

Abstract
The serotonin transporter (SERT) is the primary target for antidepressant drugs. The existence of a high affinity primary orthosteric binding site (S1) and a low affinity secondary site (S2) has been described and their relation to antidepressant pharmacology has been debated. Herein, structural modifications to the N-, 4, 5, and 4’-positions of (±)citalopram (1) are reported. All of the analogues were SERT-selective and demonstrated that steric bulk was tolerated at the SERT S1 site, including two dimeric ligands (15 and 51.) In addition, 8 analogues were identified with similar potencies to S-1 for decreasing the dissociation of [3H]S-1 from the S1 site, via allosteric modulation at S2. Both dimeric compounds had similar affinities for the SERT S1 site (Ki=19.7 and 30.2 nM, respectively), whereas only the N-substituted analogue, 51, was as effective as S-1 in allosterically modulating the binding of [3H]S-1 via S2.
The serotonin transporter (SERT) belongs to the Neurotransmitter:Sodium Symporter (NSS) family of transporters and serves to regulate synaptic serotonin, which plays a critical role in centrally-mediated functions including sleep, mood and appetite.1 Moreover, the SERT is the primary target for selective serotonin reuptake inhibitors (SSRIs) and tricyclic antidepressants (TCAs) that are prescribed for the treatment of anxiety and major depressive disorders. These drugs bind to the SERT and prevent the reuptake of serotonin into the cell, resulting in increased levels of synaptic serotonin, which is thought to relieve the symptoms associated with these conditions. Despite clinical success, the molecular mechanisms underlying the effectiveness of these drugs have remained elusive and further, drug-protein interactions at the molecular level that result in inhibition of serotonin reuptake have not been characterized fully.
Although small molecule structure-activity relationship (SAR) studies have led to the discovery of many effective SERT inhibitors, including S-citalopram (S-1), clomipramine, sertraline and fluoxetine, characterization of the binding domains in which these structurally divergent classes of molecules interact remains undefined. The resolution of the crystal structures of the bacterial homologue, the amino acid transporter LeuT, showed the presence of its substrate leucine and two sodium ions2 binding to a primary high affinity binding site, termed S1. The homologous S1 site for SERT has been recently characterized extensively using both molecular biology and small molecule SAR, particular through analogues of (±)citalopram (1).3–8 However, the tricyclic antidepressant clomipramine as well as the SSRIs sertraline and fluoxetine have been co-crystallized in LeuT and localized to an extracellularly located vestibule termed, S2. 9–11 Moreover, computational studies in combination with binding and ion flux experiments have revealed a role for the S2 site on LeuT in substrate binding as well.12,13 In total, these studies demonstrate the existence of S1 and S2 sites on LeuT that also exist on SERT.
Indeed, the LeuT crystal structure studies support a possible role of the extracellular vestibule-located S2 site and it has been suggested by some to be primarily responsible for the pharmacological effects of the antidepressants that crystallized therein.9–11 However, experiments with antidepressants and their analogues have challenged this assertion and have demonstrated that these drugs only bind with high affinity to the SERT S1 site, and S1 binding is thus responsible for their actions in vivo. 4,5,8,14–16 These studies have opened the door to better understand how these drugs interact at the protein level through medicinal chemistry, molecular pharmacology and computational modeling.
Although the existence of the S2 site on SERT was first described over 30 years ago,17,18 its relevance to the pharmacological actions of the SSRIs and TCAs was unknown. It was later demonstrated that selected SSRIs and TCAs as well as serotonin itself could modulate the dissociation rates of serotonin and other SERT inhibitors, in particular S-1 and imipramine, via this secondary site suggesting an allosteric role.19–22 A series of experiments were reported in which site-directed mutagenesis in the transmembrane (TM) segments 10 (TM10) and TM12 attenuated the allosteric effects of S-1, but not its binding affinity for S1, suggesting distinct binding domains.21, 23–26
Recently, a detailed molecular characterization of the S2 site in SERT was undertaken guided by computational modeling and experimentally supported by site-directed mutagenesis, Zn2+ site engineering and cysteine –reactivity assays.27 The results localize the S2 site to the vestibule extracellular to the primary binding site flanked by residues in transmembrane domains (TMs) 1, 3 and 10 from beneath and the sides as well as the extracellular loop (ECL) 4 from above.27 Interestingly, binding to the allosteric site impedes dissociation of S1 bound drug probably by a steric blockade of the exit pathway.27
The S2 site is conserved within NSS proteins from bacteria to mammals although the location is not identical to the S2 site in LeuT shown to bind antidepressants.9–11 In SERT, it is located ~13 Å above the S1 site and its role in mediating the antidepressant actions of both TCAs and SSRIs has been debated 4, 5, 8–11, 14–16, 28 as well as its role in substrate translocation. 12, 13, 29 Based on the data so far, the S2 site appears to be a larger, perhaps more promiscuous binding domain,26–28 hence we reasoned that sterically bulky analogues of 1 might bind to S2 with higher affinities than reported for S1 and in this study we explored the N-, 4-, 5-, and 4’-positions to ascertain which of these positions might be further exploited to differentiate S1 from S2 SAR. We used the previously published docking model of S-1 in S227 to help guide positions for introducing substituents that would increase the SERT:ligand contacts in S2 and thus improve binding affinity (Fig. 1). We found four suitable positions on S-1 for introducing substituents that were also synthetically accessible. We reasoned that: 1) substitution in the 4-position could protrude into the flexible extracellular loops (ECL) 2 and 4 thereby increase interactions there; 2) position 5 substituents might line up between TM10 and ECL4; 3) the addition of 4’ substituent might increase interactions with TM1 and TM8 and finally 4) the N-substitutions would be directed toward TM9 and 10 where residues have also previously been reported to be important. Since the S2 site is larger than the S1 we expected that some or most of the sterically bulky analogues of S-1 would have lower affinity for the S1 site than the parent compound.
Figure 1.

Proposed binding mode of S-1 in the S1 and S2 sites of a SERT model. A. Top view showing a snap shot of the docking of S-1 into the central (S1) binding site of SERT. The binding site is mainly composed of transmembrane domains (TM) 1, 3, 6 and 8 (grey helices and loops) The residues within the top of TM1 and from the intracellular loops 1 and 3 have been removed for clarity. The S1-bound S-1 is depicted as a space filling model in green (carbons), grey (hydrogens) and blue (nitrogen). The areas in which the N- and 5-position substitutions on S-1 might bind are indicated by the transparent spheres in green. B. Docking of S-1 into the S2 site of SERT (top view). The binding site is mainly composed of TM1, 3, 8 and 10 as well as the extracellular loops 2 and 4. The S1 docked S-1 is located deeper into the SERT (yellow). In the S2 site docked S-1 also the oxygen (red) and the F-group (cyan) are depicted. As for the S1 docked S-1, the areas of interaction for the N- and 5-CN groups on S-1 are indicated by the transparent spheres in green. The docking models are taken from Ref. 27.
To date, extensive SAR has been described for the SERT, 3–8, 14, 15, 30 however, there have been no attempts at determining SAR for the S2 site. Though, while this manuscript was being prepared, a description of binding affinities for a set of analogues of 1 at the S2 site appeared. 34 In order to discover tools that would allow further understanding of the role the S2 site plays in the structure and function of the SERT and its possible role in antidepressant action, we synthesized several series of analogues of 1. These modifications provided a structurally diverse set for initial evaluation of S2 binding. The 44 novel compounds were first tested in radiolabeled binding assays for SERT and NET, in rat brain. In addition, a set of 24 compounds reported herein and 21 previously reported compounds8 were compared to S-1 for their ability to decrease the dissociation of [3H]S-1 from the S1 site in hSERT, through allosteric modulation via S2.
Chemistry
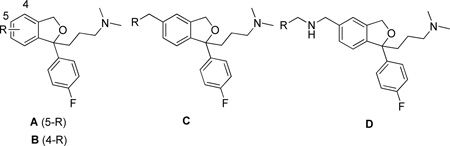
In the first series of compounds, replacement of the 5-position CN group was undertaken to assess tolerance of steric bulk and a variety of functional groups, in this position, for high affinity and selective binding to the SERT. All compounds in the present report are the racemic mixtures. As outlined in Scheme 1, the methyl ester 2 was prepared using a standard procedure to convert 1 to its carboxylic acid derivative in HCl under reflux conditions followed by esterification in MeOH using H2SO4. Compound 330 was prepared by treating 1 with H2O2 in EtOH under basic conditions. The key intermediate 4 was prepared by oxidation of 1 with an aluminum-nickel catalyst in formic acid. Another key intermediate 5 was prepared by reduction of 1 with LiAlH4 in THF. Compounds 6–10 were synthesized from 4 and the corresponding secondary amine by using Na(OAc)3BH in dichloroethane. These compounds were designed to further investigate tolerance of the SERT to a tertiary amine in the 5-position and to explore the arylpiperazine motif, which is prominent in certain 5-HT receptor ligands e.g. 5-HT1A. 31, 32 Compounds 11–15 were synthesized from 5 and the corresponding aldehydes by employing reductive amination conditions of NaBH4 in MeOH. These compounds served as an extension of the design rationale for compounds 6–10 and incorporated extended aryl ring systems including the second pharmacophore to give the dimeric compound 15. Compound 16 was prepared from 5 using HCHO and Na(OAc)3BH.
Scheme 1.

Synthesis of 5-substituted analogues of 1a
aReagents and conditions: (a) (i) 6N HCl, reflux, 12 h; (ii) H2SO4, MeOH, reflux, 5 h; (b) KOH, DMSO, 35% H2O2, EtOH, 60 °C, 2 h; (c) Ni-Al, 85% formic acid, reflux, 3 h; (d) LiAlH4, THF, reflux, 2 h; (e) corresponding secondary amine, Na(OAc)3BH, dichloroethane, RT, 12 h. (f) (i) corresponding aldehyde, MeOH, RT, 12 h; (ii) NaBH4, 30 min; (g) HCHO, Na(OAc)3BH, dichloroethane, 12 h.
To further explore 4- and additional 5–substituted analogues of 1, a strategy shown in Scheme 2 was devised, starting from compounds 1730 and 18, which were synthesized from 5- or 6-bromophthalide using a modified double Grignard reaction as previously reported.8 Suzuki coupling of 17 gave a set of 5- substituted analogues 19–21. In addition, the 4-I analogue 22 was synthesized from 18 by halogen exchange using CuI and KI, at 150 °C.
Scheme 2.

Synthesis of 4- and additional 5-substituted analogues of 1a
aReagents and conditions: (a) bis(pinacolato)diboron, potassium acetate, PdCl2(dppf)2, DMF, 105 °C, 3 h; (b) 4-nitrophenyl boronic acid, Na2CO3, Pd(PPh3)4, DME, H2O, 70–80 °C, overnight; (c) boronic acids, Na2CO3, Pd(PPh3)4, DME, H2O, 70–80 °C, overnight; (d) CuI, KI, HMPA, 150 °C, 3 h;
The synthesis of 1-(4-substituted-phenyl) analogues of 1, starting from the commercially available 5-cyanophthalide (23), is shown in Scheme 3. In this series, substitution of the 4’-F with increasingly bulky groups was investigated. Instead of the 4’-fluorophenyl group, the 4’-bromophenyl moiety was introduced by a double Grignard reaction as shown in Scheme 3 to give the diol 24. By treating with MsCl in CH2Cl2, the ring closed compound 25 was obtained. Suzuki coupling of the dihydrobenzofuran 25 gave a set of 4-substituted analogues 26–28.
Scheme 3.

Synthesis of 4’-substituted-analogues of 1a
aReagents and conditions: (a) (4-bromophenyl)magnesium bromide THF, 0 °C to RT, 3 h; (b) (3-(dimethylamino)propyl)magnesium chloride, THF, 0 °C to RT, overnight; (c) triethylamine, MsCl, dichloromethane, 0 °C, overnight; (d) boronic acids, Na2CO3, Pd(PPh3)4, DME, H2O, 70–80 °C, overnight.
In order to determine if the position of the oxygen on the dihydroisobenzofuran ring was critical for high affinity binding to SERT, synthesis of compound 37 was undertaken. Starting from the condensation of the commercially available 5-bromo-2-methoxybenzaldehyde and freshly prepared organolithium reagent 30 from 29, compound 31 was synthesized. Treatment with thionyl chloride gave 32, which was converted to the biphenyl nitrile 33. The dimethylamino side chain was introduced by alkylation using LDA to give 34. The intermediate lactone 35 was obtained, under acidic conditions, and was further reduced with LiAlH4 to give the diol 36. Ring closure with MsCl gave the final compound 37. It should be noted that for compound 37, in addition to the oxygen in the dihydroisobenzofuran ring being moved, the Br-group is now in a position analogous to position 6 on the parent compound 1.
Schemes 5 and 6 describe the synthesis of a series of N-substituted analogues of 1. Although N-demethylated analogues of 1 have been previously described,4, 7, 30 extended SAR at this position had not been investigated. Hence, it was of interest to evaluate the effect of additional steric bulk as well as several substituted aryl and heteroaryl ring systems in this position on SERT binding and to compare tolerance at the S1 vs. S2 site. The key intermediate 384, 7, 30 was synthesized by selective mono-demethylation of 1 using ACE-Cl under reflux conditions in dichloroethane. Compounds 39–51 were synthesized from 38 and the corresponding aldehydes or ketones using Na(OAc)3BH in dichloroethane. Compounds 53 and 55 could not be synthesized directly from 38 and corresponding indole aldehydes by using this procedure. We therefore used Boc protected indole aldehydes to obtain 52 and 54. Deprotection with K2CO3 in MeOH yielded 53 and 55.
Scheme 5.

Synthesis of N-substituted analogues of 1a
aReagents and conditions: (a) (i) ACE-Cl, K2CO3, DCE, reflux, 4 h; (ii) MeOH, reflux, 2 h; (b) corresponding aldehyde, Na(OAc)3BH, CH3COOH, dichloroethane, RT, 12 h; (c) K2CO3, MeOH, reflux, 2 h.
Scheme 6.

Synthesis of N-substituted analogues of 1a
aReagents and conditions: (a) 2-(1,3-dioxoisoindolin-2-yl)acetaldehyde, Na(OAc)3BH, DCE, CH3COOH, RT, 12 h (b) hydrazine, EtOH, reflux, 3 h; (c) 1,4-Cyclohexanedione monoethyleneketal, Na(OAc)3BH, CH3COOH, dichloroethane, RT, 12h ; (d) 3N HCl, diethylether, RT, 2 h; (e) indole, pyrrolidine, EtOH, reflux, 24 h.
In Scheme 6, intermediate 56 was prepared from 38 under reductive amination conditions and then deprotected to give 57 to examine the effect of a primary amine on SERT binding and to be used to append additional functional groups at this position in the future. Intermediate 58 was likewise prepared by reductive amination and hydrolysis of the ketal group using HCl in ether to give compound 59. Compound 60 was synthesized by coupling 59 with indole using pyrrolidine in EtOH. The design of this compound was inspired by the cyclohexen-4-yl indole motif previously described in a series of SERT-selective analogues33
Binding Results and Discussion
All the compounds were tested in binding assays for competition with radioligands at SERT and NET, using [3H]1 and [3H]nisoxetine in rat brain stem and frontal cortex, respectively. Compounds were first screened at a concentration of 10 µM. All compounds that displaced the radioligand by >70% were tested with full concentration curves and Ki values were calculated. These Ki values are displayed in Tables 1–3. Where percentages are reported, these compounds only displaced the radioligand by this percent at the single concentration of 10 µM and were not evaluated further. Experimental details of these assays have been previously published35 and are described in brief in the Experimental Methods section. These binding results showed that most of the 4-, 5-, 4’- and N-substituted analogues of 1 were well tolerated at the SERT and none of the compounds demonstrated high binding affinity at NET.
Table 1.
Binding Data for 4- or 5-substituted analogues of 1a
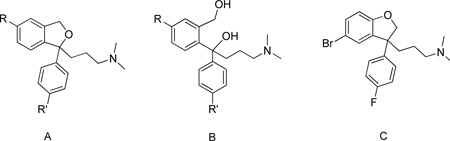
 | |||||
|---|---|---|---|---|---|
| Compd | Struc ture |
R | SERT Ki ± SEM (nM) |
NET Ki ± SEM (nM) |
Ki(NET)/Ki (SERT) |
| 1b | A | CN | 1.94±0.198 | 5950±77.4 | 3070 |
| S-1b | A | CN | 0.89±0.132 | 10500±893 | 11800 |
| 2 | A | COOCH3 | 4.17±0.482 | 64.3%e | >1000 |
| 3c | A | CONH2 | 17.7±1.80 | 123±17.7 | 7 |
| 4d | A | CHO | 4.3±0.096 | 46.1% e | >1000 |
| 5 | A | CH2NH2 | 41.9±5.73 | 33.2% e | >1000 |
| 6 | C | 18.9±0.793 | 29.3% e | >1000 | |
| 7 | C | 13.6±1.87 | >80000 | 5882 | |
| 8 | C | 3.24±0.328 | 11100±1490 | 3426 | |
| 9 | C | 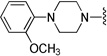 |
7.95±1.07 | 39.7% e | >1000 |
| 10 | C | 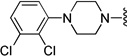 |
22.7±2.62 | 19.7% e | >1000 |
| 11 | D | 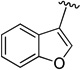 |
11. 0± 1.64 | 284±19.0 | 26 |
| 12 | D |  |
11.3±1.19 | 683±40.3 | 60 |
| 13 | D | 8.3±0.537 | 653±86.5 | 79 | |
| 14 | D | 23.6±1.54 | 2330±331 | 99 | |
| 15 | D |  |
19.7±2.80 | 66.9% e | >100 |
| 16 | C | CH2N(CH3)2 | 32.1±1.24 | 21.9% e | >1000 |
| 17b,d | A | Br | 1.04±0.126 | 28400 (global fit no SEM) |
>10,000 |
| 19 | A | 18.6±1.65 | 8280±825 | 227 | |
| 20 | A | 29.2±4.11 | 42% e | >10,000 | |
| 21 | A |  |
21.0±1.72 | 4750±860 | 226 |
| 22 | B | I | 7.05±0.404 | 11300±867 | 1603 |
| 61b | A |  |
10.4±1.34 | 3820±318 | 367 |
| 62b | A | 9.32±1.36 | 11400±1090 | 1223 | |
| 63b | A | I | 1.42±0.155 | 32500± 4630 | >10,000 |
Table 3.
Binding Data for N-substituted analogues of 1a
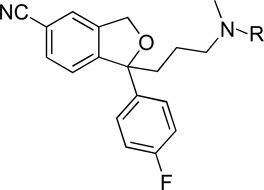 | ||||
|---|---|---|---|---|
| Compd | R | SERT Ki ± SEM (nM) |
NET Ki ± SEM (nM) |
Ki(NET)/Ki (SERT) |
| 38b | H | 5.04±0.176 | 880±12.4 | 174.6 |
| 39 | 93.4±7.83 | 69.8%c | >100 | |
| 40 | 118±8.46 | 60.4% c | >100 | |
| 41 | 61.6±6.19 | 276±41.1 | 4.5 | |
| 42 | 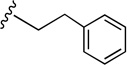 |
40.9±3.51 | 179±17.7 | 4.4 |
| 43 | 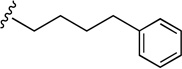 |
25.1±2.03 | 448±24.8 | 17.8 |
| 44 | 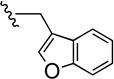 |
57.9±8.44 | 265±37.0 | 4.5 |
| 45 | 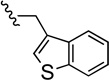 |
22.8±2.29 | 62.9% c | >100 |
| 46 | 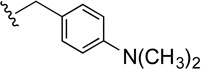 |
97.2±1.54 | 1380±162 | 14.2 |
| 47 | 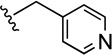 |
152±9.25 | 28.1% c | >100 |
| 48 | 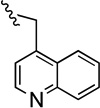 |
404±26.3 | 31.2% c | >100 |
| 49 | 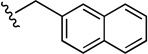 |
98.8±13.3 | 272±48.5 | 2.7 |
| 50 | 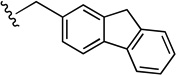 |
19.0±2.65 | 60.3% c | >100 |
| 51 | 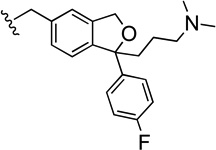 |
30.2±4.29 | 65.1% c | >100 |
| 53 | 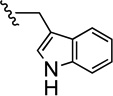 |
53.1±3.0 | 1300±152 | 24.5 |
| 55 |  |
48.5±5.24 | 5090±749 | 105 |
| 57 | 114±11.1 | 20.2% c | >100 | |
| 60 |  |
47.2±5.54 | 1020±67.4 | 21.6 |
Specifically, as seen in Table 1, electron-withdrawing groups at the 5-position (e.g. 2 and 4) retained high binding affinity at SERT. When the 5-CN group was hydrolyzed to the amide 3, SERT binding affinity decreased ~9-fold compared to 1. Reduction to the primary amine 5 resulted in additional reduction in SERT binding, which was not significantly improved in the 5-(dimethylamino)methyl analogue 16. However, further extension of dimethylamino group resulted in compounds 6–10 that exhibited similar or somewhat better binding affinity at SERT compared to their precursor 5. Moreover, further derivatization with different heterocyclic and extended aromatic ring systems in 11–15 and including the dimeric ligand 15 were generally well tolerated. Substitution of the 5-CN group with 4-nitrophenyl (19) and 4-aminophenyl (20) also retained good SERT binding affinity just slightly lower than the previously reported aniline isomer 61.8 Further extension of these compounds by introducing an E-alkenyl group in 21 was also tolerated at SERT and the additional methylene group only reduced affinity ~2-fold as compared to the previously described 62.8 Therefore, overall, substitution at the 5-postion of 1 was well tolerated at SERT. Moreover, none of the 5-substituted compounds exhibited high binding affinities to NET, although in some cases, NET binding was improved compared to 1 e.g. 3, 11, 12, 13. The 4-iodo-substitution (22) was well tolerated although displayed a somewhat lower affinity for SERT than 1 and its 5-I isomer, 63.8
In Table 2, replacement of the 4’-F group of 1 was investigated. When the 4-’F group was replaced with Br (25) binding affinity at SERT was reduced by ~6-fold, which is similar to what has previously been reported.6 The dihydroxy intermediate, 24 showed a further reduction in SERT affinities, although interestingly, SERT affinity was not abolished. Bulky substituents such as the 3-cyanophenyl in 27 and the 3-aminophenyl in 28 decreased SERT binding affinity. The alkenylphenyl substitution in 26 was similarly tolerated at SERT, but NET binding was abolished. Hence, overall increasing the steric bulk of the 4’substituent decreased binding affinity at SERT.
Table 2.
Binding Data for 5- and 4’-substituted analogues of 1a
 | ||||||
|---|---|---|---|---|---|---|
| Compd | Structure | R | R’ | SERT Ki±SEM (nM) |
NET Ki±SEM (nM) |
Ki (NET)/Ki (SERT) |
| 24 | B | CN | Br | 50.9±3.89 | 17% b | >10,000 |
| 25 | A | CN | Br | 11.4±0.74 | 45% b | |
| 26 | A | CN |  |
38.0±4.21 | 44% b | >10,000 |
| 27 | A | CN | 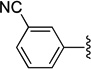 |
51.5±6.63 | 2800±353 | 54 |
| 28 | A | CN | 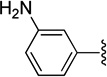 |
125±15.2 | 7020±581 | 56 |
| 37 | C | _ | _ | 681±88.2 | 4970±738 | 7.3 |
These novel analogues were assessed by radioligand binding displacement of [3H]1 (for SERT) and [3H]nisoxetine (for NET) in rat brain stem and frontal cortex, respectively.35
% Displacement at 10 µM.
Modification of the 1,3-dihydroisobenzofuran to the 2,3-dihydroisobenzofuran 37 reduced the SERT binding affinity and selectivity over NET and additional analogues were not prepared. This suggests that the position of the oxygen in the pharmacophore of 1 is important for high affinity binding at SERT. However, it should be noted that the Br-group is now in a position ortho to the Br group in 17 (e.g. position 6), which may also contribute to its lower affinity at SERT. Unfortunately, we were unable to prepare the 2,3-dihydroisobenzofuran-homologue of compound 17 for direct comparison. However, although the 6-Br homologue of 17 has not been reported, a 6-Cl analogue was compared to its 5-Cl homologue (in both cases there was a 4’-Cl rather than the 4’-F substituent in 1 and 17) and the IC50 value for inhibition of serotonin uptake was comparable (e.g. 90 v. 120 nM).30 These data support our conclusion that the position of the oxygen in the dihydroisobenzofuran ring is more critical to SERT binding than the position of the halogen substituent (e.g. positions 5 v. 6).
In Table 3, SAR of N-substitution was explored. Compound 38,4, 7, 30 the N-demethylated analogue of 1, showed high binding affinity at SERT (Ki-=5.04 nM) as previously reported. Typically N-alkyl and N-alkyl aryl analogues of 38 demonstrated lower affinity at SERT than 1, but some of these analogues showed higher binding affinity at NET (e.g., 41, 42, 43). A similar pattern was observed in several heterocyclic and aromatic substituted analogues, although several of these analogues still demonstrated binding affinities in the Ki<50 nM range for SERT. Large weakly basic substituents at the N-position yielded somewhat lower affinities at SERT (e.g. 46, 47, 48 and 57) relative to those bearing bulky neutral N-groups (e.g., 44, 45, 50), suggesting polarity is not tolerated in this region of the molecule. Interestingly, the dimeric compound 51 showed a similar binding affinity and selectivity profile to the other dimeric compound 15 demonstrating that the position of attachment of a second pharmacophore is not critical to maintain moderate affinity for the SERT S1 site.
In the second phase of this study, a subset of the compounds in this series, as well as a set of previously published analogues of 1 were tested for binding to the SERT S2 site. The S2 site is characterized as an allosteric binding site that is located in the extracellular vestibule of the SERT. Although extensive site-directed mutagenesis and other experiments at the protein level have been reported to characterize this site, to our knowledge this is the first attempt at pharmacophore identification. The measurement of S2 binding was assessed as previously reported,27 by utilizing its allosteric interactions on S1 binding and thereby measuring the inhibition of dissociation of S1 bound [3H]S-1 by the S2-binding compound. Details of these experiment can be found in the Experimental Methods section and Tables 4 and 5 legends. The times for dissociating 50% of the bound [3H]S-1 at 18 °C without the presence of an allosterically bound compound were measured to t½=16.1±1.0 min (Table 4 – compounds in this report only; data on previously reported compounds8 can be found in S.I.). If the analogues showed any allosteric binding to SERT it would be measured as an inhibition of the dissociation rate (t½) of the pre-bound [3H]S-1 (see Experimental Methods for further details). Accordingly, we tested the inhibition of [3H]S-1 dissociation by applying 30 µM of the compound after pre-incubation with [3H]S-1 and investigating its effect on [3H]S-1 dissociation measured as the change in t½ (Table 4). Examples of compounds that decreased the dissociation rate of [3H]S-1 from S1 are shown in Fig. 2A. The majority of the tested compounds inhibited [3H]S-1 dissociation although to a lesser extent than S-1 itself. Indeed, 30 µM 5-HT had no measurable effect on [3H]S-1 dissociation (data not shown).
Table 4.
Effects of the investigated analogues in inhibiting the dissociation of [3H]S-1 from hSERT WT. The relative potency of 30 µM compound in inhibiting [3H]S-1 dissociation as compared to buffer and S-1.
| Compound | [3H] S-1 dissoc. t½ (min) at 18°C |
[3H] S-1 dissoc. t½ (min) at 24°C |
24°C dissociation t½ calculated to a t½ at 18°C (min) |
|---|---|---|---|
| Control | 16.1±1.0 | 7.0±0.3 | |
| S-1 | 68±12 | 28.1±3.1 | |
| 5 | 17.4±0.8 | ||
| 8 | 16.4±0.3 | ||
| 9 | 16.3±0.1 | ||
| 10 | 24.8±1.1 | ||
| 12 | 22.0±1.2 | ||
| 13 | 22.1±1.5 | ||
| 14 | 11.4±0.2 | ||
| 15 | 21.4±0.3 | ||
| 19 | 40.2±7.0 | 97±17 | |
| 20 | 52.6±4.1 | 127±10 | |
| 21 | 23.1±2.9 | ||
| 22 | 33.2±5.8 | ||
| 24 | 24.7±5.4 | ||
| 25 | 63±13 | 151±30 | |
| 26 | 44±12 | ||
| 42 | 45.4±5.9 | 110±14 | |
| 45 | 59.6±1.0 | ||
| 46 | 41.2±2.8 | ||
| 48 | 29.0±1.3 | ||
| 49 | 60.1±1.0 | ||
| 51 | 52.3±3.2 | 127.1±6.8 | |
| 53 | 93±15 | 225±37 | |
| 55 | 58.7±10.0 | 143±24 | |
| 60 | 52.0±7.6 | ||
| 61 | 43.1±6.2 | ||
| (+)62 | 54.9±3.5 | 132.3±8.5 | |
| (−)62 | 37.3±9.5 | ||
| 63 | 34.4±6.6 |
Experiments were performed on membrane preparations from COS-7 cells transiently expressing with hSERT WT. [3H]S-1 was added until equilibrium was obtained and subsequently diluted in 12× buffer volumes containing 30 µM of the indicated compound. Dissociation were measured at 18°C to obtain a suitable t½ (<100 min). For the compounds with t½ >100 min, the temperature were raised to 24°C for a more accurate determination. Since the dissociation t½ is directly proportional to the temperature, it is possible to calculate a t½ at 18°C from the 24°C dissociation. This temperature constant is found for the effect of S-1 on [3H]S-1 dissociation at 18°C and 24°C, respectively. Values are mean±S.E. of 3–6 experiments performed in triplicate.
Table 5.
Allosteric potencies of the most potent compounds in inhibiting the dissociation of [3H]S-1 from hSERT.
| Compound | Allosteric Potencya (in µM)a |
|---|---|
| S-1 | 4.6[4.2;5.0]b |
| 20 | 3.6[3.4;3.8] |
| 25 | 4.7[3.8;5.7] |
| 42 | 5.3[3.0;9.5] |
| 51 | 4.9[4.4;5.5] |
| 53 | 3.0[2.2;4.0] |
| 55 | 5.8[4.8;7.1] |
| (+)62 | 3.4[2.4;4.8] |
The allosteric potencies are the IC50 values obtained from non-linear regression analysis of data from [3H]S-1 dissociation experiments in the presence of increasing concentrations of the indicated compounds. The dissociation rate constants (k[cmpd]) at different compound concentrations were calculated by linear regression and expressed relative to the dissociation rate constant without the presence of unlabeled ligand (kbuffer). The allosteric potency is determined as the IC50 value of the drug concentration (log[cmpd]) that impairs the dissociation rate by 50% compared with dissociation in buffer (k[cmpd]/kbuffer) and are shown as mean values calculated from means of pIC50 and the SE interval from the pIC50 ±S.E of 3–8 experiments performed in triplicate.
Data previously published in ref. 27.
Figure 2.

Characterization of allosteric potency by selected analogues of 1. A. Experiments for the inhibition of prebound [3H]S-1 dissociation by 30 µM of selected analogues of 1 at 18 °C. The addition of the compounds affects [3H]S-1 dissociation to various extents relative to [3H]S-1 dissociation without the presence of compounds (filled circles). Data are shown relative to the dissociation without compound (filled circles) and the addition of 30 µM S-1 (gray stars). The t½ (in min) for the shown dissociation experiments are listed in Table 4, data are means±SE (error bars) of 3–6 experiments. B. Determination of allosteric potency for 51. Each data point represents a dissociation rate experiments as shown in A., at different concentrations of 51. The EC50 for 51 is 4.9[4.4;5.5] µM, mean[SE interval], N=8. The dotted line is the allosteric potency of S-1 as determined previously.27 All experiments are performed on membrane preparations from COS7 cells transiently expressing hSERT.
The assay time can only be extended to 90 min, which limits the exact measurement of very long t½, i.e. above 90 min. However, dissociation follows a first order reaction directly proportional to temperature and, thus, increasing the temperature increases dissociation rate by a measurable factor. Increasing the temperature to 24 °C decreases the t½ for S-1 by a factor 2.4 (Table 4). Eight new compounds (19, 20, 25, 42, 51, 53, 55) and (+)628 had an effect on [3H]S-1 dissociation resulting in a t½ >90 min at 18 °C. To obtain an extrapolated t½ for these compounds, their allosteric effects on [3H]S-1 dissociation were measured at 24 °C (Table 4). Assuming that the temperature factor is identical for all investigated compounds, it is possible to estimate their dissociation rate at 18 °C and thereby compare the allosteric effect with the compounds measured at 18 °C (Table 4).
To further analyze the allosteric effects of the most potent compounds i.e. the compounds producing an estimated t½ >100 min at 18°C, we investigated their effects on [3H]S-1 dissociation impairment as a function of the added concentration of the compound: the allosteric potency (see Experimental Methods for details). In Fig. 2B the data for the determination of the allosteric potency of 51 is shown and compared to the allosteric potency of S-1.27 A determination of the allosteric potencies for all the compounds producing a t½ >100 min showed that they are similarly potent to S-1 at the S2 site (Table 5). Although, our most potent analogues at the S2 site have similar potencies to one another, the dimeric analogues may provide a clue to SAR separability between S1 and S2. Both dimeric compounds 15 and 51 had similar affinities for the SERT S1 site (Ki=19.7 and 30.2 nM, respectively) whereas only the N-substituted dimeric ligand, 51, was at least as effective as S-1 in allosterically modulating the binding of [3H]S-1 for the S1 site, via S2. It should also be noted that 51 had >30-fold lower affinity for S1 than S-1. One strategy may be to modify 51 to deliberately decrease its binding at the S1 site, using the SAR derived herein, and to also synthesize the S-enantiomer(s) to potentially improve binding at the S2 site, as it was previously reported that R-1 is less potent than S-1 at both the SERT S1 and S2 sites.26 In addition, herein we showed that (+)62 was more potent at S2 and than (-)62, supporting this approach for future drug design. Computational modeling using the chiral analogues and follow-up molecular studies may provide further clues as to how these compounds are binding the SERT and further investigation is underway.
In summary, analogues of the SERT-selective inhibitor, 1, in which structural modifications were made at the N-, 4, 5, and 4’-positions, were synthesized and characterized. The 44 novel compounds were first tested for displacement of radiolabeled ligands from SERT and NET in rat brain membranes to extend SAR at the SERT S1 site. All of the analogues were racemates and none showed higher affinities for binding at SERT than the parent compound. Nevertheless, most of the analogues showed Ki values of <50 nM and all of the analogues were selective for SERT over NET. These studies demonstrated that substitutions at either the N- or 5-positions was tolerated at SERT, including two dimeric ligands, compounds 15 and 51. These findings further suggest that the N- or 5-positions can be modified with relatively large and potentially multifunctional substituents, such as fluorophores, and retain high affinity binding at the SERT. We have recently confirmed this with a novel SERT fluorescent ligand in which rhodamine was extended from the 5-position of 1.36
In addition, a set of 24 new compounds and 21 previously reported compounds8 were compared to S-1 in dissociation experiments for their ability to decrease the dissociation rate of [3H]S-1 from the S1 site, via allosteric modulation at S2. We discovered 8 compounds that have similar potencies to the parent S-1 at S2, but differ in both structure and affinity for the SERT S1 site. Indeed, these data imply that analogues with sterically bulky and in some cases, multifunctional substituents appended to the terminal amine of 1 provide the basis for the design of a new series of compounds in which binding at S1 may be decreased, while binding at S2 is improved. S2-selective ligands will undoubtedly provide critical tools for future examination of its role in the structure and function of SERT and will aid in determining if binding to the S2 site is related to the therapeutic effects of the SSRI and TCA classes of antidepressant agents.
Experimental Methods
1H and 13C NMR spectra were acquired using a Varian Mercury Plus 400 spectrometer. Chemical shifts are reported and referenced according to deuterated solvent for 1H spectra (CDCl3, 7.26; (CD3)2SO, 2.50; CD3OD, 3.31), 13C spectra (CDCl3, 77.2; (CD3)2SO, 39.5; CD3OD, 49.0), 19F spectra (CFCl3, 0). Infrared spectra were recorded as a KBr thin film using a Perkin-Elmer Spectrum RZ I FT-IR spectrometer or recorded as powder using an Avatar 370 FT-IR thermo Nicolet spectrometer. Combustion analysis was performed by Atlantic Microlab, Inc. (Norcross, GA) and agrees within 0.4% of calculated values. Melting point (Mp) determinations were conducted using a Thomas-Hoover melting point apparatus and are uncorrected. Some of the compounds were highly hygroscopic, as indicated, and thus no Mp is recorded. Anhydrous solvents were purchased from Aldrich and were used without further purification, except for tetrahydrofuran, which was freshly distilled from sodium-benzophenone ketyl. All other chemicals and reagents were purchased from Aldrich Chemical Co., Combi-Blocks, TCI, America., Matrix Scientific; Lancaster Synthesis, Inc. (Alfa Aesar) and AK Scientific, Inc. Final compounds (free base) were purified by column chromatography (EMD Chemicals, Inc.; 230–400 mesh, 60 Å) or preparative thin layer chromatography (silica gel, Analtech, 1000 µm). The eluting solvent system CMA refers to CHCl3/MeOH/NH4OH in the percentage indicated where NH4OH is 0.1 %. The final products were converted into either oxalate or HBr salts. All these salts were prepared by adding oxalic acid in acetone or HBr in MeOH to the free base in alcohol (2-PrOH, EtOH, MeOH), followed by precipitation from a combination of organic solvents. Yields and reaction conditions were not optimized. All final free base compounds are colorless oils and are the racemic mixtures. Generally, yields and spectroscopic data refer to the free base. On the basis of 1H NMR, GC-MS (where obtainable), and combustion analysis data, all final compounds are >95% pure.
General Method A. Reductive amination
Amine (1 eq) and aldehyde (1 eq) were mixed in 1,2-dichloroethane (5 mL), and then treated with sodium triacetoxyborohydride (1.5 eq) and AcOH (0.2 g). The mixture was stirred at RT under an argon atmosphere for 6 h. The reaction mixture was quenched by adding 1N NaOH (5 mL), and the product was extracted with EtOAc. The EtOAc extract was washed with brine, dried over MgSO4, and concentrated in vacuo. The crude product was purified by flash column chromatography eluting with (90% CMA) to give the pure product.
General Method B. Reductive amination
To a solution of compound 5 (328 mg, 1 eq) in MeOH (10 mL) was added aldehyde (1 eq), and the reaction mixture was stirred under argon for 12 h. NaBH4 (75 mg, 2 eq) was added to the reaction mixture and stirred for 30 min. The reaction was quenched by adding 1 N NaOH (15 mL). The resulting mixture was filtered through celite, and the residue was washed with CHCl3 (50 mL). The organic layer was separated and dried over MgSO4. The solvent was removed under reduced pressure. The crude product was purified by column chromatography using 90% CMA.
General Method C. Suzuki coupling of heteroaryl bromides with boronic acid
To a suspension of boronic acid (1–1.5 eq), heteroaryl bromide (1 eq), and Na2CO3 in a mixture of solvents DME/H2O (3/1, 4 mL for 1 mmol scale reaction) was added Pd(PPh3)4 (5 mol%) under Argon. The mixture was heated at 70–80 °C overnight. The solvent was then removed under reduced pressure, and the residue was extracted with EtOAc. The organic layer was dried over MgSO4 and concentrated to a crude product, which was then purified by flash column chromatography to give the pure product.
Methyl 1-(3-(dimethylamino)propyl)-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-5-carboxylate (2)
A solution of 1 (3.24 g, 10 mmol) in 6N HCl (20 mL) was stirred at reflux for 12h. The reaction was allowed to cool to RT and neutralized to pH 7 with a saturated NaHCO3 solution, then extracted with CHCl3. The organic layer was washed with brine, dried over MgSO4, and concentrated to give the crude acid product. The crude acid was dissolved in MeOH (30 mL), H2SO4 (2mL) was added and the reaction mixture was stirred at reflux for 5 h. The reaction was allowed to cool to RT. Residual MeOH was removed in vacuo and the residue was diluted and neutralized with saturated NaHCO3 solution, and then extracted with CHCl3. The organic layer was dried over MgSO4, and concentrated to give the crude methyl ester compound, which was then purified by flash column chromatography eluting with CHCl3/MeOH, (9:1) to give the pure product (1.4 g) in 40% yield. 1H NMR (400 MHz, CDCl3) δ 7.98 (d, J = 8.4 Hz, 1H), 7.88 (s, 1H), 7.47-7.42 (m, 2H), 7.34 (d, J = 8 Hz, 1H), 7.02-6.96 (m, 2H), 5.20 (d, J = 12.8 Hz, 1H), 5.15 (d, J = 12.8 Hz, 1H), 3.91 (s, 3H), 2.25-2.15 (m, 4H), 2.14 (s, 6H), 1.52-1.25 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 166.9, 162.1 (d, J = 243.9 Hz), 149.4, 140.5 (d, J = 3.1 Hz), 139.8, 130.1, 129.6, 127.1 (d, J = 8.4 Hz), 122.9, 122.0, 115.3 (d, J = 20.6 Hz), 91.1, 71.8, 59.8, 52.4, 45.6, 39.3, 22.4; GC-MS (EI) m/z 357 (M+); The HBr salt was precipitated from MeOH and was hygroscopic; Anal. (C21H24FNO3·HBr·3/4H2O) C, H, N.
1-(3-(Dimethylamino)propyl)-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-5-carboxamide (3)30
To a solution of 1 (972 mg, 3 mmol) in EtOH (8 mL), were added KOH (1.7 g, 30 mmol), DMSO (0.2 mL) and 35% H2O2. The mixture was heated at 60 °C and stirred for 2 h. The solvent was then removed under reduced pressure. Water was added to the resultant residue and extracted with CHCl3. The organic layer was dried over MgSO4 and concentrated to give crude carboxamide, which was then purified by flash column chromatography eluting with (90% CMA) to give the pure product (0.5 g) in 49% yield. 1H NMR (400 MHz, CDCl3) δ 7.72 (d, J = 8.4 Hz, 1H), 7.67 (s, 1H), 7.46-7.42 (m, 2H), 7.33 (d, J = 7.6 Hz, 1H), 7.01-6.96 (m, 2H), 6.38 (brs, 2H), 5.18 (d, J = 12.8 Hz, 1H), 5.13 (d, J = 12.8 Hz, 1H), 3.91 (s, 3H), 2.25-2.15 (m, 4H), 2.13 (s, 6H), 1.53-1.25 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 169.5, 162.1 (d, J = 244.6 Hz), 148.4, 140.6 (d, J = 3 Hz), 140.0, 133.6, 127.1 (d, J = 13 Hz), 127.0, 122.2, 120.9, 115.4 (d, J = 20.6 Hz), 91.1, 71.8, 59.8, 45.6, 39.4, 22.4; GC-MS (EI) m/z 342 (M+); The oxalate salt was precipitated from EtOAc and was hygroscopic; Anal. (C20H23FN2O2·C2H2O4·H2O) C, H, N.
1-(3-(Dimethylamino)propyl)-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-5-carbaldehyde (4)
To a solution of 1 (3.24 g, 10 mmol) in 85% formic acid (65 mL), was added aluminum-nickel catalyst (5.71 g). The mixture was heated at 80 °C and stirred for 3 h. The reaction was allowed to cool to RT and filtered, then neutralized with saturated NaHCO3 solution. The resulting solution was extracted with CHCl3. The organic layer was dried over MgSO4 and concentrated to give crude aldehyde, which was then purified by flash column chromatography eluting with CHCl3/MeOH (19:1) to give the pure product (3.0 g) in 92% yield. 1H NMR (400 MHz, CDCl3) δ 10.0 (s, 1H), 7.81 (d, J = 8.0 Hz, 1H), 7.73 (s, 1H), 7.48-7.43 (m, 3H), 7.03-6.98 (m, 2H), 5.24 (d, J = 12.8 Hz, 1H), 5.18 (d, J = 12.8 Hz, 1H), 3.91 (s, 3H), 2.26-2.15 (m, 4H), 2.14 (s, 6H), 1.55-1.28 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 191.7, 162.2 (d, J = 244.6 Hz), 151.2, 140.5, 140.2 (d, J = 3.1 Hz), 136.6, 130.4, 127.1 (d, J = 7.7 Hz), 122.7, 122.5, 115.4 (d, J = 21.3 Hz), 91.1, 71.6, 59.7, 45.6, 39.3, 22.4; GC-MS (EI) m/z 327 (M+); The HBr salt was precipitated from MeOH and was hygroscopic; Anal. (C20H22FNO2·HBr·H2O) C, H, N.
3-(5-(Aminomethyl)-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-1-yl)-N,N-dimethylpropan-1-amine (5)37
To a suspension of LiAlH4 (759 mg, 20 mmol) in anhydrous THF (25 mL), was added 1 (3.24 g, 10 mL) in anhydrous THF (25 mL) drop-wise at 0 °C under an argon atmosphere. The reaction mixture was stirred at reflux for 4h, cooled to RT and saturated NaOH (6 mL) was carefully added to quench the excess LiAlH4. The resulting mixture was filtered, washed with H2O, extracted with EtOAc, dried over MgSO4 and concentrated to give the product (3.1 g) in 94% yield. 1H NMR (400 MHz, CDCl3) δ 7.47-7.43 (m, 2H), 7.24-7.20 (m, 2H), 7.16 (s, 1H), 6.99-6.94 (m, 2H), 5.15 (d, J = 12.0 Hz, 1H), 5.11 (d, J = 12.0 Hz, 1H), 3.86 (s, 2H), 2.24-2.15 (m, 4H), 2.13 (s, 6H), 1.55-1.25 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 162.0 (d, J = 243.9 Hz), 143.3,143.0, 141.4 (d, J = 3.1 Hz), 127.0 (d, J = 7.7 Hz), 126.8, 122.1, 120.1, 115.1 (d, J = 21.4 Hz), 91.02, 72.05, 59.9, 46.5, 45.7, 39.6, 22.6; GC-MS (EI) m/z 328 (M+); The oxalate salt was precipitated from EtOAc and was hygroscopic; Anal. (C20H25FN2O·2C2H2O4·H2O) C, H, N.
3-(1-(4-Fluorophenyl)-5-(piperidin-1-ylmethyl)-1,3-dihydroisobenzofuran-1-yl)-N,N-dimethylpropan-1-amine (6)
Compound 6 was prepared from piperidine (72 mg, 0.84 mmol) and 4 (272 mg, 0.84 mmol) according to General Method A (303 mg) in 91% yield. 1H NMR (400 MHz, CDCl3) δ 7.48-7.44 (m, 2H), 7.20-7.19 (m, 2H), 7.15 (s, 1H), 6.99-6.94 (m, 2H), 5.14 (d, J = 12.0 Hz, 1H), 5.10 (d, J = 12.0 Hz, 1H), 3.43 (s, 2H), 2.35 (brs, 4H), 2.25-2.06 (m, 10H), 1.60-1.28 (m, 8H); 13C NMR (100 MHz, CDCl3) δ 162.0 (d, J = 243.9 Hz), 143.0, 141.5 (d, J = 3.8 Hz), 139.3, 138.7, 128.7, 127.0 (d, J = 7.6 Hz), 122.0, 121.6, 115.1 (d, J = 21.3 Hz), 91.0, 72.0, 63.9, 60.0, 54.8, 45.6, 39.7, 26.2, 24.6, 22.5; 19F NMR (376 MHz, CDCl3) δ -117.10; GC-MS (EI) m/z 396 (M+); The HBr salt was precipitated from MeOH and was hygroscopic; Anal. (C25H33FN2O·2HBr·7/4H2O) C, H, N.
3-(1-(4-fluorophenyl)-5-(morpholinomethyl)-1,3-dihydroisobenzofuran-1-yl)-N,N-dimethylpropan-1-amine (7)
Compound 7 was prepared from morpholine (78 mg, 0.9 mmol) and 4 (290 mg, 0.9 mmol) according to General Method A (312 mg) in 87% yield. 1H NMR (400 MHz, CDCl3) δ 7.47-7.43 (m, 2H), 7.22-7.21 (m, 2H), 7.16 (s, 1H), 6.99-6.94 (m, 2H), 5.15 (d, J = 12.0 Hz, 1H), 5.11 (d, J = 12.4 Hz, 1H), 3.69 (t, J = 4.8 Hz, 4H), 3.47 (s, 2H), 2.42 (t, J = 4.4 Hz, 4H), 2.25-2.06 (m, 10H), 1.54-1.28 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 162.0 (d, J = 243.2 Hz), 143.4, 141.4 (d, J = 3.8 Hz), 139.5, 137.8, 128.8, 127.0 (d, J = 8.4 Hz), 122.0, 121.8, 115.2 (d, J = 21.3 Hz), 91.0, 72.0, 67.2, 63.4, 59.9, 53.9, 45.6, 39.7, 22.5; 19F NMR (376 MHz, CDCl3) δ -117.07; GC-MS (EI) m/z 398 (M+); The oxalate salt was precipitated from EtOAc and was hygroscopic; Anal. (C24H31FN2O2·2C2H2O4·2H2O) C, H, N.
3-(1-(4-Fluorophenyl)-5-((4-phenylpiperazin-1-yl)methyl)-1,3-dihydroisobenzofuran-1-yl)-N,N-dimethylpropan-1-amine (8)
Compound 8 was prepared from 1-phenylpiperazine (149 mg, 0.92 mmol) and 4 (300 mg, 0.92 mmol) according to General Method A (418 mg) in 96% yield. 1H NMR (400 MHz, CDCl3) δ 7.49-7.45 (m, 2H), 7.28-7.23 (m, 4H), 7.20 (s, 1H), 7.01-6.82 (m, 5H), 5.16 (d, J = 12.0 Hz, 1H), 5.12 (d, J = 12.0 Hz, 1H), 3.54 (s, 2H), 3.19 (t, J = 5.2 Hz, 4H), 2.59 (t, J = 5.2 Hz, 4H), 2.27−2.08 (m, 10H), 1.54-1.28 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 162.0 (d, J = 243.9 Hz), 151.6, 143.4, 141.4 (d, J = 3 Hz), 139.5, 138.0, 129.3, 128.8, 127.0 (d, J = 7.6 Hz), 122.0, 121.8, 119.9, 116.3, 115.2 (d, J = 21.4 Hz), 91.0, 72.0, 63.1, 59.9, 53.4, 49.4, 45.6, 39.7, 22.5; 19F NMR (376 MHz, CDCl3) δ -117.06; The oxalate salt was precipitated from EtOAc; Mp 114–115 °C; Anal. (C30H36FN3O·2C2H2O4·3/2H2O) C, H, N.
3-(1-(4-Fluorophenyl)-5-((4-(2-methoxyphenyl)piperazin-1-yl)methyl)-1,3-dihydroisobenzofuran-1-yl)-N,N-dimethylpropan-1-amine (9)
Compound 9 was prepared from 1-(2-methoxyphenyl)piperazine (177 mg, 0.92 mmol) and 4 (300 mg, 0.92 mmol) according to General Method A (435 mg) in 94% yield. 1H NMR (400 MHz, CDCl3) δ 7.49-7.45 (m, 2H), 7.24-7.21 (m, 2H), 7.20 (s, 1H), 7.01-6.83 (m, 6H), 5.16 (d, J = 12.0 Hz, 1H), 5.12 (d, J = 12.0 Hz, 1H), 3.85 (s, 3H), 3.56 (s, 2H), 3.08 (brs, 4H), 2.64 (brs, 4H), 2.26-2.07 (m, 10H), 1.54-1.28 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 162.0 (d, J = 243.1 Hz), 152.5, 143.3, 141.6, 141.4 (d, J = 3.1 Hz), 139.5, 138.1, 128.9, 127.0 (d, J = 7.6 Hz), 123.1, 122.1, 121.8, 121.2, 118.4, 115.2 (d, J = 20.5 Hz), 111.3, 91.0, 72.0, 63.2, 59.9, 55.5, 53.6, 50.9, 45.6, 39.7, 22.5; 19F NMR (376 MHz, CDCl3) δ -117.12; The oxalate salt was precipitated from EtOAc; Mp 160–162 °C; Anal. (C31H38FN3O2·2C2H2O4·5/2H2O) C, H, N.
3-(5-((4-(2,3-Dichlorophenyl)piperazin-1-yl)methyl)-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-1-yl)-N,N-dimethylpropan-1-amine (10)
Compound 10 was prepared from 1-(2,3-dichlorophenyl)piperazine (213 mg, 0.92 mmol) and 4 (300 mg, 0.92 mmol) according to General Method A (458 mg) in 92% yield. 1H NMR (400 MHz, CDCl3) δ 7.49-7.45 (m, 2H), 7.25-7.23 (m, 2H), 7.19 (s, 1H), 7.16-7.10 (m, 2H), 7.01–6.92 (m, 3H), 5.16 (d, J = 12.4 Hz, 1H), 5.12 (d, J = 12.4 Hz, 1H), 3.56 (s, 2H), 3.05 (brs, 4H), 2.63 (brs, 4H), 2.27-2.07 (m, 10H), 1.54-1.28 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 161.8 (d, J = 243.2 Hz), 151.3, 143.2, 141.1 (d, J = 3.0 Hz), 139.3, 137.8, 134.0, 128.6, 127.4 (d, J = 6.1 Hz), 126.8, 126.7, 124.5, 121.8, 121.6, 118.6, 114.9 (d, J =21.3 Hz), 90.7, 71.7, 62.8, 59.7, 53.3, 51.3, 45.4, 39.5, 22.3; 19F NMR (376 MHz, CDCl3) δ -116.95; The oxalate salt was precipitated from EtOAc; Mp 116–118 °C; Anal. (C30H34FN3O· 2C2H2O4·5/4H2O) C, H, N.
3-(5-(((Benzofuran-2-ylmethyl)amino)methyl)-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-1-yl)-N,N-dimethylpropan-1-amine (11)
Compound 11 was prepared from 5 (164 mg, 0.5 mmol) and benzofuran-3-carbaldehyde (73 mg, 0.5 mmol) according to General Method B (215 mg) in 94% yield. 1H NMR (400 MHz, CDCl3) δ 7.63-7.60 (m, 1H), 7.56 (s, 1H), 7.48-7.43 (m, 3H), 7.32-7.19 (m, 5H), 7.0-6.95 (m, 2H), 5.15 (d, J = 12.4 Hz, 1H), 5.11 (d, J = 12.4 Hz, 1H), 3.93 (s, 2H), 3.85 (s, 2H), 2.24-2.07 (m, 10H), 1.54-1.26 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 161.9 (d, J = 243.9 Hz), 155.7, 143.2, 142.4, 141.3, 140.1, 139.6, 127.7, 127.6, 126.9 (d, J = 7.6 Hz), 124.5, 122.6, 121.9, 121.0, 120.0, 115.0 (d, J = 21.4 Hz), 111.7, 90.9, 72.0, 59.8, 53.3, 45.5, 42.9, 39.6, 22.4; 19F NMR (376 MHz, CDCl3) δ -117.0; The HBr salt was precipitated from MeOH and was hygroscopic; Anal. (C29H31FN2O2·5/2HBr·3/4H2O) C, H, N.
3-(5-(((Benzo[b]thiophen-2-ylmethyl)amino)methyl)-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-1-yl)-N,N-dimethylpropan-1-amine (12)
Compound 12 was prepared from 5 (164 mg, 0.5 mmol) and benzo[b]thiophene-3-carbaldehyde (81 mg, 0.5 mmol) according to General Method B (208 mg) in 88% yield. 1H NMR (400 MHz, CDCl3) δ 7.87-7.84 (m, 1H), 7.81-7.78 (m, 1H), 7.48-7.43 (m, 2H), 7.39-7.31 (m, 3H), 7.20-7.27 (m, 3H), 7.0-6.95 (m, 2H), 5.15 (d, J = 12.4 Hz, 1H), 5.11 (d, J =12.4 Hz, 1H), 4.05 (s, 2H), 3.87 (s, 2H), 2.24-2.07 (m, 10H), 1.54-1.28 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 161.9 (d, J = 243.1 Hz), 143.2, 141.3 (d, J = 3.0 Hz), 140.8, 140.1, 139.6, 138.5, 135.1, 127.7, 126.9 (d, J = 7.7 Hz), 124.5, 124.1, 123.2, 123.0, 121.9, 121.9, 121.0, 115.0 (d, J = 21.4 Hz), 90.9, 71.9, 59.8, 53.4, 47.2, 45.5, 39.6, 22.4; 19F NMR (376 MHz, CDCl3) δ -116.99; The HBr salt was precipitated from MeOH; Mp 131–132 °C; Anal. (C29H31FN2OS·2HBr·7/4H2O) C, H, N.
3-(1-(4-Fluorophenyl)-5-(((naphthalen-2-ylmethyl)amino)methyl)-1,3-dihydroisobenzofuran-1-yl)-N,N-dimethylpropan-1-amine (13)
Compound 13 was prepared from 5 (164 mg, 0.5 mmol) and 2-naphthaldehyde (78 mg, 0.5 mmol) according to General Method B in 91% (213 mg) yield. 1H NMR (400 MHz, CDCl3) δ 7.84-7.80 (m, 3H), 7.77 (s, 1H), 7.43–7.49 (m, 5H), 7.22–7.27 (m, 2H), 7.20 (s, 1H), 6.95–7.0 (m, 2H), 5.16 (d, J = 12.4 Hz, 1H), 5.12 (d, J = 12.4 Hz, 1H), 3.98 (s, 2H), 3.83 (s, 2H), 2.24-2.07 (m, 10H), 1.54-1.28 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 161.9 (d, J = 243.1 Hz), 143.1, 141.3 (d, J = 3.0 Hz), 140.2, 139.5, 139.5, 137.8, 133.6, 132.8, 128.2, 127.8, 127.8, 127.8, 127.7, 126.9 (d, J = 8.4 Hz), 126.7, 126.6, 126.2, 126.1, 125.7, 121.0, 115.1 (d, J = 21.4 Hz), 90.9, 72.0, 59.8, 53.5, 53.1, 45.5, 39.6, 22.4; 19F NMR (376 MHz, CDCl3) δ -117.01; The HBr salt was precipitated from MeOH; Mp 129–130 °C; Anal. (C31H33FN2O· 2HBr·7/4H2O) C, H, N.
3-(5-((((9H-Fluoren-2-yl)methyl)amino)methyl)-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-1-yl)-N,N-dimethylpropan-1-amine (14)
Compound 14 was prepared from 5 (164 mg, 0.5 mmol) and 9H–fluorene-2-carbaldehyde (97 mg, 0.5 mmol) according to General Method B (223 mg) in 88% yield. 1H NMR (400 MHz, CDCl3) δ 7.78-7.73 (m, 2H), 7.55-7.52 (m, 2H), 7.48-7.44 (m, 2H), 7.39-7.20 (m, 7H), 7.0-6.95 (m, 2H), 5.16 (d, J = 12.4 Hz, 1H), 5.12 (d, J = 12.4 Hz, 1H), 3.89 (s, 2H), 3.88 (s, 2H), 3.83 (s, 2H), 2.24-2.07 (m, 10H), 1.54-1.28 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 161.9 (d, J = 243.8 Hz), 143.7, 143.4, 143.1, 141.7, 141.3 (d, J = 3.0 Hz), 140.8, 140.2, 139.6, 139.0, 127.7, 126.9 (d, J = 7.7 Hz), 126.9, 126.7, 125.2, 125.0, 121.9, 121.0, 119.9, 119.9, 115.1 (d, J = 21.4 Hz), 90.9, 72.0, 59.8, 53.7, 53.2, 45.5, 39.6, 37.0, 22.4; 19F NMR (376 MHz, CDCl3) δ -117.0; The HBr salt was precipitated from MeOH; Mp 138–140 °C; Anal. (C34H35FN2O·2HBr·7/4H2O) C, H, N.
3-(5-((((1-(3-(Dimethylamino)propyl)-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-5-yl)methyl)amino)methyl)-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-1-yl)-N,N-dimethylpropan-1-amine (15)
Compound 15 was prepared from 5 (164 mg, 0.5 mmol) and 4 (164 mg, 0.5 mmol) according to General Method B (294 mg) in 92% yield. 1H NMR (400 MHz, CDCl3) δ 7.47-7.42 (m, 4H), 7.25-7.17 (m, 6H), 6.94-6.99 (m, 4H), 5.14 (d, J = 12.4 Hz, 2H), 5.10 (d, J = 12.4 Hz, 2H), 3.78 (s, 4H), 2.24-2.07 (m, 20H), 1.52-1.25 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 161.9 (d, J = 243.8 Hz), 143.1, 141.3 (d, J = 3.0 Hz), 140.1, 139.6, 127.6, 126.9 (d, J = 8.4 Hz), 121.9, 120.9, 115.1 (d, J = 21.4 Hz), 90.9, 71.9, 59.8, 53.2, 45.6, 39.6, 22.5; 19F NMR (376 MHz, CDCl3) δ -116.99; The HBr salt was precipitated from MeOH; Mp 126–128 °C; Anal. (C40H47F2N3O2·7/2HBr·2H2O) C, H, N.
3-(5-((Dimethylamino)methyl)-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-1-yl)-N,N-dimethylpropan-1-amine (16)
Compound 16 was prepared from 5 (1.0 g, 3.0 mmol) and 37% aq. formaldehyde (0.5 g, 6.0 mmol) according to General Method A (413 mg) in 38% yield. 1H NMR (400 MHz, CDCl3) δ 7.48-7.44 (m, 2H), 7.22-7.20 (m, 2H), 7.15 (s, 1H), 6.99-6.94 (m, 2H), 5.15 (d, J = 12.0 Hz, 1H), 5.11 (d, J = 12.0 Hz, 1H), 3.39 (s, 2H), 2.23-2.10 (m, 16H), 1.53-1.28 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 162.0 (d, J = 243.2 Hz), 143.3, 141.5 (d, J = 3.1 Hz), 139.5, 139.0, 128.6, 127.0 (d, J = 8.3 Hz), 121.9, 121.8, 115.1 (d, J = 20.6 Hz), 91.0, 72.0, 64.4, 59.9, 59.9, 45.7, 45.6, 39.7, 22.5; GC-MS (EI) m/z 356 (M+); The oxalate salt was precipitated from acetone and was hygroscopic; Anal. (C22H29FN2O·2C2H2O4·1/2H2O) C, H, N.
3-(1-(4-Fluorophenyl)-5-(4-nitrophenyl)-1,3-dihydroisobenzofuran-1-yl)-N,N-dimethylpropan-1-amine (19)
A suspension of 178, 30 (0.38 g, 1 mmol), bis(pinacolato)diboron (0.26 g, 1 mmol), potassium acetate (0.3 g, 6 mmol), and PdCl2(dppf)2 (24 mg, 0.06 mmol) in DMF (3 mL) was degassed and heated at 105 °C for 3 h. Solvent was removed under vacuum. The residue was taken up in ether, washed in brine, dried over MgSO4 and evaporated to give the crude intermediate product (0.4 g) in 94% yield. GC-MS (EI) m/z 425 (M+).
To a suspension of this intermediate product (0.21 g, 0.5 mmol), 4-nitrophenyl iodide (0.13 g, 0.5 mmol), and Na2CO3 (0.17 g, 1.5 mmol) in a mixture of solvents DME/H2O (3 mL / 1 mL) was added Pd(PPh3)4 (7 mg, 5 mol%) under Argon. The mixture was heated at 70 °C overnight. The solvent was then removed under reduced pressure, and the residue was extracted with EtOAc. The organic layer was dried over MgSO4 and concentrated to give the crude product, which was then purified by flash column chromatography eluting with CHCl3/MeOH (5:1) to give the pure product (0.14 g) in 60% yield. 1H NMR (400 MHz, CD3OD) δ 8.30 (d, J = 8.8 Hz, 2H), 7.85 (d, J = 8.9 Hz, 2H), 7.69-7.58 (m, 5H), 7.07 (t, J = 8.8 Hz, 2H), 5.25 (d, J = 12 Hz, 2H), 3.15 (t, J = 8 Hz, 2H), 2.80 (s, 6H), 2.35-2.26 (m, 2H), 1.72-1.65 (m, 2H); 13C NMR (100 MHz, CD3OD) δ 165.0, 147.4, 147.2, 144.6, 140.4, 139.2, 127.9, 127.2, 127.0, 126.9, 123.9, 120.4, 115.1, 114.9, 90.6, 71.6, 57.8, 42.2, 37.6, 20.0; 19F NMR (376 MHz, CD3OD) δ -118.3; IR (powder) 1158, 1228 cm−1; GC-MS (EI) m/z 420 (M+); The oxalate salt was precipitated from acetone; Mp 135–138°C; Anal. (C25H25FN2O3· C2H2O4 CH3OH) C, H, N.
3-(5-(4-Aminophenyl)-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-1-yl)-N,N-dimethylpropan-1-amine (20)
Compound 20 was prepared by following the General Suzuki coupling Method C using compound 178, 30 (0.38 g, 1 mmol) and 4-(N-BOC-amino)-phenylboronic eluting with CHCl3/MeOH (5:1) to give the pure product (0.49 g) in 99% yield. The resulting syrup was treated with an HBr solution in MeOH (saturated) for 30 minutes. The reaction mixture was evaporated, neutralized with NH4OH and purified by flash column chromatography eluting with CHCl3/MeOH (5:1) to give pure product (0.12 g) in 30% yield. 1H NMR (400 MHz, CDCl3) δ 7.51-7.31 (m, 9H), 6.99 (t, J = 12 Hz, 2 H), 6.52 (br, 2H), 5.19 (d, J = 5.2 Hz, 2H), 2.31 (m, 2H), 2.21 (m, 2H), 2.19 (s, 6H), 1.54 (m, 1H), 1.48 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 162.7, 160.2, 152.0, 142.8, 141.2, 140.0, 139.5, 138.0, 135.7, 127.9, 127.1, 127.0, 126.7, 122.3, 119.7, 119.0, 115.4, 115.1, 91.0, 80.9, 72.1, 59.8, 45.4, 39.5, 28.6; 19F NMR (376 MHz, CDCl3) δ −117.1; GC-MS (EI) m/z 390 (M+); The oxalate salt was precipitated from 2-PrOH; Mp 120–122°C; Anal. (C25H27FN2O C2H2O4·4/3H2O) C, H, N.
(E)-3-(1-(4-Fluorophenyl)-5-(3-phenylprop-1-en-1-yl)-1,3-dihydroisobenzofuran-1-yl)-N,N-dimethylpropan-1-amine (21)
Compound 21 was prepared by following the General Suzuki coupling Method C using compound 178, 30 (0.5 g, 1.3 mmol) and trans-3-phenyl-1-propen-1-ylboronic acid (0.24 g, 1.5 mmol), eluting with CHCl3/MeOH (10:1) to give the pure product (0.5 g) in 92% yield. 1H NMR (400 MHz, CDCl3) δ 7.40 (m, 2H), 7.30, 7.21 (2 m, 8H), 6.99 (m, 2H), 6.42 (m, 2H), 5.08 (d, J = 7.2 Hz, 2H), 3.54 (d, J = 6.4 Hz, 2H), 3.07 (m, 2H), 2.74 (s, 6H), 2.17 (m, 2H), 1.72 (m, 1H), 1.64 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 162.7, 142.0, 140.0, 139.3, 137.8, 130.3, 130.1, 128.6, 128.5, 126.6, 126.5, 126.2, 126.2, 121.7, 118.7, 115.4, 115.2, 90.2, 71.7, 58.1, 43.0, 39.3, 37.9, 31.0, 19.6; 19F NMR (376 MHz, CDCl3) δ −116.0; GC-MS (EI) m/z 415.3 (M+); The oxalate salt was precipitated from Et2O; Mp 80–81°C; Anal. (C28H30FNO· C2H2O4·3/5H2O) C, H, N.
3-(1-(4-Fluorophenyl)-4-iodo-1,3-dihydroisobenzofuran-1-yl)-N,N-dimethylpropan-1-amine (22)
A suspension of 188 (0.38, 1 mmol), KI (2.46 g, 15 mmol) and CuI (0.95 g, 5 mmol) in HMPA (3 mL) was heated at 150 °C for 3 h. The reaction mixture was purified by flash column chromatography eluting with CHCl3/MeOH (30:1 and 5:1) to give the product (0.25 g) in 59% yield. 1H NMR (400 MHz, CD3OD) δ 7.93 (d, J = 8.0 Hz, 1H), 7.66 (d, J = 7.6 Hz, 1H), 7.55 (m, 1H), 7.30 (m, 1H), 7.06 (m, 3H), 5.03 (d, J = 8.8 Hz, 2H), 3.12(m, 2H), 2.78 (s, 6H), 2.25 (m, 2H), 1.76 (m, 1H), 1.61 (m, 1H); 13C NMR (100 MHz, CD3OD) δ 165.5, 140.0, 137.0, 129.8, 126.8, 121.6, 115.1, 114.9, 114.8, 114.6, 75.8, 57.7, 37.7, 29.5, 19.8, 19.5; 19F NMR (376 MHz, CD3OD) δ −118.1; IR (powder) 1169, 1222 cm−1; GC-MS (EI) m/z 425 (M+); The oxalate salt was precipitated from EtOAc; Mp 141–143°C; Anal. (C19H21FINO·C2H2O4) C, H, N.
4-(1-(4-Bromophenyl)-4-(dimethylamino)-1-hydroxybutyl)-3-(hydroxymethyl)benzonitrile (24)
To a cooled (0 °C) suspension of 5-cyanophthalide (23, 6.36 g, 40 mmol) in dry THF (40 mL) was added a freshly-made solution of 4-bromophenylmagnesium bromide in THF (1.0 M, 45 mL, 44 mmol) drop-wise over 30 min. The reaction was allowed to warm to RT and stirred for 3 h, then cooled to 0 °C. The second freshly made Grignard reagent (3-(dimethylamino)propyl)magnesium chloride (~50 mmol, 0.8 M, 60 mL) was added drop-wise over 30 min. The reaction mixture was allowed to warm to RT and stirred overnight. The reaction mixture was treated with aq NH4Cl solution (sat.), and extracted with ether several times. The organic layer was washed with sat. NaHCO3, brine, dried over MgSO4, and concentrated to give the crude diol product, which was then purified by flash column chromatography eluting with CHCl3/MeOH (10:1, 5:1) to give the pure product (12.4 g) in 80% yield. 1H NMR (400 MHz, CDCl3) δ 7.60, 7.40, 7.20 (3 m, 7H), 4.24 (d, J = 13 Hz, 1H), 4.14 (d, J = 7.2 Hz, 1H), 2.41 (m, 4H), 2.25 (s, 6H), 1.67 (m, 1H), 1.57 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 150.7, 146.1, 142.2, 135.7, 131.1, 130.9, 127.8, 127.1, 120.8, 118.5, 111.5, 64.0, 59.7, 44.6, 44.6, 43.4, 30.9, 22.0; GC-MS (EI) m/z 384, 386 (M-18+); The oxalate salt was precipitated from EtOAc; Mp 154–156 °C; IR (powder) 3253, 2230, 1763 cm−1; Anal. (C20H23BrN2O2·C2H2O4) C, H, N.
1-(4-Bromophenyl)-1-(3-(dimethylamino)propyl)-1,3-dihydroisobenzofuran-5-carbonitrile (25)
To a solution of 24 (11 g, 27 mmol) in CH2Cl2 at 0 °C , was added methanesulfonyl chloride (4.4 mL, 32 mmol) and triethylamine (15 mL) drop-wise. The reaction mixture was stirred overnight at RT, and then extracted from CH2Cl2. The organic layer was washed with aq NaHCO3 (sat.) and brine, dried over MgSO4, concentrated and purified by flash column chromatography eluting with CHCl3/MeOH (5:1) to give pure product (3 g) in 29% yield. 1H NMR (400 MHz, CDCl3) δ 7.62-7.23 (m, 7H), 5.19 (m, 2H), 2.28 (m, 4H), 2.17 (s, 6H), 1.47 (m, 1H), 1.38 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 149.1, 142.8, 140.2, 131.9, 131.6, 128.5, 126.8, 125.2, 124.9, 122.7, 121.5, 111.8, 105.0, 91.1, 59.3, 45.2, 45.1, 38.7, 21.9; GC-MS (EI) m/z 384, 386 (M+); The oxalate salt was precipitated from 2-PrOH; Mp 185–187 °C; Anal. (C20H21BrN2O·C2H2O4) C, H, N.
(E)-1-(3-(Dimethylamino)propyl)-1-(4-styrylphenyl)-1,3-dihydroisobenzofuran-5-carbonitrile (26)
Compound 26 was prepared by following the General Suzuki coupling Method C from 25 (0.3 g, 0.8 mmol) and 2-phenylvinyl boronic acid (0.13 g, 0.82 mmol), eluting with CHCl3/MeOH (10:1) to give the product (0.2 g) in 61% yield; 1H NMR (400 MHz, CDCl3) δ 7.81-6.98 (m, 14H), 5.21 (d, J = 6.4 Hz, 2H), 2.31 (m, 4H), 2.19 (s, 6H), 1.53 (m, 1H), 1.41 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 149.6, 143.0, 140.3, 137.2, 136.6, 131.8, 129.0, 128.7, 127.9, 127.7, 126.7, 126.5, 125.2, 122.8, 118.7, 111.6, 91.4, 71.4, 59.4, 45.2, 38.7, 22.0; GC-MS (EI) m/z 408 (M+). The oxalate salt was precipitated from acetone; Mp 129– 131°C; Anal. (C28H28N2O·3/2C2H2O4) C, H, N.
1-(4-(3-Cyanophenyl)phenyl)-1-(3-(dimethylamino)propyl)-1,3-dihydroisobenzofuran-5-carbonitrile (27)
Compound 27 was prepared by following the General Suzuki coupling Method C from 25 (0.45 g, 1.2 mmol) and 3-cyanophenyl boronic acid (0.17 g, 1.2 mmol), eluting with CHCl3/MeOH (10:1) to give the product (0.1 g) in 20% yield; 1H NMR (400 MHz, CDCl3) δ 7.81-7.46 (m, 11H), 5.22 (d, J = 3.6 Hz, 2H), 2.45 (m, 4H), 2.27 (s, 6H), 1.56 (m, 1H), 1.49 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 149.2, 143.8, 141.7, 140.2, 138.1, 132.0, 131.4, 130.8, 130.6, 129.7, 128.3, 127.3, 125.7, 125.3, 122.8, 118.8, 118.6, 113.0, 112.0, 91.1, 71.4, 58.9, 44.7, 38.7, 21.2; GC-MS (EI) m/z 407 (M+). The oxalate was precipitated from 2-PrOH; Mp 128–130 °C; Anal. (C27H25N3O·3/2 C2H2O4) C, H, N.
1-(4-(3-Aminophenyl)phenyl)1-(3-(dimethylamino)propyl)-1,3-dihydroisobenzofuran-5-carbonitrile (28)
Compound 28 was prepared by following the General Suzuki coupling Method C from 25 (0.6 g, 1.6 mmol) and 3-amino-phenyl boronic acid (0.28 g, 1.8 mmol), eluting with CHCl3/MeOH/NH4OH (10:2:1) to give the product (0.25 g) in 40% yield; 1H NMR (400 MHz, CDCl3) δ 7.60–6.64 (m, 11H), 5.21 (m, 2H), 2.26 (m, 4H), 2.16 (s, 6H), 1.51 (m, 1H), 1.38 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 149.7, 146.7, 142.6, 141.7, 140.5, 140.4, 131.9, 129.7, 127.2, 125.3, 125.2, 122.9, 118.7, 117.6, 114.2, 113.8, 111.6, 91.4, 71.4, 59.4, 45.3, 38.9, 22.1; GC-MS (EI) m/z 397 (M+). The oxalate was precipitated from acetone; Mp 189– 191 °C; Anal. (C26H27N3O·5/2 C2H2O4) C, H, N.
(5-Bromo-2-methoxyphenyl)(4-fluorophenyl)methanol (31)
To a solution of p-fluorophenyl bromide (29, 1.1 mL, 10 mmol) in anhydrous THF (10 mL) at −78 °C, n-butyllithium (4 mL, 2.5 M in hexane, 10 mmol) was added drop-wise. The reaction mixture was allowed to warm to RT and stirred for 3 h. Then a solution of 5-bromo-2-methoxybenzaldehyde (2.15 g, 10 mmol) in THF (10 mL) was added drop-wise. The mixture was stirred overnight at RT, then diluted with ice/H2O, extracted with EtOAc, washed with brine, dried over MgSO4, and concentrated to give a white solid (3 g) in 97% yield, and was used in the next step without further purification; Mp 97–99 °C; GC-MS (EI) m/z 310, 312 (M+).
4-Bromo-2-(chloro(4-fluorophenyl)methyl)-1-methoxybenzene (32)
A solution of 31 (0.31 g, 1mmol) and thionyl chloride (0.07 mL, 1mmol) in CH2Cl2 (8 mL) was stirred at reflux for 3 h. The reaction mixture was diluted with CH2Cl2, washed with brine, dried over MgSO4, and concentrated to a brown syrup (0.2 g) in 60% yield, and was used in the next step without further purification. GC-MS (EI) m/z 328, 330 (M+).
2-(5-Bromo-2-methoxyphenyl)-2-(4-fluorophenyl)acetonitrile (33)
A suspension of 32 (0.2 g, 0.6 mmol) and silver cyanide (0.081 g, 0.6 mmol) in acetonitrile (5 mL) was stirred at reflux for 5 h. The cooled reaction mixture was filtered, the filtrate was concentrated to dryness to give the crude product (0.22 g) in 99% yield. The crude product was used in the next step without further purification. GC-MS (EI) m/z 319, 321 (M+).
2-(5-Bromo-2-methoxyphenyl)-5-(dimethylamino)-2-(4-fluorophenyl)pentanenitrile (34)
To a solution of 33 (0.22 g, 0.6 mmol) in anhydrous THF (20 mL) in a cold bath (acetonitrile/dry ice −44 °C) was added LDA (0.4 mL, 2.5 M, 1 mmol). The mixture was warmed to 0 °C, and a solution of dimethylaminopropyl chloride (1 mL, 1M in ether) was added drop-wise. The resulting mixture was stirred at reflux for 1.5 h, quenched with H2O (2 mL), and evaporated. The residue was extracted with CH2Cl2, washed with H2O, dried over MgSO4, and concentrated to syrup (0.12 g) in 94% yield; 1H NMR (400 MHz, CDCl3) δ 7.63 (d, J = 2.4 Hz, 1H), 7.44 (dd, J1 = 8.8 Hz, J2 = 2.4 Hz, 1H), 7.27 (m, 2H), 6.98 (dd, J = 8.8, 8.4 Hz, 2H), 6.74 (d, J = 9.2 Hz, 1H), 3.59 (s, 3H), 2.52, 2.29 (2 m, 4H), 2.23 (s, 6H), 1.39 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 156.4, 135.8, 132.9, 132.4, 130.3, 130.1, 128.3, 128.2, 121.2, 115.6, 115.3, 114.5, 113.3, 60.6, 59.1, 45.6, 36.6, 23.6; GC-MS (EI) m/z 404, 406 (M+).
5-Bromo-3-(3-(dimethylamino)propyl)-3-(4-fluorophenyl)benzofuran-2(3H)-one (35)
A mixture of 34 (1.22 g, 3 mmol) and HBr (aq 48%, 40 mL) was stirred at reflux overnight. The cooled mixture was basified with 1N NaOH extracted with CH2Cl2, washed with H2O, dried over MgSO4, and concentrated to syrup. It was partially dissolved in ether and filtered. The filtrate (0.4 g) was concentrated and used in the next reaction without purification; yield (47%); 19F NMR (376 MHz, CDCl3) δ −114.1; GC-MS (EI) m/z 391, 393 (M+).
4-Bromo-2-(5-(dimethylamino)-2-(4-fluorophenyl)-1-hydroxypentan-2-yl)phenol (36)
A solution of 35 (0.4 g, 1 mmol) in anhydrous ether (10 mL) was added drop-wise into a suspension of LiAlH4 (0.8 g, 2 mmol) in ether (10 mL) in an ice bath. The reaction mixture was allowed to warm to RT and stirred for a total of 2.5 h, quenched with Na2SO4 (aq) and filtered. The aq layer was extracted with EtOAc, combined with ether filtrate, washed with Na2CO3 (aq), brine, dried over MgSO4, and concentrated to give the product as a clear syrup (0.34 g) in 61% yield; the product was used in the next step without further purification.
3-(5-Bromo-3-(4-fluorophenyl)-2,3-dihydrobenzofuran-3-yl)-N,N-dimethylpropan-1-amine (37)
To a solution of 36 (0.34 g, 0.87 mmol) in CH2Cl2 (15 mL) in an ice bath, was added methanesulfonyl chloride (0.1 mL, 1.3 mmol) and triethylamine (0.36 mL) drop-wise. The reaction mixture was kept on ice for 3 h, extracted from CH2Cl2, washed by aq NaOH and brine, dried over MgSO4 and concentrated. The crude product was purified by flash column chromatography eluting with CHCl3/MeOH (30:1, 10:1) to give a syrup (0.29 g) in 80% yield; 1H NMR (400 MHz, CDCl3) δ 7.31, 7.29 (2 m, 5H), 7.03 (m, 2H), 4.54 (m, 2H), 2.38 (m, 2H), 2.27 (s, 6H), 2.12 (m, 2H), 1.54 (m, 1H), 1.27 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 162.8, 160.4, 159.3, 139.9, 135.2, 131.6, 128.3, 127.8, 115.6, 115.4, 112.3, 111.8, 84.5, 59.3, 53.5, 45.0, 36.5, 22.2; 19F NMR (376 MHz, CDCl3) δ −116.3; GC-MS (EI) m/z 377, 379 (M+); The oxalate was precipitated from acetone; Mp 131–132 °C; Anal. (C19H21BrFNO 2C2H2O4) C, H, N.
1-(4-Fluorophenyl)-1-(3-(methylamino)propyl)-1,3-dihydroisobenzofuran-5-carbonitrile (38).4,7,30
Solid K2CO3 (7.0 g, 50 mmol, 5 eq) was added to a solution of 1 (3.24 g, 10 mmol) in anhydrous 1,2-dichloroethane (25 mL). α-Chloroethylchloroformate (ACE-Cl, 2.86 g, 2 eq) was added drop-wise under an argon atmosphere. After stirring the suspension at reflux for 2h, a second aliquot of ACE-Cl (2.86 g, 2 eq) was added. The reaction was stirred at reflux for an additional 2h, and then cooled to RT. The K2CO3 was filtered off and the solvent was evaporated. The remaining carbamate intermediate in the reaction mixture was destroyed by dissolving the resulting oil in MeOH (50 mL) and stirring the solution at reflux for 2h. The solvent was evaporated and the mixture was converted to the free base in CHCl3 with NH4OH. The product was purified by flash column chromatography eluting with 90% CMA to give the pure product (1.8 g) in 57% yield. 1H NMR (400 MHz, CDCl3) δ 7.57 (dd, J = 7.6, 1.2 Hz, 1H), 7.49 (s, 1H), 7.43-7.36 (m, 3H), 7.02-6.96 (m, 2H), 5.18 (d, J = 12.8 Hz, 1H), 5.13 (d, J = 12.8 Hz, 1H), 2.55 (t, J = 7.2 Hz, 2H), 2.35 (s, 3H), 2.24-2.10 (m, 2H), 1.54-1.26 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 162.1 (d, J = 245.4 Hz), 149.4, 140.4, 139.6 (d, J = 3 Hz), 132.0, 126.8 (d, J = 7.7 Hz), 125.3, 122.9, 118.7, 115.4 (d, J = 21.3 Hz, 2C), 91.2, 71.4, 51.8, 39.0, 36.3, 24.3; 19F NMR (376 MHz, CDCl3) δ −115.76; GC-MS (EI) m/z 310 (M+); The HBr salt was precipitated from MeOH; Anal. (C19H19FN2O·HBr·3/4H2O) C, H, N.
1-(3-((Cyclopropylmethyl)(methyl)amino)propyl)-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-5-carbonitrile (39)
Compound 39 was prepared from 38 (155 mg, 0.5 mmol) and cyclopropanecarbaldehyde (36 mg, 0.5 mmol) according to General Method A (171 mg) in 94% yield. 1H NMR (400 MHz, CDCl3) δ 7.59 (dd, J = 8.0, 1.2 Hz, 1H), 7.50 (s, 1H), 7.44-7.37 (m, 3H), 7.03-6.97 (m, 2H), 5.19 (d, J = 12.8 Hz, 1H), 5.14 (d, J = 12.8 Hz, 1H), 2.35 (t, J = 7.6 Hz, 2H), 2.24-2.07 (m, 7H), 1.53-1.24 (m, 2H), 0.84-0.74 (m, 1H), 0.49-0.44 (m, 2H), 0.06-0.02 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 162.2 (d, J = 244.6 Hz), 149.6, 140.5, 139.7 (d, J = 3.1 Hz), 132.0, 126.9 (d, J = 8.4 Hz), 125.3, 122.9, 118.8, 115.4 (d, J = 21.4 Hz), 111.8, 91.3, 71.4, 62.8, 57.6, 42.4, 39.3, 22.0, 8.9, 4.0; 19F NMR (376 MHz, CDCl3) δ −115.85; GC-MS (EI) m/z 364; The HBr salt was precipitated from MeOH; Anal. (C23H25FN2O·HBr·H2O) C, H, N.
1-(4-Fluorophenyl)-1-(3-(methyl(propyl)amino)propyl)-1,3-dihydroisobenzofuran-5-carbonitrile (40)
Compound 40 was prepared from 38 (155 mg, 0.5 mmol) and propionaldehyde (29 mg, 0.5 mmol) according to General Method A (162 mg) in 92% yield. 1H NMR (400 MHz, CDCl3) δ 7.60-7.57 (m, 2H), 7.50 (s, 1H), 7.44-7.36 (m, 3H), 7.03-6.97 (m, 2H), 5.19 (d, J = 12.8 Hz, 1H), 5.14 (d, J = 12.8 Hz, 1H), 2.28 (t, J = 7.6 Hz, 2H), 2.23-2.05 (m, 7H), 1.52-1.24 (m, 4H), 0.85 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 162.2 (d, J = 244.6 Hz), 149.6, 140.5, 139.8 (d, J = 3.1 Hz), 132.0, 126.9 (d, J = 7.6 Hz), 125.3, 122.9, 118.8, 115.4 (d, J = 20.6 Hz), 111.8, 91.3, 71.4, 59.9, 57.6, 42.3, 39.2, 22.0, 20.5, 12.1; 19F NMR (376 MHz, CDCl3) δ −115.86; GC-MS (EI) m/z 352; The HBr salt was precipitated from MeOH; Anal. (C25H25FN2O·5/4HBr·3/4H2O) C, H, N.
1-(3-(Butyl(methyl)amino)propyl)-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-5-carbonitrile (41)
Compound 41 was prepared from 38 (310 mg, 1.0 mmol) and butyraldehyde (137 mg, 1.0 mmol) according to General Method A in 93% (340 mg) yield. 1H NMR (400 MHz, CDCl3) δ 7.60-7.57 (m, 2H), 7.51-7.49 (m, 1H), 7.44-7.36 (m, 3H), 7.03-6.98 (m, 2H), 5.19 (d, J = 12.8 Hz, 1H), 5.14 (d, J = 12.8 Hz, 1H), 2.29-2.07 (m, 9H), 1.52-1.21 (m, 6H), 0.88 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 162.2 (d, J = 244.6 Hz), 149.6, 140.5, 139.8 (d, J = 3.1 Hz), 132.0, 126.9 (d, J = 8.4 Hz), 125.3, 122.9, 118.8, 115.4 (d, J = 21.3 Hz), 111.8, 91.3, 71.4, 57.7, 57.7, 42.3, 39.2, 29.5, 22.0, 20.8, 14.2; 19F NMR (376 MHz, CDCl3) δ −115.86; GC-MS (EI) m/z 366; The HBr salt was precipitated from MeOH; Anal. (C23H27FN2O·HBr·H2O) C, H, N.
1-(4-Fluorophenyl)-1-(3-(methyl(phenethyl)amino)propyl)-1,3-dihydroisobenzofuran-5-carbonitrile (42)
Compound 42 was prepared from 38 (155 mg, 0.5 mmol) and 2-phenylacetaldehyde (60 mg, 0.5 mmol) according to General Method A (178 mg) in 86% yield. 1H NMR (400 MHz, CDCl3) δ 7.56–7.59 (m, 1H), 7.49 (s, 1H), 7.41-7.37 (m, 2H), 7.33-7.14 (m, 5H), 7.02-6.98 (m, 2H), 5.19 (d, J = 12.8 Hz, 1H), 5.14 (d, J = 12.8 Hz, 1H), 2.74-2.69 (m, 2H), 2.55-2.51 (m, 2H), 2.38 (t, J = 7.2 Hz, 2H), 2.22 (s, 3H), 2.21-2.05 (m, 2H), 1.54-1.26 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 162.1 (d, J = 244.7 Hz), 149.6, 140.5, 140.4, 139.7, 132.0, 128.9, 128.8, 128.5, 126.8 (d, J = 7.6 Hz), 126.1, 125.3, 122.9, 118.8, 125.3, 122.9, 118.7, 115.4 (d, J = 21.3 Hz), 91.3, 71.4, 59.3, 57.3, 42.1, 39.0, 33.7, 21.9; 19F NMR (376 MHz, CDCl3) δ −115.82; The HBr salt was precipitated from MeOH; Mp 130–132 °C; Anal. (C27H27FN2O·HBr·3/2H2O) C, H, N.
1-(4-Fluorophenyl)-1-(3-(methyl(4-phenylbutyl)amino)propyl)-1,3-dihydroisobenzofuran-5-carbonitrile (43)
Compound 43 was prepared from 38 (155 mg, 0.5 mmol) and 4-phenylbutanal (74 mg, 0.5 mmol) according to General Method A in 72% (159 mg) yield. 1H NMR (400 MHz, CDCl3) δ 7.56 (d, J = 7.6 Hz, 1H), 7.49 (s, 1H), 7.42-7.33 (m, 3H), 7.28-7.24 (m, 2H), 7.20-7.14 (m, 2H), 7.02-6.97 (m, 2H), 5.18 (d, J = 12.8 Hz, 1H), 5.13 (d, J = 12.8 Hz, 1H), 2.59 (t, J = 7.2 Hz, 2H), 2.35 (s, 3H), 2.29-2.23 (m, 4H), 2.20-2.06 (m, 5H), 1.62-1.25 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 162.1 (d, J = 244.7 Hz), 149.6, 140.5, 139.7 (d, J = 3 Hz), 132.0, 128.5, 128.4, 126.9 (d, J = 7.7 Hz), 125.8, 125.3, 122.9, 118.8, 115.4 (d, J = 21.4 Hz), 91.3, 71.4, 57.7, 57.6, 42.3, 39.2, 36.0, 29.6, 27.1, 22.0; 19F NMR (376 MHz, CDCl3) δ −115.84; The HBr salt was precipitated from MeOH; Anal. (C29H31FN2O·HBr·5/4H2O) C, H, N.
1-(3-((Benzofuran-2-yl-methyl)(methyl)amino)propyl)-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-5-carbonitrile (44)
Compound 44 was prepared from 38 (155 mg, 0.5 mmol) and benzofuran-3-carbaldehyde (73 mg, 0.5 mmol) according to General Method A (210 mg) in 95% yield. 1H NMR (400 MHz, CDCl3) δ 7.65-7.62 (m, 1H), 7.54–7.57 (m, 1H), 7.49-7.46 (m, 2H), 7.38-7.17 (m, 6H), 7.02-6.96 (m, 2H), 5.09–5.17 (m, 2H), 3.55 (s, 2H), 2.35 (t, J = 7.2 Hz, 2H), 2.25-2.05 (m, 5H), 1.62-1.25 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 162.0 (d, J = 244.6 Hz), 155.4, 149.5, 142.9, 140.3, 139.6 (d, J = 3 Hz), 131.8, 128.0, 128.4, 126.7 (d, J = 7.6 Hz), 125.2, 124.3, 122.7, 122.4, 120.5, 118.7, 115.3 (d, J = 21.4 Hz), 111.7, 111.4, 91.2, 71.3, 56.8, 51.5, 42.2, 38.9, 22.0; 19F NMR (376 MHz, CDCl3) δ −115.83; The HBr salt was precipitated from MeOH; Mp 118–120 °C; Anal. (C28H25FN2O2·HBr·7/5H2O) C, H, N.
1-(3-((Benzo[b]thiophen-3-yl-methyl)(methyl)amino)propyl)-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-5-carbonitrile (45)
Compound 45 was prepared from 38 (160 mg, 0.52 mmol) and benzo[b]thiophene-3-carbaldehyde (84 mg, 0.52 mmol) according to General Method A (216 mg) in 91% (216 mg) yield. 1H NMR (400 MHz, CDCl3) δ 7.92-7.84 (m, 2H), 7.55-7.52 (m, 1H), 7.46 (s, 1H), 7.36-7.21 (m, 6H), 7.01-6.96 (m, 2H), 5.16-5.08 (m, 2H), 3.65 (s, 2H), 2.36 (t, J = 6.8 Hz, 2H), 2.21-2.04 (m, 5H), 1.54-1.29 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 166.6 (d, J = 244.7 Hz), 149.6, 140.8, 140.4, 139.8 (d, J = 3.0 Hz), 139.1, 132.0, 126.8 (d, J = 7.6 Hz), 125.3, 124.4, 123.9, 122.9, 122.8, 115.4 (d, J = 21.4 Hz), 111.8, 91.3, 71.4, 57.2, 56.5, 42.5, 39.0, 22.1; 19F NMR (376 MHz, CDCl3) δ −115.87; The HBr salt was precipitated from MeOH; Mp 128–130 °C; Anal. (C28H25FN2OS·5/4HBr·1/2H2O) C, H, N.
1-(3-((4-(Dimethylamino)benzyl)(methyl)amino)propyl)-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-5-carbonitrile (46)
Compound 46 was prepared from 38 (155 mg, 0.5 mmol) and 4-(dimethylamino)benzaldehyde (75 mg, 0.5 mmol) according to General Method A (200 mg) in 90% yield. 1H NMR (400 MHz, CDCl3) δ 7.57 (dd, J = 7.6, 1.2 Hz, 1H), 7.50-7.48 (m, 1H), 7.42-7.38 (m, 2H), 7.35(d, J = 8.0 Hz, 1H), 7.11-6.98 (m, 2H), 7.70-6.65 (m, 2H), 5.18 (d, J = 13.2 Hz, 1H), 5.13 (d, J = 13.2 Hz, 1H), 3.32 (s, 2H), 2.93 (s, 6H), 2.29 (t, J = 7.2 Hz, 2H), 2.22-2.08 (m, 5H), 1.54-1.28 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 162.1 (d, J = 244.6 Hz), 149.9, 149.6, 140.5, 139.8, 132.0, 130.1, 126.9 (d, J = 8.4 Hz), 126.8, 125.3, 122.9, 118.8, 115.4 (d, J = 21.4 Hz), 112.5, 111.7, 91.3, 71.4, 61.8, 56.9, 42.2, 40.9, 39.1, 22.1; 19F NMR (376 MHz, CDCl3) δ −115.88; The HBr salt was precipitated from MeOH; Mp 184–185 °C; Anal. (C28H30FN3O·2HBr·5/2H2O) C, H, N.
1-(4-Fluorophenyl)-1-(3-(methyl(pyridin-4-yl)amino)propyl)-1,3-dihydroisobenzofuran-5-carbonitrile (47)
Compound 47 was prepared from 38 (190 mg, 0.62 mmol) and isonicotinaldehyde (66 mg, 0.62 mmol) according to General Method A (234 mg) in 94% yield. 1H NMR (400 MHz, CDCl3) δ 8.53-8.50 (m, 2H), 7.60-7.58 (m, 1H), 7.50 (brs, 1H), 7.43-7.35 (m, 3H), 7.21 (d, J = 6.0 Hz, 2H), 7.03-6.98 (m, 2H), 5.18 (d, J = 13.2 Hz, 1H), 5.13 (d, J = 13.2 Hz, 1H), 3.41 (s, 2H), 2.34 (t, J = 7.2 Hz, 2H), 2.26-2.09 (m, 7H), 1.55-1.30 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 162.3 (d, J = 244.6 Hz), 150.0, 149.6, 148.8, 140.5, 139.7, 132.1, 126.9 (d, J = 7.6 Hz), 125.5, 123.9, 122.9, 118.8, 115.6 (d, J = 20.5 Hz), 112.0, 91.3, 71.5, 61.4, 57.5, 42.5, 39.0, 22.2; GC-MS (EI) m/z 401 (M+); The oxalate salt was precipitated from EtOAc; Anal. (C25H24FN3O·2C2H2O4·5/4H2O) C, H, N.
1-(4-Fluorophenyl)-1-(3-(methyl(quinolin-4-ylmethyl)amino)propyl)-1,3-dihydroisobenzofuran-5-carbonitrile (48)
Compound 48 was prepared from 38 (110 mg, 0.35 mmol) and quinoline-4-carbaldehyde (55 mg, 0.35 mmol) according to General Method A (140 mg) in 89% yield. 1H NMR (400 MHz, CDCl3) δ 8.84 (d, J = 4.4 Hz, 1H), 8.21-8.18 (m, 1H), 8.12 (d, J = 2.1 Hz, 1H), 7.73-7.69 (m, 1H), 7.55-7.46 (m, 3H), 7.36-7.20 (m, 4H), 7.0-6.95 (m, 2H), 5.11 (s, 2H), 3.86 (d, J = 14.0 Hz, 1H), 3.82 (d, J = 14.0 Hz, 1H), 2.41 (t, J = 6.8 Hz, 2H), 2.18 (s, 3H), 2.16-2.08 (m, 2H), 1.55-1.30 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 162.1 (d, J = 244.6 Hz), 150.3, 149.5, 148.5, 145.0, 140.4, 139.7, 132.0, 130.1, 129.3, 127.8, 126.8 (d, J = 7.6 Hz), 126.3, 125.3, 124.4, 122.7, 121.5, 118.8, 115.4 (d, J = 21.3 Hz), 111.8, 91.2, 71.39, 59.7, 57.5, 42.5, 39.0, 22.0; 19F NMR (376 MHz, CDCl3) δ −115.74; The HBr salt was precipitated from MeOH; Mp 150–152 °C; Anal. (C29H26FN3O·11/4HBr·3/2H2O) C, H, N.
1-(4-Fluorophenyl)-1-(3-(methyl(naphthalen-2-ylmethyl)amino)propyl)-1,3-dihydroisobenzofuran-5-carbonitrile (49)
Compound 49 was prepared from 38 (190 mg, 0.6 mmol) and 2-naphthaldehyde (94 mg, 0.6 mmol) according to General Method A (148 mg) in 92% yield. 1H NMR (400 MHz, CDCl3) δ 7.84-7.81 (m, 1H), 7.79-7.76 (m, 1H), 7.67 (s, 1H), 7.53 (dd, J = 7.6, 1.2 Hz, 1H), 7.48-7.30 (m, 7H), 7.01-6.96 (m, 2H), 5.15-5.08 9m, 2H), 3.57 (s, 2H), 2.36 (t, J = 6.8 Hz, 2H), 2.24-2.08 (m, 5H), 1.57-1.31 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 162.1 (d, J = 244.7 Hz), 149.6, 140.4, 139.8 (d, J = 3.8 Hz), 132.0, 133.4, 132.8, 131.9, 128.0, 127.8, 127.5, 127.4, 126.9 (d, J = 8.4 Hz), 126.1, 125.7, 125.3, 122.9, 115.4 (d, J = 21.4 Hz), 111.8, 91.3, 71.4, 62.7, 57.3, 42.6, 39.1, 22.2; 19F NMR (376 MHz, CDCl3) δ −115.83; The HBr salt was precipitated from MeOH; Mp 130–132 °C; Anal. (C30H27FN2O·5/4HBr·3/4H2O) C, H, N.
1-(3-(((9H-Fluoren-2-yl)methyl)(methyl)amino)propyl)-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-5-carbonitrile (50)
Compound 50 was prepared from 38 (140 mg, 0.45 mmol) and 9H–fluorene-2-carbaldehyde (87 mg, 0.45 mmol) according to General Method A (202 mg) in 92% yield. 1H NMR (400 MHz, CDCl3) δ 7.77 (d, J = 7.2 Hz, 1H), 7.66 (d, J = 7.6 Hz, 1H), 7.46-7.24 (m, 8H), 7.02-6.97 (m, 2H), 5.15 (d, J = 13.2 Hz, 1H), 5.11 (d, J = 13.2 Hz, 1H), 3.85 (s, 2H), 3.48 (s, 2H), 2.36 (t, J = 7.2 Hz, 2H), 2.25-2.08 (m, 5H), 1.58-1.31 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 162.1 (d, J = 244.7 Hz), 149.6, 143.5, 143.4, 141.7, 140.8, 140.5, 139.8 (d, J = 3.1 Hz), 132.0, 127.8, 126.9, 126.9 (d, J = 8.4 Hz), 125.8, 125.3, 125.2, 122.9, 120.0, 119.9, 119.6, 118.8, 118.8, 115.4 (d, J = 21.4 Hz), 111.8, 91.3, 71.4, 62.7, 57.3, 42.5, 39.1, 36.9, 22.2; 19F NMR (376 MHz, CDCl3) δ −115.80; GC-MS (EI) m/z 366; The HBr salt was precipitated from MeOH; Anal. (C33H29FN2O·5/4HBr·3/4H2O) C, H, N.
1-(3-(((1-(3-(Dimethylamino)propyl)-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-5-yl)methyl)(methyl)amino)propyl)-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-5-carbonitrile (51)
Compound 51 was prepared from 38 (310 mg, 1.0 mmol) and 1-(3-(dimethylamino)propyl)-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-5-carbaldehyde (327 mg, 1.0 mmol) according to General Method A (546 mg) in 88% yield. 1H NMR (400 MHz, CDCl3) δ 7.57 (d, J = 7.6 Hz, 1H), 7.48-7.37 (m, 5H), 7.34 (d, J = 8.0 Hz, 2H), 7.21-7.12 (m, 2H), 7.08 (s, 1H), 7.02-6.95 (m, 4H), 5.16-5.06 (m, 4H), 3.37 (s, 2H), 2.31 (t, J = 7.2 Hz, 2H), 2.24-2.09 (m, 12H), 1.54-1.27 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 162.1 (d, J = 245.4 Hz), 161.9 (d, J = 243.9), 149.5, 143.1, 141.3, 141.3, 140.5, 139.7 (d, J = 3.0 Hz), 139.3, 139.0, 132.0, 128.4, 126.9 (d, J = 7.7 Hz), 126.8 (d, J = 7.6 Hz), 125.3, 121.6, 121.6, 118.8, 115.4 (d, J = 21.4 Hz), 115.0 (d, J = 21.3 Hz), 114.9, 111.8, 91.3, 90.9, 71.9, 71.4, 62.2, 59.8, 57.4, 42.2, 39.6, 39.1, 22.4, 22.1; 19F NMR (376 MHz, CDCl3) δ −115.75, −116.93; The HBr salt was precipitated from MeOH; Mp 146–148 °C; Anal. (C39H41F2N3O2·2HBr·7/4H2O) C, H, N.
tert-Butyl-3-(((3-(5-Cyano-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-1-yl)propyl)(methyl)amino)methyl)-1H-indole-1-carboxylate (52)
Compound 52 was prepared from 38 (420 mg, 1.35 mmol) and tert-butyl 3-formyl-1H–indole-1-carboxylate (synthesis described in S.I.; 245 mg, 1.35 mmol) according to General Method A (630 mg) in 87% yield. 1H NMR (400 MHz, CDCl3) δ 8.13 (s, 1H), 7.65 (d, J = 7.2 Hz, 1H), 7.55 (dd, J = 8, 1.2 Hz, 1H), 7.48-7.17 (m, 7H), 6.96–7.01 (m, 2H), 5.16 (d, J = 12.8 Hz, 1H), 5.12 (d, J = 12.8 Hz, 1H), 3.54 (s, 2H), 2.35 (t, J = 7.2 Hz, 2H), 2.15 (s, 3H), 1.66 (s, 9H), 1.54-1.30 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 162.2 (d, J = 244.6 Hz), 149.8, 140.5, 132.0, 127.0, 126.9, 124.9, 124.6, 123.0, 122.6, 120.2, 118.9, 118.6, 115.5 (d, J = 21.4 Hz), 111.9, 91.4, 71.5, 57.2, 53.4, 42.6, 39.1, 28.4, 22.2.
1-(3-(((1H-Indol-3-yl)methyl)(methyl)amino)propyl)-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-5-carbonitrile (53)
Potassium carbonate (276 mg, 2 mmol) was added to a solution of 52 (539 mg, 1 mmol) in MeOH (10 mL). The solution was stirred at reflux for 1h, allowed to cool to RT, and the solvent was removed under reduced pressure. Water was added to the crude mixture and extracted with CHCl3. The organic layer was dried over MgSO4, and concentrated to give the crude deprotected compound, which was then purified by flash column chromatography eluting with CHCl3/EtOAc/MeOH (5:5:1) to give the pure product (285 mg) in 65% yield. 1H NMR (400 MHz, CDCl3) δ 8.13 (s, 1H), 7.67 (d, J = 8.0 Hz, 1H), 7.53 (m, 1H), 7.46 (s, 1H), 7.38-7.34 (m, 3H), 7.28-7.18 (m, 2H), 7.12-7.08 (m, 2H), 7.01-6.96 (m, 2H), 5.15 (d, J = 12.8 Hz, 1H), 5.11 (d, J = 12.8 Hz, 1H), 3.66 (s, 2H), 2.40-2.36 (m, 2H), 2.23-2.08 (m, 5H), 1.58-1.32 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 162.0 (d, J = 244.6 Hz), 149.5, 140.3, 139.7, 136.2, 131.8, 127.9, 126.7 (d, J = 8.4 Hz), 125.1, 122.8, 119.5,119.5, 118.7, 115.3 (d, J = 20.6 Hz), 111.6, 111.1, 91.2, 71.3, 56.6, 52.4, 91.6, 42.1, 38.9, 21.9; 19F NMR (376 MHz, CDCl3) δ −115.89; The HBr salt was precipitated from MeOH; Mp 144–145 °C; Anal. (C28H26FN3O·9/4HBr) C, H, N.
tert-Butyl-3-(((3-(5-Cyano-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-1-yl)propyl)(methyl)amino)methyl)-5-methoxy-1H-indole-1-carboxylate (54)
Compound 54 was prepared from 38 (360 mg, 1.2 mmol) and tert-butyl 3-formyl-5-methoxy-1H–indole-1-carboxylate (synthesis described in S.I.; 330 mg, 1.2 mmol) according to General Method A (621 mg) in 91% yield. 1H NMR (400 MHz, CDCl3) δ 8.0 (s, 1H), 7.55 (d, J = 8.0 Hz, 1H), 7.45 (s, 1H), 7.40-7.13 (m, 5H), 6.90–7.01 (m, 3H), 5.14 (d, J = 12.8 Hz, 1H), 5.11 (d, J = 12.8 Hz, 1H), 3.73 (s, 3H), 3.50 (s, 2H), 2.34 (t, J = 7.2 Hz, 2H), 2.23-2.08 (m, 5H), 1.65 (s, 9H), 1.56-1.28 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 162.2 (d, J = 244.7 Hz), 155.8, 149.7, 140.5, 139.9, 132.0, 126.9 (d, J = 8.4 Hz), 125.4, 123.0, 118.9, 118.4, 116.0 , 115.5 (d, J = 21.4 Hz), 112.9, 104.0, 91.4, 71.5, 57.1, 55.8, 53.7, 42.5, 39.2, 28.5, 22.2.
1-(4-Fluorophenyl)-1-(3-(((5-methoxy-1H-indol-3-yl)methyl)(methyl)amino)propyl)-1,3-dihydroisobenzofuran-5-carbonitrile (55)
Compound 55 was prepared from 54 (570 mg, 1 mmol) and K2CO3 (176 mg, 2 mmol) according to the method described for 53 (286 mg) in 61% yield. 1H NMR (400 MHz, CDCl3) δ 8.02 (s, 1H), 7.56-7.52 (m, 1H), 7.46 (s, 1H), 7.39-7.34 (m, 2H), 7.31-7.25 (m, 2H), 7.13-7.07 (m, 2H), 7.01-6.96 (m, 2H), 6.87 (dd, J = 8.4, 2.4 Hz, 1H), 5.15 (d, J = 12.8 Hz, 1H), 5.11 (d, J = 12.8 Hz, 1H), 3.77 (s, 2H), 2.46-2.36 (m, 2H), 2.26-2.10 (m, 5H), 1.60-1.33 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 162.1 (d, J = 244.6 Hz), 154.1, 149.4, 140.3, 139.6 (d, J = 3.9 Hz), 131.9, 131.5, 128.3, 126.8 (d, J = 7.6 Hz), 125.2, 125.1, 122.8, 118.8, 115.4 (d, J = 21.4 Hz), 112.1, 101.4, 91.2, 71.3, 56.4, 55.9, 53.0, 41.8, 38.9, 21.7; The HBr salt was precipitated from MeOH; Mp 151–152 °C; Anal. (C29H28FN3O2·2HBr·H2O) C, H, N.
1-(3-((2-(1,3-Dioxoisoindolin-2-yl)ethyl)(methyl)amino)propyl)-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-5-carbonitrile (56)
Compound 56 was prepared from 38 (310 mg, 1 mmol) and 2-(1,3-dioxoisoindolin-2-yl)acetaldehyde (synthesis described in S.I.; 189 mg, 1 mmol) according to General Method A in 96% (463 mg) yield. 1H NMR (400 MHz, CDCl3) δ 7.78 (dd, J = 5.2, 2.8 Hz, 2H), 7.69 (dd, J = 4.8, 2.8 Hz, 1H) 7.55 (dd, J = 8.0, 1.2 Hz, 1H), 7.46 (s, 1H), 7.33-7.24 (m, 3H), 6.98-6.93 (m, 2H), 5.14 (d, J = 12.8 Hz, 1H), 5.09 (d, J = 12.8 Hz, 1H), 3.75 (t, J = 6.8 Hz, 2H), 2.62-2.52 (m, 2H), 2.33 (t, J = 6.4 Hz, 2H), 2.17 (s, 3H), 2.11-1.95 (m, 2H), 1.43-1.15 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 168.6, 162.2 (d, J = 244.6 Hz), 149.7, 140.5, 139.6 (d, J = 3.1 Hz), 134.1, 132.3, 132.0, 126.9 (d, J = 7.6 Hz), 125.4, 123.3, 122.99, 118.9, 115.5 (d, J = 21.3 Hz), 111.8, 91.3, 71.5, 57.4, 55.0, 42.1, 38.7, 36.1, 23.0; 19F NMR (376 MHz, CDCl3) δ −115.01; Mp 134–135 °C; Anal. (C25H27FN2O· C2H2O4·4/3H2O) C, H, N.
1-(3-((2-Aminoethyl)(methyl)amino)propyl)-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-5-carbonitrile (57)
To a solution of 56 (300 mg, 0.62 mmol) in EtOH was added hydrazine (80 mg, 2.5 mmol), and the mixture was stirred at reflux for 3h. The cooled reaction mixture was evaporated under vacuum. The solid residue was partitioned between CHCl3 and 20% K2CO3 solution, and the organic layer was collected and dried over MgSO4. The solvent was removed under vacuum to give pure product in 95% (208 mg) yield. 1H NMR (400 MHz, CDCl3) δ 7.60 (d, J = 8.0 Hz, 1H), 7.50 (s, 1H), 7.43-7.37 (m, 3H), 7.03-6.98 (m, 2H), 5.19 (d, J = 12.8 Hz, 1H), 5.14 (d, J = 12.8 Hz, 1H), 2.71 (t, J = 6.4 Hz, 2H), 2.332.28 (m, 4H), 2.24-2.05 (m, 5H), 1.52-1.24 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 162.0 (d, J = 244.6 Hz), 149.5, 140.3, 139.6 (d, J = 3.0 Hz), 131.9, 126.9 (d, J = 8.4 Hz), 125.2, 122.8, 118.7, 115.3 (d, J = 21.3 Hz), 111.7, 91.1, 71.3, 60.5, 57.8, 42.0, 39.5, 39.0, 21.9; 19F NMR (376 MHz, CDCl3) δ −115.80; GC-MS (EI) m/z 323 ((M+-CH2NH2); The HBr salt was precipitated from MeOH; Anal. (C21H24FN3O·2HBr·5/4H2O) C, H, N.
1-(4-Fluorophenyl)-1-(3-(methyl(1,4-dioxaspiro[4.5]decan-8-yl)amino)propyl)-1,3-dihydroisobenzofuran-5-carbonitrile (58)
Compound 58 was prepared from 38 (850 mg, 2.75 mmol) and 1,4-dioxaspiro[4.5]decan-8-one (390 mg, 2.75 mmol) according to General Method A (1.16 g) in 94% yield. 1H NMR (400 MHz, CDCl3) δ 7.61-7.57 (m, 1H), 7.51-7.49 (m, 1H), 7.43-7.36 (m, 3H), 7.03-6.98 (m, 2H), 5.20 (d, J = 12.4 Hz, 1H), 5.14 (d, J = 12.4 Hz, 1H), 3.92 (s, 2H), 3.73 (s, 2H), 2.39-2.35 (m, 2H), 2.22-2.08 (m, 5H), 1.92-1.21 (m, 11H) ; GC-MS (EI) m/z 450 (M+).
1-(4-Fluorophenyl)-1-(3-(methyl(4-oxocyclohexyl)amino)propyl)-1,3-dihydroisobenzofuran-5-carbonitrile (59)
To a solution of 58 (1.16 g, 2.6 mmol) in anhydrous ether (8 mL), was added 3N HCl (4 mL). The mixture was stirred at RT for 12 h. The solvent was then removed under reduced pressure. Water was added to the resultant residue and extracted with CHCl3. The organic layer was dried over MgSO4 and concentrated. Crude product was purified by flash column chromatography eluting with CHCl3:MeOH (9:1) to give the pure product (770 mg) in 73% yield. 1H NMR (400 MHz, CDCl3) δ 7.62-7.59 (m, 1H), 7.51-7.49 (m, 1H), 7.44-7.38 (m, 3H), 7.04-6.98 (m, 2H), 5.20 (d, J = 12.8 Hz, 1H), 5.15 (d, J = 12.8 Hz, 1H), 2.78-2.71 (m, 1H), 2.46-2.10 (m, 11H), 2.01-1.92 (m, 2H), 1.78-1.69 9m, 2H), 1.54-1.24 (m, 2H) ; GC-MS (EI) m/z 406 (M+).
1-(3-((4-(1H-Indol-3-yl)cyclohex-3-en-1-yl)(methyl)amino)propyl)-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-5-carbonitrile (60)
To a solution of 59 (770 mg, 1.9 mmol) and indole (222 mg, 1.9 mmol) in EtOH (15 mL), was added pyrrolidine (405 mg, 5.7 mmol). The mixture was stirred at reflux for 12 h. The reaction was allowed to cool to RT, and then solvent was removed under reduced pressure. Water was added to the resultant residue and extracted with CHCl3. The organic layer was dried over MgSO4 and concentrated. Crude product was purified by flash column chromatography eluting with CHCl3:MeOH (9:1)) to give the pure product (500 mg) in 52 % yield. 1H NMR (400 MHz, CDCl3) δ 8.11 (brs, 1H), 7.88 (d, J = 7.6 Hz, 1H), 7.58 (t, J = 7.2 Hz, 1H), 7.50 (s, 1H), 7.50-7.45 (m, 4H), 7.22-7.12 (m, 3H), 7.03-6.98 (m, 2H), 6.21 (brm, 1H), 5.21 (d, J = 12.8 Hz, 1H), 5.14 (d, J = 12.8 Hz, 1H), 2.85-1.95 (m, 12H), 1.75-1.25 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 162.3 (d, J = 244.6 Hz), 149.6, 140.6, 139.8, 137.0, 132.1, 132.1, 131.3, 127.0, 127.0, 125.4, 123.1, 122.4, 121.2, 120.9, 120.7, 120.2, 118.9, 118.7, 115.6 (d, J = 21.4 Hz), 91.4, 71.5, 59.3, 53.6, 39.3, 37.8, 29.5, 27.5, 25.9, 22.6; 19F NMR (376 MHz, CDCl3) δ −115.76; The HBr salt was precipitated from MeOH; Mp 184–185 °C; Anal. (C33H32FN3O.3/2HBr) C, H, N.
Radioligand Binding Assay for SERT
Brains from male Sprague-Dawley rats weighing 200–225 g (Taconic Labs) were removed, midbrain was dissected and rapidly frozen. Membranes were prepared by homogenizing tissues in 20 volumes (w/v) of 50 mM Tris, 120 mM NaCl, 5 mM KCl pH 7.4 at 25°C (experimental buffer), using a Brinkman Polytron (setting 6 for 20 sec.) and centrifuged at 20,000 × g for 10 min at 4°C. The resulting pellet was resuspended in buffer and recentrifuged. The final pellet was resuspended in cold buffer to a concentration of 15 mg/ml (original wet weight).
Ligand binding experiments were conducted in assay tubes containing 0.5 mL of buffer for 60 min at 25°C (RT). Each tube contained 1.4nM [3H]1 (specific activity 80 Ci/mMol) PerkinElmer Life Science, and 1.5 mg midbrain tissue. Nonspecific binding was determined using 10 µM fluoxetine (Sigma). Incubations were terminated by rapid filtration through Whatman GF/B filters (presoaked in 0.3% polyethylenimine in water to reduce non-specific binding) using a Brandel cell harvester (Brandel Instruments Gaithersburg, MD). The filters were washed twice with 5mL cold buffer and transferred to scintillation vials. Beckman Ready Safe (3.0 mL) was added and the vials were counted the next day using a Beckman 6000 liquid scintillation counter (Beckman Coulter Instruments, Fullerton, California). Data were analyzed by using GraphPad Prism software (San Diego, California).
Radioligand Binding Assay for NET
Rat brain membranes were prepared as described for SERT. Ligand binding experiments were conducted in assay tubes containing 0.5 mL buffer for 180 min at 0–4°C. Each tube contained 0.5 nM [3H]Nisoxetine (PerkinElmer Life Science) and 8 mg frontal cortex tissue (original wet weight). Nonspecific binding was determined using 1uM desipramine (Sigma). The rest of the procedure was identical to that described for SERT.
SERT S2 displacement assays
Membrane Preparation
Membranes were prepared from COS-7 cells 2 days after transient transfection with the cDNA of the human SERT inserted into the pUbi1z expression vector38 using the Lipo2000 transfection protocol (Invitrogen) as described previously39 after detachment with 10 mM EDTA in PBS, cells were lysed with one ultrasound burst (Branson Sonifier with microtip) in membrane buffer (120 mM NaCl, 5 mM KCl, 1.2 mM CaCl2, 1.2 mM Mg Cl2, 25 mM HEPES, pH 7.5), pelleted (4900 ×G, 10’ Sigma SK 15 swing-out rotor) and resuspended in membrane buffer containing 0.3 M sucrose. The membranes were stored at −80 °C until use. The freeze-thaw procedure of the membranes results in a decreased Bmax for [3H]S-1 to approximately 40% of the freshly prepared membranes (data not shown).
[3H]S-1 dissociation rate assay
[3H]S-1 60 Ci/mmol was custom synthesized by Ubichem Pharma Service, Hungary. Dissociation rates were measured as previously described.27 In brief, COS-7 cell membranes expressing SERT were preincubated in membrane buffer for 30 min with 15 nM [3H]S-1 at 0 0C. Dissociation was initiated by diluting the samples 12X with 20 °C membrane buffer containing 0.1 µM paroxetine (note that 0.1 µM paroxetine has no allosteric effect on [3H]S-1 dissociation at the indicated concentration.)23 The dissociation was assessed over 5 – 90 minutes and stopped by rapid filtration of the samples through GF/B filters using a Tomtec cell harvester and washed for 20 sec with ice-cold 0.2 M NaCl. The amount of bound [3H]S-1 was determined using a Wallac microplate scintillation counter. Non-specific binding was determined with 0.1 µM paroxetine at 37 °C for 90 minutes to allow dissociation of all [3H]S-1 bound to the orthosteric site in hSERT. Dissociation rates were determined in at least three independent experiments for all the drugs tested.
Data Calculations
Receptor-ligand complexes dissociate according to first-order kinetics (B=B0e-kt), allowing calculation of the dissociation rate constant “koff” expressed as the half-life t½=ln2/k from log-transformed plots of residual binding versus time of dissociation. Dissociation rates were expressed as the half-lives of the receptor-ligand complexes after calculating “koff” from the binding data for the individual curves using non-linear regression analysis.
Allosteric potency was determined for some of the drugs, and was calculated as previously described.25 The calculated dissociation rate constants (k[drug]) at different drug concentrations were expressed relative to the dissociation rate constant without the presence of the drug (kbuffer ). The allosteric potency was determined as the drug concentration, which impairs the dissociation rate by 50% compared with dissociation in membrane buffer containing 0.1 µM paroxetine. IC50 values were calculated from concentration effect curves of normalized dissociation ratios (k[drug]/kbuffer) versus log[drug] and are shown as mean values calculated from means pIC50 and the S.E. interval from pIC50± S.E. All data were subjected to linear and non-linear regression analysis using Prism version 5.0 (GraphPad Software inc., San Diego, CA) and performed on data sets for at least three independent experiments.
Supplementary Material
Scheme 4.

Synthesis of a 2,3-dihydrobenzofuran analoguea
aReagents and conditions: (a) n-BuLi, THF, −78 °C, 3 h; (b) 5-bromo-2-methoxybenzaldehyde, THF, RT, overnight; (c) SOCl2, CH2Cl2, reflux, 3 h; (d) AgCN, CH3CN, reflux, 5 h; (e) 3-chloro-N,N-dimethylpropan-1-amine, LDA, THF, reflux 1.5 h; (f) HBr/H2O, reflux, overnight; (g) LiAlH4, ether, 0 °C to RT, 2.5 h; (h) MsCl, NEt3, 0 °C, 3 h.
Acknowledgment
This work was supported by the NIDA Intramural Research Program (AHN and JLK). Danish Independent Research Council – Sapere Aude (CJL), and the Lundbeck Foundation (CJL). BAK was supported by an NIH visiting postdoctoral fellowship, PZ was supported by an NIH Intramural Research Training Award (IRTA) postdoctoral fellowship and GCC was supported by a post-baccalaureate IRTA fellowship. The authors would like to thank Dr. Lei Shi, Weill Medical School of Cornell University for advising us on the S-1 docking models in the design of Figure 1, Dr. Vivek Kumar (NIDA-IRP) for his critical reading of an earlier version of this manuscript, Dr. Dwight Williams (NIDA-IRP summer student) for his contributions to the synthesis of several intermediates described herein and Bente Bennike (UC) for excellent technical assistance.
Abbreviations
- NSS
Neurotransmitter:Sodium Symporter
- ACE-Cl
1-chloroethyl chloroformate
- SERT
serotonin transporter
- NET
norephinephrine transporter
- LeuT
Leucine Transporter
- S1
primary SERT binding site
- S2
secondary SERT binding site
- SSRI
Selective Serotonin Reuptake Inhibitor
- TCA
Tricyclic antidepressant
Footnotes
Supporting Information Available
Elemental analysis results and a supplemental table of data for compounds not described in the present manuscript are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Kristensen AS, Andersen J, Jorgensen TN, Sorensen L, Eriksen J, Loland CJ, Stromgaard K, Gether U. SLC6 neurotransmitter transporters: structure, function, and regulation. Pharmacol. Rev. 2011;63:585–640. doi: 10.1124/pr.108.000869. [DOI] [PubMed] [Google Scholar]
- 2.Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl--dependent neurotransmitter transporters. Nature. 2005;437:215–223. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- 3.Sorensen L, Andersen J, Thomsen M, Hansen SM, Zhao X, Sandelin A, Stromgaard K, Kristensen AS. Interaction of Antidepressants with the Serotonin and Norepinephrine Transporters: Mutational Studies of the S1 Substrate Binding Pocket. J. Biol. Chem. 2012;287:43694–43707. doi: 10.1074/jbc.M112.342212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Andersen J, Stuhr-Hansen N, Zachariassen L, Toubro S, Hansen SM, Eildal JN, Bond AD, Bogeso KP, Bang-Andersen B, Kristensen AS, Stromgaard K. Molecular determinants for selective recognition of antidepressants in the human serotonin and norepinephrine transporters. Proc. Natl. Acad. Sci. U. S. A. 2011;108:12137–12142. doi: 10.1073/pnas.1103060108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andersen J, Olsen L, Hansen KB, Taboureau O, Jorgensen FS, Jorgensen AM, Bang-Andersen B, Egebjerg J, Stromgaard K, Kristensen AS. Mutational mapping and modeling of the binding site for (S)-citalopram in the human serotonin transporter. J. Biol. Chem. 2010;285:2051–2063. doi: 10.1074/jbc.M109.072587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andersen J, Taboureau O, Hansen KB, Olsen L, Egebjerg J, Stromgaard K, Kristensen AS. Location of the antidepressant binding site in the serotonin transporter: importance of Ser-438 in recognition of citalopram and tricyclic antidepressants. J. Biol. Chem. 2009;284:10276–10284. doi: 10.1074/jbc.M806907200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eildal JN, Andersen J, Kristensen AS, Jorgensen AM, Bang-Andersen B, Jorgensen M, Stromgaard K. From the selective serotonin transporter inhibitor citalopram to the selective norepinephrine transporter inhibitor talopram: synthesis and structure-activity relationship studies. J. Med. Chem. 2008;51:3045–3048. doi: 10.1021/jm701602g. [DOI] [PubMed] [Google Scholar]
- 8.Zhang P, Cyriac G, Kopajtic T, Zhao Y, Javitch JA, Katz JL, Newman AH. Structure-activity relationships for a novel series of citalopram (1-(3-(dimethylamino)propyl)-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-5-carbon itrile) analogues at monoamine transporters. J. Med. Chem. 2010;53:6112–6121. doi: 10.1021/jm1005034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singh SK, Yamashita A, Gouaux E. Antidepressant binding site in a bacterial homologue of neurotransmitter transporters. Nature. 2007;448:952–956. doi: 10.1038/nature06038. [DOI] [PubMed] [Google Scholar]
- 10.Zhou Z, Zhen J, Karpowich NK, Goetz RM, Law CJ, Reith ME, Wang DN. LeuT-desipramine structure reveals how antidepressants block neurotransmitter reuptake. Science. 2007;317:1390–1393. doi: 10.1126/science.1147614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou Z, Zhen J, Karpowich NK, Law CJ, Reith ME, Wang DN. Antidepressant specificity of serotonin transporter suggested by three LeuT-SSRI structures. Nat. Struct. Mol. Biol. 2009;16:652–657. doi: 10.1038/nsmb.1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao Y, Terry DS, Shi L, Quick M, Weinstein H, Blanchard SC, Javitch JA. Substrate-modulated gating dynamics in a Na+-coupled neurotransmitter transporter homologue. Nature. 2011;474:109–113. doi: 10.1038/nature09971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quick M, Shi L, Zehnpfennig B, Weinstein H, Javitch JA. Experimental conditions can obscure the second high-affinity site in LeuT. Nat. Struct. Mol. Biol. 2012;19:207–211. doi: 10.1038/nsmb.2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koldso H, Severinsen K, Tran TT, Celik L, Jensen HH, Wiborg O, Schiott B, Sinning S. The two enantiomers of citalopram bind to the human serotonin transporter in reversed orientations. J. Am. Chem. Soc. 2010;132:1311–1322. doi: 10.1021/ja906923j. [DOI] [PubMed] [Google Scholar]
- 15.Sinning S, Musgaard M, Jensen M, Severinsen K, Celik L, Koldso H, Meyer T, Bols M, Jensen HH, Schiott B, Wiborg O. Binding and orientation of tricyclic antidepressants within the central substrate site of the human serotonin transporter. J. Biol. Chem. 2010;285:8363–8374. doi: 10.1074/jbc.M109.045401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Henry LK, Field JR, Adkins EM, Parnas ML, Vaughan RA, Zou MF, Newman AH, Blakely RD. Tyr-95 and Ile-172 in transmembrane segments 1 and 3 of human serotonin transporters interact to establish high affinity recognition of antidepressants. J. Biol. Chem. 2006;281:2012–2023. doi: 10.1074/jbc.M505055200. [DOI] [PubMed] [Google Scholar]
- 17.Wennogle LP, Meyerson LR. Serotonin modulates the dissociation of [3H]imipramine from human platelet recognition sites. Eur. J. Pharmacol. 1982;86:303–307. doi: 10.1016/0014-2999(82)90333-8. [DOI] [PubMed] [Google Scholar]
- 18.Plenge P, Mellerup ET. Antidepressive drugs can change the affinity of [3H]imipramine and [3H]paroxetine binding to platelet and neuronal membranes. Eur J. Pharmacol. 1985;119:1–8. doi: 10.1016/0014-2999(85)90314-0. [DOI] [PubMed] [Google Scholar]
- 19.Plenge P, Mellerup ET, Laursen H. Affinity modulation of [3H]imipramine, [3H]paroxetine and [3H]citalopram binding to the 5-HT transporter from brain and platelets. Eur. J. Pharmacol. 1991;206:243–250. doi: 10.1016/s0922-4106(05)80025-2. [DOI] [PubMed] [Google Scholar]
- 20.Plenge P, Mellerup ET. An affinity-modulating site on neuronal monoamine transport proteins. Pharmacol. Toxicol. 1997;80:197–201. doi: 10.1111/j.1600-0773.1997.tb00396.x. [DOI] [PubMed] [Google Scholar]
- 21.Chen F, Larsen MB, Neubauer HA, Sanchez C, Plenge P, Wiborg O. Characterization of an allosteric citalopram-binding site at the serotonin transporter. J. Neurochem. 2005;92:21–28. doi: 10.1111/j.1471-4159.2004.02835.x. [DOI] [PubMed] [Google Scholar]
- 22.Zhong H, Haddjeri N, Sanchez C. Escitalopram, an antidepressant with an allosteric effect at the serotonin transporter-a review of current understanding of its mechanism of action. Psychopharmacology (Berl) 2012;219:1–13. doi: 10.1007/s00213-011-2463-5. [DOI] [PubMed] [Google Scholar]
- 23.Plenge P, Gether U, Rasmussen SG. Allosteric effects of R- and S-citalopram on the human 5-HT transporter: evidence for distinct high- and low-affinity binding sites. Eur. J. Pharmacol. 2007;567:1–9. doi: 10.1016/j.ejphar.2007.03.055. [DOI] [PubMed] [Google Scholar]
- 24.Zhong H, Hansen KB, Boyle NJ, Han K, Muske G, Huang X, Egebjerg J, Sanchez C. An allosteric binding site at the human serotonin transporter mediates the inhibition of escitalopram by R-citalopram: kinetic binding studies with the ALI/VFL-SI/TT mutant. Neurosci. Lett. 2009;462:207–212. doi: 10.1016/j.neulet.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 25.Neubauer HA, Hansen CG, Wiborg O. Dissection of an allosteric mechanism on the serotonin transporter: a cross-species study. Mol. Pharmacol. 2006;69:1242–1250. doi: 10.1124/mol.105.018507. [DOI] [PubMed] [Google Scholar]
- 26.Chen F, Larsen MB, Sanchez C, Wiborg O. The S-enantiomer of R,Scitalopram, increases inhibitor binding to the human serotonin transporter by an allosteric mechanism. Comparison with other serotonin transporter inhibitors. Eur. Neuropsychopharmacol. 2005;15:193–198. doi: 10.1016/j.euroneuro.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 27.Plenge P, Shi L, Beuming T, Te J, Newman AH, Weinstein H, Gether U, Loland CJ. Steric hindrance mutagenesis in the conserved extracellular vestibule impedes allosteric binding of antidepressants to the serotonin transporter. J. Biol. Chem. 2012;287:39316–39326. doi: 10.1074/jbc.M112.371765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sarker S, Weissensteiner R, Steiner I, Sitte HH, Ecker GF, Freissmuth M, Sucic S. The high-affinity binding site for tricyclic antidepressants resides in the outer vestibule of the serotonin transporter. Mol. Pharmacol. 2010;78:1026–1035. doi: 10.1124/mol.110.067538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang H, Gouaux E. Substrate binds in the S1 site of the F253A mutant of LeuT, a neurotransmitter sodium symporter homologue. EMBO Rep. 2012;13:861–8666. doi: 10.1038/embor.2012.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bigler AJ, Bogeso KP, Toft A, Hansed V. Quantitative structure-activity relationships in a series of selective 5-HT uptake inhibitors. Eur. J. Med. Chem. -Chim. Ther. 1977;12:289–295. [Google Scholar]
- 31.Siracusa MA, Salerno L, Modica MN, Pittalà V, Romeo G, Amato ME, Nowak M, Bojarski AJ, Mereghetti I, Cagnotto A, Mennini T. Synthesis of new arylpiperazinylalkylthiobenzimidazole, benzothiazole, or benzoxazole derivatives as potent and selective 5-HT1A serotonin receptor ligands. J. Med. Chem. 2008;51:4529–4538. doi: 10.1021/jm800176x. [DOI] [PubMed] [Google Scholar]
- 32.Newman AH, Grundt P, Cyriac G, Deschamps JR, Taylor M, Kumar R, Ho D, Luedtke RR. N-(4-(4-(2,3-Dichloro- or 2-methoxyphenyl)piperazin-1-yl)butyl)heterobiarylcarboxamides with functionalized linking chains as high affinity and enantioselective D3 receptor antagonists. J. Med Chem. 2009;52:2559–2570. doi: 10.1021/jm900095y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deskus JA, Epperson JR, Sloan CP, Cipollina JA, Dextraze P, Qian-Cutrone J, Gao Q, Ma B, Beno BR, Mattson GK, Molski TF, Krause RG, Taber MT, Lodge NJ, Mattson RJ. Conformationally restricted homotryptamines 3. Indole tetrahydropyridines and cyclohexenylamines as selective serotonin reuptake inhibitors. Bioorg. Med. Chem. Lett. 2007;17:3099–3104. doi: 10.1016/j.bmcl.2007.03.040. [DOI] [PubMed] [Google Scholar]
- 34.Bogeso K, Sanchez C. The Discovery of Citalopram and its Refinement to Escitalopram. In: Fischer C, Ganellin R, Rotella DP, editors. Analogue-based Drug Discovery III. 1st ed. Weinheim, Germany: Wiley-VCH Verlag GmbH & Co; 2013. pp. 269–294. [Google Scholar]
- 35.Xu L, Izenwasser S, Katz JL, Kopajtic T, Klein-Stevens C, Zhu N, Lomenzo SA, Winfield L, Trudell ML. Synthesis and biological evaluation of 2-substituted 3beta-tolyltropane derivatives at dopamine, serotonin, and norepinephrine transporters. J. Med. Chem. 2002;45:1203–1210. doi: 10.1021/jm010453u. [DOI] [PubMed] [Google Scholar]
- 36.Zhang P, Jorgensen TJ, Loland CJ, Newman AH. A Rhodamine-Labeled Citalopram Analogue as a High-Affinity Fluorescent Probe for the Serotonin Transporter. Bioorg. Med. Chem. Lett. 2013;23:323–326. doi: 10.1016/j.bmcl.2012.10.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Biessen EA, Horn AS, Robillard GT. Partial purification of the 5-hydroxytryptamine-reuptake system from human blood platelts using citalopram-derived affinity resin. Biochemistry. 1990;29:3349–3354. doi: 10.1021/bi00465a029. [DOI] [PubMed] [Google Scholar]
- 38.Rasmussen SG, Gether U. Purification and fluorescent labeling of the human serotonin transporter. Biochemistry. 2005;44:3494–3505. doi: 10.1021/bi048022b. [DOI] [PubMed] [Google Scholar]
- 39.Beuming T, Bergmann ML, Shi L, Gracia L, Raniszewska K, Newman AH, Javitch JA, Weinstein H, Gether U, Loland CJ. Mapping of the Binding Sites for Cocaine and Dopamine in the Dopamine Transporter. Nature Neurosci. 2008;11:780–789. doi: 10.1038/nn.2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.