

Abstract
Significance: Autophagy is emerging as an important pathway in many biological processes and diseases. This review summarizes the current progress on the role of autophagy in renal physiology and pathology. Recent Advances: Studies from renal cells in culture, human kidney tissues, and experimental animal models implicate that autophagy regulates many critical aspects of normal and disease conditions in the kidney, such as diabetic nephropathy and other glomerular diseases, tubular injuries, kidney development and aging, cancer, and genetic diseases associated with the kidney. Critical Issues: The importance of autophagy in the kidney has just started to be elucidated. How the process of autophagy is altered in the pathogenesis of kidney diseases and how this alteration is beneficial or detrimental to kidney functions still need to be fully understood. Future Directions: Investigations that uncover the precise mechanism and regulation of autophagy in various kidney diseases may lead to new strategies for therapeutic modulation. Antioxid. Redox Signal. 20, 519–537.
Introduction
Autophagy is a tightly regulated system in which the endogenous cellular protein aggregates and damaged organelles are degraded via the lysosomal pathway. Autophagy was originally known as a catabolic process to provide nutrients and energy to cells under starvation through recycling of endogenous materials. Recently emerging body of evidence indicates that autophagy plays important roles in many biological processes and diseases, including immunity and autoimmunity, inflammation, development and aging, neurodegenerative diseases, and cancer.
There are three types of autophagy, namely macroautophagy, microautophagy and chaperone-mediated autophagy (CMA) (Fig. 1). Macroautophagy, generally referred simply as autophagy, is the most studied and the major type of autophagy discussed in this review. Autophagy initiates with the formation of autophagophores around substances which will be sequestered. Autophagophores elongate and expand, and finally form double-membrane vacuoles, called autophagosomes with sequestered materials inside. Subsequently, outer membranes of autophagosomes fuse with lysosomes or endosomes to form single-membrane autolysosomes, which contain lysosomal enzymes to digest the enclosed substrates. Autophagy is a multiple-step process involving many proteins complexes. The UNC-51-like kinase (ULK) complex (ULK1/2-Atg13-FIP200-Atg101) is responsible for autophagy initiation, the class III phosphoinositide-3 kinase (PI3K) complex (Vps34-Vps15-Beclin1-Barkor) for vesicle nucleation and phagophore formation, followed by Atg12-Atg5-Atg16L complex and lipidated LC3 (LC3-II) for elongation and closure of membrane to form an autophagosome (19, 47, 101). Autophagic flux refers to the entire autophagic process, including the formation of autophagosomes and delivery of cargo to lysosomes via fusion and subsequent degradation (Fig. 1A). LC3-II has been frequently used as a marker to assess autophagy. However, given that autophagy represents a dynamic process, the assessment of autophagic activity requires coordinate use of different methods for monitoring autophagy. Guidelines for the use and interpretation of the methods for monitoring autophagy are discussed in detail in a recent review by Klionsky et al. (43). In contrast to autophagy, substrates of microautophagy are engulfed by direct invagination of the lysosomal membranes into tubulovesicular structures, without the formation of autophagosomes, and are degraded (53) (Fig. 1B). In CMA, proteins containing KFERQ motif are selectively recognized by the cytosolic chaperone, heat shock cognate protein of 70 kDa (hsc70), and in turn, the hsc70/substrate protein complex binds to the lysosomal membrane via lysosome-associated membrane protein 2A (LAMP-2A), leading to unfolding of the substrate and translocation inside the lysosome for subsequent degradation (34) (Fig. 1C).
FIG. 1.

Schematic illustration of the molecular process of autophagy. (A) Macroautophagy (generally referred as autophagy) sequesters and degrades cellular organelles and protein aggregates through the sequential formation of autophagophores, autophagosomes and autolysosomes. (B) Microautophagy degrades cellular organelles and protein aggregates by direct lysosomal engulfment. (C) CMA selectively degrades proteins containing KFERQ motif through the cooperation of heat shock cognate protein of 70 kDa (hsc70) and LAMP-2A. CMA, chaperone-mediated autophagy, LAMP, lysosome-associated membrane protein. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Research into the role of autophagy in kidney physiology and pathology remains still a largely understudied field. We are just beginning to appreciate the complexity of the autophagic pathway owing to a growing body of evidence implicating the importance of autophagy in both the maintenance of kidney homeostasis and disease pathogenesis. Much of the current insight stems from research in renal cells in culture, human kidney tissues, and experimental animal models. These will be discussed in this review. The regulation and function of autophagy is likely cell type and context specific. Here, we will focus on studies in three resident renal cell types, glomerular mesangial cells, and podocytes, which participate in the vital functions of glomerular filtration, and renal tubular epithelial cells (Fig. 2). These highly specialized cell types are targets of many forms of kidney injury and diseases, and highlighted in this review include acute kidney injury (AKI), diabetic nephropathy and other glomerular diseases and renal fibrosis, tubular injury, oxidative stress, cancer, and genetic diseases associated with the kidney, as well as kidney development and aging.
FIG. 2.
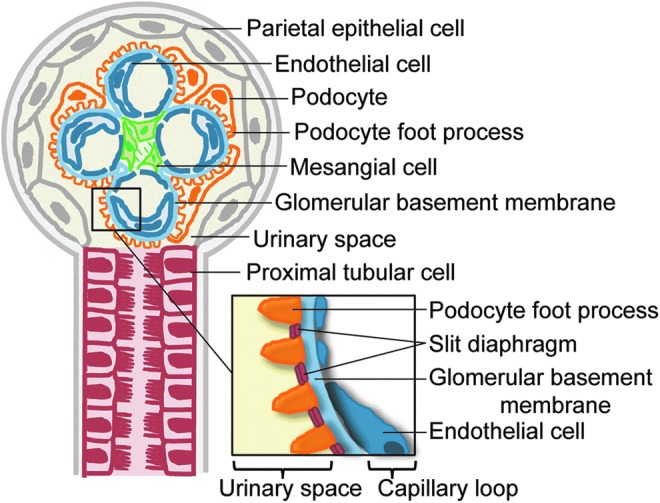
Structure of the glomerulus and proximal tubule. The resident renal cells and components of the glomerulus and proximal tubule are shown. Approximately 180 L of renal plasma is filtered by the glomerulus daily. Filtration begins with the glomerular endothelial cells which functions as part of the glomerular filtration unit (see inset), that also includes the glomerular basement membrane and the podocytes with foot processes, between which are proteins comprising the slit diaphragm. The resultant filtrate from glomerulus flows through the tubules with reabsorption and secretion of ions, carbonhydrates, amino acids, and eventual elimination of urine. Under normal condition, the ultrafiltrate is virtually free of plasma protein. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Autophagy in Glomerular Mesangial Cells
Glomerular mesangial cells are specialized contractile pericytes, unique to the kidney, and located in the centrilobular region called the mesangium, providing structural support for the glomerular tuft as well as forming a functional unit, together with adjacent glomerular capillary endothelial cells and podocytes, to regulate glomerular filtration. Mesangial cells also function as the primary producer of the extracellular matrix that constitutes the mesangium, and are important in maintenance of mesangial matrix homeostasis. They are also major targets of a number of glomerular diseases, such as IgA nephropathy and diabetic nephropathy. In response to injury and progressive kidney disease, mesangial cells proliferate, and produce excessive extracellular matrix, leading to the development of glomerulosclerosis and kidney fibrosis.
The role of autophagy in mesangial cells was not recognized, until very recently. To date, there have been few studies examining autophagy in mesangial cells. Wang and colleagues reported that autophagy is induced in mesangial cells following exposure to the heavy metal cadmium, an environmental toxin which accumulates in the kidneys and causes nephrotoxicity (91). They noted that cadmium induced both autophagy and apoptosis in mesangial cells. However, pharmacological blockade of autophagy resulted in increased cell viability without affecting apoptosis, suggesting that autophagy plays a role in cell death in mesangial cells exposed to cadmium. Moreover, cadmium induced autophagic cell death through a calcium-extracellular signal-regulated kinase dependent pathway (91) and, in part, through increased reactive oxygen species (ROS) production and activation of glycogen synthase kinase-3β (GSK-3β) to stimulate autophagy in mesangial cells (92) (Fig. 3A).
FIG. 3.

Autophagy in glomerular mesangial cells. (A) Autophagy contributes to cadmium-induced mesangial cell death through calcium-ERK and ROS-GSK-3β. (B) TGF-β1-induced autophagy promotes mesangial cell survival by inhibiting apoptosis and inducing degradation of p27Kip1 during prolonged serum deprivation. (C) Autophagy negatively regulates matrix production in mesangial cells by promoting degradation of intracellular Col-I. ERK, extracellular signal-regulated kinase; GSK-3β, glycogen synthase kinase-3β; ROS, reactive oxygen species; Col-I, type I collagen; TGF-β1, transforming growth factor-β1. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Hence, autophagy can lead to cell death in response to stress. Excessive autophagic activity leads to type II programmed cell death that is morphologically distinct from apoptosis, or type I programmed cell death. Autophagy can also serve as a protective mechanism against cell death during nutrient deprivation, and cells undergo apoptotic cell death when autophagy is blocked. Thus, autophagy can have opposing effects on cell survival, and the functional role of autophagy may depend on the specific stimulus and context. We recently reported that autophagy contributed to survival of mesangial cells. Under the condition of serum deprivation, autophagy was induced by transforming growth factor-β 1 (TGF-β1) in mesangial cells, and autophagy enhanced cell survival by inhibiting mesangial cells from undergoing apoptosis (17). Both transient knockdown of LC3 by siRNA and LC3 gene deletion (LC3−/−) in mesangial cells abrogated TGF-β1 rescue from serum deprivation-induced apoptosis. TGF-β1 also enhanced cell cycle progression under serum deprivation conditions through up-regulation of cyclin D1 and E, while downregulating cyclin-dependent kinase inhibitor p27, to promote mesangial cell survival. We also showed that the induction of autophagy by TGF-β1 in mesangial cells was mediated via TGF-β-activated kinase 1 (TAK1) and the PI3K-Akt dependent pathway (17) (Fig. 3B). These findings provide support for the cytoprotective role of autophagy in mesangial cells.
The balance between synthesis and degradation of mesangial matrix is crucial for the maintenance of tissue homeostasis. Mesangial cells produce and regulate turnover of its extracellular matrix. Collagens are the main components of the glomerular mesangium and type I collagen (Col-I) produced by mesangial cells is the major type associated with disease states. Our recent studies unveiled a novel role of autophagy in negatively regulating the matrix production in mesangial cells by promoting the degradation of intracellular Col-I (38). In the kidneys of mice deficient in autophagic protein Beclin 1, through heterozygous deletion of beclin 1 (beclin 1+/−), there was significantly increased collagen deposition compared to littermate controls. Mesangial cells isolated from beclin 1+/− mice or transfected with Beclin 1 siRNA expressed higher basal level of Col-I, and treatment with autophagy inhibitor Bafilomycin A1, but not proteasome inhibitor MG132, increased Col-I protein levels which colocalized with LC3 as well as lysosomal marker LAMP-1. Accordingly, treatment with trifluoperazine, an inducer of autophagy, resulted in decreased Col-I protein levels induced by TGF-β1, without alterations in Col-I α1 mRNA (Fig. 3C). Our studies also confirmed that Col-I and aggregated, insoluble procollagen I undergo intracellular degradation through autophagy (38).
We also explored the role of autophagy in reducing Col-I expression by treatment with low-dose carbon monoxide (CO) which we had previously shown to exert antifibrotic effects in a model of kidney fibrosis induced by unilateral ureteral obstruction (UUO) (89). These studies demonstrated that CO induced autophagy in the kidneys of mice exposed to low-dose CO and in mesangial cells treated with CO-releasing molecule 2 (CORM-2) (38). Treatment with CORM-2 in wild-type mesangial cells also reduced Col-I protein stimulated by TGF-β1, whereas these CORM-2 effects were abrogated in autophagy-deficient beclin 1+/− mesangial cells, suggesting that CO suppresses accumulation Col-I protein induced by TGF-β1, at least, in part, through induction of autophagy (Fig. 3C).
Interestingly, our investigation also showed that TGF-β1 induced autophagy in mesangial cells, and this was mediated via the mitogen-activated protein kinase kinase 3 (MKK3) signaling pathway. TGF-β1 is well-known as an inducer of collagen synthesis, and we previously demonstrated the importance of TAK1-MKK3-p38 signaling in TGF-β1-induced collagen production in mesangial cells (37, 90), indicating the vital role of this signaling cascade in controlling the level of Col-I in mesangial cells (Fig. 3C). TGF-β1 is a prototypic multifunctional cytokine. The dual functions of TGF-β1, as both an inducer of Col-I synthesis and an inducer of autophagy and Col-I degradation, underscore the multifunctionality of TGF-β1.
In summary, these studies suggest that autophagy may constitute an adaptive mechanism to glomerular injury by inhibiting apoptosis and promoting mesangial cell survival. The findings also implicate a novel role of autophagy as a cytoprotective mechanism to negatively regulate and prevent excess collagen accumulation in the glomeruli, and hold promise of a new therapeutic target to mitigate pathogenesis of glomerulosclerosis and prevent kidney fibrosis. Indeed, further research in the future is needed to help to broaden and deepen our understanding of the role of autophagy in mesangial cells.
Autophagy in Podocytes
Podocytes are highly differentiated epithelial cells lining the outer aspect of the glomerular basement membrane, and have characteristic foot processes that interdigitate with foot processes of adjacent podocytes. Filtration slits are formed between the interdigitating foot processes of neighboring podocytes and are covered by the slit diaphragm. The podocyte foot processes and slit diaphragm play a major role in the selective permeability of the glomerular filtration barrier (Fig. 2). Podocyte injury that results in disruption of this complex molecular architecture, either from alterations in the slit diaphragm components or effacement of foot processes, is typically associated with marked proteinuria. Terminally differentiated podocytes are vulnerable to various kinds of injury, and the loss of podocytes is considered a key feature of progressive glomerular disease. Autophagy is a fundamental cellular homeostatic process that cells use to degrade and recycle cellular proteins and remove damaged organelles. This self-repair mechanism is especially important in postmitotic cells, such as podocytes which, like neurons but unlike the other cell types forming the glomerular filtration unit, have a very limited capacity for cell division and replacement. Evidence suggest that podocytes exhibit a high basal level of autophagy as seen from LC3 staining of normal rat kidney tissue (1), and in green fluorescent protein (GFP)-LC3 transgenic mice (27) and GFP-γ-aminobutyric acid a receptor-associated protein (GABARAP) transgenic mice (80) compared to other cell types in the kidney. GABARAP has recently been characterized as a homolog of LC3. Abundance of autophagosomes in podocytes was confirmed by electron microscopy (27). Moreover, blockade of autophagy with lysosomal inhibitor chloroquine resulted in accumulation of LC3-II and GFP-LC3 positive autophagosomes in GFP-LC3 transgenic podocytes, indicating a high rate of autophagic flux (27).
The regulation of autophagy in podocytes is just beginning to be elucidated. Class III PI3K vacuolar protein sorting 34 (Vps34) is generally thought to play an important role in the initiation of autophagy. Mice with podocyte-specific deletion of Vps34 developed early proteinuria, progressive glomerulosclerosis, and renal failure by 9 weeks (3). Podocytes from these knockout mice displayed a phenotype of impaired autophagic flux with accumulation of enlarged vacuoles and elevated levels of LC3, LAMP-1, and LAMP-2, suggesting that Vps34 participates in maintaining autophagic flux in podocytes (Fig. 4D).
FIG. 4.

Autophagy in podocytes. (A) Atg5 gene deletion in podocytes (Atg5Δpodocyte) increases susceptibility to injury induced by PAN or adriamycin (ADR). (B) Autophagy is activated by ER stress induced by PHN or tunicamycin (TM). (C) Autophagy prevents angiotensin II (ANG-II)-induced apoptosis in podocytes. (D) Vps34 gene deletion in podocytes (Vps34Δpodocyte) inhibits formation of autophagophore and the mTOR pathway, resulting in accumulation of autophagosomes (APs) and autolysosomes (ALs) in podocytes. (E) Mtor gene deletion (MtorΔpodocyte) or chronic use of rapamycin disrupt the recycling of lysosomes from autolysosomes, resulting in the accumulation of APs and ALs in podocytes. (F) PRR gene deletion in podocytes (PRRΔpodocyte) inhibits acidification of lysosomes by inactivating H+-ATPase, resulting in accumulation of APs and unacidified ALs. Vps34, vacuolar protein sorting 34; PRR, prorenin receptor; PAN, puromycin aminonucleoside; PHN, passive Heymann nephritis; ER, endoplasmic reticulum. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
The mechanistic target of rapamycin (mTOR), a serine/threonine kinase, is the classical pathway that regulates autophagic activity. It functions through its association with two distinct protein complexes mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). In general, mTOR negatively regulates autophagy. Nutrient starvation induces autophagy primarily through inhibition of mTORC1. How mTOR regulates autophagy in podocytes is still not well understood. Interestingly, recent study in mice with podocyte-selective knockout of the Mtor gene (Mtor pod-KO) suggests that inhibition of mTOR disrupts autophagic flux in podocytes. These mice developed proteinuria at 3 weeks of age, and progressive podocyte damage with foot process effacement, widespread vacuolization, and ultimately end-stage kidney failure by 5 weeks of age (13). Increased glomerular LC3-II expression and accumulation of autophagosomes, autolysosomes, and damaged mitochondria were seen in podocytes. Similarly, in immortalized human podocytes, prolonged treatment with mTOR inhibitor rapamycin (also known as sirolimus) induced accumulation of autophagosomes and autolysosomes (13). Almost complete colocalization of GFP-LC3 puncta (autophagosomes) with lysosomes was noted, indicating incomplete autophagy due to a block in the final steps of clearance and recycling of membrane components and lysosomal reformation. Spatial coupling of mTOR and autolysosomes has been observed in podocytes (58). Furthermore, a recent report showed that mTOR regulated both the initiation and termination of autophagy. Inhibition of mTOR activated autophagy, but prolonged starvation reactivated mTOR with degradation of autolysosomal products, and mTOR reactivation was required to regenerate functional lysosomes and completion of autophagic process (103). However, in Mtor pod-KO mice or prolonged rapamycin treatment, mTOR activity is blocked at both points. Thus, mTOR inhibition activates autophagy, but mTOR reactivation is also blocked, resulting in disruption of autophagic flux (Fig. 4E). Development of de novo or worsening proteinuria is well-recognized in patients with chronic use of rapamycin. Given that disruption of the autophagic pathway may play a role in the pathogenesis of proteinuria, therapy with mTOR inhibitors can be a double-edged sword with both favorable and unfavorable consequences.
Recently, two groups simultaneously reported that autophagic flux is impaired in podocytes with prorenin receptor (PRR/ATP6ap2) deletion (61, 66). The podocyte-specific, conditional, PRR-knockout (cKO) mice developed nephrotic syndrome within 2–3 weeks after birth, and eventually all were dead by the 4th week (61, 66). Electron microscopy revealed that the cKO mice displayed progressive podocyte damage with foot process effacement and vacuolation, and podocyte cell death (61, 66). Increased LC3 and LAMP-2 staining in glomeruli of the cKO mice reflected an accumulation of autophagsomes and lysosomes in podocytes with PRR deletion (61, 66). Dramatic accumulation of ubiquitinated protein and ubiquitin-binding scaffold protein p62/sequestosome 1 (SQSTM1) further suggested a block in autophagic clearance of the ubiquitinated protein aggregates (66). In vitro, inhibiting the function of PRR using adenoviral Cre recombinase in primarily isolated floxed podocytes (66) or siRNA knockdown of PRR in conditionally immortalized podocytes (61) resulted in a disruption of the cellular actin network and an accumulation of lysosomes with impaired acidification and autolysosomes, which resembled the effects of vacuolar H+-ATPase (V-ATPase) inhibitor Bafilomycin A1. Deficiency of PRR in podocytes disrupts the glomerular filtration barrier and V-ATPase function, leading to impairment of autophagy in podocytes and cell death. Thus, PRR is critical for normal podocyte function and cell survival through maintenance of autophagic flux in podocytes by sustaining the normal function of V-ATPase and lysosomal acidification (Fig. 4F).
Autophagy in Podocyte Injury
Recent studies have shown that injuries to podocytes play a critical role in the development of many glomerular diseases. Analysis of human kidney biopsies showed evidence of increased autophagosome formation in podocytes in several glomerular diseases. Autophagic vacuoles containing aggregated ribosomes and lipid droplets and formation of autophagosomes were detected in podocytes by light and transmission electron microscopy analysis of biopsy tissues from patients with IgA nephropathy, and correlated with histopathologically more aggressive disease (71, 72). In addition, immunofluorescence studies confirmed increased LC3-positive autophagosomes in podocytes in the kidney biopsy samples from patients with membranous glomerulonephritis compared with controls from pretransplant allograft biopsies (27). Furthermore, Atg3 mRNA was significantly higher in microdissected glomeruli from patients with focal segmental glomerulosclerosis and membranous glomerulonephritis than in normal controls (pretransplant allograft biopsies) (27). These findings demonstrate upregulation of autophagy in podocytes in human proteinuric glomerular diseases.
Studies in animal models of glomerular injury inducing proteinuric diseases also demonstrated upregulation of autophagy in podocytes. Puromycin aminonucleoside (PAN)-induced nephrosis is an experimental rat model of nephrotic syndrome, akin to human minimal change disease. Increased LC3-positive autophagosomes were detected in podocytes during recovery after PAN injection, compared with control normal kidney (1). Complementary studies in cultured differentiated mouse podocytes confirmed increases in LC3-II levels and LC3 puncta during recovery from PAN-induced injury (1). Similar findings of increased LC3-II levels and glomerular LC3 immunofluorescence staining were reported in a rat model of passive Heymann nephritis (PHN), which resembles membranous nephropathy in humans, and in tunicamycin treated podocytes, through induction of endoplasmic reticulum (ER) stress (88) (Fig. 4B). Furthermore, the administration of albumin (bovine serum albumin [BSA])-overload in GFP-LC3 transgenic mice resulted in a threefold increase in GFP-LC3 positive autophagosomes (27). These data indicate involvement of autophagy in podocyte injury response in proteinuric glomerular diseases. The induction of proteinuria in mice with podocyte-specific deletion of the Atg5 gene with PAN or adriamycin resulted in more severe albuminuria, foot process effacement, loss of podocytes, and glomerulosclerosis, compared with littermate control mice (27) (Fig. 4A). The podocyte-specific Atg5 deleted mice also developed significantly higher transient albuminuria after challenge with low dose of BSA or lipopolysaccharide (27). These studies provide evidence that deficiency in autophagy enhances susceptibility to development of glomerular diseases, and that autophagy represents a stress adaptive response of podocytes that is cytoprotective against glomerular disease.
Autophagy in Renal Tubular Injury
Renal tubules, especially proximal tubules, are susceptible to different kinds of injuries from AKI, induced by nephrotoxic drugs, environmental toxins, and ischemia/reperfusion (I/R), to chronic kidney diseases (CKD), characterized by tubular atrophy and interstitial fibrosis. Autophagic vacuoles were first observed in isolated cortical tubular fragments from the rat kidney by electron microscopy in a report by Pfeifer and Guder in 1975 (65). However, the functional role of autophagy in renal tubular cells, particularly during stress conditions arising from various injuries has not been extensively studied until the recent decade. Here we discuss the current advances in our understanding of the role of autophagy in renal tubular injury during AKI and CKD.
Autophagy in nephrotoxin-induced AKI
Numerous studies have demonstrated the induction of autophagy in renal proximal tubular cells by cisplatin, a well-known nephrotoxic anticancer drug, which accumulates in the proximal tubules and causes AKI. Upregulation of autophagy before apoptosis was detected both in the renal proximal tubules of mice injected with cisplatin and in cultured proximal tubular cells treated with cisplatin (6, 29, 32, 64, 67, 81, 99). Whether the induction of autophagy in kidney tubular injury is a cell death or cell survival mechanism is debatable. Inoue and colleagues showed that cisplatin-induced apoptosis was suppressed when autophagy was suppressed with autophagy inhibitors or knockdown of Beclin 1 by siRNA in proximal tubular cells, suggesting that autophagy might promote cell death in proximal tubular cells after cisplatin treatment (29). Other studies demonstrated that autophagy inhibitors or Beclin shRNA enhanced the activation of caspases and apoptosis in cisplatin treated proximal tubular cells, suggesting that autophagy protects tubular cells from apoptosis (64, 67, 99). More recent study reported that proximal tubule-specific autophagy-deficient mice developed more severe AKI and increased apoptosis after cisplatin treatment, compared to the control mice (32, 81). Moreover, cisplatin treatment increased DNA damage, p53 and c-Jun N-terminal kinase (JNK) activation, and accumulation of toxic protein aggregates and ROS in autophagy-deficient proximal tubules (32, 81) (Fig. 5A). These results provide further support for the cytoprotective role of autophagy in cisplatin-induced AKI.
FIG. 5.

Autophagy in tubular injuries. (A) Autophagy activation, through ROS or ER stress, protects tubular cells from cisplatin or ischemia-reperfusion (I/R)-induced injuries. Mice with Atg5 or Atg7 gene deletion in PTC developed more severe tubular damage and renal dysfunction (increased BUN and SCr), compared to control littermates. P53 inhibitor or Bcl-2 inhibits autophagy induced by cisplatin in tubular cells. Repetitive hypoxic conditioning, Bcl-xL, Bcl-2, or ischemic conditioning inhibits both autophagy, apoptosis and renal dysfunction. (B) TGF-β1 overexpression in tubular cells induces autophagy, through ROS, resulting in tubular apoptosis and decomposition. (C) Autophagy is activated in a model of renal fibrosis induced by UUO, and is associated with inhibition of tubular apoptosis and interstitial fibrosis in the obstructed kidney, and reduced cell proliferation in the contralateral kidney. Bcl-xL, B-cell lymphoma-extra large; UUO, unilateral ureteral obstruction; I/R, ischemia-reperfusion; BUN, blood urea nitrogen; SCr, serum creatinine; PTC, proximal tubular cells. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
The induction of autophagy by a number of other nephrotoxins has been reported recently. Autophagy is induced in proximal tubular cells during cyclosporine-induced nephrotoxicity (62). Cyclosporine is a potent immunosuppressive drug widely used in preventing transplant rejection and treating autoimmune diseases, but its long-term use causes tubular atrophy, interstitial fibrosis, and glomerulosclerosis, and impairs renal function. Autophagy was activated via induction of ER stress by cyclosporine, and protected tubular cells from cell death (62). In addition, exposure to environmental toxins, cadmium, and arsenite, activated autophagy in the tubular cells. Cadmium is a heavy metal which is reabsorbed by proximal tubular cells, accumulates in the lysosomes of these cells, and causes nephrotoxicity. Activation of autophagy was observed in proximal tubules of mice given subtoxic doses of cadmium which did not affect the tubular functions or induce apoptosis (10). Cadmium induced both autophagy and ER stress in cultured proximal tubular cells (10). Autophagy may serve as an adaptive mechanism to remove the damaged proteins generated from ER stress. In contrast, autophagy may play a detrimental role in arsenite-induced AKI. Sodium arsenite induced autophagic cell death in renal tubular cells both in vitro and in vivo, and suppression of autophagy by 3-methyadenine (3-MA) or Atg7 knockdown attenuated cell death (40). Autophagy is also implicated in the cytotoxicity of nanomaterials in proximal tubular cells (33, 78). Cell death induced by fullerenol exposure at millimolar concentrations was associated with cytoskeleton disruption, autophagic vacuole accumulation and mitochondrial dysfunction. Furthermore, autophagy inhibitor 3-MA ameliorated loss of mitochondrial membrane potential and ATP depletion, suggesting that autophagy may contribute to fullerenol-induced cell death (33).
Autophagy in I/R-induced AKI
I/R injury is a common cause of AKI seen in the clinical setting. It inevitably occurs during kidney transplantation, and the initial extent of I/R injury significantly affects the function of graft. Activation of autophagy in tubular cells during I/R injury was demonstrated in cell cultures and in animal models of I/R injury (11, 23, 30, 31, 41, 79, 85, 93, 102), and increased autophagosomes were observed in biopsy specimens from human transplanted kidney (79). In regard to the question whether autophagy provides protection or contributes to the tubular damage from I/R injury, there are both lines of evidence.
A number of studies show both in vivo and in vitro evidence supporting for a detrimental role of autophagy in I/R injury. For instance, augmentation of antiapoptotic B-cell lymphoma-extra large (Bcl-xL) expression by intrarenal adenoviral Bcl-xL gene transfer, or overexpression of Bcl-2 in Bcl-2/GFP-LC3 double transgenic mice, protected renal tubular epithelial cells from I/R injury by inhibiting both autophagy and apoptosis (11, 30) (Fig. 5A). Ischemic conditioning by short periods of reperfusion and hypoxic preconditioning reduced both autophagy and apoptosis and tubular injury induced by I/R, and provided renoprotection against I/R (93, 102) (Fig. 5A). The inhibition of autophagy using autophagy inhibitor 3-MA or Atg7 siRNA significantly reduced ROS (hydrogen peroxide) induced cell death in proximal tubular epithelial cell line (HK-2) (79). Inhibition of autophagy flux using bafilomycin A1 also significantly reduced apoptotic cell death and caspase activity in kidneys with prolonged ischemic preservation (85).
On the other hand, Jiang and colleagues demonstrated that blocking autophagy with 3-MA or either Beclin1 siRNA or Atg5 siRNA enhanced hypoxia-induced apoptosis in cultured renal proximal tubular cells (31). In vivo inhibition of autophagy by 3-MA or chloroquine worsened renal I/R injury in mice (31). Therefore, these results support the notion that autophagy provides a protective mechanism against I/R injury. Recent studies using mice with conditional Atg5 or Atg7 gene deletion in the proximal tubule confirmed that autophagy protected the proximal tubule from I/R injury (32, 41). The autophagy-deficient mice exhibited kidney dysfunction with significantly elevated levels of blood urea nitrogen and serum creatinine than did the control mice, and had increased apoptotic cells in proximal tubules with accumulation of p62 and ubiquitin positive cytoplasmic inclusions, indicating that autophagy maintains the tubular homeostasis by removal the accumulation of harmful protein aggregates and thus ameliorated proximal tubule damage in I/R injury (41) (Fig. 5A). Taken together, these studies suggest that autophagy functions to maintain proximal tubule cell homeostasis and protects against ischemic injury. Further studies are needed to determine whether enhancing autophagy could be a novel therapeutic approach to minimize AKI.
Autophagy in tubulointerstitial fibrosis
Renal tubulointerstitial fibrosis is the hallmark of progressive CKD and end-stage kidney diseases accompanied by tubular degeneration and atrophy. TGF-β1 is arguably the most potent profibrotic cytokine known. Induction of autophagy was reported in a transgenic mouse model with tertracycline-controlled overexpression of TGF-β1 in renal tubular epithelial cells (44). Tubular overexpression of TGF-β1 induced dedifferentiation and decomposition of tubular cells by autophagy, and tubulointerstitial fibrosis, but not epithelial-to-mesenchymal transition, and no sign of apoptosis, suggesting that autophagy might mediate tubular cell death (44). Cell culture studies showed that TGF-β1 activated autophagy through ROS, and autophagy promoted apoptosis in tubular cells (97) (Fig. 5B). Increased autophagy with apoptosis and necrosis in tubules was also demonstrated in the renal fibrosis model, induced by UUO, in several studies (21, 22, 39, 50, 96). The UUO model is a well-established model of progressive renal interstitial fibrosis. The induction of autophagy was shown to precede tubular apoptosis and interstitial fibrosis and peaked after 3 days of UUO in the obstructed kidney of rats, and inhibition of autophagy with 3-MA enhanced tubular cell apoptosis and interstitial fibrosis (39) (Fig. 5C). The expression of Akt and mTOR was decreased over the first 3 days in the obstructed kidney after UUO, suggesting that Akt-mTOR signaling might regulate activation of autophagy in the UUO model (39). These studies indicate that autophagy has a beneficial role in alleviating tubular damage and kidney fibrosis.
In summary, autophagy is activated in various forms of renal tubular injury. At the current stage, the precise role of autophagy in tubular injury response and the pathogenesis of kidney fibrosis is not well understood. There have been studies that provide evidence to support a cytoprotective role of autophagy, and others that support deleterious effects of autophagy. It is plausible that it is context dependent. The difference in the types of injuries and severity of injuries may produce different outcome of autophagy, in that, a certain degree of autophagic activity can maintain tissue homeostasis, whereas excessive autophagic activity results in cell death. Future investigations, for instance by using targeted autophagic gene knockout mice, are necessary to elucidate and clarify the precise functional role of autophagy in tubular injuries.
Autophagy in Kidney Development
It has become increasingly evident that autophagy is important in many biological processes, and deficiency in autophagy leads to pathogenesis of kidney diseases. However, autophagy appears to be dispensable for kidney development. Constitutive embryonic deletion of Atg5 gene does not impair glomerular development (27). Histology and electron microscopy examination did not reveal any obvious morphological abnormality in the glomeruli of Atg5 knockout mice at E19, although there was a lack of conversion of LC3-I to LC3-II, indicating disruption of autophagic activity in the kidneys of the Atg5 knockout mice (27). Therefore, Atg5-dependent autophagy is dispensable for glomerular development. The formation of autophagosomes, as visualized by GFP-LC3 puncta in the GFP-LC3 transgenic mice, is not detectable until late podocyte differentiation in the late capillary loop stage. Therefore, autophagy is dispensable for normal podocyte differentiation, even though podocytes exhibit a high level of basal autophagy in adult mice (27, 55). The glomeruli and podocytes of newborn podocyte-specific Atg5 knockout mice also appeared normal by histological and ultrastructural analysis, and Atg5 deletion did not affect the number of developing nephrons (27). Examination of tubular-specific Atg5 knockout mouse embryos at E19.5 also revealed no discernible tubular abnormalities, indicating that autophagy is dispensable for renal tubule development (51). These studies indicate that Atg5-dependent autophagy is dispensable for kidney development, although they do not exclude the possibility of compensation by an Atg5-independent autophagy, other autophagy pathways, such as CMA, or other proteolytic processes, such as proteasome-mediated degradative pathways.
Autophagy in Kidney Homeostasis and Aging
Although autophagy may be dispensable for kidney development, data suggest that autophagy is vital for the maintenance of kidney tissue homeostasis and aging. Autophagy is generally believed to be responsible for maintaining cellular homeostasis in postmitotic cells, such as podocytes, which cannot be self-renewed through cell division. Podocytes exhibit a high level of basal autophagy that could serve as a mechanism for their maintenance of cellular homeostasis. Although the podocyte-specific Atg5 knockout mice develop normally, by 8–12 months of age, they developed mild proteinuria, and ultrastructural analysis showed signs of retardation in the turnover of organelles with an accumulation of enlarged rough ER and abnormal membranous structures in podocytes (27) (Fig. 6A). These changes were accompanied by enhanced activity of the proteasomal machinery, compensating for autophagy deficiency, resulting in no significant accumulation of poly-ubiquitinated proteins. Inhibition of proteasome activity with bortezomib injection induced increased proteinuria in the podocyte-specific Atg5 knockout mice compared to the control littermates (27). However, this compensatory mechanism through the proteasome pathway waned with aging, and by age 20–24 months, significantly reduced proteasomal activity was observed in the glomeruli of podocyte-specific Atg5 knockout mice (27). These mice developed more severe proteinuria, with foot process effacement, proteinaceous casts, tubular dilation, and vacuolar degeneration, compared to the control mice, and displayed features typical of aging cells, such as damaged mitochondria and lipofuscin accumulation (27). Both ER stress and the accumulation of ubiquitinated protein aggregates and oxidized proteins ultimately resulted in podocyte loss and glomerulosclerosis (27) (Fig. 6A). Therefore, these studies underscore the importance of basal autophagy and compensatory proteasome pathway in the maintenance of glomerular homeostasis and function, and with aging, the deficiency in autophagy and proteasome pathways leads to proteinuria, loss of podocytes and development of glomerulosclerosis.
FIG. 6.

Autophagy in kidney homeostasis and aging. (A) Autophagy maintains podocyte homeostasis. Autophagy deficiency is compensated by elevated proteasome activity in podocytes with Atg5 gene deletion in adult mice (ages 8–12 months of age), resulting in mild proteinuria. In aged Atg5Δpodocyte mice (20–24 months of age) compensatory proteasome activitydecreases, leading to severe proteinuria, loss of podocytes, and glomerulosclerosis. (B) Autophagy maintains tubular homeostasis. Double knockout of Atg5 in PTC and DTC results in kidney dysfunction. (C) Autophagy protects against aging-associated CKD. Activation of autophagy by calorie restriction protects aging kidney from hypoxia-induced oxidative stress and mitochondrial damage, and attenuates aging-associated kidney dysfunction. CKD, chronic kidney diseases; DTC, distal tubular cells. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Unlike podocytes, tubules display a low level of basal autophagy under normal conditions, as only a few GFP-LC3 positive autophagosomes were detected in the tubules in GFP-LC3 transgenic mice (51). This finding may imply that autophagy is not crucial for preservation of normal physiological function in tubules. However, data from mice with tubule-specific deletion of Atg5 gene indicate to the contrary. Mice with Atg5 knockout in proximal tubules (Atg5flox/flox;KAP-Cre+) gradually developed deformed mitochondria and accumulation of cytosolic inclusions, leading to proximal tubular cell hypertrophy and eventual degeneration, not observed in the wildtype control mice (41). They also developed mild glycosuria and amino aciduria, but no urinary albumin excretion, and serum urea nitrogen remained normal up to 9 months of age (41). Mice with Atg5 knockout in distal tubules (Atg5flox/flox;Ksp-Cre) also displayed a significant accumulation of p62/SQSTM1 and oxidative stress markers in the Atg5 deficient distal tubules, but no significant alteration in kidney function (assessed by serum creatinine and urea nitrogen levels) up to 12 months of age, compared to control littermates (51). Atg5 deletion in the entire tubule system in doxycycline-inducible kidney tubule-specific Atg5 knockout mice (Atg5flox/flox;Pax8.rtTA;tetO.Cre) resulted in accumulation of p62/SQSTM1 throughout the tubular segments, and at 5 months after induction, in aging-dependent manner, there was a mild but significant increase in serum creatinine (51). Therefore, while Atg5 deficiency solely in proximal or distal tubular cells did not cause significant renal dysfunction, Atg5 deficiency in all tubule segments caused impairment of kidney function, suggesting tubular autophagy is important in preservation of kidney function (Fig. 6B). Moreover, Atg5 deficiency in tubules induced more severe ischemic kidney injury, with rapid accumulation of p62/SQSTM1- and ubiquitin-positive inclusions in the tubules in response to I/R injury, and accumulation of damaged mitochondria, tubular cell apoptosis, and impaired kidney function (51). Taken together, these studies suggest that autophagy is important for maintaining homeostasis of kidney tubular cells, and that autophagy deficiency promotes aging-associated tubular injury and increases susceptibility to ischemic kidney injury.
Kidney is a target organ during the process of aging and progressive decline in kidney function occurs with aging. Increased incidence of CKD is seen with aging. Impaired autophagy may contribute to aging. Decrease in autophagic activity with aging was observed in the kidneys of mice and rats (14, 45). Reduced expression of Atg7 mRNA and LC3 protein with aging was associated with increase in p62/SQSTM1 and polyubiquitin aggregates in the kidneys of aged mice and rats (14, 45). Mitochondrial dysfunction and oxidative stress are features of aging, and accordingly, the mitochondrial DNA oxidative damage marker 8-hydrodeoxyguanosine was increased in the kidneys of aged mice and rats, with evidence of mitochondrial cristae enlargement and disintegration by transmission electron microscopy (14, 45). If decreased autophagic activity contributes to aging and age-related disease, activation of autophagy may be antiaging and protect the kidney against aging, by engulfing and effectively removing aging-associated mitochondrial damage and reducing oxidative stress. A recent study demonstrated that calorie restriction increases autophagic activity, via silent information regulator T1 (SIRT1)-forkhead box O3 (FoxO3) axis, and protected aging kidney from hypoxia-induced oxidative stress and mitochondrial damage, and attenuated aging-associated kidney dysfunction in aged mice (45) (Fig. 6C). Therefore, restoring autophagic activity during aging could be a promising strategy to attenuate aging-associated tissue damage and preserve the function of renal cells in aged kidneys.
Autophagy in Diabetic Nephropathy
Diabetic nephropathy is a devastating complication in patients with type 1 and type 2 diabetes and is associated with increased morbidity and mortality, and significant economic burden. It is the leading cause of end-stage kidney disease in the United States. Development of progressive albuminuria seen in diabetic nephropathy is indicative of damage of the glomerular filtration system. Histological changes include mesangial matrix expansion, thickening of glomerular and tubular basement membrane, diffuse or nodular glomerulosclerosis, hyalinosis, and tubulointersitial fibrosis. Studies suggest that the pathogenesis of diabetic nephropathy is associated with impaired autophagic activity. Evidence of inhibition of cellular autophagy was first observed in streptozotocin (STZ)-induced diabetes in rats by electron microscopy analysis in the 1990s (2, 25, 26). The volume and number of autophagic vacuoles were significantly lower in proximal (2, 25) and distal tubules (26) of diabetic rats. The inhibition of autophagy was partially reversed by insulin treatment or islet transplantation in the diabetic rats (25, 26). More recent studies showed that the level of p62/SQSTM1 was significantly elevated, an indicator of impaired autophagy, in the kidneys of mice with STZ-induced diabetes mellitus (86) and Wistar fatty (fa/fa) rats (42), which are models of type 1 and type 2 diabetes, respectively. In addition, swollen mitochondria with disrupted cristae, indicative of mitochondrial damage, were also observed in the kidneys of the Wistar fatty (fa/fa) rats (42). Taken together, these studies indicate impairment of autophagy in diabetic nephropathy.
CMA was also inhibited in the kidneys of STZ-induced diabetes in rats (76). The abundance of CMA substrates with KFERQ motif was increased in the renal cortex of diabetic rats. The levels of M2 isoform of pyruvate kinase (M2PK) and GAPDH, two representative proteins degraded by CMA, were elevated in the diabetic rats. Moreover, the lysosomal enrichment of molecular chaperone hsc-73 was significantly reduced in the renal cortex of these diabetic rats compared with pair-fed controls (76). Thus, there was a decrease in proteins which regulate CMA and an increase in proteins degraded by this pathway, indicating that CMA was inhibited in diabetes and may contribute to the accumulation of certain proteins in diabetic-induced renal hypertrophy.
Recent work in experimental diabetes induced by STZ in rats has shown that treatment with rapamycin, an inhibitor of mTORC1, prevents the development of diabetic nephropathy (18, 52, 56, 57, 68, 100). This implicates a potential pathogenic role of mTOR pathway in diabetic nephropathy. The nutrient-sensing mTOR pathway is a well-established inhibitor of autophagy, and nutrient starvation induces autophagy primarily through inhibition of mTORC1. Enhanced mTORC1 activity is seen in human and experimental diabetic nephropathy (18, 24, 28, 52, 56, 57, 68). Studies in mice with podocyte-specific knockout of tuberous sclerosis complex 1 (Tsc1) gene showed that podocyte-specific mTORC1 activation recapitulated many features of diabetic nephropathy, including podocyte injury, glomerular basement membrane thickening, mesangial expansion, and proteinuria (24, 28). Tuberous sclerosis complex 1 (TSC1) is an upstream negative regulator of mTORC1, and loss of TSC1 results in activation of the mTORC1 pathway. Conversely, reduction of mTORC1 activity in podocytes by deletion of one allele of Raptor gene in diabetic mice prevented the development of diabetic nephropathy (24, 28). RAPTOR is a regulatory-associated protein of mTOR and an essential component of mTORC1, thus RAPTOR deficiency inhibits TORC1 activity. These data suggest that diabetes-induced mTORC1 activation in podocytes contributes to podocyte injury and proteinuria, and thereby plays a crucial role in the development of diabetic nephropathy.
One of the major upstream regulators of mTOR is AMP-activated protein kinase (AMPK), a critical energy sensor. Glucose is a metabolic fuel that regulates cellular energy status. AMPK is activated in energy-depleted states, such as those induced by cellular stressors, including hypoxia, heat-shock, ischemia, and glucose deprivation. Activation of AMPK inhibits mTOR pathway, and thereby induces autophagy (Fig. 7A). In the high glucose milieu, studies showed that AMPK phosphorylation and activity were reduced in the renal cortex of kidneys from STZ-induced diabetic rats and db/db mice, which are models of type 1 and type 2 diabetes, respectively (9, 15, 36, 46, 75, 82). AMPK activators, resveratrol (9, 15, 36), metformin (9, 15, 36, 46, 75, 82), and 5-aminoimidazole-4-carboxamide-1β-riboside (AICAR) (46) attenuated renal hypertrophy, lipid accumulation and albumin excretion in diabetic kidneys. Thus, inhibition of AMPK activity contributes to alterations in the diabetic kidney, such as renal hypertrophy, and AMPK may be an attractive therapeutic target for reversing diabetic kidney injury.
FIG. 7.

Autophagy in kidney diseases. (A) Activation of autophagy ameliorates DN. Autophagic activity is inhibited in DN due to aberrant energy sensing pathways. Reactivation of autophagy, for instance by caloric restriction, reduces oxidative stress and protects against DN. (B) Autophagy in PKD and NC is impaired. Basal level of autophagy can be altered through aberrant activation of mTOR or HIF-1α, and the autophagic flux is impaired in PKD. Whether autophagy protects against apoptosis in cyst lining tubular cells and cystogenesis is unknown. Mitophagy is impaired in NC through impairment of autophagic flux, and leads to tubular cell apoptosis, ROS production, and ATP reduction. (C) Autophagy can promote cell survival and tumor growth, or autophagic cell death in RCC. Loss of miR-204, through inactivation of VHL tumor suppressor gene, induces autophagy and tumor growth. In VHL-deficient RCC cells, STF-62247, or sphingosine kinase 2 inhibitor inhibits tumor growth through autophagic cell death. PI3K/mTOR inhibition induces apoptosis and autophagy. HIF-1α, hypoxia-inducible factor-1α; PI3K, phosphoinositide-3 kinase; PKD, polycystic kidney disease; VHL, von Hippel-Lindau; RCC, renal cell carcinoma; NC, nephropathic cystinosis; DN, diabetic nephropathy. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
SIRT1 is an NAD+-dependent deacetylase that functions also as an intracellular energy sensor, to detect the concentration of NAD+ and controls in vivo metabolic changes under caloric restriction and starvation. Similar to AMPK, SIRT1 is activated under low-energy conditions, and negatively regulates mTOR, and thereby induces autophagy. Like AMPK, treatment with resveratrol, metformin, and AICAR can increase SIRT1 activity, though some may not be direct activators of SIRT1. It has been suggested that the similarities occur because AMPK and SIRT1 both regulate each other and share many common target molecules. Reduced expression and activity of SIRT1 have been reported in glomeruli of diabetic patients and in the kidneys from experimental type 1 and type 2 diabetic animals (12, 36, 48, 94). Increasing SIRT1 activity, for instance by using SIRT1 activators, such as resveratrol, ameliorates kidney injury in both type 1 and type 2 experimental diabetes (36, 48, 94, 95, 106), reduces high glucose-mediated oxidative stress and senescence in mesangial cells (36, 48, 94, 95, 106), and protects podocytes from advanced glycation end product (AGE)-induced apoptosis (12) (Fig. 7A).
Implication of Kidney Autophagy in Genetic Diseases and Cancer
Dysregulation of autophagy is implicated in several genetic diseases involving the kidney. Polycystic kidney disease (PKD) is a genetic disorder characterized by the formation of numerous fluid-filled cysts that results in progressive kidney enlargement with the normal structure being replaced by cysts, which finally leads to loss of kidney function and kidney failure. Increased autophagosome formation was detected by electron microscopy and LC3-II expression in massively enlarged kidneys of two rodent models of PKD, the Cy/Cy Han:SPRD rat model with features of autosomal dominant PKD (ADPKD), the most common inherited renal disorder, and the cpk/cpk mouse model of autosomal recessive PKD (4). The increase in LC3-II in the kidneys of Cy/Cy rats and cpk/cpk mice was associated with increased hypoxia-inducible factor-1α (HIF-1α). Given that hypoxia induces autophagy, and cyst formation in PKD results in localized areas of hypoxia, it is plausible that activation of HIF-1α induces autophagy in PKD (Fig. 7B). Interestingly, Bafilomycin A1 treatment did not induce a further increase in LC3-II in the kidneys of cpk/cpk mice, suggesting a defect in autophagy in PKD resulting from impaired autophagic flux due to a block of autophagosome-lysosome fusion and subsequent autolysosomal degradation (4). Studies suggest aberrant activation of mTOR in cystic epithelia in patients with ADPKD and in mouse models that are associated with progressive enlargement of the kidneys (74). Rapamycin, which inhibits mTOR signaling and thereby inducing autophagy, has been shown to be highly effective in reducing cystogenesis in several rodent models of PKD, suggesting that the mTOR pathway plays a critical role in cyst formation and growth (74, 83). However, in contrast to the results of preclinical studies, a recent study in patients with ADPKD and early CKD found that treatment with sirolimus (rapamycin) for 18 months did not halt polycystic kidney growth (73). A separate study reported that treatment with mTOR inhibitor everolimus for up to 2 years slowed the increase in total kidney volume but did not slow the progression of renal impairment in patients with ADPKD (87). Whether autophagy plays a cytoprotective role for cell survival of the tubular cells lining the cysts, as apoptosis worsens cyst formation in PKD, or autophagy impairment plays a pathogenic role in PKD requires further investigation.
Two genetic disorders, namely TSC and von Hippel-Lindau (VHL) disease, are also known to be associated with development of renal cystic disease, and in both, the role of autophagic pathway has been implicated. TSC is an autosomal dominant genetic disorder characterized by benign tumors in multiple organs, including the kidney, and hyperactivation of mTORC1 have been reported in TSC-associated tumors. Inhibition of autophagy by downregulation of Atg5 or Beclin 1, or downregulation of autophagy substrate p62/SQSTM1 decreased TSC tumorigenesis, suggesting that autophagy is a critical component of tumorigenesis in TSC (63). VHL disease is an autosomal dominant syndrome characterized by germline mutations that inactivate the VHL tumor suppressor gene, and is associated with an increased incidence of malignant carcinomas, including renal cell carcinomas (RCCs). A recent study showed that VHL induces tumor-suppressing miR-204 which targets LC3B to inhibit autophagy, and the loss of VHL-miR204 in clear cell RCC results in upregulation of LC3B-mediated autophagy which is necessary for tumor growth (54) (Fig. 7C). In VHL-deficient RCC cells, the compound STF-62247, identified through small molecule screening, induced autophagic cell death and reduced tumor growth (84). STF-62247 activated autophagy, as assessed by increased level of LC3-II and formation of LC3 puncta and autophagic vacuoles, and inhibition of autophagy with 3-MA or knockdown of Atg5 reduced the cytotoxic effects of STF-62247 in VHL-deficient cells compared to VHL-positive RCC cells, suggesting that the autophagy pathway is critical for STF-62247 induced cell death in VHL-deficient RCC cells (84). A recent study demonstrated that a sphingosine kinase 2 inhibitor induced autophagic cell death in another RCC cell line (A498) and delayed tumor growth in immunodeficient mice bearing A498 xenografts (5) (Fig. 7C). These findings suggest that the autophagic pathway is an ideal target for therapy in RCCs. However, studies have shown that induction of autophagy can promote either cell survival or death in RCC cells. Inhibition of mTOR pathway induced autophagy in several RCC cell lines (7, 49, 104, 105), but autophagy inhibitor chloroquine promoted cell death by preventing autophagy and inducing RIP kinase- and ROS-mediated necroptosis in RCC cell line (RCC4) and suppressed xenograft growth (7), whereas PI3K/mTOR inhibitor suppressed cell growth and induced apoptosis in RCC cells (786-0) and concomitantly activated autophagy that protected against apoptosis (49, 104) (Fig. 7C). Rapamycin and its analogs, such as temsirolimus and everolimus, have been used for the treatment in patients with RCCs, but despite promising clinical activity in advanced RCC, their effects are below expectation. Given that these agents primarily inhibit mTORC1, the possibility of compensatory activation of mTORC2 was examined by using a small molecule compound Ku0063794 which inhibits both mTOR complexes. Compared to temsirolimus, Ku0063794 was more effective in decreasing the viability and growth of RCC cells, but there was no difference in the inhibition of tumor growth in a xenograft model (105).
Cystinosis is an autosomal recessive disorder of lysosomal storage due to mutations of CTNS gene, which encodes the lysosomal cystine transporter called cystinosin, causing defective lysosomal membrane transport resulting in the accumulation of cystine in all organs and tissues. Among the three forms of cystinosis, nephropathic cystinosis (NC) is the most severe type and causes proximal tubular dysfunction of Fanconi syndrome and kidney failure in children. Accumulation of cystine is not the sole reason responsible for NC, as the amount of intracellular cystine does not always correlate with the severity of renal tubular dysfunction, and additional processes, such as apoptosis, diminished intracellular ATP, and mitochondrial injury have been proposed in the pathogenesis of NC (59). An impaired autophagic flux has been suggested in renal proximal tubular epithelial (RPTE) cells from patients with NC. Higher ratio of LC3-II/LC3-I and increased numbers of autophagosomes, morphologically abnormal mitochondria, and abnormal patterns of mitochondrial autophagy (mitophagy) with a high number of autophagic vacuoles and fewer mitochondria were detected in RPTE cells from NC patients compared to those of normal controls (70). In addition, increased basal level of p62/SQSTM1 was detected in RPTE cells in vitro and in proximal tubules in vivo in kidney biopsies of patients with NC (69). Interestingly, starvation induced a further increase in p62/SQSTM1 in RPTE cells from patients with NC, suggesting that autophagic flux is blocked in NC (69). Immunostaining showed increased colocalization among LC3, p62/SQSTM1, lysosomal marker LAMP-2 and mitochondrial marker protein ATP5H. These findings imply that a defect in mitophagy contributed to reduced ATP generation, increased ROS production, and increased apoptosis in NC (70) (Fig. 7B).
Autophagy and Oxidative Stress in Kidney
It is generally believed that ROS induces autophagy, and autophagy eliminates the oxidized proteins and dysfunctional organelles, such as damaged mitochondria, to mitigate oxidative stress and restores the cellular ROS balance. The association of oxidative stress and autophagy has been demonstrated in kidney aging, injuries, and diseases. In one hand, ROS is a mediator of activation of autophagy in renal cells. For instance, autophagy was activated through ROS in cadmium-treated mesangial cells that led to autophagic cell death (92) (Fig. 3A), and in angiotensin-II-stimulated mouse podocytes which protected podocytes from angiotensin-II-induced apoptosis (98) (Fig. 4C). Therefore, ROS-induced activation of autophagy could result in different outcomes that can promote either cell death or cell survival.
On the other hand, deficiency in autophagy induces oxidative stress in kidney, as autophagy blockade leads to accumulation of oxidized proteins and damaged mitochondria. Increased mitochondria-derived ROS results in reduced cell viability in Atg5 deficient proximal tubular cells exposed to cisplatin compared with control cells, suggesting that autophagy may alleviate AKI induced by cisplatin through removing ROS-producing mitochondria (81). Oxidized proteins and damaged mitochondria accumulate in podocytes of aged Atg5 knockout mice, and autophagy is essential for reducing oxidative stress in aged kidneys (27). Activation of autophagy with calorie restriction reduced oxidative stress-induced tissue damage and attenuated renal dysfunction in aged mice (45). In RCC cells, blockade of autophagy with chloroquine impeded the clearance of ROS-producing mitochondria through mitophagy in the presence of mTOR inhibitor CCI-779, which ultimately caused cell death of RCC cells (7).
In diabetic nephropathy, excessive mitochondrial ROS inducing oxidative stress plays a pivotal role in its pathogenesis (20, 77). Impairment of autophagy was noted in diabetic kidneys (2, 8, 26, 42, 86). Excessive generation of mitochondrial ROS was proposed as the initiator of kidney damage, which activates four major pathways, including increased flux through the polyol pathways, PKC activation, increased AGE formation, and increased hexosamine pathway, and ultimately leads to the pathophysiological changes in diabetic kidneys (8). Autophagy might be able to ameliorate diabetic nephropathy by reducing oxidative stress. Some autophagy activators, for example SIRT1, negatively correlated with increased oxidative stress and pathological changes in diabetic kidneys (48, 94). SIRT1 reduced ROS production and high glucose-mediated oxidative stress in mesangial cells (36, 48, 94, 95, 106) and SIRT1 activator resveratrol ameliorated kidney injury in both type 1 and type 2 experimental diabetes (36, 48, 94, 95, 106). The renoprotective effects of SIRT1 in diabetic nephropathy may be, at least in part, mediated through reducing mitochondrial ROS production by removing dysfunctional mitochondria (mitophagy) (16, 60). Indeed, a relative normal-looking pool of mitochondria was observed concordantly with numerous of autophagosomes in proximal tubular cells of diabetic Wistar fatty rats subjected to dietary restriction (42) (Fig. 7A). In addition, the CMA has been demonstrated to participate in eliminating the harmful oxidized soluble protein (35).
Conclusion
Autophagy, a conserved and important “self-eating” pathway, serves as an essential mechanism to maintain homeostasis of glomeruli and tubules, and implicated in various aspects of kidney injuries, aging and diseases. Autophagy is indispensable for stress adaption in kidney injuries by removal of protein aggregates and damaged organelles, and promotes cell survival. However, autophagy can also contribute to cell death by autophagic cell death or enhancing apoptosis. While implications of cytoprotective roles of autophagy in the kidney suggest that promoting autophagy may be a promising therapeutic strategy, in certain circumstances, autophagy may be deleterious. Future investigations are needed that will further advance our understanding of the role of autophagy in the kidney and guide potential therapies.
Abbreviations Used
- 3-MA
3-methyadenine
- ADPKD
autosomal dominant PKD
- AGE
advanced glycation end product
- AICAR
5-aminoimidazole-4-carboxamide-1β-riboside
- AKI
acute kidney injury
- AMPK
AMP-activated protein kinase
- Atg
autophagy-related gene
- Bcl-xL
B-cell lymphoma-extra large
- BSA
bovine serum albumin
- BUN
blood urea nitrogen
- CKD
chronic kidney diseases
- cKO
conditional, PRR-knockout
- CMA
chaperone-mediated autophagy
- Col-I
type I collagen
- CORM-2
CO-releasing molecule 2
- DN
diabetic nephropathy
- DTC
distal tubular cell
- ER
endoplasmic reticulum
- ERK
extracellular signal-regulated kinase
- FoxO3
forkhead box O3
- GABARAP
GFP-γ-aminobutyric acid a receptor-associated protein
- GFP
green fluorescent protein
- GSK-3β
glycogen synthase kinase-3β
- HIF-1α
hypoxia-inducible factor-1α
- I/R
ischemia-reperfusion
- JNK
c-Jun N-terminal kinase
- LAMP
lysosome-associated membrane protein
- LC3
microtubule-associated protein 1 light chain 3
- MG132
carbobenzoxy-Leu-Leu-leucinal proteasome inhibitor
- MKK3
mitogen-activated protein kinase kinase kinase 3
- mTOR
mechanistic target of rapamycin
- mTORC
mTOR complex
- NC
nephropathic cystinosis
- PAN
puromycin aminonucleoside
- PHN
passive Heymann nephritis
- PI3K
phosphoinositide-3 kinase
- PKD
polycystic kidney disease
- PRR
prorenin receptor
- PTC
proximal tubular cells
- RCC
renal cell carcinoma
- ROS
reactive oxygen species
- RPTE
renal proximal tubular epithelial
- SCr
serum creatinine
- SIRT1
silent information regulator T1
- SQSTM1
sequestosome 1
- TAB
TAK1 binding protein
- TAK1
TGF-β-activated kinase 1
- TGF-β1
transforming growth factor-β1
- TSC
tuberous sclerosis complex
- Tsc1
tuberous sclerosis complex 1
- ULK
UNC-51-like kinase
- UUO
unilateral ureteral obstruction
- VHL
von Hippel-Lindau
- Vps
vacuolar protein sorting
References
- 1.Asanuma K, Tanida I, Shirato I, Ueno T, Takahara H, Nishitani T, Kominami E, and Tomino Y. MAP-LC3, a promising autophagosomal marker, is processed during the differentiation and recovery of podocytes from PAN nephrosis. FASEB J 17: 1165–1167, 2003 [DOI] [PubMed] [Google Scholar]
- 2.Barbosa Junior Ade A, Zhou H, Hultenschmidt D, Totovic V, Jurilj N, and Pfeifer U. Inhibition of cellular autophagy in proximal tubular cells of the kidney in streptozotocin-diabetic and uninephrectomized rats. Virchows Arch B Cell Pathol Incl Mol Pathol 61: 359–366, 1992 [DOI] [PubMed] [Google Scholar]
- 3.Bechtel W, Helmstadter M, Balica J, Hartleben B, Kiefer B, Hrnjic F, Schell C, Kretz O, Liu S, Geist F, Kerjaschki D, Walz G, and Huber TB. Vps34 deficiency reveals the importance of endocytosis for podocyte homeostasis. J Am Soc Nephrol 24: 727–743, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belibi F, Zafar I, Ravichandran K, Segvic AB, Jani A, Ljubanovic DG, and Edelstein CL. Hypoxia-inducible factor-1alpha (HIF-1alpha) and autophagy in polycystic kidney disease (PKD). Am J Physiol Renal Physiol 300: F1235–F1243, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beljanski V, Knaak C, and Smith CD. A novel sphingosine kinase inhibitor induces autophagy in tumor cells. J Pharmacol Exp Ther 333: 454–464, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bolisetty S, Traylor AM, Kim J, Joseph R, Ricart K, Landar A, and Agarwal A. Heme oxygenase-1 inhibits renal tubular macroautophagy in acute kidney injury. J Am Soc Nephrol 21: 1702–1712, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bray K, Mathew R, Lau A, Kamphorst JJ, Fan J, Chen J, Chen HY, Ghavami A, Stein M, DiPaola RS, Zhang D, Rabinowitz JD, and White E. Autophagy suppresses RIP kinase-dependent necrosis enabling survival to mTOR inhibition. PLoS One 7: e41831, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes 54: 1615–1625, 2005 [DOI] [PubMed] [Google Scholar]
- 9.Chang CC, Chang CY, Wu YT, Huang JP, Yen TH, and Hung LM. Resveratrol retards progression of diabetic nephropathy through modulations of oxidative stress, proinflammatory cytokines, and AMP-activated protein kinase. J Biomed Sci 18: 47, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chargui A, Zekri S, Jacquillet G, Rubera I, Ilie M, Belaid A, Duranton C, Tauc M, Hofman P, Poujeol P, El May MV, and Mograbi B. Cadmium-induced autophagy in rat kidney: an early biomarker of subtoxic exposure. Toxicol Sci 121: 31–42, 2011 [DOI] [PubMed] [Google Scholar]
- 11.Chien CT, Shyue SK, and Lai MK. Bcl-xL augmentation potentially reduces ischemia/reperfusion induced proximal and distal tubular apoptosis and autophagy. Transplantation 84: 1183–1190, 2007 [DOI] [PubMed] [Google Scholar]
- 12.Chuang PY, Dai Y, Liu R, He H, Kretzler M, Jim B, Cohen CD, and He JC. Alteration of forkhead box O (foxo4) acetylation mediates apoptosis of podocytes in diabetes mellitus. PLoS One 6: e23566, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cina DP, Onay T, Paltoo A, Li C, Maezawa Y, De Arteaga J, Jurisicova A, and Quaggin SE. Inhibition of MTOR disrupts autophagic flux in podocytes. J Am Soc Nephrol 23: 412–420, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cui J, Bai XY, Shi S, Cui S, Hong Q, Cai G, and Chen X. Age-related changes in the function of autophagy in rat kidneys. Age (Dordr) 34: 329–339, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ding DF, You N, Wu XM, Xu JR, Hu AP, Ye XL, Zhu Q, Jiang XQ, Miao H, Liu C, and Lu YB. Resveratrol attenuates renal hypertrophy in early-stage diabetes by activating AMPK. Am J Nephrol 31: 363–374, 2010 [DOI] [PubMed] [Google Scholar]
- 16.Ding WX. and Yin XM. Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol Chem 393: 547–564, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ding Y, Kim JK, Kim SI, Na HJ, Jun SY, Lee SJ, and Choi ME. TGF-{beta}1 protects against mesangial cell apoptosis via induction of autophagy. J Biol Chem 285: 37909–37919, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Flaquer M, Lloberas N, Franquesa M, Torras J, Vidal A, Rosa JL, Herrero-Fresneda I, Grinyo JM, and Cruzado JM. The combination of sirolimus and rosiglitazone produces a renoprotective effect on diabetic kidney disease in rats. Life Sci 87: 147–153, 2010 [DOI] [PubMed] [Google Scholar]
- 19.Fleming A, Noda T, Yoshimori T, and Rubinsztein DC. Chemical modulators of autophagy as biological probes and potential therapeutics. Nat Chem Biol 7: 9–17, 2011 [DOI] [PubMed] [Google Scholar]
- 20.Forbes JM, Coughlan MT, and Cooper ME. Oxidative stress as a major culprit in kidney disease in diabetes. Diabetes 57: 1446–1454, 2008 [DOI] [PubMed] [Google Scholar]
- 21.Forbes MS, Thornhill BA, and Chevalier RL. Proximal tubular injury and rapid formation of atubular glomeruli in mice with unilateral ureteral obstruction: a new look at an old model. Am J Physiol Renal Physiol 301: F110–F117, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Forbes MS, Thornhill BA, Minor JJ, Gordon KA, Galarreta CI, and Chevalier RL. Fight-or-flight: murine unilateral ureteral obstruction causes extensive proximal tubular degeneration, collecting duct dilatation, and minimal fibrosis. Am J Physiol Renal Physiol 303: F120–F129, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Funk JA. and Schnellmann RG. Persistent disruption of mitochondrial homeostasis after acute kidney injury. Am J Physiol Renal Physiol 302: F853–F864, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Godel M, Hartleben B, Herbach N, Liu S, Zschiedrich S, Lu S, Debreczeni-Mor A, Lindenmeyer MT, Rastaldi MP, Hartleben G, Wiech T, Fornoni A, Nelson RG, Kretzler M, Wanke R, Pavenstadt H, Kerjaschki D, Cohen CD, Hall MN, Ruegg MA, Inoki K, Walz G, and Huber TB. Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. J Clin Invest 121: 2197–2209, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Han K, Lehringer-Polzin M, Zhou H, and Pfeifer U. Cellular autophagy in proximal tubules of early diabetic rats following insulin treatment and islet transplantation. Virchows Arch B Cell Pathol Incl Mol Pathol 61: 367–373, 1992 [DOI] [PubMed] [Google Scholar]
- 26.Han K, Zhou H, and Pfeifer U. Inhibition and restimulation by insulin of cellular autophagy in distal tubular cells of the kidney in early diabetic rats. Kidney Blood Press Res 20: 258–263, 1997 [DOI] [PubMed] [Google Scholar]
- 27.Hartleben B, Godel M, Meyer-Schwesinger C, Liu S, Ulrich T, Kobler S, Wiech T, Grahammer F, Arnold SJ, Lindenmeyer MT, Cohen CD, Pavenstadt H, Kerjaschki D, Mizushima N, Shaw AS, Walz G, and Huber TB. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J Clin Invest 120: 1084–1096, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Inoki K, Mori H, Wang J, Suzuki T, Hong S, Yoshida S, Blattner SM, Ikenoue T, Ruegg MA, Hall MN, Kwiatkowski DJ, Rastaldi MP, Huber TB, Kretzler M, Holzman LB, Wiggins RC, and Guan KL. mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J Clin Invest 121: 2181–2196, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Inoue K, Kuwana H, Shimamura Y, Ogata K, Taniguchi Y, Kagawa T, Horino T, Takao T, Morita T, Sasaki S, Mizushima N, and Terada Y. Cisplatin-induced macroautophagy occurs prior to apoptosis in proximal tubules in vivo. Clin Exp Nephrol 14: 112–122, 2010 [DOI] [PubMed] [Google Scholar]
- 30.Isaka Y, Suzuki C, Abe T, Okumi M, Ichimaru N, Imamura R, Kakuta Y, Matsui I, Takabatake Y, Rakugi H, Shimizu S, and Takahara S. Bcl-2 protects tubular epithelial cells from ischemia/reperfusion injury by dual mechanisms. Transplant Proc 41: 52–54, 2009 [DOI] [PubMed] [Google Scholar]
- 31.Jiang M, Liu K, Luo J, and Dong Z. Autophagy is a renoprotective mechanism during in vitro hypoxia and in vivo ischemia-reperfusion injury. Am J Pathol 176: 1181–1192, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang M, Wei Q, Dong G, Komatsu M, Su Y, and Dong Z. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int 82: 1271–1283, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson-Lyles DN, Peifley K, Lockett S, Neun BW, Hansen M, Clogston J, Stern ST, and McNeil SE. Fullerenol cytotoxicity in kidney cells is associated with cytoskeleton disruption, autophagic vacuole accumulation, and mitochondrial dysfunction. Toxicol Appl Pharmacol 248: 249–258, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaushik S. and Cuervo AM. Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol 22: 407–417, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kiffin R, Bandyopadhyay U, and Cuervo AM. Oxidative stress and autophagy. Antioxid Redox Signal 8: 152–162, 2006 [DOI] [PubMed] [Google Scholar]
- 36.Kim MY, Lim JH, Youn HH, Hong YA, Yang KS, Park HS, Chung S, Koh SH, Shin SJ, Choi BS, Kim HW, Kim YS, Lee JH, Chang YS, and Park CW. Resveratrol prevents renal lipotoxicity and inhibits mesangial cell glucotoxicity in a manner dependent on the AMPK-SIRT1-PGC1alpha axis in db/db mice. Diabetologia 56: 204–217, 2013 [DOI] [PubMed] [Google Scholar]
- 37.Kim SI, Kwak JH, Zachariah M, He Y, Wang L, and Choi ME. TGF-beta-activated kinase 1 and TAK1-binding protein 1 cooperate to mediate TGF-beta1-induced MKK3-p38 MAPK activation and stimulation of type I collagen. Am J Physiol Renal Physiol 292: F1471–F1478, 2007 [DOI] [PubMed] [Google Scholar]
- 38.Kim SI, Na HJ, Ding Y, Wang Z, Lee SJ, and Choi ME. Autophagy promotes intracellular degradation of type I collagen induced by transforming growth factor (TGF)-beta1. J Biol Chem 287: 11677–11688, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim WY, Nam SA, Song HC, Ko JS, Park SH, Kim HL, Choi EJ, Kim YS, Kim J, and Kim YK. The role of autophagy in unilateral ureteral obstruction rat model. Nephrology (Carlton) 17: 148–159, 2011 [DOI] [PubMed] [Google Scholar]
- 40.Kimura A, Ishida Y, Wada T, Hisaoka T, Morikawa Y, Sugaya T, Mukaida N, and Kondo T. The absence of interleukin-6 enhanced arsenite-induced renal injury by promoting autophagy of tubular epithelial cells with aberrant extracellular signal-regulated kinase activation. Am J Pathol 176: 40–50, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kimura T, Takabatake Y, Takahashi A, Kaimori JY, Matsui I, Namba T, Kitamura H, Niimura F, Matsusaka T, Soga T, Rakugi H, and Isaka Y. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J Am Soc Nephrol 22: 902–913, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kitada M, Takeda A, Nagai T, Ito H, Kanasaki K, and Koya D. Dietary restriction ameliorates diabetic nephropathy through anti-inflammatory effects and regulation of the autophagy via restoration of Sirt1 in diabetic Wistar fatty (fa/fa) rats: a model of type 2 diabetes. Exp Diabetes Res 2011: 908185, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, Ahn HJ, Ait-Mohamed O, Ait-Si-Ali S, Akematsu T, Akira S, Al-Younes HM, Al-Zeer MA, Albert ML, Albin RL, Alegre-Abarrategui J, Aleo MF, Alirezaei M, Almasan A, Almonte-Becerril M, Amano A, Amaravadi R, Amarnath S, Amer AO, Andrieu-Abadie N, Anantharam V, Ann DK, Anoopkumar-Dukie S, Aoki H, Apostolova N, Arancia G, Aris JP, Asanuma K, Asare NY, Ashida H, Askanas V, Askew DS, Auberger P, Baba M, Backues SK, Baehrecke EH, Bahr BA, Bai XY, Bailly Y, Baiocchi R, Baldini G, Balduini W, Ballabio A, Bamber BA, Bampton ET, Banhegyi G, Bartholomew CR, Bassham DC, Bast RC, Jr, Batoko H, Bay BH, Beau I, Bechet DM, Begley TJ, Behl C, Behrends C, Bekri S, Bellaire B, Bendall LJ, Benetti L, Berliocchi L, Bernardi H, Bernassola F, Besteiro S, Bhatia-Kissova I, Bi X, Biard-Piechaczyk M, Blum JS, Boise LH, Bonaldo P, Boone DL, Bornhauser BC, Bortoluci KR, Bossis I, Bost F, Bourquin JP, Boya P, Boyer-Guittaut M, Bozhkov PV, Brady NR, Brancolini C, Brech A, Brenman JE, Brennand A, Bresnick EH, Brest P, Bridges D, Bristol ML, Brookes PS, Brown EJ, Brumell JH, Brunetti-Pierri N, Brunk UT, Bulman DE, Bultman SJ, Bultynck G, Burbulla LF, Bursch W, Butchar JP, Buzgariu W, Bydlowski SP, Cadwell K, Cahova M, Cai D, Cai J, Cai Q, Calabretta B, Calvo-Garrido J, Camougrand N, Campanella M, Campos-Salinas J, Candi E, Cao L, Caplan AB, Carding SR, Cardoso SM, Carew JS, Carlin CR, Carmignac V, Carneiro LA, Carra S, Caruso RA, Casari G, Casas C, Castino R, Cebollero E, Cecconi F, Celli J, Chaachouay H, Chae HJ, Chai CY, Chan DC, Chan EY, Chang RC, Che CM, Chen CC, Chen GC, Chen GQ, Chen M, Chen Q, Chen SS, Chen W, Chen X, Chen YG, Chen Y, Chen YJ, Chen Z, Cheng A, Cheng CH, Cheng Y, Cheong H, Cheong JH, Cherry S, Chess-Williams R, Cheung ZH, Chevet E, Chiang HL, Chiarelli R, Chiba T, Chin LS, Chiou SH, Chisari FV, Cho CH, Cho DH, Choi AM, Choi D, Choi KS, Choi ME, Chouaib S, Choubey D, Choubey V, Chu CT, Chuang TH, Chueh SH, Chun T, Chwae YJ, Chye ML, Ciarcia R, Ciriolo MR, Clague MJ, Clark RS, Clarke PG, Clarke R, Codogno P, Coller HA, Colombo MI, Comincini S, Condello M, Condorelli F, Cookson MR, Coombs GH, Coppens I, Corbalan R, Cossart P, Costelli P, Costes S, Coto-Montes A, Couve E, Coxon FP, Cregg JM, Crespo JL, Cronje MJ, Cuervo AM, Cullen JJ, Czaja MJ, D'Amelio M, Darfeuille-Michaud A, Davids LM, Davies FE, De Felici M, de Groot JF, de Haan CA, De Martino L, De Milito A, De Tata V, Debnath J, Degterev A, Dehay B, Delbridge LM, Demarchi F, Deng YZ, Dengjel J, Dent P, Denton D, Deretic V, Desai SD, Devenish RJ, Di Gioacchino M, Di Paolo G, Di Pietro C, Diaz-Araya G, Diaz-Laviada I, Diaz-Meco MT, Diaz-Nido J, Dikic I, Dinesh-Kumar SP, Ding WX, Distelhorst CW, Diwan A, Djavaheri-Mergny M, Dokudovskaya S, Dong Z, Dorsey FC, Dosenko V, Dowling JJ, Doxsey S, Dreux M, Drew ME, Duan Q, Duchosal MA, Duff K, Dugail I, Durbeej M, Duszenko M, Edelstein CL, Edinger AL, Egea G, Eichinger L, Eissa NT, Ekmekcioglu S, El-Deiry WS, Elazar Z, Elgendy M, Ellerby LM, Eng KE, Engelbrecht AM, Engelender S, Erenpreisa J, Escalante R, Esclatine A, Eskelinen EL, Espert L, Espina V, Fan H, Fan J, Fan QW, Fan Z, Fang S, Fang Y, Fanto M, Fanzani A, Farkas T, Farre JC, Faure M, Fechheimer M, Feng CG, Feng J, Feng Q, Feng Y, Fesus L, Feuer R, Figueiredo-Pereira ME, Fimia GM, Fingar DC, Finkbeiner S, Finkel T, Finley KD, Fiorito F, Fisher EA, Fisher PB, Flajolet M, Florez-McClure ML, Florio S, Fon EA, Fornai F, Fortunato F, Fotedar R, Fowler DH, Fox HS, Franco R, Frankel LB, Fransen M, Fuentes JM, Fueyo J, Fujii J, Fujisaki K, Fujita E, Fukuda M, Furukawa RH, Gaestel M, Gailly P, Gajewska M, Galliot B, Galy V, Ganesh S, Ganetzky B, Ganley IG, Gao FB, Gao GF, Gao J, Garcia L, Garcia-Manero G, Garcia-Marcos M, Garmyn M, Gartel AL, Gatti E, Gautel M, Gawriluk TR, Gegg ME, Geng J, Germain M, Gestwicki JE, Gewirtz DA, Ghavami S, Ghosh P, Giammarioli AM, Giatromanolaki AN, Gibson SB, Gilkerson RW, Ginger ML, Ginsberg HN, Golab J, Goligorsky MS, Golstein P, Gomez-Manzano C, Goncu E, Gongora C, Gonzalez CD, Gonzalez R, Gonzalez-Estevez C, Gonzalez-Polo RA, Gonzalez-Rey E, Gorbunov NV, Gorski S, Goruppi S, Gottlieb RA, Gozuacik D, Granato GE, Grant GD, Green KN, Gregorc A, Gros F, Grose C, Grunt TW, Gual P, Guan JL, Guan KL, Guichard SM, Gukovskaya AS, Gukovsky I, Gunst J, Gustafsson AB, Halayko AJ, Hale AN, Halonen SK, Hamasaki M, Han F, Han T, Hancock MK, Hansen M, Harada H, Harada M, Hardt SE, Harper JW, Harris AL, Harris J, Harris SD, Hashimoto M, Haspel JA, Hayashi S, Hazelhurst LA, He C, He YW, Hebert MJ, Heidenreich KA, Helfrich MH, Helgason GV, Henske EP, Herman B, Herman PK, Hetz C, Hilfiker S, Hill JA, Hocking LJ, Hofman P, Hofmann TG, Hohfeld J, Holyoake TL, Hong MH, Hood DA, Hotamisligil GS, Houwerzijl EJ, Hoyer-Hansen M, Hu B, Hu CA, Hu HM, Hua Y, Huang C, Huang J, Huang S, Huang WP, Huber TB, Huh WK, Hung TH, Hupp TR, Hur GM, Hurley JB, Hussain SN, Hussey PJ, Hwang JJ, Hwang S, Ichihara A, Ilkhanizadeh S, Inoki K, Into T, Iovane V, Iovanna JL, Ip NY, Isaka Y, Ishida H, Isidoro C, Isobe K, Iwasaki A, Izquierdo M, Izumi Y, Jaakkola PM, Jaattela M, Jackson GR, Jackson WT, Janji B, Jendrach M, Jeon JH, Jeung EB, Jiang H, Jiang JX, Jiang M, Jiang Q, Jiang X, Jimenez A, Jin M, Jin S, Joe CO, Johansen T, Johnson DE, Johnson GV, Jones NL, Joseph B, Joseph SK, Joubert AM, Juhasz G, Juillerat-Jeanneret L, Jung CH, Jung YK, Kaarniranta K, Kaasik A, Kabuta T, Kadowaki M, Kagedal K, Kamada Y, Kaminskyy VO, Kampinga HH, Kanamori H, Kang C, Kang KB, Kang KI, Kang R, Kang YA, Kanki T, Kanneganti TD, Kanno H, Kanthasamy AG, Kanthasamy A, Karantza V, Kaushal GP, Kaushik S, Kawazoe Y, Ke PY, Kehrl JH, Kelekar A, Kerkhoff C, Kessel DH, Khalil H, Kiel JA, Kiger AA, Kihara A, Kim DR, Kim DH, Kim EK, Kim HR, Kim JS, Kim JH, Kim JC, Kim JK, Kim PK, Kim SW, Kim YS, Kim Y, Kimchi A, Kimmelman AC, King JS, Kinsella TJ, Kirkin V, Kirshenbaum LA, Kitamoto K, Kitazato K, Klein L, Klimecki WT, Klucken J, Knecht E, Ko BC, Koch JC, Koga H, Koh JY, Koh YH, Koike M, Komatsu M, Kominami E, Kong HJ, Kong WJ, Korolchuk VI, Kotake Y, Koukourakis MI, Kouri Flores JB, Kovacs AL, Kraft C, Krainc D, Kramer H, Kretz-Remy C, Krichevsky AM, Kroemer G, Kruger R, Krut O, Ktistakis NT, Kuan CY, Kucharczyk R, Kumar A, Kumar R, Kumar S, Kundu M, Kung HJ, Kurz T, Kwon HJ, La Spada AR, Lafont F, Lamark T, Landry J, Lane JD, Lapaquette P, Laporte JF, Laszlo L, Lavandero S, Lavoie JN, Layfield R, Lazo PA, Le W, Le Cam L, Ledbetter DJ, Lee AJ, Lee BW, Lee GM, Lee J, Lee JH, Lee M, Lee MS, Lee SH, Leeuwenburgh C, Legembre P, Legouis R, Lehmann M, Lei HY, Lei QY, Leib DA, Leiro J, Lemasters JJ, Lemoine A, Lesniak MS, Lev D, Levenson VV, Levine B, Levy E, Li F, Li JL, Li L, Li S, Li W, Li XJ, Li YB, Li YP, Liang C, Liang Q, Liao YF, Liberski PP, Lieberman A, Lim HJ, Lim KL, Lim K, Lin CF, Lin FC, Lin J, Lin JD, Lin K, Lin WW, Lin WC, Lin YL, Linden R, Lingor P, Lippincott-Schwartz J, Lisanti MP, Liton PB, Liu B, Liu CF, Liu K, Liu L, Liu QA, Liu W, Liu YC, Liu Y, Lockshin RA, Lok CN, Lonial S, Loos B, Lopez-Berestein G, Lopez-Otin C, Lossi L, Lotze MT, Low P, Lu B, Lu Z, Luciano F, Lukacs NW, Lund AH, Lynch-Day MA, Ma Y, Macian F, MacKeigan JP, Macleod KF, Madeo F, Maiuri L, Maiuri MC, Malagoli D, Malicdan MC, Malorni W, Man N, Mandelkow EM, Manon S, Manov I, Mao K, Mao X, Mao Z, Marambaud P, Marazziti D, Marcel YL, Marchbank K, Marchetti P, Marciniak SJ, Marcondes M, Mardi M, Marfe G, Marino G, Markaki M, Marten MR, Martin SJ, Martinand-Mari C, Martinet W, Martinez-Vicente M, Masini M, Matarrese P, Matsuo S, Matteoni R, Mayer A, Mazure NM, McConkey DJ, McConnell MJ, McDermott C, McDonald C, McInerney GM, McKenna SL, McLaughlin B, McLean PJ, McMaster CR, McQuibban GA, Meijer AJ, Meisler MH, Melendez A, Melia TJ, Melino G, Mena MA, Menendez JA, Menna-Barreto RF, Menon MB, Menzies FM, Mercer CA, Merighi A, Merry DE, Meschini S, Meyer CG, Meyer TF, Miao CY, Miao JY, Michels PA, Michiels C, Mijaljica D, Milojkovic A, Minucci S, Miracco C, Miranti CK, Mitroulis I, Miyazawa K, Mizushima N, Mograbi B, Mohseni S, Molero X, Mollereau B, Mollinedo F, Momoi T, Monastyrska I, Monick MM, Monteiro MJ, Moore MN, Mora R, Moreau K, Moreira PI, Moriyasu Y, Moscat J, Mostowy S, Mottram JC, Motyl T, Moussa CE, Muller S, Munger K, Munz C, Murphy LO, Murphy ME, Musaro A, Mysorekar I, Nagata E, Nagata K, Nahimana A, Nair U, Nakagawa T, Nakahira K, Nakano H, Nakatogawa H, Nanjundan M, Naqvi NI, Narendra DP, Narita M, Navarro M, Nawrocki ST, Nazarko TY, Nemchenko A, Netea MG, Neufeld TP, Ney PA, Nezis IP, Nguyen HP, Nie D, Nishino I, Nislow C, Nixon RA, Noda T, Noegel AA, Nogalska A, Noguchi S, Notterpek L, Novak I, Nozaki T, Nukina N, Nurnberger T, Nyfeler B, Obara K, Oberley TD, Oddo S, Ogawa M, Ohashi T, Okamoto K, Oleinick NL, Oliver FJ, Olsen LJ, Olsson S, Opota O, Osborne TF, Ostrander GK, Otsu K, Ou JH, Ouimet M, Overholtzer M, Ozpolat B, Paganetti P, Pagnini U, Pallet N, Palmer GE, Palumbo C, Pan T, Panaretakis T, Pandey UB, Papackova Z, Papassideri I, Paris I, Park J, Park OK, Parys JB, Parzych KR, Patschan S, Patterson C, Pattingre S, Pawelek JM, Peng J, Perlmutter DH, Perrotta I, Perry G, Pervaiz S, Peter M, Peters GJ, Petersen M, Petrovski G, Phang JM, Piacentini M, Pierre P, Pierrefite-Carle V, Pierron G, Pinkas-Kramarski R, Piras A, Piri N, Platanias LC, Poggeler S, Poirot M, Poletti A, Pous C, Pozuelo-Rubio M, Praetorius-Ibba M, Prasad A, Prescott M, Priault M, Produit-Zengaffinen N, Progulske-Fox A, Proikas-Cezanne T, Przedborski S, Przyklenk K, Puertollano R, Puyal J, Qian SB, Qin L, Qin ZH, Quaggin SE, Raben N, Rabinowich H, Rabkin SW, Rahman I, Rami A, Ramm G, Randall G, Randow F, Rao VA, Rathmell JC, Ravikumar B, Ray SK, Reed BH, Reed JC, Reggiori F, Regnier-Vigouroux A, Reichert AS, Reiners JJ, Jr, Reiter RJ, Ren J, Revuelta JL, Rhodes CJ, Ritis K, Rizzo E, Robbins J, Roberge M, Roca H, Roccheri MC, Rocchi S, Rodemann HP, Rodriguez de Cordoba S, Rohrer B, Roninson IB, Rosen K, Rost-Roszkowska MM, Rouis M, Rouschop KM, Rovetta F, Rubin BP, Rubinsztein DC, Ruckdeschel K, Rucker EB, 3rd, Rudich A, Rudolf E, Ruiz-Opazo N, Russo R, Rusten TE, Ryan KM, Ryter SW, Sabatini DM, Sadoshima J, Saha T, Saitoh T, Sakagami H, Sakai Y, Salekdeh GH, Salomoni P, Salvaterra PM, Salvesen G, Salvioli R, Sanchez AM, Sanchez-Alcazar JA, Sanchez-Prieto R, Sandri M, Sankar U, Sansanwal P, Santambrogio L, Saran S, Sarkar S, Sarwal M, Sasakawa C, Sasnauskiene A, Sass M, Sato K, Sato M, Schapira AH, Scharl M, Schatzl HM, Scheper W, Schiaffino S, Schneider C, Schneider ME, Schneider-Stock R, Schoenlein PV, Schorderet DF, Schuller C, Schwartz GK, Scorrano L, Sealy L, Seglen PO, Segura-Aguilar J, Seiliez I, Seleverstov O, Sell C, Seo JB, Separovic D, Setaluri V, Setoguchi T, Settembre C, Shacka JJ, Shanmugam M, Shapiro IM, Shaulian E, Shaw RJ, Shelhamer JH, Shen HM, Shen WC, Sheng ZH, Shi Y, Shibuya K, Shidoji Y, Shieh JJ, Shih CM, Shimada Y, Shimizu S, Shintani T, Shirihai OS, Shore GC, Sibirny AA, Sidhu SB, Sikorska B, Silva-Zacarin EC, Simmons A, Simon AK, Simon HU, Simone C, Simonsen A, Sinclair DA, Singh R, Sinha D, Sinicrope FA, Sirko A, Siu PM, Sivridis E, Skop V, Skulachev VP, Slack RS, Smaili SS, Smith DR, Soengas MS, Soldati T, Song X, Sood AK, Soong TW, Sotgia F, Spector SA, Spies CD, Springer W, Srinivasula SM, Stefanis L, Steffan JS, Stendel R, Stenmark H, Stephanou A, Stern ST, Sternberg C, Stork B, Stralfors P, Subauste CS, Sui X, Sulzer D, Sun J, Sun SY, Sun ZJ, Sung JJ, Suzuki K, Suzuki T, Swanson MS, Swanton C, Sweeney ST, Sy LK, Szabadkai G, Tabas I, Taegtmeyer H, Tafani M, Takacs-Vellai K, Takano Y, Takegawa K, Takemura G, Takeshita F, Talbot NJ, Tan KS, Tanaka K, Tang D, Tanida I, Tannous BA, Tavernarakis N, Taylor GS, Taylor GA, Taylor JP, Terada LS, Terman A, Tettamanti G, Thevissen K, Thompson CB, Thorburn A, Thumm M, Tian F, Tian Y, Tocchini-Valentini G, Tolkovsky AM, Tomino Y, Tonges L, Tooze SA, Tournier C, Tower J, Towns R, Trajkovic V, Travassos LH, Tsai TF, Tschan MP, Tsubata T, Tsung A, Turk B, Turner LS, Tyagi SC, Uchiyama Y, Ueno T, Umekawa M, Umemiya-Shirafuji R, Unni VK, Vaccaro MI, Valente EM, Van den Berghe G, van der Klei IJ, van Doorn W, van Dyk LF, van Egmond M, van Grunsven LA, Vandenabeele P, Vandenberghe WP, Vanhorebeek I, Vaquero EC, Velasco G, Vellai T, Vicencio JM, Vierstra RD, Vila M, Vindis C, Viola G, Viscomi MT, Voitsekhovskaja OV, von Haefen C, Votruba M, Wada K, Wade-Martins R, Walker CL, Walsh CM, Walter J, Wan XB, Wang A, Wang C, Wang D, Wang F, Wang G, Wang H, Wang HG, Wang HD, Wang J, Wang K, Wang M, Wang RC, Wang X, Wang YJ, Wang Y, Wang Z, Wang ZC, Wansink DG, Ward DM, Watada H, Waters SL, Webster P, Wei L, Weihl CC, Weiss WA, Welford SM, Wen LP, Whitehouse CA, Whitton JL, Whitworth AJ, Wileman T, Wiley JW, Wilkinson S, Willbold D, Williams RL, Williamson PR, Wouters BG, Wu C, Wu DC, Wu WK, Wyttenbach A, Xavier RJ, Xi Z, Xia P, Xiao G, Xie Z, Xu DZ, Xu J, Xu L, Xu X, Yamamoto A, Yamashina S, Yamashita M, Yan X, Yanagida M, Yang DS, Yang E, Yang JM, Yang SY, Yang W, Yang WY, Yang Z, Yao MC, Yao TP, Yeganeh B, Yen WL, Yin JJ, Yin XM, Yoo OJ, Yoon G, Yoon SY, Yorimitsu T, Yoshikawa Y, Yoshimori T, Yoshimoto K, You HJ, Youle RJ, Younes A, Yu L, Yu SW, Yu WH, Yuan ZM, Yue Z, Yun CH, Yuzaki M, Zabirnyk O, Silva-Zacarin E, Zacks D, Zacksenhaus E, Zaffaroni N, Zakeri Z, Zeh HJ, 3rd, Zeitlin SO, Zhang H, Zhang HL, Zhang J, Zhang JP, Zhang L, Zhang MY, Zhang XD, Zhao M, Zhao YF, Zhao Y, Zhao ZJ, Zheng X, Zhivotovsky B, Zhong Q, Zhou CZ, Zhu C, Zhu WG, Zhu XF, Zhu X, Zhu Y, Zoladek T, Zong WX, Zorzano A, Zschocke J, and Zuckerbraun B. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8: 445–544, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koesters R, Kaissling B, Lehir M, Picard N, Theilig F, Gebhardt R, Glick AB, Hahnel B, Hosser H, Grone HJ, and Kriz W. Tubular overexpression of transforming growth factor-beta1 induces autophagy and fibrosis but not mesenchymal transition of renal epithelial cells. Am J Pathol 177: 632–643, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kume S, Uzu T, Horiike K, Chin-Kanasaki M, Isshiki K, Araki S, Sugimoto T, Haneda M, Kashiwagi A, and Koya D. Calorie restriction enhances cell adaptation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouse aged kidney. J Clin Invest 120: 1043–1055, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee MJ, Feliers D, Mariappan MM, Sataranatarajan K, Mahimainathan L, Musi N, Foretz M, Viollet B, Weinberg JM, Choudhury GG, and Kasinath BS. A role for AMP-activated protein kinase in diabetes-induced renal hypertrophy. Am J Physiol Renal Physiol 292: F617–F627, 2007 [DOI] [PubMed] [Google Scholar]
- 47.Levine B, Mizushima N, and Virgin HW. Autophagy in immunity and inflammation. Nature 469: 323–335, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li C, Cai F, Yang Y, Zhao X, Wang C, Li J, Jia Y, Tang J, and Liu Q. Tetrahydroxystilbene glucoside ameliorates diabetic nephropathy in rats: involvement of SIRT1 and TGF-beta1 pathway. Eur J Pharmacol 649: 382–389, 2010 [DOI] [PubMed] [Google Scholar]
- 49.Li H, Jin X, Zhang Z, Xing Y, and Kong X. Inhibition of autophagy enhances apoptosis induced by the PI3K/AKT/mTor inhibitor NVP-BEZ235 in renal cell carcinoma cells. Cell Biochem Funct 2012[Epub ahead of print]; DOI: 10.1002/cbf.2917 [DOI] [PubMed] [Google Scholar]
- 50.Li L, Zepeda-Orozco D, Black R, and Lin F. Autophagy is a component of epithelial cell fate in obstructive uropathy. Am J Pathol 176: 1767–1778, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu S, Hartleben B, Kretz O, Wiech T, Igarashi P, Mizushima N, Walz G, and Huber TB. Autophagy plays a critical role in kidney tubule maintenance, aging and ischemia-reperfusion injury. Autophagy 8: 826–837, 2012 [DOI] [PubMed] [Google Scholar]
- 52.Lloberas N, Cruzado JM, Franquesa M, Herrero-Fresneda I, Torras J, Alperovich G, Rama I, Vidal A, and Grinyo JM. Mammalian target of rapamycin pathway blockade slows progression of diabetic kidney disease in rats. J Am Soc Nephrol 17: 1395–1404, 2006 [DOI] [PubMed] [Google Scholar]
- 53.Mijaljica D, Prescott M, and Devenish RJ. Microautophagy in mammalian cells: revisiting a 40-year-old conundrum. Autophagy 7: 673–682, 2011 [DOI] [PubMed] [Google Scholar]
- 54.Mikhaylova O, Stratton Y, Hall D, Kellner E, Ehmer B, Drew AF, Gallo CA, Plas DR, Biesiada J, Meller J, and Czyzyk-Krzeska MF. VHL-regulated MiR-204 suppresses tumor growth through inhibition of LC3B-mediated autophagy in renal clear cell carcinoma. Cancer Cell 21: 532–546, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, and Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 15: 1101–1111, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mori H, Inoki K, Masutani K, Wakabayashi Y, Komai K, Nakagawa R, Guan KL, and Yoshimura A. The mTOR pathway is highly activated in diabetic nephropathy and rapamycin has a strong therapeutic potential. Biochem Biophys Res Commun 384: 471–475, 2009 [DOI] [PubMed] [Google Scholar]
- 57.Nagai K, Matsubara T, Mima A, Sumi E, Kanamori H, Iehara N, Fukatsu A, Yanagita M, Nakano T, Ishimoto Y, Kita T, Doi T, and Arai H. Gas6 induces Akt/mTOR-mediated mesangial hypertrophy in diabetic nephropathy. Kidney Int 68: 552–561, 2005 [DOI] [PubMed] [Google Scholar]
- 58.Narita M, Young AR, Arakawa S, Samarajiwa SA, Nakashima T, Yoshida S, Hong S, Berry LS, Reichelt S, Ferreira M, Tavare S, Inoki K, and Shimizu S. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science 332: 966–970, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nesterova G. and Gahl WA. Cystinosis: the evolution of a treatable disease. Pediatr Nephrol 28: 51–59, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Novak I. Mitophagy: a complex mechanism of mitochondrial removal. Antioxid Redox Signal 17: 794–802, 2012 [DOI] [PubMed] [Google Scholar]
- 61.Oshima Y, Kinouchi K, Ichihara A, Sakoda M, Kurauchi-Mito A, Bokuda K, Narita T, Kurosawa H, Sun-Wada GH, Wada Y, Yamada T, Takemoto M, Saleem MA, Quaggin SE, and Itoh H. Prorenin receptor is essential for normal podocyte structure and function. J Am Soc Nephrol 22: 2203–2212, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pallet N, Bouvier N, Legendre C, Gilleron J, Codogno P, Beaune P, Thervet E, and Anglicheau D. Autophagy protects renal tubular cells against cyclosporine toxicity. Autophagy 4: 783–791, 2008 [DOI] [PubMed] [Google Scholar]
- 63.Parkhitko A, Myachina F, Morrison TA, Hindi KM, Auricchio N, Karbowniczek M, Wu JJ, Finkel T, Kwiatkowski DJ, Yu JJ, and Henske EP. Tumorigenesis in tuberous sclerosis complex is autophagy and p62/sequestosome 1 (SQSTM1)-dependent. Proc Natl Acad Sci U S A 108: 12455–12460, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Periyasamy-Thandavan S, Jiang M, Wei Q, Smith R, Yin XM, and Dong Z. Autophagy is cytoprotective during cisplatin injury of renal proximal tubular cells. Kidney Int 74: 631–640, 2008 [DOI] [PubMed] [Google Scholar]
- 65.Pfeifer U. and Guder WG. Stimulation of cellular autophagy by parathyroid hormone and cyclic adenosine 3',5': monophosphate in isolated tubular fragments from the rat's kidney cortex. Virchows Arch B Cell Pathol 19: 51–67, 1975 [DOI] [PubMed] [Google Scholar]
- 66.Riediger F, Quack I, Qadri F, Hartleben B, Park JK, Potthoff SA, Sohn D, Sihn G, Rousselle A, Fokuhl V, Maschke U, Purfurst B, Schneider W, Rump LC, Luft FC, Dechend R, Bader M, Huber TB, Nguyen G, and Muller DN. Prorenin receptor is essential for podocyte autophagy and survival. J Am Soc Nephrol 22: 2193–2202, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rovetta F, Stacchiotti A, Consiglio A, Cadei M, Grigolato PG, Lavazza A, Rezzani R, and Aleo MF. ER signaling regulation drives the switch between autophagy and apoptosis in NRK-52E cells exposed to cisplatin. Exp Cell Res 318: 238–250, 2011 [DOI] [PubMed] [Google Scholar]
- 68.Sakaguchi M, Isono M, Isshiki K, Sugimoto T, Koya D, and Kashiwagi A. Inhibition of mTOR signaling with rapamycin attenuates renal hypertrophy in the early diabetic mice. Biochem Biophys Res Commun 340: 296–301, 2006 [DOI] [PubMed] [Google Scholar]
- 69.Sansanwal P. and Sarwal MM. p62/SQSTM1 prominently accumulates in renal proximal tubules in nephropathic cystinosis. Pediatr Nephrol 27: 2137–2144, 2012 [DOI] [PubMed] [Google Scholar]
- 70.Sansanwal P, Yen B, Gahl WA, Ma Y, Ying L, Wong LJ, and Sarwal MM. Mitochondrial autophagy promotes cellular injury in nephropathic cystinosis. J Am Soc Nephrol 21: 272–283, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sato S, Kitamura H, Adachi A, Sasaki Y, and Ghazizadeh M. Two types of autophagy in the podocytes in renal biopsy specimens: ultrastructural study. J Submicrosc Cytol Pathol 38: 167–174, 2006 [PubMed] [Google Scholar]
- 72.Sato S, Yanagihara T, Ghazizadeh M, Ishizaki M, Adachi A, Sasaki Y, Igarashi T, and Fukunaga Y. Correlation of autophagy type in podocytes with histopathological diagnosis of IgA nephropathy. Pathobiology 76: 221–226, 2009 [DOI] [PubMed] [Google Scholar]
- 73.Serra AL, Poster D, Kistler AD, Krauer F, Raina S, Young J, Rentsch KM, Spanaus KS, Senn O, Kristanto P, Scheffel H, Weishaupt D, and Wuthrich RP. Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N Engl J Med 363: 820–829, 2010 [DOI] [PubMed] [Google Scholar]
- 74.Shillingford JM, Murcia NS, Larson CH, Low SH, Hedgepeth R, Brown N, Flask CA, Novick AC, Goldfarb DA, Kramer-Zucker A, Walz G, Piontek KB, Germino GG, and Weimbs T. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci U S A 103: 5466–5471, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Soetikno V, Sari FR, Sukumaran V, Lakshmanan AP, Harima M, Suzuki K, Kawachi H, and Watanabe K. Curcumin decreases renal triglyceride accumulation through AMPK-SREBP signaling pathway in streptozotocin-induced type 1 diabetic rats. J Nutr Biochem 24: 796–802, 2012 [DOI] [PubMed] [Google Scholar]
- 76.Sooparb S, Price SR, Shaoguang J, and Franch HA. Suppression of chaperone-mediated autophagy in the renal cortex during acute diabetes mellitus. Kidney Int 65: 2135–2144, 2004 [DOI] [PubMed] [Google Scholar]
- 77.Stanton RC. Oxidative stress and diabetic kidney disease. Curr Diab Rep 11: 330–336, 2011 [DOI] [PubMed] [Google Scholar]
- 78.Stern ST, Zolnik BS, McLeland CB, Clogston J, Zheng J, and McNeil SE. Induction of autophagy in porcine kidney cells by quantum dots: a common cellular response to nanomaterials? Toxicol Sci 106: 140–152, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Suzuki C, Isaka Y, Takabatake Y, Tanaka H, Koike M, Shibata M, Uchiyama Y, Takahara S, and Imai E. Participation of autophagy in renal ischemia/reperfusion injury. Biochem Biophys Res Commun 368: 100–106, 2008 [DOI] [PubMed] [Google Scholar]
- 80.Takagi-Akiba M, Asanuma K, Tanida I, Tada N, Trejo JA, Nonaka K, Asanuma E, Kominami E, Ueno T, and Tomino Y. Doxorubicin-induced glomerulosclerosis with proteinuria in GFP-GABARAP transgenic mice. Am J Physiol Renal Physiol 302: F380–F389, 2012 [DOI] [PubMed] [Google Scholar]
- 81.Takahashi A, Kimura T, Takabatake Y, Namba T, Kaimori J, Kitamura H, Matsui I, Niimura F, Matsusaka T, Fujita N, Yoshimori T, Isaka Y, and Rakugi H. Autophagy guards against cisplatin-induced acute kidney injury. Am J Pathol 180: 517–525, 2012 [DOI] [PubMed] [Google Scholar]
- 82.Takiyama Y, Harumi T, Watanabe J, Fujita Y, Honjo J, Shimizu N, Makino Y, and Haneda M. Tubular injury in a rat model of type 2 diabetes is prevented by metformin: a possible role of HIF-1alpha expression and oxygen metabolism. Diabetes 60: 981–992, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tao Y, Kim J, Schrier RW, and Edelstein CL. Rapamycin markedly slows disease progression in a rat model of polycystic kidney disease. J Am Soc Nephrol 16: 46–51, 2005 [DOI] [PubMed] [Google Scholar]
- 84.Turcotte S, Chan DA, Sutphin PD, Hay MP, Denny WA, and Giaccia AJ. A molecule targeting VHL-deficient renal cell carcinoma that induces autophagy. Cancer Cell 14: 90–102, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Turkmen K, Martin J, Akcay A, Nguyen Q, Ravichandran K, Faubel S, Pacic A, Ljubanovic D, Edelstein CL, and Jani A. Apoptosis and autophagy in cold preservation ischemia. Transplantation 91: 1192–1197, 2011 [DOI] [PubMed] [Google Scholar]
- 86.Vallon V, Rose M, Gerasimova M, Satriano J, Platt KA, Koepsell H, Cunard R, Sharma K, Thomson SC, and Rieg T. Knockout of Na-glucose transporter SGLT2 attenuates hyperglycemia and glomerular hyperfiltration but not kidney growth or injury in diabetes mellitus. Am J Physiol Renal Physiol 304: F156–F167, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Walz G, Budde K, Mannaa M, Nurnberger J, Wanner C, Sommerer C, Kunzendorf U, Banas B, Horl WH, Obermuller N, Arns W, Pavenstadt H, Gaedeke J, Buchert M, May C, Gschaidmeier H, Kramer S, and Eckardt KU. Everolimus in patients with autosomal dominant polycystic kidney disease. N Engl J Med 363: 830–840, 2010 [DOI] [PubMed] [Google Scholar]
- 88.Wang L, Hong Q, Lv Y, Feng Z, Zhang X, Wu L, Cui S, Hou K, Su H, Huang Z, Wu D, and Chen X. Autophagy can repair endoplasmic reticulum stress damage of the passive Heymann nephritis model as revealed by proteomics analysis. J Proteomics 75: 3866–3876, 2012 [DOI] [PubMed] [Google Scholar]
- 89.Wang L, Lee JY, Kwak JH, He Y, Kim SI, and Choi ME. Protective effects of low-dose carbon monoxide against renal fibrosis induced by unilateral ureteral obstruction. Am J Physiol Renal Physiol 294: F508–F517, 2008 [DOI] [PubMed] [Google Scholar]
- 90.Wang L, Ma R, Flavell RA, and Choi ME. Requirement of mitogen-activated protein kinase kinase 3 (MKK3) for activation of p38alpha and p38delta MAPK isoforms by TGF-beta 1 in murine mesangial cells. J Biol Chem 277: 47257–47262, 2002 [DOI] [PubMed] [Google Scholar]
- 91.Wang SH, Shih YL, Ko WC, Wei YH, and Shih CM. Cadmium-induced autophagy and apoptosis are mediated by a calcium signaling pathway. Cell Mol Life Sci 65: 3640–3652, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang SH, Shih YL, Kuo TC, Ko WC, and Shih CM. Cadmium toxicity toward autophagy through ROS-activated GSK-3beta in mesangial cells. Toxicol Sci 108: 124–131, 2009 [DOI] [PubMed] [Google Scholar]
- 93.Wu HH, Hsiao TY, Chien CT, and Lai MK. Ischemic conditioning by short periods of reperfusion attenuates renal ischemia/reperfusion induced apoptosis and autophagy in the rat. J Biomed Sci 16: 19, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wu L, Zhang Y, Ma X, Zhang N, and Qin G. The effect of resveratrol on FoxO1 expression in kidneys of diabetic nephropathy rats. Mol Biol Rep 39: 9085–9093, 2012 [DOI] [PubMed] [Google Scholar]
- 95.Xu Y, Nie L, Yin YG, Tang JL, Zhou JY, Li DD, and Zhou SW. Resveratrol protects against hyperglycemia-induced oxidative damage to mitochondria by activating SIRT1 in rat mesangial cells. Toxicol Appl Pharmacol 259: 395–401, 2012 [DOI] [PubMed] [Google Scholar]
- 96.Xu Y, Ruan S, Wu X, Chen H, Zheng K, and Fu B. Autophagy and apoptosis in tubular cells following unilateral ureteral obstruction are associated with mitochondrial oxidative stress. Int J Mol Med 31: 628–636, 2013 [DOI] [PubMed] [Google Scholar]
- 97.Xu Y, Yang S, Huang J, Ruan S, Zheng Z, and Lin J. Tgf-beta1 induces autophagy and promotes apoptosis in renal tubular epithelial cells. Int J Mol Med 29: 781–790, 2012 [DOI] [PubMed] [Google Scholar]
- 98.Yadav A, Vallabu S, Arora S, Tandon P, Slahan D, Teichberg S, and Singhal PC. ANG II promotes autophagy in podocytes. Am J Physiol Cell Physiol 299: C488–C496, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yang C, Kaushal V, Shah SV, and Kaushal GP. Autophagy is associated with apoptosis in cisplatin injury to renal tubular epithelial cells. Am J Physiol Renal Physiol 294: F777–F787, 2008 [DOI] [PubMed] [Google Scholar]
- 100.Yang Y, Wang J, Qin L, Shou Z, Zhao J, Wang H, Chen Y, and Chen J. Rapamycin prevents early steps of the development of diabetic nephropathy in rats. Am J Nephrol 27: 495–502, 2007 [DOI] [PubMed] [Google Scholar]
- 101.Yang Z. and Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol 12: 814–822, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yeh CH, Hsu SP, Yang CC, Chien CT, and Wang NP. Hypoxic preconditioning reinforces HIF-alpha-dependent HSP70 signaling to reduce ischemic renal failure-induced renal tubular apoptosis and autophagy. Life Sci 86: 115–123, 2009 [DOI] [PubMed] [Google Scholar]
- 103.Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F, Hailey DW, Oorschot V, Klumperman J, Baehrecke EH, and Lenardo MJ. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 465: 942–946, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhan YH, Liu J, Qu XJ, Hou KZ, Wang KF, Liu YP, and Wu B. beta-Elemene induces apoptosis in human renal-cell carcinoma 786-0 cells through inhibition of MAPK/ERK and PI3K/Akt/mTOR signalling pathways. Asian Pac J Cancer Prev 13: 2739–2744, 2012 [DOI] [PubMed] [Google Scholar]
- 105.Zhang H, Berel D, Wang Y, Li P, Bhowmick NA, Figlin RA, and Kim HL. A comparison of Ku0063794, a dual mTORC1 and mTORC2 inhibitor, and temsirolimus in preclinical renal cell carcinoma models. PLoS One 8: e54918, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhang S, Cai G, Fu B, Feng Z, Ding R, Bai X, Liu W, Zhuo L, Sun L, Liu F, and Chen X. SIRT1 is required for the effects of rapamycin on high glucose-inducing mesangial cells senescence. Mech Ageing Dev 133: 387–400, 2012 [DOI] [PubMed] [Google Scholar]