: 10.1016/j.bbapap.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Svetlitchnyi V, Peschel C, Acker G, Meyer O. J Bacteriol. 2001;183:5134. doi: 10.1128/JB.183.17.5134-5144.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Drennan CL, Heo JY, Sintchak MD, Schreiter E, Ludden PW. Proc Natl Acad Sci USA. 2001;98:11973. doi: 10.1073/pnas.211429998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jeoung JH, Dobbek H. Science. 2007;318:1461. doi: 10.1126/science.1148481. [DOI] [PubMed] [Google Scholar]
- 8.Lindahl PA, Munck E, Ragsdale SW. J Biol Chem. 1990;265:3873. [PubMed] [Google Scholar]
- 9.Spangler NJ, Lindahl PA, Bandarian V, Ludden PW. J Biol Chem. 1996;271:7973. doi: 10.1074/jbc.271.14.7973. [DOI] [PubMed] [Google Scholar]
- 10.Kumar M, Lu WP, Liu LF, Ragsdale SW. J Am Chem Soc. 1993;115:11646. [Google Scholar]
- 11.Lu WP, Jablonski PE, Rasche M, Ferry JG, Ragsdale SW. J Biol Chem. 1994;269:9736. [PubMed] [Google Scholar]
- 12.Parkin A, Seravalli J, Vincent KA, Ragsdale SW, Armstrong FA. J Am Chem Soc. 2007;129:10328. doi: 10.1021/ja073643o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fraser DM, Lindahl PA. Biochemistry. 1999;38:15706. doi: 10.1021/bi990398f. [DOI] [PubMed] [Google Scholar]
- 14.Lindahl PA, Ragsdale SW, Munck E. J Biol Chem. 1990;265:3880. [PubMed] [Google Scholar]
- 15.Amara P, Mouesca JM, Volbeda A, Fontecilla-Camps JC. Inorg Chem. 2011;50:1868. doi: 10.1021/ic102304m. [DOI] [PubMed] [Google Scholar]
- 16.Seravalli J, Kumar M, Lu WP, Ragsdale SW. Biochemistry. 1995;34:7879. doi: 10.1021/bi00024a012. [DOI] [PubMed] [Google Scholar]
- 17.Dobbek H, Svetlitchnyi V, Liss J, Meyer O. J Am Chem Soc. 2004;126:5382. doi: 10.1021/ja037776v. [DOI] [PubMed] [Google Scholar]
- 18.Ha SW, Korbas M, Klepsch M, Meyer-Klaucke W, Meyer O, Svetlitchnyi V. J Biol Chem. 2007;282:10639. doi: 10.1074/jbc.M610641200. [DOI] [PubMed] [Google Scholar]
- 19.Feng J, Lindahl PA. J Am Chem Soc. 2004;126:9094. doi: 10.1021/ja048811g. [DOI] [PubMed] [Google Scholar]
- 20.Ensign SA, Hyman MR, Ludden PW. Biochemistry. 1989;28:4973. doi: 10.1021/bi00438a011. [DOI] [PubMed] [Google Scholar]
- 21.Anderson ME, Lindahl PA. Biochemistry. 1994;33:8702. doi: 10.1021/bi00195a011. [DOI] [PubMed] [Google Scholar]
- 22.Anderson ME, Derose VJ, Hoffman BM, Lindahl PA. J Am Chem Soc. 1993;115:12204. [Google Scholar]
- 23.Ragsdale SW, Clark JE, Ljungdahl LG, Lundie LL, Drake HL. J Biol Chem. 1983;258:2364. [PubMed] [Google Scholar]
- 24.Vincent KA, Armstrong FA. Inorg Chem. 2005;44:798. doi: 10.1021/ic048519+. [DOI] [PubMed] [Google Scholar]
- 25.Armstrong FA, Hirst J. Proc Natl Acad Sci USA. 2011;108:14049. doi: 10.1073/pnas.1103697108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Woolerton TW, Sheard S, Pierce E, Ragsdale SW, Armstrong FA. Energy Environ Sci. 2011;4:2393. [Google Scholar]
- 27.Woolerton TW, Sheard S, Chaudhary YS, Armstrong FA. Energy Environ Sci. 2012;5:7470. [Google Scholar]
- 28.Seravalli J, Ragsdale SW. Biochemistry. 2008;47:6770. doi: 10.1021/bi8004522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Diekert GB, Thauer RK. J Bacteriol. 1978;136:597. doi: 10.1128/jb.136.2.597-606.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Drake HL, Hu SI, Wood HG. J Biol Chem. 1980;255:7174. [PubMed] [Google Scholar]
- 31.Lindahl PA. Angew Chem, Int Ed. 2008;47:4054. doi: 10.1002/anie.200800223. [DOI] [PubMed] [Google Scholar]
- 32.Anderson ME, Lindahl PA. Biochemistry. 1996;35:8371. doi: 10.1021/bi952902w. [DOI] [PubMed] [Google Scholar]
- 33.Hu ZG, Spangler NJ, Anderson ME, Xia JQ, Ludden PW, Lindahl PA, Munch E. J Am Chem Soc. 1996;118:830. [Google Scholar]
- 34.Dobbek H, Svetlitchnyi V, Gremer L, Huber R, Meyer O. Science. 2001;293:1281. doi: 10.1126/science.1061500. [DOI] [PubMed] [Google Scholar]
- 35.Doukov TI, Iverson TM, Seravalli J, Ragsdale SW, Drennan CL. Science. 2002;298:567. doi: 10.1126/science.1075843. [DOI] [PubMed] [Google Scholar]
- 36.Darnault C, Volbeda A, Kim EJ, Legrand P, Vernede X, Lindahl PA, Fontecilla-Camps JC. Nat Struct Biol. 2003;10:271. doi: 10.1038/nsb912. [DOI] [PubMed] [Google Scholar]
- 37.Kung Y, Doukov TI, Seravalli J, Ragsdale SW, Drennan CL. Biochemistry. 2009;48:7432. doi: 10.1021/bi900574h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.DeRose VJ, Telser J, Anderson ME, Lindahl PA, Hoffman BM. J Am Chem Soc. 1998;120:8767. [Google Scholar]
- 39.Gong W, Hao B, Wei Z, Ferguson DJ, Tallant T, Krzycki JA, Chan MK. Proc Natl Acad Sci USA. 2008;105:9558. doi: 10.1073/pnas.0800415105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen JY, Huang S, Seravalli J, Gutzman H, Swartz DJ, Ragsdale SW, Bagley KA. Biochemistry. 2003;42:14822. doi: 10.1021/bi0349470. [DOI] [PubMed] [Google Scholar]
- 41.Grahame DA, Stadtman TC. J Biol Chem. 1987;262:3706. [PubMed] [Google Scholar]
- 42.Ensign SA, Bonam D, Ludden PW. Biochemistry. 1989;28:4968. doi: 10.1021/bi00438a010. [DOI] [PubMed] [Google Scholar]
- 43.Jeoung JH, Dobbek H. J Am Chem Soc. 2009;131:9922. doi: 10.1021/ja9046476. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.