

Abstract
Anaplasma phagocytophilum invades neutrophils to cause the emerging infection, human granulocytic anaplasmosis. Here, we provide a focused review of the A. phagocytophilum invasin-host cell receptor interactions that promote bacterial entry and the degradative and membrane traffic pathways that the organism exploits to route nutrients to the organelle in which it resides. Because its obligatory intracellular nature hinders knock out-complementation approaches, we also discuss current methods used to study A. phagocytophilum gene function and the potential benefit of applying novel tools that have advanced studies of other obligate intracellular bacterial pathogens.
INTRODUCTION
Anaplasma phagocytophilum is a tick-borne, obligate intracellular bacterium of the family Anaplasmataceae that infects granulocytes, bone marrow progenitor cells, and endothelial cells of various mammalian species, including humans [1, 2]. A. phagocytophilum is unusual in its ability to flourish within neutrophils, which are important effector cells of microbial killing. Infection of humans by A. phagocytophilum causes human granulocytic anaplasmosis (HGA), an emerging infectious disease first detected in 1994 [3]. Cases of HGA have also been documented in Europe and Asia. Though incidence is on the rise, HGA remains an underreported disease. HGA is an acute febrile infection accompanied by many non-specific symptoms including chills, headache, malaise, and myalgia. Clinical manifestations include leukopenia, thrombocytopenia, and elevations in serum hepatic aminotransferases. The disease is generally self-limiting in healthy individuals, though up to 50% of symptomatic patients require hospitalization [1]. Potential complications include rhabdomyolysis, pneumonitis, shock, seizures, hemorrhage, and increased susceptibility to potentially fatal secondary infections. Risk for complications and death is greater for individuals having pre-existing immunocompromise, the elderly, and when antibiotic therapy is delayed [1, 4].
A. phagocytophilum’s obligatory intracellular nature is predicated on its need to parasitize host cell nutrients. Following invasion, the bacterium resides in a host cell-derived vacuole that it remodels into a protective niche [1]. Membrane traffic is rerouted to the A. phagocytophilum-occupied vacuole (ApV) to deliver vital nutrients and host cell membrane material. This review describes our current understanding of the molecular events involved in A. phagocytophilum cellular invasion and molecular parasitism. We also discuss the challenges of studying the organism and conclude with a survey of recent advances in the genetic manipulation of other obligate intracellular bacteria and their potential applications to studying A. phagocytophilum.
UNLOCKING THE DOORS: A. phagocytophilum cellular invasion
A. phagocytophilum must attach to and enter host cells in order to survive (Figure 1). This process is facilitated by multiple bacterial adhesins/invasins that cooperatively recognize host cell receptors and initiate signaling cascades to promote pathogen internalization.
Figure 1.
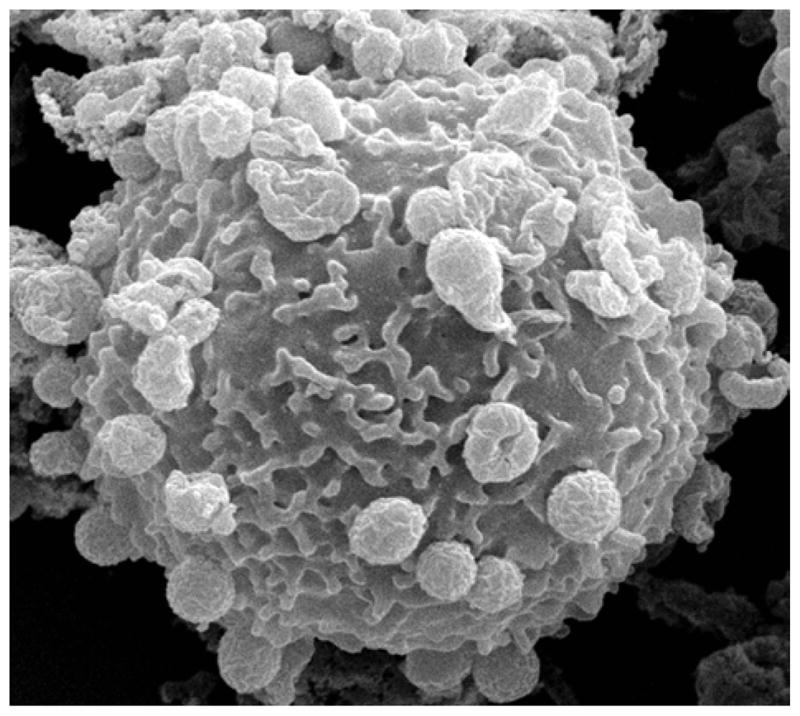
Scanning electron micrograph of a human neutrophil with numerous A. phagocytophilum bacteria attached to the host cell surface.
Host cell receptors
The most well characterized A. phagocytophilum receptor is P-selectin glycoprotein ligand-1 (PSGL-1). Engaging PSGL-1 is critical for infection of human neutrophils, bone marrow progenitors, and promyelocytic HL-60 cells [5, 6] and initiates a Syk- and ROCK1-dependent signaling cascade that promotes bacterial uptake [7]. PSGL-1 is capped by an O-glycan that is terminally decorated with sialyl Lewis x, a tetrasaccharide that includes α2,3-sialic acid and α1,3-fucose [8]. The bacterium cooperatively binds three structural determinants of human PSGL-1: (i) PSGL-1 N-terminal peptide, (ii) α1,3-fucose of sLex, and (iii) α2,3-sialic acid of sLex [9]. Binding to α2,3-sialic acid promotes cellular entry, but is dispensable for adhesion whereas recognition of PSGL-1 and α1,3-fucose are important for binding and invasion [5, 6, 10]. A. phagocytophilum infection of murine neutrophils does not involve PSGL-1 because murine PSGL-1 lacks the DFLPE peptide that is found in the human PSGL-1 N-terminus and is critical for A. phagocytophilum binding [9, 10]. α2,3-sialic acid and α1,3-fucose are important and necessary, respectively, for A. phagocytophilum infection of murine neutrophils [10]. α1,3-fucose but not sialic acid is essential for A. phagocytophilum to colonize ixodid ticks [11]. Therefore, α1,3-fucose is a unifying determinant that A. phagocytophilum targets to infect its natural murine and arthropod reservoirs and accidental human hosts.
A. phagocytophilum adhesion and invasion also occur through PSGL-1-independent routes that involve β2 integrin and lipid rafts. Differences in receptor binding occur when A. phagocytophilum-neutrophil interactions are examined under shear flow conditions similar to those in a blood vessel [12]. Whereas the pathogen solely engages PSGL-1 under static conditions [5, 6, 13], it binds both PSGL-1 and β2 integrin under flow [12]. Lipid rafts are important signaling platforms and contain enriched amounts of glycophosphatidylinositol (GPI)-anchored proteins and caveolin-1. GPI-anchored proteins are required for infection, and caveolin-1 colocalizes with early A. phagocytophilum vacuoles, suggesting that the bacterium enters host cells at lipid rafts [14]. PSGL-1 is found in lipid rafts, and β2 integrin mobilization into lipid rafts has been linked to bacterial pathogenesis [15, 16]. Prolonged cultivation in α1,3-fucosyltransferase- and sialyltransferase-defective cell lines selects for A. phagocytophilum organisms that no longer rely on sLex, PSGL-1, or Syk for entry [13, 17, 18]. Whether this enriched subpopulation consists of phenotypic or genotypic variants that target β2 integrin, lipid rafts, or other receptors is unknown.
A. phagocytophilum surface proteins implicated in infection
Several A. phagocytophilum outer membrane proteins (OMPs) have been indicted in mediating attachment to and invasion of mammalian host cells. The most thoroughly dissected are OmpA and Asp14 (14-kDa A. phagocytophilum surface protein). Both are transcriptionally induced during the tick transmission bloodmeal and are upregulated when the bacterium engages sLex-capped PSGL-1, suggesting their importance in establishing infection [19, 20]. The OmpA predicted N-terminal extracellular domain, specifically amino acids 19–74, binds α2,3-sialic acid of sLex. Bacterial entry (but not attachment to host cells) is significantly impaired by OmpA antibody and is antagonized by recombinant forms of OmpA or its extracellular domain. Competitive inhibition of A. phagocytophilum binding to sLex-capped PSGL-1 by recombinant OmpA mimics the inhibition afforded by a monoclonal antibody specific for α2,3-sialic acid of sLex [20]. Asp14 is also critical for cellular invasion but not adhesion. The Asp14 invasion domain maps to the protein’s C-terminal 12 to 24 amino acids. While its receptor is unknown, it is clear that Asp14 functions in cooperation with OmpA to promote optimal A. phagocytophilum entry. Treating host cells with recombinant OmpA or Asp14 prior to bacterial addition results in a 57 to 65% reduction in infection, whereas pretreatment with both proteins results in a 90% reduction [19]. This result also implies that OmpA and Asp14 promote infection through complementary signaling pathways.
Asp55, Asp62, and APH1235 are other A. phagocytophilum proteins implicated in adhesion and invasion. Antibodies specific for Asp55 and Asp62 significantly inhibit A. phagocytophilum infection of host cells [21]. Since the Asp55 and Asp62 receptors are undefined, it is difficult to interpret if this inhibition is specific. APH1235 is induced during transmission feeding of infected ticks and when non-infectious RC bacteria convert to the infectious DC form. Reports are conflicting as to whether APH1235 is exposed on the bacterial surface [22, 23]. However, propagating A. phagocytophilum in HL-60 cell culture in the presence of APH1235 antibody significantly reduces bacterial load [22].
Msp2 (P44) is the bacterium’s major surface protein and consists of conserved N- and C-terminal domains and a central hypervariable region. The A. phagocytophilum genome carries a repertoire of 113 p44 (msp2) paralogs and a single expression site, which enables the organism to utilize gene conversion to vary the P44 (Msp2) protein that it expresses. This phenomenon and its role in antigenic variation has been reported and reviewed elsewhere [1, 4, 24–26]. Evidence suggests that P44 (Msp2) is also involved in cellular invasion. Recombinant Msp2 (P44), the specific paralog of which was not clarified in the referenced study, binds myeloid cells and thereby inhibits A. phagocytophilum attachment and internalization [27]. It also antagonizes bacterial binding to α1,3-fucosylated PSGL-1 [27], indicating that a specific paralog or perhaps a group of structurally related Msp2 (P44) proteins may recognize α1,3-fucose and/or PSGL-1 N-terminal peptide. The latter possibility is more likely since A. phagocytophilum more frequently expresses individual Msp2 (P44) paralogs during cultivation in human myeloid cells and more frequently expresses certain clades of Msp2 (P44) paralogs during infection of specific mammalian hosts [24, 28–30]. If Msp2 (P44) is involved in cellular invasion, then its receptor binding domain is presumably located in a conserved region. Alternatively, it could exist in the hypervariable region if it is a conformational binding determinant and is structurally conserved. Pretreatment of A. phagocytophilum with monoclonal antibodies specific for epitopes in the Msp2 (P44) N-terminal conserved region or the hypervariable region of a specific paralog, Msp2 (P44)-18, block bacterial binding to or replication in human myeloid cells, respectively [31].
ORDERING ROOM SERVICE: A. phagocytophilum selectively routes degradative and membrane trafficking pathways to its vacuole
Nutritional virulence, as coined by Abu Kwaik and Bumann means that, “without proper nutritional resources for survival/proliferation in the host, bacterial pathogens do not cause disease [32].” A. phagocytophilum is auxotrophic for 16 amino acids and requires cholesterol for intracellular survival [33–36]. It must not only parasitize these essential nutrients but also must route them to its organelle. A series of recent key findings have begun to illuminate A. phagocytophilum nutritional virulence strategies.
Exploiting autophagy
Autophagy is a eukaryotic cellular homeostasis process that digests unwanted intracellular objects, including damaged organelles. It also targets intracellular pathogens and is therefore an important arm of the innate immune response. Autophagosome formation is controlled by the sequential assembly of autophagy-related (ATG) proteins and is initiated by the formation of a complex that includes ATG14 and BECN1 (Beclin 1) [37]. The complex promotes formation of a structure called the omegasome, from which the phagophore forms to elongate and enclose cytoplasmic contents with the help of a pair of ubiquitin-like conjugation systems, one of which includes the protein, LC3 [37, 38].
The ApV resembles the early autophagosome based on association of double-lipid bilayer membranes, Beclin 1, and LC3. Early in infection, clumps of Beclin1 and LC3 form, signifying the induction of autophagy, and these proteins localize to the bacterial inclusion. However, rather than clear the infection, autophagy is exploited by A. phagocytophilum as a clever way to pirate amino acids. The T4SS effector, Ats-1 (A. phagocytophilum translocated substrate 1) is secreted from the organism across the inclusion membrane into the cytoplasm where it binds both Beclin 1 and ATG14 to induce omegasome formation. The isolation membrane elongates to envelope cytoplasmic content in a double-membraned, LC3-decorated autophagosome. Autophagosomes are targeted to the ApV, where they subsequently fuse to deliver autophagic body-like vesicles into the vacuole’s lumen. Supplementation of excessive amino acids partially overrides the growth inhibition of A. phagocytophilum that occurs in the presence of the autophagic pathway inhibitor, 3-methyl-adenine [39]. How the autophagic-like bodies are broken down to yield free amino acids within the ApV lumen is unclear, but it has been speculated that bacterial surface-localized proteases may be responsible. Support for this hypothesis is provided by the observation that treatment of A. phagocytophilum infected host cells with a membrane-permeable serine-protease inhibitor retards infection [39]. Interestingly, Ats-1 is a dual-function effector, as it is also imported into the mitochondria where it interferes with apoptosis induction [40].
Recent evidence suggests the involvement of mono- and polyubiquitinated proteins in autophagy [41, 42]. Inclusion bodies that are too large for degradation in the 26S proteasome are targets for selective autophagy and contain polyubiquitinated proteins and LC3 [42]. Additionally, labeling of intravacuolar bacteria with monoubiquitin has been shown to target bacteria for destruction in the autophagosome [41], though some bacteria such as Salmonella enterica subvert this process [43]. Interestingly, while the ApV is decorated with LC3, it is not decorated with polyubiquitinated proteins. Rather, ApVs stain positive for monoubiquitin in mammalian and, to a lesser extent, tick host cells. De novo bacterial protein synthesis is important for continued association of monoubiquitin with the ApV suggesting this is a bacterial-mediated process [44]. Monoubiquitinated proteins play various other roles in the cell including directing endocytic traffic [45]. Thus, whether Ap uses monoubiquitinated proteins specifically to aid in autophagosome fusion remains unknown.
Cholesterol acquisition
The A. phagocytophilum genome does not contain any genes for synthesis of lipid A or the full complement required for peptidoglycan synthesis [33, 34]. As such, the bacterium stabilizes its outer membrane by incorporating cholesterol. A lack of genes related to cholesterol synthesis or modification obligates it to hijack cholesterol from its mammalian host [34]. Indeed, exogenous cholesterol is taken up by the bacterium [34], endogenous cholesterol synthesis is upregulated in infected cells, and free (unesterified) cholesterol is enriched in the A. phagocytophilum-occupied vacuole (ApV) [35]. Both extraction of cholesterol from the membranes of host cell-free bacteria with methyl-β-cyclodextrin (MβCD) and addition of the structural but poorly functional cholesterol derivative, NBD-cholesterol, significantly reduce infectivity [34], suggesting that cholesterol is also important for A. phagocytophilum virulence.
Host leukocyte cholesterol is either synthesized in the smooth endoplasmic reticulum (ER) or acquired mostly through low-density lipoprotein receptor (LDLR)-mediated endocytosis. Culture of infected cells in lipoprotein-deficient serum, treatment of host cells with an LDLR antibody and pharmacological inhibitors of the LDLR pathway, but not inhibitors of cholesterol synthesis, significantly reduce A. phagocytophilum infection and replication, implicating LDLR-mediated endocytosis in cholesterol acquisition by the bacterium. A. phagocytophilum infection increases LDLR mRNA stability, resulting in a marked increase in LDLR protein compared to uninfected cells [35], providing a functional explanation for the increased cholesterol levels associated with infection. In LDLR-mediated endocytosis, cholesterol esters are hydrolyzed in acidified endosomes while free cholesterol is trafficked in a vesicular compartment containing the cholesterol-binding protein Niemann-Pick type C1 (NPC1) to the ER, sometimes by way of the trans-Golgi network (TGN). In the ER, excess free cholesterol is esterified and stored or trafficked back to the plasma membrane [46]. NPC1 and NPC2 are upregulated in A. phagocytophilum-infected cells and localize to and in the ApV, respectively. siRNA knockdown of NPC1 significantly reduces infection and cholesterol levels in purified bacteria, indicating the importance of this pathway in cholesterol acquisition. Treatment of host cells with U18666A, a compound that blocks free cholesterol egress from the endosomal pathway to the ER or TGN to produce an NPC1-deficient phenotype, impedes infection. During infection and subsequent increase in intracellular cholesterol, ER-resident sterol regulatory element-binding proteins (SREBPs), key transcriptional regulators maintaining cholesterol homeostasis in the cell, are not activated, suggesting that the ApV intercepts NPC1 vesicles downstream of the U18666A target site and upstream of their delivery to the ER [36]. Furthermore, siRNA knockdown of VAMP4, a protein that is important for delivery of NPC1 vesicles to the TGN and localizes to the ApV [46], only marginally reduces infection and does not alter A. phagocytophilum cholesterol uptake [36]. These studies indicate a novel mechanism whereby the pathogen hijacks free cholesterol from NPC1 vesicles en route to the TGN and/or ER. Lipid droplets may be an additional cholesterol source for A. phagocytophilum [35]. Perlipin, a lipid droplet-associated phosphoprotein that is important for lipolysis and cholesterol synthesis, is transcriptionally upregulated in A. phagocytophilum infected cells and siRNA knockdown of perilipin inhibits bacterial infection and growth [47].
A. phagocytophilum’s dependence on cholesterol indicates that patients with hypercholesterolemia may experience more severe infection [48]. Indeed, a high cholesterol diet facilitates increased infection of apolipoprotein E-deficient (apoE−/−) mice [49]. HGA is more commonly associated with older individuals, contrary to the median age of persons inflicted with other tick-borne pathogens. Older individuals generally have weakened immune systems and elevated levels of blood cholesterol. Thus, A. phagocytophilum’s cholesterol dependence may at least partially explain the increase in median age. It has been speculated that therapeutics aimed at lowering plasma cholesterol levels could aid control of HGA in this population [35, 48, 49].
Selective targeting of Rab GTPases
Rab GTPases are master regulators of membrane dynamics on organelles. The Rab family consists of nearly 70 members, each of which is involved in controlling a defined vesicular transport step [50, 51]. A. phagocytophilum selectively targets a subset of Rab GTPases to its vacuole [52]. Doing so provides a means of molecular camouflage and likely contributes to the bacterium satisfying its obligatory intracellular nutritional requirements for amino acids and cholesterol. It also conceivably provides a continuous supply of host membrane material to enable of expansion of the vacuolar membrane such that it can accommodate the growing intravacuolar bacterial population. Rab1, Rab4A, Rab10, Rab11A, Rab14, Rab22A, and Rab35 localize to the ApV [52] (Table 1). These Rabs direct vesicular traffic associated with clathrin-dependent endocytic recycling (Rab4A, Rab35) [53], clathrin-independent endocytic recycling (Rab10, Rab11A, Rab14, Rab22A) [51, 53], the endoplasmic reticulum (ER; Rab1, Rab10) [54], and the trans-Golgi network (TGN; Rab10, Rab11A, Rab14, Rab22A) [50, 51, 53]. This phenomenon is driven by nascent bacterial protein synthesis and is critical for the ApV to evade lysosomal fusion [52].
Table 1.
Rab GTPases that are recruited to the A. phagocytophilum-occupied vacuole
| Rab GTPasea | Site(s) of Actionb | Transport Function(s) |
|---|---|---|
| Rab1 | ERc | ER to Golgi, Golgi to ER, IC to PM |
| Rab4A | Clathrin-dependent recycling endosomes | Rapid endocytic recycling |
| Rab10 | ER Clathrin-independent recycling endosomes TGN |
Dynamic ER tubules Endocytic recycling TGN to PM TGN to RE |
| Rab11A | Clathrin-independent recycling endosomes TGN |
Endocytic recycling TGN to PM |
| Rab14 | Clathrin-independent recycling endosomes TGN |
Endocytic recycling TGN to EE |
| Rab22A | Clathrin-independent recycling endosomes TGN |
Endocytic recycling EE to TGN |
| Rab35 | Clathrin-dependent recycling endosomes | Endocytic recycling |
Rab GTPase localization data from Huang et al (2010)
Site of Action and Transport Function data summarized from references by Grant & Donaldson (2009), Stenmark (2009), Liu & Storrie (2012), and English & Voeltz (2013)
ER, endoplasmic reticulum; IC, pre-Golgi intermediate compartment; PM, plasma membrane; RE, recycling endosomes; TGN, trans-Golgi network; EE, early endosome
From the exclusivity of the pathways that Rab1 and Rab4A regulate [53], it is clear that the ApV intercepts ER traffic and clathrin-dependent recycling endosomes. Rab35 localizes with Rab4 on clathrin-dependent recycling endosomes and is found along with Rab10 and Rab22A on tubular recycling endosomes of the clathrin-independent recycling pathway [53]. Since the frequency of Rab35 localization to the ApV both pronouncedly exceeds that of Rab4 and is comparable to the frequencies by which Rab10 and Rab22A associate with the ApV [52], it can be inferred that the ApV also co-opts clathrin-independent endocytic recycling. Proteins involved in amino acid uptake and cholesterol both traffic through recycling endosomes [53].
Rab10, Rab11A, Rab14, and Rab22A regulate endocytic recycling and also mediate transport of vesicles from (Rab10, Rab11A, Rab14) and to (Rab22A) the TGN [53]. VAMP4 and syntaxin-16, two SNARE proteins that are important for cargo transport between recycling endosomes and the TGN [46], also localize to the ApV membrane [36]. Thus, the ApV may intercept RE-to-TGN traffic and/or may hijack TGN-derived vesicles. Neither Rab10 and Rab14 recruitment to the ApV nor A. phagocytophilum growth is impaired by the Golgi apparatus destabilizing agent, Brefeldin A [52], which implies that an intact Golgi apparatus is not required for either phenomena. Indeed, cis-Golgi fragmentation has been observed in cells that are heavily infected with A. phagocytophilum [36], suggesting that the pathogen itself promotes Golgi destabilization, at least under conditions of high bacterial load. Golgi fragments, particularly those derived from the TGN, would be rich in cholesterol. Chlamydia trachomatis induces Golgi fragmentation and routes lipid-rich TGN-derived vesicles to its inclusion, a process that is important for chlamydial survival and generating infectious progeny [55]. Thus, thorough examination of whether A. phagocytophilum targets the TGN is warranted.
Rab proteins cycle between an inactive cytosolic GDP-bound form and an active GTP-bound form that associates with organelle membranes to regulate membrane fusion events [51]. In line with normal Rab membrane cycling, GTP- but not GDP-bound forms of Rab1, Rab4A, and Rab11A [52], as well as Rab14, Rab22A, and Rab35 (Huang and Carlyon, unpublished observations) associate with the ApV. Strikingly, GTP-bound, GDP-bound, and guanine nucleotide-free forms of Rab10 localize to the ApV with comparable efficiencies, an observation that suggests an A. phagocytophilum protein may target Rab10 to the ApV [52].
HELP IS ON THE WAY: Promising tools for elucidating A. phagocytophilum gene function
A major hindrance to fully characterizing A. phagocytophilum virulence mechanisms is the bacterium’s intractability to traditional genetic manipulation methods. For instance, due to its obligate intracellular nature knocking out a gene that is essential for cellular invasion or intracellular survival would prohibit the mutant’s selection. In addition, its reduced genome size could affect both the stability and length of clonal passages of genomic insertions. However, in line with the proverbial saying, “necessity is the mother of invention,” scientists have developed methods that circumvent and even nullify the challenges that obligate intracellular bacteria present to genetic manipulation. Here we summarize what has been used to study A. phagocytophilum and discuss additional genetic tools developed through the study of other obligate intracellular bacteria that would potentially benefit A. phagocytophilum research. For a more detailed review of these methods, see Beare et al [56].
Tools used to study A. phagocytophilum gene function
Nearly a decade ago, it was demonstrated that A. phagocytophilum could be transformed using the Himar1 transposon system when cassettes encoding fluorescent proteins and antibiotic resistance were stably inserted into the bacterium’s chromosome [57, 58]. Since then, the Himar1 system has been applied to study other obligate intracellular bacteria including A. marginale, Ehrlichia chaffeensis, Rickettsia prowazekii, and Coxiella burnetii [57, 59–61]. Himar1 cassette-mediated disruption of the dihydrolipoamide dehydrogenase 1 (LPDA1) gene (aph0065) enabled researchers to identify LPDA1 as an important A. phagocytophilum immunopathological molecule [62]. Transcriptional profiling of an A. marginale Himar1 transformant that exhibited a slow growth phenotype revealed a gene network that was transcriptionally altered in trans by the Himar1 insertion [63]. While both examples highlight the Himar1 transposon system as an important tool for the field and a first step toward the development of a system for studying Anaplasma spp. gene function, the latter example also denotes that, because of the random nature of Himar1 transposition, unwanted off target effects can result. Moreover, because Himar1 cannot be used for site-specific gene disruption, libraries of Himar1 mutants must be laboriously screened to first identify a discernable phenotype, next the mutant must be sequenced to identify the disrupted gene, and only then can testable hypotheses in regards to the disrupted gene’s function be formed. Furthermore, random insertion into genomic regions that are critical for infection of host cells, replication, or genome maintenance would be missed because the resulting transgenic populations would be unable to invade or survive in host cells and would consequently die or would be outgrown by host cells and other more fit A. phagocytophilum organisms.
Tools developed through study of other obligate intracellular bacteria
Systems that enable stable, site-specific insertions into the chromosome or that effectively deliver antisense oligonucleotides to modulate bacterial gene expression are needed to efficiently elucidate A. phagocytophilum gene function. Several promising tools developed for studying other obligate intracellular bacteria satisfy these criteria. Homologous recombination by the modified mobile group II intron (TargeTron) method was used to insert cassettes into constitutive and differentially expressed genes and intergenic regions of the Ehrlichia chaffeensis chromosome [60]. Though the mutants survived for only eight days, this study proved that site-specific mutagenesis of Anaplasmataceae bacteria is achievable. Self-replicating plasmids for transforming A. phagocytophilum would enable genetic manipulation of the bacterium without targeting or disrupting the bacterial genome itself and would potentially generate more stable transformants. Like A. phagocytophilum, plasmids have not been found in R. prowazekii. Yet, researchers were able to transform R. prowazekii with a replicating plasmid from the less pathogenic species, R. amblyomii [64], marking the first plasmid to be stably maintained in R. prowazekii. A shuttle plasmid for transforming C. trachomatis has also been developed [65].
The formulation of an axenic growth medium for C. burnetii has greatly accelerated the development of genetic techniques for this organism [66]. Because C. burnetii colonies can be grown on semi-solid media, isogenic clones can be selected, enabling recovery of mutants carrying site-directed insertions, inducible expression systems, and targeted gene deletions [59, 67]. Host cell-free growth has also recently been achieved for C. trachomatis [68], which is a harbinger that, once the nutritional requirements of an obligate intracellular bacterium are understood, a medium that supports axenic growth can be developed.
A very promising method for rapidly modulating obligate intracellular bacterial gene expression is to use polyamidoamine (PAMAM) dendrimers to deliver DNA into bacteria that are actively growing inside eukaryotic host cells [69, 70]. Indeed, highly efficient knockdown of gene expression was achieved within six hours of incubation of PAMAM dendrimer-antisense oligonucleotide complexes with C. trachomatis infected cells [70]. This approach is especially appealing for studying genes that are critical for cellular invasion or intracellular survival in A. phagocytophilum and other obligate intracellular bacteria because it circumvents having to generate a mutation that is potentially fatal or would result in less fit organisms that could not be recovered. Dendrimers were also used to efficiently deliver a 7.5-kb plasmid into C. pneumoniae inclusions inside host cells. The chlamydiae took up the plasmid and maintained the plasmid for five passages, resulting in heterologous GFP-expressing organisms that expressed all the plasmid’s genes [69]. It has yet to be demonstrated whether an isogenic population can be obtained following dendrimer-mediated transformation, but given its high efficiency, it is certainly plausible that clonal organisms could be isolated from a transformant population via limiting dilution.
CONCLUSIONS
The invasion and nutrient parasitism mechanisms described here highlight many of A. phagocytophilum’s clever virulence tactics. Though these strategies have been thoroughly researched, knockout-complementation and other genetic approaches will provide proof of principle, examine non-redundancy of function, and further fine-tune understanding of these and other virulence mechanisms. To accomplish this, it will be critical to explore new tools, such as those that have been effectively used to study other obligate intracellular bacterial pathogens. Both the development of an axenic media for cell-free cultivation of A. phagocytophilum and using dendrimers to knockdown target gene expression in actively growing organisms are particularly promising.
Figure 2.

A. phagocytophilum cellular invasion. The infectious dense-cored form of the bacterium utilizes multiple surface proteins to cooperatively bind PSGL-1 at an N- terminal amino acid sequence and α2,3-sialic acid and α1,3-fucose of the sLex tetrasaccharide that caps PSGL-1. Binding to PSGL-1 initiates a signaling cascade that involves Syk and phosphorylation of ROCK1 and promotes bacterial internalization. A. phagocytophilum also binds β2-integrin, but the relevance of this interaction is only detectable under conditions of shear flow. The pathogen binds at lipid rafts enriched for caveolin-1. Bacterial adherence to and infection of host cells from which GPI-anchored proteins have been cleaved is significantly impaired. OmpA, specifically a domain contained with amino acids 19–74, binds to α2,3-sialic acid and is the only A. phagocytophilum invasin for which a receptor is known. Asp14, specifically a domain contained with amino acids 100–124, is also important for invasion. Msp2 (P44), or a least a paralog thereof, may be involved in binding to PSGL-1. ???, unidentified A. phagocytophilum adhesins/invasins or host cell receptors.
Acknowledgments
We apologize to our colleagues whose papers we could not cite due to space limitation. This work was supported by funding from NIH/NIAID grant 2R56 AI067283. Scanning electron microscopy was performed in the VCU Department of Anatomy and Neurobiology Microscopy Facility, supported, in part, with funding from NIH-NINDS Center core grant 5P30NS047463.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Carlyon JA. Establishing intracellular infection: modulation of host cell functions (Anaplasmataceae) In: Palmer GH, Azad A, editors. Intracellular Pathogens II: Rickettsiales. ASM Press; Washington, D. C: 2012. [Google Scholar]
- 2.Rikihisa Y. Anaplasma phagocytophilum and Ehrlichia chaffeensis: subversive manipulators of host cells. Nature reviews Microbiology. 2010;8:328–339. doi: 10.1038/nrmicro2318. [DOI] [PubMed] [Google Scholar]
- 3.Chen SM, Dumler JS, Bakken JS, Walker DH. Identification of a granulocytotropic Ehrlichia species as the etiologic agent of human disease. Journal of clinical microbiology. 1994;32:589–595. doi: 10.1128/jcm.32.3.589-595.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rikihisa Y. Mechanisms of obligatory intracellular infection with Anaplasma phagocytophilum. Clinical Microbiology Reviews. 2011;24:469–489. doi: 10.1128/CMR.00064-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goodman JL, Nelson CM, Klein MB, Hayes SF, Weston BW. Leukocyte infection by the granulocytic ehrlichiosis agent is linked to expression of a selectin ligand. J Clin Invest. 1999;103:407–412. doi: 10.1172/JCI4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herron MJ, Nelson CM, Larson J, Snapp KR, Kansas GS, Goodman JL. Intracellular parasitism by the human granulocytic ehrlichiosis bacterium through the P-selectin ligand, PSGL-1. Science. 2000;288:1653–1656. doi: 10.1126/science.288.5471.1653. [DOI] [PubMed] [Google Scholar]
- 7.Thomas V, Fikrig E. Anaplasma phagocytophilum specifically induces tyrosine phosphorylation of ROCK1 during infection. Cellular microbiology. 2007;9:1730–1737. doi: 10.1111/j.1462-5822.2007.00908.x. [DOI] [PubMed] [Google Scholar]
- 8.Zarbock A, Ley K, McEver RP, Hidalgo A. Leukocyte ligands for endothelial selectins: specialized glycoconjugates that mediate rolling and signaling under flow. Blood. 2011;118:6743–6751. doi: 10.1182/blood-2011-07-343566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yago T, Leppanen A, Carlyon JA, Akkoyunlu M, Karmakar S, Fikrig E, Cummings RD, McEver RP. Structurally distinct requirements for binding of P-selectin glycoprotein ligand-1 and sialyl Lewis x to Anaplasma phagocytophilum and P-selectin. J Biol Chem. 2003;278:37987–37997. doi: 10.1074/jbc.M305778200. [DOI] [PubMed] [Google Scholar]
- 10.Carlyon JA, Akkoyunlu M, Xia L, Yago T, Wang T, Cummings RD, McEver RP, Fikrig E. Murine neutrophils require alpha1,3-fucosylation but not PSGL-1 for productive infection with Anaplasma phagocytophilum. Blood. 2003;102:3387–3395. doi: 10.1182/blood-2003-02-0621. [DOI] [PubMed] [Google Scholar]
- 11.Pedra JH, Narasimhan S, Rendic D, DePonte K, Bell-Sakyi L, Wilson IB, Fikrig E. Fucosylation enhances colonization of ticks by Anaplasma phagocytophilum. Cellular microbiology. 2010;12:1222–1234. doi: 10.1111/j.1462-5822.2010.01464.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schaff UY, Trott KA, Chase S, Tam K, Johns JL, Carlyon JA, Genetos DC, Walker NJ, Simon SI, Borjesson DL. Neutrophils exposed to A. phagocytophilum under shear stress fail to fully activate, polarize, and transmigrate across inflamed endothelium. Am J Physiol Cell Physiol. 2010;299:C87–96. doi: 10.1152/ajpcell.00165.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reneer DV, Kearns SA, Yago T, Sims J, Cummings RD, McEver RP, Carlyon JA. Characterization of a sialic acid- and P-selectin glycoprotein ligand-1-independent adhesin activity in the granulocytotropic bacterium Anaplasma phagocytophilum. Cellular microbiology. 2006;8:1972–1984. doi: 10.1111/j.1462-5822.2006.00764.x. [DOI] [PubMed] [Google Scholar]
- 14.Lin M, Rikihisa Y. Obligatory intracellular parasitism by Ehrlichia chaffeensis and Anaplasma phagocytophilum involves caveolae and glycosylphosphatidylinositol-anchored proteins. Cellular microbiology. 2003;5:809–820. doi: 10.1046/j.1462-5822.2003.00322.x. [DOI] [PubMed] [Google Scholar]
- 15.Bumba L, Masin J, Fiser R, Sebo P. Bordetella adenylate cyclase toxin mobilizes its beta2 integrin receptor into lipid rafts to accomplish translocation across target cell membrane in two steps. PLoS Pathog. 2010;6:e1000901. doi: 10.1371/journal.ppat.1000901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abbal C, Lambelet M, Bertaggia D, Gerbex C, Martinez M, Arcaro A, Schapira M, Spertini O. Lipid raft adhesion receptors and Syk regulate selectin-dependent rolling under flow conditions. Blood. 2006;108:3352–3359. doi: 10.1182/blood-2006-04-013912. [DOI] [PubMed] [Google Scholar]
- 17.Reneer DV, Troese MJ, Huang B, Kearns SA, Carlyon JA. Anaplasma phagocytophilum PSGL-1-independent infection does not require Syk and leads to less-efficient AnkA delivery. Cellular microbiology. 2008 doi: 10.1111/j.1462-5822.2008.01168.x. In press. [DOI] [PubMed] [Google Scholar]
- 18.Sarkar M, Reneer DV, Carlyon JA. Sialyl-Lewis x-independent infection of human myeloid cells by Anaplasma phagocytophilum strains HZ and HGE1. Infect Immun. 2007;75:5720–5725. doi: 10.1128/IAI.00905-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kahlon A, Ojogun N, Ragland SA, Troese MJ, Seidman D, Ottens AK, Mastronunzio JE, Truchan HK, Walker NJ, Borjesson DL, Fikrig E, Carlyon JA. Anaplasma phagocytophilum Asp14 (APH_0248) is an invasin that interacts with mammalian host cells via its C-terminus to facilitate infection. Infection and Immunity. 2013 doi: 10.1128/IAI.00932-12. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ojogun N, Kahlon A, Ragland SA, Troese MJ, Mastronunzio JE, Walker NJ, Viebrock L, Thomas RJ, Borjesson DL, Fikrig E, Carlyon JA. Anaplasma phagocytophilum outer membrane protein A interacts with sialylated glycoproteins to promote infection of mammalian host cells. Infection and Immunity. 2012 doi: 10.1128/IAI.00654-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ge Y, Rikihisa Y. Identification of novel surface proteins of Anaplasma phagocytophilum by affinity purification and proteomics. J Bacteriol. 2007;189:7819–7828. doi: 10.1128/JB.00866-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mastronunzio JE, Kurscheid S, Fikrig E. Postgenomic analyses reveal development of infectious Anaplasma phagocytophilum during transmission from ticks to mice. Journal of bacteriology. 2012;194:2238–2247. doi: 10.1128/JB.06791-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Troese MJ, Kahlon A, Ragland SA, Ottens AK, Ojogun N, Nelson KT, Walker NJ, Borjesson DL, Carlyon JA. Proteomic analysis of Anaplasma phagocytophilum during infection of human myeloid cells identifies a protein that is pronouncedly upregulated on the infectious dense-cored cell. Infection and Immunity. 2011;79:4696–4707. doi: 10.1128/IAI.05658-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Foley JE, Nieto NC, Barbet A, Foley P. Antigen diversity in the parasitic bacterium Anaplasma phagocytophilum arises from selectively-represented, spatially clustered functional pseudogenes. PloS one. 2009;4:e8265. doi: 10.1371/journal.pone.0008265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rejmanek D, Foley P, Barbet A, Foley J. Antigen variability in Anaplasma phagocytophilum during chronic infection of a reservoir host. Microbiology. 2012;158:2632–2641. doi: 10.1099/mic.0.059808-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rejmanek D, Foley P, Barbet A, Foley J. Evolution of antigen variation in the tick-borne pathogen Anaplasma phagocytophilum. Molecular biology and evolution. 2012;29:391–400. doi: 10.1093/molbev/msr229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park J, Choi KS, Dumler JS. Major surface protein 2 of Anaplasma phagocytophilum facilitates adherence to granulocytes. Infect Immun. 2003;71:4018–4025. doi: 10.1128/IAI.71.7.4018-4025.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sarkar M, Troese MJ, Kearns SA, Yang T, Reneer DV, Carlyon JA. Anaplasma phagocytophilum MSP2(P44)-18 predominates and is modified into multiple isoforms in human myeloid cells. Infect Immun. 2008;76:2090–2098. doi: 10.1128/IAI.01594-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Troese MJ, Sarkar M, Galloway NL, Thomas RJ, Kearns SA, Reneer DV, Yang T, Carlyon JA. Differential expression and glycosylation of anaplasma phagocytophilum major surface protein 2 paralogs during cultivation in sialyl Lewis x-deficient host cells. Infect Immun. 2009;77:1746–1756. doi: 10.1128/IAI.01530-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang X, Rikihisa Y, Lai TH, Kumagai Y, Zhi N, Reed SM. Rapid sequential changeover of expressed p44 genes during the acute phase of Anaplasma phagocytophilum infection in horses. Infect Immun. 2004;72:6852–6859. doi: 10.1128/IAI.72.12.6852-6859.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang X, Kikuchi T, Rikihisa Y. Two monoclonal antibodies with defined epitopes of P44 major surface proteins neutralize Anaplasma phagocytophilum by distinct mechanisms. Infection and Immunity. 2006;74:1873–1882. doi: 10.1128/IAI.74.3.1873-1882.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abu Kwaik Y, Bumann D. Microbial quest for food in vivo: ‘Nutritional virulence’ as an emerging paradigm. Cellular microbiology. 2013 doi: 10.1111/cmi.12138. [DOI] [PubMed] [Google Scholar]
- 33.Dunning Hotopp JC, Lin M, Madupu R, Crabtree J, Angiuoli SV, Eisen JA, Seshadri R, Ren Q, Wu M, Utterback TR, Smith S, Lewis M, Khouri H, Zhang C, Niu H, Lin Q, Ohashi N, Zhi N, Nelson W, Brinkac LM, Dodson RJ, Rosovitz MJ, Sundaram J, Daugherty SC, Davidsen T, Durkin AS, Gwinn M, Haft DH, Selengut JD, Sullivan SA, Zafar N, Zhou L, Benahmed F, Forberger H, Halpin R, Mulligan S, Robinson J, White O, Rikihisa Y, Tettelin H. Comparative genomics of emerging human ehrlichiosis agents. PLoS genetics. 2006;2:e21. doi: 10.1371/journal.pgen.0020021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin M, Rikihisa Y. Ehrlichia chaffeensis and Anaplasma phagocytophilum lack genes for lipid A biosynthesis and incorporate cholesterol for their survival. Infect Immun. 2003;71:5324–5331. doi: 10.1128/IAI.71.9.5324-5331.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xiong Q, Lin M, Rikihisa Y. Cholesterol-dependent anaplasma phagocytophilum exploits the low-density lipoprotein uptake pathway. PLoS Pathog. 2009;5:e1000329. doi: 10.1371/journal.ppat.1000329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xiong Q, Rikihisa Y. Subversion of NPC1 pathway of cholesterol transport by Anaplasma phagocytophilum. Cellular microbiology. 2012;14:560–576. doi: 10.1111/j.1462-5822.2011.01742.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baxt LA, Garza-Mayers AC, Goldberg MB. Bacterial subversion of host innate immune pathways. Science. 2013;340:697–701. doi: 10.1126/science.1235771. [DOI] [PubMed] [Google Scholar]
- 38.Lilienbaum A. Relationship between the proteasomal system and autophagy. International journal of biochemistry and molecular biology. 2013;4:1–26. [PMC free article] [PubMed] [Google Scholar]
- 39.Niu H, Xiong Q, Yamamoto A, Hayashi-Nishino M, Rikihisa Y. Autophagosomes induced by a bacterial Beclin 1 binding protein facilitate obligatory intracellular infection. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:20800–20807. doi: 10.1073/pnas.1218674109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Niu H, Kozjak-Pavlovic V, Rudel T, Rikihisa Y. Anaplasma phagocytophilum Ats-1 is imported into host cell mitochondria and interferes with apoptosis induction. PLoS Pathog. 2010;6:e1000774. doi: 10.1371/journal.ppat.1000774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dupont N, Temime-Smaali N, Lafont F. How ubiquitination and autophagy participate in the regulation of the cell response to bacterial infection. Biology of the cell/under the auspices of the European Cell Biology Organization. 2010;102:621–634. doi: 10.1042/BC20100101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kraft C, Peter M, Hofmann K. Selective autophagy: ubiquitin-mediated recognition and beyond. Nature cell biology. 2010;12:836–841. doi: 10.1038/ncb0910-836. [DOI] [PubMed] [Google Scholar]
- 43.Mesquita FS, Thomas M, Sachse M, Santos AJ, Figueira R, Holden DW. The Salmonella deubiquitinase SseL inhibits selective autophagy of cytosolic aggregates. PLoS Pathog. 2012;8:e1002743. doi: 10.1371/journal.ppat.1002743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang B, Ojogun N, Ragland SA, Carlyon JA. Monoubiquitinated proteins decorate the Anaplasma phagocytophilum-occupied vacuolar membrane. FEMS immunology and medical microbiology. 2012;64:32–41. doi: 10.1111/j.1574-695X.2011.00873.x. [DOI] [PubMed] [Google Scholar]
- 45.Collins CA, Brown EJ. Cytosol as battleground: ubiquitin as a weapon for both host and pathogen. Trends Cell Biol. 2010;20:205–213. doi: 10.1016/j.tcb.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 46.Urano Y, Watanabe H, Murphy SR, Shibuya Y, Geng Y, Peden AA, Chang CC, Chang TY. Transport of LDL-derived cholesterol from the NPC1 compartment to the ER involves the trans-Golgi network and the SNARE protein complex. Proc Natl Acad Sci U S A. 2008;105:16513–16518. doi: 10.1073/pnas.0807450105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Manzano-Roman R, Almazan C, Naranjo V, Blouin EF, Kocan KM, de la Fuente J. Expression of perilipin in human promyelocytic cells in response to Anaplasma phagocytophilum infection results in modified lipid metabolism. Journal of medical microbiology. 2008;57:159–163. doi: 10.1099/jmm.0.47504-0. [DOI] [PubMed] [Google Scholar]
- 48.Rikihisa Y. Molecular events involved in cellular invasion by Ehrlichia chaffeensis and Anaplasma phagocytophilum. Vet Parasitol. 2010;167:155–166. doi: 10.1016/j.vetpar.2009.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xiong Q, Wang X, Rikihisa Y. High-cholesterol diet facilitates Anaplasma phagocytophilum infection and up-regulates macrophage inflammatory protein-2 and CXCR2 expression in apolipoprotein E-deficient mice. J Infect Dis. 2007;195:1497–1503. doi: 10.1086/514819. [DOI] [PubMed] [Google Scholar]
- 50.Liu S, Storrie B. Are Rab proteins the link between Golgi organization and membrane trafficking? Cellular and molecular life sciences: CMLS. 2012;69:4093–4106. doi: 10.1007/s00018-012-1021-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol. 2009;10:513–525. doi: 10.1038/nrm2728. [DOI] [PubMed] [Google Scholar]
- 52.Huang B, Hubber A, McDonough JA, Roy CR, Scidmore MA, Carlyon JA. The Anaplasma phagocytophilum-occupied vacuole selectively recruits Rab-GTPases that are predominantly associated with recycling endosomes. Cellular microbiology. 2010;12:1292–1307. doi: 10.1111/j.1462-5822.2010.01468.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grant BD, Donaldson JG. Pathways and mechanisms of endocytic recycling. Nat Rev Mol Cell Biol. 2009;10:597–608. doi: 10.1038/nrm2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.English AR, Voeltz GK. Rab10 GTPase regulates ER dynamics and morphology. Nature cell biology. 2013;15:169–178. doi: 10.1038/ncb2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Heuer D, Rejman Lipinski A, Machuy N, Karlas A, Wehrens A, Siedler F, Brinkmann V, Meyer TF. Chlamydia causes fragmentation of the Golgi compartment to ensure reproduction. Nature. 2009;457:731–735. doi: 10.1038/nature07578. [DOI] [PubMed] [Google Scholar]
- 56.Beare P, Sandoz K, Omsland A, Rockey D, Heinzen R. Advances in Genetic Manipulation of Obligate Intracellular Bacterium. Frontiers in Microbiology. 2011;2 doi: 10.3389/fmicb.2011.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Felsheim RF, Chavez AS, Palmer GH, Crosby L, Barbet AF, Kurtti TJ, Munderloh UG. Transformation of Anaplasma marginale. Vet Parasitol. 2010;167:167–174. doi: 10.1016/j.vetpar.2009.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Felsheim RF, Herron MJ, Nelson CM, Burkhardt NY, Barbet AF, Kurtti TJ, Munderloh UG. Transformation of Anaplasma phagocytophilum. BMC Biotechnol. 2006;6:42. doi: 10.1186/1472-6750-6-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Beare PA, Gilk SD, Larson CL, Hill J, Stead CM, Omsland A, Cockrell DC, Howe D, Voth DE, Heinzen RA. Dot/Icm type IVB secretion system requirements for Coxiella burnetii growth in human macrophages. MBio. 2011;2:e00175–00111. doi: 10.1128/mBio.00175-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cheng C, Nair AD, Indukuri VV, Gong S, Felsheim RF, Jaworski D, Munderloh UG, Ganta RR. Targeted and random mutagenesis of Ehrlichia chaffeensis for the identification of genes required for in vivo infection. PLoS Pathog. 2013;9:e1003171. doi: 10.1371/journal.ppat.1003171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Welch MD, Reed SC, Lamason RL, Serio AW. Expression of an epitope-tagged virulence protein in Rickettsia parkeri using transposon insertion. PloS one. 2012;7:e37310. doi: 10.1371/journal.pone.0037310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen G, Severo MS, Sakhon OS, Choy A, Herron MJ, Felsheim RF, Wiryawan H, Liao J, Johns JL, Munderloh UG, Sutterwala FS, Kotsyfakis M, Pedra JH. Anaplasma phagocytophilum dihydrolipoamide dehydrogenase 1 affects host-derived immunopathology during microbial colonization. Infect Immun. 2012;80:3194–3205. doi: 10.1128/IAI.00532-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pierle SA, Hammac GK, Palmer GH, Brayton KA. Transcriptional pathways associated with the slow growth phenotype of transformed Anaplasma marginale. BMC Genomics. 2013;14:272. doi: 10.1186/1471-2164-14-272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wood DO, Hines A, Tucker AM, Woodard A, Driskell LO, Burkhardt NY, Kurtti TJ, Baldridge GD, Munderloh UG. Establishment of a replicating plasmid in Rickettsia prowazekii. PloS one. 2012;7:e34715. doi: 10.1371/journal.pone.0034715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang Y, Kahane S, Cutcliffe LT, Skilton RJ, Lambden PR, Clarke IN. Development of a transformation system for Chlamydia trachomatis: restoration of glycogen biosynthesis by acquisition of a plasmid shuttle vector. PLoS Pathog. 2011;7:e1002258. doi: 10.1371/journal.ppat.1002258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Omsland A, Cockrell DC, Howe D, Fischer ER, Virtaneva K, Sturdevant DE, Porcella SF, Heinzen RA. Host cell-free growth of the Q fever bacterium Coxiella burnetii. Proc Natl Acad Sci U S A. 2009;106:4430–4434. doi: 10.1073/pnas.0812074106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Beare PA, Larson CL, Gilk SD, Heinzen RA. Two systems for targeted gene deletion in Coxiella burnetii. Applied and environmental microbiology. 2012;78:4580–4589. doi: 10.1128/AEM.00881-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Omsland A, Sager J, Nair V, Sturdevant DE, Hackstadt T. Developmental stage-specific metabolic and transcriptional activity of Chlamydia trachomatis in an axenic medium. Proc Natl Acad Sci U S A. 2012;109:19781–19785. doi: 10.1073/pnas.1212831109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gerard HC, Mishra MK, Mao G, Wang S, Hali M, Whittum-Hudson JA, Kannan RM, Hudson AP. Dendrimer-enabled DNA delivery and transformation of Chlamydia pneumoniae. Nanomedicine: nanotechnology, biology, and medicine. 2013 doi: 10.1016/j.nano.2013.04.004. [DOI] [PubMed] [Google Scholar]
- 70.Mishra MK, Gerard HC, Whittum-Hudson JA, Hudson AP, Kannan RM. Dendrimer-enabled modulation of gene expression in Chlamydia trachomatis. Molecular pharmaceutics. 2012;9:413–421. doi: 10.1021/mp200512f. [DOI] [PubMed] [Google Scholar]