

Modified nucleosides in the anticodon sequence of tRNAs play important roles in reading of the genetic code. This paper describes the identification of a new modified nucleoside, 5-cyanomethyl uridine, in the anticodon wobble position of tRNAs from archaea. It also addresses questions on (1) the role of this new nucleoside in codon recognition in archaea and (2) its prevalence in tRNAs from different archaeal clades.
Keywords: 5-cyanomethyl uridine, tRNA modification, anticodon wobble position, codon reading properties, archaea
Abstract
Most archaea and bacteria use a modified C in the anticodon wobble position of isoleucine tRNA to base pair with A but not with G of the mRNA. This allows the tRNA to read the isoleucine codon AUA without also reading the methionine codon AUG. To understand why a modified C, and not U or modified U, is used to base pair with A, we mutated the C34 in the anticodon of Haloarcula marismortui isoleucine tRNA (tRNA2Ile) to U, expressed the mutant tRNA in Haloferax volcanii, and purified and analyzed the tRNA. Ribosome binding experiments show that although the wild-type tRNA2Ile binds exclusively to the isoleucine codon AUA, the mutant tRNA binds not only to AUA but also to AUU, another isoleucine codon, and to AUG, a methionine codon. The G34 to U mutant in the anticodon of another H. marismortui isoleucine tRNA species showed similar codon binding properties. Binding of the mutant tRNA to AUG could lead to misreading of the AUG codon and insertion of isoleucine in place of methionine. This result would explain why most archaea and bacteria do not normally use U or a modified U in the anticodon wobble position of isoleucine tRNA for reading the codon AUA. Biochemical and mass spectrometric analyses of the mutant tRNAs have led to the discovery of a new modified nucleoside, 5-cyanomethyl U in the anticodon wobble position of the mutant tRNAs. 5-Cyanomethyl U is present in total tRNAs from euryarchaea but not in crenarchaea, eubacteria, or eukaryotes.
INTRODUCTION
Of the 16 four-codon boxes in the genetic code, the AUN box (N=U, C, A, or G) is unique in that three of the four codons AUU, AUC, and AUA specify one amino acid (isoleucine), whereas the fourth codon, AUG, specifies another amino acid (methionine). This three-to-one distribution of codons within a four-codon box is different from essentially all other codon boxes in which all four codons either specify the same amino acid or are split two to two, codons ending in pyrimidines specifying one amino acid, and codons ending in purines specifying a different amino acid (Khorana 1968; Nirenberg 1968).
Archaea and bacteria use two different isoleucine tRNAs (tRNA1Ile and tRNA2Ile) to read the three isoleucine codons. tRNA1Ile with the anticodon GAU reads AUU and AUC following the Wobble Hypothesis (Crick 1966), whereas tRNA2Ile reads the remaining isoleucine codon AUA without misreading the methionine codon AUG (Harada and Nishimura 1974; Köhrer et al. 2008; Ikeuchi et al. 2010; Mandal et al. 2010). Most interestingly, in both kingdoms, tRNA2Ile has almost always C*AU as the anticodon sequence, C*, a modified C in the anticodon wobble position, being agmatidine in archaea (Ikeuchi et al. 2010; Mandal et al. 2010) and lysidine in bacteria (Muramatsu et al. 1988; Grosjean and Björk 2004). Thus, C* can form a base pair with A of the AUA but not with G of the AUG codon. This raises the question of why archaea and bacteria have evolved a mechanism to use a modified C instead of a U or a modified U to base pair exclusively with A.
In an attempt to answer the preceding question, we mutated the C in the anticodon of the Haloarcula marismortui tRNA2Ile gene to U (U34 mutant), expressed the mutant tRNA in Haloferax volcanii, and purified it. The mutant tRNA was found to be a poorer substrate for the haloarchaeal isoleucyl-tRNA synthetase (IleRS) in vivo and in vitro. The mutant tRNA was, however, a substrate for Escherichia coli IleRS, and this allowed us to aminoacylate it with isoleucine and study its codon reading properties using H. marismortui ribosomes. In contrast to wild-type Ile-tRNA2Ile, which binds only to the AUA codon, the U34 mutant tRNA binds not only to AUA but also to AUU, another isoleucine codon, and AUG, a methionine codon. To determine whether the U34 mutant derived from H. marismortui tRNA1Ile species would have similar coding properties, we also mutated G34 to U34 in the anticodon of tRNA1Ile. The U34 mutant of tRNA1Ile also bound to AUA, AUU, and AUG.
Analyses of the U34 mutant tRNAs showed that U34 in the anticodon wobble position is modified to 5-cyanomethyl U (cnm5U), a new modified nucleoside. We, therefore, investigated (1) whether some other naturally occurring tRNAs in H. volcanii contain this modified nucleoside; and (2) how widespread the occurrence of this modified nucleoside is in tRNAs from other archaeal organisms and from other kingdoms. Mass spectrometric analyses of nucleosides present in total tRNAs indicate that cnm5U is present in total tRNA from haloarchaea and Methanococcus maripaludis but not in total tRNA from Sulfolobus solfactaricus, Saccharomyces cerevisiae, and Escherichia coli, suggesting its possible presence only in tRNAs from euryarchaea.
RESULTS
Expression of H. marismortui wild-type and mutant isoleucine tRNAs in H. volcanii and growth phenotype of transformants
Previously, we described the purification of H. marismortui tRNA2Ile and showed that it contained agmatidine in the anticodon wobble position and bound to the isoleucine codon AUA but not to the methionine codon AUG (Mandal et al. 2010). In this work, we mutated the C in the anticodon wobble position of the tRNA2Ile gene to U (Fig. 1A) and expressed the U34 mutant tRNA in H. volcanii. In a parallel experiment, the G34 in the anticodon wobble position of tRNA1Ile was also mutated to U34 (Fig. 1B) and expressed in H. volcanii.
FIGURE 1.

H. marismortui isoleucine tRNA mutants containing 5-cyanomethyl uridine at the wobble position. Cloverleaf structures of tRNA2Ile (A) and tRNA1Ile (B) from H. marismortui. The anticodon changes at the wobble position from C+ to U* and G to U*, respectively, are indicated. (C+) agmatidine; (U*) 5-cyanomethyl uridine (cnm5U). Growth of H. volcanii cells transformed with plasmids for overexpression of H. marismortui tRNA2Ile wild-type and the tRNA2Ile U34 mutant (C), and H. marismortui tRNA1Ile wild-type and the tRNA1Ile U34 mutant (D). The empty vector (pWL201) is shown as a control in C and D.
H. volcanii cells were transformed with pWL201-derived expression vectors carrying either the H. marismortui wild-type tRNA2Ile gene, mutant tRNA2Ile gene, wild-type tRNA1Ile gene, or the mutant tRNA1Ile gene; the transformants were plated on media containing 4 μg/mL of mevinolin. Colonies of good size appeared 5–7 d after plating; however, transformants containing either of the mutant tRNA genes were always delayed by ∼2 d. Also, although plasmids carrying the empty pWL201 vector or either of the wild-type tRNA genes yielded ∼107 transformants/μg of plasmid DNA, those carrying either of the mutant tRNA genes yielded only 105–106 transformants/μg of plasmid DNA. Additionally, when individual freshly isolated colonies of similar size were used to inoculate liquid media containing mevinolin and growth was monitored over a period of several days, there was a consistent lag of 1–2 d in growth of transformants carrying the mutant tRNA genes (Fig. 1C,D), although the transformants eventually grow at a rate similar those carrying the wild-type tRNA genes. Taken together, these findings suggest that H. volcanii cells expressing the mutant tRNAs are less fit and require a phase of adaptation.
Purification of U34 mutants of tRNA2Ile and tRNA1Ile
As for the wild-type tRNAs, the mutant tRNAs were purified by hybrid selection using a biotinylated DNA oligonucleotide bound to streptavidin-sepharose, followed by native polyacrylamide gel electrophoresis (Suzuki and Suzuki 2007; Mandal et al. 2010). Figure 2A shows that the purified mutant tRNA2Ile is essentially homogeneous and consists of a major band and a minor band (Fig. 2A, left panel, lane 3), both of which hybridize to a DNA oligonucleotide probe complementary to H. marismortui tRNA2Ile (Fig. 2A, right panel, lane 3). The yield of purified tRNA from a 12-L culture of H. volcanii was about 4 A260 units and represented ∼0.4% of total tRNA. The U34 mutant of H. marismortui tRNA1Ile was purified similarly and also yielded a homogeneous product (Supplemental Fig. S1).
FIGURE 2.
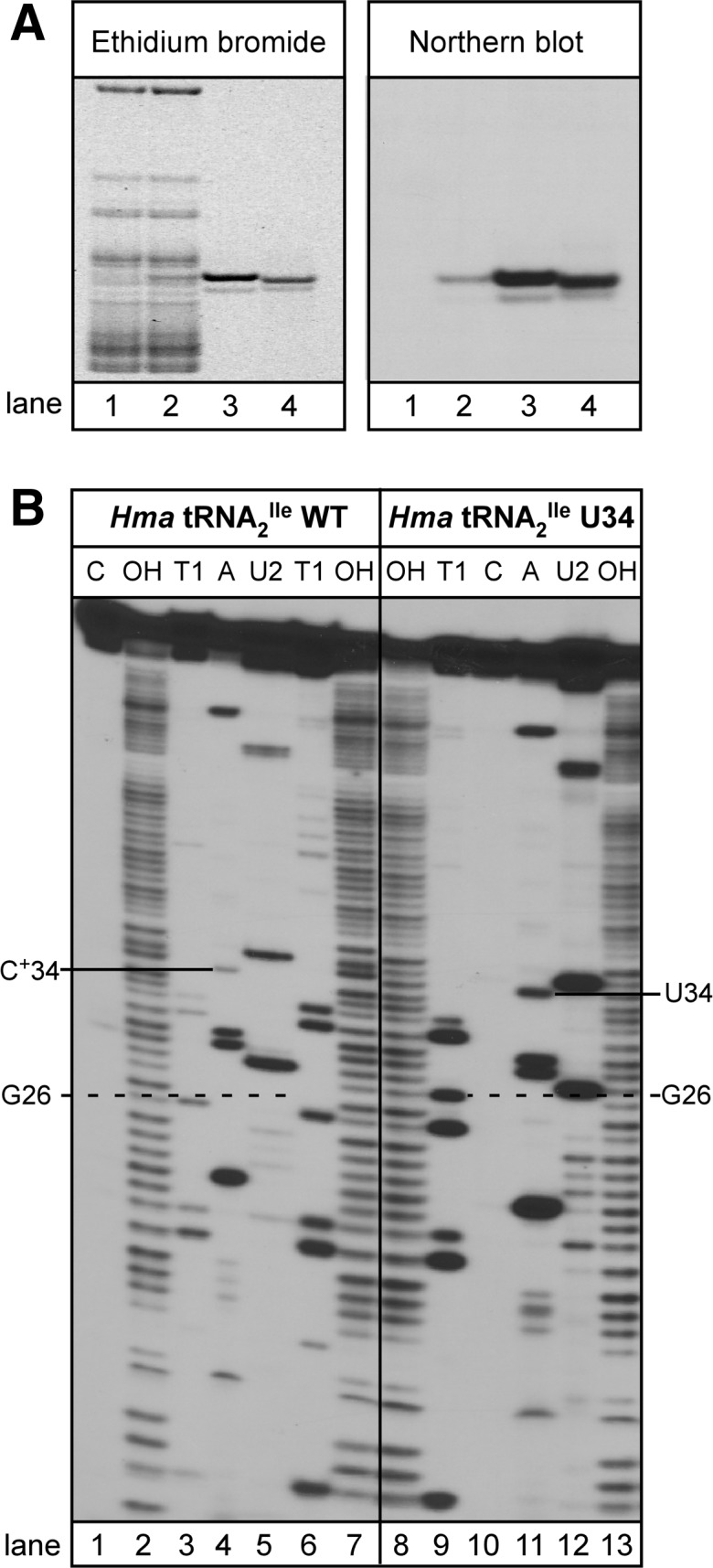
Purification and characterization of the H. marismortui tRNA2Ile U34 mutant. (A) Overexpression and purification of the H. marismortui tRNA2Ile U34 mutant. Native PAGE analysis of total tRNA from H. volcanii (lane 1), total tRNA from H. volcanii overexpressing the H. marismortui tRNA2Ile U34 mutant (lane 2), and purified H. marismortui tRNA2Ile U34 mutant (lane 3). Purified H. marismortui wild-type tRNA2Ile is shown as a control (lane 4). One-tenth A260 unit of total tRNA and 0.01 A260 of purified tRNA were applied per lane. tRNAs are visualized by ethidium bromide staining (left panel) or Northern blot analysis using a probe specific for H. marismortui tRNA2Ile (right panel). (B) Characterization of purified H. marismortui wild-type and U34 mutant tRNA2Ile. The homogeneity of purified tRNAs was confirmed by partial RNase T1 (T1 lanes), RNase A (A lanes), RNase U2 (U2 lanes) digests of 5′-32P-labeled tRNA. Two different concentrations of RNase T1 were used in lanes 3 and 6. (Lane OH) partial alkali hydrolysis; (Lane C) undigested tRNA control. 32P-labeled fragments were separated by denaturing PAGE and visualized by autoradiography.
Additional evidence for homogeneity of the purified tRNA was derived from partial RNase T1, RNase A, and RNase U2 digests of 5′-32P-labeled tRNA (Donis-Keller et al. 1977; Simoncsits et al. 1977; Lockard et al. 1978). All the 32P-labeled bands present in the RNase T1 digest were as expected for the mutant tRNA and were essentially the same as that for the wild-type tRNA (Fig. 2B, cf. lanes 3 and 6 to lane 9). The only exception was an RNase T1 cut at position 26 of the mutant tRNA, which is absent in RNase T1 digests of the wild-type tRNA. This is most likely due to a partial lack of modification of G to N2N2-dimethyl G at position 26 in the mutant tRNA. The cleavage patterns seen in digests with RNase A and RNase U2 are also essentially identical between the mutant and the wild-type tRNAs and are, as expected, based on the tRNA sequence (Fig. 2B, cf. lane 4 to lane 11, lane 5 to lane 12). Because partial digestions with RNases A and U2 were carried out in the absence of urea, digestion with these enzymes is more limited compared to digestion with RNase T1 in the presence of 7 M urea. Basically, similar results were obtained for RNase T1 and RNase A digests of 5′-32P-labeled wild-type and U34 mutant of tRNA1Ile (Supplemental Fig. S2).
The U34 mutant tRNA2Ile and tRNA1Ile overexpressed in H. volcanii are poor substrates for aminoacylation by H. volcanii IleRS
To determine the aminoacylation status of the H. marismortui mutant tRNAs, which are overexpressed in H. volcanii, we isolated total tRNA from a small scale culture of H. volcanii under acidic conditions, separated tRNAs from aminoacyl-tRNAs on an acid urea polyacrylamide gel, and detected wild-type and mutant H. marismortui tRNA2Ile by RNA blot hybridization (Varshney et al. 1991; Köhrer and RajBhandary 2008). Both the mutant and the wild-type tRNA2Ile were aminoacylated rather poorly (20%–25% and 25%–30%, respectively), based on phosphorimager analysis of the bands, in H. volcanii (Fig. 3A, top panel). For the U34 mutant derived from tRNA1Ile, the corresponding numbers were ∼60% for the mutant and >90% for the wild-type tRNA (Fig. 3B, top panel). Control experiments using H. volcanii tRNA2Ile and tRNA1Ile probes on the same blots show that the endogenous wild-type H. volcanii tRNA2Ile and tRNA1Ile are aminoacylated essentially quantitatively (Fig. 3A,B, bottom panels).
FIGURE 3.

State of in vivo aminoacylation of H. marismortui wild-type and U34 mutant tRNA2Ile (A) and wild-type and U34 mutant H. marismortui tRNA1Ile (B). Total tRNA was isolated under acidic conditions and analyzed by acid urea PAGE followed by Northern hybridization using probes specific for H. marismortui tRNA2Ile (A, top panel) and H. marismortui tRNA1Ile (B, top panel), respectively. The same blots were stripped and rehybridized using probes specific for the endogenous H. volcanii tRNA2Ile (A, bottom panel) and H. volcanii tRNA1Ile (B, bottom panel). Note that the H. volcanii tRNA1Ile-specific probe also picks up the overexpressed H. marismortui tRNA1Ile because of the high levels of overexpression of H. marismortui tRNAs and close sequence similarity between the overexpressed and the endogenous tRNA1Ile. (ac) tRNA isolated under acidic conditions; (OH−) tRNA after deacylation by base-treatment.
In vitro aminoacylation of the purified H. marismortui mutant tRNA2Ile and tRNA1Ile using IleRS present in S10, S30, or S100 extracts of H. volcanii showed that the mutant tRNAs were poor substrates for the archaeal IleRS (data not shown). The mutant tRNAs could, however, be aminoacylated essentially quantitatively with 3H-Ile using an excess of purified E. coli IleRS (Supplemental Fig. S3).
Codon-reading properties of the U34 mutant tRNA2Ile and tRNA1Ile
Purified wild-type and mutant tRNA2Ile and mutant tRNA1Ile were aminoacylated with 3H-Ile using E. coli IleRS, and the 3H-Ile-tRNAs were used for studying oligonucleotide-dependent binding to H. marismortui ribosomes. The oligonucleotides used were AUGAUA, AUGAUC, AUGAUU, and AUGAUG. Figure 4 shows the results. As previously published (Mandal et al. 2010), the wild-type Ile-tRNA2Ile binds to AUA but not to AUU, AUC, or AUG (Fig. 4A). In contrast, the mutant tRNA2Ile binds best to AUA, but it also binds to AUU, another isoleucine codon, and to AUG, a methionine codon (AUA>AUG>AUU) (Fig. 4B). The codon-dependent binding of the tRNA to the ribosome is dependent in all cases on concentration of the oligonucleotide. The codon binding properties of mutant tRNA1Ile are the same as those of the mutant tRNA2Ile (AUA>AUG>AUU) (Fig. 4C).
FIGURE 4.

Ribosome binding of U34 mutants of H. marismortui tRNA2Ile and tRNA1Ile. Template-dependent binding of purified 3H-Ile-tRNA2Ile wild-type (A) and 3H-Ile-tRNA2Ile U34 (B) to ribosomes isolated from H. marismortui. Oligonucleotides used were AUGAUA, AUGAUC, AUGAUG, and AUGAUU. (C) Binding of purified 3H-Ile-tRNA2Ile U34 and 3H-Ile-tRNA1Ile U34 to ribosomes isolated from H. volcanii; the mRNA concentration was 1 mM; the AUGUUU and “no mRNA” controls are also shown in C. Results from three independent experiments are shown.
Analysis of nucleotide in the anticodon wobble position of the mutant tRNA
In view of the codon binding properties of the U34 mutant tRNAs, a key question of interest is whether U34 in the mutant tRNA is modified in H. volcanii; if so, what is the nature of the modification? For this, we partially digested the mutant tRNA2Ile randomly with alkali, labeled the 5′-hydroxyl groups of the fragments generated with 32P using T4 polynucleotide kinase, and separated the 5′-32P-labeled fragments on a denaturing polyacrylamide gel (Stanley and Vassilenko 1978; Gupta and Randerath 1979; Kuchino et al. 1979; RajBhandary 1980). Figure 5A shows part of the pattern obtained for the wild-type and the mutant tRNAs. The digest of wild-type tRNA shows a pronounced shift in the electrophoretic mobility of fragments, which are produced by cleavage of phosphodiester bonds between nucleotides 33/34 and 34/35, and which differ by one nucleotide. This is due to the presence of the positively charged agmatidine at position 34 (Mandal et al. 2010). As expected, such a shift in mobility between neighboring fragments is not observed in digests of the U34 mutant tRNA.
FIGURE 5.

5′-32P-labeling of random alkali fragments (A), and 2D-TLC analysis of the wobble base nucleotide in U34 mutants of H. marismortui tRNA2Ile (B) and tRNA1Ile (C). Purified tRNAs were cleaved randomly with alkali; fragments were 5′-end labeled with 32P and separated by gel electrophoresis. The 5′-end nucleotide of the fragment corresponding to U*34 was analyzed by nuclease P1 digestion and 2D-TLC. Nonradioactive markers pA, pC, pG, and pU were visualized by UV-shadowing and are indicated by circles. (pU*) position of cnm5U.
For identification of the 5′-terminal nucleotides of the fragments, the 32P-labeled bands corresponding to nucleotides 32–38 in the anticodon loop of the wild-type and mutant tRNA were eluted from the gel, cleaved with nuclease P1, and the 32P-labeled 5′-terminal nucleotide of each fragment was identified by thin layer chromatography (Nishimura 1979; Silberklang et al. 1979). Figure 5B shows the two-dimensional thin layer chromatography pattern obtained for U34 in the mutant tRNA. The 5′-32P-labeled nucleotide migrates very close to, but not identical with, pU. Thus, U34 in the mutant tRNA contains a base modification. A similar analysis of the U34 mutant derived from H. marismortui tRNA1Ile shows that U34 in this mutant tRNA also carries the same base modification (Fig. 5C).
Mass spectrometric identification of the modified U as 5-cyanomethyl U (cnm5U)
To identify nucleosides that differed between wild-type and the U34 mutant H. marismortui tRNA2Ile, purified tRNA2Ile was enzymatically hydrolyzed, and the nucleosides were resolved by reversed-phase HPLC. Comparison of HPLC profiles showed the presence of a nucleoside present exclusively in the mutant tRNA2Ile (retention time 7.9 min) (Fig. 6, vertical arrow). Subsequent analysis of both samples by liquid chromatography-coupled quadrupole time-of-flight mass spectrometry (LC-QTOF) revealed a species with m/z 284.0872 (Fig. 7A), again only in the mutant tRNA. In the same high-resolution spectrum, we also identified an ion with m/z 152.0451, which is consistent with the loss of ribose from a precursor species with m/z 284.0872 (Fig. 7A). Similar analysis of a chemically synthesized 5-cyanomethyl-2′-deoxyuridine standard (Fig. 8B; Sakata et al. 1980; Kuwahara et al. 2006) revealed an ion with an identical m/z of 152.0451 (Fig. 7C).
FIGURE 6.
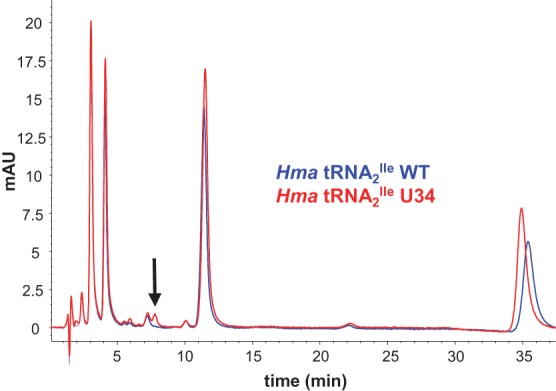
Identification of a unique ribonucleoside species in the U34 mutant tRNA2Ile. Purified H. marismortui wild-type and the U34 mutant thereof were digested to ribonucleosides and resolved by HPLC. The UV (260 nm) absorption patterns of the two digests were overlaid, and a unique peak was identified in the mutant sample, highlighted by the arrow.
FIGURE 7.
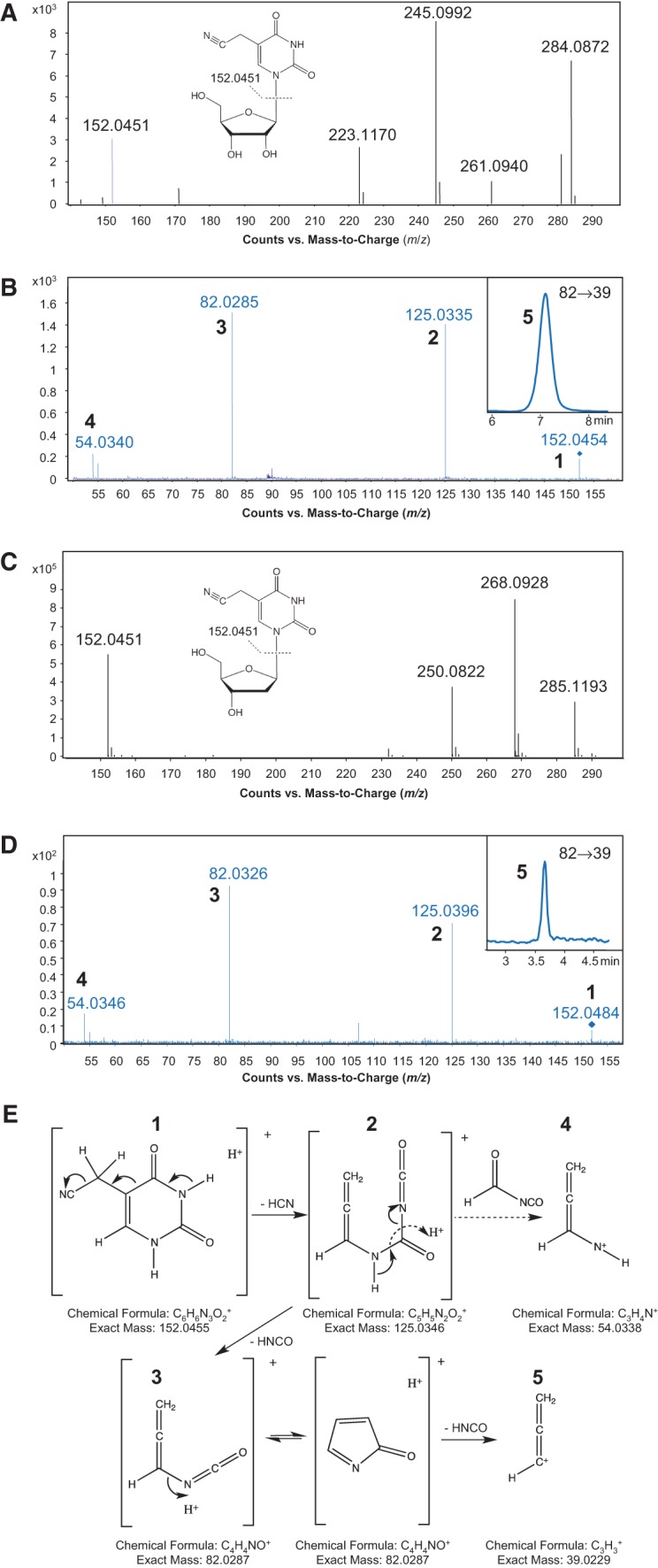
Structural elucidation of 5-cyanomethyl uridine. (A,C) High mass-accuracy mass spectra of the unknown ribonucleoside species (A) found in the purified H. marismortui tRNA2Ile U34 mutant and the 5-cyanomethyl-2′-deoxyuridine synthetic standard (C). The parent ions have m/z values of 284.0872 (A) and 268.0928 (C), and the deglycosylated base ions have identical m/z values of 152.0451 (A,C). (B,D) Pseudo-MS3 mass spectra of the unknown ribonucleoside species (B) and the 5-cyanomethyl-2′-deoxyuridine synthetic standard (D). Following in-source fragmentation of the parent ribonucleoside, the deglycosylated base ion was subjected to CID, with the product ions (numbered 1–5) in close agreement with the theoretical fragmentation pathway shown in panel E. The MRM chromatogram of the m/z 82→39 transition (species 5) is shown in the insets of B and D. (E) Proposed fragmentation pathway for 5-cyanomethyl uracil, with each numbered fragment along with its formula and exact mass.
FIGURE 8.
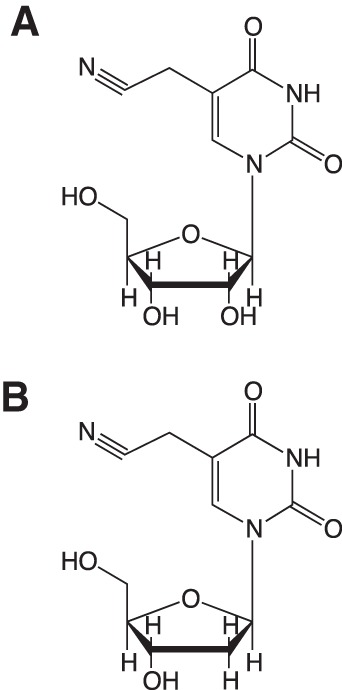
Chemical structures of 5-cyanomethyl uridine (A) and 5-cyanomethyl-2′-deoxyuridine (B).
The mass spectral data for the modified ribonucleoside identified in this study suggested a chemical formula of C11H13N3O6 (calculated m/z 284.0877), consistent with a U modified with methylene and cyano functional groups. Comparison of the UV absorption spectra of the modified U (isolated by HPLC), thymidine, and the synthetic 5-cyanomethyl-2′-deoxyuridine showed that they all had a λmax at 267 nm (Supplemental Fig. S4), suggesting that the cyano group is not attached directly to the ring, and the modified U most likely has a –CH2–C≡N side chain. Since most tRNA anticodon wobble modifications on U occur at the 5-position of the base (Yokoyama and Nishimura 1995), one candidate structure for the modified U is 5-cyanomethyl uridine (cnm5U) (Fig. 8A).
This structure was confirmed by tandem mass spectrometric analysis of the modified U in comparison with the synthetic 5-cyanomethyl-2′-deoxyuridine. As shown in Figure 7B and D, both the modified U and 5-cyanomethyl-2′-deoxyuridine were subjected to pseudo-MS3 fragmentation analysis by LC-QTOF. In this analysis, the nucleoside is deglycosylated in-source, and the resulting protonated base was selected by Q1 for further fragmentation by collision induced dissociation (CID) in the collision cell. In the high-resolution tandem mass spectra thus obtained, nucleobases from both the modified U (Fig. 7B) and the synthetic 5-cyanomethyl-2′-deoxyuridine (Fig. 7D) generated identical fragmentation patterns with m/z values of 125, 82, and 54, which are consistent with the proposed fragmentation pathway shown in Figure 7E. If the cyanomethyl group was present at position 5 of U, and not position 6, then CID fragmentation should also produce the allene species 5 (m/z 39) shown in Figure 7E. Although this species was not apparent by QTOF analysis due to low sensitivity, analysis of the protonated base ion by in-source fragmentation of the modified U and the 5-cyanomethyl-2′-deoxyuridine standard by tandem triple-quadruple (QqQ) mass spectrometry revealed a species with m/z 39 for both nucleosides (Fig. 7B,D, insets). Altogether, our data identify cnm5U as the unique modified U in the mutant H. marismortui tRNA2Ile. A similar mass spectrometric analysis of purified mutant H. marismortui tRNA1Ile also confirmed the presence of cnm5U in this tRNA (data not shown).
cnm5U is present in total tRNAs from haloarchaea and M. maripaludis but not in tRNAs from S. solfataricus, S. cerevisiae, and E. coli
The identification of cnm5U as a new modified nucleoside in mutant H. marismortui tRNA2Ile and tRNA1Ile expressed in H. volcanii raised the question of whether it is present in total tRNA from H. volcanii and from other organisms. To identify cnm5U in endogenous tRNAs in H. volcanii, we analyzed total tRNA digests with or without expression of the H. marismortui U34 mutant tRNA2Ile by QTOF. To reduce background and improve detection, ribonucleosides in total tRNA digests were separated by HPLC, and fractions eluting in the region of cnm5U were collected and subjected to LC-QTOF. We found cnm5U in total tRNA with or without expression of the U34 mutant H. marismortui tRNA2Ile, suggesting the presence of cnm5U in endogenous tRNAs in H. volcanii (data not shown). We also developed a multiple reaction-monitoring (MRM) method based on the fragmentation information from the LC-QTOF analysis of 5-cyanomethyl-2′-deoxyuridine and cnm5U, using a tandem triple-quadrupole mass spectrometer (Fig. 9A); and using this approach, we observe a distinct peak corresponding to cnm5U in total tRNA isolated from wild-type H. volcanii cells (Fig. 9B).
FIGURE 9.

cnm5U in total tRNAs from haloarchaea, M. maripaludis, S. solfataricus, S. cerevisiae, and E. coli. Nucleoside detection was performed, either by monitoring m/z 284.2→125 transition on an Agilent 6510 triple-quadrupole mass spectrometer (A–F) or by extracted ion chromatogram for the nucleoside m/z 284.2 on an Agilent 6520 QTOF mass spectrometer (G,H).
We subsequently analyzed total tRNA from Halobacterium salinarum, H. marismortui, M. maripaludis, S. solfataricus, S. cerevisiae, and E. coli for the presence of cnm5U. In the case of H. salinarum, H. marismortui, M. maripaludis, and S. solfataricus, total tRNA digests were directly analyzed using the MRM method (Fig. 9C–F); in the case of S. cerevisiae and E. coli, HPLC fractions eluting at the cnm5U retention time were collected and the prepurified fractions were subjected to LC-QTOF analysis (Fig. 9G,H). In all cases, purified cnm5U from the U34 mutant tRNA2Ile and total tRNAs from H. volcanii were used as positive control. Within the detection limits of the instrument, we found cnm5U in total tRNA from haloarchaea and M. maripaludis but not from S. solfactaricus, S. cerevisiae, and E. coli.
DISCUSSION
To answer the question why most bacterial and archaeal isoleucine tRNAs use lysidine or agmatidine in the anticodon wobble position to base pair exclusively with A but not with G, we have reported here the expression, purification, and analysis of the codon binding properties of mutants of H. marismortui tRNA2Ile and tRNA1Ile, in which the anticodon wobble position has been mutated to U. The U34 mutants of H. marismortui tRNA2Ile and tRNA1Ile bind not only to the isoleucine codons AUA and AUU but also to the methionine codon AUG. Binding to AUG could lead to misreading of AUG and insertion of isoleucine in place of methionine into proteins. The extent of misreading of the AUG codon in H. volcanii would, however, depend upon several factors, including the extent of aminoacylation of the mutant tRNA, its concentration in vivo, its relative affinity for the AUG codon on the ribosome, and competition by the endogenous H. volcanii elongator Met-tRNAMet, which normally binds to the AUG codon. The findings that transformants carrying the genes for the U34 mutants of tRNA2Ile or tRNA1Ile have a growth disadvantage compared with those carrying the wild-type tRNA2Ile or tRNA1Ile (Fig. 1C,D) suggest that misreading of AUG also occurs in vivo. This would explain why nearly all archaea and bacteria use a modified C and not U or a modified U in the anticodon wobble position to read the isoleucine codon AUA. Although agmatidine and lysidine, both modified derivatives of C, base pair specifically with A of AUA, similar derivatives of U have so far not been found in nature. It is likely that such U derivatives would have tautomeric structures in which both the N3 and O4 could act as H-bond acceptors and, therefore, be incapable of forming stable base pairs with A on the ribosome.
Our results do not, however, rule out the possibility that some archaeal or bacterial organism will use U or a different modified U in the anticodon wobble position to read the isoleucine codon AUA without also reading the methionine codon AUG. For example, most eukaryotic organisms encode an isoleucine tRNA (tRNA2Ile) that has a U modified to Ψ in the anticodon wobble position (Senger et al. 1997). The Ψ in this tRNA is thought to base pair with A of the AUA codon but not with G of the AUG codon. Also, it is known that two archaeal organisms, a nanoarchaeon and a korachaeon (Waters et al. 2003; Randau et al. 2005; Elkins et al. 2008), and some bacteria including Mycoplasma mobile, Bifidobacterium adolescentis, and a mutant strain of B. subtilis encode tRNAIle with a UAU in the anticodon (Fabret et al. 2011). It has been shown that the U in the anticodon wobble position of tRNAIle from M. mobile and the mutant tRNAIle from B. subtilis is not modified, and these tRNAs read the isoleucine codon AUA without misreading the methionine codon AUG to any significant extent in vivo and in vitro (Köhrer et al. 2013; Taniguchi et al. 2013). It is possible that in these few organisms, the ribosomes and the tRNAIle sequences have coevolved in such a way that U or a modified U base pairs much better with A than with G.
Characterization of the U34 mutants of H. marismortui tRNA2Ile and tRNA1Ile led to the discovery of cnm5U, a new modified nucleoside in H. volcanii. Mass spectrometric analysis of nucleosides derived from total tRNAs isolated from H. volcanii, H. salinarum, H. marismortui, and M. maripaludis has shown that this modified nucleoside is also present in endogenous tRNAs from these organisms but not in total tRNAs isolated from S. solfactaricus (a crenarchaeon), S. cerevisiae, or E. coli. Thus, cnm5U may be present mostly in tRNAs from euryarchaea.
Ribosome binding experiments with the U34 mutant of H. marismortui tRNA2Ile show that it binds to AUA, AUU, and AUG (AUA>AUG>AUU) with minimal binding, if any, to AUC (Fig. 4B,C). The corresponding mutant of tRNA1Ile also binds to AUA, AUU, and AUG but not to AUC (Fig. 4C). These results suggest that cnm5U in the anticodon wobble position of a tRNA forms base pairs with U, A, or G on the mRNA.
The codons of a four-codon box in the genetic code Table are, in general, read by at least two tRNAs. For example, in eukaryotes a tRNA with inosine in the anticodon wobble position reads codons ending in U, C, and A, whereas another tRNA with C in the anticodon wobble position reads codons ending in G (Björk 1998). In contrast, in bacteria such as E. coli, a tRNA with 5′-carboxymethoxy U (cmo5U) in the anticodon wobble position reads codons ending in U, A, and G, whereas another tRNA with G in the anticodon wobble position reads codons ending in U and C (Murao et al. 1970; Yokoyama and Nishimura 1995). Similarly, in B. subtilis, a tRNA with 5′-methoxy U (mo5U) reads codons ending in U, A, and G, and a tRNA with G in the anticodon reads codons ending in U and C (Murao et al. 1976; Yokoyama and Nishimura 1995). Information on the codon recognition patterns and properties of archaeal tRNAs is, however, quite limited. Except for the classical work of Gupta on nucleotide sequences of H. volcanii tRNAs and identification of many of the modified bases in these tRNAs (Gupta 1984, 1986) and identification of genes encoding many of the tRNA modifying enzymes using bioinformatic analyses (Phillips and de Crecy-Lagard 2011), there has been very little work done with purified tRNAs from archaea and on the codon recognition properties of archaeal tRNAs. The results we have obtained with two mutant haloarchaeal tRNAs containing cnm5U in the anticodon wobble position suggest that the codon recognition properties of cnm5U, which base pairs with U, A, or G, parallel those of cmo5U in E. coli and mo5U in B. subtilis. With the knowledge that other tRNAs in H. volcanii contain cnm5U (this work), it becomes important now to purify and identify these tRNAs and establish their codon recognition properties using H. volcanii ribosomes.
Finally, in spite of the fact that H. volcanii tRNAs have been among the best studied among the archaea in terms of nucleotide sequences and base modifications, our recent work on two haloarchaeal tRNAs, tRNA2Ile and a mutant derived from it, led to the discovery of two new modified nucleosides, agmatidine and 5-cyanomethyl uridine in H. volcanii and other haloarchaea. With so many other archaeal organisms, whose tRNAs have not been purified or studied to any extent, most likely there are many modified nucleosides with interesting structures and functions that remain to be discovered (Phillips and de Crecy-Lagard 2011). The methods for tRNA purification and analysis used here combined with the increased power and sophistication of mass spectral techniques and instrumentation should significantly facilitate this discovery.
MATERIALS AND METHODS
General
Strain H. volcanii WFD11 and plasmids pUCsptProM and pWL201 (Lam and Doolittle 1989; Nieuwlandt and Daniels 1990) were kindly provided by Drs. John R. Palmer and Charles J. Daniels (Department of Microbiology, Ohio State University). H. marismortui ATCC 43,049 was kindly provided by Dr. Peter Moore (Department of Chemistry, Yale University). The E. coli strains used in this work, E. coli XL1-blue and E. coli GM2163, have been described before (Ramesh and RajBhandary 2001). General manipulations of H. marismortui, H. volcanii, and E. coli were performed according to standard procedures (DasSarma and Fleischmann 1995; Sambrook and Russell 2001). RNase T1, RNase A, RNase U2, nuclease P1, and snake venom phosphodiesterase I were from Sigma; T4 polynucleotide kinase (T4-PNK), antarctic phosphatase, calf intestinal phosphatase, and inorganic pyrophosphatase were from New England Biolabs; DNA and RNA oligonucleotides were from IDT; and oligonucleotides used for cloning, detection, and purification of tRNAs are listed in Supplemental Table S1.
Cloning and overexpression of H. marismortui tRNA2Ile and tRNA1Ile (WT and the U34 mutant)
The gene for H. marismortui tRNA2Ile was first cloned into the vector pUCsptProM (Ramesh and RajBhandary 2001). For this, the gene for H. marismortui tRNA2Ile was PCR amplified from H. marismortui genomic DNA and inserted into the XbaI and BamHI sites of plasmid pUCsptProM downstream from the tRNALys promoter to generate pUCHmaIle2WT. The C34 to U34 mutation in the anticodon of tRNA2Ile was generated by QuikChange mutagenesis (Stratagene) using pUCHmaIle2WT as template. A HindIII/EcoRI fragment containing the wild-type tRNA2Ile gene or its U34 mutant, including the tRNALys promoter, was subcloned into the shuttle plasmid pWL201 to generate pWLHmaIle2WT and pWLHmaIle2MUT. Similarly, pWLHmaIle1WT and pWLHmaIle1MUT carrying the H. marismortui wild-type tRNA1Ile gene or its U34 mutant were generated. All initial cloning was done in E. coli XL-1 blue. After confirming the sequences of the desired wild-type and mutant tRNA genes, the respective plasmids were used to transform E. coli GM2163 (dam−). Plasmid DNA isolated from this strain was then used to transform H. volcanii as described (DasSarma and Fleischmann 1995; Ramesh and RajBhandary 2001). Transformants were grown in H. volcanii medium containing 4 µg/mL mevinolin (US Biologicals).
Polyacrylamide gel electrophoresis (PAGE) and Northern blot analysis of tRNA
Total RNA was extracted under acidic conditions using Trizol (Invitrogen) as described before (Köhrer et al. 2008). RNA was resuspended in 10 mM sodium acetate pH 5.0 and stored at −80°C. tRNAs were analyzed by acid urea PAGE (Varshney et al. 1991; Köhrer and RajBhandary 2008) or native PAGE as indicated, followed by Northern blotting (Köhrer and RajBhandary 2008). tRNAs were visualized by hybridization using 32P-labeled DNA oligonucleotides according to standard procedures (Sambrook and Russell 2001). Oligonucleotides were 5′-end labeled with γ-[32P]-ATP (3000 Ci/mmol; PerkinElmer) using T4-PNK. Northern blots were analyzed by autoradiography followed by PhosphorImaging using Imagequant software.
Purification of overexpressed H. marismortui tRNA2Ile and tRNA1Ile (WT and U34 mutant) from H. volcanii
H. volcanii transformants were grown at 37°C to an OD600 of 3–3.5, and crude RNA was isolated by acid guanidinium thiocyanate–phenol–chloroform extraction (Chomczynski and Sacchi 2006). Ribosomal RNA was removed by precipitation with 1 M LiCl yielding total tRNA. Purification of overexpressed isoleucine tRNAs was done as described earlier for tRNA2Ile (Suzuki and Suzuki 2007; Mandal et al. 2010), involving hybrid selection of the desired tRNA using a biotinylated DNA oligonucleotides immobilized to a streptavidin resin. The biotinylated oligonucleotide for tRNA2Ile purification was complementary to nucleotides 54–73 of the tRNA; the biotinylated oligonucleotide for tRNA1Ile purification was complementary to nucleotides 39–58 of the tRNA. Affinity purification was followed by electrophoresis of the enriched tRNA on native 10%–12.5% polyacrylamide gels. After elution from the gel and extensive dialysis against 5 mM ammonium acetate pH 5.5, the tRNA was concentrated by evaporation, precipitated with ethanol, and the precipitate was washed several times with 70% ethanol. Typically, about 4.0 A260 units of the U34 mutant of tRNA2Ile were obtained from approximately 1000 A260 of total tRNA; about 5.5 A260 units of the U34 mutant of tRNA1Ile were obtained from about 400 A260 of total tRNA. The purity of the tRNAs was assessed by PAGE, in vitro aminoacylation with isoleucine, and partial digestion of 5′-32P-labeled tRNAs with RNases T1, A, and U2.
In vitro aminoacylation of tRNA
One hundredth to 1.0 A260 units of tRNA were aminoacylated in vitro with L-isoleucine as described below using purified E. coli IleRS or S10, S30, or S100 extracts prepared from H. volcanii. Reaction mixtures using purified E. coli IleRS contained 50 mM 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (Hepes) pH 7.5, 10 mM MgCl2, 5 mM ATP, 0.1 μg/μL BSA, 5 μM [3H]-isoleucine (American Radiolabeled Chemicals), and 0.05 μM of IleRS. Alternatively, in vitro aminoacylations were carried using S10, S30, or S100 extracts from H. volcanii in a reaction containing 10 mM Hepes pH 7.5, 2.5 M KCl, 50 mM Mg(OAc)2, 5 mM ATP, 5 μM [3H]-isoleucine, and ∼0.5 μg/μL extract. At various time points, aliquots were removed and analyzed by precipitation with TCA followed by liquid scintillation counting of TCA-precipitable counts. Background (obtained from reactions run without tRNA) was subtracted from all values unless otherwise noted. Quantitative aminoacylation of purified H. marismortui isoleucine tRNAs for ribosome binding assays were carried out with purified E. coli IleRS; for this, reactions were allowed to proceed for 2 h, and inorganic pyrophosphatase was added at a final concentration of 0.04 units/μL. After aminoacylation, tRNAs were extracted with acid phenol and precipitated with ethanol.
Isolation of H. marismortui and H. volcanii ribosomes
Ribosomes from H. marismortui and H. volcanii were isolated as described (Mandal et al. 2010) using the basic procedure of Wittman and coworkers (Shevack et al. 1985) with minor modifications. Cells from a 2 L culture grown to midlog phase (OD600 ∼3.5) were suspended in ice-cold 10 mM Hepes pH 7.5, 100 mM Mg(OAc)2, 3.4 M KCl and 6 mM 2-mercaptoethanol (“high-salt ribosome buffer”), and lysed by French Press (Constant Cell Disruption System; 10,000 Psi, 2 passages). The ribosomes were then prepared as described, resuspended in “high-salt ribosome buffer,” divided into aliquots, quick frozen, and stored at −80°C.
Binding of aminoacylated tRNAs to ribosomes
The mRNA oligonucleotides (AUGAUA, AUGAUC, AUGAUU, AUGAUG, and AUGUUU) used in the ribosome binding experiments were stored at a stock concentration of 10 mM. The standard reaction (10 μL) contained 50 mM Tris-HCl pH 7.5, 50 mM NH4Cl, 5 mM Mg(OAc)2, 3 mM 2-mercaptoethanol, 2.1 M KCl, 1.2 A260 units of H. marismortui ribosomes, RNA oligonucleotides (0–1.5 mM), and ∼1.6 pmol [3H]-labeled tRNA (10,000 cpm). Alternatively, reactions (20 μL) contained 50 mM Hepes pH 7.5, 50 mM NH4Cl, 50 mM Mg(OAc)2, 3 mM 2-mercaptoethanol, 2.1 M KCl, 1 A260 unit of H. volcanii ribosomes, RNA oligonucleotides (0–1.5 mM), and ∼1.6 pmol [3H]-labeled tRNA (10,000 cpm). Incubation was at room temperature for 1 h. The reaction was stopped by adding 0.5 mL wash buffer [50 mM Tris-HCl or Hepes pH 7.5, 50 mM NH4Cl, 5 mM Mg(OAc)2, 2.1 M KCl], and the mixture was filtered through a nitrocellulose filter (HA 0.45 μm, Millipore) prewashed with the same buffer. The filters were washed several times with wash buffer, dried, and counted for radioactivity in a liquid scintillation counter.
5′-32P-end labeling and partial digestion of tRNA
tRNA (0.01 A260) was dephosphorylated with 1 unit of calf intestinal phosphatase in a 50-μL reaction containing 50 mM Tris-HCl pH 7.9 and 1 mM EDTA. Before adding enzyme, the mixture was incubated for 2 min at 65°C and then for 5 min at 37°C. After dephosphorylation for 30 min at 37°C, the reaction mixture was incubated with 5 mM nitrilotriacetic acid (Sigma) for 30 min at room temperature, and the phosphatase was inactivated by heat for 2 min at 65°C. Dephosphorylated tRNAs were labeled with 32P at the 5′ terminus in a 50-μL reaction containing 70 mM Tris-HCl pH 7.5, 10 mM MgCl2, 5 mM dithiothreitol (DTT), 10 pmol of γ-[32P]-ATP, and 20 U of T4-PNK. After 30 min of incubation at 37°C, the 32P-labeled tRNA was precipitated with a 2.5 volume of ethanol, 0.2 M ammonium acetate pH 5.2, and 10 µg glycogen (Ambion). The precipitate was washed with 70% ethanol. 32P-labeled tRNA was separated by electrophoresis on a 6% denaturing polyacrylamide gel, and the band corresponding to the full-length tRNA was excised and eluted in 10 mM Tris-HCl pH 7.5 and 5 mM MgCl2. The eluted radiolabeled tRNA was further precipitated and washed as above and then dissolved in sterile water.
For partial alkali hydrolysis and partial RNase digestions, 20,000 cpm of 32P-labeled tRNA and 0.01 A260 units of total H. marismortui tRNA were used in reaction volumes of 7.5–10 μL. The partial alkali hydrolysis reaction was in 33 mM Na2CO3/NaHCO3 buffer pH 9.2 for 2–3 min at 95°C. The partial RNase T1 digestion was in 50 mM Tris-HCl pH 7.5, 7 M urea and 5–10 U of RNase T1 and incubation was for 10 min at 50°C. Partial digestion with RNase U2 (0.002 unit) was in 50 mM ammonium acetate pH 4.5 for 5 min at 37°C. Partial digestion with RNase A (0.002–0.005 unit) was in 10 mM Tris-HCl (pH 8.0), 1 mM EDTA for 5 min at 37°C. After the incubation, samples were quick frozen, lyophilized to dryness, and then dissolved in formamide loading buffer. The samples were separated by electrophoresis on a 10% denaturing polyacrylamide gel; gels were dried and then used for autoradiography.
Nuclease P1 digests of 5′-32P-end labeled fragments of tRNA
Purified tRNAs (0.01 A260) were partially hydrolyzed with alkali as described above. The mixture was evaporated to dryness, and the residue was dissolved in 10 μL water. Digested tRNA (0.001 A260) was incubated with 5 units of T4-PNK in a 20-μL reaction containing 70 mM Tris-HCl pH 7.5, 10 mM MgCl2, 5 mM DTT, and 2.5 pmol γ-[32P]-ATP for 5 min at 37°C. The 5′- 32P -labeled fragments were separated by 10% denaturing PAGE, and bands corresponding to the anticodon base U34 (for mutant tRNA2Ile and tRNA1Ile) and bases preceding and following U34 were eluted with buffer A (20 mM Tris-HCl pH 7.5, 10 mM MgCl2), precipitated with ethanol, washed, dissolved in sterile water, and digested with nuclease P1 in 50 mM ammonium acetate buffer pH 5.0 overnight at 37°C. The incubation mixtures were quick frozen on dry ice and lyophilized under vacuum. Nucleotides were dissolved in sterile water and lyophilized to dryness three times to remove any ammonium acetate prior to thin layer chromatography.
Thin layer chromatography
Cellulose F plates (20 × 20 cm) with fluorescent background used for two-dimensional thin layer chromatography (2D-TLC) analyses were obtained from Merck. The solvent system for the first and second dimension were as follows: (1) isobutyric acid:concentrated ammonia:water (66:1:33); and (2) isopropanol:concentrated HCl:water (70:15:15). The first dimension was run in Solvent 1 for ∼16 h; the plates were then dried overnight. The second dimension was run in Solvent 2 for ∼28 h. Plates were dried and analyzed by autoradiography. Nonradioactive nucleotides were used as markers and visualized by UV shadowing.
Enzymatic hydrolysis of tRNA and HPLC analysis of modified ribonucleosides
Purified H. marismortui tRNA2Ile and tRNA1Ile (WT and U34 mutants) and total tRNA from H. volcanii, H. salinarum, H. marismortui, M. maripaludis, S. solfataricus, S. cerevisiae, and E. coli were digested to nucleosides using nuclease P1, snake venom phosphodiesterase I, and antarctic phosphatase as described in detail by Crain (Crain 1990). After digestion, nucleosides were passed through a centrifugal filter (Amicon Ultra; 10K) for removal of enzymes.
Nucleosides were then separated by HPLC using an Agilent 1100 HPLC and a Thermo Scientific Hypersil GOLD aQ reverse-phase column (150 × 2.1 mm, 3 µm) eluted with the following gradient of water to acetonitrile containing 8 mM ammonium acetate at a flow rate of 0.3 mL/min and 36°C: 0–18 min, 0%; 18–23 min, 0%–1%; 23–28 min, 1%–6%; 28–30 min, 6%; 30–40 min, 6%–100%; 40–50 min, 100%; the HPLC eluent was monitored at 260 nm using a diode-array detector.
Liquid chromatography-coupled mass spectrometric analysis of ribonucleosides
Ribonucleosides identified by HPLC were structurally characterized by LC-MS analysis. Initial analysis of the unknown ribonucleoside was performed using an Agilent 1290 UPLC equipped with diode array detectors and a Thermo Scientific Hypersil GOLD aQ C18 reverse-phase column (100 × 1 mm, 1.9 µm particle size) that was coupled to an Agilent 6510 QTOF high resolution mass spectrometer with an electrospray ionization source operated in positive ion mode with the following parameters: drying gas temperature, 325°C; drying gas flow, 8 L/min; nebulizer, 25 psi; and capillary voltage, 3500 V. The QTOF was operated in MS mode, with 140 V fragmentor voltage and with mass range m/z 100–1000. HPLC resolution was performed with the same gradient as before of water to acetonitrile at a flow rate of 70 µL/min and 36°C: 0–18 min, 0%; 18–23 min, 0%–1%; 23–28 min, 1%–6%; 28–30 min, 6%; 30–40 min, 6%–100%; 40–50 min, 100%. The unknown species with m/z 284.0872 eluted at 6.6 min.
Collision induced dissociation (CID) experiments were performed using an Agilent 1200 HPLC with the same Thermo Scientific Hypersil GOLD aQ reverse-phase column coupled to the QTOF mass spectrometer. HPLC resolution was performed isocratically with 8 mM ammonium acetate at a flow rate of 70 µL/min for 20 min and 36°C. The unknown species with m/z 284.0872 eluted at 4.27 min and 5-cyanomethyl-2′-deoxyuridine eluted at 3.56 min. The initial CID analysis of 5-cyanomethyl-2′-deoxyuridine and the unknown molecular species was performed in targeted MS/MS mode on m/z 268.0928 or m/z 284.08771, respectively, and a fragmentor voltage of 140 V or 90 V, respectively, with collision energies of 5 V, 10 V, or 15 V used to optimize product ion formation. In-source fragmentation and subsequent CID analysis of protonated base was used to perform pseudo-MS3 analysis of both 5-cyanomethyl-2′-deoxyuridine and the unknown molecular species. In this analysis, a fragmentor voltage 300 V was used in targeted MS/MS on the m/z 152.04545 fragment of 5-cyanomethyl-2′-deoxyuridine, with collision energies of 5 V, 10 V, or 15 V. For the unknown species, a fragmentor voltage of 160 V was used for targeted MS/MS on the m/z 152.04545 fragment of the unknown ribonucleoside.
Subsequent fragmentation analysis by triple-quadrupole (QqQ) mass spectrometry was performed by MRM analysis using an Agilent 1100 HPLC with a Thermo Scientific Hypersil GOLD aQ column (150 × 2.1 mm, 3 µm particle size) coupled to an Agilent 6410 QqQ mass spectrometer. For analysis of 5-cyanomethyl-2′-deoxyuridine and the purified unknown species, ribonucleosides were eluted isocratically with 8 mM ammonium acetate at a flow rate of 0.2 mL/min for 20 min and 36°C. The QqQ mass spectrometer with an electrospray ionization source was operated in positive ion mode with the following parameters: drying gas temperature, 325°C; drying gas flow, 8 L/min; nebulizer, 30 psi; and capillary voltage, 4000 V. The first and third quadrupoles (Q1 and Q3) were set to unit resolution and the monitored ion transitions. The m/z of the transmitted parent ion, m/z of the monitored product ion, fragmentor voltage, and collision energy, respectively, for 5-cyanomethyl-2′-deoxyuridine were as follows: 268.2→152.1, 90 V, 5 V; 152.1→125.1, 140 V, 5 V; 125.1→82.1, 150 V, 10 V; 82.1→39, 160 V, 15 V. For cnm5U, the parameters were: 284.2→152.1, 90 V, 5 V; 152.1→125.1, 140 V, 5 V; 125.1→82.1, 150 V, 10 V; 82.1→39, 170 V, 15 V. All MRM produced peaks at the same retention time (transition 82.1→39 shown in Fig. 7B, inset), suggesting they are from the same parent compound. Detection of cnm5U in purified tRNA or total tRNA was achieved by resolving enzymatic hydrolysates on the reversed-phase HPLC column with a gradient of water to acetonitrile containing 8 mM ammonium acetate at a flow rate of 0.2 mL/min and 36°C: 0–15 min, 0%; 15–16 min, 0%–100%; 16–20 min, 100%; 20–21 min, 100%–0%. The QqQ mass spectrometer was operated in positive ion mode with the following parameters: gas temperature, 325°C; gas flow, 10 L/min; nebulizer, 20 psi; and capillary voltage, 4000 V. The m/z of the transmitted parent ion, m/z of the monitored product ion, fragmentor voltage, and collision energy were as follows: 284.2→125.1, 110 V, 15 V; 152.1→125.1, 140 V, 10 V; 152.1→82.1, 140 V, 15 V.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank Annmarie McInnis for her usual cheerfulness and help with preparing this manuscript. This work was supported by grants from the US National Institutes of Health (GM17151 to U.L.R.; GM22854 to D. Söll; ES017010 to P.C.D.); the Singapore-MIT Alliance for Research and Technology sponsored by the National Research Foundation of Singapore (P.C.D.); and the Department of Energy (DE-FG36-08GO88055 to P.B.).
REFERENCES
- Björk GR 1998. Modified nucleosides at positions 34 and 37 of tRNAs and their predicted coding capacities. In Modification and editing of RNA (ed. Grosjean H, Benne R), pp. 577–581 American Society for Microbiology, Washington, DC [Google Scholar]
- Chomczynski P, Sacchi N 2006. The single-step method of RNA isolation by acid guanidinium thiocyanate–phenol–chloroform extraction: Twenty-something years on. Nat Protoc 1: 581–585 [DOI] [PubMed] [Google Scholar]
- Crain PF 1990. Preparation and enzymatic hydrolysis of DNA and RNA for mass spectrometry. Methods Enzymol 193: 782–790 [DOI] [PubMed] [Google Scholar]
- Crick FH 1966. Codon–anticodon pairing: The wobble hypothesis. J Mol Biol 19: 548–555 [DOI] [PubMed] [Google Scholar]
- DasSarma S, Fleischmann EM 1995. Archaea: A laboratory manual—halophiles. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- Donis-Keller H, Maxam AM, Gilbert W 1977. Mapping adenines, guanines, and pyrimidines in RNA. Nucleic Acids Res 4: 2527–2538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkins JG, Podar M, Graham DE, Makarova KS, Wolf Y, Randau L, Hedlund BP, Brochier-Armanet C, Kunin V, Anderson I, et al. 2008. A korarchaeal genome reveals insights into the evolution of the Archaea. Proc Natl Acad Sci 105: 8102–8107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabret C, Dervyn E, Dalmais B, Guillot A, Marck C, Grosjean H, Noirot P 2011. Life without the essential bacterial tRNAIle2–lysidine synthetase TilS: A case of tRNA gene recruitment in Bacillus subtilis. Mol Microbiol 80: 1062–1074 [DOI] [PubMed] [Google Scholar]
- Grosjean H, Björk GR 2004. Enzymatic conversion of cytidine to lysidine in anticodon of bacterial tRNAIle—an alternative way of RNA editing. Trends Biochem Sci 29: 165–168 [DOI] [PubMed] [Google Scholar]
- Gupta R 1984. Halobacterium volcanii tRNAs. Identification of 41 tRNAs covering all amino acids, and the sequences of 33 class I tRNAs. J Biol Chem 259: 9461–9471 [PubMed] [Google Scholar]
- Gupta R 1986. Transfer RNAs of Halobacterium volcanii: Sequences of five leucine and three serine transfer RNAs. Syst Appl Microbiol 7: 102–105 [Google Scholar]
- Gupta RC, Randerath K 1979. Rapid print-readout technique for sequencing of RNA's containing modified nucleotides. Nucleic Acids Res 6: 3443–3458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada F, Nishimura S 1974. Purification and characterization of AUA specific isoleucine transfer ribonucleic acid from Escherichia coli B. Biochemistry 13: 300–307 [DOI] [PubMed] [Google Scholar]
- Ikeuchi Y, Kimura S, Numata T, Nakamura D, Yokogawa T, Ogata T, Wada T, Suzuki T 2010. Agmatine-conjugated cytidine in a tRNA anticodon is essential for AUA decoding in archaea. Nat Chem Biol 6: 277–282 [DOI] [PubMed] [Google Scholar]
- Khorana HG 1968. Nucleic acid synthesis in the study of the genetic code. In Nobel lectures. Elsevier, Amsterdam [Google Scholar]
- Köhrer C, RajBhandary UL 2008. The many applications of acid urea polyacrylamide gel electrophoresis to studies of tRNAs and aminoacyl-tRNA synthetases. Methods 44: 129–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhrer C, Srinivasan G, Mandal D, Mallick B, Ghosh Z, Chakrabarti J, RajBhandary UL 2008. Identification and characterization of a tRNA decoding the rare AUA codon in Haloarcula marismortui. RNA 14: 117–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhrer C, Mandal D, Gaston KW, Grosjean H, Limbach PA, RajBhandary UL 2013. Life without tRNAIle-lysidine synthetase: Translation of the isoleucine codon AUA in Bacillus subtilis lacking the canonical tRNA2Ile. Nucleic Acids Res 10.1093/nar/gkt1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchino Y, Kato M, Sugisaki H, Nishimura S 1979. Nucleotide sequence of starfish initiator tRNA. Nucleic Acids Res 6: 3459–3469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwahara M, Nagashima J, Hasegawa M, Tamura T, Kitagata R, Hanawa K, Hososhima S, Kasamatsu T, Ozaki H, Sawai H 2006. Systematic characterization of 2′-deoxynucleoside-5′-triphosphate analogs as substrates for DNA polymerases by polymerase chain reaction and kinetic studies on enzymatic production of modified DNA. Nucleic Acids Res 34: 5383–5394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam WL, Doolittle WF 1989. Shuttle vectors for the archaebacterium Halobacterium volcanii. Proc Natl Acad Sci 86: 5478–5482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockard RE, Alzner-Deweerd B, Heckman JE, MacGee J, Tabor MW, RajBhandary UL 1978. Sequence analysis of 5′[32P] labeled mRNA and tRNA using polyacrylamide gel electrophoresis. Nucleic Acids Res 5: 37–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal D, Köhrer C, Su D, Russell SP, Krivos K, Castleberry CM, Blum P, Limbach PA, Soll D, RajBhandary UL 2010. Agmatidine, a modified cytidine in the anticodon of archaeal tRNAIle, base pairs with adenosine but not with guanosine. Proc Natl Acad Sci 107: 2872–2877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramatsu T, Nishikawa K, Nemoto F, Kuchino Y, Nishimura S, Miyazawa T, Yokoyama S 1988. Codon and amino-acid specificities of a transfer RNA are both converted by a single post-transcriptional modification. Nature 336: 179–181 [DOI] [PubMed] [Google Scholar]
- Murao K, Saneyoshi M, Harada F, Nishimura S 1970. Uridin-5-oxy acetic acid: A new minor constituent from E. coli valine transfer RNA I. Biochem Biophys Res Commun 38: 657–662 [DOI] [PubMed] [Google Scholar]
- Murao K, Hasegawa T, Ishikura H 1976. 5-methoxyuridine: A new minor constituent located in the first position of the anticodon of tRNAAla, tRNAThr, and tRNAVal from Bacillus subtilis. Nucleic Acids Res 3: 2851–2860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieuwlandt DT, Daniels CJ 1990. An expression vector for the archaebacterium Haloferax volcanii. J Bacteriol 172: 7104–7110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nirenberg M 1968. The genetic code. In Nobel lectures, Elsevier, Amsterdam [Google Scholar]
- Nishimura S 1979. Chromatographic mobilities of modified nucleotides. In tRNA: Structure, properties, and recognition (ed. Schimmel PR, Söll D, Abelson JN), pp. 551–552 Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- Phillips G, de Crecy-Lagard V 2011. Biosynthesis and function of tRNA modifications in Archaea. Curr Opin Microbiol 14: 335–341 [DOI] [PubMed] [Google Scholar]
- RajBhandary UL 1980. Recent developments in methods for RNA sequencing using in vitro 32P-labeling. Fed Proc 39: 2815–2821 [PubMed] [Google Scholar]
- Ramesh V, RajBhandary UL 2001. Importance of the anticodon sequence in the aminoacylation of tRNAs by methionyl-tRNA synthetase and by valyl-tRNA synthetase in an Archaebacterium. J Biol Chem 276: 3660–3665 [DOI] [PubMed] [Google Scholar]
- Randau L, Pearson M, Söll D 2005. The complete set of tRNA species in Nanoarchaeum equitans. FEBS Lett 579: 2945–2947 [DOI] [PubMed] [Google Scholar]
- Sakata S, Shibuya S, Machida H, Yoshino H, Hirota K, Senda S, Ikeda K, Mizuno Y 1980. Synthesis and antiherpesviral activity of 5-C-substituted uracil nucleosides. Nucleic Acids Symp Ser 8: s39–s42 [PubMed] [Google Scholar]
- Sambrook J, Russell DW 2001. Molecular cloning: A laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- Senger B, Auxilien S, Englisch U, Cramer F, Fasiolo F 1997. The modified wobble base inosine in yeast tRNAIle is a positive determinant for aminoacylation by isoleucyl-tRNA synthetase. Biochemistry 36: 8269–8275 [DOI] [PubMed] [Google Scholar]
- Shevack A, Gewitz HS, Hennemann B, Yonath A, Wittmann HG 1985. Characterization and crystallization of ribosomal particles from Halobacterium marismortui. FEBS Lett 184: 68–71 [Google Scholar]
- Silberklang M, Gillum AM, RajBhandary UL 1979. Use of in vitro 32P labeling in the sequence analysis of nonradioactive tRNAs. Methods Enzymol 59: 58–109 [DOI] [PubMed] [Google Scholar]
- Simoncsits A, Brownlee GG, Brown RS, Rubin JR, Guilley H 1977. New rapid gel sequencing method for RNA. Nature 269: 833–836 [DOI] [PubMed] [Google Scholar]
- Stanley J, Vassilenko S 1978. A different approach to RNA sequencing. Nature 274: 87–89 [DOI] [PubMed] [Google Scholar]
- Suzuki T, Suzuki T 2007. Chaplet column chromatography: Isolation of a large set of individual RNAs in a single step. Methods Enzymol 425: 231–239 [DOI] [PubMed] [Google Scholar]
- Taniguchi T, Miyauchi K, Nakane D, Miyata M, Muto A, Nishimura S, Suzuki T 2013. Decoding system for the AUA codon by tRNAIle with the UAU anticodon in Mycoplasma mobile. Nucleic Acids Res 41: 2621–2631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshney U, Lee CP, RajBhandary UL 1991. Direct analysis of aminoacylation levels of tRNAs in vivo. Application to studying recognition of Escherichia coli initiator tRNA mutants by glutaminyl-tRNA synthetase. J Biol Chem 266: 24712–24718 [PubMed] [Google Scholar]
- Waters E, Hohn MJ, Ahel I, Graham DE, Adams MD, Barnstead M, Beeson KY, Bibbs L, Bolanos R, Keller M, et al. 2003. The genome of Nanoarchaeum equitans: Insights into early archaeal evolution and derived parasitism. Proc Natl Acad Sci 100: 12984–12988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama S, Nishimura S 1995. Modified nucleosides and codon recognition. In tRNA: Structure, biosynthesis, and function (ed. Söll D, RajBhandary UL), pp. 207–223 American Society for Microbiology, Washington, DC [Google Scholar]