

Abstract
Opioid receptors are widely distributed in the human body and are crucially involved in numerous physiological processes. These include pain signaling in the central and the peripheral nervous system, reproduction, growth, respiration, and immunological response. Opioid receptors additionally play a major role in the gastrointestinal (GI) tract in physiological and pathophysiological conditions. This review discusses the physiology and pharmacology of the opioid system in the GI tract. We additionally focus on GI disorders and malfunctions, where pathophysiology involves the endogenous opioid system, such as opioid-induced bowel dysfunction, opioid-induced constipation or abdominal pain. Based on recent reports in the field of pharmacology and medicinal chemistry, we will also discuss the opportunities of targeting the opioid system, suggesting future treatment options for functional disorders and inflammatory states of the GI tract.
Keywords: Abdominal pain, Functional gastrointestinal disorders, Inflammatory bowel diseases, Opioid-induced bowel dysfunction, Opioid receptors and their ligands
Introduction
Opioids are broadly used medical and recreational psychoactive substances worldwide. For centuries, they have been used for acute and chronic treatment of moderate to severe pain, in particular in cancer patients. Opioids have also been administered in patients after surgical interventions to achieve sufficient post-operative pain control. Major limitations of prolonged opioid use result from severe adverse effects, including slowing of GI motility, respiratory depression, development of tolerance and physical dependence.
The analgesic effect of opioids implies their action in the central nervous system. However, opioids are also active in the periphery and this encouraged their use for therapeutic purposes also in the GI tract, like in the treatment of diarrhea or abdominal pain. Identification of opioid receptors in the GI tract and characterisation of their role in GI pathophysiology made them an attractive pharmacological target for numerous pathophysiological conditions.
In this review, we discuss the physiology and pharmacology of the opioid system, in particular in the GI tract. We additionally focus on GI disorders and malfunction, where pathophysiology is related to the endogenous opioid system, such as opioid-induced bowel dysfunction (OBD), opioid-induced constipation (OIC) or abdominal pain. Based on recent reports in pharmacology and medicinal chemistry, we will also explore the possibilities of targeting the opioid system for the treatment of functional disorders and inflammatory states of the GI tract.
Opioid receptors
The endogenous opioid system is composed of cell surface receptors [1] and their endogenous ligands [2]. Opioid receptors were divided into three major types: μ (mu, MOR), δ (delta, DOR), and κ (kappa, KOR) [2–4]. All three opioid receptor types were cloned in the 1990s, first DOR from mice [5], followed by KOR [6, 7] and MOR [8]. Over the years opioid receptors were characterized at biochemical and pharmacological levels. Further division into subtypes according to their localization, ligands and function has been proposed. However, the classification of opioid receptors into subtypes is still controversial due to unclear criteria, which would enable their proper categorization [9].
Opioid receptors as members of the G protein-coupled receptor (GPCR) family
Opioid receptors belong to the family of G protein-coupled receptors (GPCR), one of the largest protein families in mammals. Opioid receptors are integral membrane proteins, coupled to heterotrimeric Gi/o proteins. The structure of opioid receptors consists of seven hydrophobic transmembrane domains TM I–VII, three intracellular hydrophobic (i1–i3) and three extracellular (e1–e3) loops, a glycosylated amino and a carboxyl terminus (Fig. 1). The intracellular loop i3 is identical in 20–23 amino acids in all opioid receptors [10, 11]. The C-terminal end is composed of 59, 51, 47 amino acids for MOR, DOR and KOR, respectively, with highly conserved sequence (identical 10–12 amino acids for all opioid receptors) [12].
Fig. 1.
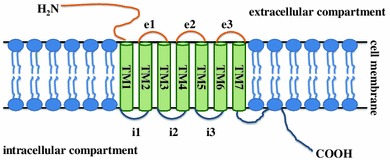
General structure of G protein-coupled receptors. Opioid receptors are integral membrane proteins, coupled to heterotrimeric G proteins. The structure of opioid receptors consists of seven hydrophobic transmembrane domains TM I–VII, three intracellular hydrophobic (i1–i3) and three extracellular (e1–e3) loops, a glycosylated amino and a carboxyl terminus
It has been demonstrated that the transmembrane domains TM V-VII are required for ligand binding in DOR [13]. The other transmembrane domain TM IV and the extracellular loop e2 are responsible for ligand binding in KOR, whereas the extracellular loop e1 is a ligand binding site in MOR [14]. Two of the intracellular loops (i1 and i3), the C-terminus receptor fragment and a transmembrane domain TM V are engaged in signal transduction pathways and participate in mediating opioid receptor–G protein interactions [15].
Activation of opioid receptors and signal transduction follow a pattern typical for all GPCRs, which is shown in Fig. 2. Following ligand binding to the opioid active site, receptor conformation changes, which activates intracellular G proteins. Each G protein consists of three subunits: α, β and γ. Opioid receptors are coupled with Gαi (existing in three forms), Gαo (existing in A and B forms) and Gαz [16]. β and γ subunits form a heterodimer, which consists of one of five different β and twelve different γ proteins [17]. The role of the Gβγ heterodimer is crucial for the function of Gα, as it enables proper conformation of Gα while ligand binding to the receptor [18].
Fig. 2.
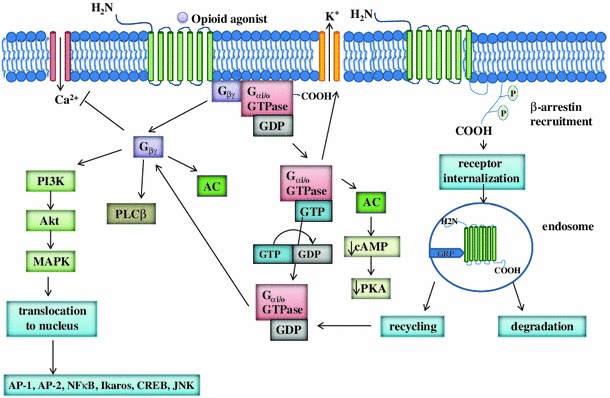
Opioid receptor-related intracellular signal transduction pathways. Opioid receptors are coupled with Gαi, Gαo and Gαz proteins. Secondary transmitters include adenylyl cyclase (AC), GPCR kinase 2/3 (GRK), phospholipase Cβ (PLCβ), and phospatidyloinositol-3-kinase (PI3K). The release of Gβγ subunit also inhibits voltage-gated Ca2+ channels (VGCC, L-type and N-type) and activates K+ channels
The canonical opioid receptor-related signaling pathway involves the dissociation of the heterotrimeric G protein into Gα and Gβγ subunits, which is followed by the Gα translocation and further interaction with Kir3 (G protein-gated inwardly rectifying K+ channel). The release of Gα subunit also inhibits adenylyl cyclase (AC) activity. The release of Gβγ subunit inhibits voltage-gated Ca2+ channels (VGCC, L-type and N-type) and causes activation of K+ channels. By inhibition of N-type VGCCs opioid receptor agonists inhibit calcium influx into the cell. Other effectors linked to Gβγ include GPCR kinase 2/3 (GRK), phospholipase Cβ (PLCβ), phospatidyloinositol-3-kinase (PI3K), adenylyl cyclase and others [3, 19]. When the Gα subunit activates the intracellular effectors, GTP hydrolyzes to GDP and Gα loses its activity and binds to subunit Gβγ—the recovered complex is inactive and can be further activated.
Many intracellular proteins, like β-arrestin, calmodulin, calnexin, filamin A, periplakin, RGS4, ribophorin I or ubiquitin interact with opioid receptors and may thus regulate opioid receptor function at a molecular level, like receptor trafficking, desensitization or endocytosis (for review see: Georgoussi et al. [20]). For example, β-arrestin is a key protein in GPCR desensitization because of blockage of protein–protein interactions, i.e. receptor promotion in clathrin-dependent manner [21]. The lack of β-arrestin prevents receptor desensitization and the development of opioid tolerance after chronic opioid treatment in vivo [22, 23].
Opioid receptor gene expression
MOR, DOR and KOR are encoded by OPRM1, OPRD1 and OPRK1 genes, respectively [24]. The opioid receptor genes are highly conserved in the sequence coding the 7-transmembrane fragment, but vary at the carboxyl and amino termini. This results in differences in the affinity to opioid ligands and distinct signaling pathways [25]. The amino acid sequences in opioid receptors are identical in roughly 60 %, which indicates their common origin [26].
The N-terminus of all opioid receptors and the transmembrane domain TMI are encoded by exon 1, while exon 2 is responsible for coding transmembrane domains TMII–IV. Distal TM domains (TMV–VII) and the intracellular C-terminus are encoded by exon 3 [27]. All genes encoding opioid receptors produce multiple mRNA isoforms, what results from alternative splicing, alternative promoters, but also various sites of polyadenylation and inclusions of non-coding sequences [28].
MOR, the major site of action of opioid drugs, is encoded by the OPRM1 gene located in chromosome 6 (6q25.2.) [29]. The OPRM1 gene possesses 23 transcription variants and the most common variant (MOR-1O) consists of 4 exons and encodes 418 amino acids [30]. The differences between OPRM1 gene variants are highly correlated with changes in dosage requirements for some exogenous opioids. These alterations result from occurring polymorphisms and include alternative splicing at both ends (3′ and 5′) of mRNA, combined with heterodimerization of the receptors. More than 100 single nucleotide polymorphisms (SNPs) were identified in OPRM1 gene, but their meaning remains in most cases unclear [25].
Several human genetic polymorphisms and their possible implications in opioid treatment, as well as the relationship between these polymorphisms and the clinical outcome have recently been discussed in an excellent review by Finco et al. [31]. The most common SNP in the OPRM1 gene is the substitution A/G (rs 1799971) on exon 1, which causes a change in the MOR protein amino acid sequence (Asn→Asp, N40D). This SNP enhances the binding affinity of β-endorphin at MOR, which causes an increased potency at the receptor [32]. However, the role of SNP A118G in antinociceptive action of MOR ligands is unclear [15, 33–35].
DOR is composed of 372 amino acids. The OPRD1 gene, which encodes DOR, is located on 1p36 and contains 3 exons [36]. Nine DOR-related SNPs, which are common in several ethnicities, have been reported [37]. Single nucleotide polymorphism A/G (rs569356) was shown to increase the activity of OPRD1 promoter, probably by enhanced binding of the transcription factor [38]. The elevated expression of OPRD1 may result in increased rewarding effect of drugs of abuse. Studies on DOR-related SNPs and their possible role in alcohol dependence and drug addiction were recently reported [39].
KOR is encoded by OPRK1 gene, which is located on 8q11.2. The major transcription variant possesses four exons (5′ is non-coding) and encodes a protein with 380 amino acids [40]. Several SNPs were classified as SNPs related to alcohol dependence. The insertion/deletion (indel) with a net addition of 830 base pairs 1986 bp upstream of the translational start site in OPRK1 reduces promoter activity by about half and is associated with alcohol dependence [41].
Opioid receptors in the central nervous system
A precise quantification of opioid receptor mRNA expression using absolute quantitative real-time reverse transcriptase PCR (AQ rt RT-PCR), together with numerous immunohistochemistry studies revealed a wide distribution of opioid receptors in the central nervous system (CNS) [42]. The highest expression of MOR was observed in cerebellum, caudate nucleus and nucleus accumbens. DOR were identified in hippocampus, cerebral cortex, putamen, caudate nucleus, nucleus accumbens and temporal lobe. The highest expression of KOR was detected in caudate nucleus, nucleus accumbens, hypothalamic nuclei and putamen.
In the CNS opioid receptors are expressed in pain-modulating descending pathways, involving locus coeruleus, medulla, and periaqueductal gray area and are mainly involved in pain signaling and antinociception. Opioid receptors also occur in midbrain, limbic and cortical structures and may thus modulate a wide range of other functions, including memory and stress response.
Activation of opioid receptors attenuates neuronal activity by pre- and postsynaptic mechanisms, which include the release of inhibitory neurotransmitters and changes in neuronal excitability [43]. Opioid receptors and their ligands are involved in an intense cross-talk with other endogenous systems. Activation of MOR, which is most crucial in pain relief, activates central dopamine reward pathways and may be involved in euphoria. Other adverse side-effects of MOR activation in the CNS, in particular upon prolonged administration of MOR agonists, include addiction, depression, anxiety, and sedation. These were characterized in many previous reports.
Distribution of opioid receptors in the gastrointestinal tract
In the periphery, opioid receptors are widely distributed in neuronal and non-neuronal tissues, including neuroendocrine, immune and ectodermal cells [10]. In the GI tract they are present in smooth muscle cells and at the terminals of sympathetic and sensory peripheral neurons. It has been shown that opioid receptors are synthesized in the dorsal root ganglion and transported centrally and peripherally to the nerve terminals [44].
The distribution of MOR in the enteric nervous system (ENS) was summarized by Sternini et al. [45]. As shown using autoradiography and radiolabeled agonists and antagonists ([3H]dihydromorphine, [3H]naloxone, and [3H]loperamide), MOR are expressed in the submucosal plexus and the myenteric plexus and longitudinal muscle of ileum from various species (including rat, guinea pig, pig, human). Some differences in distribution of MOR among studied species were also reported (for review see: [46]).
The expression of DOR in murine enteric neurons was assessed using enhanced green fluorescent protein (GFP), inserted into OPRD1 gene [47]. The product of insertion, a 80-kDa protein DOReGFP, was detected in esophagus, gastric corpus and antrum, and small and large intestine. Further studies revealed that DOReGFP is expressed in neuropeptide Y (NPY)-positive secretomotor and vasodilator neurons in the submucosal plexus of the small bowel. Moreover, DOReGFP is also present in excitatory motoneurons and interneurons of the myenteric plexus, which express SP and choline acetyltransferase, and in inhibitory interneurons and motoneurons expressing nitric oxide synthase. DOR is also found in nitrergic myenteric neurons in the mouse colon.
ΚORs are highly expressed in the periphery, among others in epidermal keratinocytes and dermal fibroblasts, as well as in nerve terminals of joints, muscles and viscera. The expression of KOR was also detected in dorsal and trigeminal root ganglia [48]. In the GI tract, KORs were localized to myenteric and submucosal neurons, fibres in muscle layer, blood vessels and mucosa in rats [49]. Furthermore, AQ rt RT-PCR revealed the presence of KOR in the liver [47].
Importantly, the opioid receptors were also found in high amounts on lymphocytes and macrophages [42].
Physiological role of opioid receptors in the gastrointestinal tract
Many structural and functional components are responsible for the proper function of the GI tract, including ENS, GI smooth muscle cells, the intestinal mucosa and blood vessels. The ENS consists of two plexus—the myenteric and the submucosal plexus. The localization of the myenteric plexus between longitudinal and circular muscles predestines its involvement in the GI motor activity and its stimulation increases peristalsis. The submucosal plexus controls local secretion and absorption activity [50]. Opioid receptors, which are expressed in the myenteric and the submucosal plexus play a major role in the regulation of the GI transit and mucosal transport of fluids and electrolytes and maintaining GI homeostasis.
Opioids affect primarily neuronal excitability in the enteric circuitry via interaction with major transmitters in the ENS, such as acetylcholine (ACh), SP, neurokinin A (NKA), ATP, vasoactive intestinal peptide (VIP), NPY or 5-hydroxytryptamine (5-HT) [51]. Results from three different studies furthermore suggested an interaction between opioid and glutamate receptors in the guinea pig ileum [52–54]. A recent study by Iwata et al. [55] suggested that the nitrergic pathway may also be involved in the opioid-induced actions in the GI tract. It was shown that the contraction of the mouse ileum induced by morphine administration was inhibited by tetrodotoxin and NG-nitro-l-arginine, indicating that the potential mechanism of morphine action may be associated with the inhibition of nitric oxide (NO) release from inhibitory nerves.
The major effects of opioid receptor agonists in the GI tract are reduction of tonic/segmental contractions and impairment of peristalsis by inhibition of the release of ACh and SP, as well as decrease of GI secretion by inhibition of the activity of ACh and VIP containing neurons [56] (Fig. 3). The intestinal mucosa has the ability to absorb dietary nutrients, water and electrolytes. The active absorption of Na+ and secretion of Cl− across the intestinal mucosa is critical for maintenance of the water-electrolyte balance, defense against bacterial infections and digestive processes [57]. Opioids reduce epithelial secretion and promote water and electrolyte absorption mainly by activation of DOR and MOR [58]. Opioid receptor-mediated increase of cyclic nucleotide concentration stimulates Cl− secretion and inhibits Na+/Cl− absorption [59].
Fig. 3.
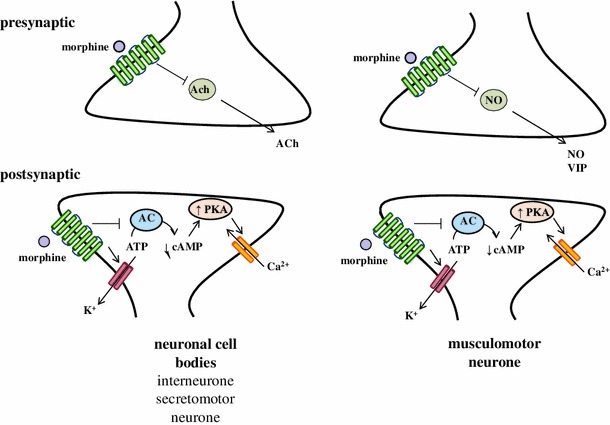
Interaction of opioids with neurotransmitters in the enteric nervous system. Opioids reduce tonic/segmental contractions and impair peristalsis by inhibition of the release of ACh and SP. The decrease of GI secretion is caused by the inhibition of the activity of ACh and VIP containing neurons
In consequence, opioids induce stationary motor patterns: inhibit relaxation of the lower esophageal sphincter, decrease propulsion of smooth muscles in the small and large intestine, increase pyloric and anal sphincter tonus, delay gastric emptying and oral-cecal transit time, and enhance absorption of fluids from intestinal contents. Because of these involvements, the effects of opioid treatment result in nausea, vomiting, altered fluid dynamics and increased GI transit time, constipation, abdominal discomfort or pain (as summarized in Fig. 4).
Fig. 4.
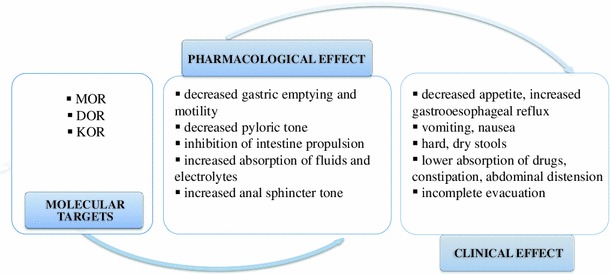
Pharmacological and clinical effects of opioids
Interestingly, opioid receptor agonists can modulate the GI function through centrally mediated actions at the sites which are not protected by blood brain barrier (BBB). For example, it was demonstrated that the activation of MOR located in the medial subnucleus of the tractus salitarius (mNTS) affects the GI motor function. The microinjection of MOR agonists (at doses 1–10 fmol) into mNTS decreased the intragastric pressure and phasic contractions, and inhibited gastric motility [60]. The actions of MOR agonists were associated with the suppression of local GABA activity, which is known to decrease gastric tone and motility [61]. In another study, low doses of MOR agonists (30–300 fmol) microinjected into mNTS area) affected gastric motility by decreasing intragastric pressure and phonic contractions. The inhibitory effect of MOR agonists in mNTS was absent following vagotomy or pretreatment with a selective MOR antagonist. This suggests the involvement of opioid receptors and their ligands in vagovagal reflexes through the release of endogenous opioids in the mNTS area.
Oligomerization of opioid receptors
Similarly to other GPCR proteins, the opioid receptors can heteromerize under physiological and pathophysiological conditions in order to form functional dimers, heterodimers and oligomers [62–64]. Opioid receptors can interact with each other and can also form complexes with other proteins. For example, DOR can exist as homodimers and in presence of an agonist the dissociation of DOR complex occurs. KOR and DOR can also heterodimerize, but their activity is decreased, when they form a complex [11]. The role of heterodimerization of opioid receptors remains unclear, because highly selective ligands for these heterodimers are not available [65]. However, it was suggested that heterooligomerization might induce changes in receptor-related signaling and alter ligand binding [66, 67].
It was suggested that the presence of MOR and cannabinoid type 1 receptor (CB1R) heterodimers may result in altered antinociceptive action of opioids. For example, the administration of Δ 9-tetrahydrocannabinol (THC), a CB1 agonist enhanced the antinociceptive action of morphine due to the formation of a MOR-CB1 complex [68, 69].
Opioid receptor ligands
Endogenous opioids
Endogenous opioid peptides are low-molecular compounds, which are produced in the CNS and peripheral tissues, like adrenal glands. Endogenous opioid peptides derive from three precursor proteins: proopiomelanocortin (POMC), prodynorphin (PDYN) and proenkephalin (PENK) (Table 1). POMC is a precursor for α- and β-endorphin and non-opioid peptides, such as adrenocorticotropic hormone (ACTH), α- and β-melanocyte-stimulating hormone (MSH), corticotropin-like intermediate peptide (CLIP) and β-lipotropin (β-LPH). Dynorphin A and B and neomorphins derive from PDYN. PENK is a precursor for enkephalins ([Leu5]enkephalin, [Met5]enkephalin, [Met5]enkephalin-Arg6-Gly7-Leu8, and [Met5]enkephalin-Arg6-Phe7). Bovine adrenal medulla (BAM) peptide and peptides E and F are further products formed from PENK [70].
Table 1.
Sequences and affinity of endogenous and exogenous opioid peptides
| Precursor | Peptide | Sequence | Receptor affinity |
|---|---|---|---|
| Pro-opiomelanocortin | β-endorphin | Tyr-Gly-Gly-Phe-Met-Thr-Ser-Glu-Lys-Ser-Gln-Thr-Pro-Leu-Val-Thr-Leu-Phe-Lys-Asn-Ala-Ile-Ile-Lys-Asn-Ala-Tyr-Lys-Lys-Gly-Glu | MOR > DOR > KOR |
| Pro-enkephalin | [Met5]Enkephalin | Tyr-Gly-Gly-Phe-Met | DOR > MOR ≫ KOR |
| [Leu5]Enkephalin | Tyr-Gly-Gly-Phe-Leu | ||
| Pro-dynorphin | Dynorphin A | Tyr-Gly-Gly-Phe-Leu-Arg-Arg-Ile-Arg-Pro-Lys-Trp-Asp-Asn-Gln | KOR ≫ MOR > DOR |
| Dynorphin B | Tyr-Gly-Gly-Phe-Leu-Arg-Arg-Gln-Phe-Lys-Val-Val-Thr | ||
| α-neomorphin | Tyr-Gly-Gly-Phe-Leu-Arg-Lys-Tyr-Pro-Lys | ||
| β-neomorphin | Tyr-Gly-Gly-Phe-Leu-Arg-Lys-Tyr-Pro | ||
| Unknown | Endomorphin-1 | Tyr-Pro-Trp-Phe-NH2 | MOR |
| Endomorphin-2 | Tyr-Pro-Phe–Phe-NH2 | MOR | |
| κ-casein | Casoxin 4 | Tyr-Pro-Ser-Tyr-OCH3 | MOR |
| β-conglycinin | Soymorphin-5 | Tyr-Pro-Phe-Val-Val | MOR |
| Soymorphin-6 | Tyr-Pro-Phe-Val-Val-Asn | ||
| Soymorphin-7 | Tyr-Pro-Phe-Val-Val-Asn-Ala |
Opioid peptides are an important link between the neuroendocrine and immune systems, and their immunomodulatory effect may play a significant clinical role in immune-mediated diseases.
Endogenous opioid peptides have been reported to inhibit the release of neurotransmitters, such as ACh, dopamine, norepinephrine in the CNS and in the periphery. There are many studies reporting the effects of endogenous opioids on human body, like control of nociception, mood, and cardiovascular functions, which have been extensively reviewed elsewhere [71–73]. Here, we discuss the effect of endogenous opioids in the GI tract and their possible role in the GI physiology and function.
In the GI tract the endogenous opioid peptides are present both in neurons and endocrine cells of the mucosa [11, 74]. Under physiological conditions, endogenous opioid peptides exert inhibitory effect on gastric emptying and intestinal motility. Opioid peptides also decrease biliary, pancreatic and intestinal secretions [75].
Enkephalins
Enkephalins are short peptides, which are produced mainly by the pituitary, adrenal glands and the pancreas. In the GI tract enkephalins are also formed in gastric and intestinal endocrine cells [76]. There are two enkephalins, which play a major role in molecular signaling through opioid receptors, [Leu5]enkephalin and [Met5]enkephalin. Both peptides are potent DOR agonists, and additionally possess some affinity at MOR. The major physiological effect of enkephalins is antinociception and inhibition of pain signaling in the CNS and the periphery, including the GI tract [11, 74].
Enkephalins were also found to be synthesized in leukocytes and may thus participate in inflammatory response. Owczarek et al. [77] found recently that the serum level of [Met5]enkephalin were decreased in patients with inflammatory bowel diseases (IBD) in comparison to healthy volunteers. Higher levels of [Met5]enkephalin were found in colonic biopsies collected from inflammatory lesions from patients with IBD, compared to biopsies from non-inflamed colon.
DAMGO, a synthetic MOR agonist, which structure is based on enkephalin, was studied in a rat model of visceral pain (i.p. and i.c.v. injections of 2 % acetic acid) and compared to morphine. I.c.v. injection of DAMGO was more potent than morphine injection in the reduction of pain. Moreover, administration of naloxone methiodide, a peripherally acting antagonist, attenuated the antinociceptive action of DAMGO and morphine [78]. In another study DAMGO was reported to induce suppression of enhanced excitability of colon dorsal root ganglion neurons from rats with chronic visceral hyperalgesia, an animal model used to characterize mechanisms related to irritable bowel syndrome (IBS), induced by i.c. injection of acetic acid [79].
Endomorphins
Endomorphin-1 (EM-1, Tyr-Pro-Trp-Phe-NH2) and EM-2 (Tyr-Pro-Phe-Phe-NH2) are two endogenous tetrapeptides isolated by Zadina et al. [80] from the bovine frontal cortex and human brain. EMs possess the highest affinity at MOR of all known endogenous agonists.
There are several reports on the activity of EMs in the GI tract. It was demonstrated that EM-1 and EM-2 inhibited striated and smooth muscle response in the esophagus (with exception of lower esophageal sphincter) in a naloxone-reversible manner [81]. In the guinea pig ileum, EMs decreased the release of ACh in LMMP preparations [82, 83].
Furthermore, EMs inhibited longitudinal muscle contractions in the mouse distal colon and evoked contractile response in circular muscle of proximal and mid colon. It was also observed that β-FNA (3 × 10−6) and naloxonazine (10−6) abolished EM-1-induced contractions, suggesting the involvement of MOR receptor subtypes in contractile responses induced by EM-1 [84].
Some studies suggest an immunomodulatory role of EMs in the GI tract. The effect of EM-2 on rat peritoneal macrophage function was investigated by Azuma et al. [85], who showed that EM-2 inhibited the production of cytokines, including TNF-α, IL-10 and IL-12. In addition, EM-2 increased IL-1β production in phorbol 12-mirystate 13-acetate (PMA)-stimulated macrophages and inhibited chemotaxis.
EMs, similarly to other endogenous opioid peptides, are prone to rapid degradation and, therefore, studies on their role in the GI tract are limited. Recently, several new EM analogs with improved pharmacological profile and biodistribution were reported (for review see: [86]). One of the novel analogs, obtained by the attachment of lactose to the N-terminus of EM via a succinamic acid spacer, showed improved membrane permeability and increased metabolic stability [87]. Interestingly, the adverse effects of this modified EM on stool hydration, measured using a castor oil-induced diarrhea assay and GI motility, assessed using a charcoal GI transit test were less significant compared to morphine administration. Furthermore, delayed transit was not observed in rodents treated with the new compound. The attractive additional benefit of this modified EM was that it may be administered orally.
Other opioids
Peptides and their derivatives
Soymorphins. The soymorphins, mainly soymorphin-5, -6, and -7 (Table 1), are MOR agonists, which were shown to suppress food intake and intestinal motility after oral administration in a naloxone-dependent manner [88]. The inhibitory potency of soymorphins on the GI transit was assessed using selective agonists and antagonists of 5HT1A (WAY100135), D2 (raclopride) and GABAB (saclofen) receptors. The obtained results suggest that soymorphins inhibit small intestinal motility through the release of serotonin and activation of 5-HT1A receptors. Then, dopamine is released and acts via D2 receptors. Finally, GABA is released, which acts through GABAB receptors.
Casoxin 4. Casoxin 4 is a tetrapeptide MOR antagonist, which was isolated from the κ-casein fraction of bovine milk (Table 1). It was demonstrated that casoxin-4 reversed morphine-induced inhibition of electrically induced contraction in isolated small intestine in both mice and guinea pigs [89]. The MOR component was more prominent in the guinea pig ileum, while KOR and DOR components were predominant in the mouse ileum. Casoxin 4 after oral administration failed to attenuate the inhibitory effect of morphine in the murine small intestine. [90].
Alkaloids and diterpenes
Morphine. Morphine is a classical opioid analgesic commonly used for the treatment of acute and chronic pain. Its major site of action is MOR, but it also displays a minor affinity at KOR. Morphine can cross the BBB and act in the CNS. Therefore, its prolonged and repetitive administration may cause tolerance, nausea, or sedation [91]. Here we focus on the beneficiary and adverse effects of morphine in the GI tract.
Morphine delays GI transit in a MOR-dependent manner, but it remains a matter of debate whether this effect is mediated by MOR in the CNS, the periphery or both. Chronic administration of morphine (0.05 mg/kg, s.c.) in healthy human volunteers caused delayed colonic transit time. Interestingly, the administration of naloxone-3-glucuronide (0.16 mg/kg, p.o.), a naloxone metabolite, reversed the effect of morphine without any impact on analgesia [92]. Naloxone-3-glucuronide is not absorbed into systemic circulation, there is no penetration through the colonic-mucosal blood barrier. This suggests that the action of opioids in the GI tract is mediated mainly by peripheral receptors. Highly polar naloxone derivatives peripherally antagonize the effect of morphine-delayed GI motility in the perfused isolated rat colon [93]. However, some studies imply that only the CNS is involved.
The development of tolerance following repeated exposition to morphine is well defined at the cellular and molecular level, but is poorly understood in vivo, in particular in terms of GI function. Chronic administration of morphine may produce tolerance in the upper GI tract, specifically in the circular muscle in the ileum, but not in the colon [94]. The lack of tolerance to morphine observed in the colon may underlie constipation and the development of the opioid-induced bowel dysfunction (OBD).
It was suggested that a transport protein, glycoprotein P, might be involved in the development of opioid tolerance, but its role has not been clearly identified [95–97]. Okura et al. [98] showed that repeated administration of morphine in rats reduced intestinal absorption of morphine, subsequently decreasing its antinociceptive effects. The decrease in absorption was related, at least partially, to the stimulation of glycoprotein P-mediated efflux. The up-regulation of glycoprotein P may thus contribute to the development of opioid tolerance to morphine and oxycodone after oral administration. Therefore, the design of opioids without glycoprotein P substrate activity might be a key to avoid the development of tolerance during their chronic administration.
Morphine and other alkaloids may be involved in the immune response mediated by the opioid receptors in the GI tract. Peng et al. [99] showed that s.c. implementation of a morphine slow release pellet suppressed cholera toxin-specific production of IgA and IgG in fragment cultures of ileal segments, Peyer’s patches and mesenteric lymph nodes. It was also found that the effect of morphine in gut-associated lymphoid tissue was mediated through a TGF-β, a putative IgA switch factor in the GI tract-dependent pathway. The inhibition of TGF-β by morphine was reversed by naltrexone, which confirms an involvement of opioid receptors in immune responses in the GI tract.
Furthermore, morphine may be involved in the development of bacterial infections, induced by Streptococcus pneumonia, Toxoplasma gondii, Klebisella pneumonia, Candida albicans and other bacterial strains. Interestingly, these infections promoted by morphine were shown to be dependent on MOR [100]. Feng et al. demonstrated that implementation of 75 mg slow release morphine pellet in mice was a potent enhancer of an oral infection with Salmonella typhimurium, which is used to induce a murine model of typhoid fever and causes gastroenteritis in humans. Morphine administered via minipumps (at doses 1–25 mg/kg/day) did not sensitize to Salmonella infection and inhibited GI transit more potently than the morphine pellets [101].
Chronic administration of morphine in mice may cause alterations in virulence expression in Pseudomonas aeruginosa and lead to lethal gut-derived sepsis [102]. The expression of virulent phenotype against intestinal epithelium (strain of Pseudomonas aeruginosa disrupting protein PA-I lectin) in response to morphine may be principally mediated by MOR. However, since a peripherally restricted MOR antagonist MNTX did not delay chemotaxis after Pseudomonas challenge, it is possible that morphine may also act on other receptors, including non-opioid.
Interestingly, Glattard et al. [103] hypothesized that endogenous morphine, which is secreted from human neutrophils following stimulation by lipopolysaccharide (LPS) and IL-8 in presence of Ca2+, may be involved in inflammatory responses. They have also shown that the endogenous morphine level is elevated in patients with sepsis.
Salvinorin A. Salvinorin A (SA), a diterpene isolated from the Mexican plant Salvia divinorum, is a selective KOR agonist, which displays significant inhibitory and anti-inflammatory effects in the GI tract. It was observed that in physiological conditions SA inhibited cholinergic twitch contractions in mouse and guinea pig small and large intestine in a KOR- and CB1-dependent manner [104, 105]. Furthermore, SA reversed ileal hypermotility induced by croton oil [106] or endotoxin [107] administration in mice. Interestingly, in this latter model the regulatory action of SA on epithelial barrier function was mediated via NO-related pathways. Recently our group showed that SA exhibits anti-inflammatory and anti-nociceptive effects in murine models of intestinal inflammation [108].
The pharmacology of SA in the GI tract was recently reviewed [109].
Opioid system in pathophysiological conditions of the GI tract
Diarrhea
The inhibitory effects of opioids in the GI tract, such as inhibition of neuronal activity, reduced propulsion and delay of GI transit have been used for centuries for example to treat diarrhea. Diarrhea is a change of normal bowel movement characterized by an increase in the water content, volume, or frequency of stools.
The classical anti-diarrheal agent, loperamide (Fig. 5), is an agonist of a putative MOR subtype, which is expressed in peripheral organs [110]. Loperamide is widely used in patients with digestive disorders and after radiotherapy and chemotherapy to control diarrhea. It has poor capacity for BBB penetration at concentrations required for anti-diarrheal effect [56]. In the GI tract loperamide causes intestinal relaxation, which is triggered by the opening of ATP-sensitive potassium channels. The activation of K+ channels triggers cAMP-PKA signaling pathways, which induce hyperpolarization of cell membranes and relaxation of smooth muscles. Opening of ATP-sensitive channels also reduces concentration of intracellular Ca2+, which is similar to that observed in OIC [111]. Finally, loperamide injected intrathecally induces MOR-mediated analgesia [112].
Fig. 5.
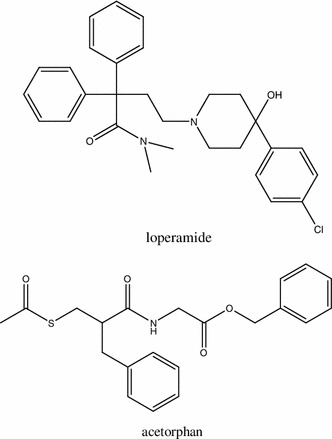
Structures of anti-diarrheal agents, loperamide and acetorphan
Acetorphan (Racecadotril) (Fig. 5) is another drug used in the treatment of acute or chronic diarrhea. Similarly to loperamide, acetorphan does not cross BBB. However, it displays a different mechanism of action, since it does not affect gut motility [113]. The anti-diarrheal effect of acetorphan results from inhibition of enkephalinases, proteolytic enzymes degrading endogenous opioids present in the GI tract. Acetorphan increases concentration of endogenous opioids in the GI tract and reduces secretion of electrolytes and water into the gut lumen.
The possible application of opioids in different types of diarrhea has been suggested. The role of opioid receptors and their ligands in murine allergic diarrhea has been studied by Duncker et al. [114]. They observed that the ovalbumin (OVA)-induced allergic diarrhea was improved by DAMGO and U50’488, synthetic agonists of MOR and KOR, respectively. Moreover, DAMGO decreased concentration of IFN-γ, IL-4, IL-5 and IL-10 after ex vivo stimulation of mesenteric lymphocytes. In comparison with U50’488, DAMGO did not decrease plasma level of mouse mast cell protease-1 (MMCP-1), which is a marker of mast cell degranulation or total plasma IgE. Interestingly, thymoquinone–lipophilic compound in hexanic extract from Nigella sativa (Black cumin) also exhibited beneficial effects in allergic diarrhea by activation of opioid receptors. The thymoquinone exhibited anti-inflammatory and anti-cancer properties and was involved in alleviation of allergic asthma [115–117]. The results of the study suggest that opioid receptor-mediated modulation of GI and immune systems may become a target for future therapies aiming at alleviation of allergy-based diarrhea symptoms.
Opioid agonists, such as trimebutine or loperamide are commonly used for treatment of symptoms in diarrhea-predominant IBS (IBS-D). However, their pharmacological profile seems less favourable compared to 5-HT3 antagonists (5-HT3RAs), like ramosetron, alosetron and cilansetron. As shown by Hirata et al. [118], 5-HT3RAs increased colonic nociceptive threshold in non-stressed rats, and also inhibited restraint-induced colonic hyperalgesia and diarrhea. The latter effect was not achieved in rats treated with loperamide [118].
Opioid-induced bowel dysfunction
Chronic administration of opioids, in particular at high doses, may cause several adverse side effects, mainly originating in the GI tract (for review see: McNicol [119]). The major opioid-related group of GI disorders, described as OBD, affects up to 10–20 % of adolescents and adults around the world [120].
Several ailments characterize OBD, such as constipation, abdominal pain, bloating and gastro-esophageal reflux. The chronic occurrence of OBD symptoms may cause nausea, fecal impaction, vomiting and critical disturbances in absorption of concomitant drugs. Vomiting and nausea may lead to further complications, including pneumonia, while mitigated oral intake can be associated with malnutrition. Abdominal distention associated with OBD may affect respiratory function and delay wound healing [121].
One of the most common side effects resulting from chronic administration of opioids is constipation. It is defined as a delay in frequency of intestine movements and is often accompanied by other symptoms, such as hard and dry stools, incomplete bowel movements, or straining during defecation. Since constipation is not always related to OBD, an additional term for this ailment, OIC, was introduced [75]. OIC is an adverse side effect of opioid administration, but some additional factors may influence its development, e.g. metabolic disorders, including diabetes mellitus, hypokalemia, hypercalcemia, and hypothyreodism, advanced age, low-fluid intake, or reduced physical activity ([122], for review see: [123]).
The mechanism of OIC is complex and implies many factors, but the activation of opioid receptors in the periphery is critical. In general, opioids induce constipation through disruption of neurotransmission between enteric neurons and their targets—smooth muscles and epithelial cells [44]. The activation of opioid receptors results in a depression of peristaltic contractions, but also in an increase of GI muscle activity, like increase of resting muscle tones of sphincters, non-propulsive patterns and spasms. In addition, opioids increase biliary and internal anal sphincter tones. Finally, suppression of ongoing discharge in secretomotor neurons in the ENS, resulting in the inhibition of basal epithelial secretion and increased absorption of fluids from the intestine, which evoke dry and hard stools, may also contribute to development of OIC [124].
The treatment of OIC involves the use of stimulant laxatives, stool softeners and osmotic agents, but this classical therapy is often not effective. Lubiprostone (Fig. 6), which is used in clinical conditions to suppress the symptoms of OIC and constipation predominant-IBS, is a bicyclic fatty acid of prostone group and derives from prostaglandin E1 [125]. Lubiprostone reverses inhibitory action of opioids on mucosal secretion in human small intestine by promoting transcellular movement of Cl− from serosal to luminal area in mucosal epithelia, which enhances fluid secretion [126]. The driving force for this increased transport of Cl– into intestinal lumen is provided by Na/K pump, with Cl− entering the cell across basolateral membrane through Na–K–2Cl co-transporter and other channels on apical membrane. It was shown that lubiprostone evokes Cl− secretion via cystic fibrosis transmembrane conductance regulator (CFTR) receptors and cAMP signaling in human T84 colon cancer cell line. Furthermore, the transport of Cl− is dose-dependent and inhibited by a CFTR inhibitor [127]. Lubiprostone thus increases mucosal secretion and liquidity of bowel contents and enables the avoidance or resolution of OIC by chloride channels without influencing the opioid receptors [128], which was confirmed in the studies on mice and guinea pigs [129]. However, therapy with lubiprostone is associated with side effects including nausea, diarrhea, abdominal pain and bloating [130].
Fig. 6.
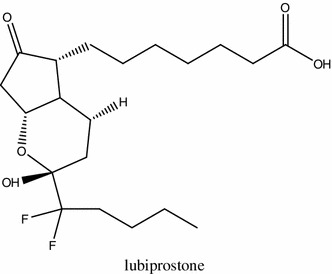
Structure of lubiprostone
Postoperative ileus
Postoperative ileus (POI) is a transient cessation of coordinated bowel function following surgical interventions [131]. POI is a common complication, which occurs mainly after abdominal, orthopaedic, or cardiac surgery and affects the whole GI tract. POI may be triggered by symphatetic reflexes, inhibitory humoral agents, release of noradrenaline from gut wall, anaesthesic agents and inflammation. Furthermore, POI is often induced by enteric inflammatory response and recruitment of leukocytes to muscularis of intestine wall, which are responsible for production of NO, a major inhibitory neurotransmitter in the GI tract. The levels of prostaglandins, upon cyclooxygenase-2 (COX-2) and inducible NO synthase (iNOS) activation, as well as cytokines, including TNF-α, IL-1β and IL-6 may also increase in POI [132].
The clinical symptoms of POI are similar to those in OBD, including abdominal distention, lack of intestinal movements, and accumulation of gas and fluids in the intestine. The inhibition of gut motility occurs immediately after surgery, persists for 2–3 days and resolves spontaneously. However, delayed GI recovery may lead to clinically relevant complications, such as poor nutritional intake, delayed wound healing, infections or pulmonary dysfunction [133] and impact the time of patient hospitalization.
Opioid agonists, which are used for the treatment of post-operative pain, may negatively contribute to POI by stimulation of MOR in the GI tract and inhibition of intestinal motility [134], as well as activation of iNOS and increase in NO release from phagocytes. The major purpose of peripherally acting MOR antagonist (PAMORA) administration is the mitigation of adverse side effects of endogenous and exogenous opioids in periphery with maintenance of analgesic effect in the CNS, mainly due to a low BBB permeability at therapeutic concentrations (Fig. 7). There are two drugs on the market that are used in the management of POI after resection of intestine, alvimopan and methylnatrexone (MNTX) (Fig. 8). Both were approved by the Food and Drug Administration (FDA) and the European Medicine Agency in 2008 for the treatment of OBD and POI. The critical difference between both drugs is in their utility: alvimopan is applied to treat opioid-naïve, while MNTX opioid-treated patients. Furthermore, alvimopan can be administrated only up to 8 days due to the possibility of myocardial events [135].
Fig. 7.
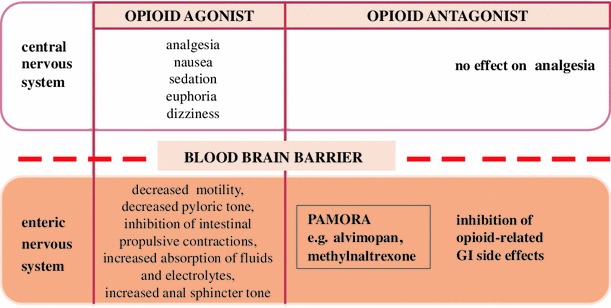
Principle of action of peripherally acting MOR antagonists (PAMORA)
Fig. 8.
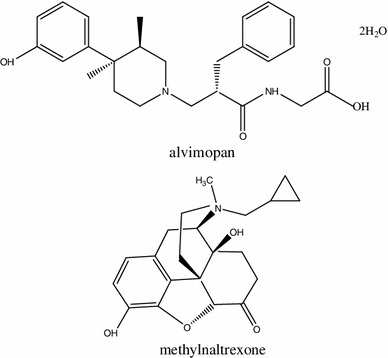
Structures of clinically used peripherally acting MOR antagonists (PAMORA): alvimopan and methylnaltrexone
Alvimopan. Alvimopan is approximately 200 times more potent at peripheral than central MOR [136]. Acute administration of alvimopan is used to accelerate the time to recovery of upper and lower GI after abdominal surgery. Chronic administration at low doses reverses OBD and OIC [137]. Furthermore, alvimopan reduces opioid-induced nausea and vomiting [138].
Earlier clinical trials with alvimopan were reviewed by Marderstein et al. [139] and the most recent ones are summarized in Table 2.
Table 2.
Summary of clinical trials for alvimopan
| Study design | No. of patients | Participants | Dosea | Conclusions | References |
|---|---|---|---|---|---|
| DB, R, P-C | 522 | Patients with OBD and non-cancer chronic pain with morphine administration >30 mg/day | 0.5/1 mg QD, BID, 6 weeks | Restoration of GI function and attenuation of OBD symptoms | [140] |
| DB, R, P-C | 168 | Patients with OBD, non-cancer pain and opioid treatment | 0.5/1 mg QD, 3 weeks | No effect on opioid-induced analgesia | [141] |
| DB, R, P-C | 518 | Patients with OBD and non-cancer pain | 0.5/1 mg QD, BID, 12 weeks | No effect on the requirement for opioid medication | [142] |
| DB, R, P-C | 485 | Patients with OBD and non-cancer pain | 0.5/1 mg QD, BID, 12 weeks | Attenuation of OBD symptoms | [143] |
| DB, R, P-C | 469 | Patients after surgery | 6/12 mg >2 h before surgery, then BID, max. 1 week | Acceleration in recovery of GI function | [144] |
| DB, R, PG, P-C | 615 | Patients after surgery | 6/12 mg >2 h before surgery, then BID, max. 1 week | Acceleration in recovery of GI transit in patients after laparotomy | [145] |
| DB, R, PG, P-C | 911 | Patients after surgery | 6/12 mg >2 h before surgery, then BID, max. 1 week | Potential benefit in bowel resection patients who received i.v. patient-controlled analgesia | [146] |
| R, PG, P-C | 78 | Patients after surgery | 1/6 mg >2 h before surgery, then BID | Shorter duration of hospitalization | [147] |
| DB, R, P-C | 654 | Patients after surgery | 12 mg 30–90 before surgery, then BID, max. 1 week | Reduction in POI–related morbidity without compromising opioid analgesia | [148] |
| DB, R, P-C | 519 | Patients after surgery | 12 mg >2 h before surgery then BID, max. 1 week | Improvement in lower GI recovery in women | [149] |
aIn all trials alvimopan was administered p.o.
DB double-blind, R randomized, PG parallel group, P-C placebo-controlled, QD once daily, BID twice daily
In 2008 a randomized, double-blind, placebo-controlled, dose-finding study was performed on 522 non-cancer humans with <3 spontaneous bowel movements per week and other complications associated with chronic opioid administration (>30 mg of morphine/day), which were maintained over 6 weeks [140]. After 3 weeks all patients treated with alvimopan (0.5 or 1.0 mg once or twice a day, p.o.) exhibited improvement in spontaneous bowel movements and all additional complications.
The post hoc analysis of four randomized, double-blind, placebo controlled, phase III trials showed a beneficial impact of alvimopan on GI recovery in 1409 patients after bowel resection [150]. Of patients orally treated with alvimopan (12 mg at least 30 min, but no longer than 5 h before surgical intervention), 80 % exhibited GI recovery on or before 5 postoperative days. Moreover, the GI recovery and discharge from hospital were improved in comparison to placebo-controlled patients.
The randomized, placebo controlled phase III trial of alvimopan for OIC treatment in 512 patients with non-chronic pain demonstrated that oral administration of alvimopan once or twice a day improved bowel movements in comparison with placebo group [142]. Moreover, treatment for 12 weeks was tolerant and safe for all patients treated with alvimopan. The administration of additional drugs, e.g. laxatives, was not required.
The length of stay in hospital of patients with laparoscopic partial colectomies, in which standard postoperative treatment was aided by alvimopan (12 mg before and 6 mg twice a day, for max. 7 days after surgery) was significantly shorter compared with control group [151, 152].
Finally, the large-scale report of alvimopan used in treatment of 3525 patients after open or laparoscopic bowel resection revealed that the administration of alvimopan reduced the length and costs of hospitalization [153]. Lower incidence of GI morbidity, mortality and intensive care unit (ICU) stay were also reported.
In 2008 Schmidt et al. [136] evaluated the action of alvimopan in the presence of the COX-2 inhibitor DFU in a rodent model of POI. Alvimopan (10 mg/kg, s.c.) reversed morphine (1 mg/kg)-induced delay in the GI transit, but had no effect on transit in control animals. In addition, the morphine-induced delay of the GI transit was not observed in mice pretreated with alvimopan. The co-administration of alvimopan with DFU inhibited the immunosuppressive action of morphine, suggesting a new strategy for the treatment of inflammatory response in postoperative inflamed GI tissues.
Recently, Vaughan-Shaw et al. [154] performed a meta-analysis, which comprises three clinical studies mentioned above [144, 145, 150]. This meta-analysis showed that 12 mg of alvimopan p.o. given 2 h before surgery and than twice a day until discharge significantly accelerates recovery of GI tract and reduces time to hospital discharge in patients undergoing open abdominal surgery enrolled in an Accelerated Recovery Program.
Methylnatrexone (MNTX). MNTX, similarly to alvimopan, antagonizes MOR located in the intestine without any effect on opioid receptors present in the CNS. MNTX is less selective at MOR than alvimopan, as it also binds at KOR [155].
The clinical trials for MNTX are summarized in Table 3. In a randomized, parallel-group, double-blind, placebo-controlled trial of 48 healthy human volunteers, MNTX (0.3 mg/kg/day, s.c.) did not induce any changes in the GI transit in comparison to placebo [156]. Moreover, MNTX did not reverse the anti-motility action of codeine (120 mg/kg/day, for 5 consecutive days).
Table 3.
Clinical trials for methylnaltrexone
| Study design | No. of patients | Participants | Drug dose | Route of administration | Conclusions | References |
|---|---|---|---|---|---|---|
| DB, R, PG, P-C | 48 | Healthy humans | 0.3 mg/kg, 1 week | s.c., p.o. | No effect on delayed GI transit | [156] |
| DB, R, P-C | 12 | |||||
| DB, R, P-C | 11 | 0.09 mg/kg morphine + 0.3 mg/kg MNTX | i.v. | Attenuation in delayed gastric emptying | [157] | |
| DB, R, P-C | 14 | 0.05 mg/kg morphine i.v., 19.2 mg/kg MNTX | p.o. | Prevention and treatment of OIC | [158] | |
| DB, R, P-C | 137 | Patients with nonmalignant pain, chronic opioid administration | 12 mg, 4 weeks | s.c. | Relief in OIC after 2 doses | [159] |
| DB, R, P-C, PG | 469 | 4 weeks | s.c. | Improvement in constipation symptoms over 1 month | [160] | |
| R, P-C | 460 | 12 mg QD or QOD, 4 weeks | s.c. | Relief of OIC | [161] | |
| DB, PG, P-C | n1:515, n2: 533 | Patients after surgery | 12 mg or 24 mg, max. 10 days | i.v. | Safe and well-tolerated drug | [162] |
| DB, R, PG, P-C | 33 | 12 mg QD, 4–7 days | s.c. | Safe and well-tolerated drug in OIC treatment | [163] | |
| DB, R, P-C | 133 | Patients with advanced illness and OIC | 0.15 mg/kg, QOD, 2 weeks | s.c. | Improved constipation distress | [164] |
| DB, R, P-C | 154 | 0.15–0.30 mg/kg, 4 months | s.c. | Defecation after 30 min. | [165] | |
| DB, R, P-C | 133 | 0.15 mg/kg, 2 weeks | s.c. | Defecation after 4 h | [124] | |
| DB, R, P-C, PG | 66 | 1/3/5 mg/kg, 1–3 weeks | s.c. | Reversal of OIC at dose = or >5 mg | [166] | |
| DB | 82 | 0.15 mg/kg, 1 month | s.c. | Improvement in OIC | [167] | |
| DB, R, P-C | 22 | Patients with methadone induced constipation | 0.015/0.05/0.1/0.2 mg/kg, 2 days | i.v. | Reversal of slowing of oral cecal-transit time | [168] |
| DB, R, PG, P-C | n1:154, n2:133 | OIC | 0.15/0.30 mg/kg, | s.c. | Safe and well-tolerated drug | [169] |
DB double-blind, R randomized, PG parallel group, P-C placebo-controlled, QD once daily, BID twice daily
The clinical efficiency of MNTX was evaluated in patients with OIC and chronic, non-malignant pain in a double-blind, randomized, placebo controlled study [159]. In this trial patients received MNTX (12 mg, s.c.), every day, every other day (alternatively with placebo) or placebo alone for 4 weeks. Roughly 40 % of patients reported rescued free bowel movements (RFBM) after ≥2 of four doses of MNTX, while 30 % had more than 3 RFBMs per week of MNTX administration. In another study with a larger group of patients (460) exhibiting OIC in advanced illness, 34 % of patients had RFBMs within 4 h after the first dose of MNTX [161]. In the placebo group only 10 % patients had RFBMs. The adverse effects during MNTX treatment were minimal—the most common was abdominal pain and the drug was safe and tolerated by all patients.
Another randomized, double-blind, parallel group, placebo-controlled study revealed no improvement of life quality in postcolectomy patients treated with MNTX or placebo [162]. The drug was intravenously administered in two doses (12 or 24 mg) 90 min after surgery and every 6 h for 24 h or 10 days. The primary efficiency end point was assessed as time from surgery to first intestine movement, and according to length of hospitalization there were no differences between all groups. The adverse effects were similar to those observed in other studies, including abdominal pain, nausea or vomiting. The additional outcome of this study was that the dose of 24 mg was safe and well-tolerated by patients.
Finally, in another study, a cohort of 469 patients was divided into three groups: placebo, MNTX every day and MNTX alternating with placebo for 4 weeks [160]. The improvement in constipation was noted for both groups subcutaneously treated with MNTX.
Recently, Garten et al. [170] reported that MNTX was administered to an infant with paralytic ileus. The neonate was 8 days after surgery and treated with fentanyl (2 μg/kg/h). The first i.v. administration of MNTX (0.15 mg/kg) resolved intestinal dismobility after 15 min. The neonate received 5 doses of MNTX and the intestinal transit was improved without any adverse side effects.
Antanals. Three novel candidates for PAMORA-type drugs, designated antanal-1, antanal-2 and antanal-2A have recently been reported by our group [171]. The antanals were shown to exhibit selective antagonist activity at MOR in the GI tract in vivo and in vitro. The antanals did not across BBB after i.p. administration in mice and may thus become valuable drug templates for the design of future PAMORA-type therapeutics.
Irritable bowel syndrome
IBS involves a dysregulation of interactions between central and peripheral nervous system, so called brain-gut axis [172]. By many, IBS is linked to disturbances in gut microbiota [173]. This disorder is associated with abdominal discomfort or pain associated with changed bowel habits for at least 3 days per month in the previous 3 months, with the absence of another organic disease [174]. In USA, 5–10 % of population suffer from IBS with prevalence between 20 and 39 years of age [175]. The prevalence of IBS is 10–20 % worldwide [176]. IBS patients report numerous extragastrointestinal symptoms such as fibromyalgia, irritable urinary bladder, changes in libido and energy levels [172].
The several types of IBS can be mentioned, they are associated with changes in colorectal motility. Patients with IBS suffer from altered bowel habits, ranging from diarrhea, constipation, alternating diarrhea and constipation or normal bowel habits. Patients with a clinically prominent gastrocolonic reflex exert increased postprandial colonic contractions. Predominant constipation IBS (IBS-C) is characteristic for patients with increased colonic contractions. There is also diarrhea-predominant IBS (IBS-D), which involves patients with reduced colonic contractions and alternating or mixed IBS subtypes (IBS-A). [177].
The treatment of IBS is still symptomatic, without a well-defined first-line therapy. One of the drugs applied to treat IBS is trimebutine (Fig. 9), which is a weak MOR agonist, but its clinical efficiency is unclear. Recently, the effect of trimebutine molecule modified with NO2-arginine residue (NO2-Arg-Trim) was investigated in a rodent model of IBS [178]. NO2-Arg-Trim displayed significantly more potent analgesic activity than trimebutine in healthy and post-colitis rats. The treatment with NO2-Arg-Trim also increased expression of genes involved in pain and inflammatory processes, including TNF-α, IL-1β, COX2 and iNOS, in tissue preparations from post-colitis rodents.
Fig. 9.
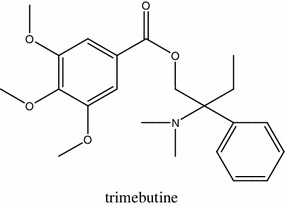
Structure of trimebutine
Linaclotide is a drug, which is useful in treatment of IBS and chronic constipation. Linaclotide is a peptide agonist of guanylate cyclase, which is important in active transport of Cl− into the intestinal lumen via CFTR channels. This drug causes increased stool water content and then relief in constipation [179]. The therapy for IBS was recently reviewed by Olden [180].
Inflammatory bowel diseases
Inflammatory bowel diseases (IBD) comprise two idiopathic ailments—Crohn’s disease (CD) and ulcerative colitis (UC). It is estimated that 1–2 % of the population has IBD and it is more common in women than men. It has been suggested that IBD involves a dysregulation of the immune response in the intestine evoked by commensal bacteria, and a genetic and an environmental predisposition was reported. The development of IBD may also imply a neural component, such as colonic nerve damage, changes in mucosal innervation and alterations in neuropeptide expression, e.g. SP, VIP and CRH [181]. The major therapeutic goals in IBD patients are the control of inflammation and the treatment of symptoms, which include abdominal pain and altered bowel movements [182].
Abdominal pain is a common symptom of IBD with a multifactorial etiology, described as a cramping sensation, varying in intensity and with exacerbations [179]. There are two types of abdominal pain—somatic, which is musculoskeletal and visceral—caused by stretching of the viscera and obstruction or widely affected inflammation. The development of visceral pain is associated with hypersensitivity of the primary sensory neurons in GI tract, which is subsequently has consequences in CNS. The changes in intrinsic sensory neurons properties and in gene expression regulation of nociceptive specific proteins lead to sensitization of primary afferent neurons. These changes contribute to increased production of pro-inflammatory molecules occurring via neurogenic inflammation, which are also involved in swelling, edema and vasodilation [183].
Since immune cells express opioid receptors, opioids may be involved in the regulation of inflammatory processes, with MOR ligands playing the most significant immunomodulatory role. Cabot et al. [184] showed that inflammation may increase the expression of POMC mRNA in immune cells, what results in elevated β-endorphin production and antinociceptive action. MOR agonists, DALDA and DAMGO, administered s.c., improved colitis in mice [182]. Furthermore, MOR−/− mice were more prone to inflammation progress than wild type animals [182]. It was also demonstrated that opioids regulate the release of pro-inflammatory cytokines (e.g. IL-12, IL-6, TNF-α) from peritoneal macrophages in mice [185]. The murine models of colitis proved that MOR exert anti-inflammatory effect on colon because of regulation of T cell proliferation and cytokine production [182]. The upregulation of MOR occurs during IBD with plausible beneficial effect on accelerated intestinal transit and duration of the inflammatory process.
It can be helpful in prevention overt pathological intestinal inflammation [186].
Goldsmith et al. [187] reported that the administration of the MOR agonist, DALDA protected against DSS-induced bowel injury in mice by promoting Stat3 phosphorylation in intestinal epithelial cells, which led to an increased expression of cytoprotective genes (Reg3b, Ccnd1, Cox2, myc), enterocyte proliferation and enhanced wound healing. DALDA may thus be useful in the treatment of diseases associated with intestinal barrier damage, e.g. IBD or radiation-induced damage.
Recent data suggest that blockade of MOR may also alleviate inflammation and, therefore, MOR antagonists have become an attractive target for drug design in the field of IBD. The anti-inflammatory action of the MOR antagonist, naltrexone in the mouse model of DSS-induced colitis was first described by Matters et al. [188]. Administration of naltrexone decreased the over-expression of pro-inflammatory cytokines IL-6 and IL-12 and improved mucosal structure. Jan et al. [189] showed that the administration of naloxone significantly inhibited endotoxin-induced activation of NF-κB in RAW264.7 cell culture, which is a major intracellular pathway involved in the expression of pro-inflammatory molecules. The proposed explanation of naloxone on NF-κB was mediated mainly by L-type calcium channels than opioid receptors. Interestingly, the administration of morphine enhanced the effect of naloxone as an anti-inflammatory compound.
To the best of our knowledge, there is only one clinical trial using opioids as an anti-inflammatory drug. In the randomized double-blind placebo controlled study, adult patients were treated with naltrexone (4.5 mg) or placebo for 12 weeks [190]. Colonoscopies were performed for all patients before and after the study and the results were reported according to the CD activity index (CDAI) scoring system. A significant improvement in GI mucosal inflammation was observed in patients after naltrexone therapy. The side effects of naltrexone treatment included insomnia, diarrhea and abdominal pain.
Sepsis
Bacterial sepsis, which is quite common in patients after surgical intervention in comparison with controls without surgery, is an important and unsolved problem in medicine, which affects, among others, the function of the GI tract.
It seems likely that opioid receptors may be involved in the development of sepsis and that morphine and other opioids may act as cofactors in its precipitation. Hilburger et al. [191] observed that morphine administration in slow-release pellets in mice caused the escape of Gram-negative and enteric bacteria (e.g. Proteus mirabilis, Escherichia coli, Enterococcus faecalis) from GI tract to the liver, the spleen and peritoneum, and led to septic state. Moreover, the application of naltrexone blocked the effect of morphine, indicating that opioid receptors are involved in sepsis development.
The cecal ligation and perforation (CLP) is used as an animal model of bacterial peritonitis, comparable with human sepsis. Topcu et al. [192] showed that the administration of fentanyl caused significant antitransit effects in the presence of systemic inflammation in rats. Furthermore, a higher antitransit effect of fentanyl was observed in rats with CLP than in the sham group. It was suggested that the peritoneal inflammation evokes sensitization of opioid receptors located in the myenteric and the submucosal plexuses in peripheral or central nerve terminals and increased the effects induced by administration of exogenous opioids, in particular MOR and DOR-selective. This is in good agreement with the study by Nardi et al. [193], who showed that opioid receptor agonists fentanyl and tramadol alleviated pain in rat CLP model. However, adverse side effects occurring after their administration, such as alteration of cardiovascular parameters and high mortality, did not allow for their chronic use.
Opioid receptors are a possible pharmacological target for the treatment of sepsis. Tang et al. [194], using rat CLP model, showed that DADLE (5 mg/kg, i.p.), a synthetic analog of [Leu5]enkephalin, protected against lethal endotoxemia in a DOR-dependent manner. In addition, concurrent and delayed treatment of rats with DADLE (10−6 M) suppressed LPS-induced apoptosis and necrosis. DADLE inhibited signal transduction in macrophages after LPS stimulation via modulation of MAPK and NFκB pathways and decreased concentration of TNF-α, IFN-γ, Il-1β in serum. The most important observation from the clinical point of view was that DADLE inhibited the release of HMBG1, a late pro-inflammatory cytokine which binds to DNA and is responsible for stimulation of genes involved in inflammatory response, from macrophages even 4 h after the onset of inflammation. This observation may encourage novel treatment strategies of sepsis.
Conclusion and future perspectives
The most anticipated goal of contemporary drug discovery is the development of a personalized therapy, which requires good knowledge of the treated disease at a molecular and genetic level and involves a careful selection of drugs, depending on the molecular target in the cells. The opioid receptor-based personalized therapy would primarily aim at the inhibition of molecular pathways responsible for the adverse side effects of opioid ligands, such as development of tolerance or OBD. Consequently, opioid peptide gene therapy was extensively studied in the last few years. The delivery of genes or their fragments encoding enkephalins, β-endorphin or EMs was already validated in numerous animal models [195, 196].
Conventional treatment of many GI disorders and malfunctions is limited to pure overcoming of their symptoms and associated with adverse side effects of drugs used. For example, currently available treatment for the intestinal inflammation, based on 5-aminosalicylate, corticosteroids and immunomodulators involves attenuation of inflammatory reaction and plain maintenance of this condition. Peripherally restricted opioids, which would act directly and indirectly on immune cells, might become important tools in the modulation of the immune system response and alleviation of the inflammatory state.
Other novel diagnostic and treatment strategies for GI disorders, implied by the presence of opioid receptors and their ligands in the GI tract and their crucial role in GI physiology and pathophysiology, are currently under investigation.
Acknowledgments
Supported by the Iuventus Plus program of the Polish Ministry of Science and Higher Education (0119/IP1/2011/71 to JF) and the National Natural Science Foundation of China (NSFC) Research Fund for International Young Scientists (81250110087 to JF).
Conflict of interest
The authors declare that they have no conflict of interest.
Abbreviations
- 5-HT
5-Hydroxytryptamine
- AC
Adenylyl cyclase
- AQ rt RT-PCR
Absolute quantitative real-time reverse transcriptase polymerase chain reaction
- BAM
Bovine adrenal medulla peptide
- BBB
Blood brain barrier
- CB1R
Cannabinoid receptor type 1
- CD
Crohn’s disease
- CFTR
Cystic fibrosis transmembrane conductance regulator
- CLP
Cecal ligation and perforation
- CNS
Central nervous system
- DADLE
[d-Ala2, d-Leu5]-Enkephalin
- DALDA
Tyr-d-Arg-Phe-Lys-NH2
- DAMGO
[d-Ala2, N-MePhe4, Gly-ol]-Enkephalin
- DFU
5,5-Dimethyl-3-(3-fluorophenyl)-4-(4-methylsulphonyl)-phenyl-2(5H)-furanone
- DOR
δ-Opioid receptor
- DSS
Dextran sulfate sodium
- EM
Endomorphin
- ENS
Enteric nervous system
- GI
Gastrointestinal
- GRK2/3
GPCR kinase 2/3
- IBD
Inflammatory bowel diseases
- KOR
κ-Opioid receptor
- LMMP
Longitudinal muscle-myenteric plexus
- LPH
Lipotropin
- MMCP-1
Mouse mast cell protease-1
- mNTS
Medial subnucleus of the tractus salitarius
- MNTX
Methylnaltrexone
- MOR
μ-opioid receptor
- MSH
Melanocyte-stimulating hormone
- OBD
Opioid-induced bowel dysfunction
- OIC
Opioid-induced constipation
- OPRD1
δ-opioid receptor gene
- OPRK1
κ-opioid receptor gene
- OPRM1
μ-opioid receptor gene
- PAMORA
Peripherally acting μ-opioid receptor antagonist
- PDYN
Prodynorphin
- PENK
Proenkephalin
- PI3K
Phosphoinositide 3-kinase
- PLCβ
Phospholipase β
- PMA
Phorbol 12-mirystate 13-acetate
- POI
Postoperative ileus
- POMC
Proopiomelanocortin
- RFBM
Rescue-free bowel movements
- RGS4
Regulator of G-protein signaling 4
- SA
Salvinorin A
- SNP
Single nucleotide polymorphism
- TNBS
2,4,6-trinitrobenzene sulfonic acid
- UC
Ulcerative colitis
- VGCC
Voltage-gated calcium channel
References
- 1.Pert CB, Snyder SH. Opiate receptor: demonstration in nervous tissue. Science. 1973;179:1011–1014. doi: 10.1126/science.179.4077.1011. [DOI] [PubMed] [Google Scholar]
- 2.Lord JA, Waterfield AA, Hughes J, Kosterlitz HW. Endogenous opioid peptides: multiple agonists and receptors. Nature. 1977;267:495–499. doi: 10.1038/267495a0. [DOI] [PubMed] [Google Scholar]
- 3.Dupre DJ, Robitaille M, Rebois RV, Hebert TE. The role of Gbetagamma subunits in the organization, assembly, and function of GPCR signaling complexes. Annu Rev Pharmacol Toxicol. 2009;49:31–56. doi: 10.1146/annurev-pharmtox-061008-103038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Piros ET, Hales TG, Evans CJ. Functional analysis of cloned opioid receptors in transfected cell lines. Neurochem Res. 1996;21:1277–1285. doi: 10.1007/BF02532368. [DOI] [PubMed] [Google Scholar]
- 5.Evans CJ, Keith DE, Jr, Morrison H, Magendzo K, Edwards RH. Cloning of a delta opioid receptor by functional expression. Science. 1992;258:1952–1955. doi: 10.1126/science.1335167. [DOI] [PubMed] [Google Scholar]
- 6.Li S, Zhu J, Chen C, Chen YW, Deriel JK, Ashby B, et al. Molecular cloning and expression of a rat kappa opioid receptor. Biochem J. 1993;295:629–633. doi: 10.1042/bj2950629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen Y, Mestek A, Liu J, Yu L. Molecular cloning of a rat kappa opioid receptor reveals sequence similarities to the mu and delta opioid receptors. Biochem J. 1993;295:625–628. doi: 10.1042/bj2950625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fukuda K, Kato S, Mori K, Nishi M, Takeshima H. Primary structures and expression from cDNAs of rat opioid receptor delta- and mu-subtypes. FEBS Lett. 1993;327:311–314. doi: 10.1016/0014-5793(93)81011-n. [DOI] [PubMed] [Google Scholar]
- 9.Dietis N, Rowbotham DJ, Lambert DG. Opioid receptor subtypes: fact or artifact? Br J Anaesth. 2011;107:8–18. doi: 10.1093/bja/aer115. [DOI] [PubMed] [Google Scholar]
- 10.Zollner C, Stein C. Opioids. Handb Exp Pharmacol. 2007;177:31–63. doi: 10.1007/978-3-540-33823-9_2. [DOI] [PubMed] [Google Scholar]
- 11.Jordan BA, Cvejic S, Devi LA. Opioids and their complicated receptor complexes. Neuropsychopharmacology. 2000;23:S5–S18. doi: 10.1016/S0893-133X(00)00143-3. [DOI] [PubMed] [Google Scholar]
- 12.Chaturvedi K, Christoffers KH, Singh K, Howells RD. Structure and regulation of opioid receptors. Biopolymers. 2000;55:334–346. doi: 10.1002/1097-0282(2000)55:4<334::AID-BIP1006>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 13.Befort K, Tabbara L, Bausch S, Chavkin C, Evans C, Kieffer B. The conserved aspartate residue in the third putative transmembrane domain of the delta-opioid receptor is not the anionic counterpart for cationic opiate binding but is a constituent of the receptor binding site. Mol Pharmacol. 1996;49:216–223. [PubMed] [Google Scholar]
- 14.Surratt CK, Johnson PS, Moriwaki A, Seidleck BK, Blaschak CJ, Wang JB, et al. mu opiate receptor. Charged transmembrane domain amino acids are critical for agonist recognition and intrinsic activity. J Biol Chem. 1994;269:20548–20553. [PubMed] [Google Scholar]
- 15.Chan AS, Law PY, Loh HH, Ho PN, Wu WM, Chan JS, et al. The first and third intracellular loops together with the carboxy terminal tail of the delta-opioid receptor contribute toward functional interaction with Galpha16. J Neurochem. 2003;87:697–708. doi: 10.1046/j.1471-4159.2003.02040.x. [DOI] [PubMed] [Google Scholar]
- 16.Reisine T, Law SF, Blake A, Tallent M. Molecular mechanisms of opiate receptor coupling to G proteins and effector systems. Ann N Y Acad Sci. 1996;780:168–175. doi: 10.1111/j.1749-6632.1996.tb15121.x. [DOI] [PubMed] [Google Scholar]
- 17.Clark MJ, Neubig RR, Traynor JR. Endogenous regulator of G protein signaling proteins suppress Galphao-dependent, mu-opioid agonist-mediated adenylyl cyclase supersensitization. J Pharmacol Exp Ther. 2004;310:215–222. doi: 10.1124/jpet.103.064824. [DOI] [PubMed] [Google Scholar]
- 18.Law PY, Wong YH, Loh HH. Molecular mechanisms and regulation of opioid receptor signaling. Annu Rev Pharmacol Toxicol. 2000;40:389–430. doi: 10.1146/annurev.pharmtox.40.1.389. [DOI] [PubMed] [Google Scholar]
- 19.Piros ET, Prather PL, Law PY, Evans CJ, Hales TG. Voltage-dependent inhibition of Ca2+ channels in GH3 cells by cloned mu- and delta-opioid receptors. Mol Pharmacol. 1996;50:947–956. [PubMed] [Google Scholar]
- 20.Georgoussi Z, Georganta EM, Milligan G. The other side of opioid receptor signalling: regulation by protein–protein interaction. Curr Drug Targets. 2012;13:80–102. doi: 10.2174/138945012798868470. [DOI] [PubMed] [Google Scholar]
- 21.Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by beta-arrestins. Science. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- 22.Maguma H, Thayne K, Taylor DA. Characteristics of tolerance in the guinea pig ileum produced by chronic in vivo exposure to opioid versus cannabinoid agonists. Biochem Pharmacol. 2010;80:522–532. doi: 10.1016/j.bcp.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 23.Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, Caron MG. Mu-opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature. 2000;408:720–723. doi: 10.1038/35047086. [DOI] [PubMed] [Google Scholar]
- 24.Fernandez Robles CR, Degnan M, Candiotti KA. Pain and genetics. Curr Opin Anaesthesiol. 2012;25:444–9. [DOI] [PubMed]
- 25.Levran O, Yuferov V, Kreek MJ. The genetics of the opioid system and specific drug addictions. Hum Genet. 2012;131:823–842. doi: 10.1007/s00439-012-1172-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raynor K, Kong H, Mestek A, Bye LS, Tian M, Liu J, et al. Characterization of the cloned human mu opioid receptor. J Pharmacol Exp Ther. 1995;272:423–428. [PubMed] [Google Scholar]
- 27.Chadzinska M. Opioid system and innate immunity. Comparative studies. I. Opioids and opioid receptors. Adv Cell Biol. 2007;34:251–262. [Google Scholar]
- 28.Cruz-Gordillo P, Fedrigo O, Wray GA, Babbitt CC. Extensive changes in the expression of the opioid genes between humans and chimpanzees. Brain Behav Evol. 2010;76:154–162. doi: 10.1159/000320968. [DOI] [PubMed] [Google Scholar]
- 29.Sehgal N, Smith HS, Manchikanti L. Peripherally acting opioids and clinical implications for pain control. Pain Physician. 2011;14:249–258. [PubMed] [Google Scholar]
- 30.Zhang H, Luo X, Kranzler HR, Lappalainen J, Yang BZ, Krupitsky E, et al. Association between two mu-opioid receptor gene (OPRM1) haplotype blocks and drug or alcohol dependence. Hum Mol Genet. 2006;15:807–819. doi: 10.1093/hmg/ddl024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Finco G, Pintor M, Sanna D, Orru G, Musu M, De CF, et al. Is target opioid therapy within sight? Minerva Anestesiol. 2012;78:462–472. [PubMed] [Google Scholar]
- 32.Kosarac B, Fox AA, Collard CD. Effect of genetic factors on opioid action. Curr Opin Anaesthesiol. 2009;22:476–482. doi: 10.1097/ACO.0b013e32832e34c9. [DOI] [PubMed] [Google Scholar]
- 33.Chavkin C, McLaughlin JP, Celver JP. Regulation of opioid receptor function by chronic agonist exposure: constitutive activity and desensitization. Mol Pharmacol. 2001;60:20–25. doi: 10.1124/mol.60.1.20. [DOI] [PubMed] [Google Scholar]
- 34.Klepstad P, Rakvag TT, Kaasa S, Holthe M, Dale O, Borchgrevink PC, et al. The 118 A>G polymorphism in the human mu-opioid receptor gene may increase morphine requirements in patients with pain caused by malignant disease. Acta Anaesthesiol Scand. 2004;48:1232–1239. doi: 10.1111/j.1399-6576.2004.00517.x. [DOI] [PubMed] [Google Scholar]
- 35.Walter C, Lotsch J. Meta-analysis of the relevance of the OPRM1 118A>G genetic variant for pain treatment. Pain. 2009;146:270–275. doi: 10.1016/j.pain.2009.07.013. [DOI] [PubMed] [Google Scholar]
- 36.Ashenhurst JR, Bujarski S, Ray LA. Delta and kappa opioid receptor polymorphisms influence the effects of naltrexone on subjective responses to alcohol. Pharmacol Biochem Behav. 2012;103:253–259. doi: 10.1016/j.pbb.2012.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Crist RC, Ambrose-Lanci LM, Vaswani M, Clarke TK, Zeng A, Yuan C, et al. Case-control association analysis of polymorphisms in the delta-opioid receptor, OPRD1, with cocaine and opioid addicted populations. Drug Alcohol Depend. 2013;127:122–128. doi: 10.1016/j.drugalcdep.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Arias AJ, Armeli S, Gelernter J, Covault J, Kallio A, Karhuvaara S, et al. Effects of opioid receptor gene variation on targeted nalmefene treatment in heavy drinkers. Alcohol Clin Exp Res. 2008;32:1159–1166. doi: 10.1111/j.1530-0277.2008.00735.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang H, Gelernter J, Gruen JR, Kranzler HR, Herman AI, Simen AA. Functional impact of a single-nucleotide polymorphism in the OPRD1 promoter region. J Hum Genet. 2010;55:278–284. doi: 10.1038/jhg.2010.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gelernter J, Kranzler HR. Genetics of drug dependence. Dialogues Clin Neurosci. 2010;12:77–84. doi: 10.31887/DCNS.2010.12.1/jgelernter. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Edenberg HJ, Wang J, Tian H, Pochareddy S, Xuei X, Wetherill L, et al. A regulatory variation in OPRK1, the gene encoding the kappa-opioid receptor, is associated with alcohol dependence. Hum Mol Genet. 2008;17:1783–1789. doi: 10.1093/hmg/ddn068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peng J, Sarkar S, Chang SL. Opioid receptor expression in human brain and peripheral tissues using absolute quantitative real-time RT-PCR. Drug Alcohol Depend. 2012;124:223–228. doi: 10.1016/j.drugalcdep.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Holzer P. Opioid antagonists for prevention and treatment of opioid-induced gastrointestinal effects. Curr Opin Anaesthesiol. 2010;23:616–622. doi: 10.1097/ACO.0b013e32833c3473. [DOI] [PubMed] [Google Scholar]
- 44.Wood JD, Galligan JJ. Function of opioids in the enteric nervous system. Neurogastroenterol Motil. 2004;16(Suppl 2):17–28. doi: 10.1111/j.1743-3150.2004.00554.x. [DOI] [PubMed] [Google Scholar]
- 45.Sternini C, Patierno S, Selmer IS, Kirchgessner A. The opioid system in the gastrointestinal tract. Neurogastroenterol Motil. 2004;16(Suppl 2):3–16. doi: 10.1111/j.1743-3150.2004.00553.x. [DOI] [PubMed] [Google Scholar]
- 46.DeHaven-Hudkins DL, DeHaven RN, Little PJ, Techner LM. The involvement of the mu-opioid receptor in gastrointestinal pathophysiology: therapeutic opportunities for antagonism at this receptor. Pharmacol Ther. 2008;117:162–87. [DOI] [PubMed]
- 47.Poole DP, Pelayo JC, Scherrer G, Evans CJ, Kieffer BL, Bunnett NW. Localization and regulation of fluorescently labeled delta opioid receptor, expressed in enteric neurons of mice. Gastroenterology. 2011;141:982–991. doi: 10.1053/j.gastro.2011.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Phan NQ, Lotts T, Antal A, Bernhard JD, Stander S. Systemic kappa opioid receptor agonists in the treatment of chronic pruritus: a literature review. Acta Derm Venereol. 2012;92:555–560. doi: 10.2340/00015555-1353. [DOI] [PubMed] [Google Scholar]
- 49.Bagnol D, Mansour A, Akil H, Watson SJ. Cellular localization and distribution of the cloned mu and kappa opioid receptors in rat gastrointestinal tract. Neuroscience. 1997;81:579–591. doi: 10.1016/s0306-4522(97)00227-3. [DOI] [PubMed] [Google Scholar]
- 50.Kurz A, Sessler DI. Opioid-induced bowel dysfunction: pathophysiology and potential new therapies. Drugs. 2003;63:649–671. doi: 10.2165/00003495-200363070-00003. [DOI] [PubMed] [Google Scholar]
- 51.Holzer P. Opioids and opioid receptors in the enteric nervous system: from a problem in opioid analgesia to a possible new prokinetic therapy in humans. Neurosci Lett. 2004;361:192–195. doi: 10.1016/j.neulet.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 52.Donnerer J, Liebmann I. Stimulus-evoked opioid inhibition in guinea-pig longitudinal muscle-myenteric plexus strip is modulated by NMDA receptors. Neurosci Lett. 2007;419:74–77. doi: 10.1016/j.neulet.2007.03.053. [DOI] [PubMed] [Google Scholar]
- 53.Patierno S, Zellalem W, Ho A, Parsons CG, Lloyd KC, Tonini M, et al. N-methyl-D-aspartate receptors mediate endogenous opioid release in enteric neurons after abdominal surgery. Gastroenterology. 2005;128:2009–2019. doi: 10.1053/j.gastro.2005.03.042. [DOI] [PubMed] [Google Scholar]
- 54.Donnerer J, Liebmann I. Evidence for opioid-induced release of glutamate in guinea pig longitudinal muscle-myenteric plexus strip. Neurosci Lett. 2009;462:118–120. doi: 10.1016/j.neulet.2009.06.086. [DOI] [PubMed] [Google Scholar]
- 55.Iwata H, Tsuchiya S, Nakamura T, Yano S. Morphine leads to contraction of the ileal circular muscle via inhibition of the nitrergic pathway in mice. Eur J Pharmacol. 2007;574:66–70. doi: 10.1016/j.ejphar.2007.06.029. [DOI] [PubMed] [Google Scholar]
- 56.De LA, Coupar IM. Insights into opioid action in the intestinal tract. Pharmacol Ther. 1996;69(2):103–115. doi: 10.1016/0163-7258(95)02053-5. [DOI] [PubMed] [Google Scholar]
- 57.Brock C, Olesen SS, Olesen AE, Frokjaer JB, Andresen T, Drewes AM. Opioid-induced bowel dysfunction: pathophysiology and management. Drugs. 2012;72:1847–1865. doi: 10.2165/11634970-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 58.Barrett KE. New insights into the pathogenesis of intestinal dysfunction: secretory diarrhea and cystic fibrosis. World J Gastroenterol. 2000;6:470–474. doi: 10.3748/wjg.v6.i4.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Barrett KE, Keely SJ. Chloride secretion by the intestinal epithelium: molecular basis and regulatory aspects. Annu Rev Physiol. 2000;62:535–572. doi: 10.1146/annurev.physiol.62.1.535. [DOI] [PubMed] [Google Scholar]
- 60.Herman MA, Gillis RA, Vicini S, Dretchen KL, Sahibzada N. Tonic GABAA receptor conductance in medial subnucleus of the tractus solitarius neurons is inhibited by activation of mu-opioid receptors. J Neurophysiol. 2012;107:1022–1031. doi: 10.1152/jn.00853.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Herman MA, Alayan A, Sahibzada N, Bayer B, Verbalis J, Dretchen KL, et al. micro-Opioid receptor stimulation in the medial subnucleus of the tractus solitarius inhibits gastric tone and motility by reducing local GABA activity. Am J Physiol Gastrointest Liver Physiol. 2010;299:G494–G506. doi: 10.1152/ajpgi.00038.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vilardaga JP, Bunemann M, Feinstein TN, Lambert N, Nikolaev VO, Engelhardt S, et al. GPCR and G proteins: drug efficacy and activation in live cells. Mol Endocrinol. 2009;23:590–599. doi: 10.1210/me.2008-0204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gomez MP, Nasi E. Light transduction in invertebrate hyperpolarizing photoreceptors: possible involvement of a Go-regulated guanylate cyclase. J Neurosci. 2000;20:5254–5263. doi: 10.1523/JNEUROSCI.20-14-05254.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Law PY, Erickson-Herbrandson LJ, Zha QQ, Solberg J, Chu J, Sarre A, et al. Heterodimerization of mu- and delta-opioid receptors occurs at the cell surface only and requires receptor-G protein interactions. J Biol Chem. 2000;280:11152–11164. doi: 10.1074/jbc.M500171200. [DOI] [PubMed] [Google Scholar]
- 65.Berger AC, Whistler JL. Morphine-induced mu opioid receptor trafficking enhances reward yet prevents compulsive drug use. EMBO Mol Med. 2011;3:385–397. doi: 10.1002/emmm.201100144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Milligan G. G protein-coupled receptor dimerisation: molecular basis and relevance to function. Biochim Biophys Acta. 2007;1768:825–835. doi: 10.1016/j.bbamem.2006.09.021. [DOI] [PubMed] [Google Scholar]
- 67.Milligan G, Parenty G, Stoddart LA, Lane JR. Novel pharmacological applications of G-protein-coupled receptor-G protein fusions. Curr Opin Pharmacol. 2007;7:521–526. doi: 10.1016/j.coph.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 68.Al-Hasani R, Bruchas MR. Molecular mechanisms of opioid receptor-dependent signaling and behavior. Anesthesiology. 2011;115:1363–1381. doi: 10.1097/ALN.0b013e318238bba6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rios C, Gomes I, Devi LA. mu opioid and CB1 cannabinoid receptor interactions: reciprocal inhibition of receptor signaling and neuritogenesis. Br J Pharmacol. 2006;148:387–395. doi: 10.1038/sj.bjp.0706757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Holzer P. Opioid receptors in the gastrointestinal tract. Regul Pept. 2009;155:11–17. doi: 10.1016/j.regpep.2009.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bunchorntavakul C, Reddy KR. Pruritus in chronic cholestatic liver disease. Clin Liver Dis. 2012;16:331–346. doi: 10.1016/j.cld.2012.03.010. [DOI] [PubMed] [Google Scholar]
- 72.Headrick JP, Pepe S, Peart JN. Non-analgesic effects of opioids: cardiovascular effects of opioids and their receptor systems. Curr Pharm Des. 2012;18:6090–6100. doi: 10.2174/138161212803582360. [DOI] [PubMed] [Google Scholar]
- 73.Husain S, Abdul Y, Potter DE. Non-Analgesic Effects of Opioids: neuroprotection in the Retina. Curr Pharm Des. 2012;18:6101–6108. doi: 10.2174/138161212803582441. [DOI] [PubMed] [Google Scholar]
- 74.Kromer W. Endogenous and exogenous opioids in the control of gastrointestinal motility and secretion. Pharmacol Rev. 1988;40:121–162. [PubMed] [Google Scholar]
- 75.Panchal SJ, Muller-Schwefe P, Wurzelmann JI. Opioid-induced bowel dysfunction: prevalence, pathophysiology and burden. Int J Clin Pract. 2007;61:1181–1187. doi: 10.1111/j.1742-1241.2007.01415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Collins S, Verma-Gandhu M. The putative role of endogenous and exogenous opiates in inflammatory bowel disease. Gut. 2006;55:756–757. doi: 10.1136/gut.2005.084418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Owczarek D, Cibor D, Mach T, Ciesla A, Pierzchala-Koziec K, Salapa K, et al. Met-enkephalins in patients with inflammatory bowel diseases. Adv Med Sci. 2011;56:158–164. doi: 10.2478/v10039-011-0051-x. [DOI] [PubMed] [Google Scholar]
- 78.Al-Khrasani M, Lacko E, Riba P, Kiraly K, Sobor M, Timar J, et al. The central versus peripheral antinociceptive effects of mu-opioid receptor agonists in the new model of rat visceral pain. Brain Res Bull. 2012;87:238–243. doi: 10.1016/j.brainresbull.2011.10.018. [DOI] [PubMed] [Google Scholar]
- 79.Xu GY, Winston JH, Chen JD. Electroacupuncture attenuates visceral hyperalgesia and inhibits the enhanced excitability of colon specific sensory neurons in a rat model of irritable bowel syndrome. Neurogastroenterol Motil. 2009;21:1302–e125. [DOI] [PubMed]
- 80.Zadina JE, Hackler L, Ge LJ, Kastin AJ. A potent and selective endogenous agonist for the mu-opiate receptor. Nature. 1997;386:499–502. doi: 10.1038/386499a0. [DOI] [PubMed] [Google Scholar]
- 81.Storr M, Geisler F, Neuhuber WL, Schusdziarra V, Allescher HD. Endomorphin-1 and -2, endogenous ligands for the mu-opioid receptor, inhibit striated and smooth muscle contraction in the rat oesophagus. Neurogastroenterol Motil. 2000;12:441–448. doi: 10.1046/j.1365-2982.2000.00220.x. [DOI] [PubMed] [Google Scholar]
- 82.Nishiwaki H, Saitoh N, Nishio H, Takeuchi T, Hata F. Relationship between muscarinic autoinhibition and the inhibitory effect of morphine on acetylcholine release from myenteric plexus of guinea pig ileum. Jpn J Pharmacol. 1998;77:271–278. doi: 10.1254/jjp.77.271. [DOI] [PubMed] [Google Scholar]
- 83.Nishiwaki H, Saitoh N, Nishio H, Takeuchi T, Hata F. Inhibitory effect of endomorphin-1 and -2 on acetylcholine release from myenteric plexus of guinea pig ileum. Jpn J Pharmacol. 1998;78:83–86. doi: 10.1254/jjp.78.83. [DOI] [PubMed] [Google Scholar]
- 84.Yu Y, Cui Y, Wang X, Lai LH, Wang CL, Fan YZ, et al. In vitro characterization of the effects of endomorphin 1 and 2, endogenous ligands for mu-opioid receptors, on mouse colonic motility. Biochem Pharmacol. 2007;73:1384–1393. doi: 10.1016/j.bcp.2007.01.011. [DOI] [PubMed] [Google Scholar]
- 85.Azuma Y, Ohura K. Endomorphin-2 modulates productions of TNF-alpha, IL-1beta, IL-10, and IL-12, and alters functions related to innate immune of macrophages. Inflammation. 2002;26:223–232. doi: 10.1023/a:1019766602138. [DOI] [PubMed] [Google Scholar]
- 86.Janecka A, Perlikowska R, Gach K, Wyrebska A, Fichna J. Development of opioid peptide analogs for pain relief. Curr Pharm Des. 2010;16:1126–1135. doi: 10.2174/138161210790963869. [DOI] [PubMed] [Google Scholar]
- 87.Varamini P, Mansfeld FM, Blanchfield JT, Wyse BD, Smith MT, Toth I. Synthesis and biological evaluation of an orally active glycosylated endomorphin-1. J Med Chem. 2012;55:5859–5867. doi: 10.1021/jm300418d. [DOI] [PubMed] [Google Scholar]
- 88.Kaneko K, Iwasaki M, Yoshikawa M, Ohinata K. Orally administered soymorphins, soy-derived opioid peptides, suppress feeding and intestinal transit via gut mu(1)-receptor coupled to 5-HT(1A), D(2), and GABA(B) systems. Am J Physiol Gastrointest Liver Physiol. 2010;299:G799–G805. doi: 10.1152/ajpgi.00081.2010. [DOI] [PubMed] [Google Scholar]
- 89.Chiba H, Tani F, Yoshikawa M. Opioid antagonist peptides derived from kappa-casein. J Dairy Res. 1989;56:363–366. doi: 10.1017/s0022029900028818. [DOI] [PubMed] [Google Scholar]
- 90.Patten GS, Head RJ, Abeywardena MY. Effects of casoxin 4 on morphine inhibition of small animal intestinal contractility and gut transit in the mouse. Clin Exp Gastroenterol. 2011;4:23–31. doi: 10.2147/CEG.S16161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Stefano GB, Ptacek R, Kuzelova H, Kream RM. Endogenous morphine: up-to-date review 2011. Folia Biol. 2012;58:49–56. [PubMed] [Google Scholar]
- 92.Netzer P, Sendensky A, Wissmeyer MP, Baumeler S, Batista C, Scheurer U, et al. The effect of naloxone-3-glucuronide on colonic transit time in healthy men after acute morphine administration: a placebo-controlled double-blinded crossover preclinical volunteer study. Aliment Pharmacol Ther. 2008;28:1334–1341. doi: 10.1111/j.1365-2036.2008.03855.x. [DOI] [PubMed] [Google Scholar]
- 93.Reber P, Brenneisen R, Flogerzi B, Batista C, Netzer P, Scheurer U. Effect of naloxone-3-glucuronide and N-methylnaloxone on the motility of the isolated rat colon after morphine. Dig Dis Sci. 2007;52:502–507. doi: 10.1007/s10620-006-9563-9. [DOI] [PubMed] [Google Scholar]
- 94.Ross GR, Gabra BH, Dewey WL, Akbarali HI. Morphine tolerance in the mouse ileum and colon. J Pharmacol Exp Ther. 2008;327:561–572. doi: 10.1124/jpet.108.143438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Aquilante CL, Letrent SP, Pollack GM, Brouwer KL. Increased brain P-glycoprotein in morphine tolerant rats. Life Sci. 2000;66:L47–L51. doi: 10.1016/s0024-3205(99)00599-8. [DOI] [PubMed] [Google Scholar]
- 96.King M, Su W, Chang A, Zuckerman A, Pasternak GW. Transport of opioids from the brain to the periphery by P-glycoprotein: peripheral actions of central drugs. Nat Neurosci. 2001;4:268–274. doi: 10.1038/85115. [DOI] [PubMed] [Google Scholar]
- 97.Yousif S, Saubamea B, Cisternino S, Marie-Claire C, Dauchy S, Scherrmann JM, et al. Effect of chronic exposure to morphine on the rat blood-brain barrier: focus on the P-glycoprotein. J Neurochem. 2008;107:647–657. doi: 10.1111/j.1471-4159.2008.05647.x. [DOI] [PubMed] [Google Scholar]
- 98.Anastassopoulos KP, Chow W, Tapia CI, Baik R, Ackerman SJ, Biondi D, et al. Economic study on the impact of side effects in patients taking oxycodone controlled-release for noncancer pain. J Manag Care Pharm. 2012;18:615–626. doi: 10.18553/jmcp.2012.18.8.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Peng X, Cebra JJ, Adler MW, Meissler JJ, Jr, Cowan A, Feng P, et al. Morphine inhibits mucosal antibody responses and TGF-beta mRNA in gut-associated lymphoid tissue following oral cholera toxin in mice. J Immunol. 2001;167:3677–3681. doi: 10.4049/jimmunol.167.7.3677. [DOI] [PubMed] [Google Scholar]
- 100.Breslow JM, Feng P, Meissler JJ, Pintar JE, Gaughan J, Adler MW, et al. Potentiating effect of morphine on oral Salmonella enterica serovar Typhimurium infection is mu-opioid receptor-dependent. Microb Pathog. 2010;49:330–335. doi: 10.1016/j.micpath.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Feng P, Rahim RT, Cowan A, Liu-Chen LY, Peng X, Gaughan J, et al. Effects of mu, kappa or delta opioids administered by pellet or pump on oral Salmonella infection and gastrointestinal transit. Eur J Pharmacol. 2006;534:250–257. doi: 10.1016/j.ejphar.2006.01.048. [DOI] [PubMed] [Google Scholar]
- 102.Babrowski T, Holbrook C, Moss J, Gottlieb L, Valuckaite V, Zaborin A, et al. Pseudomonas aeruginosa virulence expression is directly activated by morphine and is capable of causing lethal gut-derived sepsis in mice during chronic morphine administration. Ann Surg. 2012;255:386–393. doi: 10.1097/SLA.0b013e3182331870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Glattard E, Welters ID, Lavaux T, Muller AH, Laux A, Zhang D, et al. Endogenous morphine levels are increased in sepsis: a partial implication of neutrophils. PLoS ONE. 2010;5:e8791. doi: 10.1371/journal.pone.0008791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Capasso R, Borrelli F, Capasso F, Siebert DJ, Stewart DJ, Zjawiony JK, et al. The hallucinogenic herb Salvia divinorum and its active ingredient salvinorin A inhibit enteric cholinergic transmission in the guinea-pig ileum. Neurogastroenterol Motil. 2006;18:69–75. doi: 10.1111/j.1365-2982.2005.00725.x. [DOI] [PubMed] [Google Scholar]
- 105.Fichna J, Schicho R, Andrews CN, Bashashati M, Klompus M, McKay DM, et al. Salvinorin A inhibits colonic transit and neurogenic ion transport in mice by activating kappa-opioid and cannabinoid receptors. Neurogastroenterol Motil. 2009;21:1326-e128. [DOI] [PubMed]
- 106.Capasso R, Borrelli F, Cascio MG, Aviello G, Huben K, Zjawiony JK, et al. Inhibitory effect of salvinorin A, from Salvia divinorum, on ileitis-induced hypermotility: cross-talk between kappa-opioid and cannabinoid CB(1) receptors. Br J Pharmacol. 2008;155:681–689. doi: 10.1038/bjp.2008.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Fichna J, Dicay M, Hirota SA, Traboulsi D, Macdonald JA, Janecka A, et al. Differential effects of salvinorin A on endotoxin-induced hypermotility and neurogenic ion transport in mouse ileum. Neurogastroenterol Motil. 2011;23:583-e212. [DOI] [PubMed]
- 108.Fichna J, Dicay M, Lewellyn K, Janecka A, Zjawiony JK, MacNaughton WK, et al. Salvinorin A has antiinflammatory and antinociceptive effects in experimental models of colitis in mice mediated by KOR and CB1 receptors. Inflamm Bowel Dis. 2012;18:1137–1145. doi: 10.1002/ibd.21873. [DOI] [PubMed] [Google Scholar]
- 109.Fichna J, Schicho R, Janecka A, Zjawiony JK, Storr M. Selective natural kappa opioid and cannabinoid receptor agonists with a potential role in the treatment of gastrointestinal dysfunction. Drug News Perspect. 2009;22:383–392. doi: 10.1358/dnp.2009.22.7.1400219. [DOI] [PubMed] [Google Scholar]
- 110.Laugsand EA, Kaasa S, Klepstad P. Management of opioid-induced nausea and vomiting in cancer patients: systematic review and evidence-based recommendations. Palliat Med. 2011;25:442–453. doi: 10.1177/0269216311404273. [DOI] [PubMed] [Google Scholar]
- 111.Chen W, Chung HH, Cheng JT. Opiate-induced constipation related to activation of small intestine opioid mu2-receptors. World J Gastroenterol. 2012;18:1391–1396. doi: 10.3748/wjg.v18.i12.1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kumar R, Reeta KH, Ray SB. Antinociceptive effect of intrathecal loperamide: role of mu-opioid receptor and calcium channels. Eur J Pharmacol. 2012;696:77–82. doi: 10.1016/j.ejphar.2012.09.022. [DOI] [PubMed] [Google Scholar]
- 113.Primi MP, Bueno L, Baumer P, Berard H, Lecomte JM. Racecadotril demonstrates intestinal antisecretory activity in vivo. Aliment Pharmacol Ther. 1999;13:3–7. doi: 10.1046/j.1365-2036.13.s6.3.x. [DOI] [PubMed] [Google Scholar]
- 114.Duncker SC, Philippe D, Martin-Paschoud C, Moser M, Mercenier A, Nutten S. Nigella sativa (Black Cumin) seed extract alleviates symptoms of allergic diarrhea in mice, involving opioid receptors. PLoS One. 2012;7:e39841. doi: 10.1371/journal.pone.0039841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Isik F, Tunali AT, Yarat A, Genc Z, Pisiriciler R, Caliskan-Ak E, et al. Protective effects of black cumin (Nigella sativa) oil on TNBS-induced experimental colitis in rats. Dig Dis Sci. 2011;56:721–730. doi: 10.1007/s10620-010-1333-z. [DOI] [PubMed] [Google Scholar]
- 116.Boskabady MH, Mohsenpoor N, Takaloo L. Antiasthmatic effect of Nigella sativa in airways of asthmatic patients. Phytomedicine. 2010;17:707–713. doi: 10.1016/j.phymed.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 117.Banerjee S, Padhye S, Azmi A, Wang Z, Philip PA, Kucuk O, et al. Review on molecular and therapeutic potential of thymoquinone in cancer. Nutr Cancer. 2010;62:938–946. doi: 10.1080/01635581.2010.509832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hirata T, Keto Y, Nakata M, Takeuchi A, Funatsu T, Akuzawa S, et al. Effects of serotonin 5-HT3 receptor antagonists on stress-induced colonic hyperalgesia and diarrhoea in rats: a comparative study with opioid receptor agonists, a muscarinic receptor antagonist and a synthetic polymer. Neurogastroenterol Motil. 2008;20:557–565. doi: 10.1111/j.1365-2982.2007.01069.x. [DOI] [PubMed] [Google Scholar]
- 119.McNicol E, Horowicz-Mehler N, Fisk RA, Bennett K, Gialeli-Goudas M, Chew PW, et al. Management of opioid side effects in cancer-related and chronic noncancer pain: a systematic review. J Pain. 2003;4:231–256. doi: 10.1016/s1526-5900(03)00556-x. [DOI] [PubMed] [Google Scholar]
- 120.Gwee KA. Irritable bowel syndrome in developing countries–a disorder of civilization or colonization? Neurogastroenterol Motil. 2005;17:317–324. doi: 10.1111/j.1365-2982.2005.00627.x. [DOI] [PubMed] [Google Scholar]
- 121.Pappagallo M. Incidence, prevalence, and management of opioid bowel dysfunction. Am J Surg. 2001;182:11S–18S. doi: 10.1016/s0002-9610(01)00782-6. [DOI] [PubMed] [Google Scholar]
- 122.Deibert P, Xander C, Blum HE, Becker G. Methylnaltrexone: the evidence for its use in the management of opioid-induced constipation. Core Evid. 2010;4:247–258. doi: 10.2147/ce.s8556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Leppert W. The role of opioid receptor antagonists in the treatment of opioid-induced constipation: a review. Adv Ther. 2010;27:714–730. doi: 10.1007/s12325-010-0063-0. [DOI] [PubMed] [Google Scholar]
- 124.Thomas J, Karver S, Cooney GA, Chamberlain BH, Watt CK, Slatkin NE, et al. Methylnaltrexone for opioid-induced constipation in advanced illness. N Engl J Med. 2008;358:2332–2343. doi: 10.1056/NEJMoa0707377. [DOI] [PubMed] [Google Scholar]
- 125.Jakab RL, Collaco AM, Ameen NA. Lubiprostone targets prostanoid signaling and promotes ion transporter trafficking, mucus exocytosis, and contractility. Dig Dis Sci. 2012;57:2826–2845. doi: 10.1007/s10620-012-2352-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Norimatsu Y, Moran AR, Macdonald KD. Lubiprostone activates CFTR, but not ClC-2, via the prostaglandin receptor (EP(4)) Biochem Biophys Res Commun. 2012;426:374–379. doi: 10.1016/j.bbrc.2012.08.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ao M, Venkatasubramanian J, Boonkaewwan C, Ganesan N, Syed A, Benya RV, et al. Lubiprostone activates Cl- secretion via cAMP signaling and increases membrane CFTR in the human colon carcinoma cell line, T84. Dig Dis Sci. 2011;56:339–351. doi: 10.1007/s10620-010-1495-8. [DOI] [PubMed] [Google Scholar]
- 128.Sun X, Wang X, Wang GD, Xia Y, Liu S, Qu M, et al. Lubiprostone reverses the inhibitory action of morphine on mucosal secretion in human small intestine. Dig Dis Sci. 2011;56:330–338. doi: 10.1007/s10620-010-1515-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Fei G, Raehal K, Liu S, Qu MH, Sun X, Wang GD, et al. Lubiprostone reverses the inhibitory action of morphine on intestinal secretion in guinea pig and mouse. J Pharmacol Exp Ther. 2010;334:333–340. doi: 10.1124/jpet.110.166116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Chamberlain SM, Rao SS. Safety evaluation of lubiprostone in the treatment of constipation and irritable bowel syndrome. Expert Opin Drug Saf. 2012;11:841–850. doi: 10.1517/14740338.2012.708732. [DOI] [PubMed] [Google Scholar]
- 131.Kalff JC, Schraut WH, Billiar TR, Simmons RL, Bauer AJ. Role of inducible nitric oxide synthase in postoperative intestinal smooth muscle dysfunction in rodents. Gastroenterology. 2000;118:316–327. doi: 10.1016/s0016-5085(00)70214-9. [DOI] [PubMed] [Google Scholar]
- 132.Bauer AJ, Boeckxstaens GE. Mechanisms of postoperative ileus. Neurogastroenterol Motil. 2004;16:54–60. doi: 10.1111/j.1743-3150.2004.00558.x. [DOI] [PubMed] [Google Scholar]
- 133.Beard TL, Leslie JB, Nemeth J. The opioid component of delayed gastrointestinal recovery after bowel resection. J Gastrointest Surg. 2011;15:1259–1268. doi: 10.1007/s11605-011-1500-3. [DOI] [PubMed] [Google Scholar]
- 134.Thompson M, Magnuson B. Management of postoperative ileus. Orthopedics. 2012;35:213–217. doi: 10.3928/01477447-20120222-08. [DOI] [PubMed] [Google Scholar]
- 135.Pfab F, Nowak-Machen M, Napadow V, Fleckenstein J. Alternatives to prokinetics to move the pylorus and colon. Curr Opin Clin Nutr Metab Care. 2012;15:166–173. doi: 10.1097/MCO.0b013e32834f3000. [DOI] [PubMed] [Google Scholar]
- 136.Schmidt J, Stoffels B, Nazir A. haven-Hudkins DL, Bauer AJ. Alvimopan and COX-2 inhibition reverse opioid and inflammatory components of postoperative ileus. Neurogastroenterol Motil. 2008;20:689–699. doi: 10.1111/j.1365-2982.2007.01078.x. [DOI] [PubMed] [Google Scholar]
- 137.Delaney CP, Weese JL, Hyman NH, Bauer J, Techner L, Gabriel K, et al. Phase III trial of alvimopan, a novel, peripherally acting, mu opioid antagonist, for postoperative ileus after major abdominal surgery. Dis Colon Rectum. 2005;48:1114–1125. doi: 10.1007/s10350-005-0035-7. [DOI] [PubMed] [Google Scholar]
- 138.Apfel CC, Jalota L. Can central antiemetic effects of opioids counter-balance opioid-induced nausea and vomiting? Acta Anaesthesiol Scand. 2010;54:129–131. doi: 10.1111/j.1399-6576.2009.02133.x. [DOI] [PubMed] [Google Scholar]
- 139.Marderstein EL, Delaney CP. Management of postoperative ileus: focus on alvimopan. Ther Clin Risk Manag. 2008;4:965–973. doi: 10.2147/tcrm.s3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Webster L, Jansen JP, Peppin J, Lasko B, Irving G, Morlion B, et al. Alvimopan, a peripherally acting mu-opioid receptor (PAM-OR) antagonist for the treatment of opioid-induced bowel dysfunction: results from a randomized, double-blind, placebo-controlled, dose-finding study in subjects taking opioids for chronic non-cancer pain. Pain. 2008;137:428–440. doi: 10.1016/j.pain.2007.11.008. [DOI] [PubMed] [Google Scholar]
- 141.Paulson DM, Kennedy DT, Donovick RA, Carpenter RL, Cherubini M, Techner L, et al. Alvimopan: an oral, peripherally acting, mu-opioid receptor antagonist for the treatment of opioid-induced bowel dysfunction—a 21-day treatment-randomized clinical trial. J Pain. 2005;6:184–192. doi: 10.1016/j.jpain.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 142.Jansen JP, Lorch D, Langan J, Lasko B, Hermanns K, Kleoudis CS, et al. A randomized, placebo-controlled phase 3 trial (Study SB-767905/012) of alvimopan for opioid-induced bowel dysfunction in patients with non-cancer pain. J Pain. 2011;12:185–193. doi: 10.1016/j.jpain.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 143.Irving G, Penzes J, Ramjattan B, Cousins M, Rauck R, Spierings EL, et al. A randomized, placebo-controlled phase 3 trial (Study SB-767905/013) of alvimopan for opioid-induced bowel dysfunction in patients with non-cancer pain. J Pain. 2011;12:175–184. doi: 10.1016/j.jpain.2010.06.013. [DOI] [PubMed] [Google Scholar]
- 144.Wolff BG, Michelassi F, Gerkin TM, Techner L, Gabriel K, Du W, et al. Alvimopan, a novel, peripherally acting mu opioid antagonist: results of a multicenter, randomized, double-blind, placebo-controlled, phase III trial of major abdominal surgery and postoperative ileus. Ann Surg. 2004;240:728–734. doi: 10.1097/01.sla.0000141158.27977.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Viscusi ER, Goldstein S, Witkowski T, Andonakakis A, Jan R, Gabriel K, et al. Alvimopan, a peripherally acting mu-opioid receptor antagonist, compared with placebo in postoperative ileus after major abdominal surgery: results of a randomized, double-blind, controlled study. Surg Endosc. 2006;20:64–70. doi: 10.1007/s00464-005-0104-y. [DOI] [PubMed] [Google Scholar]
- 146.Buchler MW, Seiler CM, Monson JR, Flamant Y, Thompson-Fawcett MW, Byrne MM, et al. Clinical trial: alvimopan for the management of post-operative ileus after abdominal surgery: results of an international randomized, double-blind, multicentre, placebo-controlled clinical study. Aliment Pharmacol Ther. 2008;28:312–325. doi: 10.1111/j.1365-2036.2008.03696.x. [DOI] [PubMed] [Google Scholar]
- 147.Taguchi A, Sharma N, Saleem RM, Sessler DI, Carpenter RL, Seyedsadr M, et al. Selective postoperative inhibition of gastrointestinal opioid receptors. N Engl J Med. 2001;345:935–940. doi: 10.1056/NEJMoa010564. [DOI] [PubMed] [Google Scholar]
- 148.Ludwig K, Enker WE, Delaney CP, Wolff BG, Du W, Fort JG, et al. Gastrointestinal tract recovery in patients undergoing bowel resection: results of a randomized trial of alvimopan and placebo with a standardized accelerated postoperative care pathway. Arch Surg. 2008;143:1098–1105. doi: 10.1001/archsurg.143.11.1098. [DOI] [PubMed] [Google Scholar]
- 149.Herzog TJ, Coleman RL, Guerrieri JP, Jr, Gabriel K, Du W, Techner L, et al. A double-blind, randomized, placebo-controlled phase III study of the safety of alvimopan in patients who undergo simple total abdominal hysterectomy. Am J Obstet Gynecol. 2006;195:445–453. doi: 10.1016/j.ajog.2006.01.039. [DOI] [PubMed] [Google Scholar]
- 150.Ludwig K, Viscusi ER, Wolff BG, Delaney CP, Senagore A, Techner L. Alvimopan for the management of postoperative ileus after bowel resection: characterization of clinical benefit by pooled responder analysis. World J Surg. 2010;34:2185–2190. doi: 10.1007/s00268-010-0635-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Itawi EA, Savoie LM, Hanna AJ, Apostolides GY. Alvimopan addition to a standard perioperative recovery pathway. JSLS. 2011;15:492–498. doi: 10.4293/108680811X13176785204076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Poston S, Broder MS, Gibbons MM, Maclaren R, Chang E, Vandepol CJ, et al. Impact of alvimopan (entereg) on hospital costs after bowel resection: results from a large inpatient database. P T. 2011;36:209–220. [PMC free article] [PubMed] [Google Scholar]
- 153.Delaney CP, Craver C, Gibbons MM, Rachfal AW, VandePol CJ, Cook SF, et al. Evaluation of clinical outcomes with alvimopan in clinical practice: a national matched-cohort study in patients undergoing bowel resection. Ann Surg. 2012;255:731–738. doi: 10.1097/SLA.0b013e31824a36cc. [DOI] [PubMed] [Google Scholar]
- 154.Vaughan-Shaw PG, Fecher IC, Harris S, Knight JS. A meta-analysis of the effectiveness of the opioid receptor antagonist alvimopan in reducing hospital length of stay and time to GI recovery in patients enrolled in a standardized accelerated recovery program after abdominal surgery. Dis Colon Rectum. 2012;55:611–620. doi: 10.1097/DCR.0b013e318249fc78. [DOI] [PubMed] [Google Scholar]
- 155.Bader S, Jaroslawski K, Blum HE, Becker G. Opioid-induced constipation in advanced illness: safety and efficacy of methylnaltrexone bromide. Clin Med Insights Oncol. 2011;5:201–211. doi: 10.4137/CMO.S4867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wong BS, Rao AS, Camilleri M, Manabe N, McKinzie S, Busciglio I, et al. The effects of methylnaltrexone alone and in combination with acutely administered codeine on gastrointestinal and colonic transit in health. Aliment Pharmacol Ther. 2010;32:884–893. doi: 10.1111/j.1365-2036.2010.04422.x. [DOI] [PubMed] [Google Scholar]
- 157.Murphy DB, Sutton JA, Prescott LF, Murphy MB. Opioid-induced delay in gastric emptying: a peripheral mechanism in humans. Anesthesiology. 1997;87:765–770. doi: 10.1097/00000542-199710000-00008. [DOI] [PubMed] [Google Scholar]
- 158.Yuan CS, Foss JF, Osinski J, Toledano A, Roizen MF, Moss J. The safety and efficacy of oral methylnaltrexone in preventing morphine-induced delay in oral-cecal transit time. Clin Pharmacol Ther. 1997;61:467–475. doi: 10.1016/S0009-9236(97)90197-1. [DOI] [PubMed] [Google Scholar]
- 159.Michna E, Weil AJ, Duerden M, Schulman S, Wang W, Tzanis E, et al. Efficacy of subcutaneous methylnaltrexone in the treatment of opioid-induced constipation: a responder post hoc analysis. Pain Med. 2011;12:1223–1230. doi: 10.1111/j.1526-4637.2011.01189.x. [DOI] [PubMed] [Google Scholar]
- 160.Iyer SS, Randazzo BP, Tzanis EL, Schulman SL, Zhang H, Wang W, et al. Effect of subcutaneous methylnaltrexone on patient-reported constipation symptoms. Value Health. 2011;14:177–183. doi: 10.1016/j.jval.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 161.Michna E, Blonsky ER, Schulman S, Tzanis E, Manley A, Zhang H, et al. Subcutaneous methylnaltrexone for treatment of opioid-induced constipation in patients with chronic, nonmalignant pain: a randomized controlled study. J Pain. 2011;12:554–562. doi: 10.1016/j.jpain.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 162.Yu CS, Chun HK, Stambler N, Carpenito J, Schulman S, Tzanis E, et al. Safety and efficacy of methylnaltrexone in shortening the duration of postoperative ileus following segmental colectomy: results of two randomized, placebo-controlled phase 3 trials. Dis Colon Rectum. 2011;54:570–578. doi: 10.1007/DCR.0b013e3182092bde. [DOI] [PubMed] [Google Scholar]
- 163.Anissian L, Schwartz HW, Vincent K, Vincent HK, Carpenito J, Stambler N, et al. Subcutaneous methylnaltrexone for treatment of acute opioid-induced constipation: phase 2 study in rehabilitation after orthopedic surgery. J Hosp Med. 2012;7:67–72. doi: 10.1002/jhm.943. [DOI] [PubMed] [Google Scholar]
- 164.Chamberlain BH, Cross K, Winston JL, Thomas J, Wang W, Su C, et al. Methylnaltrexone treatment of opioid-induced constipation in patients with advanced illness. J Pain Symptom Manage. 2009;38:683–690. doi: 10.1016/j.jpainsymman.2009.02.234. [DOI] [PubMed] [Google Scholar]
- 165.Slatkin N, Thomas J, Lipman AG, Wilson G, Boatwright ML, Wellman C, et al. Methylnaltrexone for treatment of opioid-induced constipation in advanced illness patients. J Support Oncol. 2009;7:39–46. [PubMed] [Google Scholar]
- 166.Portenoy RK, Thomas J, Moehl Boatwright ML, Tran D, Galasso FL, Stambler N, et al. Subcutaneous methylnaltrexone for the treatment of opioid-induced constipation in patients with advanced illness: a double-blind, randomized, parallel group, dose-ranging study. J Pain Symptom Manage. 2008;35:458–68. [DOI] [PubMed]
- 167.Lipman AG, Karver S, Cooney GA, Stambler N, Israel RJ. Methylnaltrexone for opioid-induced constipation in patients with advanced illness: a 3-month open-label treatment extension study. J Pain Palliat Care Pharmacother. 2011;25:136–145. doi: 10.3109/15360288.2011.573531. [DOI] [PubMed] [Google Scholar]
- 168.Yuan CS, Foss JF, O’connor M, Osinski J, Karrison T, Moss J, et al. Methylnaltrexone for reversal of constipation due to chronic methadone use: a randomized controlled trial. JAMA. 2000;283:367–372. doi: 10.1001/jama.283.3.367. [DOI] [PubMed] [Google Scholar]
- 169.Garnock-Jones KP, McKeage K. Methylnaltrexone. Drugs. 2010;70:919–28. [DOI] [PubMed]
- 170.Garten L, Degenhardt P, Buhrer C. Resolution of opioid-induced postoperative ileus in a newborn infant after methylnaltrexone. J Pediatr Surg. 2011;46:e13–e15. doi: 10.1016/j.jpedsurg.2010.10.015. [DOI] [PubMed] [Google Scholar]
- 171.Fichna J, Gach K, Perlikowska R, Cravezic A, Bonnet JJ, do-Rego JC, et al. Novel endomorphin analogues with antagonist activity at the mu-opioid receptor in the gastrointestinal tract. Regul Pept. 2010;162:109–14. [DOI] [PubMed]
- 172.Fichna J, Storr MA. Brain-gut interactions in IBS. Front Pharmacol. 2012;3:127. [DOI] [PMC free article] [PubMed]
- 173.Farup PG, Sperber AD, Simren M. Irritable bowel syndrome. Gastroenterol Res Pract. 2012;2012:612479. doi: 10.1155/2012/612479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Andrews EB, Eaton SC, Hollis KA, Hopkins JS, Ameen V, Hamm LR, et al. Prevalence and demographics of irritable bowel syndrome: results from a large web-based survey. Aliment Pharmacol Ther. 2005;22:935–942. doi: 10.1111/j.1365-2036.2005.02671.x. [DOI] [PubMed] [Google Scholar]
- 175.Wilkins T, Pepitone C, Alex B, Schade RR. Diagnosis and management of IBS in adults. Am Fam Physician. 2012;86:419–426. [PubMed] [Google Scholar]
- 176.Philpott H, Gibson P, Thien F. Irritable bowel syndrome—an inflammatory disease involving mast cells. Asia Pac Allergy. 2011;1:36–42. doi: 10.5415/apallergy.2011.1.1.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Alaradi O, Barkin JS. Irritable bowel syndrome: update on pathogenesis and management. Med Princ Pract. 2002;11:2–17. doi: 10.1159/000048654. [DOI] [PubMed] [Google Scholar]
- 178.Distrutti E, Mencarelli A, Renga B, Caliendo G, Santagada V, Severino B, et al. A nitro-arginine derivative of trimebutine (NO2-Arg-Trim) attenuates pain induced by colorectal distension in conscious rats. Pharmacol Res. 2009;59:319–329. doi: 10.1016/j.phrs.2009.01.008. [DOI] [PubMed] [Google Scholar]
- 179.Lee N, Wald A. Linaclotide: evidence for its potential use in irritable bowel syndrome and chronic constipation. Core Evid. 2012;7:39–47. doi: 10.2147/CE.S25240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Olden KW. Targeted therapies for diarrhea-predominant irritable bowel syndrome. Clin Exp Gastroenterol. 2012;5:69–100. doi: 10.2147/CEG.S29023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Gross KJ, Pothoulakis C. Role of neuropeptides in inflammatory bowel disease. Inflamm Bowel Dis. 2007;13:918–932. doi: 10.1002/ibd.20129. [DOI] [PubMed] [Google Scholar]
- 182.Philippe D, Dubuquoy L, Groux H, Brun V, Chuoi-Mariot MT, Gaveriaux-Ruff C, et al. Anti-inflammatory properties of the mu opioid receptor support its use in the treatment of colon inflammation. J Clin Invest. 2003;111:1329–1338. doi: 10.1172/JCI16750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Srinath AI, Walter C, Newara MC, Szigethy EM. Pain management in patients with inflammatory bowel disease: insights for the clinician. Therap Adv Gastroenterol. 2012;5:339–357. doi: 10.1177/1756283X12446158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Cabot PJ, Carter L, Gaiddon C, Zhang Q, Schafer M, Loeffler JP, et al. Immune cell-derived beta-endorphin. Production, release, and control of inflammatory pain in rats. J Clin Invest. 1997;100:142–148. doi: 10.1172/JCI119506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Tomassini N, Renaud FL, Roy S, Loh HH. Mu and delta receptors mediate morphine effects on phagocytosis by murine peritoneal macrophages. J Neuroimmunol. 2003;136:9–16. doi: 10.1016/s0165-5728(02)00463-0. [DOI] [PubMed] [Google Scholar]
- 186.Philippe D, Chakass D, Thuru X, Zerbib P, Tsicopoulos A, Geboes K, et al. Mu opioid receptor expression is increased in inflammatory bowel diseases: implications for homeostatic intestinal inflammation. Gut. 2006;55:815–823. doi: 10.1136/gut.2005.080887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Goldsmith JR, Uronis JM, Jobin C. Mu opioid signaling protects against acute murine intestinal injury in a manner involving Stat3 signaling. Am J Pathol. 2011;179:673–683. doi: 10.1016/j.ajpath.2011.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Matters GL, Harms JF, McGovern C, Fitzpatrick L, Parikh A, Nilo N, et al. The opioid antagonist naltrexone improves murine inflammatory bowel disease. J Immunotoxicol. 2008;5:179–187. doi: 10.1080/15476910802131469. [DOI] [PubMed] [Google Scholar]
- 189.Jan WC, Chen CH, Hsu K, Tsai PS, Huang CJ. L-type calcium channels and mu-opioid receptors are involved in mediating the anti-inflammatory effects of naloxone. J Surg Res. 2011;167:e263–e272. doi: 10.1016/j.jss.2010.03.039. [DOI] [PubMed] [Google Scholar]
- 190.Smith JP, Bingaman SI, Ruggiero F, Mauger DT, Mukherjee A, McGovern CO, et al. Therapy with the opioid antagonist naltrexone promotes mucosal healing in active Crohn’s disease: a randomized placebo-controlled trial. Dig Dis Sci. 2011;56:2088–2097. doi: 10.1007/s10620-011-1653-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Hilburger ME, Adler MW, Truant AL, Meissler JJ, Jr, Satishchandran V, Rogers TJ, et al. Morphine induces sepsis in mice. J Infect Dis. 1997;176:183–188. doi: 10.1086/514021. [DOI] [PubMed] [Google Scholar]
- 192.Topcu I, Ekici NZ, Isik R, Sakarya M. The effects of tramadol and fentanyl on gastrointestinal motility in septic rats. Anesth Analg. 2006;102:876–881. doi: 10.1213/01.ane.0000196506.28780.94. [DOI] [PubMed] [Google Scholar]
- 193.Nardi GM, Bet AC, Sordi R, Fernandes D, Assreuy J. Opioid analgesics in experimental sepsis: effects on physiological, biochemical, and haemodynamic parameters. Fundam Clin Pharmacol. 2012 doi: 10.1111/j.1472-8206.2012.01041.x. [DOI] [PubMed] [Google Scholar]
- 194.Tang CW, Feng WM, Du HM, Bao Y, Zhu M. Delayed administration of D-Ala2-D-Leu5-enkephalin, a delta-opioid receptor agonist, improves survival in a rat model of sepsis. Tohoku J Exp Med. 2011;224:69–76. doi: 10.1620/tjem.224.69. [DOI] [PubMed] [Google Scholar]
- 195.Goins WF, Cohen JB, Glorioso JC. Gene therapy for the treatment of chronic peripheral nervous system pain. Neurobiol Dis. 2012;48:255–270. doi: 10.1016/j.nbd.2012.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Fink DJ, Wechuck J, Mata M, Glorioso JC, Goss J, Krisky D, et al. Gene therapy for pain: results of a phase I clinical trial. Ann Neurol. 2011;70:207–212. doi: 10.1002/ana.22446. [DOI] [PMC free article] [PubMed] [Google Scholar]