

SUMMARY
CarD from Mycobacterium tuberculosis (Mtb) is an essential protein thought to be involved in stringent response through downregulation of rRNA and ribosomal protein genes. CarD interacts with the β-subunit of RNAP and this interaction is vital for Mtb’s survival during the persistent infection state. We have determined the crystal structure of CarD in complex with the RNAP β-subunit β1 and β2 domains at 2.1 Å resolution. The structure reveals the molecular basis of CarD/RNAP interaction, providing a basis to further our understanding of RNAP regulation by CarD. The structural fold of the CarD N-terminal domain is conserved in RNAP interacting proteins such as TRCF-RID and CdnL, and displays similar interactions to the predicted homology model based on the TRCF/RNAP β1 structure. Interestingly, the structure of the C-terminal domain, which is required for complete CarD function in vivo, represents a novel DNA binding fold.
INTRODUCTION
Tuberculosis (TB) is a global health threat responsible for approximately two million deaths annually (www.who.int/tb). The treatment for the primary causative agent of TB, Mycobacterium tuberculosis (Mtb), is challenging due to the emergence of multi-drug (MDR-TB) and extensively-drug resistant (XDR-TB) strains. As most of the antibiotics currently used for Mtb therapy are potent only against replicating bacteria, mycobacteria are able to survive in the host in a non-replicating, persistent, or chronic state. Identifying new drugs that can target Mtb during the persistent stages of infection is very important (Gupta et al., 2009; Raman et al., 2008; Sacchettini et al., 2008).
In Mtb, the gene product of Rv3583c, annotated CarD, is required for persistence and has been identified as an essential protein in vitro and in vivo during normal growth conditions as well as under genotoxic stress and nutrient deprivation (Stallings et al., 2009). Microarray studies have shown that not only is Rv3583c upregulated in response to oxidative stress, DNA damage, and starvation, but also that depletion of Mtb CarD results in loss of transcriptional regulation of rRNA and ribosomal components, indicating its involvement in stringent response (Stallings et al., 2009). Mtb CarD can be used to complement E. coli DksA protein, which regulates stringent response alongside the main stringent response element hyperphosphorylated guanine nucleotide ((p)ppGpp), suggesting that the two proteins are functional homologs. DksA directly interacts with the RNA polymerase (RNAP) through the RNAP secondary channel and potentiates the effect of (p)ppGpp (Paul et al., 2004b; Srivatsan and Wang, 2008). Mtb produces (p)ppGpp, but does not have a DksA homolog.
The first and best studied CarD protein is from Myxococcus xanthus, and is a transcription regulator involved in carotenogenesis. M. xanthus CarD interacts with RNAP, CarG, and the carQRS promoter DNA through its N- and C-terminal domains (Nicolas et al., 1996; Penalver-Mellado et al., 2006). Mtb CarD shares only a 30% sequence homology with the N-terminus of M. xanthus CarD, and the C-terminal domain is not similar, suggesting that Mtb CarD does not contain the HMGI-like DNA binding domain (AT hook DNA binding motif sequence) found in M. xanthus CarD (Figure S1). Bacterial two hybrid assays and immunoprecipitation experiments have shown that Mtb CarD associates with the RNAP β-subunit (Stallings et al., 2009; Weiss et al., 2012). All CarD and CarD N-terminal like (CdnL) proteins belong to the Transcription-Repair Coupling Factor (TRCF) family of proteins, and share sequence and structural homology with the TRCF RNAP Interacting Domain (RID). Also, they are thought to interact with RNAP in a homologous manner as TRCF. The previously determined crystal structure of the Tth TRCF-RID/Taq RNAP β1 complex (Westblade et al., 2010) and the homology models generated for CarD-RNAP interaction based on this structure (Weiss et al., 2012) predicted a similar set of interactions of CarD with RNAP. However, the mechanism CarD uses to regulate RNAP function and transcription, and the role of the Mtb CarD C-terminal domain are unknown. The CarD/RNAP interaction is crucial for Mtb’s stringent response, viability, and resistance to oxidative stress, and loss of the CarD/RNAP interaction sensitizes Mtb to the anti-TB drug rifampicin emphasizing the importance of understanding this protein-protein interaction.
Here, we report the crystal structure of Mtb CarD complexed with the Mtb RNAP β-subunit lobe domains at 2.1 Å resolution. The CarD/RNAP β structure reveals that the RNAP CarD binding site is located on the β-subunit arm of the RNAP claws, specifically on the solvent exposed surface of the β1 domain, and is far from the catalytic center of the RNAP. The structure not only provides insight into the molecular basis of RNAP interaction with Mtb CarD, but also with other CarD family proteins and CarD homologs. The structural basis for the RNAP regulation through CarD interaction, which is distinct from the DksA regulation mechanism, is presented by comparing the uncomplexed Mtb RNAP-β and CarD/RNAP-β complex structures. While the structural fold of the CarD N-terminal domain is conserved among other CarD, CdnL, and TRCF-RID domains, the C-terminal domain structure has not been identified in any other structure in the PDB to date. We show that Mtb CarD is a DNA binding protein with a novel DNA binding domain and that it exhibits a non-sequence specific DNA binding mode.
RESULTS and DISCUSSION
Structure determination of Mtb RpoBtr and the CarD/RNAP complex
E. coli expression plasmids for the full length Mtb RNAP β-subunit (RpoB) and several truncations (based on the secondary structure predictions) were made to test for recombinant protein expression. One truncation containing the β-lobes (consisting of residues 47 to 433) of RpoB (referred as RpoBtr) yielded soluble protein and when co-expressed with Mtb CarD resulted in complex formation, and it was chosen for subsequent crystallographic studies. The β1 (residues 47 to 172 and 375 to 428, corresponding to the Taq-β residues 1–130 and 334–395 of the TRCF/β1 structure) and β2 (residues 177 to 370) domains contained within this truncation are important for RNAP regulation. They form the β-arm of the RNAP claws that cover the DNA/RNA hybrid and dsDNA in the transcription complex. Regulation through these domains often occurs through interaction with various regulatory proteins, such as TRCF and sigma70 (Vassylyev et al., 2002; Westblade et al., 2010). RpoB does not have any dispensable regions, and the archaebacterial split site, which maps around residue 570 of RpoB, is not contained within the β-subunit truncation used in this work. Crystals of RpoBtr were determined to be in the P212121 space group with two molecules in the ASU. The RpoBtr structure was solved by single-wavelength anomalous diffraction using Se-Met derived crystals to a resolution of 2.9 Å. Subsequently, the resolution was improved to 2.5 Å for native (non Se-Met) RpoBtr crystals. The structure was refined to Rwork = 21% and Rfree = 26%, with excellent stereochemistry (Table 1).
Table 1.
Data Collection and refinement statistics
| Se-Met RpoBtr | Native RpoBtr | CarD/RpoBtr complex | |
|---|---|---|---|
| Data collection | |||
| Space group | P212121 | P212121 | C2221 |
| Cell dimensions | |||
| a, b, c (Å) | 52.8, 124.1, 135.5 | 53.1, 123.9, 135.6 | 49.1, 128.9, 225.5 |
| α, β, γ (0) | 90,90,90 | 90,90,90 | 90,90,90 |
| Wavelength (Å) | 0.979 | 0.979 | 0.979 |
| Resolution (Å) | 2.8 (2.79) | 2.5 (2.45) | 2.1 (2.11) |
| Completeness (%) | 98.5 (90.3) | 99.5 (97) | 98.1 (92) |
| Redundancy | 3.1 (3.1) | 7.0 (6.3) | 4.8 (4.6) |
| I/Iσ | 13.9 (2.4) | 15.1 (1.9) | 10.1 (2.0) |
| Rsym (%) | 5.28 (53.4) | 5.45 (65.7) | 7.8 (73.14) |
| Refinement | |||
| Resolution | 48.6–2.79 | 49–2.45 | 37.6–2.11 |
| No. reflections | 22552 | 33551 | 40747 |
| Rwork/Rfree | 0.24/0.28 | 0.21/0.26 | 0.20/0.23 |
| No. atoms | |||
| Protein | 5844 | 6097 | 4203 |
| Water | 19 | 126 | 305 |
| B-factors | |||
| Protein | 95.2 | 46.4 | 31.4 |
| Water | 57.1 | 47.3 | 45.0 |
|
| |||
| R.m.s deviations | |||
| Bond lengths (Å) | 0.010 | 0.003 | 0.005 |
| Bond angles (°) | 1.52 | 0.68 | 0.87 |
Highest resolution shell values are given in parenthesis.
Full length CarD bound to RpoBtr was crystallized after co-expression and purification of the complex. The crystals belonged to the C2221 space group with a single copy of the heterodimer in the ASU. The structure of the CarD/RpoBtr complex (hereafter referred to as the CarD/RNAP complex) was determined by molecular replacement, using the uncomplexed RpoBtr β1 and β2 domain structures as two individual search molecules. After locating RpoBtr using molecular replacement and initial refinement, there was clear electron density |Fo|-|Fc| that was unaccounted for (Figure S2A). This extra electron density belonged to the protein, and the entire atomic model of CarD was manually built into the difference electron density map. The crystal structure of the complex was refined to Rwork = 20% and Rfree = 23% using diffraction data to 2.1 Å resolution.
Overall structure of Mtb CarD
Mtb CarD belongs to the α + β protein class (SCOP)(Murzin et al., 1995). The structure is composed of two distinct domains: an all β-stranded N-terminal domain (residues 1–49) and an all α-helical C-terminal domain (residues 63–160) (Figures 1A and 2A). The N- and C-terminal domains are connected by a six residue twisted α-helix (α1) and an eight residue loop. The N-terminal domain has a Tudor-like fold (Selenko et al., 2001) consisting of four anti-parallel β-strands. Residues Thr26, Ile27, Lys28, and Gly29 which lie on the β-turn connecting the β2 and β3-strands were the only residues disordered in the N-terminal domain.
Figure 1. Ribbon representation of the Mtb CarD/RNAP β1-β2 domain complex structure.

RpoBtr is represented by orange ribbons; CarD is represented by blue ribbons. (A) Overall structure of the complex. β1 and β2 domains of RNAP β-subunit, β4-strands of each protein and N- and C-terminals of each chain are labeled. The inter-domain bridging β-sheet of RpoBtr (β7 and β15) is also indicated. (B) Zoom at the CarD-RNAP interface. Direct and water mediated H-bonding interactions between side chains and backbone-backbone interactions are shown. Hydrogen bonds and non-bonded contacts between RpoBtr and CarD are formed by the residues located on the β4-strands of both proteins, on the loop connecting α12 and α13 of the RNAP β1 domain, and on the turn between the β1 and β2-strands of CarD. H-bonds are represented by dashed lines and water molecules are shown in red spheres. For the distances refer to Table 2. See also Figure S2.
Figure 2. Ribbon representation of Mtb CarD.
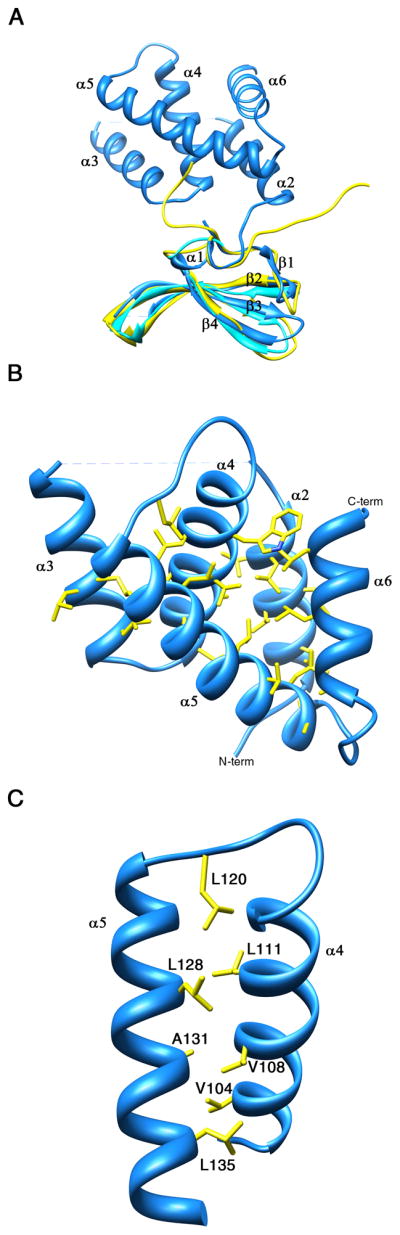
(A) Superposition of Mtb CarD N-terminal domain structure (blue) with the Tth CdnL-NMR structure (yellow) and the Tth TRCF-RID structure (cyan). The RNAP interacting β4-strand is labeled. The RMSD over the Cα backbone is 1.1 Å. (B) Ribbon representation of the CarD C-terminal domain. The helices contain mostly hydrophobic amino acids at their helix-helix interfaces, generating a hydrophobic core. The remainder of the structure is omitted for clarity. (C) The leucine zipper present between α4 and α5 in the C-terminal domain of CarD is formed by residues Leu120, Leu111, Leu128, Leu135, and surrounded by hydrophobic residues Val104, Val108 and Ala131. The rest of the structure is omitted for clarity. The leucine zipper lies inside the hydrophobic core of CarD and is not involved in dimerization or DNA interaction. In both panels, hydrophobic residues are shown with yellow sticks. See also Figure S1.
The structure of the CarD N-terminal domain is conserved among RNAP interacting proteins such as TRCF-RID and Thermus thermophilus (Tth) CdnL (CarD N-terminal Like protein involved in cell division). Superposition of the Mtb CarD N-terminal domain structure with the Tth CdnL N-terminal domain (PDB ID: 2LQK) (Gallego-Garcia et al., 2012) and Tth TRCF-RID structures (PDB ID: 3MLQ) (Westblade et al., 2010) gives an RMSD of 1.1 Å over the Cα backbone for residues 1–49 (Figure 2A).
The CarD C-terminal domain is comprised of an α-helical bundle of five α-helices that contains an unexpected internal leucine zipper between helices α4 and α5 (Figures 2B and 2C). Helices α4 and α5 interact only through hydrophobic and van der Waals interactions, afforded by the leucine zipper. Helices α2 and α3 are positioned parallel to each other, whereas α4, α5, and α6 pack orthogonal to each other. Helices α3 and α4 are connected by a γ-turn, while α4–α5 and α5–α6 are connected by β-turns (Figure 2B). The loop connecting α2 and α3, spanning residues His78 to Asn83, is completely disordered in the structure. The helices contain mostly hydrophobic amino acids at their helix-helix interfaces, generating a hydrophobic core. There are just two polar interactions inside this compact bundle, the hydrogen bonds between Lys95 (α3)-Glu106 (α4) (2.7 A) and Arg132 (α5)-Asp155 (α6) (2.9 Å).
The three helix bundle of α3, α4, and α5 is involved in DNA binding (see DNA binding studies on Mtb CarD section for details). The structure of this three helix bundle is unlike any other DNA binding protein in the PDB, including all of the other HTH motifs and leucine zipper domains. DNA binding proteins with classical HTH motifs usually insert their second (recognition) α-helix in the major groove for base-specific DNA interaction. Leucine zippers containing DNA binding proteins usually dimerize through the hydrophobic leucine zipper region, while also interacting with the major groove of the DNA; however, this is not the case for CarD. The DNA interacting region of Mtb CarD is mapped to the N-termini of α3 and α5, the C-terminus of α4, and the β-turn connecting α4 and α5 (Figure 5). The leucine zipper motif of CarD appears to stabilize the conformation of α4 and α5 inside the hydrophobic core, and is not involved in dimerization or DNA interaction per se (Figure 2C).
Figure 5. DNA binding activity of Mtb CarD determined by EMSA.

(A) CarD interaction with upstream DNA of 16S rRNA gene (313 bp). Lane 1: MW (molecular weight) marker. Lane 2: DNA probe, no protein. Lanes 3–7: 4.4–13.8 μM CarD. (B) Interaction of CarD domains and a well known non-DNA binding protein with 16S rRNA upstream DNA probe. Lane 1: InhA (10 μM). Lane 2: CarD1–53 (10 μM). Lane 3: CarD1–74 (10 μM). Lanes 4–5: CarD61–162 (5 and 10 μM). Lanes 6–7: CarD83–162 (5 and 10 μM). Lane 8: CarD full length (10 μM). Lane 9: 16S rRNA upstream DNA probe, no protein. Lane 10: MW marker. (C) EMSA experiments with CarD mutant proteins. Mutation of Arg and Lys residues to Ala significantly reduced the DNA binding activity of Mtb CarD. R87A-R88A-K90A showed the greatest effect. Lanes 1, 8, 15: 16S rRNA upstream DNA probe, no protein. Lanes 2–7, 9–14, 16–21: 0–44 μM mutant CarD protein (as labeled on the gel). Lanes 22–23: 11 and 22 μM native CarD. (D, E) Mutations are mapped on the ribbon representation and electrostatic potential surface of CarD. R87-R88-K90 are red, K125-R126-K130 are dark orange, and R114–R118 are light orange. Gel imaging was done using the Bio-Rad Chemidoc XRS+ molecular imager, by excitation at 255 nm and emission at 520 nm. Electrostatic potential surface calculations were done with PyMol (The PyMOL Molecular Graphics System, Version 0.99rc6, Schrödinger, LLC) using APBS as the macromolecular electrostatics calculation program(Baker et al., 2001).
Neither the AT-hook DNA binding motif of M. xanthus CarD, nor any other recognizable DNA binding motif is present in the Mtb CarD protein sequence. Structural similarity searches of the CarD C-terminal domain structure, using the PDBeFold, VAST, and DALI servers against the PDB and SCOP databases, did not identify any significant structural homologs. The structural alignment scores were well below the threshold of significance (VAST score <5.5, VAST -log(p)-value <4.0, and Q-score <0.49) for each alignment program(Gibrat et al., 1996; Holm and Rosenstrom, 2010; Krissinel and Henrick, 2004). It has been observed that the VAST hits do not share any common functional or structural features with Mtb CarD, besides being α-helical proteins.
Overall structure of Mtb RNAP β1–β2 domains
The RpoBtr structure comprises the β1 and β2 domains of the Mtb RNAP β-subunit (corresponding to the protrusion and lobe domains, respectively, of eukaryotic RNAP II) that form the RNAP claws, together, with the β′ subunit. RNAP interacts with the transcription bubble non-template strand, especially with the G+2 base, through the β2 domain residues, and these interactions are critical for sequence-specific promoter recognition of RNAP along with the transcription bubble formation and stability (Zhang et al., 2012).
The RpoBtr β1 domain aligns well with the β1 domain from the TRCF/β1 structure (RMSD 1.04 Å). However, superposition of RpoBtr β1-domain with the E. coli, Tth, and Taq RNAP β-lobe structures gives an RMSD of approximately 10 Å over the Cα atoms of the β2 domain (Figures S3A and S3B). β-lobes are known to have conformational flexibility (Tagami et al., 2010), and the relative conformation adopted by the β1 and β2 domains of RpoBtr has not been observed in any other bacterial core or holo RNAP structure. The two molecules in the ASU of the uncomplexed RpoBtr structure, RpoBtr_A and RpoBtr_B, are also in different conformations. When the β1 domains of RpoBtr_A and RpoBtr_B are aligned, the RMSD of the Cαs of the β2 domains is 5.2 Å (over 191 atom pairs). The conformational difference observed in the β1–β2 domain-domain orientation can be explained by rotation around the hinge axis centered on the two stranded anti-parallel β-sheet connecting the two domains (Figure S3C).
The RNAP β-subunits are structurally highly conserved among different kingdoms, even though sequence conservation is low (Lane and Darst, 2010; Severinov et al., 1996). As expected, the secondary and the tertiary structure of the Mtb RNAP β1–β2 domains are almost identical to the E. coli and Tth RNAP β1–β2 domains (Figures S3A and S3B). The β1 domain (residues 47 to 172 and 375 to 428) consists of four anti-parallel β-strands flanked by five α-helices on one side and one α-helix and a β-hairpin from the other side (Figure 3A). The β2 domain (residues 177 to 370) is composed of four anti-parallel β-strands flanked by seven α-helices. The two domains are connected by a two stranded anti-parallel β-sheet (the β7 strand and β15 strand), positioned like a bridge (Figure 3A). In contrast to other bacterial RNAPs, RpoBtr has an additional twelve residue β-hairpin connecting α11 and the bridge strand β15 on the β2 domain.
Figure 3. Conformational differences in RpoBtr in the uncomplexed and complexed forms.

(A) Ribbon representation of an uncomplexed RpoBtr molecule. The secondary structure assignments were done with PDBsum server (Laskowski, 2009) (B) The conformational differences observed in the β1–β2 domain-domain orientation between uncomplexed RpoBtr (green) and RpoBtr complexed with CarD (orange). Superposition of the β1 domains yields an RMSD of 2.8 Å over the Cαs of the β2 domains. The hinge axis centered on the two-stranded anti-parallel β-sheet (β7 and β15) bridging the two domains is also shown. (C) Local conformational changes of the RNAP β1 domain residues at the CarD/RNAP interface. β1-E404, S143, and E140 change conformation to interact with CarD-H13, R47, T45, and Y11. (D) Local conformational changes of the RNAP β1 domain residues at the β1–β2 domain interface. E396 and R392 of the β1 domain make additional water mediated interactions with the β2 domain residues P277 and G278 in the CarD/RNAP complex. The CarD residues are shown in blue. Coloring of RpoBtr is the same as in (B). Molecular visualization and analysis, including RMSD calculations, were performed with the UCSF Chimera package (Pettersen et al., 2004). See also Figure S3.
Overall structure of the CarD/RNAP-β1–β2 complex
The CarD binding site of RNAP is located at the solvent exposed surface of the β1 domain, which is approximately 70 Å away from the RNAP active site Mg+2 (based on the Tth EC, PDB ID: 2O5I) (Figure 4A). Despite the long distance between the binding site and the active site, this domain serves as an interaction module for various regulatory proteins including sigma factors at different stages of transcription, and is important for RNAP DNA binding and open complex stability (Trinh et al., 2006; Vassylyev et al., 2002).
Figure 4. Superposition of the Mtb CarD/RNAP complex structure with the bacterial elongation (EC) and initiation complex (IC) structures.
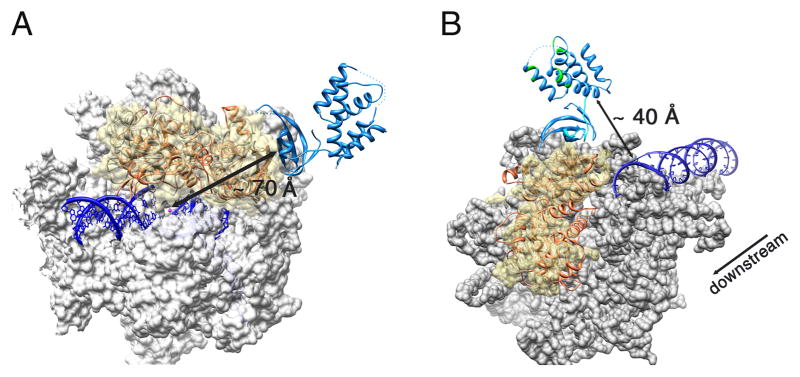
The Mtb CarD/RNAP complex is colored as previously (RpoBtr is orange and CarD is blue) (A) Superposition of the Mtb CarD/RNAP complex structure with the Tth RNAP EC structure (PDB ID: 2O5I). Tth RNAP is gray, the DNA duplex and DNA-RNA hybrid are dark blue, the active site Mg+2 is shown as a magenta sphere. The molecular surface of the α and β′ subunits is shown in gray, where the β1–β2 domains of the Tth RNAP are represented as ribbons under a transparent yellow surface. CarD binds to the solvent exposed surface of the β1 domain, ~ 70 Å away from the catalytic center. (B) Superposition of the Mtb CarD/RNAP complex structure with the Taq RNAP IC structure (PDB ID: 1L9Z). The molecular surface of the Taq RNAP is gray except for the β1–β2 domains which are represented as a transparent yellow surface. The DNA duplex is colored dark blue. The flexible α1 and eight residue loop of CarD are colored cyan. CarD’s DNA interacting patches as determined by EMSA are colored green. The direction of transcription is also indicated. The CarD C-terminal DNA interaction domain lies in close proximity to the downstream end of the dsDNA in the initiation complex (~40 Å).
At the CarD-RNAP interface, the primary four-stranded β-sheet of the CarD N-terminal domain forms an extended eight-stranded β-sheet with the β1 domain of the RNAP β-subunit (see Figure S2 for a stereo image). Specifically, the β4-strand of CarD comprising residues Leu44 to Pro49 forms an anti-parallel β-sheet with the β4-strand residues Thr138 to Gln144 of RpoBtr (1:1 heterodimer) (Figures 1A and S2B). This results in a mixed β-sheet in the 1↑ 2↓ 3↑ 4↓ 4′↑ 3′↓ 2′↑ 1′↓ topology. Surprisingly, association of RNAP with CarD results in only 500 Å2 (otherwise solvent exposed) buried surface area, which is below average (1500–2000 Å2) for heteromeric protein-protein complexes(Kleanthous, 2000). While the buried surface is relatively small, it is rich in intermolecular hydrogen bonds. There are eight hydrogen bonds and sixty-nine non-bonded contacts between RpoBtr and CarD formed by the residues located on the β4-strands of both proteins, on the loop connecting α12 and α13 of the RNAP β1 domain, and on the turn between the β1 and β2-strands of CarD. Specifically, β1-Ile141 interacts with CarD-Arg47 (2.8 Å), and β1-Ser143 interacts with CarD-Thr45 (2.7 Å) through four backbone-backbone hydrogen bonds (Figure 1B). Interestingly, the side chain specific hydrogen bonding interactions are present only between β1-Lys142:CarD-His14 (2.9 Å), β1-Glu140:CarD-Tyr11 (2.4 Å), β1-Thr138:CarD-Asn52 (2.8 Å), and β1-Gln144:CarD-Gly42 (3.0 Å) (Figure 1B). The intermolecular interface is also stabilized by electrostatic, hydrophobic, and van der Waals interactions (Figure S2). In fact, electrostatic forces contribute significantly to the CarD/RNAP interaction as altering the local charge distribution at the interface was reported to abolish CarD/RNAP interaction completely (Weiss et al., 2012). A more detailed analysis of the intermolecular contacts is provided in Table 2.
Table 2.
Details of the intermolecular interactions between Mtb β1 domain residues and Mtb CarD. See also Figure S4.
| RNAP β1 | CarD | Distance (Å) | Interaction type |
|---|---|---|---|
| Thr138 | Asn52 | 2.8 | H-bond and van der Waals |
| Pro49 | 3.5 | van der Waals | |
| Val56 | 3.9 | van der Waals | |
| Gly139 | Pro49 | 3.5 | van der Waals |
| Arg47 | 3.5 | van der Waals | |
| Glu140 | Arg47 | 3.4 | van der Waals |
| Val48 | 3.8 | van der Waals | |
| Tyr11 | 2.4 | H-bond and van der Waals | |
| Val56 | 3.6 | van der Waals | |
| Ile141 | Arg47 | 2.8 | H-bond and van der Waals |
| Thr45 | 3.4 | van der Waals | |
| Val46 | 3.2 | van der Waals, hydrophobic | |
| Arg25 | 3.9 | van der Waals | |
| Lys142 | Thr45 | 3.3 | van der Waals |
| His14 | 2.9 | H-bond and van der Waals | |
| Ser143 | Thr45 | 2.8 | H-bond and van der Waals |
| Leu44 | 3.6 | van der Waals | |
| Gln144 | Leu44 | 3.8 | van der Waals |
| Asp43 | 3.8 | van der Waals | |
| Gly42 | 3.0 | H-bond and van der Waals | |
| Glu404 | His14 | 3.8 | van der Waals |
| His13 | 3.4 | van der Waals | |
| Ala405 | His13 | 3.7 | van der Waals |
| His14 | 3.7 | van der Waals | |
| Thr407 | His14 | 3.3 | van der Waals |
In contrast to the structural model generated by homology modeling (based on Tth TRCF-RID/β1 structure), mutagenesis and two-hybrid assays (Weiss et al., 2012), which suggested that β1-Glu132 interacts with both Arg25 and Arg47 directly through hydrogen bonding and that these residues are critical for intermolecular interaction, we observed from the Mtb CarD/RNAP structure that β1-Glu132 is not in direct contact with CarD-Arg25 and CarD-Arg47 (5.0 Å and 6.1 Å, respectively). Arg25 interacts with β1-Ile141 only through van der Waals interactions and does not appear crucial for CarD/RNAP interaction. Similarly, Glu132 and Arg47 interact only through a water molecule in the CarD/RNAP crystal structure, and Arg47 is engaged in other hydrogen bonding and van der Waals interactions with β1-Ile141, β1-Glu140, and β1-Gly139 (Table 2). Therefore, loss of the CarD/RNAP interaction, as suggested by two-hybrid assays, upon E132R, R25E, and R47E mutations, should be due to these factors rather than the disruption of the direct interaction between Glu132-Arg25 and Glu132-Arg47.
Comparison of Mtb CarD/RNAP and Tth TRCF-RID/β1 complex structures reveals that CarD and TRCF-RID display a similar set of interactions with RNAP, even though there is no sequence conservation between the CarD β4-(43DLTVRVP49) and TRCF β4- (358EGKLYLP364) strands that interact with the RNAP β1 domain (except for the last proline residues) (Figure S4A). CarD residues Tyr11, His13, and His14, located on the turn between β1 and β2 strands, also interact with the RNAP-β1 domain, which was not observed in the TRCF-RID/β1 structure. We have tested the contribution of Y11 and H14 to the CarD/RNAP interaction by generating CarD-Y11A-H14A mutant, and comparing the thermal stability of CarD-Y11A-H14A/RpoBtr and CarD-wt/RpoBtr complexes by ThermoFluor (DSF) experiments (Kopec and Schneider, 2011; Madhurantakam et al., 2012). The thermal denaturation profiles suggested that the CarD-Y11A-H14A/RpoBtr complex is less stable than the CarD-wt/RpoBtr complex (TmC11A–14A=37.9 ± 0.1 °C vs. TmCwt=39.2± 0.1 °C) (Figure S4B), which was also supported by the size exclusion chromatography (data not shown), consistent with our structure that these residues are involved in CarD/RNAP interaction. On the other hand, the salt bridge interaction observed in the TRCF-RID/β1 structure between residues RNAP Glu110 and TRCF Tyr362 and Arg341 (Westblade et al., 2010) is not present in CarD/RNAP structure.
DNA binding studies on Mtb CarD
Mtb CarD is classified as a CdnL protein due to the lack of a DNA binding motif in its protein sequence. It has been proposed that CdnL proteins do not interact with DNA directly (Garcia-Moreno et al., 2010). We have tested whether Mtb CarD can interact with DNA by EMSA.
Because CarD is required for stringent response in mycobacteria, we tested CarD binding to ribosomal protein and rRNA operons. The 200–300 bp upstream of the rpsH, 16S, 23S, and 5S rRNA genes were amplified for gel shift assays. Our results showed a clear shift of electrophoretic mobility between the free DNA and the CarD-bound DNA for these probes (Figure 5A). The gel shift assays done with various random DNA probes as well as DNAse footprinting experiments (data not shown) suggested that Mtb CarD does not show a sequence preference for DNA interaction, indicating a non-specific DNA binding mode.
To elucidate the Mtb CarD-DNA interaction further, four different N- and C- terminally truncated CarD variants (CarD61–162, CarD83–162, CarD1–53, CarD1–74) were cloned and expressed to test each domain’s DNA binding activity. CarD61–162 and CarD83–162 contain α-helical C-terminal domain and exhibited a gel shift, though with different mobilities, which could be due to the charge, size and shape differences of the two constructs. EMSA results verified that the C-terminal domain is the DNA interaction domain (Figure 5B). The N-terminal domain is not involved in DNA interaction and is required only for RNAP interaction as observed from the CarD/RNAP structure.
Electrostatic potential surface calculations on the Mtb CarD structure revealed a single positively charged patch in the C-terminal domain formed by helices α3, α4, and α5 (Figures 5D and 5E). The basic residues contributing to this positively charged surface, i.e. Arg87-Arg88-Lys90 on α3, Arg114-Arg118 on α4, and Lys125-Arg126-Lys130 on α5, were mutated to alanine and subjected to EMSA. As anticipated, mutation of all the aforementioned Arg and Lys residues to Ala significantly reduced the DNA binding activity of CarD, with the R87A-R88A-K90A mutation located on the solvent exposed surface of α3, showing the greatest effect (Figure 5C), suggesting that the CarD-DNA interaction is mainly electrostatically driven, as expected.
Sequence independent DNA binding modes are commonly seen in bacterial nucleoid associated proteins, which are involved in chromosome compaction and structuring, DNA replication, repair, and transcription (Basu et al., 2009). The M. xanthus CarD protein, which has affinity for AT-rich DNA sequences, and the M. xanthus CdnL protein, which does not have a DNA binding sequence motif, were both localized to the nucleoid, but this localization was proposed to occur through protein-protein interactions with RNAP (Elias-Arnanz et al., 2010; Garcia-Moreno et al., 2010). It is plausible that Mtb CarD also localizes to the nucleoid in the same manner as the M. xanthus CdnL and M. xanthus CarD proteins, but considering the sequence independent DNA binding activity, we suggest that this localization might be provided by the DNA-binding ability of CarD rather than by associating and tailing with RNAP. The C-terminal domain, and thereby the DNA binding activity of Mtb CarD, is crucial for mycobacterial viability because CarD depletion cannot be complemented with the RID domain alone (Weiss et al., 2012).
Conformational changes in RNAP upon CarD binding
The conformation of the β1 and β2 domains observed in the CarD/RNAP complex differs from the conformations observed in the uncomplexed RpoBtr structure. Superposition of the uncomplexed RpoBtr_A and RpoBtr_B β1 domains with the CarD/RpoBtr β1 domain structure gives an RMSD of 6.8 A and 2.8 A, respectively, over the Cαs of the β2 domains (Figure 3B). In this context, the conformation of RpoBtr in complex with CarD is closer to the conformation of the uncomplexed RpoBtr_B molecule. It was reported in the TRCF/β1 structure that the RNAP β4-strand undergoes a ‘register shift’ with respect to the β3-strand in the complex structure (Westblade et al., 2010). In contrast, CarD does not cause a register shift or conformational rearrangement in the β4-strand upon RNAP binding.
The CarD/RNAP β1 domain interaction causes local conformational changes primarily in the nearby RNAP side chains that are propagated through the water mediated network of interactions and transferred to the β1–β2 domain interface and β2 domain residues. In particular, in the CarD/RNAP complex, β1-Glu404, Ser143, and Glu140 change conformation to interact with CarD-His13, Arg47, Thr45, and Tyr11, respectively (Figure 3C). The side chain of β1-Lys142 also moves 1.4 Å and loses direct H-bonding interaction with β1-Ile406, instead forming hydrogen bonds with CarD-His14 and β1-Glu140. Consecutively, the β1-domain residues Glu396 and Arg392, located at the β1–β2 domain interface of RpoBtr, adopt different conformations in CarD/RNAP complex and make additional water mediated interactions with the β2 domain residues Pro277 and Gly278 (Figure 3D). This can explain the particular conformation adopted by the two domains in the complex structure.
RNAP regulation by CarD
The CarD/RNAP structure indicates that CarD and the functional homolog DksA regulate RNAP through different mechanisms. DksA is proposed to bind to the RNAP secondary channel, very close to the active site, to coordinate to a (p)ppGpp-bound active site Mg2+ ion through its coiled-coil Asp residues, and stabilize the (p)ppGpp-RNAP complex(Perederina et al., 2004). In contrast, CarD interacts with the β1 domain of the β-subunit, approximately 70 Å away from the active site, through its Tudor-fold N-terminal domain. It’s interesting that even though CarD and DksA do not share sequence and structural homology, CarD can complement DksA function in a ΔDksA E. coli strain (Stallings et al., 2009). Furthermore, DksA is not a DNA binding protein, whereas we’ve shown that CarD can interact with DNA. Whether CarD functions synergistically with (p)ppGpp the same way as DksA needs to be determined experimentally.
It is not known whether CarD regulates RNAP function during transcription initiation or elongation, or has any effect on the rate of transcription. Based on our structural data, we propose that CarD might be involved in RNAP regulation in three different ways. The first is by inducing conformational changes in the β-lobes and affecting the open complex stability and the downstream non-specific DNA binding activity of RNAP. This can explain how Mtb CarD can complement DksA, which destabilizes the open complex together with (p)ppGpp during stringent response in E. coli (Paul et al., 2004a; Stallings et al., 2009). Bacterial RNAP β1 and β2 domains (equivalently eukaryotic RNAP II protrusion and lobe domains) are involved in various processes during transcription such as downstream DNA binding and selection of the transcription initiation site, formation and stabilization of the open complex, maintaining the proper transcription bubble via downstream DNA gripping, keeping the template and non-template strand-separated DNA in place during transcription initiation, and covering the DNA/RNA hybrid inside the RNAP active-site channel (Figures 4A and 4B)(Lane and Darst, 2010; Murakami et al., 2002; Nechaev et al., 2000; Trautinger and Lloyd, 2002; Trinh et al., 2006) Therefore, conformational changes in this region may likely alter critical interactions of RNAP with DNA and DNA/RNA hybrid. To test if CarD interaction with the β1 domain would affect RNAP’s DNA binding affinity, we compared the non-specific DNA binding activity of RpoBtr in both the uncomplexed form and in complex with CarD by EMSA. Our results suggest that the CarD/RpoBtr complex has a higher affinity than the uncomplexed RpoBtr for the same DNA probe (Figure 6). We propose that CarD might affect the DNA binding affinity of the β-lobes, and the affinity change of RpoBtr for DNA may result primarily from the conformational changes of the β-lobes induced by CarD interaction.
Figure 6. Comparison of the DNA binding activity of RpoBtr by EMSA.
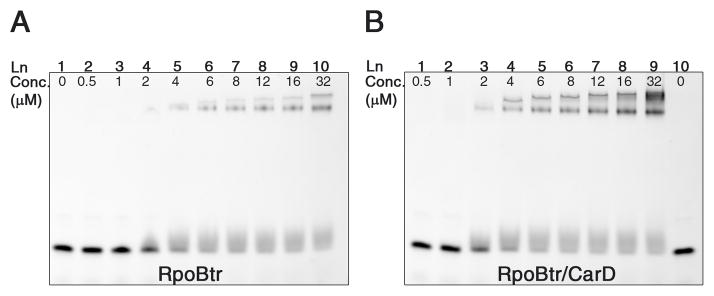
(A) in the uncomplexed form (B) in complex with CarD. The CarD/RpoBtr complex has higher affinity than the uncomplexed RpoBtr for the same DNA probe. (A) Lane 1: dsDNA probe, no protein. Lanes 2–10: 0.5 μM-32 μM RpoBtr. (B) Lanes 1–9: 0.5 μM-32 μM RpoBtr/CarD complex. Lane 10: dsDNA probe, no protein.
Overlay of the Taq RNAP initiation complex and CarD/RNAP complex structures show that the CarD C-terminal DNA interaction domain lies in close proximity (< 40 Å) to the downstream end of dsDNA in the initiation complex (Figure 4B), suggesting that CarD may interact with the promoter DNA together with RNAP during initiation. The CarD C-terminal domain is connected to the CarD N-terminal domain by a twisted α1-helix and a short loop and may adopt a different relative conformation in solution than the one observed in the crystal structure. Therefore, a second possibility is that this domain can either function as an anchor on DNA to hold CarD in place and strengthen the CarD/RNAP interaction or it can have a direct functional role in transcription regulation such as promoter selection and binding. The role of the C-terminal domain on CarD function is currently under investigation.
Another possible mechanism is allosteric regulation by inducing conformational changes around the RNAP active site despite the distance of the CarD binding site from the RNAP catalytic center. In fact, mutations at the interface that weaken the CarD/RNAP interaction were reported to make Mtb more susceptible to rifampicin (Rif), which binds to the RNAP active site and inhibits transcription elongation. This suggests that CarD/RNAP interaction is able to induce conformational changes not only in the β1–β2 domains but also in the Rif binding pocket, causing Rif to bind more weakly to RNAP. The clinically isolated Rif resistant Mtb strains carrying mutations on distant β residues, such as Val170, which do not interact directly with Rif, affect the conformation of the Rif binding pocket, and alter the affinity of RNAP for the drug (Campbell et al., 2001). Similarly, CarD interaction with the β-lobes may result in a reduced affinity of RNAP for Rif. A more complete understanding of the effect of CarD on RNAP both structurally and functionally must await the solution of the full length RNAP/CarD structure.
The CarD/RNAP structure presented here reveals the molecular basis of this protein-protein interaction and provides insights into RNAP regulation by CarD. EMSA experiments revealed an unexpected DNA binding activity for Mtb CarD which is required for complete in vivo function and mycobacterial viability, and is provided by a distinct domain not associated with RNAP interaction. Determination of the CarD/DNA complex and RNAP/CarD/DNA ternary complex crystal structures are needed to further characterize the transcriptional regulation by CarD.
EXPERIMENTAL PROCEDURES
Generation of expression constructs and cloning
Rv3583c, encoding the Mtb CarD protein, and DNA encoding the Mtb RNAP β-subunit (Rv0667) residues 47–433 (labeled RpoBtr), were amplified from Mtb H37Rv genomic DNA by PCR. The genes were inserted into pET15b and pET30b (Novagen) expression vectors using the NdeI-BamHI and NdeI-HindIII restriction sites. The pET15b construct contained an N-terminal 6X-His tag and labeled RpoBtr-NHis, while the pET30b construct had a stop codon at the end of the gene sequence, generating an untagged protein (CarD-notag). DNA encoding the full length and truncated CarD proteins (CarD1–74, CarD1–53, CarD61–162 and CarD83–162) were amplified from Mtb H37Rv genomic DNA by PCR with the NdeI-HindIII restriction sites and inserted into the pET28b vector (Novagen). CarD_R87A-R88A-K90A, CarD_R114A-R118A, and CarD_K125A-R126A-K130A plasmids were generated using a site directed mutagenesis kit (Stratagene). The sequence of each construct was verified by DNA sequencing. The primers used in this study are provided in Table S1.
Expression and purification
Expression plasmids for the uncomplexed RpoBtr, native and mutant CarD proteins were transformed to E. coli BL21(DE3) cells, and recombinant protein expression was induced with 1 mM IPTG. For co-expression of Mtb RNAP β1–β2 domains and Mtb CarD, the plasmids RpoBtr-NHis and CarD-notag were cotransformed into E. coli Rosetta2(DE3)pLysS cells, and expression was induced with 0.75 mM IPTG. Proteins were extracted with French press and purified by metal affinity and size exclusion chromatography. The RpoBtr-NHis:CarD complex eluted as a single peak from the size exclusion column. Co-elution of RpoBtr and CarD from the IMAC and size-exclusion columns was verified by SDS-PAGE. Finally, the purified proteins were concentrated to 10 mg/ml and stored at −80 ºC for further use. Details of the expression and purification process are provided in Supplemental Experimental Procedures.
Crystallization
RpoBtr-NHis-SeMet and native protein crystals were obtained using hanging-drop vapor diffusion method by incubating 2 μl of purified protein solution with 2 μl of crystallization solution (0.1 M MgCl2, 0.1 M Hepes 7.5, 10% (w/v) PEG4000, and 0.2 M potassium citrate tribasic monohydrate, 20% (w/v) PEG3350, respectively) at 16 ºC. The RpoBtr-NHis:CarD complex was crystallized by mixing 2 μl of protein solution with 2 μl of mother liquor (2% (v/v) tacsimate pH 5.0, 0.1 M sodium citrate tribasic dihydrate pH 5.6, 14% (w/v) PEG3350) by hanging-drop vapor diffusion. Crystals were cryo-protected with 20% (v/v) ethylene glycol and flash frozen prior to data collection. Data was collected at the Advanced Light Source (ALS - Lawrance Berkeley National Laboratory) and at the Advanced Photon Source (APS beamlines 23ID and 19ID-Argonne National Laboratory) at 0.979 Å.
Data collection and structure determination
The structure of the Mtb RNAP β-subunit β1–β2 domains was solved by single-wavelength anomalous diffraction (SAD) using RpoBtr-NHis-SeMet crystals. Crystals belonged to the P212121 space group and diffracted to 2.9 Å. Subsequently, resolution was improved to 2.5 Å by diffraction data obtained from native (non-SeMet) RpoBtr-NHis crystals. Refinement and iterative manual model building was performed with Phenix (Adams et al., 2010) and COOT (Emsley et al., 2010), and the final model had Rwork and Rfree values of 0.21 and 0.26, respectively.
The RpoBtr-NHis:CarD complex crystals belonged to the C2221 space group and the diffraction data to 2.1 Å resolution was processed with Denzo/Scalepack. The structure was solved by MR using the RpoBtr β1 and β2 domains as two different search ensembles (Phaser, CCP4) (McCoy et al., 2007; Winn et al., 2011). After locating one copy of RpoBtr in the ASU, CarD was built into the additional |Fo-Fc| density manually. The final model included one RpoBtr:CarD complex in the ASU, and the structure was refined with Phenix Refine to a Rwork= 0.20 and an Rfree= 0.23. Data collection and processing statistics are provided in Table 1. Details of the data collection and structure determination are provided in Supplemental Experimental Procedures.
EMSA assays
For electrophoretic mobility shift assays (EMSA), DNA 200–300 bp upstream of the rpsH, 16S, 23S and 5S rRNA promoters were amplified from H37Rv genomic DNA by PCR (for primers see Table S1) and purified by gel-extraction. 40 ηg of dsDNA was incubated with different amounts of protein (0–8μg) at room temperature for 30 min in 25 mM Tris pH 7.5, 50 mM NaCl. As a negative control, a known non-DNA binding protein, enoyl-ACP reductase InhA from Mtb, was used to confirm that binding of Mtb CarD to DNA is protein specific. The mixture was loaded on a pre-cast 10% non-denaturing polyacrylamide gel and the gel was run at a constant voltage (120 V) with pre-chilled 0.5X TBE (89 mM Tris base, 89 mM boric acid, 1 mM EDTA, pH 8.0) buffer at 4 ºC. After the run was completed, the gel was stained with 1X Syber green (Invitrogen) DNA stain solution for 30 min in the dark and imaged (Jing et al., 2003).
ThermoFluor measurements
Differential scanning fluorimetry (DSF) experiments were carried out with 1 μM of CarD/RpoBtr or CarD-Y11A-H14A/RpoBtr complex in 200 mM Tris pH 7.5, 100 mM NaCl buffer, in the presence of 5X Sypro orange dye (Molecular Probes), in a 20 μl reaction volume. The temperature of the samples were changed from 25 to 95 °C at a heating rate of 0.5 °C/min, and the fluorescence was monitored by Mx3005P qPCR instrument (Agilent). The melting point (Tm) was calculated as the lowest point of the first derivative plot (DeSantis et al., 2012).
Supplementary Material
Highlights.
Crystal structure of the CarD-RNAP complex
CarD binds to the β1 domain of the RNAP β-subunit, away from the active center
Structural basis of RNAP regulation mechanism by CarD
Mtb CarD is a DNA binding protein with a novel DNA binding domain
Acknowledgments
This work is supported by grants to Sacchettini, J.C. by the Welch foundation, grant A-0015 and by the TB Structural Genomics grant NIH P01AIO95208. We would like to thank the beamline staff of APS-19 ID, APS-23-ID, and Dr. Li-Wei Hung (ALS) for data collection; to Tracey Musa and Amir Safi for careful reading of the manuscript. Authors declare no competing financial interests.
Footnotes
ACCESSION NUMBERS
The coordinates for the uncomplexed RpoBtr and RpoBtr/CarD complex have been deposited in the Protein Data Bank with the entry numbers 4KBJ and 4KBM, respectively.
Supplemental Information includes four figures, one table and supplemental experimental procedures.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta crystallographica Section D, Biological crystallography. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu D, Khare G, Singh S, Tyagi A, Khosla S, Mande SC. A novel nucleoid-associated protein of Mycobacterium tuberculosis is a sequence homolog of GroEL. Nucleic acids research. 2009;37:4944–4954. doi: 10.1093/nar/gkp502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell EA, Korzheva N, Mustaev A, Murakami K, Nair S, Goldfarb A, Darst SA. Structural mechanism for rifampicin inhibition of bacterial rna polymerase. Cell. 2001;104:901–912. doi: 10.1016/s0092-8674(01)00286-0. [DOI] [PubMed] [Google Scholar]
- DeSantis K, Reed A, Rahhal R, Reinking J. Use of differential scanning fluorimetry as a high-throughput assay to identify nuclear receptor ligands. Nuclear receptor signaling. 2012;10:e002. doi: 10.1621/nrs.10002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias-Arnanz M, Padmanabhan S, Murillo FJ. The regulatory action of the myxobacterial CarD/CarG complex: a bacterial enhanceosome? FEMS microbiology reviews. 2010;34:764–778. doi: 10.1111/j.1574-6976.2010.00235.x. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta crystallographica Section D, Biological crystallography. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallego-Garcia A, Mirassou Y, Elias-Arnanz M, Padmanabhan S, Jimenez MA. NMR structure note: N-terminal domain of Thermus thermophilus CdnL. Journal of biomolecular NMR. 2012;53:355–363. doi: 10.1007/s10858-012-9648-z. [DOI] [PubMed] [Google Scholar]
- Garcia-Moreno D, Abellon-Ruiz J, Garcia-Heras F, Murillo FJ, Padmanabhan S, Elias-Arnanz M. CdnL, a member of the large CarD-like family of bacterial proteins, is vital for Myxococcus xanthus and differs functionally from the global transcriptional regulator CarD. Nucleic acids research. 2010;38:4586–4598. doi: 10.1093/nar/gkq214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibrat JF, Madej T, Bryant SH. Surprising similarities in structure comparison. Current opinion in structural biology. 1996;6:377–385. doi: 10.1016/s0959-440x(96)80058-3. [DOI] [PubMed] [Google Scholar]
- Gupta RK, Thakur TS, Desiraju GR, Tyagi JS. Structure-based design of DevR inhibitor active against nonreplicating Mycobacterium tuberculosis. Journal of medicinal chemistry. 2009;52:6324–6334. doi: 10.1021/jm900358q. [DOI] [PubMed] [Google Scholar]
- Holm L, Rosenstrom P. Dali server: conservation mapping in 3D. Nucleic acids research. 2010;38:W545–549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing D, Agnew J, Patton WF, Hendrickson J, Beechem JM. A sensitive two-color electrophoretic mobility shift assay for detecting both nucleic acids and protein in gels. Proteomics. 2003;3:1172–1180. doi: 10.1002/pmic.200300438. [DOI] [PubMed] [Google Scholar]
- Kleanthous C. Protein-protein recognition. Oxford ; New York: Oxford University Press; 2000. [Google Scholar]
- Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta crystallographica Section D, Biological crystallography. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- Lane WJ, Darst SA. Molecular evolution of multisubunit RNA polymerases: sequence analysis. Journal of molecular biology. 2010;395:671–685. doi: 10.1016/j.jmb.2009.10.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA. PDBsum new things. Nucleic acids research. 2009;37:D355–359. doi: 10.1093/nar/gkn860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. Journal of applied crystallography. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami KS, Masuda S, Campbell EA, Muzzin O, Darst SA. Structural basis of transcription initiation: an RNA polymerase holoenzyme-DNA complex. Science. 2002;296:1285–1290. doi: 10.1126/science.1069595. [DOI] [PubMed] [Google Scholar]
- Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. Journal of molecular biology. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- Nechaev S, Chlenov M, Severinov K. Dissection of two hallmarks of the open promoter complex by mutation in an RNA polymerase core subunit. J Biol Chem. 2000;275:25516–25522. doi: 10.1074/jbc.M002511200. [DOI] [PubMed] [Google Scholar]
- Nicolas FJ, Cayuela ML, Martinez-Argudo IM, Ruiz-Vazquez RM, Murillo FJ. High mobility group I(Y)-like DNA-binding domains on a bacterial transcription factor. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:6881–6885. doi: 10.1073/pnas.93.14.6881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul BJ, Barker MM, Ross W, Schneider DA, Webb C, Foster JW, Gourse RL. DksA: a critical component of the transcription initiation machinery that potentiates the regulation of rRNA promoters by ppGpp and the initiating NTP. Cell. 2004a;118:311–322. doi: 10.1016/j.cell.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Paul BJ, Ross W, Gaal T, Gourse RL. rRNA transcription in Escherichia coli. Annual review of genetics. 2004b;38:749–770. doi: 10.1146/annurev.genet.38.072902.091347. [DOI] [PubMed] [Google Scholar]
- Penalver-Mellado M, Garcia-Heras F, Padmanabhan S, Garcia-Moreno D, Murillo FJ, Elias-Arnanz M. Recruitment of a novel zinc-bound transcriptional factor by a bacterial HMGA-type protein is required for regulating multiple processes in Myxococcus xanthus. Molecular microbiology. 2006;61:910–926. doi: 10.1111/j.1365-2958.2006.05289.x. [DOI] [PubMed] [Google Scholar]
- Perederina A, Svetlov V, Vassylyeva MN, Tahirov TH, Yokoyama S, Artsimovitch I, Vassylyev DG. Regulation through the secondary channel--structural framework for ppGpp-DksA synergism during transcription. Cell. 2004;118:297–309. doi: 10.1016/j.cell.2004.06.030. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. Journal of computational chemistry. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Raman K, Yeturu K, Chandra N. targetTB: a target identification pipeline for Mycobacterium tuberculosis through an interactome, reactome and genome-scale structural analysis. BMC systems biology. 2008;2:109. doi: 10.1186/1752-0509-2-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacchettini JC, Rubin EJ, Freundlich JS. Drugs versus bugs: in pursuit of the persistent predator Mycobacterium tuberculosis. Nature reviews Microbiology. 2008;6:41–52. doi: 10.1038/nrmicro1816. [DOI] [PubMed] [Google Scholar]
- Selenko P, Sprangers R, Stier G, Buhler D, Fischer U, Sattler M. SMN tudor domain structure and its interaction with the Sm proteins. Nature structural biology. 2001;8:27–31. doi: 10.1038/83014. [DOI] [PubMed] [Google Scholar]
- Severinov K, Mustaev A, Kukarin A, Muzzin O, Bass I, Darst SA, Goldfarb A. Structural modules of the large subunits of RNA polymerase. Introducing archaebacterial and chloroplast split sites in the beta and beta’ subunits of Escherichia coli RNA polymerase. The Journal of biological chemistry. 1996;271:27969–27974. doi: 10.1074/jbc.271.44.27969. [DOI] [PubMed] [Google Scholar]
- Srivatsan A, Wang JD. Control of bacterial transcription, translation and replication by (p)ppGpp. Current opinion in microbiology. 2008;11:100–105. doi: 10.1016/j.mib.2008.02.001. [DOI] [PubMed] [Google Scholar]
- Stallings CL, Stephanou NC, Chu L, Hochschild A, Nickels BE, Glickman MS. CarD is an essential regulator of rRNA transcription required for Mycobacterium tuberculosis persistence. Cell. 2009;138:146–159. doi: 10.1016/j.cell.2009.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagami S, Sekine S, Kumarevel T, Hino N, Murayama Y, Kamegamori S, Yamamoto M, Sakamoto K, Yokoyama S. Crystal structure of bacterial RNA polymerase bound with a transcription inhibitor protein. Nature. 2010;468:978–982. doi: 10.1038/nature09573. [DOI] [PubMed] [Google Scholar]
- Trautinger BW, Lloyd RG. Modulation of DNA repair by mutations flanking the DNA channel through RNA polymerase. EMBO J. 2002;21:6944–6953. doi: 10.1093/emboj/cdf654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinh V, Langelier MF, Archambault J, Coulombe B. Structural perspective on mutations affecting the function of multisubunit RNA polymerases. Microbiology and molecular biology reviews : MMBR. 2006;70:12–36. doi: 10.1128/MMBR.70.1.12-36.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassylyev DG, Sekine S, Laptenko O, Lee J, Vassylyeva MN, Borukhov S, Yokoyama S. Crystal structure of a bacterial RNA polymerase holoenzyme at 2.6 A resolution. Nature. 2002;417:712–719. doi: 10.1038/nature752. [DOI] [PubMed] [Google Scholar]
- Weiss LA, Harrison PG, Nickels BE, Glickman MS, Campbell EA, Darst SA, Stallings CL. The Interaction of CarD with RNAP Mediates Mycobacterium tuberculosis Viability, Rifampicin Resistance, and Pathogenesis. Journal of bacteriology. 2012 doi: 10.1128/JB.00879-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westblade LF, Campbell EA, Pukhrambam C, Padovan JC, Nickels BE, Lamour V, Darst SA. Structural basis for the bacterial transcription-repair coupling factor/RNA polymerase interaction. Nucleic acids research. 2010;38:8357–8369. doi: 10.1093/nar/gkq692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, et al. Overview of the CCP4 suite and current developments. Acta crystallographica Section D, Biological crystallography. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Feng Y, Chatterjee S, Tuske S, Ho MX, Arnold E, Ebright RH. Structural basis of transcription initiation. Science. 2012;338:1076–1080. doi: 10.1126/science.1227786. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.