

Abstract
Significance: The autophagy and inflammasome pathways are ancient innate immune mechanisms for controlling invading pathogens that are linked by mutual regulation. In addition to controlling the metabolic homeostasis of the cell through nutrient recycling, the “self-eating” process of autophagy is also responsible for the degradation of damaged organelles, cells, and pathogens to protect the integrity of the organism. As a cytosolic pathogen recognition receptor (PRR) complex, the inflammasome both induces and is induced by autophagy through direct interactions with autophagy proteins or through the effects of secondary molecules, such as mitochondrial reactive oxygen species and mitochondrial DNA. Recent Advances: While the molecular mechanisms of inflammasome activation and regulation are largely unknown, much of the current knowledge has been established through investigation of the role of autophagy in innate immunity. Likewise, regulatory proteins in the NOD-like receptor family, which includes inflammasome PRRs, are able to stimulate autophagy in response to the presence of a pathogen. Critical Issues: Many of the newly uncovered links between autophagy and inflammasomes have raised new questions about the mechanisms controlling inflammasome function, which are highlighted in this review. Future Directions: Our basic understanding of the mutual regulation of inflammasomes and autophagy will be essential for designing therapeutics for chronic inflammatory diseases, especially those for which autophagy and inflammasome genes have already been linked. Antioxid. Redox Signal. 20, 495–506.
Introduction
As an increasingly appreciated component of the innate immune response, the Nod-like receptor (NLR) family of pathogen recognition receptors (PRRs) initiates a quick and potent inflammatory cytokine response to pathogen associated molecular patterns (PAMPs) and danger associated molecular patterns (DAMPs) detected in phagocytes. PAMPs are conserved features of bacterial and viral pathogens and DAMPs are byproducts of cell death or increased membrane permeability, which can arise due to invasion by a pathogen or by tissue damage. NLRs contain a protein binding domain (pyrin or CARD domain), a central nucleotide-binding and oligomerization domain (NACHT, NOD, NB-ARC), and a leucine-rich repeat (LRR) domain, which senses ligands or regulates NLR function. At least 22 NLR family members exist, but very few have been characterized as genuine PRRs, while the other members either regulate innate immune responses or do not have a known function (82). The NLRs sense PAMPs and DAMPs in the cytosol and form a signaling platform protein complex called the inflammasome, which consists of an oligomerized NLR bound to caspase-1 directly or by means of the apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) adapter protein (Fig. 1). The number of NLR oligomer subunits and the actual structure of the inflammasome have not been elucidated in a high resolution crystal structure, but it has been estimated that the NLRC4/NAIP5 inflammasome forms a disc-shaped structure of 12 monomers of each protein as visualized by electron microscopy (EM) (36), and a similar structure was observed for oliogomers of NLRP1 by EM (10). The initiation steps of inflammasome formation are not clear, but since the inflammasome is only detectable upon treatment with a PAMP or DAMP (60), it likely forms upon activation of an NLR after a ligand is sensed.
FIG. 1.

The inflammasome pathway. The four steps of inflammasome activation are summarized here and described in detail in the main text. NLR proteins detect cytosolic PAMPs and DAMPs in step one, which induces the assembly of the inflammasome complex in step two. In step three, the inflammasome activates the cleavage of pro-caspase-1 to the p10 and p20 subunits of active caspase-1 enzyme. Finally, active caspase-1 cleaves pro-IL-1β to its secreted 17 kDa form in step four. DAMP, danger associated molecular pattern; PAMP, pathogen associated molecular pattern; NLR, Nod-like receptor. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Inflammasome complexes induce the cleavage of pro-caspase-1 to the p10 and p20 subunits of active caspase-1 enzyme, which in turn cleaves the presynthesized pro-IL-1β cytokine into its active 17 kDa form that is then secreted from the cell (Fig. 1). While caspase-1 is able to cleave itself in an inflammasome complex in vitro (24), the mechanism and the possible roles of additional proteases in vivo remain unknown. IL-1β release requires two PRR activating signals: signal one activates the NFκB-dependent expression of pro-IL-1β, usually through toll-like receptor (TLR) activation, and signal two activates caspase-1 to cleave pro-IL-1β through NLR-mediated inflammasome formation. The mechanism of IL-1β release after cleavage is unclear, although several noncanonical models have been proposed, as previously reviewed (23). IL-18 and cell death by pyroptosis also follow inflammasome activation and have also previously been reviewed (64). Very recently, it was shown that the caspase-1 knockout mouse previously used to determine the role of caspase-1 in inflammasome activation also had a truncation mutation in the caspase-11 gene, which was subsequently shown to regulate noncanonical inflammasome activation leading to pyroptosis (4, 8, 34, 47, 76). Although caspase-11 is not required for inflammasome responses to some sterile stimulants, such as K+ efflux, it is required for responses to gram-negative bacterial infections (4, 8, 34, 76). Early work has hinted at potential molecular mechanisms of caspase-11-mediated regulation of inflammasome activation, but additional work is required to determine what the targets of caspase-11 are and how they might mediate pyroptosis.
Phagocytes are the only cell type known to contain inflammasomes and these same cells rely upon autophagy to monitor for the presence of pathogens. Autophagy is a “self-eating” process by which a cell collects protein aggregates, bacteria, viruses, dying cells, and damaged organelles into a double-membrane compartment called an autophagosome, which fuses with the lysosome to degrade the contents for recycling and antigen presentation. Of the three types of autophagy, macroautophagy is the most relevant to inflammasome studies and will be referred to as “autophagy” in this review. Autophagy ensures an energetic homeostasis in the cell by providing nutrients during metabolic stress and also serves a protective role by removing damaged and damaging components of the cytosol. Given these important roles, it is not surprising that autophagy is a tightly regulated process that is linked to other major pathways, such as phagocytosis, biosynthesis, and innate immune signaling. Autophagy is normally kept to a minimum in unstressed cells, but it is quickly enhanced during nutrient deprivation, infection, and cellular damage.
Autophagy occurs in four phases: induction, nucleation, elongation, and fusion, with points for regulation at each step (Fig. 2) (49). During the induction step, repression of autophagy is relieved by one of many possible initiation signaling cascades involving class I phosphatidylinositol 3 kinase/Akt/molecular target of rapamycin (PI3K/Akt/mTOR), 5′-adenosine monophosphate activated protein kinase (AMPK), elongation initiation factor 2α (eIF2α), tumor protein 53 (p53), c-Jun N-terminal kinase 1 (JNK1), inositol-phosphate requiring enzyme 1 (IRE-1), inositol-triphosphate receptor (IP3R), or intracellular calcium. Vesicle nucleation occurs during the second step, which results in the assembly of a phagophore, the double membrane vesicle that will engulf cytosolic cargo. The phagophore begins to form upon the assembly of a protein complex consisting of the lipid kinase Vps34 and the regulatory proteins Vps15 and Beclin1, which initiates Vps34 enzymatic activity to phosphorylate phosphatidylinositols to generate phosphatidylinositol 3-phosphate (PI3P). The presence of PI3Ps recruits FYVE and PX domain containing proteins to the membrane, labeling it as an autophagophore. The autophagophore is elongated during the third step of autophagy via a ubiquitin E3 ligase complex consisting of autophagy proteins (Atg) Atg5, Atg12, and Atg16L1, which conjugates phosphatidylethanolamine (PE) to microtubule-associated protein 1A/1B, light chain 3 (LC3). LC3-PE remains associated with the inner and outer membranes of the phagophore as it extends in length until it has completely engulfed its cargo to become an autophagosome. During the final fusion step of autophagy, the autophagosome fuses with the lysosome to create an autolysosome, which degrades its contents via hydrolases and exports the remaining materials to the cytoplasm for reuse.
FIG. 2.

The autophagy pathway. As described in the text in further detail, autophagy induction occurs by inhibition of mTor, which results in reduced phosphorylation of the ULK1/2/Atg13/FIP200 complex in step one. In step two, vesicle nucleation occurs upon formation of the Beclin/Vps34/Vps15 complex, which phosphorylates PI to PI3P and labels the membrane as an autophagophore. The phagophore membrane is elongated during step three by conjugation of PE to LC3 by the Atg5/Atg12/Atg16L complex. The resulting autophagophore fuses with the lysosome in step 4 to digest the cargo in an autolysosome. Atg, autophagy protein; LC3, microtubule-associated protein 1A/1B, light chain 3; mTOR, molecular target of rapamycin; PE, phosphatidylethanolamine; PI3P, phosphatidylinositol 3-phosphate. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Both autophagy and inflammsome proteins have been genetically linked to human autoinflammatory diseases, suggesting that these processes might be functionally related. Indeed, recent work has uncovered exciting new regulatory connections between autophagy and inflammasome activation, which is the focus of this review. Autophagy is required for inflammasome activation and inflammasome complexes are turned off upon targeted degradation in autophagolysosomes. Consequently, NLR proteins bind and regulate autophagy proteins to initiate autophagophore formation. These topics and the unanswered questions they contain will be discussed in detail in the following sections of this review.
The Inflammasomes
The NLRs which have been shown to form inflammasomes in response to PAMPs or DAMPs in macrophage cells are NLRP1 (Nalp1, Nlrp1b), NLRP3 (Cias1, Nalp3), NLRC4 (Ipaf), NLRP7 (NOD12), NLRP12 (monarch-1), and absent in melanoma 2 (AIM2) (PYHIN). Although absent in melanoma 2 (AIM2) is structurally unrelated to the other NLRs, it is functionally related and will be discussed below. NLRP1 has been shown to respond to anthrax lethal toxin (LeTx) and the muramyl dipeptide (MDP) moiety of peptidoglycans (7, 18, 25, 31, 56), although experiments with bone-marrow derived macrophage (BMDM) from the first NLRP1 knockout mouse have recently shown that NLRP1 is only required for the LeTx inflammasome response and not the MDP response, which instead requires NLRP3 (53). Mechanistically, LeTx directly cleaves NLRP1, which activates NLRP1 to form inflammasomes, activate caspase-1, and cleave IL-1β (18, 31). Both N-terminal and FIIND domain cleavage sites have been reported for NLRP1, although only cleavage at the N-terminal site has been shown to be LeTx-dependent (18, 31, 56) (Fig. 3). NLRP1 has recently been reported to respond to reduced ATP levels in the cytoplasm in response to nutrient deprivation, but NLRP1 cleavage was not detectable under these stimulation conditions (57). The NLRP1 inflammasome does not include ASC (25, 67) possibly because NLRP1 has a pyrin domain that could bind to the pyrin domain of Caspase-1 independently of ASC. Interestingly, ASC is required for the response to MDP (69), further supporting a role for NLRP3 and not NLRP1 in MDP stimulation.
FIG. 3.

Post-translational modifications regulate inflammasomes. (a) NLRP1 is cleaved in the N-terminus or FIIND domain in the presence of LeTx to activate the NLRP1 inflammasome (17, 30). (b) Deubiquitination of NLRP3 by BRCC3 is required for NLRP3 inflammasome activation (44, 57, 71). (c) Phosphorylation of serine 533 by PKCδ is required for NLRC4 inflammasome activation (72). (d) Ubiquitination of ASC leads to p62-directed autophagosomal degradation of the AIM2/ASC inflammasome (84). BRCC3, BRCA1-BRCA2-containing complex 3; LeTx, anthrax lethal toxin; ASC, apoptosis-associated speck-like protein containing a caspase recruitment domain. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
NLRP3, the most well-characterized NLR family member, is activated by a wide range of pathogens and sterile DAMP signals, including K+ efflux, MDP, monosodium urate (MSU), cholesterol crystals, mitochondrial reactive oxygen species (mtROS), Listeria monocytogenes, Candida albicans, Salmonella typhimurium, and influenza virus, among others (29). The role of NLRP3 in caspase-1 activation and IL-1β secretion in response to these signals has been evaluated with the NLRP3 and ASC knockout mice, which both had defects for all tested stimuli (29, 41, 44, 69), indicating that NLRP3 utilizes the ASC adapter. Moreover, NLRP3 has also been shown to bind ASC (3, 21), and ASC foci are visible by immunofluorescence microscopy upon NLRP3 stimulation (71). Given the structurally diverse range of NLRP3 stimulants, it is not surprising that the molecular mechanism of NLRP3 activation remains unclear and will be discussed further in the following sections. While much effort has focused on defining the potential activating factors of NLRP3, relatively less is known about what signals regulate turning off active NLRP3. Recent evidence suggests that intracellular Ca2+, cyclic AMP (cAMP), and nitric oxide (NO) negatively regulate NLRP3 activation through binding or modification of NLRP3 (39, 51, 54). However, Ca2+, cAMP, and NO levels are controlled by several pathways and thus, the negative regulation of NLRP3 is likely a complex balance of converging signals.
NLRC4 is stimulated by intracellular flagellin and type III secretion systems from bacteria, including S. typhimurium, Shigella flexneri, and Escherichia coli (28, 62, 63). NLRC4 utilizes ASC (59, 71), Naip5, and Naip2 adapter proteins depending on the bacterial stimulus (29). While NLRC4 can directly detect the N-terminus of flagellin, Naip5 association is required to respond to the C-terminus of flagellin (52, 99). The Naip2 adapter protein detects a rod component of type III secretion systems and binds NLRC4 to activate inflammasome formation (52, 99). The ASC adapter protein is important but not essential for all types of NLRC4 inflammasomes, as ASC deficient BMDMs have a partial defect in caspase-1 activity and IL-1β secretion upon flagellin stimulation, suggesting that ASC enhances the activity of the NLRC4 inflammasome (9, 62, 99). However, ASC binds NLRC4 (33) and ASC deficient BMDMs have a complete defect in caspase-1 cleavage and NLRC4 inflammasome foci formation upon infection with S. typhimurium or Legionella pneumophila (12, 59, 71). As a further complication, caspase-1 dependent cell death, or pyroptosis, is intact in ASC deficient BMDMs upon infection with Pseudomonas aeruginosa, L. monocytogenes, or S. flexneri (30, 81, 90). Thus, the role of ASC in NLRC4 activation might depend on the context of inflammasome stimulation. Activation of NLRC4 during S. typhimurium infection also requires phosphorylation at serine 533 (S533) between the NACHT and LRR domains, which is likely targeted for phosphorylation by the PKCδ kinase (73) (Fig. 3). This is the first evidence of the phosphorylation-mediated regulation of inflammasomes and it will be of interest to determine whether additional NLRs are regulated by phosphorylation and dephosphorylation.
Recent work has identified two new NLRs as pathogen sensors; NLRP12 detects Yersinia pestis and NLRP7 detects acylated lipopeptides from bacteria (48, 95). NLRP12 binds ASC (96) and is required for IL-1β secretion and caspase-1 activity in response to Y. pestis infection in vivo and in cultured phagocytes, although NLRP3 was also required in these experiments (95). The ligand for the NLRP12 inflammasome activation is unknown, but the Y. pestis type III secretion system is required (95). NLRP7 also detects specific PAMPs from intracellular bacterial pathogens. The acylated lipopeptides that activate NLRP7 in human macrophage THP-1 cells are from Mycoplasma spp., which are common contaminants in cell cultures. ASC foci are also colocalized with NLRP7 upon stimulation, suggesting an inflammasome forms (48). Notably, since lipopeptides were recognized by TLR2, no additional priming of the cells was required for IL-1β release, making mycoplasma contamination a threat to inflammasome studies performed in cell culture (48). Significantly less is known about the NLRP12 and NLRP7 inflammasomes in comparison to NLRP3 and subsequent studies will provide more insight into the mechanisms and roles of their activation.
AIM2 is structurally unique from other NLRs because it has a HIN-200 domain instead of an LRR, and similarly to NLRP1, AIM2 has a pyrin domain at its N-terminus (82). AIM2 recognizes cytosolic double-stranded DNA (dsDNA) from bacteria and viruses, and utilizes the ASC adapter protein as shown by ASC knockout mice and ASC foci formation upon AIM2 stimulation (26, 43, 75). Although it is unclear what the molecular determinants are for distinguishing bacterial DNA molecules in the cytosol, genetic evidence clearly shows that AIM2 is required for IL-1β secretion upon infection of BMDMs with Mycobacterium tuberculosis and Francisella tularensis in addition to the viral pathogens, such as vaccinia virus and cytomegalovirus (50, 75, 79). AIM2 is also partially responsible for IL-1β secretion in response to L. monocytogenes, which is also detected by NLRP3 (50, 75). Notably, in vivo infections of AIM2 knockout mice with mouse cytomegalovirus revealed that early control of virus replication is AIM2-dependent (75), highlighting the critical role of inflammasomes in innate immunity in vivo.
Autophagy Studies Uncover Regulation of Inflammasomes by ROS
Deletion or depletion of autophagy by genetic or pharmacological means has a clear enhancing effect on IL-1β secretion upon inflammasome stimulation or LPS treatment conditions. Specifically, IL-1β secretion was apparently increased upon inhibition of autophagy by 3-methyladenine (3MA) treatment, expression of the Atg4B dominant negative mutant, deletion of LC3B, deletion of Atg16L1, or depletion of Beclin1 (Fig. 1) (38, 66, 80, 85, 100). A study utilizing the Atg16L1 knockout mouse model system has found that the TIR-domain-containing adapter-inducing interferon-β (TRIF) TLR adapter protein, K+ efflux, and ROS are required for inflammasome activation (80). ROS is produced by NADPH oxidases or mitochondria in response to phagocytosis and TLR stimulation (68, 97). Although initial reports suggested that NADPH oxidases might be important for ROS production during inflammasome stimulation in the human THP-1 cell line (20, 86), studies examining PBMCs from human patients lacking the p22phox, p47phox, or NOX2 subunits of the NADPH oxidases conclusively showed that NADPH oxidases were not required for IL-1β secretion (93, 94). These conflicting results might be due to differences between primary and transformed cells, since primary BMDM from the gp91 NADPH oxidase subunit knockout mouse were also competent for IL-1β secretion in a subsequent study (86). Although NADPH oxidase-derived ROS is not required for inflammasome activation, ROS from an alternative source is required since treatment with a general ROS scavenger reduced IL-1β production in NADPH oxidase deficient human PBMCs and mouse bone-marrow derived dendritic cells (38, 93, 94). This left mitochondria as the likely source of ROS required for inflammasome activation, but it was not clear how autophagy was able to regulate its production in response to inflammasome stimulants.
An important clue came when blockage of autophagy by treatment of macrophage cells with 3MA (PI3K inhibitor) resulted in the production of mitochondrial ROS (mtROS) and NLRP3-dependent IL-1β secretion in the absence of traditional inflammasome stimulants (Fig. 4) (38, 100). This suggested that when damaged mitochondria were not cleared by autophagy, they released mtROS, which stimulated NLRP3 inflammasomes. If all traditional NLRP3 stimulants induce mitochondrial damage, then the mtROS model would present a unified mechanism for the activation of NLRP3 by a structurally diverse set of stimulants. The mtROS model of NLRP3 inflammasome activation was further supported by the colocalization of NLRP3 with mitochondrial markers, suggesting that the inflammasome activation site is physically located near the source of mtROS (100). Mitochondria function was specifically important for mtROS production and IL-1β secretion because shRNA-mediated depletion of voltage dependent anion channel 1 (VDAC1), which is important for mitochondrial function and mtROS production (88), reduced caspase-1 cleavage and IL-1β secretion in response to sterile NLRP3 stimuli (100). Notably, this effect was not observed for NLCR4 or AIM2 stimuli, making mtROS a specific requirement of NLRP3 inflammasome activation, although the role of mtROS in additional inflammasomes must also be examined further (87, 100). High levels of Bcl-2, which partially blocks VDAC, reduces mtROS production, and blocks apoptosis, also reduced NLRP3-inflammasome activation in BMDMs and an immortalized human macrophage cell line (87, 100). In contrast, it has previously been shown that a Bcl-2 overexpression in the human THP-1 macrophage cell line has lower levels of caspase-1 activity and IL-1β secretion in response to MDP and ATP stimulation (10, 100), making it likely that the role of Bcl-2 may differ between cell lines or that MDP and ATP stimulation is unique from other NLRP3 stimulants. Although the role of Bcl-2 in THP-1 cells was initially investigated in the context of NLRP1 stimulation, the response to MDP and ATP treatment used in this study has since been shown to be NLRP3-dependent and not NLRP1-dependent in knockout mice (53), limiting the known requirement of mtROS to NLRP3-inflammasomes.
FIG. 4.
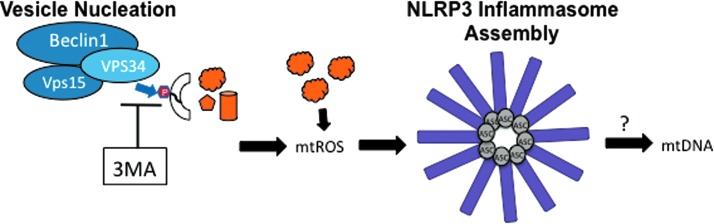
Activation of the NLRP3 inflammasome by 3MA treatment. Treatment with the Vps34 inhibitor, 3MA, blocks autophagic vesicle nucleation, which results in the accumulation of damaged mitochondria (38, 100). The mtROS produced by damaged mitochondria activates the NLRP3 inflammasome (2, 32, 40, 66), which may induce the release of mtDNA (66, 87). 3MA, 3-methyladenine; mtDNA, mitochondrial DNA; mtROS, mitochondrial reactive oxygen species. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
A major advance in understanding the molecular mechanism of the mtROS-mediated activation of NLRP3 was achieved in experiments using mice deficient for autophagophore formation and elongation due to the knockout of LC3 (LC3−/−) or the depletion of Beclin1 (beclin1+/−) (66). BMDMs from these mice had elevated caspase-1 activation and IL-1β secretion upon stimulation of the NLRP3 inflammasome with ATP, which causes a K+ efflux in the cell through the P2X7 channel (46). An increase in swollen and damaged mitochondria was visible by EM, confirming that mitochondrial damage resulted from K+ efflux due to ATP treatment (2, 32, 40, 66). Remarkably, BMDMs lacking mitochondrial DNA (mtDNA), called ρ0 cells, were unable to secrete IL-1β in response to NLRP3 stimuli and DNAseI treatment reduced caspase-1 activation and IL-1β secretion in normal BMDM, suggesting that mtDNA is involved in NLRP3 inflammasome activation (66, 87). In a series of elegantly designed experiments, it was revealed that mtDNA is released to the cytosol upon stimulation with ATP in an mtROS and NLRP3-dependent manner (66), indicating that caspase-1 activation is downstream from NLRP3-mediated translocation of mtDNA to the cytosol (Fig. 4). Since mtDNA release to the cytosol in response to ATP was impaired in BMDMs from either ASC or NLRP3 knockout mice, the inflammasome itself is likely involved in mtDNA release (66). This is in contrast to a more recent model proposed by Shimada et al., in which NLRP3 is activated after it directly binds to mtDNA released upon mitochondrial damage due to apoptosis triggered by NLRP3 stimulants (87). Shimada et al. argue that mtDNA was not detected in NLRP3 KO BMDM upon ATP treatment because the mtDNA was degraded in the absence of NLRP3-binding, although this possibility has not been tested. Intriguingly, oxidized nucleoside 8-hydroxy-guanosine (8-OH-dG), a marker for oxidized mtDNA, was detectable in endogenous NLRP3 immunoprecipitations and was even capable of blocking IL-1β production when it was added in excess to BMDM by competitively binding to endogenous NLRP3 (87). This strongly supports a role for oxidized mtDNA in NLRP3 inflammasome activation, although it is not clear whether NLRP3 may also facilitate mtDNA release before binding (Fig. 4).
The roles of other PRRs have also been examined in mtROS and mtDNA production. First, while the AIM2-inflammasome is also activated by exogenous mtDNA, endogenous mtDNA did not directly bind endogenous AIM2 in BMDM and ATP-dependent IL-1β production was AIM2 independent (66, 87). Furthermore, AIM2 activation by dsDNA was not affected by an inhibitor of mtROS production, AIM2 stimulation did not change mitochondrial membrane potential, and Bcl-2 did not affect AIM2 stimulation (66, 87). Thus, cytosolic mtDNA activates NLRP3 specifically. Secondly, TLRs have also been shown to induce mtROS production by a separate mechanism, which is important for clearance of intracellular bacterial pathogens (97). Upon stimulation of TLR1, 2, or 4, the TRAF6 signaling adapter protein translocates to the mitochondria and interacts with evolutionarily conserved signaling intermediate in Toll pathways (ECSIT), resulting in the ubiquitination of ECSIT and the increased ROS and mtROS (97). Cells must be primed by TLR activation to induce pro-IL-1β expression before NLR stimulation in inflammasome studies, making it likely that TLR signalling contributes to some mtROS production. Indeed, both overnight and 6 h stimulation of TLR4 with LPS lead to the production of ROS and mtROS (97), which are commonly used priming conditions, for inflammasome activation. Since TLR signaling induces mtROS production (84, 97), and ROS induces autophagy (17), it is possible to hypothesize that TLR signaling initiates and limits inflammasome activation through the mtROS-mediated activation of NLRP3, the NFκB-mediated expression of pro-IL-1β, and the autophagy-mediated degradation of pro-IL1β.
Regulation of Inflammasomes by Autophagosomal Degradation
In addition to its role as a regulator of NLRP3-inflammasome activation, autophagy also negatively regulates inflammasomes through newly discovered autophagy-dependent degradation of inflammasome proteins and IL-1β (38, 85). Studying the negative regulation of inflammasome activation is important for understanding how this potent signal is “turned off” to avoid acute tissue damage. Not surprisingly, many NLRs and autophagy factors are linked to autoimmune and autoinflammatory diseases (74, 83), which are characterized by high levels of inflammatory cytokines. As a negative regulatory mechanism of inflammasome activation, pro-IL1β is targeted to autophagosomes for degradation in response to TLR stimulation (38). Specifically, it has been shown that IL-1β is sequestered in the LC3-positive autophagosomes upon TLR stimulation and pro-IL-1β protein levels decreased when autophagy was induced by rapamycin (Fig. 5) (38). This suggests that TLR stimulation induces both pro-IL-1β expression and degradation by autophagy, thereby limiting the amount of available pro-IL-1β protein in the absence of NLR stimulation. A recent report suggests that mature IL-1β uses the autophagy machinery for secretion in a noncanonical secretory pathway (22). However, more evidence is required to vigorously evaluate this hypothesis.
FIG. 5.

Augophagosomal degradation negatively regulates inflammasomes. TLR activation results in the sequestration of pro-IL-1β to autophagosomal compartments, leading to reduced levels of pro-IL-1β (38). In parallel, ASC is targeted for K63-liked ubiquitination upon AIM2 stimulation, which results in localization of ASC to autophagosomes (85). To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Inflammasomes are also negatively regulated by autophagy upstream from IL-1β secretion. It has been shown that a portion of ASC-containing inflammasomes is redirected towards autophagosomes and autophagolysosomes upon NLRP3 or AIM2 stimulation in THP-1 and primary human macrophage cells (85). Mechanistically, ASC localization to autophagosomes was dependent on both Beclin-1 and p62, a protein that specifically recruits ubiquitinated proteins to autophagosomes for degradation (Fig. 5) (85). ASC and ASC-containing inflammasome complexes could be recruited to autophagosomes by p62 since the K63-ubiquitination of ASC was detectable upon AIM2 stimulation (Fig. 3) (85). However, many new questions will need to be answered to elucidate the molecular mechanism of this potential negative regulation of inflammasomes. The ubiquitin ligase that modifies ASC and the trigger for ubiquitination are unknown. However, ASC protein levels did not change upon AIM2 stimulation and AIM2 levels actually increased at the timepoints in this study, making the significance of ASC localization to the autophagosome unclear. Perhaps the degradation of ASC-containing inflammasomes is balanced by increased protein expression, or proportionally very few inflammasomes are recruited to autophagosomes under stimulation conditions. Since experimental evidence is lacking, further investigation into the molecular mechanisms, regulation, and purpose of targeting inflammasomes to autophagosomes is imperative for determining how autophagy may work as a potential ‘off switch’ for activated inflammasomes.
In addition to ASC, NLRP3 is also ubiquitinated (45, 58, 72), although binding to p62 has not been reported so it is not known whether NLRP3 can be independently recruited to autophagosomes. NLRP3 is ubiquitinated in the LRR domain with a mix of lysine 63 (K63) and lysine 48 (K48) linked chains by unknown ubiquitin ligases (Fig. 3) (72). Ubiquitinated NLRP3 is inactive because its deubiquitination is required for inflammasome activation and is triggered by TLR activation, mtROS, and ATP (45, 58, 72). The deubiquitinase responsible for NLRP3 activation, BRCA1-BRCA2-containing complex 3 (BRCC3), is a member of the BRCC36-containing isopeptidase (BRISC) deubiquitination complex, which has been suggested to specifically cleave K63-linked ubiquitin chains and not K48-linked chains (15, 16, 27), making the mechanism of K48-linked ubiquitin removal unclear. Although K48-linked ubiquitinated proteins are often targeted for proteasomal degradation (27), MG132 proteasomal inhibitor treatment did not affect ubiquitinated NLRP3 protein levels in the presence of a BCC3 inhibitor (72), suggesting that ubiquitination of NLRP3 does not lead to proteasomal degradation of NLRP3. Thus, the mechanism of inhibition of NLRP3 by ubiquitination remains unknown.
NLRs Upregulate Autophagy
The positive and negative regulation of inflammasomes by autophagy is complemented by NLR-mediated control of autophagy (Fig. 6). NLRX1, a resident mitochondria protein (65), enhances autophagy during viral infection through an interaction with mitochondrial Tu translation elongation factor (TUFM) (55). TUFM likely increases autophagy through its interaction with the Atg5-Atg12 complex, which is an essential component of the vesicle elongation step of autophagy and is unable to bind NLRX1 on its own (Fig. 6) (55). Although it is not clear how the NLRX1/TUFM interaction with Atg5-Atg12 elevates autophagy, the resulting decrease in vesicular stomatitis virus (VSV) production in NLRX1 and Atg5 knockout mouse embryonic fibroblast (MEF) cells suggests that NLRX1 and autophagy are proviral factors during VSV infection (55). NLRX1 knockout MEFs had a larger decrease in VSV replication than Atg5−/− MEFS, which might be due to the proposed negative regulation of RIG-I-like helicase (RLH) signaling through the mitochondrial antiviral signaling (MAVS) adapter protein by NLRX1 (55, 98). This function has been called into question because different methods of knocking out NLRX1 in mice have given conflicting results. While two strains of NLRX1−/− mice have intact RLH/MAVS signaling and virus replication, a third strain has a defect in RLH/MAVS signaling and enhanced virus replication (5, 77, 89, 98). The latter strain was used to study the role of NLRX1 in TUFM-mediated autophagy enhancement, so it will be important to evaluate whether this effect is consistent in the other two NLRX1−/− strains as well.
FIG. 6.

Regulation of autophagy by NOD2 and NLRX1. NLRX1 binds TUFM to upregulate autophagy via the TUFM/Atg5-Atg12 interaction (55). Upon MDP stimulation, NOD2 binds Atg16L to recruit the autophagy machinery to the site of bacterial infection where autophagosomes may assist in bacterial clearance (42). MDP, muramyl dipeptide; TUFM, mitochondrial Tu translation elongation factor. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
In addition to NLRX1, NOD2 also positively regulates autophagy. NOD2 is an NLR family member that utilizes the receptor interacting protein 2 (Rip2) adapter protein kinase to form a protein complex resembling the inflammasome, called a nodosome, which forms upon stimulation with MDP. Instead of activating caspase-1, the NOD2 nodosome activates NFκB, resulting in inflammatory cytokine production (6). Both NOD2 and Atg16L1 have been identified by several groups as the genes with risk alleles for Crohn's disease, a chronic inflammatory bowel disease characterized by high levels of inflammatory cytokines (1, 13, 37, 61, 78). A T300A mutation in the N-terminus of the WD repeats of Atg16L1 is associated with Crohn's disease in the presence or absence of NOD2 LRR domain point mutations R702W, G908R, or L1007C(frameshift) (6, 74). Recently, NOD2 and Atg16L1 have been functionally linked in a mechanism of bacterial pathogen clearance, which may explain why they are both genetically linked to Crohn's disease (14, 42, 70, 92). NOD2 induces autophagophore formation upon MDP stimulation by binding and recruiting Atg16L1 to the bacterial entry site of the plasma membrane to initiate autophagic destruction of the bacterial pathogen (Fig. 6) (42). Strikingly, dendritic cells from Crohn's patients with NOD2 or Atg16L1 variant alleles have wild type levels of autophagy in response to a TLR1/2 ligand, but have significantly reduced autophagy levels in response to MDP, a NOD2 stimulant (14). Consequently, cells from Crohn's patients have a defect in lysosomal destruction of bacterial pathogens (14, 92). The resulting higher load of bacteria in cells from Crohn's patients may contribute to higher levels of inflammatory cytokines, including IL-6 and IL-1β, which are detected in Crohn's patients' tissues. The increase of IL-1β cytokine production in Crohn's patients could be due to either increased pro-IL-1β protein levels or increased caspase-1 activity. Consistent with a role for NOD2 in Crohn's disease, the enhanced IL-1β secretion in Crohn's patients' cells in response to MDP is due to increased pro-IL1β mRNA levels and not due to caspase-1 activity, which remains unchanged when compared to healthy patients (70). NOD2 is constitutively expressed in immune cells and inducibly expressed in intestinal epithelial cells upon pro-inflammatory stimulation (6, 35). Thus, a potential mechanism for the trigger that controls NOD2-mediated autophagy could be the induction of NOD2 expression. Since RIP2 is ubiquitinated and phosphorylated to regulate NOD2 activation, additional post-transcriptional mechanisms may also regulate NOD2 protein levels (19, 91). As a potential mechanism for disease pathogenesis, NOD2-mediated autophagy may serve as a model for future studies since many additional autophagy and NLR proteins are also linked to chronic inflammatory diseases with unknown mechanisms.
Conclusions
The intersection of inflammasomes and autophagy is an exciting area of research with many unanswered questions that should be addressed by future investigation. Of course, for every host defense there is an opposing pathogen offense, making it likely that bacterial or viral proteins may exist that target or exploit the newly discovered links between autophagy and inflammasome regulation to avoid detection or destruction. Thus, as we uncover new mechanisms of regulation between the ancient innate immune pathways of inflammasomes and autophagy, we may also find novel host-pathogen interactions that may be targeted therapeutically. Moreover, treatments for chronic inflammatory diseases in which NLRs and autophagy proteins are implicated will rely on these findings as well.
Abbreviations Used
- 3MA
3-methyladenine
- 8-OH-dG
oxidized nucleoside 8-hydroxy-guanosine
- AIM2
absent in melanoma 2
- AMPK
5′-adenosine monophosphate activated protein kinase
- ASC
apoptosis-associated speck-like protein containing a caspase recruitment domain
- Atg
autophagy protein
- BMDM
bone marrow derived macrophage cells
- BRCC3
BRCA1-BRCA2-containing complex 3
- BRISC
BRCC36-containing isopeptidase
- cAMP
cyclic AMP
- DAMP
danger associated molecular pattern
- dsDNA
double-stranded DNA
- ECSIT
evolutionarily conserved signaling intermediate in Toll pathways
- eIF2α
elongation initiation factor 2α
- EM
electron microscopy
- IP3R
inositol-triphosphate receptor
- IRE-1
inositol-phosphate requiring enzyme 1
- JNK1
c-Jun N-terminal kinase 1
- LC3
microtubule-associated protein 1A/1B, light chain 3
- LeTx
anthrax lethal toxin
- LRR
leucine-rich repeat
- MAVS
mitochondrial antiviral signaling
- MDP
muramyl dipeptide
- MEF
mouse embryonic fibroblast
- mtDNA
mitochondrial DNA
- mTOR
molecular target of rapamycin
- mtROS
mitochondrial reactive oxygen species
- NLR
Nod-like receptor
- NO
nitric oxide
- p53
tumor protein 53
- PAMP
pathogen associated molecular pattern
- PE
phosphatidylethanolamine
- PI3K
class I phosphatidylinositol 3 kinase
- PI3P
phosphatidylinositol 3-phosphate
- PRR
pathogen recognition receptor
- Rip2
receptor interacting protein 2
- RLH
RIG-I like helicase
- TLR
toll-like receptor
- TRIF
TIR-domain-containing adapter-inducing interferon-β
- TUFM
mitochondrial Tu translation elongation factor
- VDAC1
voltage dependent anion channel 1
- VSV
vesicular stomatitis virus
Acknowledgments
This work was partly supported by 1F32AI096698 (M.R.); CA082057, CA31363, CA115284, DE019085, AI073099, AI083025, HL110609, GRL, the Hastings Foundation, and the Fletcher Jones Foundation (J.U.J.).
References
- 1.Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls Nature 447: 661–678, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adinolfi E, Callegari MG, Ferrari D, Bolognesi C, Minelli M, Wieckowski MR, Pinton P, Rizzuto R, and Di Virgilio F.Basal activation of the P2X7 ATP receptor elevates mitochondrial calcium and potential, increases cellular ATP levels, and promotes serum-independent growth. Mol Biol Cell 16: 3260–3272, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, and Tschopp J. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity 20: 319–325, 2004 [DOI] [PubMed] [Google Scholar]
- 4.Akhter A, Caution K, Abu Khweek A, Tazi M, Abdulrahman BA, Abdelaziz DHA, Voss OH, Doseff AI, Hassan H, Azad AK, Schlesinger LS, Wewers MD, Gavrilin MA, and Amer AO. Caspase-11 promotes the fusion of phagosomes harboring pathogenic bacteria with lysosomes by modulating actin polymerization. Immunity 37: 35–47, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allen IC, Moore CB, Schneider M, Lei Y, Davis BK, Scull MA, Gris D, Roney KE, Zimmermann AG, Bowzard JB, Ranjan P, Monroe KM, Pickles RJ, Sambhara S, and Ting JP-Y. NLRX1 Protein attenuates inflammatory responses to infection by interfering with the RIG-I-MAVS and TRAF6-NF-kappa-B signaling pathways. Immunity 34: 854–865, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Billmann-Born S, Lipinski S, Böck J, Till A, Rosenstiel P, and Schreiber S. The complex interplay of NOD-like receptors and the autophagy machinery in the pathophysiology of Crohn disease. Eur J Cell Biol 90: 593–602, 2011 [DOI] [PubMed] [Google Scholar]
- 7.Boyden ED. and Dietrich WF. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat Genet 38: 240–244, 2006 [DOI] [PubMed] [Google Scholar]
- 8.Broz P, Ruby T, Belhocine K, Bouley DM, Kayagaki N, Dixit VM, and Monack DM. Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature 490: 288–291, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Broz P, von Moltke J, Jones JW, Vance RE, and Monack DM. Differential requirement for caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe 8: 471–483, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bruey J-M, Bruey-Sedano N, Luciano F, Zhai D, Balpai R, Xu C, Kress CL, Bailly-Maitre B, Li X, Osterman A, Matsuzawa S-i, Terskikh AV, Faustin B, and Reed JC. Bcl-2 and Bcl-XL regulate proinflammatory caspase-1 activation by interaction with NALP1. Cell 129: 45–56, 2007 [DOI] [PubMed] [Google Scholar]
- 11.This reference has been deleted.
- 12.Case CL. and Roy CR. Asc modulates the function of NLRC4 in response to infection of macrophages by Legionella pneumophila. mBio 2: e00117–11, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cho JH. The genetics and immunopathogenesis of inflammatory bowel disease. Nat Rev Immunol 8: 458–466, 2008 [DOI] [PubMed] [Google Scholar]
- 14.Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, Ferguson DJP, Campbell BJ, Jewell D, and Simmons A. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med 16: 90–97, 2010 [DOI] [PubMed] [Google Scholar]
- 15.Cooper EM, Boeke JD, and Cohen RE. Specificity of the BRISC deubiquitinating enzyme is not due to selective binding to Lys63-linked polyubiquitin. J Biol Chem 285: 10344–10352, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cooper EM, Cutcliffe C, Kristiansen TZ, Pandey A, Pickart CM, and Cohen RE. K63-specific deubiquitination by two JAMM/MPN+ complexes: BRISC-associated Brcc36 and proteasomal Poh1. EMBO J 28: 621–631, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cruz CM, Rinna A, Forman HJ, Ventura ALM, Persechini PM, and Ojcius DM. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J Biol Chem 282: 2871–2879, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.D'Osualdo A, Weichenberger CX, Wagner RN, Godzik A, Wooley J, and Reed JC. CARD8 and NLRP1 undergo autoproteolytic processing through a ZU5-like domain. PLoS One 6: e27396, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Damgaard RB, Nachbur U, Yabal M, Wong WW, Fiil BK, Kastirr M, Rieser E, Rickard JA, Bankovacki A, Peschel C, Ruland J, Bekker-Jensen S, Mailand N, Kaufmann T, Strasser A, Walczak H, Silke J, Jost PJ, and Gyrd-Hansen M. The ubiquitin ligase XIAP recruits LUBAC for NOD2 signaling in inflammation and innate immunity. Mol Cell 46: 746–758, 2012 [DOI] [PubMed] [Google Scholar]
- 20.Dostert C, Pétrilli V, van Bruggen R, Steele C, Mossman BT, and Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320: 674–677, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duncan JA, Bergstralh DT, Wang Y, Willingham SB, Ye Z, Zimmermann AG, and Ting JP. Cryopyrin/NALP3 binds ATP/dATP, is an ATPase, and requires ATP binding to mediate inflammatory signaling. Proc Natl Acad Sci U S A 104: 8041–8046, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, and Deretic V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. EMBO J 30: 4701–4711, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eder C. Mechanisms of interleukin-1β release. Immunobiology 214: 543–553, 2009 [DOI] [PubMed] [Google Scholar]
- 24.Elliott JM, Rouge L, Wiesmann C, and Scheer JM. Crystal structure of procaspase-1 zymogen domain reveals insight into inflammatory caspase autoactivation. J Biol Chem 284: 6546–6553, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Faustin B, Lartigue L, Bruey J-M, Luciano F, Sergienko E, Bailly-Maitre B, Volkmann N, Hanein D, Rouiller I, and Reed JC. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol Cell 25: 713–724, 2007 [DOI] [PubMed] [Google Scholar]
- 26.Fernandes-Alnemri T, Yu J-W, Datta P, Wu J, and Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458: 509–513, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Finley D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu Rev Biochem 78: 477–513, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Franchi L, Amer A, Body-Malapel M, Kanneganti TD, Ozoren N, Jagirdar R, Inohara N, Vandenabeele P, Bertin J, Coyle A, Grant EP, and Nunez G. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin-1beta in salmonella-infected macrophages. Nat Immunol 7: 576–582, 2006 [DOI] [PubMed] [Google Scholar]
- 29.Franchi L, Muñoz-Planillo R, and Núñez G. Sensing and reacting to microbes through the inflammasomes. Nat Immunol 13: 325–332, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Franchi L, Stoolman J, Kanneganti TD, Verma A, Ramphal R, and Nunez G. Critical role for Ipaf in Pseudomonas aeruginosa-induced caspase-1 activation. Eur J Immunol 37: 3030–3039, 2007 [DOI] [PubMed] [Google Scholar]
- 31.Frew BC, Joag VR, and Mogridge J. Proteolytic processing of nlrp1b is required for inflammasome activity. PLoS Pathog 8: e1002659, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garcia-Marcos M, Fontanils U, Aguirre A, Pochet S, Dehaye JP, and Marino A. Role of sodium in mitochondrial membrane depolarization induced by P2X7 receptor activation in submandibular glands. FEBS Lett 579: 5407–5413, 2005 [DOI] [PubMed] [Google Scholar]
- 33.Geddes BJ, Wang L, Huang WJ, Lavellee M, Manji GA, Brown M, Jurman M, Cao J, Morgenstern J, Merriam S, Glucksmann MA, DiStefano PS, and Bertin J. Human CARD12 is a novel CED4/Apaf-1 family member that induces apoptosis. Biochem Biophys Res Commun 284: 77–82, 2001 [DOI] [PubMed] [Google Scholar]
- 34.Gurung P, Malireddi RKS, Anand PK, Demon D, Walle LV, Liu Z, Vogel P, Lamkanfi M, and Kanneganti T-D. TRIF-mediated caspase-11 production integrates TLR4- and Nlrp3 inflammasome-mediated host defense against enteropathogens. J Biol Chem 287: 34474–34483, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gutierrez O, Pipaon C, Inohara N, Fontalba A, Ogura Y, Prosper F, Nunez G, and Fernandez-Luna JL. Induction of Nod2 in myelomonocytic and intestinal epithelial cells via nuclear factor-kappa B activation. J Biol Chem 277: 41701–41705, 2002 [DOI] [PubMed] [Google Scholar]
- 36.Halff EF, Diebolder CA, Versteeg M, Schouten A, Brondijk THC, and Huizinga EG. Formation and structure of a NAIP5-NLRC4 inflammasome induced by direct interactions with conserved N- and C-terminal regions of flagellin. J Biol Chem 287: 38460–38472, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, Albrecht M, Mayr G, De La Vega FM, Briggs J, Gunther S, Prescott NJ, Onnie CM, Hasler R, Sipos B, Folsch UR, Lengauer T, Platzer M, Mathew CG, Krawczak M, and Schreiber S. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet 39: 207–211, 2007 [DOI] [PubMed] [Google Scholar]
- 38.Harris J, Hartman M, Roche C, Zeng SG, O'Shea A, Sharp FA, Lambe EM, Creagh EM, Golenbock DT, Tschopp J, Kornfeld H, Fitzgerald KA, and Lavelle EC. Autophagy controls IL-1 secretion by targeting Pro-IL-1 for degradation. J Biol Chem 286: 9587–9597, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hernandez-Cuellar E, Tsuchiya K, Hara H, Fang R, Sakai S, Kawamura I, Akira S, and Mitsuyama M. Cutting edge: nitric oxide inhibits the NLRP3 inflammasome. J Immunol 189: 5113–5117, 2012 [DOI] [PubMed] [Google Scholar]
- 40.Hewison J. and MacKenzie A. A key role for redox signaling in rapid P2X 7 receptor-induced IL-1. J Immunol 180: 8410–8420, 2008 [DOI] [PubMed] [Google Scholar]
- 41.Hise AG, Tomalka J, Ganesan S, Patel K, Hall BA, Brown GD, and Fitzgerald KA. An essential role for the NLRP3 inflammasome in host defense against the human fungal pathogen Candida albicans. Cell Host Microbe 5: 487–497, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Homer CR, Kabi A, Marina-García N, Sreekumar A, Nesvizhskii AI, Nickerson KP, Chinnaiyan AM, Núñez G, and McDonald C. A dual role for receptor-interacting protein kinase 2 (RIP2) kinase activity in nucleotide-binding oligomerization domain 2 (NOD2)-dependent autophagy. J Biol Chem 287: 25565–25576, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, and Fitzgerald KA. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458: 514–518, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA, and Latz E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol 9: 847–856, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Juliana C, Fernandes-Alnemri T, Kang S, Farias A, Qin F, and Alnemri ES. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem 287: 36617–36622, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kanneganti TD, Lamkanfi M, Kim YG, Chen G, Park JH, Franchi L, Vandenabeele P, and Nunez G. Pannexin-1-mediated recognition of bacterial molecules activates the cryopyrin inflammasome independent of Toll-like receptor signaling. Immunity 26: 433–443, 2007 [DOI] [PubMed] [Google Scholar]
- 47.Kayagaki N, Warming S, Lamkanfi M, Walle LV, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, Zhang J, Lee WP, Roose-Girma M, and Dixit VM. Non-canonical inflammasome activation targets caspase-11. Nature 479: 117–121, 2011 [DOI] [PubMed] [Google Scholar]
- 48.Khare S, Dorfleutner A, Bryan NB, Chawon Y, Radian AD, de Almeida L, Rojanasakul Y, and Stehlik C. An NLRP7-containing inflammasome mediates recognition of microbial lipopeptides in human macrophages. Immunity 36: 464–476, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim HJ, Lee S, and Jung JU. When autophagy meets viruses: a double-edged sword with functions in defense and offense. Semin Immunopathol 32: 323–341, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim S, Bauernfeind F, Ablasser A, Hartmann G, Fitzgerald KA, Latz E, and Hornung V. Listeria monocytogenes is sensed by the NLRP3 and AIM2 inflammasome. Eur J Immunol 40: 1545–1551, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim YM, Talanian RV, Li J, and Billiar TR. Nitric oxide prevents IL-1beta and IFN-gamma-inducing factor (IL-18) release from macrophages by inhibiting caspase-1 (IL-1beta-converting enzyme). J Immunol 161: 4122–4128, 1998 [PubMed] [Google Scholar]
- 52.Kofoed EM. and Vance RE. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature 477: 592–595, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kovarova M, Hesker PR, Jania L, Nguyen M, Snouwaert JN, Xiang Z, Lommatzsch SE, Huang MT, Ting JP-Y, and Koller BH. NLRP1-dependent pyroptosis leads to acute lung injury and morbidity in mice. J Immunol 189: 2006–2016, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee GS, Subramanian N, Kim AI, Aksentijevich I, Goldbach-Mansky R, Sacks DB, Germain RN, Kastner DL, and Chae JJ. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature 492: 123–127, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lei Y, Wen H, Yu Y, Taxman DJ, Zhang L, Widman DG, Swanson KV, Wen K-W, Damania B, Moore CB, Giguère PM, Siderovski DP, Hiscott J, Razani B, Semenkovich CF, Chen X, and Ting JP-Y. The mitochondrial proteins NLRX1 and TUFM form a complex that regulates type I interferon and autophagy. Immunity 36: 933–946, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Levinsohn JL, Newman ZL, Hellmich KA, Fattah R, Getz MA, Liu S, Sastalla I, Leppla SH, and Moayeri M. Anthrax lethal factor cleavage of nlrp1 is required for activation of the inflammasome. PLoS Pathog 8: e1002638, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liao KC. and Mogridge J. Activation of the Nlrp1b inflammasome by reduction of cytosolic ATP. Infect Immun 81: 570–579, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lopez-Castejon G, Luheshi NM, Compan V, High S, Whitehead RC, Flitsch SL, Kirov A, Prudovsky I, Swanton E, and Brough D. Deubiquitinases regulate the activity of caspase-1 and IL-1beta secretion via assembly of the inflammasome. J Biol Chem 288: 2721–2733, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, Roose-Girma M, Erickson S, and Dixit VM. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 430: 213–218, 2004 [DOI] [PubMed] [Google Scholar]
- 60.Martinon F, Burns K, and Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 10: 417–426, 2002 [DOI] [PubMed] [Google Scholar]
- 61.Massey D. and Parkes M. Common pathways in Crohn's disease and other inflammatory diseases revealed by genomics. Gut 56: 1489–1492, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, Miller SI, and Aderem A. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat Immunol 7: 569–575, 2006 [DOI] [PubMed] [Google Scholar]
- 63.Miao EA, Mao DP, Yudkovsky N, Bonneau R, Lorang CG, Warren SE, Leaf IA, and Aderem A. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci U S A 107: 3076–3080, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Miao EA, Rajan JV, and Aderem A. Caspase-1-induced pyroptotic cell death. Immunol Rev 243: 206–214, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Moore CB, Bergstralh DT, Duncan JA, Lei Y, Morrison TE, Zimmermann AG, Accavitti-Loper MA, Madden VJ, Sun L, Ye Z, Lich JD, Heise MT, Chen Z, and Ting JP-Y. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature 451: 573–577, 2008 [DOI] [PubMed] [Google Scholar]
- 66.Nakahira K, Haspel JA, Rathinam VAK, Lee S-J, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, Fitzgerald KA, Ryter SW, and Choi AMK. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12: 222–230, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nour AM, Yeung YG, Santambrogio L, Boyden ED, Stanley ER, and Brojatsch J. Anthrax lethal toxin triggers the formation of a membrane-associated inflammasome complex in murine macrophages. Infect Immun 77: 1262–1271, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ogier-Denis E, Mkaddem SB, Vandewalle A. NOX enzymes and TLR signaling. Semin Immunopathol 30: 291–300, 2008 [DOI] [PubMed] [Google Scholar]
- 69.Pan Q, Mathison J, Fearns C, Kravchenko VV, Da Silva Correia J, Hoffman HM, Kobayashi KS, Bertin J, Grant EP, Coyle AJ, Sutterwala FS, Ogura Y, Flavell RA, and Ulevitch RJ. MDP-induced interleukin-1beta processing requires Nod2 and CIAS1/NALP3. J Leukoc Biol 82: 177–183, 2007 [DOI] [PubMed] [Google Scholar]
- 70.Plantinga TS, Crişan TO, Oosting M, van de Veerdonk FL, de Jong DJ, Philpott DJ, van der Meer JWM, Girardin SE, Joosten LAB, and Netea MG. Crohn's disease-associated ATG16L1 polymorphism modulates pro-inflammatory cytokine responses selectively upon activation of NOD2. Gut 60: 1229–1235, 2011 [DOI] [PubMed] [Google Scholar]
- 71.Proell M, Gerlic M, Mace PD, Reed JC, and Riedl SJ. The CARD plays a critical role in ASC foci formation and inflammasome signaling. Biochem J 449: 613–621, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Py BF, Kim MS, Vakifahmetoglu-Norberg H, and Yuan J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell 49: 331–338, 2012 [DOI] [PubMed] [Google Scholar]
- 73.Qu Y, Misaghi S, Izrael-Tomasevic A, Newton K, Gilmour LL, Lamkanfi M, Louie S, Kayagaki N, Liu J, Kömüves L, Cupp JE, Arnott D, Monack D, and Dixit VM. Phosphorylation of NLRC4 is critical for inflammasome activation. Nature 490: 539–542, 2012 [DOI] [PubMed] [Google Scholar]
- 74.Ramjeet M, Hussey S, Philpott DJ, and Travassos LH. “Nodophagy”: new crossroads in Crohn disease pathogenesis. Gut microbes 1: 307–315, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rathinam VAK, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L, Vanaja SK, Monks BG, Ganesan S, Latz E, Hornung V, Vogel SN, Szomolanyi-Tsuda E, and Fitzgerald KA. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol 11: 395–402, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rathinam VAK, Vanaja SK, Waggoner L, Sokolovska A, Becker C, Stuart LM, Leong JM, and Fitzgerald KA. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell 150: 606–619, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rebsamen M, Vazquez J, Tardivel A, Guarda G, Curran J, and Tschopp J. NLRX1/NOD5 deficiency does not affect MAVS signalling. Cell Death Differ 18: 1387, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, Green T, Kuballa P, Barmada MM, Datta LW, Shugart YY, Griffiths AM, Targan SR, Ippoliti AF, Bernard EJ, Mei L, Nicolae DL, Regueiro M, Schumm LP, Steinhart AH, Rotter JI, Duerr RH, Cho JH, Daly MJ, and Brant SR. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet 39: 596–604, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Saiga H, Kitada S, Shimada Y, Kamiyama N, Okuyama M, Makino M, Yamamoto M, and Takeda K. Critical role of AIM2 in Mycobacterium tuberculosis infection. Int Immunol 24: 637–644, 2012 [DOI] [PubMed] [Google Scholar]
- 80.Saitoh T, Fujita N, Jang MH, Uematsu S, Yang B-G, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, Tanaka K, Kawai T, Tsujimura T, Takeuchi O, Yoshimori T, and Akira S. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 456: 264–268, 2008 [DOI] [PubMed] [Google Scholar]
- 81.Sauer JD, Pereyre S, Archer KA, Burke TP, Hanson B, Lauer P, and Portnoy DA. Listeria monocytogenes engineered to activate the Nlrc4 inflammasome are severely attenuated and are poor inducers of protective immunity. Proc Natl Acad Sci U S A 108: 12419–12424, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schroder K. and Tschopp J. The inflammasomes. Cell 140: 821–832, 2010 [DOI] [PubMed] [Google Scholar]
- 83.Shaw PJ, McDermott MF, and Kanneganti TD. Inflammasomes and autoimmunity. Trends Mol Med 17: 57–64, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shi C-S. and Kehrl JH. MyD88 and Trif target Beclin 1 to trigger autophagy in macrophages. J Biol Chem 283: 33175–33182, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA, Sher A, and Kehrl JH. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol 13: 255–263, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shimada K, Crother TR, Karlin J, Chen S, Chiba N, Ramanujan VK, Vergnes L, Ojcius DM, and Arditi M. Caspase-1 dependent IL-1β secretion is critical for host defense in a mouse model of Chlamydia pneumoniae lung infection. PLoS One 6: e21477, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, Rentsendorj A, Vargas M, Guerrero C, Wang Y, Fitzgerald KA, Underhill DM, Town T, and Arditi M. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36: 401–414, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shoshan-Barmatz V. and Ben-Hail D. VDAC, a multi-functional mitochondrial protein as a pharmacological target. Mitochondrion 12: 24–34, 2012 [DOI] [PubMed] [Google Scholar]
- 89.Soares F, Tattoli I, Wortzman ME, Arnoult D, Philpott DJ, and Girardin SE. NLRX1 does not inhibit MAVS-dependent antiviral signalling. Innate Immun 2012. [Epub ahead of print]; DOI: 10.1177/1753425912467383 [DOI] [PubMed] [Google Scholar]
- 90.Suzuki T, Franchi L, Toma C, Ashida H, Ogawa M, Yoshikawa Y, Mimuro H, Inohara N, Sasakawa C, and Núñez G. Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophages. PLoS Pathog 3: e111, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tigno-Aranjuez JT. and Abbott DW. Ubiquitination and phosphorylation in the regulation of NOD2 signaling and NOD2-mediated disease. Biochim Biophys Acta 1823: 2022–2028, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Travassos LH, Carneiro LAM, Ramjeet M, Hussey S, Kim Y-G, Magalhães JG, Yuan L, Soares F, Chea E, Le Bourhis L, Boneca IG, Allaoui A, Jones NL, Núñez G, Girardin SE, and Philpott DJ. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol 11: 55–62, 2010 [DOI] [PubMed] [Google Scholar]
- 93.van Bruggen R, Koker MY, Jansen M, van Houdt M, Roos D, Kuijpers TW, and van den Berg TK. Human NLRP3 inflammasome activation is Nox1-4 independent. Blood 115: 5398–5400, 2010 [DOI] [PubMed] [Google Scholar]
- 94.van de Veerdonk FL, Smeekens SP, Joosten LA, Kullberg BJ, Dinarello CA, van der Meer JW, and Netea MG. Reactive oxygen species-independent activation of the IL-1beta inflammasome in cells from patients with chronic granulomatous disease. Proc Natl Acad Sci U S A 107: 3030–3033, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Vladimer GI, Weng D, Paquette SWM, Vanaja SK, Rathinam VAK, Aune MH, Conlon JE, Burbage JJ, Proulx MK, Liu Q, Reed G, Mecsas JC, Iwakura Y, Bertin J, Goguen JD, Fitzgerald KA, and Lien E. The NLRP12 inflammasome recognizes Yersinia pestis. Immunity 37: 96–107, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang L, Manji GA, Grenier JM, Al-Garawi A, Merriam S, Lora JM, Geddes BJ, Briskin M, DiStefano PS, and Bertin J. PYPAF7, a novel PYRIN-containing Apaf1-like protein that regulates activation of NF-kappa B and caspase-1-dependent cytokine processing. J Biol Chem 277: 29874–29880, 2002 [DOI] [PubMed] [Google Scholar]
- 97.West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, Walsh MC, Choi Y, Shadel GS, and Ghosh S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 472: 476–480, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Xia X, Cui J, Wang HY, Zhu L, Matsueda S, Wang Q, Yang X, Hong J, Songyang Z, Chen ZJ, and Wang R-F. NLRX1 negatively regulates TLR-induced NF-kappaB signaling by targeting TRAF6 and IKK. Immunity 34: 843–853, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhao Y, Yang J, Shi J, Gong Y-N, Lu Q, Xu H, Liu L, and Shao F. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477: 596–600, 2011 [DOI] [PubMed] [Google Scholar]
- 100.Zhou R, Yazdi AS, Menu P, and Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature 469: 221–225, 2010 [DOI] [PubMed] [Google Scholar]