

Abstract
Nanoparticles have recently emerged as a promising class of carriers for the co-delivery of multiple drugs. Combination therapies of small-molecule drugs are common in clinical practice and it is anticipated that packaging into single macromolecular carriers will enable drug release in precisely balanced ratios and rates, and in selectively targeted tissues and cells. This vast level of pharmacological control is intriguing, especially from the perspective of tailoring personalized treatments with maximized therapeutic synergy for individual patients. Here, we discuss promising formulations and opportunities to employ advanced screening tools and new animal models of disease that can improve chances for successful clinical translation.
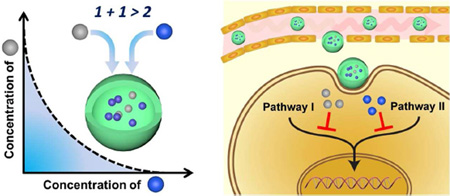
Combination drug therapy is the co-administration of two or more drugs to a patient, and is standard clinical practice in the treatment of many classes of cancer and infectious disease.1 These multi-drug regimens are usually designed to achieve therapeutic synergy, or a medicinal effect that is greater than the sum of each drug treatment alone. Small-molecule drugs have been the mainstay of such strategies, but nanoparticle-formulated drugs have now come of age in the clinic and are inspiring innovative investigations in multidrug delivery. Nanoparticle therapies provide improved drug solubility, reduced systemic toxicity, longer circulation times in the blood, controllable release profiles, and the potential to target specific cells and tissues.2 The first clinical treatments were approved in the 1990s as liposome-based therapies (e.g., Doxil) for cancer, and >25 nanotherapies have since been clinically tested, with many more in industrial preclinical development.2 The first multi-drug nanotherapies are now undergoing phase II/III clinical trials for cancer.
This convergence of nanomaterials and combination therapy is compelling in the context of personalized medicine practices that aim to prescribe a drug treatment regimen optimized for each patient.3 Aided by technological breakthroughs in affordable, high-throughput gene sequencing as well as computational and systems biology, genetic profiles of patients, diseased tissues, and pathogens are now being factored into the diagnosis and treatment process to interfere selectively with cellular pathways that cause harm and to spare healthy cells. This significant step forward in disease management is only at the early stages of implementation, but it is tempting to think that nanomedicine will offer clinicians the ability to fine-tune the pharmacological properties of multi-drug cocktails with cell-specific targeting in the near future. Clinical nanomedicines still need substantial optimization, but rapidly developing combinatorial drug screening tools and advanced animal models may help to maximize their clinical potency in ways that were not previously possible.
Nanoparticle Designs for Co-Delivery of Drugs
Nanoparticle formulations offer several advantages for multidrug delivery compared to combinations of free drugs. Controlled release from nanocarriers can normalize the pharmacokinetics, biodistribution, and stability of chemically dissimilar drugs that independently have disparate pharmacological behaviors. Thus, long-circulating formulations can continuously release drugs at controlled ratios or allow independent tuning of release rates of each drug in ways that would simply not be feasible with rapidly clearing free drugs. In addition, stimulus- responsive, targeted carriers in development can co-release drugs in the same organ, tissue, or cell, to increase efficacy and to reduce toxicity from off-target exposure.
Nanoparticle formulations in clinical use include polymers, liposomes, micelles, and proteins, in which a drug is either encapsulated or conjugated to internal domains of the carrier. For coformulation of two drugs, chemical and physical differences must be considered, as drugs span a wide range of characteristics from hydrophobic small molecules (e.g., most inhibitors and chemotherapies) to large hydrophilic macromolecules (e.g., antibodies and nucleic acids). Hydrophobic drugs like paclitaxel naturally partition to hydrophobic domains of nanoparticles whereas hydrophilic drugs typically require physical entrapment or chemical conjugation to prevent rapid release. To expedite clinical implementation, it is also necessary to use carriers composed of FDA-approved materials, which often include lipids and a variety of biodegradable polymers like hydrophilic polyethylene glycol (PEG) and polyvinyl alcohol (PVA) as well as hydrophobic polylactic acid (PLA), polyglycolic acid (PGA), and PLA-PGA co-polymers (PLGA). Within these constraints, scientists have devised unique formulations that allow both high drugloading capacity and co-delivery of diverse combinations of drugs (see Figure 1).
Figure 1.

Nanoparticle designs for co-delivery of multiple drugs.
Liposomes have been designed to encapsulate diverse hydrophilic and hydrophobic chemotherapy pairs, including irinotecan/floxuridine (CPX-1, Celator), cytarabine/daunorubicin (CPX-351), and paclitaxel/tanespimycin.4–6 These drug combinations have been used clinically as small molecules but have limited efficacy due to mismatched clearance rates and/or poor solubility that requires co-administration with toxic solvents. Drug ratios can be readily tuned in these 100–300-nm nanoparticles, and sustained release maintains specific synergistic ratios in the blood over 24 h after injection (Figure 2a).5 The drug combination CPX-351 is now in phase III clinical trials after showing markedly lower toxicity than the free drug combination as well as improved efficacy for acute myeloid leukemia in preclinical and clinical studies (Figure 2b), and CPX-1 showed promising results in phase II trials for colorectal cancer.4
Figure 2.

Combination drug therapy with cytarabine/daunorubicin (CPX-351). (a) Plasma drug concentration of cytarabine and daunorubicin after IV co-injection as CPX-351 nanoparticles or as free drugs to mice. Inset: circulating plasma cytarabine:daunorubicin molar ratios for CPX-351 calculated from absolute plasma concentrations. (b) Survival of BDF-1 mice bearing P388 leukemia tumors at day 55 following IV treatment on days 1, 4, and 7 with saline or liposome co-encapsulated cytarabine and daunorubicin at different drug molar ratios. Formulations were dosed at their maximum tolerated dose (MTD) with the exception of CPX-351 (5:1 molar drug ratio), which was dosed at 0.8 of its MTD. Adapted with permission from ref 5. Copyright 2009 Elsevier.
Compared with liposomes and micelles composed of lipids, micelles prepared from synthetic polymers have more tunable sizes, drug-loading capacities, and release rates. Widely utilized copolymers of amphiphilic PLGA-PEG spontaneously assemble into micelles and have been screened across a large parameter space by Farokhzad, Langer, and coworkers; the lead candidate is an aptamer-targeted 100-nm nanoparticle containing the hydrophobic chemotherapeutic docetaxel (BIND-014) that has shown promising results in phase I clinical trials for a variety of cancers.7 The same group has now co-encapsulated docetaxel with chemotherapeutic Pt(IV) coordination complexes.8 The Pt(IV) complexes are hydrophilic and thus do not match the chemical properties of docetaxel for which the system was developed, so it was first conjugated to a hydrophobic PLA polymer prior to co-assembly in micelles, and the combination drug showed substantially higher toxicity toward cultured cancer cells compared with single-drug nanoparticles. Fahmy and coworkers used a different approach to co-formulate two very dissimilar drugs, a hydrophobic inhibitor of transforming growth factor β (TGF-β) and the hydrophilic protein interleukin-2, in the core of a 120-nm liposomal nanoparticle.9 Unlike a conventional liposome with an aqueous core, the core was a hydrophilic PEG gel. To equalize their association in the same matrix, the hydrophobic drug was complexed with the cyclic carbohydrate cyclodextrin prior to co-encapsulation. These particles showed significantly improved efficacy as tumor immunomodulators compared to free drug combinations.
Nanoparticle formulations in which drugs themselves serve as carriers provide a number of interesting opportunities; when compounded through top-down fabrication or solvent-mediated self-assembly, insoluble drugs can form homogenous colloids with tunable sizes and shapes that simply are not possible with standard FDA-approved carriers. In this issue of ACS Nano, Mitragotri and coworkers show the ability to combine three chemically disparate drugs into the same formulation in a self-assembled carrier-free system: the core is a rod-shaped 500-nm × 50-nm nanoparticle composed of the hydrophobic drug camptothecin, to which the therapeutic antibody trastuzumab (anti-HER2) is adsorbed, along with the hydrophilic drug doxil.10 The combined treatment synergistically inhibited the growth of cultured breast cancer cells and all of the components exhibited fluorescence in distinct spectral bands, allowing independent visualization of target binding.
In the near term, there is a drive to create more compact formulations than the current typical size near 100 nm in order to enhance biodistribution and targeting for solid tumors. Compact particles penetrate into tumor tissue more effectively, can enhance total tumor accumulation, and allow possible excretion through renal filtration rather than slower hepatobiliary clearance, which can reduce the chance of liver toxicity.11,12 However engineering on this size range is difficult because of the lower drug-carrying capacity per particle and the difficulty in maintaining a homogeneous size distribution. In addition, surface fouling plays a bigger role: adsorption by serum proteins alters the total size and charge more than for larger particles, drastically altering distribution and circulation times. Cheng and coworkers have recently produced silica nanoparticles in this range with much improved tumor penetration and accumulation.12 While not yet FDA-approved, silica particles are intriguing because of the expectation of biocompatibility and their capacity to bind and deliver both hydrophobic and hydrophilic small molecules as well as macromolecules.13,14
Balancing Synergy and Toxicity
For combination drug therapy it is hoped that the net effect of two drugs is synergistic, or greater than the sum of effects of each drug alone. There are many formalisms to describe synergy; one of the simplest is the combination index (CI),15 defined for the pair of drugs A and B as:
where IC50 is the drug concentration that inhibits a cellular function or behavior (e.g., cell growth) by 50%, for each individual drug or for the drug given as an A–B pair. Thus, if A and B act synergistically, smaller doses of both A and B are required to lead to the same cellular effect, so IC50 (A or B)pair < IC50 (A or B) and CI < 1 . For drugs that act additively (independently), CI is near 1, and for those that act antagonistically, CI > 1. This only describes a specific drug pair ratio; a half-inhibitory dose can be theoretically reached with any ratio between A and B and synergy can be complexly manifested across this continuum; thus a full combinatorial analysis is needed to determine the optimum combination (see Figure 3).
Figure 3.

Interactions between drugs A and B in the inhibition of a cellular effect, demonstrating synergy, additivity, or antagonism. Concentrations are defined relative to the minimum inhibitory concentration (MIC) at which the cellular effect is 100% inhibited. If A and B are similar drugs, their combined effect at equal concentrations is the same effect of one of the drugs in double the dose. For example, 0.5 MIC of drug A combined with 0.5 MIC of drug B (+ in the figure) is equivalent to 1 MIC of drug A or 1 MIC of drug B in an additive drug pair. Examples of different patterns of antagonism are depicted. Adapted with permission from ref 16. Copyright 2009 Nature Publishing Group.
Combination therapies may also show equally diverse modes of interaction in yielding toxic side effects. For cancer and other diseases of host tissue (as opposed to infectious diseases from foreign pathogens), this is a major concern because drugs interfere with healthy tissues that share the same physiological machinery as the diseased tissue. Thus, the therapeutic window, the range of drug concentrations separating therapeutic effects from toxic effects, can be small and variable between individuals. However, it has been predicted and empirically verified that selectivity and synergy are intrinsically correlated in drug combinations.17 That is, if two drugs work together synergistically to disrupt a process in diseased cells, this drug ratio will often have just an additive or antagonistic interaction in normal cells, widening the therapeutic window (see Figure 4). This is a consequence of biological complexity in organisms: all cells in a human share the same set of available proteins, but their differential levels of expression and diverse implementation is what distinguishes each cell type. Thus, as more drugs interfere with more targets, greater cell selectivity can emerge. This selectivity is expected to be further enhanced through targeted nanoparticle delivery due to a narrower range of cell types exposed, a key reason why clinical toxicity rates are lower for nanoparticle-formulated drugs.18 In addition, slow release and prolonged circulation allow synergistic dosing within the therapeutic window over a longer duration of treatment in comparison with free drugs, which often rapidly clear from circulation.
Figure 4.

In vitro screening to measure interactions between drugs A and B and to predict therapeutic windows. (a) Cell function is measured across a two-dimensional range of drug dosages in a multiwall plate. This can be performed for target cells (b, Test assay) and cells that are off target (c, Control assay). By determining the differences between the Test and Control (Ctrl), the therapeutic window can be evaluated (d) over which only target cells will be strongly impacted by the treatment. Adapted with permission from ref 17. Copyright 2009 Nature Publishing Group.
Drug Synergy Assessment and Discovery: Systems Biology and In Vitro Screening
Synergistic drug pairs are rare (4–10%)19 and are most effectively discovered through mechanistic insight and high-throughput screening. Systems biology has provided much of the theory for how we understand combination treatments and how to predict combination protein targets de novo for disease intervention. We now view these targets as components of networks in cells that channel signals toward an emergent property regulated at the gene or protein level.
Because of network interconnectivity and redundancy, simply shutting off a single protein node with a drug inhibitor is not sufficient to have a sustained effect, as the cell can compensate by rerouting the signal through altered protein expression or through mutations, resulting in resistance. Drug combinations are thus much more effective (synergistic and less disposed to resistance) if they block parallel pathways that feed into the same cellular behavior. In addition, drugs can act synergistically by enhancing the uptake or reducing the elimination or degradation of another drug (e.g., by blocking drug efflux pumps).20
In vitro screening on cultured cells is the standard empirical test for drug synergy and also helps establish dosing. Cells are typically grown in multiwell plates and exposed to a pair of drugs across a wide range of concentrations and ratios (Figure 4a). While automated liquid-handling instruments, microfluidic cell systems,21 and cell microarrays22 have made drug pair screening manageable, costs grow rapidly as more drugs and more cell types are added. Screening for synergy of combinations of three or more drugs is expected to improve cellular selectivity but is very expensive. However, a surprising finding is that, at least in bacteria, three or more drugs in combination appear to act only through pairwise interactions, no matter their mechanism.23 That is, drugs A, B, and C may show complicated pairwise synergy or antagonism in the 2-drug combinations A–B, B–C, or A–C, but when all 3 are combined (A–B–C) no higher order interactions arise such that the pairwise interactions are entirely sufficient to predict cell response. This counterintuitive finding of biological simplicity has only been verified for bacterial systems and it is not clear how this will translate to more complex eukaryotic cells or multicellular organisms, but it greatly simplifies drug synergy studies in bacteria.
Screening nanoparticle-based therapeutics presents a unique challenge because of limited similarity between mechanisms of nanoparticle drug delivery in vitro and in vivo. Unlike small-molecule drugs, nanoparticles tend to remain compartmentalized in the bloodstream for long periods of time and eventually accumulate in the liver, spleen, and lymphatics, and there are two distinguishable but convolved mechanisms by which they delivery drugs: slow release during systemic circulation and local release after target binding or uptake. Controlled systemic release is likely the most relevant mechanism in human subjects, but modeling this as an isolated effect in cultured cells is difficult due to uptake through endocytosis or phagocytosis, and because of effects like particle sedimentation that are not expected to be relevant in vivo. Delivery after target binding is simple to study in cultured cells, however we have poor understanding of its mechanisms in vivo for diseases such as cancer. Culture systems that mimic the vasculature and microenvironments of tissues have been developed but are not yet sufficient for wide screening of drug combination formulations.24
Screening is also performed to assay toxic effects in “healthy” cells, using a panel of cells thought to represent the most susceptible tissues (Figure 4b–c). Here, targeted nanotherapies may have a distinct advantage: whereas the susceptible targets for small-molecule drugs are vast in number, resulting in diverse toxicological outcomes in vivo (e.g., cardiotoxicity, hepatotoxicity) for which in vitro screening is not yet possible, only a select number of tissues and microenvironments will be directly exposed to nanoparticle drugs through uptake mechanisms. Thus, it may be possible to recapitulate these environments with just a handful of culture systems to predict clinical toxicity.
Microenvironment Considerations
In vitro screening is typically performed on cell monolayers, but cells in human tissue exist in three-dimensional microenvironments where cell-cell interactions, cell-matrix interactions, and gradients of soluble factors play important roles in dictating responses to drugs.24 This is particularly relevant for tumor tissue, for which protein expression patterns are widely distributed across tumor cells, leading to heterogeneous drug responses. For example, hypoxic (low oxygen) microenvironments in tumors are thought to harbor quiescent tumor cell populations that survive drug treatment and repopulate the tumor.25 Heath and coworkers screened the response of glioblastoma tumor cells toward promising drug inhibitors of the protein mTOR and observed that hypoxia-induced alterations in protein expression resulted in cells that do not respond to this therapy and thus may require specialized therapeutic interventions.26
It is not known if nanoparticles can be effective treatments for hypoxic tumor microenvironments, as hypoxia increases with distance from blood vessels, inversely with the penetration depth of macromolecular drugs, yet vascular permeability has been found to correlate with hypoxia in certain tumors.25 However, it may be possible to modulate the microenvironments available to nanotherapies; drugs that inhibit vascular dysfunction such as the antibody bevacizumab (an inhibitor of vascular endothelial growth factor receptor, VEGFR) can increase tumor penetration of small nanoparticles (12 nm) through vascular normalization, and improve outcomes for certain cancer patients treated with chemotherapeutics.25 Microenvironments that are more directly relevant for nanoparticles are proximal to blood vessels and include the “microenvironment of metastasis” in breast tumors, which involves a cell–cell interaction loop between tumor cells and macrophages.27 However recapitulating such a complex microenvironment in vitro for ratiometric screening of candidate nanoparticle drugs that knock down this loop is a major challenge.
Preclinical Animal Models
In vitro tests allow the determination of ideal drug ratios, but animal models of disease are the most critical tools to facilitate clinical translation. Unfortunately, a widely cited reason for the failure of a large fraction of drugs in the clinic is the disparity between human disease and models of disease in small animals, particularly mice.28 Indeed, nanotherapy efficacy in the clinic has been modest and the major advantage has been reduced toxicity rates, despite outstanding efficacy in preclinical studies. Targeting efficiency may be a central problem; whereas some tissues such as tumors in mice typically accumulate 1% of a systemic dose of a targeted macromolecule (and even up to 20% in some cases), it is estimated that <0.01% of antibodies reach their targets in human tumors.29–31 A major problem is that we have a limited understanding of targeting in humans and do not know how to evaluate and to employ animal models that reflect appropriate human characteristics. This is especially true for tumors, for which intratumoral delivery is mediated by the enhanced permeability and retention (EPR) effect resulting from hyperpermeable vasculatures and insufficient lymphatic drainage. Evidence of the EPR effect in human patients is surprisingly sparse32,33 and outcomes of targeted delivery mediated by the EPR effect are often conflicting in mouse tumor models.34
Common animal models lack a variety of significant characteristics that are known to be critical in human disease. For example, mouse tumors are typically grown through subcutaneous injection of highly passaged cell lines in inbred immune-compromised mice, models that do not mirror important microenvironment or immunological components of human disease, its slow development, or the genetic heterogeneity between patients and within tumors.28 Nanoparticle targeting studies need to take advantage of a wealth of new and promising disease models. A recently emerging practice is the use of patient-derived cells and tissues for drug screening: tumors isolated from patients and grown in mice retain components of the human microenvironment and the heterogenous, patient-specific biology and, most importantly, reproducibly respond to drug treatments in the same way as the original patient.35 But even these models do not retain a native immune system. Spontaneous or induced tumor models in immunocompetent mice (e.g., PyMT-MMTV36) can fill this void, but these still do not express human antigens. While there is no ideal system, these models should be appropriately implemented to avoid the common pitfalls that have precluded the clinical success of so many other therapies in previous studies.
Clinical Outlook
As combination drug nanoparticles advance in precision during the age of personalized medicine, it is reasonable to expect the emergence of effective multidrug treatments that were previously limited by both lack of efficacy and concern for toxicity. A clinician could prescribe individually formulated combination treatments based on tumor tissue expression of actionable and druggable genes with mechanistic knowledge from patient-derived tumor xenograft models. Single-formulation cocktails could be designed to provide long-duration exposure to synergistic drug combinations, allow staged release to “prime” cells toward potent drugs or to inhibit side effects, and allow high efficiency local delivery to specific disease microenvironments. Critical insights are needed to proceed toward these goals: we need to understand mechanisms by which systemically administered nanoparticles function in humans, to understand the contexts in which in vitro screens and animal studies are clinically relevant, and to define key attributes of current nanoformulations that limit cellular and tissue selectivity. To increase the relevance of ongoing studies, it is important for the nanotechnology community to make use of unique resources that are becoming available in screening technologies, animal models, genomic data, and bioinformatics and systems maps of drug targets, both for validating new nanoparticle combination therapies and for ensuring that the mechanistic rationale behind these new treatment paradigms are patient-centric to improve both quality and longevity of life.
Acknowledgments
This work was supported by the NIH (R00CA153914) and seed funds from the UIUC ACES FIRE program.
Footnotes
Conflict of Interest. The authors declare no competing financial interest.
References
- 1.Woodcock J, Griffin JP, Behrman RE. Development of Novel Combination Therapies. N. Engl. J. Med. 2011;364:985–987. doi: 10.1056/NEJMp1101548. [DOI] [PubMed] [Google Scholar]
- 2.Wang AZ, Langer R, Farokhzad OC. Nanoparticle Delivery of Cancer Drugs. Ann. Rev. Med. 2012;63:185–198. doi: 10.1146/annurev-med-040210-162544. [DOI] [PubMed] [Google Scholar]
- 3.Al-Lazikani B, Banerji U, Workman P. Combinatorial Drug Therapy for Cancer in the Post-Genomic Era. Nat. Biotechnol. 2012;30:1–13. doi: 10.1038/nbt.2284. [DOI] [PubMed] [Google Scholar]
- 4.Batist G, Gelmon KA, Chi KN, Miller WH, Chia SKL, Mayer LD, Swenson CE, Janoff AS, Louie AC. Safety, Pharmacokinetics, and Efficacy of CPX-1 Liposome Injection in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2009;15:692–700. doi: 10.1158/1078-0432.CCR-08-0515. [DOI] [PubMed] [Google Scholar]
- 5.Tardi P, Johnstone S, Harasyrn N, Xie SW, Harasyrn T, Zisman N, Harvie P, Bermudes D, Mayer L. In Vivo Maintenance of Synergistic Cytarabine:Daunorubicin Ratios Greatly Enhances Therapeutic Efficacy. Leuk. Res. 2009;33:129–139. doi: 10.1016/j.leukres.2008.06.028. [DOI] [PubMed] [Google Scholar]
- 6.Katragadda U, Fan W, Wang YZ, Teng Q, Tan C. Combined Delivery of Paclitaxel and Tanespimycin via Micellar Nanocarriers: Pharmacokinetics, Efficacy and Metabolomic Analysis. PLoS ONE. 2013;8:e58619. doi: 10.1371/journal.pone.0058619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hrkach J, Von Hoff D, Ali MM, Andrianova E, Auer J, Campbell T, De Witt D, Figa M, Figueiredo M, Horhota A, et al. Preclinical Development and Clinical Translation of a PSMA-Targeted Docetaxel Nanoparticle with a Differentiated Pharmacological Profile. Sci. Transl. Med. 2012;4 doi: 10.1126/scitranslmed.3003651. 128ra39. [DOI] [PubMed] [Google Scholar]
- 8.Kolishetti N, Dhar S, Valencia PM, Lin LQ, Karnik R, Lippard SJ, Langer R, Farokhzad OC. Engineering of Self-Assembled Nanoparticle Platform for Precisely Controlled Combination Drug Therapy. Proc. Natl. Acad. Sci. U.S.A. 2010;107:17939–17944. doi: 10.1073/pnas.1011368107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park J, Wrzesinski SH, Stern E, Look M, Criscione J, Ragheb R, Jay SM, Demento SL, Agawu A, Limon PL, et al. Combination Delivery of TGF-Beta Inhibitor and IL-2 by Nanoscale Liposomal Polymeric Gels Enhances Tumour Immunotherapy. Nat. Mater. 2012;11:895–905. doi: 10.1038/nmat3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mitragotri S, Barua S. Synergistic Targeting of Cell Membrane, Cytoplasm, and Nucleus of Cancer Cells using Rod-Shaped Nanoparticles. ACS Nano. 2013;7 doi: 10.1021/nn403913k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi HS, Liu W, Liu F, Nasr K, Misra P, Bawendi M, Frangioni JV. Design Considerations for Tumor-Targeted Nanoparticles. Nat. Nanotech. 2010;5:42–47. doi: 10.1038/nnano.2009.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tang L, Fan TM, Borst LB, Cheng JJ. Synthesis and Biological Response of Size- Specific, Monodisperse Drug-Silica Nanoconjugates. ACS Nano. 2012;6:3954–3966. doi: 10.1021/nn300149c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meng HA, Liong M, Xia TA, Li ZX, Ji ZX, Zink JI, Nel AE. Engineered Design of Mesoporous Silica Nanoparticles to Deliver Doxorubicin and P-Glycoprotein siRNA to Overcome Drug Resistance in a Cancer Cell Line. ACS Nano. 2010;4:4539–4550. doi: 10.1021/nn100690m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ashley CE, Carnes EC, Phillips GK, Padilla D, Durfee PN, Brown PA, Hanna TN, Liu JW, Phillips B, Carter MB, et al. The Targeted Delivery of Multicomponent Cargos to Cancer Cells by Nanoporous Particle-Supported Lipid Bilayers. Nat. Mater. 2011;10:389–397. doi: 10.1038/nmat2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chou TC, Talalay P. Quantitative Analysis of Dose-Effect Relationships: The Combined Effects of Multiple Drugs or Enzyme Inhibitors. Adv. Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 16.Yeh PJ, Hegreness MJ, Aiden AP, Kishony R. Drug Interactions and the Evolution of Antibiotic Resistance. Nat. Rev. Microbiol. 2009;7:460–466. doi: 10.1038/nrmicro2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lehar J, Krueger AS, Avery W, Heilbut AM, Johansen LM, Price ER, Rickles RJ, Short GF, Staunton JE, Jin XW, et al. Synergistic Drug Combinations Tend To Improve Therapeutically Relevant Selectivity. Nat. Biotechnol. 2009;27:659–666. doi: 10.1038/nbt.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi JJ, Xiao ZY, Kamaly N, Farokhzad OC. Self-Assembled Targeted Nanoparticles: Evolution of Technologies and Bench to Bedside Translation. Acc. Chem. Res. 2011;44:1123–1134. doi: 10.1021/ar200054n. [DOI] [PubMed] [Google Scholar]
- 19.Cokol M, Chua HN, Tasan M, Mutlu B, Weinstein ZB, Suzuki Y, Nergiz ME, Costanzo M, Baryshnikova A, Giaever G, et al. Systematic Exploration of Synergistic Drug Pairs. Mol. Syst. Biol. 2011;7:544. doi: 10.1038/msb.2011.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jia J, Zhu F, Ma XH, Cao ZWW, Li YXX, Chen YZ. Mechanisms of Drug Combinations: Interaction and Network Perspectives. Nat. Rev. Drug Discov. 2009;8:111–128. doi: 10.1038/nrd2683. [DOI] [PubMed] [Google Scholar]
- 21.Shi QH, Qin LD, Wei W, Geng F, Fan R, Shin YS, Guo DL, Hood L, Mischel PS, Heath JR. Single-Cell Proteomic Chip for Profiling Intracellular Signaling Pathways in Single Tumor Cells. Proc. Natl. Acad. Sci. U.S.A. 2012;109:419–424. doi: 10.1073/pnas.1110865109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wood KC, Konieczkowski DJ, Johannessen CM, Boehm JS, Tamayo P, Botvinnik OB, Mesirov JP, Hahn WC, Root DE, Garraway LA, et al. MicroSCALE Screening Reveals Genetic Modifiers of Therapeutic Response in Melanoma. Sci. Signaling. 2012;5 doi: 10.1126/scisignal.2002612. rs4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wood K, Nishida S, Sontag ED, Cluzel P. Mechanism-Independent Method for Predicting Response to Multidrug Combinations in Bacteria. Proc. Natl. Acad. Sci. U.S.A. 2012;109:12254–12259. doi: 10.1073/pnas.1201281109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zervantonakis IK, Hughes-Alford SK, Charest JL, Condeelis JS, Gertler FB, Kamm RD. Three-Dimensional Microfluidic Model for Tumor Cell Intravasation and Endothelial Barrier Function. Proc. Natl. Acad. Sci. U.S. A. 2012;109:13515–13520. doi: 10.1073/pnas.1210182109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D, Jain RK. Normalization of the Vasculature for Treatment of Cancer and Other Diseases. Physiol. Rev. 2011;91:1071–1121. doi: 10.1152/physrev.00038.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wei W, Shi QH, Remacle F, Qin LD, Shackelford DB, Shin YS, Mischel PS, Levine RD, Heath JR. Hypoxia Induces a Phase Transition Within a Kinase Signaling Network in Cancer Cells. Proc. Natl. Acad. Sci. U.S. A. 2013;110:E1352–E1360. doi: 10.1073/pnas.1303060110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roussos ET, Condeelis JS, Patsialou A. Chemotaxis in Cancer. Nat. Rev. Cancer. 2011;11:573–587. doi: 10.1038/nrc3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kopetz S, Lemos R, Powis G. The Promise of Patient-Derived Xenografts: The Best Laid Plans of Mice and Men. Clin. Cancer Res. 2012;18:5160–5162. doi: 10.1158/1078-0432.CCR-12-2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olafsen T, Kenanova VE, Sundaresan G, Anderson AL, Crow D, Yazaki PJ, Li L, Press MF, Williams LE, Wong JYC, et al. Optimizing Radiolabeled Engineered Anti-p185(HER2) Antibody Fragments for In Vivo Imaging. Cancer Res. 2005;65:5907–5916. doi: 10.1158/0008-5472.CAN-04-4472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schmidt MM, Wittrup KD. A Modeling Analysis of the Effects of Molecular Size and Binding Affinity on Tumor Targeting. Mol. Cancer Ther. 2009;8:2861–2871. doi: 10.1158/1535-7163.MCT-09-0195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carver LA, Schnitzer JE. Caveolae: Mining Little Caves for New Cancer Targets. Nat. Rev. Cancer. 2003;3:571–581. doi: 10.1038/nrc1146. [DOI] [PubMed] [Google Scholar]
- 32.Prabhakar U, Maeda H, Jain RK, Sevick-Muraca EM, Zamboni W, Farokhzad OC, Barry ST, Gabizon A, Grodzinski P, Blakey DC. Challenges and Key Considerations of the Enhanced Permeability and Retention Effect for Nanomedicine Drug Delivery in Oncology. Cancer Res. 2013;73:2412–2417. doi: 10.1158/0008-5472.CAN-12-4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chrastina A, Massey KA, Schnitzer JE. Overcoming In Vivo Barriers to Targeted Nanodelivery. Wiley Interdisciplinary Rev.–Nanomed. Nanobiotechnol. 2011;3:421–437. doi: 10.1002/wnan.143. [DOI] [PubMed] [Google Scholar]
- 34.Huang XH, Peng XH, Wang YQ, Wang YX, Shin DM, El-Sayed MA, Nie SM. A Reexamination of Active and Passive Tumor Targeting by Using Rod-Shaped Gold Nanocrystals and Covalently Conjugated Peptide Ligands. ACS Nano. 2010;4:5887–5896. doi: 10.1021/nn102055s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Joo KM, Kim J, Jin J, Kim M, Seol HJ, Muradov J, Yang H, Choi YL, Park WY, Kong DS, et al. Patient-Specific Orthotopic Glioblastoma Xenograft Models Recapitulate the Histopathology and Biology of Human Glioblastomas In Situ. Cell Reports. 2013;3:260–273. doi: 10.1016/j.celrep.2012.12.013. [DOI] [PubMed] [Google Scholar]
- 36.Lin EY, Jones JG, Li P, Zhu UY, Whitney KD, Muller WJ, Pollard JW. Progression to Malignancy in the Polyoma Middle T Oncoprotein Mouse Breast Cancer Model Provides a Reliable Model for Human Diseases. Am. J. Pathol. 2003;163:2113–2126. doi: 10.1016/S0002-9440(10)63568-7. [DOI] [PMC free article] [PubMed] [Google Scholar]