

Group I introns are cis-splicing ribozymes that excise themselves from RNA primary transcripts. These ribozymes can be converted into trans-splicing ribozymes. In the absence of a 5′-terminal extended guide sequence (EGS), the Azoarcus group I intron ribozyme favored the same splice sites as the Tetrahymena ribozyme on a given substrate mRNA in vitro. Further, the Azoarcus ribozyme was most efficient with a trans-splicing design that resembled the secondary structure context of the natural cis-splicing Azoarcus ribozyme, the anticodon stem of tRNAIle.
Keywords: group I intron, ribozyme, Azoarcus, trans-splicing
Abstract
Group I introns are ribozymes (catalytic RNAs) that excise themselves from RNA primary transcripts by catalyzing two successive transesterification reactions. These cis-splicing ribozymes can be converted into trans-splicing ribozymes, which can modify the sequence of a separate substrate RNA, both in vitro and in vivo. Previous work on trans-splicing ribozymes has mostly focused on the 16S rRNA group I intron ribozyme from Tetrahymena thermophila. Here, we test the trans-splicing potential of the tRNAIle group I intron ribozyme from the bacterium Azoarcus. This ribozyme is only half the size of the Tetrahymena ribozyme and folds faster into its active conformation in vitro. Our results showed that in vitro, the Azoarcus and Tetrahymena ribozymes favored the same set of splice sites on a substrate RNA. Both ribozymes showed the same trans-splicing efficiency when containing their individually optimized 5′ terminus. In contrast to the previously optimized 5′-terminal design of the Tetrahymena ribozyme, the Azoarcus ribozyme was most efficient with a trans-splicing design that resembled the secondary structure context of the natural cis-splicing Azoarcus ribozyme, which includes base-pairing between the substrate 5′ portion and the ribozyme 3′ exon. These results suggested preferred trans-splicing interactions for the Azoarcus ribozyme under near-physiological in vitro conditions. Despite the high activity in vitro, however, the splicing efficiency of the Azoarcus ribozyme in Escherichia coli cells was significantly below that of the Tetrahymena ribozyme.
INTRODUCTION
Group I introns are catalytic RNAs (ribozymes) that do not require the spliceosome for their removal from primary transcripts. Instead, these ribozymes fold into catalytically active structures that undergo two successive transphosphorylation reactions, facilitating their own excision and the ligation of the two flanking exons (Kruger et al. 1982; for review, see Vicens and Cech 2006). These natural cis-splicing ribozymes can be converted into trans-acting ribozymes by removing their 5′ exon and modifying the ribozyme 5′ terminus. In this format, trans-acting group I intron ribozymes can cleave a substrate RNA (Inoue et al. 1985), insert or remove one or several nucleotides (Bell et al. 2002; Johnson et al. 2005), replace the 3′ portion of the substrate RNA with the 3′ exon of the ribozyme (Inoue et al. 1985; Sullenger and Cech 1994), or replace the 5′ portion of the substrate RNA with the 5′ exon of the ribozyme (Alexander et al. 2005). These replacement reactions may be useful for therapeutic applications by either repairing the sequence of genetically mutated mRNAs (Sullenger and Cech 1994) or by inserting the coding sequence for a toxic peptide into substrate mRNAs that indicate a disease state (Ayre et al. 1999; Song and Lee 2006). Most previous work has focused on the reaction that replaces the mRNA 3′ portion; this reaction is also the focus of the present study.
Trans-splicing group I intron ribozymes recognize the target site on the substrate RNA by base-pairing, forming the P1 helix with the ribozyme's 5′ terminus (Fig. 1). The only sequence requirement in this helix is that a uracil at the splice site of the substrate is base-paired to a guanine in the ribozyme, the 5′-terminal nucleotide of the internal guide sequence (IGS) (Doudna et al. 1989). Factors that affect the efficiency of the trans-splicing reaction include the accessibility of the target site in the substrate RNA and the strength of the base-pairing interaction between ribozyme IGS and substrate RNA (Jones et al. 1996; Meluzzi et al. 2012).
FIGURE 1.

Secondary structure representations of trans-splicing group I intron ribozymes from (A) Tetrahymena and (B) Azoarcus. The 5′ terminus of the trans-splicing ribozymes (black) is truncated at or near the splice site G:U base pair (filled circle) such that the P1 helix (P1) is formed between the ribozyme 5′ terminus and the target site on the substrate (red). This helix is extended on the Azoarcus ribozyme construct by a P1 helix extension (P1ex) to facilitate a similar ribozyme-substrate complex stability as with the Tetrahymena ribozyme. A 5′-terminal guanosine was added for better in vitro transcription yields. Filled triangles indicate the 5′ splice sites, whereas empty triangles denote the 3′ splice sites. Empty squares denote nucleotides that differ between splice sites. (C) Schematic of a trans-splicing reaction. The colors for substrate and ribozyme 3′ exon are as in A and B, whereas sequences are denoted by solid lines (with the exception of the splice site G:U pair), and the ribozyme is denoted by a solid black line. During the trans-splicing reaction, the ribozyme 3′ exon (blue) replaces the 3′ portion of the substrate.
The efficiency of the trans-splicing reaction can be improved by increasing the substrate-ribozyme interaction via an extended guide sequence (EGS). The EGS is an elongation of the 5′ terminus of the trans-splicing ribozyme and has been tested on the group I intron ribozymes from Tetrahymena thermophila (subgroup IC1), Didymium (subgroup IE), and Fuligo (subgroup IC1) (Köhler et al. 1999; Byun et al. 2003; Fiskaa et al. 2006; Olson and Muller 2012). Despite the improvements mediated by the EGS, the trans-splicing efficiency remains a limiting factor for potential therapeutic applications of these ribozymes (Fiskaa and Birgisdottir 2010). Notably, all of these previous studies employed the same EGS design principle, which was originally developed for the Tetrahymena ribozyme. Because different group I introns may prefer different EGSs, it may be possible to obtain higher trans-splicing efficiencies by optimizing the EGSs for ribozymes other than the Tetrahymena ribozyme.
To date, only four different group I introns have been tested for their trans-splicing potential replacing the substrate 3′ portion (Sullenger and Cech 1994; Lundblad et al. 2004; Fiskaa et al. 2006), compared to the more than 16,000 known group I introns (Zhou et al. 2008). Of these four, the Tetrahymena ribozyme achieved the highest trans-splicing efficiencies in vitro, is understood at the most detailed biochemical level, and remains the only group I intron tested for mRNA repair / replacement trans-splicing in vivo (Michel and Westhof 1990; Lehnert et al. 1996; Kuo and Piccirilli 2001; Koduvayur and Woodson 2004; Shi et al. 2012). Therefore, to achieve efficient trans-splicing, it is promising to analyze group I intron ribozymes from different species and identify specific designs that increase their respective trans-splicing efficiencies.
The group I intron ribozyme from Azoarcus BH72 is an attractive candidate for trans-splicing experiments. It belongs to the IC3 group I intron family and is located in the anticodon stem–loop of tRNAIle (Reinhold-Hurek and Shub 1992). With a length of 205 nt, it is about twofold smaller than the Tetrahymena ribozyme (Fig. 1A,B). The Azoarcus ribozyme folds into its active conformation faster than the Tetrahymena ribozyme in vitro (Russell and Herschlag 2001; Rangan et al. 2004; Mitra et al. 2011; Sinan et al. 2011), and it can mediate trans-ligation reactions at high concentrations of magnesium, ribozyme, and at elevated temperature (Chowrira et al. 1993; Hayden and Lehman 2006; Burton and Lehman 2010). The structure of the Azoarcus ribozyme is known at the highest detail of all group I intron ribozymes, for different stages of the splicing pathway (Adams et al. 2004a,b; Lipchock and Strobel 2008). Due to its fast folding kinetics and its ability to react in trans, the Azoarcus ribozyme is a promising candidate to test its potential for trans-splicing under near-physiological in vitro conditions and in cells.
Here, we characterize the trans-splicing potential of the group I intron ribozyme from Azoarcus at near-physiological in vitro conditions and in Escherichia coli cells. The results show that in vitro, the Azoarcus ribozyme favors the same splice sites on a model substrate mRNA as the Tetrahymena ribozyme. When both ribozymes are designed with their preferred secondary structure interactions, they achieve similar trans-splicing efficiencies in vitro. Importantly, the favored secondary structure context of the Azoarcus ribozyme is different from that of previously established trans-splicing ribozymes and reflects the natural cis-splicing context of the Azoarcus ribozyme. These results suggested how the Azoarcus ribozyme could be used for trans-splicing reactions under near-physiological in vitro conditions. In E. coli cells, the trans- and cis-splicing activities of the Azoarcus ribozyme were significantly lower than the Tetrahymena ribozyme activities.
RESULTS
To analyze the potential of the Azoarcus ribozyme for trans-splicing applications we proceeded in three steps. First, we compared the pattern of favored splice sites on a model mRNA between trans-splicing Azoarcus and Tetrahymena ribozymes. Second, we searched and identified an efficient secondary structure design for ribozyme-substrate interactions of trans-splicing Azoarcus ribozymes, which is different from that of the Tetrahymena ribozyme. Third, we tested the activity of trans-splicing Azoarcus ribozymes in E. coli cells.
Splice site preferences by trans-splicing Azoarcus and Tetrahymena ribozymes
To test whether the Azoarcus ribozyme could access the same splice sites as the Tetrahymena ribozyme, we determined the accessible splice sites on a model substrate mRNA, the chloramphenicol acetyl transferase (CAT) mRNA. Splice sites were identified using an RT-PCR assay that specifically amplifies trans-splicing products from a reaction with ribozymes containing a randomized substrate recognition sequence (Jones et al. 1996). The randomized substrate recognition sequence enabled the pool of ribozymes to splice at any accessible U in the substrate RNA because the phylogenetically conserved G•U pair in the P1 helix is the only requirement for ribozyme activity (Doudna et al. 1989). The P1 helix of the Azoarcus ribozyme was elongated by 3 nt beyond the splice site such that both Azoarcus and Tetrahymnena ribozymes formed similarly stable six-base-pair P1 duplexes with the substrate (Fig. 1B). After RT-PCR, cloning, and sequencing of trans-splicing products, the identified splice sites for 56 trans-splicing events of the Azoarcus ribozyme were mapped on the CAT mRNA and compared to 56 trans-splice sites of the Tetrahymena ribozyme, in experiments conducted side-by-side (Fig. 2A; Meluzzi et al. 2012). Overall, the splice site patterns of the Azoarcus and Tetrahymena ribozymes were very similar. The region between position ∼200 and ∼400 reported few splicing events for both ribozymes. To identify additional splice sites in this region, we modified the assay by using a nested 5′ PCR primer (Fig. 2A, gray symbols). Comparison of all splice sites between both ribozymes showed no significant difference in the profile, suggesting that the Azoarcus and Tetrahymena ribozymes share the same splice site preference pattern.
FIGURE 2.
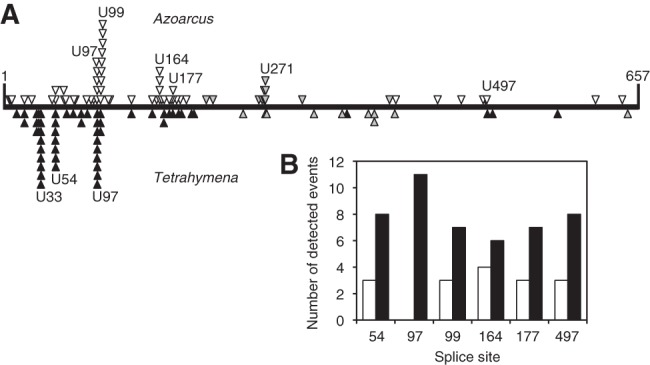
Experimental comparison of favored splice sites and trans-splicing efficiencies between Tetrahymena and Azoarcus. (A) Patterns of splicing events along the sequence of CAT mRNA substrate (horizontal line) between positions 1 and 657. The events detected for the Azoarcus ribozyme (above the line; open triangles) are compared to the events recorded for the Tetrahymena ribozyme (below the line; closed triangles) (from Meluzzi et al. 2012). Open and closed triangles denote splicing events that were detected with RT-PCR primers covering the entire length of the CAT mRNA. Gray triangles denote splicing events that were detected with a nested 5′ PCR primer to identify splice sites at least 200 nt downstream from the 5′ terminus of the CAT mRNA. All identified splicing products are reported here, which includes 10 non-U splice sites for the Azoarcus ribozyme and seven non-U splice sites for the Tetrahymena ribozyme. The positions of all individual splice sites are listed in Supplemental Table S1. (B) Comparison of trans-splicing efficiencies of Azoarcus ribozymes (white columns) and Tetrahymena ribozymes (black columns) when targeted to six specific splice sites on CAT mRNA.
The previous assay reported the pattern of favored splice sites but not the relative reaction efficiencies of both ribozymes. To estimate the relative efficiencies of the two trans-splicing ribozymes, they were reacted at equimolar concentrations in the same reaction vessel, with an excess of CAT mRNA. Therefore, the number of splicing events detected for each ribozyme revealed the relative reaction efficiency of the two ribozymes (Fig. 2B). The splicing products of the two ribozymes were discriminated by introducing a single point mutation into one of the ribozyme 3′ exons, 26 nt downstream from the splice site. Because this mutation became part of the product, the sequence of the trans-splicing products reported which of the two ribozymes had generated the product. Six representative splice sites were chosen, some of which were favored by the Tetrahymena ribozyme in the previous assay, whereas others were favored by Azoarcus ribozyme (see Supplemental Table S1). To target these six splice sites, the IGSs of the ribozymes had six specific sequences complementary to the six target sites on the CAT mRNA. After the trans-splicing reaction, reverse transcription, and cloning, 10–13 splicing products from each of the six reactions were sequenced. At all six splice sites, at least 10 sequences reported splicing at the targeted splice site (>80%), and at most two sequences (<20%) reported mis-splicing at a different splice site. For both ribozymes, a total of three mis-splicing events were detected, suggesting a similar, low error rate under these conditions (∼9%). Importantly, the Tetrahymena ribozyme gave rise to more splicing products than the Azoarcus ribozyme on all six tested splice sites (Fig. 2B). This outcome was not due to the point mutation in the ribozyme 3′ exons because the result was the same when the point mutation in the ribozyme 3′ exon was switched between the ribozymes. These results indicated that the Tetrahymena ribozyme trans-spliced more efficiently than the Azoarcus ribozyme in the absence of optimized ribozyme-substrate contacts.
Design of optimal ribozyme-substrate contacts for the Azoarcus ribozyme
The extended guide sequence (EGS) is an extension of the ribozyme 5′ terminus that can improve the trans-splicing efficiency and specificity. This was originally demonstrated with the group I intron ribozyme from Tetrahymena (Köhler et al. 1999), where the optimal design of the EGS includes an extension of the P1 helix by 2–4 base pairs (P1 extension), followed by an internal loop and a duplex with at least 5 base pairs between the ribozyme 5′ terminus and the substrate RNA. The internal loop region of the EGS may also form a P10 duplex with the 3′ exon, which can benefit the trans-splicing efficiency (Fig. 3A; Köhler et al. 1999; Byun et al. 2003; Olson and Muller 2012). Subsequent studies found that this “classical design” also increases the trans-splicing efficiencies of the group I introns from Fuligo and Didymium (Lundblad et al. 2004; Fiskaa et al. 2006). To test whether the “classical design” of ribozyme-substrate contacts also increases the trans-splicing efficiency of the Azoarcus ribozyme, we generated Azoarcus and Tetrahymena ribozyme variants that formed a P1 extension, an internal loop, and a 5′ duplex (Supplemental Fig. S1). Because the formation of a P10 duplex does not always increase trans-splicing efficiency (Olson and Muller 2012), we tested several constructs for each ribozyme and splice site with either a strong, a weak, or no predicted P10 duplex. Three splice sites were tested on the CAT mRNA (sites 97, 177, and 258, with position 1 being the A of the AUG start codon) to reduce a possible bias from the choice of splice sites. These variations in splice site, P10 duplex, and P1 extension length generated seven reaction contexts for each of the two ribozymes. Three of the seven Tetrahymena ribozyme constructs gave by far the highest trans-splicing efficiency, with a conversion of ∼20% substrate to product, all on splice site 258 (Fig. 3B). In contrast, the three most efficient reactions of the Azoarcus ribozyme achieved efficiencies only up to 7%. The low efficiency of trans-splicing with all Azoarcus ribozyme constructs compared to the Tetrahymena ribozyme constructs suggested that the “classical design” was not well suited for the Azoarcus ribozyme.
FIGURE 3.

“Classical design” for trans-splicing group I intron ribozymes. (A) Secondary structure of the Azoarcus ribozyme (black) with a 5′-terminal extended guide sequence (EGS; green), and 3′ exon (blue), formed with a pre-mRNA (red). The positions of the P1 helix (P1), P1 extension (P1ex), internal loop, and 5′ duplex are indicated. Each empty square symbolizes a position where nucleotide identities change between splice sites while maintaining the secondary structure. Individual constructs differ by nucleotide insertions, deletions, and mutations (see Supplemental Fig. S1). (B) Autoradiogram of 5′ radiolabeled trans-splicing products separated by denaturing 5% polyacrylamide gel electrophoresis. The targeted splice site (97, 177, or 258) and the designed number of base pairs (bp) in the P10 helix (bp in P10) are indicated on top of the autoradiogram. The side of the autoradiogram indicates the size of markers in the first and last lane (M), which correspond to full-length CAT mRNA substrate (Sub), the expected trans-splicing product (Prod), and the 5′ fragments produced by the first step of splicing (5′-Frag), at splice sites 97, 177, and 258. The length of the trans-splicing products is the same for all splice sites because the length of the ribozyme 3′ exons was covaried with the splice sites. The quantitations of trans-splicing efficiencies are shown below the autoradiogram, as percentage of substrate converted to product. Columns for the Azoarcus ribozyme (white) and the Tetrahymena ribozymes (gray) are in the same order as the lanes in the autoradiogram. Error bars denote standard deviations from three experiments.
An alternative design for trans-splicing Azoarcus ribozymes is a simple elongation of the P1 duplex past the splice site, termed the P1 extension (Fig. 4A). We expected that the length of this P1 extension had an optimum because lengthening the P1 extensions would not only increase the strength of the ribozyme-substrate duplex but would also reduce the splice site specificity (Herschlag 1991) and may prevent conformational changes of the ribozyme between the two catalytic steps of splicing (Cech 1990). To test this design principle and identify optimal P1 extension lengths, we analyzed a total of 32 Azoarcus ribozyme constructs with P1 extension lengths between 3 and 63 base pairs, at the three splice sites 97, 177, or 258 (Fig. 4B). Again, none of these constructs exceeded a trans-splicing efficiency of 7%. Although there was no clear optimal length of the P1 extension, the most robust trans-splicing appeared at a P1 length of 16–18 base pairs, where all constructs had a trans-splicing efficiency between 3.5% and 7%. Because these efficiencies were significantly below the trans-splicing efficiencies achieved with the classic design principle on the Tetrahymena ribozyme, these results suggested that elongated P1 helices alone (the “P1 extension design”) are not sufficient to generate efficient trans-splicing Azoarcus ribozymes.
FIGURE 4.

“P1 extension design” for the trans-splicing Azoarcus ribozyme. (A) Secondary structure of the Azoarcus ribozyme (black), with a 5′-terminal extension of variable length (green). Depending on the length of this 5′ extension, the P1 extension helices (P1ex) are formed with corresponding lengths, stacked upon the P1 helix (P1). The ribozyme 3′ exon is in blue. (B) Autoradiogram of 5′ radiolabeled trans-splicing products separated by denaturing polyacrylamide gel electrophoresis. The targeted splice site (97, 177, or 258) and the number of base pairs in the P1 extension are indicated above the autoradiogram. Size markers in the first and last lane indicate the position of full-length CAT mRNA substrate (Sub), the expected trans-splicing product (Prod), and the 5′ fragments produced by the first step of splicing (5′-Frag), at splice sites 97, 177, and 258. The length of the trans-splicing products is the same for all splice sites because the length of the ribozyme 3′ exons was covaried with the splice sites. The quantitations of trans-splicing efficiencies are shown below the autoradiogram, as percentage of substrate converted to product. The scale is the same as in Figures 3 and 5 to facilitate comparison. Error bars denote standard deviations from three experiments.
In a third design, we tested whether trans-splicing Azoarcus ribozymes would benefit from a secondary structure context similar to their native structural context near the splicing junction, the anticodon stem of tRNAIle (Fig. 5A; Reinhold-Hurek and Shub 1992; Adams et al. 2004a; Rangan et al. 2004). This anticodon stem contains five base pairs between the 5′ exon and the 3′ exon, 3 nt distant from the 5′ and 3′ splice site, respectively. In the context of trans-splicing Azoarcus ribozymes, this means that a sequence in the 3′ exon of the ribozyme needs to form base pairs with the 5′ portion of the substrate. In addition to the anticodon stem, the natural cis-splicing context of the Azoarcus ribozyme also forms a P1 extension helix before the first step of splicing (Adams et al. 2004a). Because the optimal length of the P1 extension helix for the trans-splicing reaction was unclear, three to four different P1 extension lengths were tested for each splice site (97, 177, 258). The choice of these P1 extension lengths was based on the highest efficiency in the previous experiment, with a preference for short P1 extensions (Fig. 4B). Satisfyingly, two of the 11 constructs resulted in trans-splicing efficiencies of ∼20%, similar to the best trans-splicing efficiencies of the Tetrahymena ribozyme with the “classical design” (Fig. 3B). This suggested that the “anticodon stem design” is a promising strategy to improve trans-splicing by the Azoarcus ribozyme. Because both the Tetrahymena ribozyme with the “classical EGS” and the Azoarcus ribozyme with the “anticodon stem design” showed high activity for only one splice site (see Figs. 3B, 5B), we suspected that the sequences flanking the splice sites might inhibit trans-splicing on other splice sites, which has been observed for the Tetrahymena ribozyme (Meluzzi et al. 2012). This inhibition could have been masking the general trend that the Azoarcus ribozyme favors the “anticodon stem design.”
FIGURE 5.

Effect of the “anticodon stem design” on the in vitro trans-splicing efficiency with Azoarcus ribozymes. (A) Schematic for the secondary structure, with a focus on interactions between substrate mRNA (red), ribozyme 5′ extension (green), and ribozyme 3′ exon (blue). The secondary structure is based on the model describing the Azoarcus ribozyme crystal structure (Adams et al. 2004a). The positions of the P1 helix (P1), P1 extension (P1ex), and anticodon stem are indicated. (B) Autoradiogram of 5′ radiolabeled trans-splicing products separated by denaturing polyacrylamide gel electrophoresis. The targeted splice site and the designed number of base pairs in the P1 extension (P1ex) are indicated for each ribozyme variant. Size markers in the first and last lane indicate the position of full-length CAT mRNA substrate (Sub), the expected trans-splicing product (Prod), and the 5′ fragments produced by the first step of splicing (5′-Frag) at splice sites 97, 177, and 258. The length of the trans-splicing products is the same for all splice sites because the length of the ribozyme 3′ exon was covaried with the splice sites. The quantitations of trans-splicing efficiencies are shown below the autoradiogram, as percentage of substrate converted to product. The scale is the same as in Figures 3 and 4 to facilitate comparison. Error bars denote standard deviations from three experiments.
To test the “anticodon stem design” for trans-splicing Azoarcus ribozymes without interference from flanking sequences, the trans-splicing reactions were repeated with short substrate RNAs (30 nt) and short ribozyme 3′ exons (10 nt) (Supplemental Fig. S3). The three short substrate RNAs that were used corresponded to the three splice sites tested on the full-length RNAs (97, 177, and 258). These RNAs were long enough to establish the structural elements used by all three design principles (P1 extensions, internal loops, 5′ duplexes, P10 duplexes, anticodon stems) but short enough to minimize potentially inhibitory secondary structure formation with the flanking sequences. For each of the three substrates and for each of the three design principles, two ribozyme constructs were chosen that showed the highest trans-splicing efficiency with full-length CAT mRNA substrates and whose P1 extension lengths were short enough to fit on the short substrates. In general, the trans-splicing efficiencies were higher with these short substrates than with the full-length CAT mRNA, confirming that the flanking sequences reduced the trans-splicing efficiency on the full-length constructs (Fig. 6; cf. to efficiencies in Figs 3–5). Importantly, the “anticodon stem design” generally resulted in the highest trans-splicing efficiencies (Fig. 6). Specifically, all six constructs with the “anticodon stem design” showed efficiencies of 19%–82%, whereas only 2/6 constructs mediated efficiencies above 4% for both the “classic design” and “P1 extension design.” In only one case did the “classical design” generate a higher trans-splicing efficiency than the “anticodon stem design.” This specific construct (splice site 177, P10 length of three base pairs) had an advantage over all other constructs because it was previously optimized by an in vivo selection procedure (Olson and Muller 2012). Note that on each splice site, the two “P1 extension” constructs had the same length of the P1 extension as in the two “anticodon stem” constructs. Comparison between these six pairs of constructs confirmed the beneficial effect of the anticodon stem interaction. Together, these results confirmed that the “anticodon stem design” is the design of choice for trans-splicing Azoarcus ribozymes.
FIGURE 6.
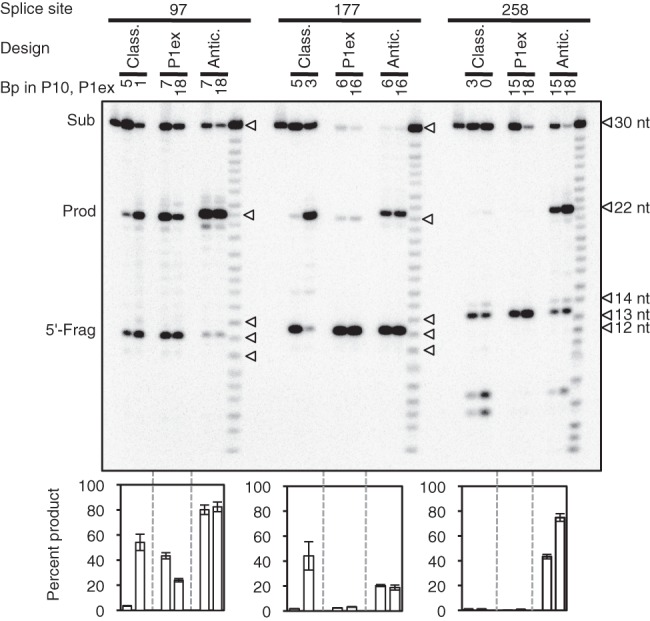
Comparison of all three designs for the in vitro trans-splicing efficiency with the Azoarcus ribozyme on short substrates (30 nt) and short ribozyme 3′ exons (10 nt). For secondary structure schematics of all individual constructs see Supplemental Figure S3. Shown is an autoradiogram of 5′ radiolabeled trans-splicing products separated by denaturing polyacrylamide gel electrophoresis. The targeted splice sites and the ribozyme designs are indicated on top of the autoradiogram. Ribozyme designs were the classical design (Class.), the P1 extension design (P1ex), and the anticodon stem design (Antic.). For each design the number of base pairs in the P10 helix (classic design) or the P1 extension (P1ex design and Antic. design) are indicated. For each splice site, the lane on the left shows unreacted substrate and the lane on the right shows an alkaline hydrolysis ladder. The position of full-length substrate (Sub), the expected trans-splicing product (Prod), and the 5′ fragments produced by the first step of splicing (5′-Frag), are indicated on the left. The length of the trans-splicing products is the same for all splice sites because the location of the splice site is the same on all short substrates. Note that the 5′ fragments (12 nt) migrate corresponding to a length of 13–14 nt because they contain a 3′ hydroxyl terminus, whereas all other labeled RNAs carry a 2′-3′ cyclic phosphate at their 3′ terminus, due to their synthesis method. Note that the 5′ fragments at splice site 258 are shifted up relative to those of splice sites 97 and 177, and that the trans-splicing products with the anticodon stem design are shifted up relative to the other two designs, due to a higher purine content in their sequence. The quantitations of trans-splicing efficiencies are shown below the autoradiogram, as percentage of substrate converted to product. The scale is larger than in Figures 3–5 due to the higher trans-splicing efficiency with short substrates. Error bars denote standard deviations from three experiments.
In vivo activity of the Azoarcus ribozyme in E. coli cells
The efficiency of trans-splicing Azoarcus ribozymes in E. coli cells was tested by expressing them from plasmids that encoded the trans-splicing ribozyme construct and its substrate, the chloramphenicol acetyl transferase (CAT) mRNA. The CAT mRNA was inactivated by a frameshift mutation and the ribozyme 3′ exon was designed to repair this mutation, such that only cells expressing an efficient trans-splicing ribozyme were able to grow on medium containing chloramphenicol (Olson and Muller 2012). Despite testing more than 20 constructs covering all design principles described in the in vitro portion of this study, no trans-splicing Azoarcus ribozyme facilitated growth on medium containing chloramphenicol (data not shown). In contrast, the Tetrahymena ribozyme with the “classical design” mediated efficient growth. To quantify the trans-splicing efficiency in cells, we measured the abundance of spliced CAT mRNA using an RT-qPCR assay (data not shown). The results paralleled the observed chloramphenicol resistance phenotypes: In E. coli cells the trans-splicing efficiency of the Tetrahymena ribozyme using the “classical” EGS was ∼10%, whereas it was <0.1% for all Azoarcus ribozyme constructs tested.
To identify the cause for the low in vivo efficiency of the Azoarcus ribozyme, several cis-splicing constructs of Azoarcus and Tetrahymena ribozymes were designed and tested for activity in E. coli cells (Fig. 7). Four of these constructs expressed CAT pre-mRNAs with the respective ribozymes inserted at splice sites 97, 177, or 258. The Tetrahymena ribozyme was inserted only at splice site 177 as a positive control. Only the Tetrahymena ribozyme construct mediated chloramphenicol resistance of E. coli cells, whereas none of the Azoarcus ribozyme constructs did. Quantitation of the cis-splicing efficiency by RT-qPCR confirmed the observed chloramphenicol resistance phenotype, showing that ∼60% of the pre-mRNAs containing the Tetrahymena ribozyme were converted to product, whereas <2% of the pre-mRNAs containing the Azoarcus ribozyme were spliced (Fig. 7, top four constructs). In vitro, however, the same cis-splicing Azoarcus ribozyme constructs that were inactive in cells, were similarly active as the Tetrahymena ribozyme (Supplemental Fig. S4). The low activity of the Azoarcus ribozyme in E. coli did not seem to be caused by a higher RNA turnover rate because the total CAT pre-mRNA levels as measured by RT-qPCR were at least 2.5-fold higher than with the Tetrahymena ribozyme constructs. Cis-splicing Azoarcus ribozyme constructs also failed to mediate chloramphenicol resistance in E. coli cells at elevated growth temperatures of 40°C and 42°C, closer to the optimal temperature of the Azoarcus ribozyme (Tanner and Cech 1996; Kuo et al. 1999). Similarly, no chloramphenicol resistance was seen when the E. coli growth medium was supplemented with 5 mM magnesium ions, the same concentration that led to efficient cis- and trans-splicing in vitro. These results indicated that the low trans-splicing activity of Azoarcus ribozymes in E. coli cells was not due to their trans-splicing context or increased turnover.
FIGURE 7.
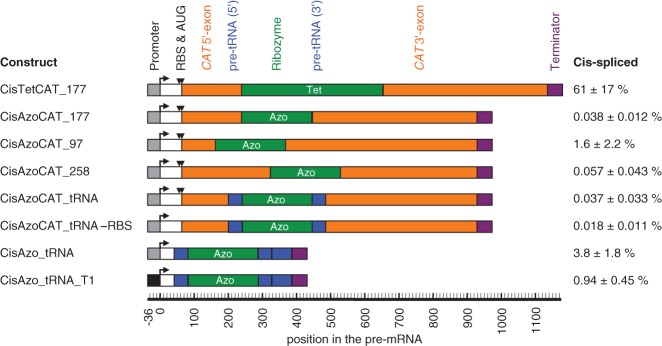
Cis-splicing efficiency of ribozyme constructs in E. coli cells. The name of each construct is given on the left, a schematic representation of the construct is given in the middle, and the observed cis-splicing efficiencies, measured by RT-qPCR, are given on the right. The names of the constructs (“Cis…”) include the ribozyme (“Tet” for Tetrahymena, and “Azo” for Azoarcus), the sequence flanking the ribozyme (CAT, CAT_tRNA, and tRNA), the splice site in the CAT mRNA contexts (177, 97, 258), the mutation inactivation of the ribosomal binding site and the start codon (-RBS) and the use of a different promoter (T1). Note that the three constructs CisAzoCAT_97, CisAzoCAT_177, and CisAzoCAT_258 contained mutations in their 3′ exon immediately downstream from the 3′ splice site to facilitate anticodon stem interactions; the construct CisAzoCAT_97 additionally contained mutations in the P1 loop to generate a P10 helix. For more details on these designs see Supplemental Figure S4. The schematics include the promoter (gray or black boxes with angled arrow), the ribosome-binding site and start codon (RBS and AUG; black triangles), the CAT exons (orange), the ribozymes (green), the flanking tRNAIle sequences (blue), and transcription terminators (purple). A second blue box indicates an extended pre-tRNAIle context. The position of each element in the pre-mRNA is given on a scale below the schematics, relative to the transcription start site. Note that the numbering of splice sites (97, 177, 258) was from the AUG start codon instead. The percentage of cis-spliced mRNA is given on the right, with errors as standard errors from three biological replicates.
To test whether the native flanking sequences of the Azoarcus ribozyme could facilitate Azoarcus ribozyme activity in E. coli cells, the construct CisAzoCAT_177 was modified by positioning the two halves of the complete 79-nt-long native tRNAIle sequence (Rangan et al. 2004) at the flanks of the Azoarcus ribozyme, thereby replacing the corresponding CAT sequence (Fig. 7, construct CisAzoCAT_tRNA). No significant cis-splicing activity was detected in E. coli cells by RT-qPCR. Because the Azoarcus ribozyme is naturally expressed as a noncoding RNA, we tested whether the translational apparatus in E. coli interfered with cis-splicing activity. To do that, the ribosome binding site and the start codon were mutated (construct CisAzoCAT_tRNA -RBS). Again, no significant activity was detected. The remaining CAT sequence was removed to test whether the exons of the CAT mRNA inhibited the Azoarcus ribozyme in E. coli cells (construct CisAzo_tRNA). Here, the cis-splicing efficiency of the Azoarcus ribozyme construct reached 3.8%, the highest in vivo activity of the Azoarcus ribozyme found in this study. When the promoter of this construct was changed from its constitutive promoter to the IPTG-inducible trc1 promoter (construct CisAzo_tRNA_T1), the cis-splicing activity dropped to ∼1% (the total CAT pre-mRNA level was increased ninefold with the trc1 promoter). This raised the possibility that the transcription complex itself might in fluence the activity of the cis-splicing Azoarcus ribozyme. In summary, the most significant hint as to why the Azoarcus ribozyme was inefficient in E. coli cells came from the comparison of different sequence environments for the cis-splicing construct, where the cis-splicing efficiency in E. coli cells varied by ∼200-fold between different sequence contexts (0.018%–3.8%), and the highest efficiency in the sequence context was closest to its natural environment in pre-tRNAIle.
DISCUSSION
In this study we explored the trans-splicing properties of the Azoarcus group I intron ribozyme. In the absence of a 5′-terminal extended guide sequence (EGS) the ribozyme favored the same splice sites as the Tetrahymena ribozyme on a given substrate mRNA in vitro. Although none of the “classical design” constructs worked well for the Azoarcus ribozyme on the full-length mRNA substrate, we found that a different design, which mimicked the natural cis-splicing context of the Azoarcus ribozyme (the anticodon stem of tRNAIle), led to improved trans-splicing efficiencies in vitro, as high as the Tetrahymena ribozyme. In contrast, the efficiency in E. coli cells of cis- and trans-splicing Azoarcus ribozymes was much lower than those of the Tetrahymena ribozyme.
The accessibility of splice sites on our model substrate, CAT mRNA, was similar between trans-splicing Azoarcus and Tetrahymena ribozymes (Fig. 2). This was consistent with a previous, less extensive comparison of splice site accessibility on a different mRNA substrate between the group I intron ribozymes from Tetrahymena and the myxomycetes Fuligo and Didymium (Fiskaa et al. 2006). Here, the three ribozymes also recognized mostly the same splice sites.
When the substrate-ribozyme interactions were optimized for the Azoarcus ribozyme constructs, they achieved similar in vitro trans-splicing efficiencies as the best Tetrahymena ribozyme constructs on the full-length CAT mRNA (cf. Figs. 3B and 5B, respectively). The results suggested that the Azoarcus ribozyme preferred a different design principle than the Tetrahymena ribozyme. The Tetrahymena ribozyme achieved up to ∼20% trans-splicing efficiency on full-length CAT mRNA with the “classical design,” which includes a short P1 extension, an internal loop that may form a P10 helix, and a 5′ duplex with the substrate (Fig. 3; Köhler et al. 1999; Byun et al. 2003; Einvik et al. 2004; Lundblad et al. 2004; Olson and Muller 2012). The Azoarcus ribozyme also achieved up to ∼20% trans-splicing efficiency but with the “anticodon stem design” (Fig. 5). This design mimicked the native secondary structure environment of the cis-splicing Azoarcus ribozyme in the anticodon stem–loop of tRNAIle, and outperformed two other tested design principles on the short substrate RNAs (Fig. 6). A similar finding was made for the Anabaena group I intron ribozyme, where the anticodon stem also stabilized the very short P1 helix and possibly also prevented the formation of inhibitory interactions in a cis-splicing context (Zaug et al. 1993). The failure of the P1 extension and classical designs to mediate efficient splicing indicated that the Azoarcus ribozyme, like the Anabaena ribozyme, required specific additional contacts immediately adjacent to the splice site. These contacts, comprising the anticodon stem, were not present in either the P1 extension or classical design principles (cf. Figs. 3A, 4A, 5A). These results suggested a more general framework for designing efficient trans-splicing ribozymes from group I introns from different species: No single design at the splice site works well for all trans-splicing group I intron ribozymes. Instead, the secondary structure designs for group I intron ribozymes from different species should try to mimic the natural secondary structure context of each ribozyme.
The in vitro trans-splicing conditions of this study had two important differences from the trans-reaction conditions of previous in vitro studies with the Azoarcus ribozyme. Previous studies used high Mg2+ concentrations (10–100 mM) and high temperature (48°C–60°C, with the optimum around 60°C) (Chowrira et al. 1993, 1995; Tanner and Cech 1996; Kuo et al. 1999; Chauhan et al. 2005; Hayden et al. 2005; Sinan et al. 2011). Under these conditions, the Azoarcus ribozyme is able to catalyze an impressive variety of trans-reactions. By repetitively splicing a substrate the Azoarcus ribozyme can cause an effect like RNA polymerization (Chowrira et al. 1993, 1995). By trans-ligating RNA oligomers the Azoarcus ribozyme can create active ribozymes from inactive ribozyme fragments (Hayden et al. 2005). The latter even allows for the covalent self-assembly of ribozymes from four different fragments (Hayden and Lehman 2006) and the spontaneous emergence of self-replicating sets from pools of ribozyme fragments with randomized substrate recognition sequences (Vaidya et al. 2012). In contrast, our study aimed to find designs for Azoarcus ribozymes that allowed for efficient trans-splicing at near-physiological magnesium concentrations (5 mM) and temperature (37°C). Our results identified such a design, described above. This design can now be used for trans-splicing reactions with the Azoarcus ribozyme under near-physiological in vitro conditions.
The trans-splicing efficiencies of Azoarcus ribozyme constructs in E. coli cells were much lower than the trans-splicing efficiencies of Tetrahymena ribozymes in E. coli cells of 1%–10% (Sullenger and Cech 1994; Olson and Muller 2012) and this study. Similarly, no cis-splicing Azoarcus ribozyme construct mediated chloramphenicol resistance in E. coli cells or achieved cis-splicing efficiencies above 5%, whereas a cis-splicing Tetrahymena ribozyme construct mediated chloramphenicol resistance and achieved ∼60% cis-splicing efficiency in cells (Fig. 7). For the Azoarcus ribozyme constructs, the 200-fold variation of cis-splicing efficiencies between different flanking sequences (0.018%–3.8%) suggested that the Azoarcus ribozyme was quite sensitive to flanking sequences in E. coli cells, whereas the same constructs were much less sensitive to flanking sequences in vitro (Supplemental Fig. S4).
The Azoarcus ribozyme's increased sensitivity to flanking sequences in E. coli could have been mediated by a number of different factors in the E. coli cellular environment, including (1) factors associated with the transcription complex, (2) RNA chaperones, and (3) RNA-binding proteins. First, the fourfold effect of different RNA polymerase promoters on cis-splicing efficiency (Fig. 7, cf. constructs CisAzo_tRNA and CisAzo_tRNA_T1) hinted that different factors interacting with the transcription complex in E. coli might have influenced the cotranscriptional folding such that portions of the Azoarcus ribozyme would be more prone to interactions with its flanking sequences. Second, one of the many RNA chaperones in E. coli cells (Semrad 2011) such as S12 (Coetzee et al. 1994), and StpA (Waldsich et al. 2002) could have unfolded portions of the Azoarcus ribozyme to make them accessible to inhibitory interactions with the flanking sequences. Third, proteins that bind to the group I intron ribozymes such as the tRNA-binding tyrosyl-tRNA synthetase Cyt-18 from Neurospora crassa (Akins and Lambowitz 1987; Caprara et al. 1996), CBP2 from S.cerevisiae (Weeks and Cech 1995), or I-AniI from Aspergillus nidulans (Solem et al. 2002) could have stabilized the Azoarcus ribozyme in a conformation that would be prone to interact with the flanking sequences. Although the studies cited above investigated protein interactions that increase group I intron activity, the same proteins could have inhibited the Azoarcus ribozyme because specific proteins can increase or decrease group I intron splicing efficiency depending on the identity of the group I intron ribozyme (Vicens et al. 2008). It is currently unclear which, if any, of the three discussed possibilities made the Azoarcus ribozyme more susceptible to interference from flanking sequences in E. coli cells.
MATERIALS AND METHODS
Plasmids and sequences
The sequence for the Azoarcus ribozyme (GenBank DQ103524.1) was custom synthesized (Genscript) and used as PCR template for subsequent cloning steps. The sequence encoding the chloramphenicol acetyltransferase (CAT) was derived from the plasmid pLysS (Invitrogen), including its constitutive promoter. The assay for the identification of splice site preferences on the CAT mRNA used ribozyme 3′ exons that were the first 57 nucleotides of the same α-mannosidase sequence used previously (Fiskaa et al. 2006; Meluzzi et al. 2012). Constructs were cloned into the plasmid pUC19 and sequenced. Exceptions were variants of the 5′ EGS sequence for in vitro splicing reactions and partially randomized pools for the identification of accessible splice sites on CAT mRNA to add the ribozyme 5′ terminus NNNGNN (for Azoarcus) and GNNNNN (for Tetrahymena). In these cases, the ribozyme 5′ terminus was modified during PCR by using 5′ PCR primers that carried the modified sequence, to generate the templates for transcription. The ribozyme 3′ exons for CAT mRNA repair at splice sites 97, 177, and 258 were added to the ribozyme sequences by introducing restriction sites near the 3′ splice site (BsrGI for splice site 97 and 258; EcoRI for splice site 177) and ligating the PCR products of both fragments appended with the same restriction sites. Introduction of the restriction sites required mutations in the 3′ exon that were silent with exception of splice site 177, where EcoRI was already present in CAT sequence. The resulting 3′ exon sequences are shown in Supplemental Table S2. To generate the cis-splicing constructs, the ribozyme 5′ exon sequences were linked to the ribozyme sequences by PCR of the two fragments with two 5′ phosphorylated primers and blunt end ligation. The promoter driving the expression of trans-splicing ribozymes in E. coli, trc1, was a down-regulated version of the trc promoter as described previously (Olson and Muller 2012), whereas the cis-splicing ribozymes used the promoter of CAT in the plasmid pLysS. The frameshift inactivation of the CAT gene for all in vivo experiments was a deletion of nucleotide 322 downstream from the translation start site, generated by site-directed mutagenesis.
RNAs for in vitro experiments
All RNAs for in vitro trans-splicing experiments were prepared essentially as described previously (Milligan et al. 1987; Meluzzi et al. 2012), by in vitro runoff transcription with T7 RNA polymerase. Template DNA was generated by PCR and transcribed by T7 RNA polymerase for 60 min at 37°C in 40 mM Tris/HCl pH 7.9, 2.5 mM spermidine, 26 mM MgCl2, 5 mM DTT, 0.01% Triton X-100, and 2 mM of each NTP. For some trans-splicing ribozyme constructs, the incubation time was reduced to 30 min to reduce self-cleavage at the 3′ splice site. Transcription reactions for cis-splicing constructs were performed with only 6 mM MgCl2 and 2 mM Spermidine (Michel et al. 1992), [α-32P]UTP for 1.5 h at 37°C to reduce self-cleavage. Ribozymes containing truncated (10 nt) 3′ exons were transcribed with hammerhead ribozymes appended to their 3′-ends to reduce 3′-end heterogeneity (see legend to Fig. 6). All transcripts of ribozymes and substrate were purified by denaturing polyacrylamide gel electrophoresis. The concentrations of purified RNAs were calculated from their absorption at 260 nm.
Identification of accessible splice sites
The accessible splice sites on CAT mRNA were determined as described previously (Lan et al. 1998; Meluzzi et al. 2012). GTP and substrate RNA were preincubated separately from the ribozyme for 10 min at 37°C in 50 mM MOPS pH 7.0, 5 mM MgCl2, 135 mM KCl, and 2 mM spermidine. The two solutions were mixed for a final concentration of 100 nM substrate, 10 nM ribozyme, and 200 μM GTP, and incubated for 1 h at 37°C. Reaction products were reverse transcribed using AMV reverse transcriptase. After RNA hydrolysis (for 10 min at 90°C in 200 mM sodium hydroxide), the reverse transcription products were PCR amplified using primers that bind to the 5′ end of the CAT mRNA and the ribozyme 3′ exon, nested from the primer of reverse transcription. The PCR products were cloned into pUC19 and sequenced to identify the splice sites. All of the splice sites found for the Tetrahymena ribozyme (Fig. 2A) were reported previously (Meluzzi et al. 2012), but the current study used only the subset of previously reported splice sites that was determined side-by-side with the splice sites for the Azoarcus ribozyme in this study. These were 56 splice sites for both ribozymes on the full-length CAT mRNA and an additional 12 (Azoarcus) and 10 (Tetrahymena) splice sites using a 5′ nested PCR primer, which annealed to positions 183–206 in the CAT mRNA sequence. The reactions with both Azoarcus and Tetrahymena present in the same reaction mixture were performed the same except that each ribozyme had a final concentration of 5 nM.
In vitro splicing reactions
To generate a gel shift between substrates and trans-spliced products on the full-length CAT mRNA, the 3′ exons of trans-splicing ribozymes were truncated, generating a uniform product size for all splice sites (3′ exons were 239 nt for splice site 97, 159 nt for splice site 177, and 78 nt for splice site 258). The sequences for short substrates (Supplemental Fig. S3) consisted of a 28-nt excerpt from the CAT mRNA at the splice sites 97, 177, or 258, and two 5′-terminal G's for higher in vitro transcription efficiency. The ribozyme 3′ exons for the short substrates contained seven nucleotides from the ribozyme 3′ exon used with the full-length CAT mRNA that were mutated for the anticodon stem designs and a 3′-terminal AUC. Incubation conditions for the trans-splicing reactions were the same as for the splice site identification assay, with the exception of final RNA concentrations (∼60 nM 5′-[32P]-labeled CAT mRNA, 1 μM ribozyme, and 200 μM GTP), and incubation times (24 h for the full-length substrate and 8 h for the minimal substrate). The cis-splicing reactions were incubated for 24 h and contained ∼30 nM internally [32P]-radiolabeled RNA and 200 μM GTP. Splice sites of the trans- and cis-splicing constructs (splice site 97, 177, and 258 with and without anticodon stem design) were confirmed by reverse transcription of the reaction products using AMV reverse transcriptase, PCR amplification, cloning into pUC19, and sequencing. Each trans-splicing construct showed the correct splicing product in one out of one sequencing reactions. Each cis-splicing construct, with the exception of splice site 258 without anticodon stem design were also correct in three out of three sequencing reactions. The cis-splicing Azoarcus construct targeting splice site 258 without the anticodon stem design included the omega G of the Azoarcus ribozyme in the splicing product of all three sequencing reactions. Radiolabeled samples from the splicing reactions were separated on 5% denaturing polyacrylamide gels for full-length CAT mRNAs and 12.5% denaturing polyacrylamide gels for short substrates. The product bands for splice site 177 in Figures 3–5 appear to migrate slightly faster because the product comigrates with the ribozyme of the same length (control experiments not shown). Bands were quantified on a phosphoImager (PMI, Bio-Rad) using the software Quantity One with the “lanes” method. The percentage of product formed in the in vitro trans-splicing reactions was calculated based on the fraction of the total radioactivity in the lane. The percentage of product formed in the in vitro cis-splicing reactions was calculated based on the normalized fraction of radioactivity in the lane. This normalization for cis-splicing reactions was necessary to account for the number of radiolabeled nucleotides in each internally labeled RNA fragment.
Quantitation of splicing in E. coli cells
All E. coli experiments used electrocompetent preparations of E. coli DH5α (Invitrogen). Total RNA was extracted from E. coli cells logarithmically growing with 1 mM IPTG using the Nucleospin RNA II kit (Machery-Nagel). Total RNA was reverse transcribed using Superscript III (Invitrogen). Reverse transcription products were directly used as template for quantitative PCR reactions with the Applied Biosystems qPCR master mix on the Fast 7500 RT-PCR machine (Applied Biosystems), essentially as described previously (Olson and Muller 2012). The abundance of substrate, ribozyme, and product and mRNA was calculated using the equation r = (0.5)N, where N is the experimentally determined threshold cycle of the qPCR. The percentage of spliced substrate, X, was calculated for each construct by using the equation X = 100 × [P/(S + P)], where S and P are the abundances of substrate and product, respectively. To differentiate stringently between CAT mRNA substrate and spliced product, 16 mutations were introduced into the ribozyme 3′ exon, starting 79 nt downstream from the 3′ splice site. None of the used primer pairs showed significant amount of cross amplification.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank Jing Yang (UC San Diego) for sharing her qPCR machine and for reagents; Karen E. Olson and Simpson Joseph (UC San Diego) for helpful discussions; and Zhaleh Amini for designing one of the ribozyme constructs. This work was supported by a grant from the Hellman family foundation to U.F.M.
REFERENCES
- Adams PL, Stahley MR, Gill ML, Kosek AB, Wang J, Strobel SA 2004a. Crystal structure of a group I intron splicing intermediate. RNA 10: 1867–1887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams PL, Stahley MR, Kosek AB, Wang J, Strobel SA 2004b. Crystal structure of a self-splicing group I intron with both exons. Nature 430: 45–50 [DOI] [PubMed] [Google Scholar]
- Akins RA, Lambowitz AM 1987. A protein required for splicing group I introns in Neurospora mitochondria is mitochondrial tyrosyl-tRNA synthetase or a derivative thereof. Cell 50: 331–345 [DOI] [PubMed] [Google Scholar]
- Alexander RC, Baum DA, Testa SM 2005. 5′ transcript replacement in vitro catalyzed by a group I intron-derived ribozyme. Biochemistry 44: 7796–7804 [DOI] [PubMed] [Google Scholar]
- Ayre BG, Köhler U, Goodman HM, Haseloff J 1999. Design of highly specific cytotoxins by using trans-splicing ribozymes. Proc Natl Acad Sci 96: 3507–3512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell MA, Johnson AK, Testa SM 2002. Ribozyme-catalyzed excision of targeted sequences from within RNAs. Biochemistry 41: 15327–15333 [DOI] [PubMed] [Google Scholar]
- Burton AS, Lehman N 2010. Enhancing the prebiotic relevance of a set of covalently self-assembling, autorecombining RNAs through in vitro selection. J Mol Evol 70: 233–241 [DOI] [PubMed] [Google Scholar]
- Byun J, Lan N, Long M, Sullenger BA 2003. Efficient and specific repair of sickle β-globin RNA by trans-splicing ribozymes. RNA 9: 1254–1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caprara MG, Lehnert V, Lambowitz AM, Westhof E 1996. A tyrosyl-tRNA synthetase recognizes a conserved tRNA-like structural motif in the group I intron catalytic core. Cell 87: 1135–1145 [DOI] [PubMed] [Google Scholar]
- Cech TR 1990. Self-splicing of group I introns. Annu Rev Biochem 59: 543–568 [DOI] [PubMed] [Google Scholar]
- Chauhan S, Caliskan G, Briber RM, Perez-Salas U, Rangan P, Thirumalai D, Woodson SA 2005. RNA tertiary interactions mediate native collapse of a bacterial group I ribozyme. J Mol Biol 353: 1199–1209 [DOI] [PubMed] [Google Scholar]
- Chowrira BM, Berzal-Herranz A, Burke JM 1993. Novel RNA polymerization reaction catalyzed by a group I ribozyme. EMBO J 12: 3599–3605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowrira BM, Berzal-Herranz A, Burke JM 1995. Novel system for analysis of group I 3′ splice site reactions based on functional trans-interaction of the P1/P10 reaction helix with the ribozyme's catalytic core. Nucleic Acids Res 23: 849–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coetzee T, Herschlag D, Belfort M 1994. Escherichia coli proteins, including ribosomal protein S12, facilitate in vitro splicing of phage T4 introns by acting as RNA chaperones. Genes Dev 8: 1575–1588 [DOI] [PubMed] [Google Scholar]
- Doudna JA, Cormack BP, Szostak JW 1989. RNA structure, not sequence, determines the 5′ splice-site specificity of a group I intron. Proc Natl Acad Sci 86: 7402–7406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einvik C, Fiskaa T, Lundblad EW, Johansen S 2004. Optimization and application of the group I ribozyme trans-splicing reaction. Methods Mol Biol 252: 359–371 [DOI] [PubMed] [Google Scholar]
- Fiskaa T, Birgisdottir AB 2010. RNA reprogramming and repair based on trans-splicing group I ribozymes. N Biotechnol 27: 194–203 [DOI] [PubMed] [Google Scholar]
- Fiskaa T, Lundblad EW, Henriksen JR, Johansen SD, Einvik C 2006. RNA reprogramming of α-mannosidase mRNA sequences in vitro by myxomycete group IC1 and IE ribozymes. FEBS J 273: 2789–2800 [DOI] [PubMed] [Google Scholar]
- Hayden EJ, Lehman N 2006. Self-assembly of a group I intron from inactive oligonucleotide fragments. Chem Biol 13: 909–918 [DOI] [PubMed] [Google Scholar]
- Hayden EJ, Riley CA, Burton AS, Lehman N 2005. RNA-directed construction of structurally complex and active ligase ribozymes through recombination. RNA 11: 1678–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herschlag D 1991. Implications of ribozyme kinetics for targeting the cleavage of specific RNA molecules in vivo: More isn't always better. Proc Natl Acad Sci 88: 6921–6925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue T, Sullivan FX, Cech TR 1985. Intermolecular exon ligation of the rRNA precursor of Tetrahymena: Oligonucleotides can function as 5′ exons. Cell 43(2 Pt 1): 431–437 [DOI] [PubMed] [Google Scholar]
- Johnson AK, Sinha J, Testa SM 2005. Trans insertion–splicing: Ribozyme-catalyzed insertion of targeted sequences into RNAs. Biochemistry 44: 10702–10710 [DOI] [PubMed] [Google Scholar]
- Jones JT, Lee SW, Sullenger BA 1996. Tagging ribozyme reaction sites to follow trans-splicing in mammalian cells. Nat Med 2: 643–648 [DOI] [PubMed] [Google Scholar]
- Koduvayur SP, Woodson SA 2004. Intracellular folding of the Tetrahymena group I intron depends on exon sequence and promoter choice. RNA 10: 1526–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler U, Ayre BG, Goodman HM, Haseloff J 1999. Trans-splicing ribozymes for targeted gene delivery. J Mol Biol 285: 1935–1950 [DOI] [PubMed] [Google Scholar]
- Kruger K, Grabowski PJ, Zaug AJ, Sands J, Gottschling DE, Cech TR 1982. Self-splicing RNA: Autoexcision and autocyclization of the ribosomal RNA intervening sequence of Tetrahymena. Cell 31: 147–157 [DOI] [PubMed] [Google Scholar]
- Kuo LY, Piccirilli JA 2001. Leaving group stabilization by metal ion coordination and hydrogen bond donation is an evolutionarily conserved feature of group I introns. Biochim Biophys Acta 1522: 158–166 [DOI] [PubMed] [Google Scholar]
- Kuo LY, Davidson LA, Pico S 1999. Characterization of the Azoarcus ribozyme: Tight binding to guanosine and substrate by an unusually small group I ribozyme. Biochim Biophys Acta 1489: 281–292 [DOI] [PubMed] [Google Scholar]
- Lan N, Howrey RP, Lee SW, Smith CA, Sullenger BA 1998. Ribozyme-mediated repair of sickle β-globin mRNAs in erythrocyte precursors. Science 280: 1593–1596 [DOI] [PubMed] [Google Scholar]
- Lehnert V, Jaeger L, Michel F, Westhof E 1996. New loop–loop tertiary interactions in self-splicing introns of subgroup IC and ID: A complete 3D model of the Tetrahymena thermophila ribozyme. Chem Biol 3: 993–1009 [DOI] [PubMed] [Google Scholar]
- Lipchock SV, Strobel SA 2008. A relaxed active site after exon ligation by the group I intron. Proc Natl Acad Sci 105: 5699–5704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundblad EW, Haugen P, Johansen SD 2004. Trans-splicing of a mutated glycosylasparaginase mRNA sequence by a group I ribozyme deficient in hydrolysis. Eur J Biochem 271: 4932–4938 [DOI] [PubMed] [Google Scholar]
- Meluzzi D, Olson KE, Dolan GF, Arya G, Muller UF 2012. Computational prediction of efficient splice sites for trans-splicing ribozymes. RNA 18: 590–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel F, Westhof E 1990. Modelling of the three-dimensional architecture of group I catalytic introns based on comparative sequence analysis. J Mol Biol 216: 585–610 [DOI] [PubMed] [Google Scholar]
- Michel F, Jaeger L, Westhof E, Kuras R, Tihy F, Xu MQ, Shub DA 1992. Activation of the catalytic core of a group I intron by a remote 3′ splice junction. Genes Dev 6: 1373–1385 [DOI] [PubMed] [Google Scholar]
- Milligan JF, Groebe DR, Witherell GW, Uhlenbeck OC 1987. Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res 15: 8783–8798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra S, Laederach A, Golden BL, Altman RB, Brenowitz M 2011. RNA molecules with conserved catalytic cores but variable peripheries fold along unique energetically optimized pathways. RNA 17: 1589–1603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson KE, Muller UF 2012. An in vivo selection method to optimize trans-splicing ribozymes. RNA 18: 581–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangan P, Masquida B, Westhof E, Woodson SA 2004. Architecture and folding mechanism of the Azoarcus group I pre-tRNA. J Mol Biol 339: 41–51 [DOI] [PubMed] [Google Scholar]
- Reinhold-Hurek B, Shub DA 1992. Self-splicing introns in tRNA genes of widely divergent bacteria. Nature 357: 173–176 [DOI] [PubMed] [Google Scholar]
- Russell R, Herschlag D 2001. Probing the folding landscape of the Tetrahymena ribozyme: Commitment to form the native conformation is late in the folding pathway. J Mol Biol 308: 839–851 [DOI] [PubMed] [Google Scholar]
- Semrad K 2011. Proteins with RNA chaperone activity: A world of diverse proteins with a common task—impediment of RNA misfolding. Biochem Res Int 2011: 532908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Solomatin SV, Herschlag D 2012. A role for a single-stranded junction in RNA binding and specificity by the Tetrahymena group I ribozyme. J Am Chem Soc 134: 1910–1913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinan S, Yuan X, Russell R 2011. The Azoarcus group I intron ribozyme misfolds and is accelerated for refolding by ATP-dependent RNA chaperone proteins. J Biol Chem 286: 37304–37312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solem A, Chatterjee P, Caprara MG 2002. A novel mechanism for protein-assisted group I intron splicing. RNA 8: 412–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song MS, Lee SW 2006. Cancer-selective induction of cytotoxicity by tissue-specific expression of targeted trans-splicing ribozyme. FEBS Lett 580: 5033–5043 [DOI] [PubMed] [Google Scholar]
- Sullenger BA, Cech TR 1994. Ribozyme-mediated repair of defective mRNA by targeted, trans-splicing. Nature 371: 619–622 [DOI] [PubMed] [Google Scholar]
- Tanner M, Cech T 1996. Activity and thermostability of the small self-splicing group I intron in the pre-tRNAIle of the purple bacterium Azoarcus. RNA 2: 74–83 [PMC free article] [PubMed] [Google Scholar]
- Vaidya N, Manapat ML, Chen IA, Xulvi-Brunet R, Hayden EJ, Lehman N 2012. Spontaneous network formation among cooperative RNA replicators. Nature 491: 72–77 [DOI] [PubMed] [Google Scholar]
- Vicens Q, Cech TR 2006. Atomic level architecture of group I introns revealed. Trends Biochem Sci 31: 41–51 [DOI] [PubMed] [Google Scholar]
- Vicens Q, Paukstelis PJ, Westhof E, Lambowitz AM, Cech TR 2008. Toward predicting self-splicing and protein-facilitated splicing of group I introns. RNA 14: 2013–2029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldsich C, Grossberger R, Schroeder R 2002. RNA chaperone StpA loosens interactions of the tertiary structure in the td group I intron in vivo. Genes Dev 16: 2300–2312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks KM, Cech TR 1995. Protein facilitation of group I intron splicing by assembly of the catalytic core and the 5′ splice site domain. Cell 82: 221–230 [DOI] [PubMed] [Google Scholar]
- Zaug AJ, McEvoy MM, Cech TR 1993. Self-splicing of the group I intron from Anabaena pre-tRNA: Requirement for base-pairing of the exons in the anticodon stem. Biochemistry 32: 7946–7953 [DOI] [PubMed] [Google Scholar]
- Zhou Y, Lu C, Wu QJ, Wang Y, Sun ZT, Deng JC, Zhang Y 2008. GISSD: Group I Intron Sequence and Structure Database. Nucleic Acids Res 36(Database issue): D31–D37 [DOI] [PMC free article] [PubMed] [Google Scholar]