

Abstract
Tuberculosis remains a global health emergency that calls for treatment regimens directed at new targets. Here we explored lipoamide dehydrogenase, a metabolic and detoxifying enzyme in Mycobacterium tuberculosis whose deletion drastically impairs Mtb's ability to establish infection in the mouse. Upon screening over 1,600,000 compounds, we identified N-methylpyridine 3-sulfonamides as potent and species-selective inhibitors of Mtb Lpd, affording over 1,000-fold selectivity versus the human homolog. The sulfonamides demonstrated low nanomolar affinity and bound at the lipoamide channel in an Lpd-inhibitor co-crystal. Their selectivity could be attributed, at least partially, to hydrogen bonding of the sulfonamide amide oxygen with the species-variant Arg93 in the lipoamide channel. Although potent and selective, the sulfonamides did not enter mycobacteria, as determined by their inability to accumulate in Mtb to effective levels and produce changes in intracellular metabolites. This work demonstrates that high potency and selectivity can be achieved at the lipoamide binding site of Mtb Lpd, a site different from the NAD+/NADH pocket targeted by previously reported species-selective triazaspirodimethoxybenzoyl inhibitors.
Tuberculosis (TB1) remains a global epidemic despite being preventable and treatable. In 2011 an estimated toll of 1.4 million people put TB second in line only to HIV/AIDS as the greatest killer due to a single infectious agent (1). New treatment regimens with novel targets are needed to outpace the emergence of drug resistance and deadly associations of TB with HIV and diabetes. One strategy is to target functions that the causative agent, Mycobacterium tuberculosis, needs in order to establish infection and survive host-imposed stresses. There are a few known Mtb genes that are nonessential in standard growth conditions in vitro but whose loss of function drastically impairs Mtb's ability to survive in the host. Most encode enzymes of central carbon metabolism or detoxification. Among these are lipoamide dehydrogenase, a component of PDH, BCKDH and peroxynitrite reductase (2–4) in Mtb; phosphoenolpyruvate carboxykinase, PckA, involved in the generation of phosphoenolpyruvate from oxaloacetate during gluconeogenesis (5); a thiol peroxidase, Tpx (6); and a probable nitroreductase, Acg (7), one of the most highly upregulated genes of the dos regulon. However, no TB drugs are known to target these enzymes.
Lpd is the E3 component of up to four eukaryotic and prokaryotic central metabolic complexes: PDH, KDH, BCKDH and the GCS. In Mtb Lpd serves its classic metabolic function in PDH and BCKDH. However, no KDH or GCS activities have been reported in mycobacteria. Mycobacterial Lpd is uniquely involved in detoxification of reactive oxygen and nitrogen intermediates by serving as a component of NADH-dependent peroxynitrite reductase, along with DlaT, the E2 component of PDH; a thioredoxin-like protein, AhpD; and a peroxiredoxin, AhpC (3). Mtb lacking Lpd fails to grow on carbohydrates as a sole carbon source in vitro, is highly susceptible to RNI and rapidly dies in mice (2).
The strict reliance of Mtb's virulence and survival on Lpd in vivo drew our attention to Lpd as a target for inhibitors. Bacterial enzymes having human homologs are usually viewed as unattractive targets due to possible host toxicity of their inhibition. Mycobacterial Lpd is 36% identical to the human homolog. The three-dimensional structures of the mycobacterial and human enzyme align closely (8). However, differences in the substrate binding sites allow triazaspirodimethoxybenzoyl compounds to act as potent and species-selective mycobacterial Lpd inhibitors (9). A triazaspirodimethoxybenzoyl inhibitor that was co-crystallized in Mtb Lpd's pocket adjacent to the NAD+/NADH binding site overlapped with the nicotinamide ring binding site, blocking its coordination with the FAD flavin ring, where it would prevent electron transfer between the cofactors. The compound produced no detectable inhibition of human homolog, affording at least 100-fold selectivity between the species. However, it had no growth inhibitory effect on whole mycobacteria. This was interpreted as probable lack of accumulation in the bacterial cells (9).
To continue the search for mycobacterial Lpd inhibitors, we screened a collection of over 1.6 million compounds at the Genomics Research Center at Academia Sinica, Taiwan. Here we report identification and characterization of a new class of sulfonamides as potent, species-selective inhibitors of Mtb's Lpd. A co-crystal revealed that, in contrast to the triazaspirodimethoxybenzoyls, 2-(2-amino-5-bromo-N-methylpyridine-3-sulfonamido)-N-(4-methoxyphenyl)acetamide binds at the lipoamide channel of Mtb's Lpd, exploiting a second route to species-selective inhibition.
EXPERIMENTAL PROCEDURES
Screening library
The compound library at the Genomics Research Center (GRC) of Academia Sinica contained over 1.6 million mostly drug-like structures (the GRC-2M library), consisting of diversified synthetic compounds (diversity ~86%), known drugs, named chemicals with a range of biological activities, purified plant and microbial products, and fractionated natural products. The compounds were stored in 384-well and 1536-well formats at −50 °C. For screening, one copy of the library in 100% DMSO was dispensed to 1536-well microtiter plates at a concentration of 1 mM in 50 nL using an online robotic system.
High-throughput screening of Lpd
We used a modified DTNB reductase assay (9). Lpd (100 nM) in 100 mM sodium phosphate buffer, pH 7.0 was dispensed at 4 μl/well into 1536-well clear bottom assay plates followed by GRC-2M library compounds. After 1 hr at RT, 4 μL/well of reaction mixture containing the substrate lipoamide (2 mM), NADH (200 μM), and EDTA (4 mM) was added, resulting in concentrations of 50 nM Lpd, 1 mM lipoamide, and 6.25 μM library compounds. After 5 min, the detection reagent DTNB, prepared as a 375 μM stock in 100 mM sodium phosphate buffer, pH 7.0, was added at 2 μl/well, resulting in a final concentration of 75 μM. TNB production was recorded by absorbance at 405 nm. Z' values for the screen were > 0.51. Signal normalization using the median of each plate was applied during data analysis. The histogram of the accumulating number against activity signal was plotted to decide the cutoff.
Proteins
Recombinant Mtb proteins were expressed with (His-AceE) or without fusion tags (Lpd, DlaT) in E. coli and purified as published (3, 4, 10). Native bovine liver thioredoxin reductase was purified from calf liver as reported (11). Purified recombinant human Lpd was a generous gift from Prof. M. Patel, University at Buffalo, SUNY.
Enzyme assays
IC50s were determined with serial dilutions (100 to 0.1 μM) of inhibitors by a spectrophotometric assay with DTNB, lipoamide and NADH (9) or a PDH assay (4) as described. Human Lpd was assayed with 10 μM lipoamide by the same DTNB assay. Bovine thioredoxin reductase was assayed with 100 μM NADPH and 75 μM DTNB. NADH was detected by absorbance at 340 nm or fluorescence using a Molecular Devices SpactraMax M5 plate reader. Curves were fitted using IGOR Pro (WaveMetrics, Portland, OR) Version 4.06A Carbon with the Hill equation: Y=Ymax/(1+(IC50/[I])n, where n is the Hill coefficient. Kinetic parameters were determined by NADH-lipoamide fluorimetric assay at variable substrate (0.2 to 5 mM lipoamide; 3.33 to 100 μM NADH) and increasing inhibitor (0, 0.01, 0.03, 0.1, 0.3, 0.6, 0.9 μM) concentrations in the presence of 20 μM NAD+ to relieve inhibition by NADH and obtain hyperbolic plots (10). Km and Vmax values were determined by fitting the data to a hyperbolic function V=Vmax*X/(Km+X). Ki values were determined from nonlinear regression analysis in Prism by fitting data to equation for noncompetitive inhibition V= Vmax /(1+I/Ki)*X/(Km+X) (NADH substrate) or competitive inhibition V= Vmax *X/(Km*(1+I/ Ki)+X) (lipoamide substrate).
MIC assay
MIC values on mycobacteria were determined in 96 well plates in 200 μl of Middlebrook 7H9 medium pH 6.6 with 0.2% glycerol, 0.05% Tween 80 and 10% ADN (5% BSA, 2% dextrose, 0.85% NaCl). Starting bacterial inoculum was 0.1 or 0.01 (OD580). Inhibitors were tested at 2-fold serial dilutions from 100 μM to 0.1 μM. MICs were defined as compound concentrations that inhibited bacterial growth >90%.
Crystallization and structure determination
Mtb Lpd expression and purification for crystallographic analysis were performed as reported (8, 9). Lpd (200 μM), inhibitor SL827 (300 μM) and 600 μM NADH were mixed and incubated on ice for 10 min prior to crystallization at 18°C by hanging drop vapor diffusion against a well solution containing 100 mM Tris pH 8.5, 50 mM NaCl, 19% PEG 10000 (w/v). Crystals were cryo-protected by addition of 25% glycerol and flash-cooled in liquid nitrogen. A single crystal was diffracted and data collected at the NE-CAT beamline 24-IDC at 0.979 Å to obtain a data set at 2.6 Å resolution. The structure of Lpd bound to SL827 was determined by molecular replacement using programs contained within CCP4 (12) and our previously determined structure of Lpd (8). The crystal lattice included two dimers in the asymmetric unit and a model comprising Lpd amino acids 1–464 and an additional N-terminal Ser residue that remained from the cleavage tag (Gly-Ser). This model was refined using Phenix (13) after manual rebuilding in O (14) and Coot (15). 459 water molecules and SL827 were added to the model and refined to an R/Rfree of 0.169/0.222 at 2.4 Å (Table 1). The model has excellent geometry with 91.7% and 8.3% of residues in favored and allowed regions of Ramachandran space as calculated by Procheck (16) with MolProbity scores for all atom contacts and protein geometry in the 100th and 99th percentile, respectively (17). Atomic coordinates and structure factors have been deposited in the Protein Data Bank with the accession code 4M52.
Table 1.
Data collection and refinement statistics
| Lpd-Compound SL827 | |
|---|---|
| Data collection* | |
| Space group | P21 |
| Cell dimensions | |
| a, b, c (Å) | 82.52, 111.95, 98.03 |
| α, β, γ (°) | 90, 94.43, 90 |
| Resolution (Å) | 40–2.4 (2.44–2.40) |
| Total reflections | 202954 |
| Unique reflections | 67889 (3369) |
| Wilson B | 30.0 |
| R merge | 0.097 (0.475) |
| Rpim | 0.066 (0.318) |
| I / σI | 15.5 (3.6) |
| Completeness (%) | 96.1 (95.7) |
| Redundancy | 3.0 (3.0) |
| Refinement | |
| Resolution (Å) | 40–2.4 (2.43–2.4) |
| No. reflections | 66805/2600 |
| Rwork / Rfree | 16.9(20.7)/22.2(28.6) |
| No. atoms | |
| Protein | 13900 |
| FAD | 212 |
| SL827 | 100 |
| Water | 459 |
| Average B-factors | |
| Protein (protomer A/B/C/D) | 37.2/ 39.4/37.9/41.2 |
| FAD (protomer A/B/C/D) | 30.6/35.2/30.7/33.3 |
| SL827 (protomer A/B/C/D) | 36.0/44.7/40.1/41.1 |
| Water | 38.5 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.004 |
| Bond angles (°) | 0.880 |
| MolProbity score | |
| All atom contacts | 100th percentile |
| Protein Geometry | 99th percentile |
* One crystal. *Highest-resolution shell shown in parentheses.
Determination of sulfonamide intracellular accumulation by metabolic profiling
Wild-type H37Rv Mtb was grown in 7H9 Middlebrook medium supplemented with 0.2% glycerol, 0.2% dextrose, 0.5% BSA, 0.085% NaCl, and 0.05% Tween 80 to an OD580 of 1.0. One milliliter of culture was filtered through a hydrophillic PVDF membrane (0.22 μm, Millipore GVWP02500). Both blank (no Mtb) and Mtb-bearing filters were placed atop 7H10 agar plates (with 0.2% glycerol, 0.2% dextrose and 0.085% NaCl) minus detergent. Plates were incubated at 37°C in 5% CO2. After 5 days, filters were transferred to 7H10 agar plates containing either 75μM compound or an equivolume amount of 100% DMSO (vehicle control) for 24 hours. Filters were flipped into a 1 ml ice-cold 40:40:20 solution of methanol, acetonitrile, and water. Mtb was scraped off filters and lysed via bead-beating three times. Blank filters (on agar with and without compound) were processed as reported before (2, 18, 19). Small molecule extracts were mixed 1:1 with an acetonitirile and 0.2% formic acid solvent for MS analysis. To determine compound intracellular uptake, a standard curve was generated by spiking mycobacterial small molecule extract with known concentrations of compound to correct for ion suppressive effects of lysate. To assess metabolite abundance, protein was measured using a Pierce BCA kit, and ion counts of specified metabolites were normalized to protein concentration.
RESULTS
High-throughput screening for Lpd inhibitors
After screening 1,622,277 compounds from the GRC-2M library, we selected 11,458 (~0.7% hit rate) for further evaluation based on achieving ≥25% inhibition. These primary hits included some natural products and known drugs that were previously identified to inhibit Lpd (9). The primary hits were retested in an 8-point concentration-response assay (Z' = 0.74). Compounds with IC50 values below 5 μM were selected (283 compounds). A small fraction of the hits (5%) were quinones. Because Mtb's Lpd can reduce quinones more efficiently than lipoamide (10), these were considered false positives and were excluded from further studies. Structure similarity analysis showed that many of the remaining inhibitors clustered into 4 major scaffolds: pyrimido[5,4-e][1,2,4]-triazine-5,7-diones, pyrrolo[2,3-b]quinoxaline, 1H-quinolin-2-ol, and imidazol-4-yl-sulfonyl piperidine (including the N-methylpyridine 3-sulfonamides) (Fig. 1A).
FIGURE 1.

Identification of major scaffolds of Mtb Lpd inhibitors (A) and characterization of triazinediones (B) and quinoxaline (C) compounds as Lpd inhibitors. Pure recombinant mLpd (closed circles), hLpd (open circles) or mycobacterial PDH complex (open squares) were tested against the hit compounds (structures shown in the inset) from freshly prepared stocks or after incubation of compound stocks in DMSO at RT (closed squares) for 10 days. Compounds toxicity (closed triangles) was assessed against monkey kidney Vero cell line by MTS assay.
The most potent inhibitors were triazinediones. Six of these were resupplied from an independent source, and all 6 inhibited mLpd with IC50s in the range of 1–2 μM. However, they also inhibited hLpd with comparable potency (IC50s 3–5 μM) (Fig. 1B). When Lpd was tested for inhibition as a part of the PDH complex along with E1 (AceE) and E2 (DlaT) components, potency declined by 100- to 200-fold, suggesting that Lpd in the PDH complex may not be accessible to triazinediones. Likewise, the compounds lost their ability to inhibit mammalian Lpd when tested against the bovine PDH complex. Finally, all triazinediones tested were toxic to monkey kidney Vero cells as accessed by the MTS assay, with IC50s similar to those against isolated mLpd.
Next, we explored the quinoxaline scaffold. Although these produced very little toxicity when tested from the library, 4 structurally related quinoxalines from an independent supplier only achieved 50% inhibition of mycobacterial or human Lpd at or above 100 μM, and there was little gain in potency upon storage in DMSO at RT for 10 days (Fig. 1C). MS analysis of the compound stocks did not reveal any differences between the inactive and active quinoxalines, suggesting that structural rearrangements affected their function.
Finally, we reordered 4 sulfonamide inhibitors belonging to the scaffold, which was most highly overrepresented among the hits (36%). All 4 compounds potently inhibited mLpd in isolation with Ki's in low nanomolar range and with comparable or slightly less potency against Lpd in the PDH complex (Table 2). Strikingly, at the highest concentration tested (100 μM), they did not inhibit hLpd, thus affording at least 400- and in some cases over 2700-fold species selectivity (Fig. 2A and Table 2). Kinetic analysis revealed that the compounds were noncompetitive inhibitors with respect to the NADH substrate but competitive against the lipoamide substrate (Fig. 2B, C). This mode of inhibition differed from that of the triazaspirodimetoxybenzoyl Lpd inhibitor that bound at the NAD+/NADH site (9). This suggested that the sulfonamides bind at a different site than the triazaspirodimetoxybenzoyls. To investigate this, we sought to co-crystallize the sulfonamide inhibitor SL827 with Mtb Lpd.
Table 2.
Sulfonamide inhibitors of mycobacterial Lpd.
| ID | Structure | Ki (Lpd), nM | Ki (PDH), nM | SI* |
|---|---|---|---|---|
| SL932 |
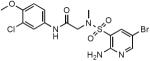
|
37±5 | 78±6 | >2700 |
| SL809 |
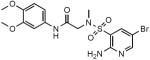
|
93±12 | 155±12 | >1075 |
| SL827 |

|
140±9 | 155±14 | >714 |
| SL917 |
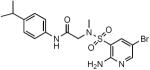
|
233±15 | 289±9 | >429 |
| SL418 |
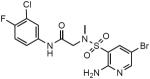
|
143±12 | 333±28 | 1300 |
Due to the solubility limit of the compounds the highest concentration tested was 100 μM. No inhibition of hLpd by SL932, SL809, SL827 and SL917 was observed at 100 μM and the SI values were calculated using 100 μM as a substitute Ki for hLpd. The resulting SI therefore reflects the lower limit of the SI value and in fact will be much greater, which is indicated by the > symbol.
FIGURE 2.

Characterization of sulfonamide Lpd inhibitors. (A) Sulfonamides SL932 (circles), SL809 (triangles), SL827 (squares) and SL917 (diamonds) potently inhibit pure recombinant mLpd (closed symbols) but not hLpd (open symbols). Sulfonamide SL827 is competitive with lipoamide (B) substrate and noncompetitive with NADH (C) substrate. DTNB assay used variable lipoamide (in (B), 0.1 to 5 mM) and NADH-lipoamide fluorimetric assay used variable NADH (in (C), 3.33 to 100 μM). Both assays were run with variable SL827 concentrations as shown. Data were fitted in Prism to a nonlinear regression function for competitive or noncompetitive inhibition. Double reciprocal Lineweaver-Burk plots are shown.
Structure of the Lpd-sulfonamide co-crystal
The Lpd protein component of the structure resembles that previously determined for Mtb Lpd in the absence of SL827 (8) and while 600 μM NADH was present in the crystallization and cryopreservation solutions no NADH was observed in the predicted NADH binding sites. SL827 occupies a deep pocket adjacent to each of the four active sites (Figure 3A), and each molecule of SL827 adopts a similar configuration up to the paramethoxyaniline ring that differs by a slight rotation due to differences in adjacent lattice contacts, presumably due to the alternative configuration of Phe99 (Figure 3B, C). The deep pocket occupied by SL827 is the presumed binding site for lipoamide and is proximal to the active site cysteine residues (Cys41 and Cys46), which adopt oxidized, disulfide-bonded configuration (Figure 3A).
FIGURE 3.

Complex between Mtb Lpd and SL827. (A) Stereo diagram of the Lpd dimer interface bound to SL827. SL827 is colored in CPK with carbon atoms in yellow. Protomers of Lpd colored in CPK, one with carbon in cyan and the other with carbon in salmon. Amino acid side chains are labeled and colored corresponding to carbon color-codes. Potential hydrogen bonds and van der Waals contacts indicated by black dashed lines with distances indicated (in A). The SL827 methoxy group points toward solvent and the bromine atom points toward the catalytic cysteine residues (Cys41 and Cys46). The FAD co-factor is labeled and located at the bottom of this view. (B) View of SL827 and select amino acids from the dimer interface illustrated in (A) with similar color-coding and selected amino acids labeled. (C) Alternative conformation of Phe99 and SL827 as observed in two of the four protomers in the crystal structure. (D) and (E) Nearly orthogonal views of SF827 as observed in (A) covered by electron density contoured at 1.2 σ (blue) and 6.0 σ (red) with the latter indicating positions of sulfur and bromine atoms, respectively. Select residues and SL827 are labeled. Electron density obtained from a simulated annealing omit map (2Fo-Fc) calculated using CNS (29). Figures generated using PyMol (30).
SL827 appears in an extended conformation that spans 17 Å from the bromine atom to the methoxy group. Interactions between SL827 and the two Lpd protomers bury approximately 825 Å2 of total surface area in each of the four binding sites. The bromine atom is less than 4 Å (Figure 3A) from side chain atoms of Pro13, His443 and Glu321 (Figure 3A) and 4.2 or 4.4 Å from side chain atoms of Leu42 and Cys41, respectively. The amino-pyridine ring is within van der Waals distance to side chain atoms from Glu321, Tyr16, Asn463, and His443. A direct hydrogen bond is observed between its amine nitrogen and the backbone carbonyl oxygen atom of Ala381 in addition to a water-mediated hydrogen bond between the amine nitrogen of the amino-pyridine ring and the side chain carboxylate of Glu448 and the backbone amide of Ala383. The sulfonamide group does not make any close contacts to the Lpd structure with the exception of a potential hydrogen bond interaction between the sulfonamide oxygen and the backbone amide of Ala383. Hydrogen bonds are observed between the amido oxygen of SL827 and the Arg93 guanidinium and the amido nitrogen of SL827 and the backbone carbonyl oxygen of Asn463 suggesting that this region of the SL827 structure is important for imparting interactions to specific groups within SL827 (see below). Although proximal to side chain atoms from Phe99, Leu100 and Phe464, the paramethoxyaniline ring is not well coordinated by the Lpd structure, perhaps consistent with the two conformations and weaker electron density observed for this segment of SL827 in the crystal structure (Figure 3B–D).
SL827 contacts with R93 are critical for its inhibitory activity
Solution of the Mtb Lpd crystal structure (8) revealed a prominent difference between the mammalian and mycobacterial Lpd structures in the architecture of the residues lining the lipoamide-binding channel. Arg93, which is conserved among mycobacterial Lpds but not eukaryotic Lpd family members, was observed in two different conformations in each monomer of the dimer, with the side chain pointing away from (`open' state) or into (`closed' state) the active site channel. Therefore we explored if contacts of SL827 with Arg93 contributed to its species selectivity by testing the ability of SL827 to inhibit the R93A mutant of mycobacterial Lpd. The Vmax of the R93A mutant is 40-fold lower than wild type when lipoamide is presented in solution, but the mutant enzyme retains the ability to accommodate lipoamide presented as attached to the E2 component of PDH. Thus, we measured inhibition by SL827 of Lpd in the PDH complex containing AceE (E1), DlaT (E2) and either WT Lpd or R93A Lpd. As predicted, SL827 was unable to efficiently inhibit R93A Lpd even at the highest concentration tested, 100 μM (Fig. 4). The Ki for SL827 increased >600-fold when R93A Lpd was substituted for WT Lpd in the PDH complex from 0.15 to 100 μM, suggesting that the contacts made between the SL827 and Arg93 are critical for correct positioning of the inhibitor in the lipoamide channel.
FIGURE 4.

SL827 inhibits Mtb PDH composed of AceE, DlaT and wt Lpd (closed circles) but not the complex composed of AceE, DlaT and R93A Lpd mutant (open circles).
Structure-activity studies of sulfonamide inhibitors
The GRC-2M library contained 299 structurally related heteroaryl sulfonamide compounds. Out of those, 127 imidazol-4-yl-sulfonyl piperidines and 14 N-methylpyridine-3-sulfonamides scored as primary hits. After the reconfirmation assay, 61 imidazol-4-yl-sulfonyl piperidines and 6 N-methylpyridine-3-sulfonamides produced IC50's below 5 μM. Imidazol-4-yl-sulfonyl piperidines were excluded from further studies because they did not inhibit PDH activity as potently as N-methylpyridine-3-sulfonamides, suggesting that they cannot compete efficiently with the lipoamide substrate when it is presented in its natural form on the DlaT protein. From the co-crystal structure, we inferred that the numerous bonds between the 2-amino-5-bromo pyridine ring of N-methylpyridine-3-sulfonamides and Lpd's side chains and the sulfonamide's perfect alignment within the lipoamide channel, allowing for hydrogen bonding of the amide oxygen with Arg93, accounts for the greater affinity of N-methylpyridine-3-sulfonamides towards Mtb Lpd and allows the compounds to efficiently compete with lipoamide in its natural milieu. In contrast, the greater length of imidazol-4-yl-sulfonyl piperidines may not allow for hydrogen bonding between the amide oxygen and Arg93 and the smaller imidazole ring may not maintain enough contacts necessary to compete with the lipoamide substrate. To further study the SAR of the N-methylpyridine-3-sulfonamides, we ordered 11 commercially available structurally related compounds. Substitutions at the meta- and para- positions on the aniline ring did not affect the potency of sulfonamides, consistent with the lack of interactions to coordinate the ring as observed in the crystal structure. Compounds with more than one substitution of the aniline ring had consistently lower IC50s, suggesting that additional side chains may help stabilize and coordinate the ring (Table 2 and Supplementary Table 1). In contrast, perturbation of the Asn463 backbone carbonyl H-bonding with the NH group of -CONH- by N-alkylation led to a 100-fold loss of potency (E114-0951 and E144-0828 in Supplementary Table 1). In addition, increasing the linker length by one carbon led to even greater loss of potency (K906-3123 in Supplementary Table 1), presumably due to the loss of hydrogen bonding with Arg93 via the amide oxygen. Thus, the amide's hydrogen bonding and its alignment within the lipoamide channel are crucial for the activity of the N-methylpyridine-3-sulfonamides.
To further expand the SAR, we tested 380 related compounds from the collection of Eli Lilly and Co through the auspices of the Lilly TB Drug Discovery Initiative. The most potent compound, SL418 (Table 2) (Ki, 143 nM) was closely related to SL827, differing only in the two side chain substitutions on the aniline ring in place of the methoxy of SL827 (Table 2). Like SL827, SL418 did not inhibit human Lpd and produced a species selectivity index of 1300.
Sulfonamide uptake by and action within intact mycobacteria
The most active sulfonamide inhibitors, SL827, SL932, SL809, SL917 and SL418, were tested for their effect on intact mycobacteria using a conventional MIC assay with the highest compound concentration at 100 μM. No growth inhibition of M. bovis BCG or M. tuberculosis ΔpanCDΔlysA was observed after 7 days in culture in the presence of inhibitors except at 100 μM. All compounds potently inhibited Lpd activity in mycobacterial lysates as measured by the PDH assay. Nor did we see any growth inhibition of M. bovis BCG when we tested the less potent sulfonamides listed in Supplementary Table. The inability of sulfonamides to phenocopy the growth inhibitory effect of lpd deletion (2) suggested that the compounds did not accumulate within mycobacteria to effective levels.
To test this, we used a metabolomic profiling platform (18–20) to determine compound uptake and its effect on Mtb's intracellular metabolites. M. tuberculosis H37Rv was grown on filters as reported (18, 19). Filters with or without Mtb were transferred to agar plates containing 75 μM SL827 for 24 hours and harvested in aqueous acetonitrile and methanol for LC-MS analysis. No Mtb-associated SL827 was observed on filters with mycobacteria compared to empty filter controls (Fig. 5), suggesting either that SL827 did not enter, was pumped out, or was rapidly converted to another product by adduction or metabolism.
FIGURE 5.

SL827 does not accumulate in Mtb. SL827 accumulation (A) and levels of intracellular metabolites (B) were determined in small molecule extracts from Mtb treated with vehicle control (black) or SL827 (white).
To test whether SL827 was effectively inhibiting Lpd, despite being inapparent in the lysate, we analyzed its impact on Mtb metabolites. Deletion of lpd in Mtb results in the accumulation of reaction substrates (pyruvate, α-ketoisovalerate, α-ketoisocaproate, α-ketomethylvalerate) of Lpd as well as their derivatives and upstream metabolites (alanine, valine, leucine and isoleucine). Pyruvate and branched chain ketoacids accumulate to levels 100- to 1000-fold above wild type (2). However, exposure of Mtb to 75 μM SL827 (Fig. 5B) produced no changes in alanine, valine and leucine levels, confirming that extracellular application of the compound did not extensively inhibit intracellular Lpd.
Absence of detectable intracellular SL827 within Mtb and lack of evidence for its impact suggested that the compound either did not penetrate the cells or was efficiently pumped out, as described for several chemical scaffolds (21, 22). Accordingly, we tested Mtb's sulfonamide susceptibility in the presence of efflux pump inhibitors verapamil and reserpine at concentrations (50 and 25 μM respectively) that produced no growth inhibitory effect on Mtb on their own. All Lpd-inhibitory sulfonamides tested on Mtb ΔpanCDΔlysA produced no growth inhibition except at the highest concentration tested, 100 μM, and the results were the same in the presence or absence of verapamil and reserpine.
DISCUSSION
Phenotypic screens are usually preferred in antibiotic discovery since they weed out compounds that fail to accumulate in the pathogen. However, phenotypic screens present their own challenges, among them that the in vitro screening conditions may differ critically from the conditions in the host (23), and it may be difficult or impossible to identify the inhibitors' molecular targets. Target-based screening is a reasonable complementary approach for molecular targets of particular interest, even if the inhibitors only provide a starting point.
Lpd is one of the few enzymes identified in Mtb that are readily susceptible to inhibition with small chemical compounds and in whose absence the pathogen becomes avirulent in the mouse. We have previously shown that it is possible to inhibit mycobacterial Lpd without inhibiting human Lpd by exploiting amino acid differences in the NAD+/NADH binding site (8, 9). Moreover, evidence is mounting that effective anti-infectives often have more than one target, as recently demonstrated for the established tuberculosis drug para-aminosalicylic acid (20) and the experimental tuberculosis pro-drug PA824, a nitroimidazo-oxazine with multiple mechanisms of action, including inhibition of cell wall biosynthesis and respiration (24) and donation of an NO-like species (25) that is likely to have multiple targets of its own (26). Lpd offers the opportunity to inhibit a single molecular target that participates in three known enzymatic complexes in Mtb that have roles in metabolism and detoxification (2, 3).
Herein we have identified a second site in mycobacterial Lpd at which a potent inhibitor binds with more than 1000-fold species selectivity. Selectivity could be attributed, at least partially, to the interaction of the sulfonamide backbone/side chains with Arg93 within the lipoamide channel. The R93A Mtb Lpd mutant protein was inhibited by the sulfonamide much less efficiently than the wild type, suggesting that those contacts are necessary to position the compound within the channel to achieve inhibition. Human Lpd encodes Leu at the same position.
However, Mtb appeared to be impermeable to the sulfonamide Lpd inhibitors. Not only did exposure of intact mycobacteria to inhibitor produce no change in Lpd substrate levels or bacterial growth rate, but we could not detect the inhibitor itself within the cells. This was unexpected, since sulfonamides were among the first drugs used to treat tuberculosis. Sulfamethoxazole and sulfanilamide, sulfonamides with little or no growth inhibitory effect on Mtb, had been assumed not to accumulate within the bacteria. However, it was recently demonstrated that these compounds do enter Mtb readily, but undergo chemical modification to inactive forms (20). Even so, the index sulfonamides were detectable, in contrast to the Lpd inhibitor studied here. However, we cannot exclude that compound SL827 may have been taken up and inactivated so rapidly that none was detected.
Domagk's discovery of the azodye Prontosil pioneered the studies on sulfonamides almost a century ago. Prontosil proved to be a prodrug that was metabolized to an active sulfonamide. Later studies suggested that sulfonamides enter bacteria via passive diffusion in a manner governed in part by the pKa of the compound and the extracellular-intracellular pH gradient (27, 28). All sulfonamide antibiotics currently in use, such as sulfisoxazole, sulfadiazine, sulfacetamide, sulfamethoxazole and sulfadoxine, have pKa values ranging from 5.0 to 6.5. In contrast, the most basic of the predicted pKa values of the sulfonamide compounds reported here range from 11.77 to 13.00, but due to their amphoteric nature, they are predicted to exist mostly as non-charged molecules at physiological pH and thus would be expected to enter by passive diffusion. Therefore, it remains unclear why we did not detect SL827 inside Mtb. It is possible that other physicochemical properties of the Lpd-inhibitory sulfonamides affect their uptake, such as polar surface area and lipophilicity. Hopefully a more extensive SAR analysis will identify analogs that preserve nanomolar potency and acquire the ability to accumulate within Mtb.
Supplementary Material
ACKNOWLEDGMENT
We thank Yan Ling for recombinant protein purification, David Warren for chemical synthesis, Madhumitha Nandakumar and Sumit Chakraborty for help with metabolomic profiling and discussion of results, Kristin Burns-Huang for helpful discussions and Prof. M. Patel (University at Buffalo, SUNY) for the generous gift of pure recombinant human Lpd. We thank the MSKCC X-ray core for use of their facility. C.D.L. is an investigator of the Howard Hughes Medical Institute.
Footnotes
This work was supported by NIH grant AI064768, the Milstein Program in Chemical Biology of Infectious Disease and the Howard Hughes Medical Institute. The collaboration was fostered by the Lilly TB Drug Discovery Initiative. The Department of Microbiology and Immunology is supported by the William Randolph Hearst Foundation.
The atomic coordinates and structure factors have been deposited in the Protein Data Bank with the accession code 4M52.
SUPPLEMENTAL INFORMATION
Supporting materials may be assessed free of charge online at http://pubs.acs.org.
Abbreviations: Mtb, Mycobacterium tuberculosis; Lpd, lipoamide dehydrogenase; hLpd, human lipoamide dehydrogenase; mLpd, mycobacterial lipoamide dehydrogenase; TB, tuberculosis; HIV, human immunodeficiency virus; PDH, pyruvate dehydrogenase complex; PNR/P, peroxynitrite reductase/peroxidase; KDH, α-ketoglutarate dehydrogenase complex; BCKDH, branched chain ketoacid dehydrogenase complex; GCS, glycine cleavage system; DlaT, dihydrolipoamide acyltransferase; AhpD, alkyl hydroperoxide reductase subunit D; RNI, reactive nitrogen intermediates; DTNB, 5,5'-dithiobis-2-nitrobenzoic acid; OD, optical density; MIC, minimum inhibitory concentration that inhibits bacterial growth by >90%; MTS, (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium); SI, selectivity index.
REFERENCES
- 1.WHO . Global tuberculosis report 2012. WHO Press; Geneva: 2012. [Google Scholar]
- 2.Venugopal A, Bryk R, Shi S, Rhee K, Rath P, Schnappinger D, Ehrt S, Nathan C. Virulence of Mycobacterium tuberculosis depends on lipoamide dehydrogenase, a member of three multienzyme complexes. Cell Host Microbe. 2011;9:21–31. doi: 10.1016/j.chom.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bryk R, Lima CD, Erdjument-Bromage H, Tempst P, Nathan C. Metabolic enzymes of mycobacteria linked to antioxidant defense by a thioredoxin-like protein. Science. 2002;295:1073–1077. doi: 10.1126/science.1067798. [DOI] [PubMed] [Google Scholar]
- 4.Tian J, Bryk R, Shi S, Erdjument-Bromage H, Tempst P, Nathan C. Mycobacterium tuberculosis appears to lack alpha-ketoglutarate dehydrogenase and encodes pyruvate dehydrogenase in widely separated genes. Mol Microbiol. 2005;57:859–868. doi: 10.1111/j.1365-2958.2005.04741.x. [DOI] [PubMed] [Google Scholar]
- 5.Marrero J, Rhee KY, Schnappinger D, Pethe K, Ehrt S. Gluconeogenic carbon flow of tricarboxylic acid cycle intermediates is critical for Mycobacterium tuberculosis to establish and maintain infection. Proc Natl Acad Sci U S A. 2010;107:9819–9824. doi: 10.1073/pnas.1000715107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hu Y, Coates AR. Acute and persistent Mycobacterium tuberculosis infections depend on the thiol peroxidase TpX. PLoS One. 2009;4:e5150. doi: 10.1371/journal.pone.0005150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hu Y, Coates AR. Mycobacterium tuberculosis acg gene is required for growth and virulence in vivo. PLoS One. 2011;6:e20958. doi: 10.1371/journal.pone.0020958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rajashankar KR, Bryk R, Kniewel R, Buglino JA, Nathan CF, Lima CD. Crystal structure and functional analysis of lipoamide dehydrogenase from Mycobacterium tuberculosis. J Biol Chem. 2005;280:33977–33983. doi: 10.1074/jbc.M507466200. [DOI] [PubMed] [Google Scholar]
- 9.Bryk R, Arango N, Venugopal A, Warren JD, Park YH, Patel MS, Lima CD, Nathan C. Triazaspirodimethoxybenzoyls as selective inhibitors of mycobacterial lipoamide dehydrogenase. Biochemistry. 2010;49:1616–1627. doi: 10.1021/bi9016186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Argyrou A, Blanchard JS. Mycobacterium tuberculosis lipoamide dehydrogenase is encoded by Rv0462 and not by the lpdA or lpdB genes. Biochemistry. 2001;40:11353–11363. doi: 10.1021/bi010575o. [DOI] [PubMed] [Google Scholar]
- 11.Holmgren A. Bovine thioredoxin system. Purification of thioredoxin reductase from calf liver and thymus and studies of its function in disulfide reduction. Journal of Biological Chemistry. 1997;252:4600–4606. [PubMed] [Google Scholar]
- 12.Collaborative Computational Project, N The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 13.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47(Pt 2):110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 15.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 16.Laskowski RA, Moss DS, Thornton JM. Main-chain bond lengths and bond angles in protein structures. J Mol Biol. 1993;231:1049–1067. doi: 10.1006/jmbi.1993.1351. [DOI] [PubMed] [Google Scholar]
- 17.Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Carvalho LP, Fischer SM, Marrero J, Nathan C, Ehrt S, Rhee KY. Metabolomics of Mycobacterium tuberculosis reveals compartmentalized co-catabolism of carbon substrates. Chem Biol. 2010;17:1122–1131. doi: 10.1016/j.chembiol.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 19.de Carvalho LP, Zhao H, Dickinson CE, Arango NM, Lima CD, Fischer SM, Ouerfelli O, Nathan C, Rhee KY. Activity-based metabolomic profiling of enzymatic function: identification of Rv1248c as a mycobacterial 2-hydroxy-3-oxoadipate synthase. Chem Biol. 2010;17:323–332. doi: 10.1016/j.chembiol.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chakraborty S, Gruber T, Barry CE, 3rd, Boshoff HI, Rhee KY. Para-aminosalicylic acid acts as an alternative substrate of folate metabolism in Mycobacterium tuberculosis. Science. 2013;339:88–91. doi: 10.1126/science.1228980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Balganesh M, Dinesh N, Sharma S, Kuruppath S, Nair AV, Sharma U. Efflux pumps of Mycobacterium tuberculosis play a significant role in antituberculosis activity of potential drug candidates. Antimicrob Agents Chemother. 2012;56:2643–2651. doi: 10.1128/AAC.06003-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rodrigues L, Villellas C, Bailo R, Viveiros M, Ainsa JA. Role of the Mmr efflux pump in drug resistance in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2013;57:751–757. doi: 10.1128/AAC.01482-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pethe K, Sequeira PC, Agarwalla S, Rhee K, Kuhen K, Phong WY, Patel V, Beer D, Walker JR, Duraiswamy J, Jiricek J, Keller TH, Chatterjee A, Tan MP, Ujjini M, Rao SP, Camacho L, Bifani P, Mak PA, Ma I, Barnes SW, Chen Z, Plouffe D, Thayalan P, Ng SH, Au M, Lee BH, Tan BH, Ravindran S, Nanjundappa M, Lin X, Goh A, Lakshminarayana SB, Shoen C, Cynamon M, Kreiswirth B, Dartois V, Peters EC, Glynne R, Brenner S, Dick T. A chemical genetic screen in Mycobacterium tuberculosis identifies carbon-source-dependent growth inhibitors devoid of in vivo efficacy. Nat Commun. 2010;1:57. doi: 10.1038/ncomms1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Manjunatha U, Boshoff HI, Barry CE. The mechanism of action of PA-824: Novel insights from transcriptional profiling. Commun Integr Biol. 2009;2:215–218. doi: 10.4161/cib.2.3.7926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singh R, Manjunatha U, Boshoff HI, Ha YH, Niyomrattanakit P, Ledwidge R, Dowd CS, Lee IY, Kim P, Zhang L, Kang S, Keller TH, Jiricek J, Barry CE., 3rd PA-824 kills nonreplicating Mycobacterium tuberculosis by intracellular NO release. Science. 2008;322:1392–1395. doi: 10.1126/science.1164571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nathan C. Microbiology. An antibiotic mimics immunity. Science. 2008;322:1337–1338. doi: 10.1126/science.1167452. [DOI] [PubMed] [Google Scholar]
- 27.Buttner D, Buttner H. pH dependency in uptake of sulfonamides by bacteria. Chemotherapy. 1980;26:153–163. doi: 10.1159/000237899. [DOI] [PubMed] [Google Scholar]
- 28.Tappe W, Zarfl C, Kummer S, Burauel P, Vereecken H, Groeneweg J. Growth-inhibitory effects of sulfonamides at different pH: dissimilar susceptibility patterns of a soil bacterium and a test bacterium used for antibiotic assays. Chemosphere. 2008;72:836–843. doi: 10.1016/j.chemosphere.2008.02.041. [DOI] [PubMed] [Google Scholar]
- 29.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 30.Delano W. The PyMOL molecular graphics system. DeLano Scientific; San Carlos CA, USA: 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.