

Abstract
RATIONALE
Rapidly performing global proteomic screens is an important goal in the post-genomic era. Correlated matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS) and fluorescent imaging of photocleavable peptide-coded random bead-arrays was evaluated as a critical step in a new method for proteomic screening that combines many of the advantages of MS with fluorescence-based microarrays.
METHODS
Small peptide-coded model bead libraries containing up to 20 different bead species were constructed by attaching peptides to 30–34 µm diameter glass, agarose or TentaGel® beads using photocleavable biotin or a custom-designed photocleavable linker. The peptide-coded bead libraries were randomly arrayed into custom gold-coated micro-well plates with 45 µm diameter wells and subjected to fluorescence and MALDI mass spectrometric imaging (MALDI-MSI).
RESULTS
Photocleavable mass-tags from individual beads in these libraries were spatially localized as ∼65 µm spots using MALDI-MSI with high sensitivity and mass resolution. Fluorescently tagged beads were identified and correlated with their matching photocleavable mass-tags by comparing the fluorescence and MALDI-MS images of the same bead-array. Post-translational modification of the peptide Kemptide was also detected on individual beads in a photocleavable peptide-coded bead-array by MALDI-MSI alone, after exposure of the beads to protein kinase A (PKA).
CONCLUSIONS
Correlated MALDI-MS and fluorescent imaging of photocleavable peptide-coded random bead-arrays can provide a basis for performing global proteomic screening. © 2013 The Authors. Rapid Communications in Mass Spectrometry published by John Wiley & Sons, Ltd.
Sequencing of the human genome has led to a new and even more ambitious goal – characterization of the human proteome. Such an endeavor involves not only understanding the function of hundreds of thousands of different proteins expressed in human cells, but also characterizing the millions of potential interactions that can occur with other cellular and extracellular molecules, including proteins, nucleic acids, lipids and small molecules. The ability to rapidly perform such massive glohbal proteomic screens would be a powerful tool in many areas of biotechnology including biomarker and drug discovery. Examples include the screening of a human protein library formatted on microarrays to discover new autoimmune and tumor antigens in patient sera that can be used in clinical diagnostics and immunotherapy,1–4 and the screening of a synthetic combinatorial peptide or peptoid library on beads against specific protein targets to discover new drugs.5–7
High-density protein microarrays (i.e. arrays of ‘bait’ proteins), first introduced in 2000 by MacBeath and Schreiber,8 contain as many as 10 000 distinct protein species (e.g. Life Technologies’ ProtoArray®). However, while protein microarrays can be queried with a particular ‘prey’ molecule of interest, allowing rapid identification of positive bait-prey interactions using fluorescence imaging, there are several limitations which have impeded their usefulness in the proteomics field including: (i) Low-Fidelity Readout – The inability of fluorescence alone to reveal the molecular details of the bait-prey interaction, including identification of hundreds of interacting prey species from a complex mixture and post-translational modifications to the bait proteins such as proteolysis, phosphorylation and glycosylation; (ii) Array Density, Reproducibility & Cost – Unlike DNA chips, whose probes can be synthesized at high density using photolithography, protein/peptide chips are printed using mechanical or piezoelectric devices8–10 resulting in relatively low array density, poor reproducibility of spot size, shape and uniformity, printing-induced damage to delicate proteins and relatively low throughput of the non-parallel printing process resulting in higher costs per microarray (for reviews, see9,11); (iii) Surface Chemistry & Assay Kinetics – Protein microarrays use a two-dimensional array format, with the proteins/peptides attached by passive adsorption or chemical linkage to a planar array surface and subsequently dried. While this design is ideal for readout of the chips, it results in potential surface/drying/printing induced denaturation of intact proteins,11 poor kinetics of the microarray bio-assay,12 and inefficient automation of a multitude of assays on the planar microarray ‘chips’.
Screening methods that employ bait molecules immobilized on micro-particles, such as beads, are also routinely used in various proteomic and diagnostic applications.12–15 Compared to printed arrays, bead-based methods allow higher flexibility in assay design, greater number of replicates, more facile assay automation in microtiter plates and near solution-phase binding kinetics that lead to improved sensitivity and accuracy. However, these methods have limited application in proteomics since they normally employ fluorescence coding which is inherently restricted by the broad spectral emissions of fluorophores. For example, flow cytometry based multiplex methods such as the Luminex (Austin, TX, USA) xMAP® technology16 are currently limited to 500 different coded beads.
In contrast to the limitations of fluorescent detection and microarrays, mass spectrometry (MS), a central tool for proteomics, has the power to detect protein modifications,17,18 perform protein identification, quantification and sequencing (e.g. using tandem mass spectrometry (MS/MS))19,20 and to detect protein–small molecule (drug) interactions.21 However, MS normally lacks the power of a microarray to localize at high density a myriad of bait molecules, and potentially interacting prey, to separate locations on a two-dimensional surface.
Recently, progress in matrix-assisted laser desorption/ionization mass spectrometric imaging (MALDI-MSI) has made it possible to achieve high two-dimensional spatial resolution on a sample such as a tissue slice.22 In this case, the instrument’s laser beam scans the surface of the specimen, collecting full spectra at each discrete ‘pixel’, and a multi-dimensional digital mass-image is created using the peak intensity at a given m/z value(s). In principle, the frequency tripled Nd-Yag laser beam at 355 nm used for most commercial MALDI instruments can be focused to less than 1 µm, much smaller than the diameter of most micro-beads commonly used for binding bait molecules and for combinatorial synthesis (10–100 µm).5,6 In addition, increasing scan speeds as well as improved detectors, image analysis software and automation make it feasible to apply MALDI-MSI to analyze bead libraries formatted as high-density two-dimensional bead-arrays.
As demonstrated here, 30–34 µm beads incorporated at high density in a random bead-array can be spatially localized using MALDI-MSI to detect photocleavable peptide mass-tags (PC-Mass-Tags) attached to each bead. Detection with both high sensitivity and high mass resolution was achieved with a variety of bead types evaluated. Furthermore, by obtaining a fluorescence image of the same bead-array, specific fluorescently labeled beads in the array were identified and correlated with their matching PC-Mass-Tags, a process which will ultimately enable the identification of positive prey interactions with the beads.
The Bead-GPS approach
The basic Bead-GPS concept is illustrated in Fig. 1. Each micro-bead in a bead library can have both a specific protein species (bait) and a photocleavable mass-tag attached (PC-Mass-Tag). The PC-Mass-Tag acts as a unique identifier (i.e. a coding agent) for the protein or other bait molecules, such as a nucleic acid, residing on the particular bead. Alternatively, as demonstrated here, the PC-Mass-Tag, such as a peptide or peptoid,7 can serve simultaneously as the bait and coding agent (without a separate bait molecule on the bead). In either case, the entire bead library is incubated with a mixture containing one or more prey molecules potentially capable of interacting with or modifying one or more of the bait. The prey are labeled with a fluorescent dye and/or coded with a PC-Mass-Tag, the latter being especially useful when multiple prey are used to query a library.
Figure 1.
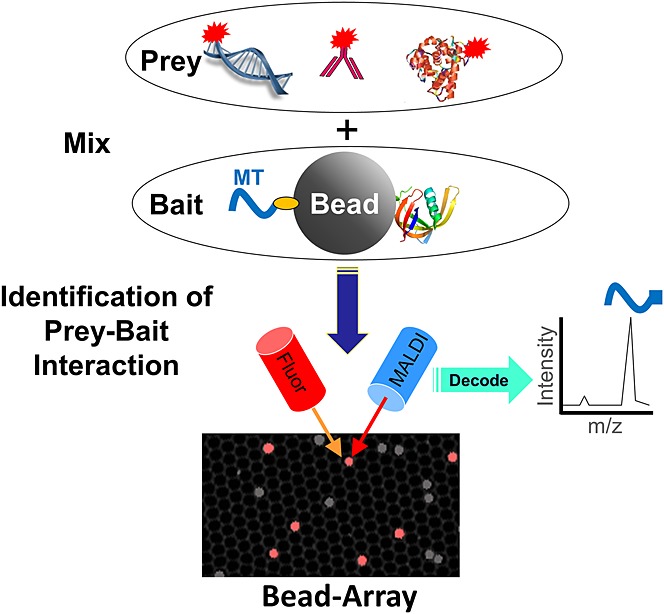
Schematic of the Bead-GPS approach. Bead-GPS facilitates the rapid identification and characterization of the interactions of a library of potentially millions of ‘bait’ molecules, such as proteins, peptides or peptoids, with one or more ‘prey’ molecules, such as serum antibodies, enzymes, DNA, drugs or drug targets. Micro-beads in a library consist of a photocleavable coding Mass-Tag (MT; orange oval is photocleavable linker), which itself can be the bait, or is accompanied by a separate bait molecule (e.g. protein shown to right of Bead). Red starbursts indicate a fluorescent label, shown attached directly to the prey for simplicity, although secondary detection of the prey is possible. Note that the image at the bottom of the figure is an actual Bead-GPS array as detected by fluorescence (gray/white shows all beads and the micro-wells, and red shows a bead sub-population carrying a specific fluorescent label).
After the prey interact with the bead library, the beads are randomly arrayed in a micro-well plate designed so that only one bead can fit in a well and the wells are ordered into a periodic two-dimensional array. Specialized ‘Sync Beads’ are also incorporated into the micro-well plate which have a specific fluorophore and specific PC-Mass-Tag attached (used for image alignment). The plate is then coated (or pre-coated) with a suitable MALDI-MS matrix compound and the array scanned by both MALDI-MSI and fluorescence. Note that the fluorescence image shown at the bottom of Fig. 1 is that of an actual Bead-GPS array in the micro-well plates, whereby the red spots are ‘Sync Beads’ labeled with the Alexa Fluor® 647 dye and the gray beads are all other beads visible via weak auto-fluorescence using the 488-nm excitation laser of the microarray scanner (the walls of the micro-wells are also visible by weak auto-fluorescence using this laser).
EXPERIMENTAL
Preparation of photocleavable (PC) biotin-labeled peptide mass-tags (PC-Mass-Tags)
Peptides of unique mass (peptides from GenScript, Piscataway, NJ, USA, and AnaSpec, Fremont, CA, USA) were chemically modified on their N-termini with photocleavable biotin (PC-Biotin) using NHS-activated (N-hydroxysuccinimidyl ester; primary amine reactive) labeling reagent23 (AmberGen, Inc., Watertown, MA, USA). The peptides were prepared at 5 mg/mL in 100 mM sodium bicarbonate and reacted overnight (with mixing) using equimolar amounts of the reagent. The resultant PC-Biotin labeled peptide mass-tags (PC-Mass-Tags) were used without further purification. Because the NHS-activated labeling reagent reacts only with primary amines, selective labeling of the N-terminus was achieved (peptides were chosen which lacked lysine residues). All peptides used for PC-Mass-Tags in the various experiments are listed in Supplementary Table S1 (Supporting Information).
Preparation of streptavidin-coated glass beads
Carboxyl-terminated, glass (silica) micro-spheres (beads), 30 µm in diameter, were purchased from Microspheres-Nanospheres (Cold Spring Harbor, NY, USA). Unless otherwise noted, beads were processed in 0.45 µm pore size spin filtration devices (400 μL capacity Ultrafree-MC Micro-Centrifuge Filter Units, Durapore PVDF Membrane; Millipore, Billerica, MA, USA). All bead washes were performed by brief vortex mixing (∼3 s) in the required solution, spinning briefly (∼15 s) in a standard micro-centrifuge (15 000 rpm) to achieve filtration, followed by discarding the filtrate and repeating as necessary.
Beads (8 mg) were washed 5× 400 μL with MES Buffer (prepared from BupH MES buffered saline packs, Thermo Fisher Scientific, Rockford, IL, USA; 0.1 M MES, pH 4.7, 0.9% NaCl [154 mM]). Beads were re-suspended in 200 μL MES Buffer and 25 μL of a 48 mM amine-terminated biotin linker solution were added (EZ-Link Amine-PEO3-Biotin, Thermo Fisher Scientific). The conjugation reaction was then initiated by the addition of 25 μL of a 100 mg/mL EDC solution prepared immediately before use (1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride powder; Thermo Fisher Scientific). The reaction was carried out for 1 h with mixing. To ensure the EDC-activated carboxyl beads retained no residual reactivity, beads were next quenched by washing (4 × 400 μL) for 5 min each with 10 mM hydroxylamine (Thermo Fisher Scientific) prepared in PBS-T followed by treatment for 30 min with mixing in 400 μL of this solution (PBS = phosphate-buffered saline = 48 mM sodium phosphate, pH 7.5, 100 mM NaCl; PBS-T contains 0.05% [v/v] Tween-20). To ensure removal of all unconjugated linker, beads were then subjected to stringent washing with PBS containing 1 M NaCl (2 × 400 μL), whereby the second wash was for 1 h with mixing. Finally, beads were washed with TBS-T (TBS = tris-buffered saline = 50 mM Tris, pH 7.5, 200 mM NaCl; TBS-T contains 0.05% [v/v] Tween-20) (4 × 400 μL).
Beads were then treated with BSA Block (1% bovine serum albumin (BSA) [w/v] in TBS-T) (2 × 400 μL) for 5 min each and coated with streptavidin (tetrameric) for 30 min using 250 μL of a 1 mg/mL solution also prepared in BSA Block. In the case where fluorescent ‘Sync Beads’ were prepared, streptavidin labeled with DyLight™ 650 (Thermo Fisher Scientific) was spiked into the aforementioned unlabeled streptavidin solution at a final concentration of 10 µg/mL. After streptavidin coating was complete, beads were washed with TBS-T (3 × 400 μL), for 5 min each with TBS containing 1 M NaCl (3 × 400 μL) and lastly with TBS (3 × 400 μL).
Streptavidin agarose beads
Unless otherwise noted, beads were processed in 0.45 µm pore size spin filtration devices (400 μL capacity Ultrafree-MC Micro-Centrifuge Filter Units, Durapore PVDF Membrane; Millipore). All bead washes were performed by brief vortex mixing (∼3 s) in the required solution, spinning briefly (∼15 s) in a standard micro-centrifuge (15 000 rpm) to achieve filtration, followed by discarding the filtrate and repeating as necessary. For non-fluorescent streptavidin agarose beads, commercially available 34 µm beads were used (Streptavidin Sepharose High Performance, GE Healthcare Life Sciences, Piscataway, NJ, USA). For fluorescent ‘Sync Beads’, fluorescent streptavidin was attached to 34 µm agarose beads as follows: Streptavidin (Thermo Fisher Scientific) was prepared to 5 mg/mL in 100 mM sodium bicarbonate and 100 mM NaCl. Then 500 μL of this solution were reacted with 2 molar equivalents of Alexa Fluor® 647 carboxylic acid, succinimidyl ester (Invitrogen, Carlsbad, CA, USA) added from a 25 mM stock (3.8 μL added; stock in anhydrous dimethylformamide (DMF)). The reaction was carried out for 30 min with mixing. To remove unreacted dye, the solution was then desalted on Illustra™ NAP™-5 G-25 Sephadex™ columns according to the manufacturer’s instructions (GE Healthcare Life Sciences) against 200 mM sodium bicarbonate and 200 mM NaCl. The column-purified fluorescently labeled streptavidin (∼1 mg/mL) was used directly without further dilution or processing. 50 μL of bead pellet volume of NHS-activated 34 µm agarose beads (NHS HP SpinTrap, GE Healthcare Life Sciences) was then washed with ice-cold 1 mM HCl (4 × 400 μL). Next, 400 μL of the aforementioned fluorescent streptavidin solution were added to the washed bead pellet and the reaction allowed to proceed for 1 h with mixing. Beads were then quenched by washing 1 × 400 μL with 200 mM glycine, 1 mM EDTA, 200 mM sodium bicarbonate and 200 mM NaCl followed by treatment for 2 × 30 min each with mixing in 400 μL of the same solution. Beads were then washed with 200 mM sodium bicarbonate and 200 mM NaCl (2 × 400 μL) followed by washing with TBS-T for 5 min each (3 × 400 μL). Beads were stored as a 20% (v/v) suspension in TBS-T at +4°C.
Loading PC-Mass-Tags to streptavidin glass or agarose beads
Peptides #1–21 listed in Supplementary Table S1 (Supporting Information) were used as PC-Mass-Tags on the glass or agarose streptavidin beads. A PC-Biotin labeled PC-Mass-Tag (250 pmol) was loaded onto approximately 45 000 streptavidin-coated glass or agarose beads (roughly 1.5 μL of bead pellet volume) using 50 μL of a 5 pmol/μL solution prepared in TBS-T. Loading of the PC-Mass-Tags was done for 30 min with mixing. For larger reactions, the PC-Mass-Tag concentration was kept constant and the volume scaled accordingly. Beads were then washed with TBS-T (3 × 400 μL) and TBS (3 × 400 μL. Beads were stored in 400 μL of TBS at +4°C or used immediately for subsequent steps.
Kinase assays on PC-Mass-Tag beads
A 2 μL pellet volume of streptavidin agarose beads (∼60 000 beads) was loaded with the Kemptide or the Tag-3.1 PC-Mass-Tag (peptides #3 and #9, respectively, in Supplementary Table S1, Supporting Information). The two bead species were then pooled, washed twice with 400 μL of 50 mM Tris, pH 7.5, 10 mM MgCl2, 0.1% (w/v) BSA and 0.01% Triton X-100 (v/v) detergent, and re-suspended in 100 μL of this same buffer. The suspension was split into two equal portions of 50 μL each. One portion of the beads (plus kinase permutation) was spiked with 2.5 ng of recombinant human protein kinase A, catalytic subunit (PKA; Millipore; added from the manufacturer’s stock) and 200 μM ATP (added from 20 mM stock prepared in water). The other portion of beads did not receive the PKA or ATP (minus kinase permutation). The reaction was allowed to proceed for 30 min at 30°C with mixing. Beads were then washed with TBS-T (3× 400 μL) and with TBS (3 × 400 μL).
Attachment of PC linker and then peptide Mass-Tags to TentaGel® beads
Mono-sized amine TentaGel® beads (30 µm, Rapp Polymere, Tübingen, Germany) were used. Unless otherwise noted, all reactions and extended mixing steps were performed in polypropylene micro-centrifuge tubes while all washes were performed in the spin filtration devices as described earlier. An exception to this rule is that all steps involving high concentrations of DMF (≥50%) were performed in micro-centrifuge tubes due to the incompatibility of the filtration devices with this solvent (washing in micro-centrifuge tubes was otherwise performed similar to as with the filtration devices described earlier, except that after spinning the beads down, the supernatant was pipetted off leaving the bead pellet behind).
Dried beads (100 mg; ∼107 beads) were washed in DMF (4 × 1 mL). Beads were then suspended in 1 mL of DMF and hydrated/swollen overnight with mixing. Beads were then washed with DMF (2 × 1 mL), 50% DMF (2 × 1 mL) and 100 mM sodium bicarbonate in 50% DMF (2 × 1 mL). Swollen/hydrated beads were re-suspended to a final 1 mL total volume (∼10 000 beads/μL) using 100 mM sodium bicarbonate in 50% DMF and were stored at +4°C.
A custom photocleavable linker was attached to the beads. The linker comprised an amine-reactive NHS ester at one end, a t-Boc-protected primary amine at the other end and a photocleavable nucleus in between (NHS-PC-tBOC Linker; AmberGen, Inc.; Fig. 2(B) shows the structure as attached to beads and after t-Boc deprotection, while Supplementary Fig. S1(A), Supporting Information, shows following Mass-Tag attachment and subsequent photocleavage). 10 mg of swollen beads was used. Beads were washed with Bicarbonate/OBG Buffer (200 mM sodium bicarbonate with 0.05% [w/v] octyl β-D-glucopyranoside detergent) (4 × 1 mL). Beads were re-suspended in 400 μL of Bicarbonate/OBG Buffer and 5 μL of a 600 mM stock of the NHS-PC-tBOC linker was added (stock in anhydrous DMF). In the case where fluorescently labeled ‘Sync Beads’ were made, 0.75 μL of a 250 μM solution of the Alexa Fluor® 647 carboxylic acid, succinimidyl ester (Invitrogen) was also added (stock in anhydrous DMF) simultaneously with the NHS-PC-tBOC Linker. The reaction was allowed to proceed for 1 h with mixing. After the reaction, beads were washed with Bicarbonate/OBG Buffer. Beads were re-suspended in 400 μL of Bicarbonate/OBG Buffer and 10 μL of a 100 mM stock of Sulfo-NHS acetate (Thermo Fisher Scientific) was added to block any remaining unreacted amines on the TentaGel® beads (stock prepared in water, immediately before use). The reaction was allowed to proceed for 30 min with mixing. After the reaction, beads were washed with Bicarbonate/OBG Buffer (1 × 400 μL), PBS-OBG (PBS containing 0.05% [w/v] octyl β-D-glucopyranoside detergent) (4 × 400 μL) and water (1 × 400 μL). Beads were re-suspended in 200 μL water and mixed with 200 μL of 85% phosphoric acid to remove the t-Boc protecting group. The reaction was allowed to proceed for 1 h with mixing. The reaction was diluted with 1 mL water, the supernatant removed from the beads and the beads washed with PBS-OBG (6 × 400 μL). The TentaGel® beads, now bearing photocleavable primary amine moieties, were stored at +4°C in 400 μL PBS-OBG or used immediately for subsequent steps.
Figure 2.
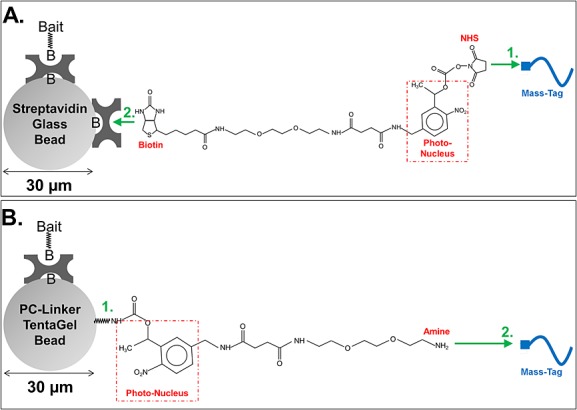
Bead configuration in Bead-GPS. (A) Mass-Tags, e.g. peptides, are end-labeled with photocleavable biotin using an NHS-activated reagent and then captured onto streptavidin-coated 30 µm glass or 34 µm agarose beads (glass beads depicted; green numbers indicate the sequence of steps). (B) A novel photocleavable primary amine-terminated linker (NHS-PC-tBOC Linker) is attached to 30 µm mono-sized TentaGel® beads, which provides through its primary amine group a substrate for combinatorial synthesis of photocleavable peptide or peptoid libraries. Alternatively, independently synthesized Mass-Tags, e.g. peptides, can be attached to the photocleavable primary amine-terminated linker on the beads. (A and B) Streptavidin (tetrameric) is attached to the bead surface by a non-cleavable biotin (indicated by ‘B’ in figure). In addition to binding the Mass-Tags in some cases (e.g. as in (A)), the streptavidin coating can also facilitate attachment of separate ‘Bait’ molecules such as whole proteins using either a non-cleavable biotin (indicated by ‘B’ in figure) or a streptavidin binding tag (not depicted). See Experimental section for more detail.
Although these photocleavable primary amine TentaGel® beads are compatible with on-bead combinatorial peptide or peptoid synthesis,7 in this work pre-synthesized peptide Mass-Tags were attached to the beads using a sulfhydryl to amine linkage (peptides #22–24 in Supplementary Table S1 (Supporting Information) were used as PC-Mass-Tags on the TentaGel® beads). For this, the BMPS linker was used (Thermo Fisher Scientific) which is a heterobifunctional cross-linker bearing an amine-reactive NHS moiety on one end and a sulfhydryl-reactive maleimide moiety on the other end. 10 mg of beads in 400 μL of PBS-OBG was combined with 14 μL of a 200 mM stock of BMPS (stock in anhydrous DMF). The reaction was allowed to proceed for 30 min with mixing. After the reaction, beads were washed with PBS-OBG (4 × 400 μL). The beads were re-suspended in 400 μL of PBS-OBG and cysteine-terminated peptides were added to a 600 μM final concentration from 4 mM stocks (peptides contained only a single, terminal cysteine; stocks in water; peptide stocks aliquoted and stored frozen at –80°C). The reaction was allowed to proceed for 30 min with mixing. Finally, beads were washed with PBS-OBG (4 × 400 μL) and re-suspended in 400 μL PBS-OBG. Beads were stored at +4°C or used immediately for subsequent steps.
Forming random bead-arrays and MALDI-MSI
Custom fiber optic bead-array micro-well plates were manufactured for AmberGen, Inc. by INCOM USA, Inc. (Charlton, MA, USA). The 1×75×25 mm plates contained ∼1 million hexagonally packed fiber optic wells created from 50 µm fibers etched to 55 µm depth. The fiber cladding yields wells of approximately 45 µm i.d. For optimal performance in MALDI-MSI, plates were coated by sputtering on a 5 nm layer of titanium followed by a 5 nm layer of gold (ThinFilms, Inc., Hillsborough, NJ, USA).
To remove air bubbles and facilitate optimal bead deposition into the micro-wells of the plates, the plates were pre-hydrated by washing in a tray of excess MS-grade water for 1 h with mixing on a rotary platform shaker. The plates were then assembled into an AHC1X24 Microarray Hybridization Cassette (ArrayIt® Corporation, Sunnyvale, CA, USA) which subdivided the plate into 24 rectangular sub-array zones, each zone measuring 7.5 mm × 6.5 mm. Then 100 μL of MS-grade water were added to each chamber of the Microarray Hybridization Cassette.
For the 30 µm glass or 34 µm agarose beads, the desired number of beads was washed in MS-grade water (4 × 400 μL) and re-suspended to the desired concentration. Then 100 μL of the bead suspension were added to each chamber of the Microarray Hybridization Cassette. The Microarray Hybridization Cassette containing the plate and beads was then agitated for 5–10 min on a rotary platform shaker. The plate was then allowed to stand undisturbed for 15 min to allow beads to settle. Finally, to fully seat the beads into the micro-wells, the Microarray Hybridization Cassette was centrifuged for 5 min at 1430 g using a properly balanced clinical swing-bucket centrifuge and a microtiter plate rotor attachment. The fluid supernatant was then pipetted out of each chamber of the Microarray Hybridization Cassette and the plates removed for washing. To eliminate beads not seated in the micro-wells, plates were washed three times for 2 min each in a tray with excess MS-grade water on a rotary platform shaker. Plates were removed from the water and bulk water allowed to run off the surface. Plates were then dried overnight at 25°C under ambient air. TentaGel® beads were deposited in essentially the same manner with the following exceptions: Prior to deposition, beads were washed with PBS-OBG (2 × 400 μL), 50% acetonitrile (6 × 400 μL), and MS-grade water (1 × 400 μL) instead of the washes detailed above. After adding the beads to the chambers of the Microarray Hybridization Cassette and shaking as described above, the plates were then immediately spun in the centrifuge as above, without any intermediate steps.
After drying the plates, fluorescence imaging of the bead-arrays was performed using a GenePix 4200A microarray scanner (Molecular Devices LLC, Sunnyvale, CA, USA). Fluorescence imaging of the bead-array was possible both from the top and the bottom (through the fiber optics) of the plates. Next, to photocleave the PC-Mass-Tags from the beads, the plates were irradiated from above for 5 min with 365 nm UV light using a Blak-Ray lamp (model XX-15; UVP, Upland, CA, USA), at a 5 cm distance (the power output under these conditions was approximately 2.6 mW/cm2 at 360 nm, 1.0 mW/cm2 at 310 nm and 0.16 mW/cm2 at 250 nm). For MALDI-MSI, the plates were then coated with a thin and uniform film of CHCA matrix (α-hydroxycinnamic acid) either using an in-house prepared nebulizing apparatus or the HTX TM-Sprayer™ (HTXImaging by HTX Technologies, LLC, Carrboro, NC, USA). For nebulizing, a LC® Sprint reusable nebulizer (Pari, Midlothian, VA, USA) was fitted with a stainless steel mask which created a 2 × 16 mm slit opening through which the mist could pass and contact the plate. By affixing the plate to a rotating mixer, the plate was cycled in and out of the matrix plume once every 2.5 s at a distance of approximately 5 mm from the slit opening. A repeating pattern was used whereby 5 cycles were performed with the nebulizer on followed by 5 cycles with the nebulizer off (to allow for drying). Using this pattern, a total of 90 cycles were performed with the nebulizer on and an equal amount with the nebulizer off. For the HTX TM-Sprayer™, typical conditions were as follows: Spray nozzle XY velocity of 650 mm/min, 12 passes, 0.1 mL/min matrix flow rate, nozzle temperature 75°C and track spacing of 3 mm, for a matrix coating of ∼0.31 mg/cm2. For both the nebulizer and HTX TM-Sprayer™, the matrix solution was 5 mg/mL CHCA in 50% acetonitrile and 0.1% trifluoroacetic acid (TFA). MALDI-MSI was performed with an model 4800 Plus TOF/TOF mass spectrometer (AB Sciex, Framingham, MA, USA) using a scan raster of 40 µm steps and 100 laser shots per pixel. Use of a smaller diameter laser beam (such as equipped with the UltrafleXtreme™ from Bruker Daltonics Inc., Billerica, MA, USA) and smaller step sizes would be expected to increase the spatial resolution (although this can come at a cost of lower sensitivity). Acquisition of the MALDI-MSI was achieved using the public domain software 4000 Series Imaging (Novartis & Applied Biosystems, Markus Stoeckli), which works with the native software on the mass spectrometer, and the images analyzed using the public domain software BioMAP (Novartis, Basel, Switzerland; Martin Rausch, MSI additions by Markus Stoeckli; see also Stoeckli et al.24). Fluorescence imaging of the plates, as detailed earlier, could also be performed after MALDI-MSI. This is particularly useful as the CHCA matrix provides some auto-fluorescence when excited with the 488-nm laser (fluorescein channel), thereby allowing visualization of the region scanned by MALDI-MSI, observed as the zone of matrix depletion.
Bulk analysis of photocleavage from TentaGel® beads
Some experiments were performed to test the ‘bulk’ photocleavage of PC-Mass-Tags from a population of prepared TentaGel® beads. In this case, a population of beads in suspension was processed and no plates or bead-arrays were involved. For this, 0.75 mg of PC-Mass-Tag conjugated TentaGel® beads (roughly 75 000 beads) was washed with MS-grade water (4 × 400 μL) and then suspended in 40 μL of water. The suspension was split into equal 20 μL portions. One portion was irradiated with 365 nm UV light using the conditions detailed earlier as for the plates, except exposure was done with the bead suspension in a thin-walled polypropylene PCR tube, through the side walls of the tube which adequately transmit the necessary light. The other portion was not irradiated with light. The beads were spun down briefly in a micro-centrifuge and the fluid supernatant (i.e. no beads) was collected. The supernatant was mixed with an equal volume of 10 mg/mL CHCA matrix in 50% acetonitrile and 0.1% TFA and 2 μL spotted onto a standard stainless steel MALDI target for standard MALDI-MS analysis on the aforementioned MALDI mass spectrometer.
Since the PC-Mass-Tag in this instance also corresponded to the VSV-G Tag binding epitope (peptide #22 in Supplementary Table S1(Supporting Information) = CRGYTDIEMNRLGK; underlined portion is VSV-Tag epitope), the remaining beads (with and without light irradiation) were probed with an anti-VSV-Tag antibody to estimate photocleavage efficiency as follows: The beads were washed with TBS-T (4 × 400 μL) and probed for 30 min with mixing using an anti-VSV-Tag-Cy3 conjugated antibody (Sigma-Aldrich, St. Louis, MO, USA) diluted to 10 µg/mL in BSA Block. Beads were then washed with TBS-T (4 × 400 μL) and water (1 × 400 μL). The beads were next embedded in polyacrylamide under a cover glass on a microscope slide for fluorescence imaging. To do so, the polyacrylamide was prepared by combining 244 μL of TBS, 57 μL of a 40% acrylamide solution (19:1 cross-linking; Bio-Rad, Hercules, CA, USA), 0.5 μL of TEMED (N,N,N’,N’-tetramethylethylenediamine; Bio-Rad), and 1 μL of 10% (w/v) ammonium persulfate. The washed bead pellet was re-suspended in 50 μL of this mixture and 25 μL of the bead suspension was placed onto a microscope slide under an 18 mm square cover glass. After the solution had polymerized, the beads were fluorescently imaged in the GenePix 4200A Microarray Scanner (Molecular Devices LLC).
RESULTS
Assembling the bead library
Some of the experiments described in this work used a bead-library constructed from 30 µm diameter streptavidin-coated glass beads or 34 µm diameter streptavidin-coated agarose beads. These streptavidin-coated beads provide a convenient method for the subsequent attachment of PC-Mass-Tags as well as proteins (or other bait) through a biotin or streptavidin binding-tag linkage (shown schematically in Fig. 2(A) for glass beads). In the case of glass beads, instead of attaching the streptavidin directly to the bead surface through a covalent bond, which is possible for example using carboxyl-terminated beads and carbodiimide chemistry (e.g. Fulton et al.16), the 30 µm glass beads (Microspheres-Nanospheres) were first derivatized with a non-cleavable biotin which allowed a non-covalent but still essentially irreversible attachment of the (tetrameric) streptavidin. This step prevented partial inactivation of the streptavidin which we found occurred with harsher chemical treatments. In the case of agarose beads, the streptavidin was directly and covalently attached to the bead surface (see Experimental section for more detail). Attachment of peptides of unique mass to the streptavidin beads was then facilitated by chemically modifying their N-termini with photocleavable biotin (PC-Biotin), using an NHS-activated (primary amine reactive) PC-Biotin labeling reagent23 (AmberGen, Inc.) (see also Fig. 2(A)).
As shown in Fig. 2(B), bead-libraries were also constructed from 30 µm diameter TentaGel® beads using a novel, custom synthesized PC-Linker. This heterobifunctional PC-Linker contains an amine-reactive NHS moiety on one end and a t-Boc-protected amine on the other (NHS-PC-tBOC Linker). The NHS-activated PC-Linker was attached to amine TentaGel® beads (Rapp Polymere), followed by removal of the t-Boc protecting group on the primary amine of the PC-Linker, resulting in photocleavable amine TentaGel® beads as shown in Fig. 2(B). Cysteine-terminated peptides were then attached to the photocleavable amine TentaGel® beads using a commercially available heterobifunctional (NHS-Maleimide) cross-linker (BMPS; Thermo-Pierce, Rockford, IL, USA). Alternatively, standard or combinatorial on-bead peptide/peptoid synthesis can be performed using the photocleavable amine TentaGel® beads as the starting point (the BMPS linker would not be needed in this case as synthesis would take place directly from the photocleavable amine). For the purpose of attaching both PC-Mass-Tags and proteins (or other bait) to photocleavable TentaGel® beads, streptavidin can also be attached to the beads using the same method described earlier for the glass beads, which allows biotinylated or streptavidin binding-tag modified proteins to be attached to the beads along with the PC-Mass-Tags (data not shown, see Fig. 2(B) for schematic).
Detection and correlation of MALDI-MSI and fluorescence signals from individual beads in a random bead-array
In order to evaluate the ability to image individual beads in a two-dimensional ordered bead-array by both MALDI-MSI and fluorescence, two different bead species were initially constructed using the streptavidin glass beads. As shown schematically in Fig. 3(A), one bead species (Bead 1) incorporated a single peptide PC-Mass-Tag and a fluorescent label, while a second bead species (Bead 2) contained two different peptide PC-Mass-Tags but no fluorescent label (see peptides #14, 7 and 9, respectively, in Supplementary Table S1, Supporting Information). The beads were then deposited randomly in a specially designed 1×25×75 mm micro-well plate with ∼1 million individual wells (∼45 µm i.d. by 55 µm deep wells) and scanned using MALDI-MSI and fluorescence. MALDI-MSI was performed in 40 µm steps in a raster scan pattern and analyzed using the BioMAP public domain software (Novartis; Martin Rausch, MSI additions by Markus Stoeckli).24
Figure 3.
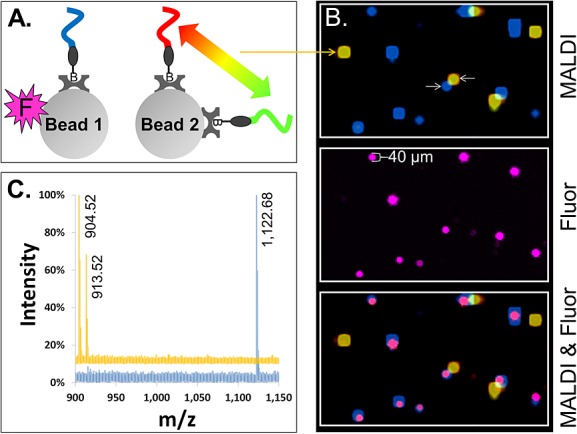
MALDI-MSI imaging of single beads carrying photocleavable peptide Mass-Tags. (A) Two species of 30 µm streptavidin glass beads were prepared, pooled and used to form a two-dimensionally ordered, random bead-array. ‘Bead 1’ carried a single photocleavable biotin peptide Mass-Tag (blue) and a fluorophore (magenta ‘F’). ‘Bead 2’ carried two different photocleavable biotin peptide Mass-Tags (red and green) but no fluorophore. (B) (‘MALDI’) Color-coded MALDI-MSI image of a 1360 by 800 µm2 region of the bead-array. Co-localization of the red and green Mass-Tags on Bead 2 appears as yellow. (‘Fluor’) Fluorescence image of same region of the bead-array, showing Bead 1. (‘MALDI & Fluor’) Synchronized MALDI-MSI and fluorescence images showing co-localization of the fluorescence marker on Bead 1 (magenta) with the expected Mass-Tag (blue). (C) Color-coded MALDI spectra are shown from the center pixel of representative beads (the beads indicated by white arrows in (B)). The blue spectrum is from Bead 1 and yellow from Bead 2. Observed monoisotopic masses of the Mass-Tags are labeled in the spectra (note that while the scaling of the spectra does not allow visual discrimination of the natural isotopes of each Mass-Tag, separated by 1 m/z, they are resolved; for example, see Fig. 4(B) ‘x-Axis Expansion’ inset).
Figure 3(B) shows the MALDI-MSI image alone (upper panel), fluorescence alone (middle panel), along with a superposition of the MALDI-MSI and fluorescence images (bottom panel) of a representative area (approximately 1360 by 800 µm). Each PC-Mass-Tag is color-coded in the image (see Fig. 3, caption). Bead species 1 clearly appears in the MALDI-MSI image as distinct blue spots. The identity of this bead species is confirmed by the correlated fluorescence spots appearing in the same positions (Fig. 3(B), bottom panel). By sampling the MALDI-MSI pixel closest to the center of each spot, it was confirmed that only the correct PC-Mass-Tag was detected above the noise level. This is exemplified in Fig. 3(C), which shows a representative MALDI-MSI spectrum of bead species 1 (blue trace; expected monoisotopic mass = 1121.63 m/z, observed = 1122.68 m/z). The non-standard micro-well plates used for the bead-arrays, compared to the standard steel MALDI targets, are believed to be the cause of the skewing of roughly 0.5 to 1 m/z between the expected and observed masses of the PC-Mass-Tags. The MALDI-MSI image of bead species 2 appears as discrete yellow spots due to the mixing of the red and green colors which in the image code for the PC-Mass-Tags attached to this bead species (Fig. 3(B), top and bottom panels). Mass spectra from the center of these spots clearly show that both of the expected PC-Mass-Tags are present on bead species 2 (expected monoisotopic mass = 903.46 and 912.45 m/z, observed 904.52 and 913.52 m/z, respectively), but the PC-Mass-Tag from bead species 1 is not detected (see Fig. 3(C), yellow trace, for representative spectrum of bead species 2).
The bead size resolved by MALDI-MSI was significantly larger than by fluoresce (65 ± 13 µm and 40 ± 4 µm, respectively), based on averaging the 15 MALDI-MSI spots and 10 fluorescent spots shown in Fig. 3(B). This result is expected due to the much larger 100 µm laser beam diameter used to acquire the MALDI-MSI data compared to the 5 µm resolution of the fluorescence scanner. The MALDI-MSI resolution still is better than 100 µm because of the smaller step size (40 µm) and the depletion scanning method used. Despite the lower resolution of the MALDI-MSI image, excellent correlation is still achieved between the MALDI-MSI image and the fluorescence image (Fig. 3(B), bottom panel).
Evaluation of a 20-member peptide-bead library
A larger library consisting of streptavidin glass beads labeled with 20 different PC-Mass-Tag peptides was constructed and scanned by both MALDI-MSI and fluorescence (Fig. 4). All 20 different bead species were identified by detection of the corresponding PC-Mass-Tags. Figure 4(A) shows a 20-color mass-image of the bead-array, and representative beads for each of the 20 species are indicated by the white circles (and labeled with observed masses), with their corresponding mass spectra shown in color-coded traces (Fig. 4(B)). Supplementary Table S2 (Supporting Information) lists the expected versus observed masses of the 20 PC-Mass-Tags from this bead library, which, as discussed earlier, were all typically offset by approximately 0.5–1 m/z. Note that even though the representative beads appear in very close proximity to other beads bearing different PC-Mass-Tags, spectral analysis of the spot center shows no cross-contamination (Fig. 4(B)). In addition, even though some of the peptides have similar masses, such as at 1106 and 1110 m/z, these peaks can be clearly resolved due to the high resolution of the MALDI mass spectrometer (resolution = mass/[full peak width at half maximum intensity] ≈ 10 000). If the x-axis of the mass spectrum is expanded, sub-component peaks arising from the presence of natural abundance isotopes, separated by 1 m/z, are also clearly observed (see inset ‘x-Axis Expansion’ in Fig. 4(B) which shows the 1106 and 1110 m/z PC-Mass-Tags with their corresponding isotopes easily resolved).
Figure 4.
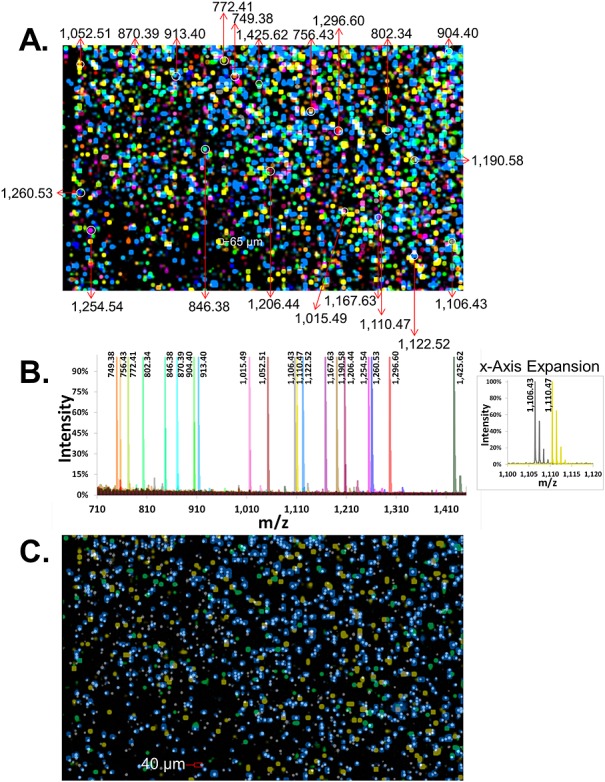
Synchronized MALDI-MSI and fluorescence imaging of 20-member Mass-Tag bead-array. (A) Color-coded MALDI-MSI image of a 5920 × 3640 µm2 region of a 20-member photocleavable Mass-Tag bead-array (photocleavable biotin peptide Mass-Tags loaded onto streptavidin glass beads). Corresponding masses (m/z) for Mass-Tags from representative beads (white circles) are indicated. (B) Color-coded overlaid MALDI-MSI spectra are shown for representative beads in the array (the beads indicated by white circles in (A)). Observed monoisotopic masses are listed. The ‘x-Axis Expansion’ inset shows a zoomed view of two distinct Mass-Tags of similar size, indicating a high mass resolution which can discriminate Mass-Tags separated by approximately 4 m/z units as well as the natural isotopes for each, spaced by 1 m/z unit. (C) One Mass-Tagged bead species in the array was labeled with fluorophore (e.g. as in Fig. 3(A)). The fluorescence image of this bead species (same region as in panel (A)) is shown (gray/white spots) synchronized with a MALDI-MSI image of its cognate Mass-Tag (blue). For simplicity, MALDI-MSI images for two other Mass-Tags on non-fluorescent bead species are also superimposed (yellow and green).
In order to demonstrate the ability to correlate MALDI and fluorescence signals arising from the same bead, a Sync Bead species was included in this library which additionally contained the fluorophore Alexa Fluor® 647 (Sync Beads were defined earlier; see ‘Bead 1’ in Fig. 3(A) for example). Figure 4(C) is a superposition of the Sync Bead fluorescence image (gray/white) and the mass-image of the corresponding PC-Mass-Tag (blue). Most significantly, a high spatial correlation between the fluorescent beads and their cognate PC-Mass-Tag is observed, with approximately 50% of the Sync Beads detected by MALDI-MSI. In addition, there is virtually no false positive detection of beads by MALDI-MSI, which would be indicated by a blue MALDI-MSI spot but no gray/white fluorescent bead. Lack of 100% bead detection by MALDI-MSI is believed to be a result of incomplete matrix coverage and/or penetration into the micro-wells. As a basis for comparison, two PC-Mass-Tags from the two most abundant non-Sync Bead species are also superimposed in Fig. 4(C) (yellow and green), and show little to no spatial correlation with the fluorescent beads (for simplicity not all 20 mass-tags are shown in Fig. 4(C)).
As a metric to quantify the alignment between the fluorescent and MALDI-MSI images, the X-Y pixel coordinates and values from the individual (aligned) digital grayscale images (one for each color) used to create Fig. 4(C) were exported in textual format using the public domain ImageJ software25 and compared. Pixels in the fluorescence and MALDI images were scored positive if they had a grayscale value of essentially 10-fold above background (i.e. >10 in grayscale images of 0–256 levels). 65% of the positive fluorescent pixels had a positive MALDI pixel for the correct PC-Mass-Tag (blue in Fig. 4(C)) at the same coordinate. Conversely, 7-fold fewer (9%) positive fluorescent pixels overlapped with pixels from either of the two PC-Mass-Tags in Fig. 4(C) which were not on the Sync Beads (yellow and green).
Measuring phosphorylation using Bead-GPS
Figure 5 illustrates results from a simple experiment designed to test the ability of Bead-GPS to screen for post-translational modifications (PTMs) mediated by a specific kinase. A model peptide (Kemptide), which serves a substrate for protein kinase A (PKA), and a control peptide (Tag-3.1) lacking phosphorylation sites, were labeled at the N-terminus using PC-Biotin and then bound to streptavidin beads as previously described (agarose beads in this case; see Supplementary Table S1 (Supporting Information) for peptide sequences). The two bead species were pooled and treated (in suspension) with and without PKA plus ATP. The beads were then arrayed and scanned by MALDI-MSI as described earlier (no fluorescence imaging in this case). The color-coded MALDI-MSI images in Figs. 5(A) and 5(B) show detection of the two different bead populations (Kemptide and control peptide beads) as discrete spots of approximately 65 µm diameter. Furthermore, without PKA treatment (Fig. 5(A)), only the unphosphorylated Kemptide is observed on the Kemptide beads (color-coded as green in figure). Figure 5(C) shows a MALDI-MSI spectrum of a representative Kemptide bead (expected unphosphorylated Kemptide mass = 771.47 m/z, observed = 772.90 m/z). Conversely, with PKA treatment, the phosphorylated Kemptide species is also observed on the Kemptide beads (Fig. 5(B) for MALDI-MSI image; yellow spots are Kemptide beads), with a precise +80.0 m/z mass shift corresponding to the expected mass shift due to the addition of a phosphate group (see Fig. 5(D) for the MALDI-MSI spectrum of a representative bead; observed phosphorylated Kemptide mass = 852.90 m/z). Note that incomplete conversion of the Kemptide by PKA results in detection of both the unphosphorylated and phosphorylated species on the same beads (Figs. 5(B) and 5(D)). No modification of the control peptide was observed (spectrum not shown), which lacked any serine or threonine phosphorylation sites and hence could not be modified.
Figure 5.
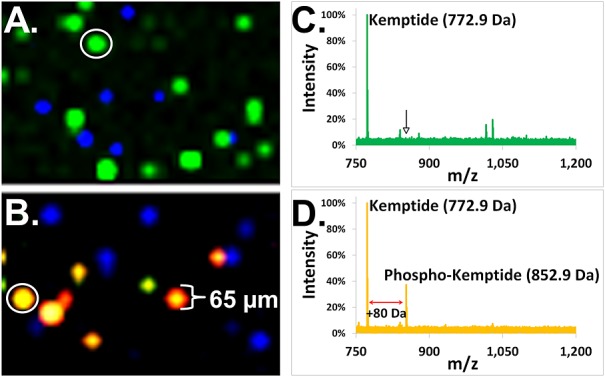
Kinase substrate profiling with Bead-GPS. Two photocleavable biotin peptide Mass-Tag bead species were prepared, one carrying a substrate for protein kinase A (PKA), Kemptide, and the other a non-substrate control peptide (lacking phosphorylation sites). Beads were pooled and the pool split. One set was left untreated while the other set was treated with PKA and ATP. A bead-array was then formed and MALDI-MSI imaging performed. (A and B) Superimposed MALDI-MSI images whereby the non-phosphorylated Kemptide is color-coded green, the non-substrate control peptide blue and the phosphorylated Kemptide red. (A) MALDI-MSI image of beads not treated with PKA. (B) MALDI-MSI image of beads treated with PKA. Note that the co-localization of the green (unphosphorylated) and red (phosphorylated) Kemptide species on the same beads appears as yellow. (C) MALDI-MSI spectrum from a representative Kemptide bead (the bead indicated by white circle in (A)) without PKA treatment. The monoisotopic mass for the unphosphorylated 772.9 m/z Kemptide is noted. The arrow indicates where the +80 m/z phosphorylated Kemptide would appear were it present. (D) MALDI-MSI spectrum from a representative Kemptide bead (the bead indicated by white circle in (B)) after PKA treatment. Due to incomplete conversion by PKA, both the unphosphorylated (772.9 m/z) and phosphorylated Kemptide (852.9 m/z), upshifted by precisely 80 m/z, were observed.
Creating TentaGel® beads with attached PC-Mass-Tags
TentaGel® beads, which consist of a co-polymer of polyethylene glycol (PEG) grafted to a polystyrene matrix (Rapp Polymere), offer several advantages over glass beads for use in proteomics. This includes their PEG ‘hydrogel’ coating which helps maintain a biocompatible, hydrated and three-dimensional environment for biomolecules, and, additionally, their compatibility and wide-spread use with combinatorial synthetic methods such as the creation of one-bead-one-compound (OBOC) libraries.26
In order to adapt such beads for use with Bead-GPS, cysteine-terminated PC-Mass-Tags (peptides) were attached to 30 µm mono-sized TentaGel® beads using the method discussed earlier and outlined in Fig. 2(B). This method employs a custom PC-Linker (NHS-PC-tBOC Linker) that allows direct covalent attachment of peptides or peptoids, or, alternatively, their direct on-bead synthesis (the latter not demonstrated here).
As a first step in evaluating these beads, the UV-photocleavage reaction was measured. While this had previously been characterized for the PC-Biotin linker,23,27 this had not yet been done for the new direct PC-Linker used with the TentaGel® beads. Therefore, both positive photo-release as well as the amount remaining on the beads was assessed using a model PC-Mass-Tag (assayed by MALDI-MS and antibody detection, respectively). Both measures showed selective photo-release of the PC-Mass-Tag (see Supplementary Fig. S1 (Supporting Information) for details). Note that a significant portion of the linker remains attached to the PC-Mass-Tag after photocleavage (Supplementary Fig. S1(A)), adding 560.57 m/z to the mass. Furthermore, two additional isotope clusters were observed by MALDI-MS corresponding to satellite peaks at +16 and –16 m/z relative to the expected mass (Supplementary Fig. S1(B)), most likely due to addition or loss of an oxygen during the photocleavage reaction, similar to earlier results obtained from photocleavable peptide-DNA conjugates synthesized using the same photo-nucleus.28
Application of Bead-GPS to TentaGel® bead libraries
A small 3-member model bead-array was created with different cysteine peptide PC-Mass-Tags attached to TentaGel® beads. Similar to Fig. 3(A), one of the bead species was additionally tagged with a fluorescent dye (‘Sync Beads’). As described previously, the beads were then deposited randomly in a specially designed 1×25×75 mm micro-well plate with ∼1 million individual wells (˜45 µm i.d. by 55 µm deep wells) and scanned using MALDI-MSI and fluorescence.
Figure 6(A) shows a fluorescence image of the bead-array, which displays both the 488-nm excited auto-fluorescence emitted by all TentaGel® beads, as well as the specific fluorescence arising from the Sync Beads (Alexa Fluor® 647 dye) excited by the 635-nm laser. Under these conditions, two distinct bead populations were observed, the Sync Beads (red in Fig. 6(A)) and the other two PC-Mass-Tag bead species (gray/white in Fig. 6(A)) which cannot be distinguished from each other on the basis of fluorescence alone. Figure 6(B) shows synchronization of this fluorescence image and the color-coded (blue, green and yellow) MALDI-MSI image obtained from the same bead-array. As seen in Fig. 6(B), the blue color-coded PC-Mass-Tag which resides on the Sync Beads aligns well with the red fluorescence arising from these beads. In contrast, the other two PC-Mass-Tags (green and yellow) align well with the non-Sync Beads (gray/white fluorescent beads). Figure 6(C) shows color-coded MALDI-MSI spectra obtained from the center of representative beads (beads indicated by white circles in Figs. 6(A) and 6(B)). As before (e.g. Supplementary Fig. S1(B)), three isotope clusters for each PC-Mass-Tag were observed, corresponding to the expected mass as well as the minus and plus oxygen species (note that due to the scaling of the spectra in Fig. 6(C), isotope clusters appear as single peaks). Monoisotopic masses for the –16 m/z (oxygen loss) species of the PC-Mass-Tags are listed in Fig. 6(C), as these are the dominant species in the bead-arrays most likely due to the dry photocleavage conditions. Observed mono-isotopic masses are 1925.64, 2199.82 and 2343.89 m/z while expected are 1925.29, 2199.36 and 2343.39 m/z, respectively.
Figure 6.
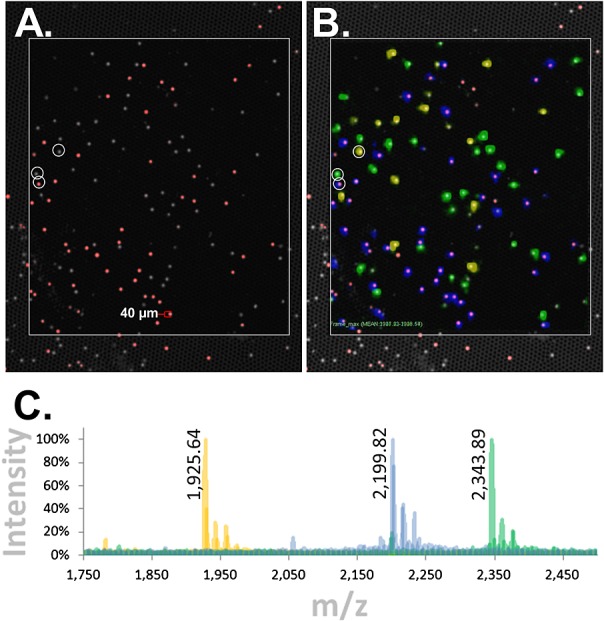
Bead-GPS array with cysteine peptides directly attached to TentaGel® beads: synchronized MALDI-MSI and fluorescence imaging. A model 3-member bead-array was created with cysteine peptide Mass-Tags attached to TentaGel® beads using the novel NHS-PC-tBOC Linker. Similar to Fig. 3(A), one of the bead species was additionally tagged with fluorescence. The bead-array was imaged by MALDI-MSI and fluorescence. (A) Fluorescence image of bead-array. Gray/white is auto-fluorescence of all TentaGel® beads when excited with the 488 nm laser of the microarray scanner (fluorescein channel; the micro-wells are also weakly visible), while red is the fluorescently tagged bead species. (B) Synchronization of fluorescence and MALDI-MSI images. The three Mass-Tags are color-coded blue, green and yellow. The blue Mass-Tag aligns with the red fluorescent beads. (C) Color-coded overlaid MALDI-MSI spectra from the center of representative beads (the beads indicated by white circles in (A) and (B)). Monoisotopic masses for the –16 m/z (oxygen loss) species of the Mass-Tags are listed as these are the dominant species in the bead-arrays. Observed monoisotopic masses are 1925.64, 2199.82 and 2343.89 m/z while expected are 1925.29, 2199.36 and 2343.39 m/z, respectively (note that due to the scaling of the spectra, monoisotopic peaks are not discernible but are resolved; for example, see Fig. 4(B) ‘x-Axis Expansion’ inset or inset in Supplementary Fig. S1 (Supporting Information)).
DISCUSSION
This work demonstrates the ability to align MALDI-MSI and fluorescence images of a random bead-array, a key feature of the Bead-GPS approach described here. Ultimately, this capability will provide the primary information to identify and quantify bait-prey interactions (i.e. hits) on individual beads in such an array. In addition, in contrast to fluorescence protein microarrays and related fluorescence-based ‘bead suspension array’ approaches,12,29 the use of MALDI-MSI provides an additional layer of information which is normally associated with classical proteomics based on MS. This includes the ability to perform detailed molecular analysis such as protein mass fingerprinting and sequencing (e.g. using tandem mass spectrometry (MS/MS)19,20), or, in the case of protein-small molecule (drug) interactions, label-free identification.21 Here, we demonstrate this ability by detecting post-translational modification of a peptide attached to specific beads in the random bead-array without the need for exogenous probes.
An additional feature of the present work is the use of PC-Linkers, which allow the coding molecules residing on the beads to remain attached to the bead, until exposure to UV light which can include the actual laser used in the MALDI instrument for matrix ionization (data not shown). Although coded beads have been widely used in the area of multiplex detection,12,29 none of these platforms approach the capabilities and/or coding capacity potentially achievable with Bead-GPS. In principle, the number of bait which can be included in the library and the number of prey molecules that can be added to the library is only limited by the number of available PC-Mass-Tags and the resolution of a mass spectrometer, which routinely exceeds 0.1 m/z. At this resolution, up to 10 000 unique mass-tags could potentially be used in the 1000–2000 m/z mass range, which is preferred range in the MS reflector mode. In the case of peptides, variable sequence and length of peptides as well as incorporation of isotope-labeled and unnatural amino acids can result in 10 000 unique masses. An even greater number of codes can be created by using a combination of two or more PC-Mass-Tags for each bead species. For example, using a pool of 1000 PC-Mass-Tags with 3 tags per bead can generate over 150 million unique combinations.
Here we demonstrated the feasibility of using glass, agarose and TentaGel® beads for Bead-GPS, although other bead types that are commonly used for libraries may also be compatible (e.g. PEGA30 acrylamide-PEG co-polymer beads). While further study is needed comparing these different bead types, several factors appear to affect the ability to obtain strong MALDI-MSI signals, well-defined spot morphology and to measure functional interaction of prey biomolecules (e.g. enzymes) with the bait on the beads. Such factors include: (i) Gel-like agarose and TentaGel® beads swell in the proper solvents by as much as 5-fold.31 As a consequence they shrink/deform upon drying, which aside from impacting spot morphology, can also leave the beads located close to the bottom of the micro-wells, making them potentially less accessible to the applied matrix and the MALDI-MS laser beam, as opposed to the rigid glass beads; (ii) TentaGel® beads exhibit high auto-fluorescence background at blue-shifted wavelengths (e.g. the fluorescein and Cy3® channels in most fluorescence imagers), which can potentially interfere with measuring fluorescence from prey molecules which interact with bait molecules residing on the bead; (iii) Surface binding capacity, which is less for solid non-porous glass beads as compared to the porous gel-type beads, plays a role in the ability to detect bait-prey interactions and record high MALDI-MSI signal-to-noise ratios; and (iv) The bead type used may be critical for functional studies such as shown previously for the case of papain and chymotrypsin, where activity was much higher on peptide libraries prepared on PEGA beads compared to TentaGel® and ArgoGel beads.32
The use of PC-Linker TentaGel® beads, as described here, was explored because such beads are compatible with the standard methods of combinatorial synthesis and one-bead-one-compound (OBOC) libraries, thereby broadening the usefulness of Bead-GPS. In particular, the typical OBOC methods of synthesis can be adopted to produce large libraries of photocleavable peptides or peptoids which can serve simultaneously as bait and coding elements for Bead-GPS. Although a large fraction of such OBOC compounds will have degenerate masses, the ability to sequence directly from the bead using MS/MS could provide a further basis for distinguishing the different coding elements/baits residing on individual beads.
One of the major advantages of Bead-GPS is its ability to decode an OBOC bead library by performing mass spectrometric imaging (i.e. MALDI-MSI) directly on the bead-array, without having to include, on a bead-by-bead basis, a separate elution and decoding step. For example, in a recent study aimed at determining kinase-substrate specificity,33 individual 300–500 µm PEGA beads identified as substrates in a 2 × 106 member bead-library were decoded by chemically cleaving the test peptide on a bead-by-bead basis. Subsequent to cleavage, decoding was completed for each individual bead-derived peptide by sequencing using partial Edman degradation mass spectrometry (PED-MS).
An additional major advantage of Bead-GPS over traditional OBOC methods is the ability to rapidly screen an entire library for positive hits (e.g. beads where a substrate-enzyme interaction occurred) without the need to physically separate individual beads. For example, in the previous work described,33 individual beads that displayed positive hits on the basis of fluorescence were physically (manually) separated with a micropipette on a Petri dish using fluorescence microscopy. Such an approach is subject to human error and can also be relatively slow, thereby preventing true high-throughput screening. In comparison, correlated MALDI-MSI and fluorescence imaging eliminates the need to physically isolate positive beads because the MALDI-MS instrument scans the entire array, and ultimately could be directed to only those beads displaying fluorescence above a critical threshold for immediate and direct identification by MS/MS. Although correlated methods were not used in the example presented for PKA, such an application should be highly feasible. For example, phosphorylation specific antibodies and fluorescent phosphorylation sensor dyes are readily available commercially. Such a phosphorylation sensor dye was reported by Martin et al.,34 and shown to effectively enable detection by fluorescence of beads containing peptides which have undergone phosphorylation.
We also note that with Bead-GPS not every bead in the library needs to be analyzed, but only those displaying fluorescence, thus dramatically shortening the time which would be needed to scan all necessary beads in the random array. Furthermore, the number of beads in the OBOC library using Bead-GPS can be comparable to those used in more conventional methods. For example, in the study cited,33 2 × 106 beads in a bead slurry were analyzed (i.e. 2 × 106 different sequences). In the case of Bead-GPS, which used much smaller beads (˜30 µm or approximately 10-fold smaller), an estimated 1 × 106 beads can be positioned in a random array on standard 25 × 75 mm microscope slide. The smaller bead size and resulting smaller pellet volume (i.e. 1000-fold smaller) also allow for much smaller biospecimen volumes to be interrogated, an important advantage when working with scarce samples.
Earlier work, particularly in the area of cleavable mass-tagging, as well as the application of MALDI-MSI to tissue analysis, provided a basis for the development of Bead-GPS. For example, photocleavable peptide-DNA conjugates have been used as hybridization probes for multiplex (non-imaging) MS detection in model genomic bio-assays.28 In a similar approach, small non-photocleavable oligonucleotide mass-tags (OMTs) were used in conjunction with MALDI-TOF to perform a multiplex assay of DNA-protein binding specificity.35
Imaging time-of-flight (TOF) secondary ion MS (SIMS), which utilizes highly focused ion beams (e.g. 150 nm), has previously been applied to obtain spatially resolved chemical information about molecules such as peptides residing on individual beads.36 In one experiment, SIMS was used to spatially resolve the target molecule angiotensin II synthesized on a 40 µm polystyrene bead after exposure to a suitable gas which clips the covalent bonds at the linking position.37 Later work also used SIMS to characterize combinatorial bead libraries.38
Improvements in MALDI-MSI techniques over the past decade have also provided increasingly detailed, spatially resolved information about the composition of endogenous and exogenous molecules such as drugs in thin tissue sections.39 In one application of tissue profiling using both MALDI-MS and beads,40 tissue sections were placed onto an array of glass beads which were embedded on a Parafilm® membrane. Stretching of the membranes results in separation of the tissue section into thousands of cell-sized pieces on the beads, which can then be individually profiled by non-imaging MALDI-MS, thereby shortening scan time and eliminating redistribution of molecules during matrix application. Later, algorithms were introduced to reconstruct mass spectrometric images from the original positions of the beads (i.e. positions prior to stretching the Parafilm® which separates the beads).41 Although intended for MALDI-MSI of tissues, model experiments were shown using peptides, whereby reconstructed ion images from dye labeled 40 µm glass beads could distinguish between a distribution of angiotensin I and II which were passively adsorbed onto two different bead species.
An analogous approach to Bead-GPS which uses several common elements but not MS for readout has also been previously reported.42 Termed PC-PRINT, the method facilitates the photocleavage release and transfer of the bait and interacting prey molecule(s), along with coding molecules, from the beads to a surface upon which the beads were placed (e.g. an activated glass slide). The transfer process forms a high-density random microarray of discrete bead-sized spots on the surface which can be fluorescently imaged in order to identify hits. In one example, PC-PRINT was used to probe protein-protein interactions between p53 (prey) and several human proteins (bait) which were cell-free expressed and captured on streptavidin-coated agarose beads (75–150 µm), including MDM and GST. In a second experiment, beads carrying photocleavably tethered proteins and quantum dots were placed in close proximity to an activated glass slide and then illuminated to transfer the proteins and coding elements to the surface to form a random, planar microarray (see Fig. 6 in Lim and Rothschild42). Based on this work, direct transfer of molecules from a random bead-array to adjacent surfaces such as the walls or bottoms of a micro-well plate should be feasible and allow direct readout of transferred molecules, including PC-Mass-Tags, using MALDI-MSI.
CONCLUSIONS
This initial work demonstrates key elements of a new approach to proteomics, termed bead-based global proteomic screening (Bead-GPS). In contrast to conventional protein microarrays8 which utilize fluorescence imaging of ‘bait’ molecules located at known positions in two-dimensional array, Bead-GPS is based on the use of correlated fluorescence and MALDI mass spectrometric images (MALDI-MSI) to analyze random bead-arrays. Here, we have demonstrated the ability to identify and correlate fluorescently tagged beads in a random bead-array with the MALDI-MS signal from photocleavable peptides which act as coding agents (PC-Mass-Tags). In addition, we have demonstrated the ability to identify individual beads in the array which undergo alterations due to enzyme-substrate interactions resulting in post-translational modifications.
In order to make further progress, much larger PC-Mass-Tag bead-libraries need to be generated and evaluated in the context of Bead-GPS. In this regard, the TentaGel® beads used here are compatible with combinatorial synthetic methods such as the creation of one-bead-one-compound (OBOC) libraries.26 In addition, a demonstration of capability to perform multiplex binding assays for individual beads in the random bead-array (e.g. antibody-antigen) needs to be performed which is essential for any global proteomic assay (e.g. see Schwenk et al.15).
Acknowledgments
This work was funded in part by the following SBIR grants from the National Institutes of Health (http://www.nih.gov) to AmberGen, Incorporated: R44CA137948, R43GM105249, R43CA161965 and R44CA114126.
Supporting Information
Additional supporting information may be found in the online version of this article at the publisher‘s web site.
REFERENCES
- [1].Feng Y, Ke X, Ma R, Chen Y, Hu G, Liu F. Clin. Chem. 2004;50:416. doi: 10.1373/clinchem.2003.023994. [DOI] [PubMed] [Google Scholar]
- [2].Quintana FJ, Hagedorn PH, Elizur G, Merbl Y, Domany E, Cohen IR. Proc. Natl. Acad. Sci. USA. 2004;(101 Suppl 2):14615. doi: 10.1073/pnas.0404848101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Robinson WH, Fontoura P, Lee BJ, de Vegvar HE, Tom J, Pedotti R, DiGennaro CD, Mitchell DJ, Fong D, Ho PP, Ruiz PJ, Maverakis E, Stevens DB, Bernard CC, Martin R, Kuchroo VK, van Noort JM, Genain CP, Amor S, Olsson T, Utz PJ, Garren H, Steinman L. Nat. Biotechnol. 2003;21:1033. doi: 10.1038/nbt859. [DOI] [PubMed] [Google Scholar]
- [4].Robinson WH, DiGennaro C, Hueber W, Haab BB, Kamachi M, Dean EJ, Fournel S, Fong D, Genovese MC, de Vegvar HE, Skriner K, Hirschberg DL, Morris RI, Muller S, Pruijn GJ, van Venrooij WJ, Smolen JS, Brown PO, Steinman L, Utz PJ. Nat. Med. 2002;8:295. doi: 10.1038/nm0302-295. [DOI] [PubMed] [Google Scholar]
- [5].Lam KS. Anticancer Drug Des. 1997;12:145. [PubMed] [Google Scholar]
- [6].Lam KS, Lebl M, Krchnak V. Chem. Rev. 1997;97:411. doi: 10.1021/cr9600114. [DOI] [PubMed] [Google Scholar]
- [7].Udugamasooriya DG, Ritchie C, Brekken RA, Kodadek T. Bioorg. Med. Chem. 2008;16:6338. doi: 10.1016/j.bmc.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].MacBeath G, Schreiber SL. Science. 2000;289:1760. doi: 10.1126/science.289.5485.1760. [DOI] [PubMed] [Google Scholar]
- [9].Stoevesandt O, Taussig MJ, He M. Expert Rev. Proteomics. 2009;6:145. doi: 10.1586/epr.09.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Bertone P, Snyder M. FEBS J. 2005;272:5400. doi: 10.1111/j.1742-4658.2005.04970.x. [DOI] [PubMed] [Google Scholar]
- [11].Fang Y, Ferrie AM. F. Lai. In: Walker JM, editor. The Proteomics Protocols Handbook. Totowa, NJ: Humana Press Inc; 2005. [Google Scholar]
- [12].Mathur A, Kelso DM. Cytometry A. 2010;77:356. doi: 10.1002/cyto.a.20841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Fritzler MJ, Fritzler ML. Curr. Med. Chem. 2006;13:2503. doi: 10.2174/092986706778201639. [DOI] [PubMed] [Google Scholar]
- [14].Wong J, Sibani S, Lokko NN, LaBaer J, Anderson KS. J. Immunol. Methods. 2009;350:171. doi: 10.1016/j.jim.2009.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Schwenk JM, Lindberg J, Sundberg M, Uhlen M, Nilsson P. Mol. Cell. Proteomics. 2007;6:125. doi: 10.1074/mcp.T600035-MCP200. [DOI] [PubMed] [Google Scholar]
- [16].Fulton RJ, McDade RL, Smith PL, Kienker LJ, Kettman JR., Jr Clin. Chem. 1997;43:1749. [PubMed] [Google Scholar]
- [17].Savitski MF, Savitski MM. Methods Mol. Biol. 2010;673:203. doi: 10.1007/978-1-60761-842-3_12. [DOI] [PubMed] [Google Scholar]
- [18].Farley AR, Link AJ. Methods Enzymol. 2009;463:725. doi: 10.1016/S0076-6879(09)63040-8. [DOI] [PubMed] [Google Scholar]
- [19].Washburn MP, Wolters D, Yates JR., 3rd Nat. Biotechnol. 2001;19:242. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- [20].Wu WW, Wang G, Baek SJ, Shen RF. J. Proteome Res. 2006;5:651. doi: 10.1021/pr050405o. [DOI] [PubMed] [Google Scholar]
- [21].Rix U, Superti-Furga G. Nat. Chem. Biol. 2009;5:616. doi: 10.1038/nchembio.216. [DOI] [PubMed] [Google Scholar]
- [22].Cazares LH, Troyer DA, Wang B, Drake RR, Semmes OJ. Anal. Bioanal. Chem. 2011;401:17. doi: 10.1007/s00216-011-5003-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Olejnik J, Sonar S, Krzymanska-Olejnik E, Rothschild KJ. Proc. Natl. Acad. Sci. USA. 1995;92:7590. doi: 10.1073/pnas.92.16.7590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Stoeckli M, Staab D, Staufenbiel M, Wiederhold KH, Signor L. Anal. Biochem. 2002;311:33. doi: 10.1016/s0003-2697(02)00386-x. [DOI] [PubMed] [Google Scholar]
- [25].Schneider CA, Rasband WS, Eliceiri KW. Nat. Methods. 2012;9:671. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Quarrell R, Claridge TD, Weaver GW, Lowe G. Mol. Diver. 1996;1:223. doi: 10.1007/BF01715526. [DOI] [PubMed] [Google Scholar]
- [27].Pandori MW, Hobson DA, Olejnik J, Krzymanska-Olejnik E, Rothschild KJ, Palmer AA, Phillips TJ, Sano T. Chem. Biol. 2002;9:567. doi: 10.1016/s1074-5521(02)00135-7. [DOI] [PubMed] [Google Scholar]
- [28].Olejnik J, Ludemann HC, Krzymanska-Olejnik E, Berkenkamp S, Hillenkamp F, Rothschild KJ. Nucleic Acids Res. 1999;27:4626. doi: 10.1093/nar/27.23.4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Schwenk JM, Gry M, Rimini R, Uhlen M, Nilsson P. J. Proteome Res. 2008;7:3168. doi: 10.1021/pr700890b. [DOI] [PubMed] [Google Scholar]
- [30].Renil M, Ferreras M, Delaisse JM, Foged NT, Meldal M. J. Peptide Scie. 1998;4:195. doi: 10.1002/(SICI)1099-1387(199805)4:3%3C195::AID-PSC141%3E3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- [31].Santini R, Griffith MC, Qi M. Tetrahedron Lett. 1998;39:8951. [Google Scholar]
- [32].Leon S, Quarrell R, Lowe G. Bioorg. Med. Chem. Lett. 1998;8:2997. doi: 10.1016/S0960-894X(98)00534-4. [DOI] [PubMed] [Google Scholar]
- [33].Trinh TB, Xiao Q, Pei D. Biochemistry. 2013;52:5645. doi: 10.1021/bi4008947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Martin K, Steinberg TH, Goodman T, Schulenberg B, Kilgore JA, Gee KR, Beechem JM, Patton WF. Combinat. Chem. High throughput Screening. 2003;6:331. doi: 10.2174/138620703106298581. [DOI] [PubMed] [Google Scholar]
- [35].Zhang L, Kasif S, Cantor AC. Proc. Natl. Acad. Sci. USA. 2007;104:3061. doi: 10.1073/pnas.0611075104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Brummel CL, Lee IN, Zhou Y, Benkovic SJ, Winograd N. Science. 1994;264:399. doi: 10.1126/science.8153627. [DOI] [PubMed] [Google Scholar]
- [37].Brummel CL, Vickerman JC, Carr SA, Hemling ME, Roberts GD, Johnson W, Weinstock J, Gaitanopoulos D, Benkovic SJ, Winograd N. Anal. Chem. 1996;68:237. doi: 10.1021/ac950998u. [DOI] [PubMed] [Google Scholar]
- [38].Enjalbal C, Maux D, Combarieu R, Martinez J, Aubagnac JL. J. Comb. Chem. 2003;5:102. doi: 10.1021/cc0200484. [DOI] [PubMed] [Google Scholar]
- [39].Chaurand P. J. Proteomics. 2012;75:4883. doi: 10.1016/j.jprot.2012.04.005. [DOI] [PubMed] [Google Scholar]
- [40].Monroe EB, Jurchen JC, Koszczuk BA, Losh JL, Rubakhin SS, Sweedler JV. Anal. Chem. 2006;78:6826. doi: 10.1021/ac060652r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Zimmerman TA, Monroe EB, Sweedler JV. Proteomics. 2008;8:3809. doi: 10.1002/pmic.200800331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lim M, Rothschild KJ. Anal. Biochem. 2008;383:103. doi: 10.1016/j.ab.2008.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found in the online version of this article at the publisher‘s web site.