

Abstract
The GXGD proteases are polytopic membrane proteins with catalytic activities against membrane-spanning substrates that require a pair of aspartyl residues1–4. Representative members of the family include preflagellin peptidase, type 4 prepilin peptidase, presenilin and signal peptide peptidase. Many GXGD proteases are important in medicine. For example, type 4 prepilin peptidase may contribute to bacterial pathogenesis5–7, and mutations in presenilin are associated with Alzheimer's disease8–10. As yet, there is no atomic-resolution structure in this protease family. Here we report the crystal structure of FlaK, a preflagellin peptidase from Methanococcus maripaludis, solved at 3.6Å resolution. The structure contains six transmembrane helices. The GXGD motif and a short transmembrane helix, helix 4, are positioned at the centre, surrounded by other transmembrane helices. The crystal structure indicates that the protease must undergo conformational changes to bring the GXGD motif and a second essential aspartyl residue from transmembrane helix 1 into close proximity for catalysis. A comparison of the crystal structure with models of presenilin derived from biochemical analysis reveals three common transmembrane segments that are similarly arranged around the active site. This observation reinforces the idea that the prokaryotic and human proteases are evolutionarily related11,12. The crystal structure presented here provides a framework for understanding the mechanism of the GXGD proteases, and may facilitate the rational design of inhibitors that target specific members of the family.
Archaeal preflagellins and bacterial type-4 prepilins, both of which are type-II (Nin -Cout) membrane proteins13,14, are synthesized with short, positively charged leader peptides5,15. They are cleaved in the membrane, at a site a few residues upstream of the hydrophobic membrane-spanning sequence, by preflagellin peptidase (PFP) and type-4 prepilin peptidase (TFPP), respectively, before being secreted and incorporated into the mature flagellum or type-4 pilus (Supplementary Fig. 1a). We have crystallized FlaK, a prototypic archaeal PFP from M. maripaludis16. The membrane protease maintained a native-like conformation throughout crystallization because both the crystallization drop and the dissolved crystals showed robust enzymatic activity (Supplementary Fig. 1b). The structure was determined by single-wavelength anomalous dispersion using a Se-Met-substituted crystal (Supplementary Fig. 2 and Supplementary Table 1).
The crystal structure shows that FlaK contains two compactly folded domains: a mostly α-helical membrane-spanning domain and a soluble domain with four anti-parallel β-strands (Fig. 1; the soluble domain is disordered in one of the two FlaK molecules in the asymmetric unit). Previous predictions correctly located all six transmembrane (TM) segments4, but the crystal structure is more complex (Supplementary Fig. 3a). One major deviation from the prediction occurs around TM4 (yellow in Fig. 1). The hydrophilic loop between TM3 and TM4 does not protrude into the cytoplasm as predicted; instead, it lowers towards the centre of the bundle of TM helices, and is followed by a short TM helix, α4. The last TM helix (α6) is also short (pink in Fig. 1), and seems unable to cross the lipid bilayer completely. The protein segments immediately after α4 and α6 form an unusual structure that protrudes sideways from the base of the TM helices. This feature does not seem to be an artefact of crystallization. A comparison between the two copies of FlaK in the asymmetric unit shows that the unusual structure is identically positioned, despite the fact that it is involved in different crystal packing interactions (Supplementary Fig. 3b). The amphipathic nature of the structure is also consistent with its position next to the TM helices, and with the possibility that it may interact peripherally with the membrane. In the big loop between α4 and α5 (including α4a), and in the carboxy-terminal segment (including α6a), all the polar side chains point downwards away from the membrane, whereas most hydrophobic side chains point up or sideways to interact either with the TM helices, or with lipids that surround the helices. Furthermore, there is a conserved asparagine on α5 (Asn 120), the side chain of which points outwards to form a hydrogen bond with the carbonyl oxygen of Gly 220 from the extended segment between α6 and α6a (Fig. 1d). If the carboxy-terminus of the protein were positioned elsewhere, Asn 120 would become unfavourably exposed to the lipid.
Figure 1. The structure of FlaK.
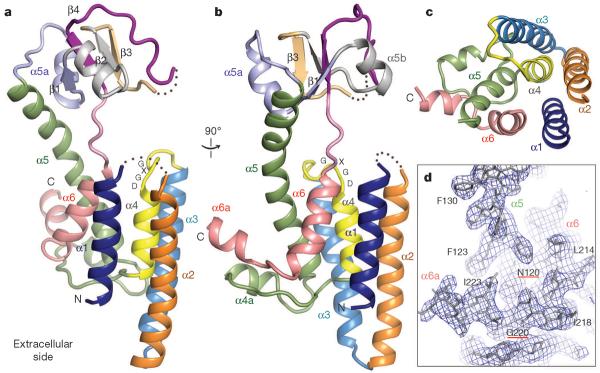
Shown here are cartoon representations of molecule A, one of the two FlaK molecules in the asymmetric unit. In molecule B, the soluble domain cannot be completely traced owing to disorder. These illustrations, as well as those in Figs 2c, 3a, b and 4b, c, were generated by PyMOL (http://www.pymol.org). a, b, Two views of the molecule from the side. The secondary structural elements and the GXGD motif are labelled. c, A view from the cytosolic side of the membrane. For clarity, the soluble domain (including part of α5) is removed. d, A portion of the final 2Fo − Fc electron density map (contoured at 1σ level) showing the position of α6a relative to TM helix α5.
To accommodate the unusual peripheral structure (α4a and α6a), and the short TM helix α6, the other TM helices must be tilted in the membrane. The tilting is required to avoid positioning charged groups such as Asp 26, Asp 49 and Asp 225 in the hydrophobic region of the membrane (Fig. 2). The tilting also makes α6 roughly perpendicular to the membrane plane, enabling it to go through the lipid bilayer with the shortest distance. A more thorough examination of the distribution of amino acids in the TM region supports the tilted model. As shown in Fig. 2c, the lower boundary of the membrane is roughly marked by a thin belt of acidic residues (red), basic residues (blue), asparagines and glutamines (green). Four tyrosine residues (Tyr 42, Tyr 50, Tyr 98 and Tyr 109) are also found within this belt. Tyrosine and tryptophan often cluster to the interface between water and lipid17. The upper boundary of the membrane probably corresponds to a plane that goes through Tyr 27, Trp 29 and Tyr 68. Although Glu 3, Tyr 4, Asn 120 and Tyr 213 appear between the two boundaries, a closer inspection shows that the polar groups on their side chains are not directly exposed to the lipid. According to the tilted model, the membrane has to be constricted around the protease. This feature was previously thought to be unique to the intramembrane proteases18–20.
Figure 2. FlaK is tilted in the membrane.
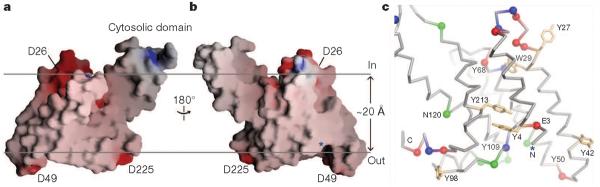
Molecule B is shown in this figure because its TM region is better defined in the electron density map, and does not contain any breaks. a, b, The molecular surface, colour-coded by electrostatic potential. The two horizontal lines roughly mark the hydrophobic belt around the membrane protease. The N terminus of the protein is labelled by a blue asterisk. c, The Cα trace of the TM region with the same orientation as in panel b. Red spheres, negatively charged residues; blue spheres, positively charged residues; green spheres, asparagines and glutamines. Tyrosines and tryptophans are shown as orange stick models (undefined side chains are not shown).
The two aspartyl residues that are essential for catalysis (Asp 18 and Asp 79) are located at the ends of TM helices α1 and α4, respectively. Despite the relatively low resolution at which the crystal structure was solved, α1 and α4 are clearly defined in the electron density map, and the experimentally determined selenium sites in both helices are consistent with the register of protein sequence with the density (Fig. 3a). The positions of α1 and α4 are almost identical in the two independently modelled FlaK molecules in the asymmetric unit of the crystal (Supplementary Fig. 3b). The spatial relationship between the two helices is, however, surprising from a functional point of view because it creates a 12Å gap between Asp 18 and Asp 79. To investigate whether the FlaK crystal structure represents a non-active conformation of the protease, we introduced a pair of cysteines (E25C and I206C) to the two TM helices (α2 and α6) that are on the opposite side of the gap (Fig. 3b). The double mutant (E25C/I206C) was proteolytically active, indicating that its conformation was not markedly perturbed by the mutations. As shown in Fig. 3c, d, the two cysteines can be readily crosslinked by 1,2-ethanedithiol dimethanesulphonate (M2M). Given its short length, the crosslinker has to lie inside the gap between α1 and α4 to bridge Cys 25 and Cys 206. Taken together, these results indicate that, in the absence of substrate, the membrane protease can adopt an inactive conformation in which the two catalytic aspartyl residues are structurally uncoupled. Consistent with this idea, we found that cross-linking between α2 and α6, which prevents movement of Asp 18 and Asp 79 towards each other, completely eliminated protease activity (Fig. 3e). Breaking the crosslinking disulphide bonds with dithiothreitol (DTT) fully restored protease activity. The observation that crosslinking in the membrane fraction is less complete than in detergent solutions indicates that FlaK may assume additional conformations in the lipid bilayer (Fig. 3c). The behaviour of FlaK is similar to that of presenilin in these regards: the human protease also switches between at least two conformations and in one conformation the two catalytic aspartates do not closely oppose each other21,22.
Figure 3. The uncoupling between Asp 18 and Asp 79.

a, The 2Fo 2 Fc map (contoured at 1σ level; blue) and the anomalous Fourier map (contoured at 4σ levels; green) around α1 and α4. b, A view from the cytosolic side of the membrane. The distance from Cγ1 of Ile 206 to Cγ of Glu 25 is ~6 Å. c, Crosslinking causes the E25C/I206C mutant protein to migrate faster in the SDS–polyacrylamide gel (to a position marked by the asterisk); adding DTT breaks the disulphide linkage. FC-12, foscholine-12. The left lane is a molecular weight marker. d, Partial proteolysis confirms the covalent linkage between Cys 25 and Cys 206. Chymotrypsin (chymo) cleaves FlaK twice between TM domains 5 and 6, generating two N-terminal fragments (N1 and N2). For crosslinked mutant protein, the N-terminal fragments (containing Cys 25) remain attached to the C-terminal fragment (containing Cys 206). e, Crosslinking between Cys 25 and Cys 206 renders the protease completely inactive; treatment with DTT fully restores activity. Wild-type (WT) FlaK is not affected by M2M and DTT. The antibody used in the western blots (panels c–e) detects the N-terminal His tag of FlaK and the C-terminal His tag of FlaB2 (substrate).
Three regions in the FlaK sequence are highly conserved (Supplementary Fig. 4). The first two are centred on Asp 18 and on the GXGD motif (Asp 79). The third region, which corresponds to the sequence around the amino terminus of TM helix α6, is shown by the crystal structure to be near the active site as well. The roles of individual residues from the conserved regions in the protease mechanism were probed by mutagenesis (Supplementary Fig. 5). Three main lessons can be learned from this analysis. First, Glu 23, Glu 25 and Asp 26, the only three acidic residues around the active site, do not seem to be essential for the binding of the positively charged leader peptide. Second, among the three glycine residues in the GXGD motif, Gly 76 is the most critical for function. Because Gly 76 is not closely packed against other residues, the large reduction of activity in the G76A mutant probably results from a partial loss of backbone flexibility in this important region. Third, most mutations from Pro 201 to Pro 208 produce a small effect; P208A, however, is different in that it markedly enhances enzymatic activity. Pro 208 is packed against TM helix α5, and points away from the active site. If the conformational change in the protease involves movement of TM helices α1, α4 and α6 (Fig. 3b), then the altered packing between α6 and α5 might have facilitated such a change.
Presenilin has nine TM segments23,24 (Fig. 4a). The last four segments (TM6–TM9) of presenilin share homology with signal peptide peptidase, and are thought to constitute the core of the protease25. Previous cysteine-scanning mutagenesis and crosslinking experiments indicated that the active site of presenilin is housed in an open hydrophilic cavity21,26, surrounded by the two TM segments (TM6 and TM7) that carry the catalytic aspartates (Asp 257 and Asp 385), and by the C-terminal TM9, which bears a conserved Pro-Ala-Leu motif22,27. The crystal structure of FlaK now shows that the active site of the prokaryotic protease has a similar architecture. As illustrated in Fig. 4a, the TM segments TM1, TM4 and TM6 of FlaK would be equivalent to presenilin's TM6, TM7 and TM9. In FlaK, there is a highly conserved leucine (Leu 11) seven residues upstream of the catalytic Asp 18 on TM1 (Supplementary Fig. 4). Leu 11 is packed against Leu 86 from TM4 (Fig. 4b). Leu 86, which is also conserved, is seven residues down-stream of the catalytic Asp 79. The residues in presenilin corresponding to Leu 11 and Leu 86 would be Leu 250 and Leu 392, respectively (seven residues away from the catalytic Asp 257 and Asp 385). Both Leu 250 and Leu 392 are highly conserved28. The fact that Asp 257 and Asp 385 are close to each other indicates that Leu 250 and Leu 392, which are on the same side of the helices as the aspartates, may also interact (Fig. 4c). In FlaK, Leu 11 makes additional contact with Tyr 213 from TM6. In presenilin, Leu 250 also seems to interact with the last TM segment because it can be readily crosslinked to many positions on TM927. Besides the packing of the three key helices, presenilin and FlaK share other features. TM7 of presenilin, like its counterpart in FlaK, also seems to contain two structural elements: an exposed region bearing the GXGD motif and a short, tightly packed hydrophobic helix26. The last TM segments in both proteases have conserved proline residues near the N terminus, and are followed by an amphipathic helix that interacts peripherally with the membrane27. It is important to note that presenilin lacks the equivalent of FlaK's TM segments 2 and 3. Therefore, other TM helices may join the central three TM segments to complete the active site. The hydrophobic domain VII, which undergoes endo-proteolysis1, must also be bound initially at the active site22.
Figure 4. Structural comparison between FlaK and presenilin-1.

a, Topology diagrams of FlaK and presenilin. The three key TM segments are highlighted in red. The grey boxes represent membrane. Arrows indicate the direction of the helices (N to C termini). b, FlaK viewed from the cytosolic side of the membrane. Asp 18, the GXGD motif and the conserved Pro 204 are shown in yellow. The two conserved leucines are shown as stick models. c, Packing of the three key TM helices in presenilin. This model is similar to the one proposed in ref. 27, but is the mirror image of another model22. Leu 250 from TM6 is hypothesized to mediate packing with TM7 (through Leu 392) and TM9.
The structure of FlaK's active site is fundamentally different from that of pepsin, a classic aspartyl protease29, in that it lacks an internal two-fold symmetry and its two catalytic aspartyl residues are not rigidly fixed. Membrane proteases have evolved unique mechanisms to conduct catalysis inside lipid bilayers. For example, rhomboid serine proteases use a surface cap to control access to a preformed and membrane-embedded Ser–His catalytic dyad30. The uncoupling between two catalytic aspartyl residues, indicated by the crystal structure described here and by earlier biochemical studies21,22, may represent a general mechanism that is widely adopted by the GXGD proteases. Such an uncoupling mechanism could potentially have an important role in regulating catalysis.
METHODS
Protein expression and purification
Many PFPs and TFPPs were screened for crystallization. Most of the genes were amplified by PCR from genomic DNAs purchased from ATCC. The genes for Pseudomonas aeruginosa PilD/PilA, E. coli BfpP and Dichelobacter nodosus FimP were gifts from S. Lory, M. Donnenberg and J. Rood, respectively. MmarC6_0338 (encoding FlaK) was amplified from the genomic DNA of M. maripaludis strain C6, and cloned into pET-28a. To facilitate removal of the N-terminal His tag, a Gly-Ser-Gly-Ser sequence was inserted between the thrombin site and FlaK sequence. Mvol_1295 (encoding FlaB2) was amplified from the genomic DNA of Methanococcus voltae strain A3 and cloned into pET-43b with a C-terminal His tag16. FlaK was expressed in E. coli Rosetta 2 (DE3) cells (Novagen), grown in Luria broth. FlaB2 was similarly overexpressed in C43(DE3) cells (Lucigen). To generate Se-Met FlaK, cells were cultured at 37 °C in M9 minimum media supplemented with Se-Met, then induced by 1 mM β-D-thiogalactopyranoside (IPTG) at an absorbance at 600 nm (A600) of 0.6, and grown at 20 °C for 16 h before collection. Cytoplasmic membranes were prepared by the spheroplast method31 and suspended in a buffer containing 50 mM sodium phosphate (pH 7.2), 500 mM NaCl, 5 mM β-mercaptoethanol and a cocktail of complete protease inhibitors (tablet without EDTA, Roche Diagnostics). For solubilization, powder of foscholine-12 (Anatrace) was added to the membrane suspension to achieve a final concentration of 1% (w/v). The His-tagged protein was eluted from a TALON metal affinity column (Clontech) in 50 mM sodium phosphate (pH 7.2), 500 mM NaCl, 200 mM imidazole, 5 mM β-mercaptoethanol and 0.1% foscholine-12. After passing through a Sephadex G-25 desalting column, the sample was cleaved by thrombin overnight at 22 °C. Finally, the protein was loaded onto a Superdex-200 column (GE Healthcare) equilibrated with 20 mM HEPES (pH 7.3), 100 mM NaCl, 1 mM TCEP and 0.1% foscholine-12. The peak fraction was pooled, concentrated to 10 mg ml−1 and dialysed against 20 mM HEPES (pH 7.3), 100 mM NaCl, 1 mM TCEP and 0.06% Cymal-6 (Anatrace) at 4 °C for 8 days. About 3 mg of purified membrane protein could be obtained for crystallization trials from 1 litre of bacterial culture.
Crystallization and structure determination
The sitting-drop method was used to prepare Se-Met FlaK crystals: 1 μl of protein solution (4 mg ml−1; the lower concentration is due to precipitation during dialysis) was mixed with 1 μl of well solution containing 30% PEG 300, 50 mM glycine (pH 9.5) and 100 mM NaCl. Needle-shaped crystals usually appeared in 2 days at 22 °C and grew to full size in 1 week. The crystals were dehydrated by equilibrating for 24 h against a well solution containing 40% PEG 300, before direct flash-freezing in liquid nitrogen. Screening and data collection were performed at the national synchrotron light source (X25 and X29) and at the advanced photon source (24-ID-C and E). All diffraction data were processed by HKL2000 (ref. 32). The structure was determined by single-wavelength anomalous dispersion33 using a highly redundant data set which was generated by merging four data sets collected at four different spots on a single Se-Met crystal at 24-ID-C. The same data set was used in refinement (Supplementary Table 1). The selenium sites and the initial phases were determined by hkl2map34. The experimental electron density map confirmed the presence of two FlaK molecules in the asymmetric unit, and clearly showed all the TM helices (Supplementary Fig. 2). The soluble domain in molecule A was visible but could not yet be traced; the soluble domain in molecule B was mostly missing. Averaging the TM regions of the two molecules by dm35 improved the clarity of the map. Modelling of the polypeptide chains using O36 was assisted by known Se sites (Supplementary Fig. 6). After rounds of model building and refinement by CNS37, the phases were sufficiently improved to allow complete tracing of the soluble domain in molecule A. The final model was refined by CNS and refmac5 (ref. 38). The electrostatic potential surfaces shown in Fig. 2a, b were generated by GRASP39.
FlaK activity assay
The enzymatic activity of FlaK was measured according to ref. 4. In brief, membranes containing overexpressed FlaK or FlaB2 were prepared using the spheroplast method, and were re-suspended in phosphate buffer. The membrane fractions were then mixed and the reaction, at 22 °C, was initiated by adding a ×5 reaction buffer containing 2.5% Triton X-100 and 100 mM HEPES (pH 7.3). The reaction was stopped by mixing with SDS–polyacrylamide gel electrophoresis sample-loading buffer. The reaction mixture was examined by western blot using an anti-His-tag antibody (Calbiochem). The purified FlaK in detergent solutions was assayed similarly. The two assays shown in Supplementary Fig. 1b were conducted at 22 °C for 120 min and 90 min, respectively: in the top panel, a large amount of protease (as indicated by the protease band) and a long incubation time were used to exclude the possibility of residual enzymatic activity in the asparagine mutants. In Supplementary Fig. 5, a shorter assay time (45 min) and a smaller amount of protease were used so that both the intact and processed FlaB2 are visible: in this setting, the amount of intact substrate is a good indicator of the reaction rate. The amount of protease used in the assay was reflected in the lower control panel, where the loading (of FlaK alone) was ten times that used in the assay, to increase the visibility of the protease. The same amount of protease was used in the assay shown in Fig. 3e, but the reaction time was longer (120 min).
Chemical crosslinking
The membrane preparation containing the E25C/I206C double mutant was suspended in a buffer containing 50mM sodium phosphate (pH 8.0) and 100mM NaCl. Crosslinking was performed either directly in the membrane suspension or in a foscholine-12-solubilized membrane fraction (0.2% foscholine-12), by treating the protein with 2mM M2M (Toronto Research Chemicals Inc.) at 22 °C for 1 h. M2M has a spacer arm length of 5.2Å40,41. The reaction was stopped by adding an equal volume of 40mM N-ethylmaleimide in 200mMHEPES (pH 7.3). DTT(final concentration 50mM)was used to break the crosslinking disulphide bond.
Supplementary Material
Acknowledgements
We thank A. Héroux, H. Robinson and A. Soares at NSLS, and J. Schuermann at APS NE-CAT for their help during data collection. X-ray diffraction data were measured at beamlines X25 and X29 at NSLS, and at 24-ID-C and 24-ID-E at APS. Financial support was principally from the US Department of Energy and from the National Institutes of Health. This work was supported by a New Scholar Award in Aging from the Ellison Medical Foundation (to Y.H.), a gift from the Neuroscience Education and Research Foundation (to Y.H.) and a pilot grant from Yale's programme in Cellular Neuroscience, Neurodegeneration, and Repair (CNNR) (to Y.H.).
Footnotes
Author Information The atomic coordinates of FlaK and structure factors have been deposited in the Protein Data Bank under accession code 3S0X. Reprints and permissions information is available at www.nature.com/reprints. The authors declare no competing financial interests. Readers are welcome to comment on the online version of this article at www.nature.com/nature.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Contributions J.H. and Y.X. purified and characterized FlaK in various detergents. J.H. obtained the high-resolution crystals of FlaK. J.H., Y.X. and Y.H. solved the crystal structure. Y.H., Y.X. and J.H. wrote the paper. Y.X. and S.L. screened many constructs and performed the initial biochemical and functional characterizations.
References
- 1.Wolfe MS, et al. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and γ-secretase activity. Nature. 1999;398:513–517. doi: 10.1038/19077. [DOI] [PubMed] [Google Scholar]
- 2.LaPointe CF, Taylor RK. The type 4 prepilin peptidases comprise a novel family of aspartic acid proteases. J. Biol. Chem. 2000;275:1502–1510. doi: 10.1074/jbc.275.2.1502. [DOI] [PubMed] [Google Scholar]
- 3.Weihofen A, Binns K, Lemberg MK, Ashman K, Martoglio B. Identification of signal peptide peptidase, a presenilin-type aspartic protease. Science. 2002;296:2215–2218. doi: 10.1126/science.1070925. [DOI] [PubMed] [Google Scholar]
- 4.Bardy SL, Jarrell KF. Cleavage of preflagellins by an aspartic acid signal peptidase is essential for flagellation in the archaeon Methanococcus voltae. Mol. Microbiol. 2003;50:1339–1347. doi: 10.1046/j.1365-2958.2003.03758.x. [DOI] [PubMed] [Google Scholar]
- 5.Lory S, Strom MS. Structure-function relationship of type-IV prepilin peptidase of Pseudomonas aeruginosa—a review. Gene. 1997;192:117–121. doi: 10.1016/s0378-1119(96)00830-x. [DOI] [PubMed] [Google Scholar]
- 6.Craig L, Pique ME, Tainer JA. Type IV pilus structure and bacterial pathogenicity. Nature Rev. Microbiol. 2004;2:363–378. doi: 10.1038/nrmicro885. [DOI] [PubMed] [Google Scholar]
- 7.Sandkvist M. Type II secretion and pathogenesis. Infect. Immun. 2001;69:3523–3535. doi: 10.1128/IAI.69.6.3523-3535.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Selkoe DJ, Wolfe MS. Presenilin: running with scissors in the membrane. Cell. 2007;131:215–221. doi: 10.1016/j.cell.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 9.Jorissen E, De Strooper B. γ-secretase and the intramembrane proteolysis of Notch. Curr. Top. Dev. Biol. 2010;92:201–230. doi: 10.1016/S0070-2153(10)92006-1. [DOI] [PubMed] [Google Scholar]
- 10.Brouwers N, Sleegers K, VanBroeckhoven C. Molecular genetics of Alzheimer's disease: an update. Ann. Med. 2008;40:562–583. doi: 10.1080/07853890802186905. [DOI] [PubMed] [Google Scholar]
- 11.Steiner H, et al. Glycine 384 is required for presenilin-1 function and is conserved in bacterial polytopic aspartyl proteases. Nature Cell Biol. 2000;2:848–851. doi: 10.1038/35041097. [DOI] [PubMed] [Google Scholar]
- 12.Rawlings ND, Morton FR, Kok CY, Kong J, Barrett AJ. MEROPS: the peptidase database. Nucleic Acids Res. 2008;36:D320–D325. doi: 10.1093/nar/gkm954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Francetic O, Buddelmeijer N, Lewenza S, Kumamoto CA, Pugsley AP. Signal recognition particle-dependent inner membrane targeting of the PulG Pseudopilin component of a type II secretion system. J. Bacteriol. 2007;189:1783–1793. doi: 10.1128/JB.01230-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bayley DP, Jarrell KF. Overexpression of Methanococcus voltae flagellin subunits in Escherichia coli and Pseudomonas aeruginosa: a source of archaeal preflagellin. J. Bacteriol. 1999;181:4146–4153. doi: 10.1128/jb.181.14.4146-4153.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kalmokoff ML, Karnauchow TM, Jarrell KF. Conserved N-terminal sequences in the flagellins of archaebacteria. Biochem. Biophys. Res. Commun. 1990;167:154–160. doi: 10.1016/0006-291x(90)91744-d. [DOI] [PubMed] [Google Scholar]
- 16.Bardy SL, Jarrell KF. FlaK of the archaeon Methanococcus maripaludis possesses preflagellin peptidase activity. FEMS Microbiol. Lett. 2002;208:53–59. doi: 10.1111/j.1574-6968.2002.tb11060.x. [DOI] [PubMed] [Google Scholar]
- 17.Killian JA, von Heijne G. How proteins adapt to a membrane–water interface. Trends Biochem. Sci. 2000;25:429–434. doi: 10.1016/s0968-0004(00)01626-1. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y, Maegawa S, Akiyama Y, Ha Y. The role of L1 loop in the mechanism of rhomboid intramembrane protease GlpG. J. Mol. Biol. 2007;374:1104–1113. doi: 10.1016/j.jmb.2007.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bondar AN, del Val C, White SH. Rhomboid protease dynamics and lipid interactions. Structure. 2009;17:395–405. doi: 10.1016/j.str.2008.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ha Y. Structure and mechanism of intramembrane protease. Semin. Cell Dev. Biol. 2009;20:240–250. doi: 10.1016/j.semcdb.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tolia A, Chavez-Gutierrez L, De Strooper B. Contribution of presenilin transmembrane domains 6 and 7 to a water-containing cavity in the γ-secretase complex. J. Biol. Chem. 2006;281:27633–27642. doi: 10.1074/jbc.M604997200. [DOI] [PubMed] [Google Scholar]
- 22.Tolia A, Horre K, De Strooper B. Transmembrane domain 9 of presenilin determines the dynamic conformation of the catalytic site of γ-secretase. J. Biol. Chem. 2008;283:19793–19803. doi: 10.1074/jbc.M802461200. [DOI] [PubMed] [Google Scholar]
- 23.Laudon H, et al. A nine-transmembrane domain topology for presenilin 1. J. Biol. Chem. 2005;280:35352–35360. doi: 10.1074/jbc.M507217200. [DOI] [PubMed] [Google Scholar]
- 24.Spasic D, et al. Presenilin-1 maintains a nine-transmembrane topology throughout the secretory pathway. J. Biol. Chem. 2006;281:26569–26577. doi: 10.1074/jbc.M600592200. [DOI] [PubMed] [Google Scholar]
- 25.Narayanan S, Sato T, Wolfe MS. A C-terminal region of signal peptide peptidase defines a functional domain for intramembrane aspartic protease catalysis. J. Biol. Chem. 2007;282:20172–20179. doi: 10.1074/jbc.M701536200. [DOI] [PubMed] [Google Scholar]
- 26.Sato C, Morohashi Y, Tomita T, Iwatsubo T. Structure of the catalytic pore of γ-secretase probed by the accessibility of substituted cysteines. J. Neurosci. 2006;26:12081–12088. doi: 10.1523/JNEUROSCI.3614-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sato C, Takagi S, Tomita T, Iwatsubo T. The C-terminal PAL motif and transmembrane domain 9 of presenilin 1 are involved in the formation of the catalytic pore of the γ-secretase. J. Neurosci. 2008;28:6264–6271. doi: 10.1523/JNEUROSCI.1163-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ponting CP, et al. Identification of a novel family of presenilin homologues. Hum. Mol. Genet. 2002;11:1037–1044. doi: 10.1093/hmg/11.9.1037. [DOI] [PubMed] [Google Scholar]
- 29.Davies DR. The structure and function of the aspartic proteinases. Annu. Rev. Biophys. Biophys. Chem. 1990;19:189–215. doi: 10.1146/annurev.bb.19.060190.001201. [DOI] [PubMed] [Google Scholar]
- 30.Wang Y, Ha Y. Open-cap conformation of intramembrane protease GlpG. Proc. Natl Acad. Sci. USA. 2007;104:2098–2102. doi: 10.1073/pnas.0611080104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaback HR, Stadtman ER. Proline uptake by an isolated cytoplasmic membrane preparation of Escherichia coli. Proc. Natl Acad. Sci. USA. 1966;55:920–927. doi: 10.1073/pnas.55.4.920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 33.Wang BC. Resolution of phase ambiguity in macromolecular crystallography. Methods Enzymol. 1985;115:90–112. doi: 10.1016/0076-6879(85)15009-3. [DOI] [PubMed] [Google Scholar]
- 34.Pape T, Schneider TR. HKL2MAP: a graphical user interface for macromolecular phasing with SHELX programs. J. Appl. Cryst. 2004;37:843–844. [Google Scholar]
- 35.Collaborative Computational Project, Number 4 The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 36.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building proteinmodels in electron density maps and the location oferrors in these models. Acta Crystallogr. A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 37.Brünger AT, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 38.Winn MD, Murshudov GN, Papiz MZ. Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 2003;374:300–321. doi: 10.1016/S0076-6879(03)74014-2. [DOI] [PubMed] [Google Scholar]
- 39.Nicholls A, Sharp KA, Honig B. Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- 40.Loo TW, Clarke DM. Determining the dimensions of the drug-binding domain of human P-glycoprotein using thiol cross-linking compounds as molecular rulers. J. Biol. Chem. 2001;276:36877–36880. doi: 10.1074/jbc.C100467200. [DOI] [PubMed] [Google Scholar]
- 41.Green NS, Reisler E, Houk KN. Quantitative evaluation of the lengths of homobifunctional protein cross-linking reagents used as molecular rulers. Protein Sci. 2001;10:1293–1304. doi: 10.1110/ps.51201. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.