

Diverse genetic and biochemical assays are used to conclude that the Z-DNA/RNA binding domain of the vaccinia E3L protein contributes to the interdiction of PKR activity, but that this function is not affected by mutations that disrupt Z-DNA binding.
Keywords: E3L, PKR, eIF2, translation
Abstract
Responding to viral infection, the interferon-induced, double-stranded RNA (dsRNA)–activated protein kinase PKR phosphorylates translation initiation factor eIF2α to inhibit cellular and viral protein synthesis. To overcome this host defense mechanism, many poxviruses express the protein E3L, containing an N-terminal Z-DNA binding (Zα) domain and a C-terminal dsRNA-binding domain (dsRBD). While E3L is thought to inhibit PKR activation by sequestering dsRNA activators and by directly binding the kinase, the role of the Zα domain in PKR inhibition remains unclear. Here, we show that the E3L Zα domain is required to suppress the growth-inhibitory properties associated with expression of human PKR in yeast, to inhibit PKR kinase activity in vitro, and to reverse the inhibitory effects of PKR on reporter gene expression in mammalian cells treated with dsRNA. Whereas previous studies revealed that the Z-DNA binding activity of E3L is critical for viral pathogenesis, we identified point mutations in E3L that functionally uncouple Z-DNA binding and PKR inhibition. Thus, our studies reveal a molecular distinction between the nucleic acid binding and PKR inhibitory functions of the E3L Zα domain, and they support the notion that E3L contributes to viral pathogenesis by targeting PKR and other components of the cellular anti-viral defense pathway.
INTRODUCTION
Viruses rely on the host cell translational machinery for the synthesis of viral proteins. One strategy that host cells use to limit viral replication is to inhibit cellular protein synthesis. Virus infection initiates an anti-viral immune response that results in the production of interferons, which, in turn, induce expression of a variety of anti-viral proteins including the double-stranded RNA (dsRNA)–dependent protein kinase PKR. PKR consists of two dsRNA-binding domains (dsRBD) at the N terminus and a Ser/Thr kinase domain at the C terminus (Fig. 2A, right). Binding of dsRNA, a product of viral replication or gene expression, to the dsRBDs promotes oligomerization of PKR, leading to kinase domain dimerization and autophosphorylation. Following this activation, PKR is competent to phosphorylate its substrate, the α subunit of eukaryotic translation initiation factor eIF2 on Ser51 (Dever et al. 1993; Dar et al. 2005; Dey et al. 2005a). Phosphorylation of eIF2α converts eIF2 from a substrate to an inhibitor of its guanine nucleotide exchange factor eIF2B, thereby preventing the recycling of inactive eIF2–GDP to functional eIF2–GTP. The resulting inhibition of translation initiation impairs both cellular and viral protein synthesis, and thus blocks virus replication.
FIGURE 2.
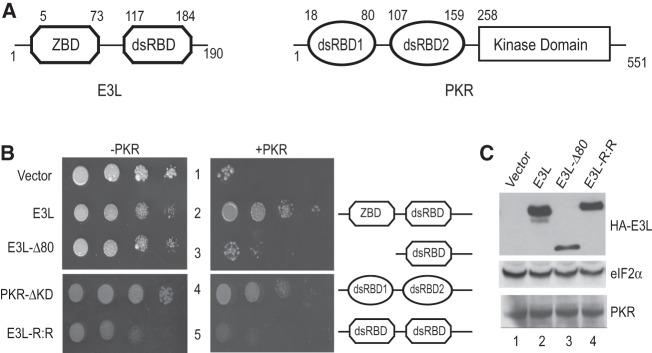
Genetic evidence that the Z-DNA binding domain of E3L is essential for PKR inhibition in yeast. (A) Domain architecture of variola E3L and human PKR. (ZBD) Z-DNA binding domain; (dsRBD) dsRNA-binding domain. (B) Transformants of yeast strains J110 (−PKR) and H2544 (+PKR) bearing empty vector pEMBLyex4 or plasmids that express the indicated E3L protein under the control of a GAL–CYC1 promoter were grown to saturation, and 5 µL of serial dilutions (of OD600 = 1.0, 0.1, 0.01, and 0.001) was spotted on SCGal (10% galactose) medium and incubated for 3 d at 30°C. (C) Immunoblot analysis of E3L and PKR. WCEs were prepared from the yeast transformants described in B, and 5 µg of total protein extracts was subjected to SDS-PAGE followed by immunoblot analysis using monoclonal antibodies directed against PKR and the HA-epitope to detect N-terminally HA-tagged E3L or polyclonal antisera against yeast eIF2α.
To enhance their success, viruses have developed a variety of mechanisms to subvert the host cell anti-viral defenses. Viral inhibitors of PKR target various stages in PKR activation and function including dsRNA binding, kinase dimerization and autophosphorylation, and eIF2α phosphorylation (for review, see Garcia et al. 2007). The adenovirus VA RNAs and Epstein–Barr virus EBER RNAs bind to the dsRBDs of PKR and block kinase activation (Kitajewski et al. 1986; Mathews and Shenk 1991; Sharp et al. 1993), while dsRNA-binding proteins including the poxvirus E3L protein (Chang et al. 1992), the rotavirus NSP3 protein (Langland et al. 1994), and the reovirus virus σ3 protein (Beattie et al. 1995; Yue and Shatkin 1997) are thought to sequester dsRNA and thereby prevent PKR activation. In addition to the E3L protein, many poxviruses also encode a pseudosubstrate inhibitor of PKR (Bratke et al. 2013). For example, the vaccinia virus K3L protein (Davies et al. 1992; Carroll et al. 1993; Kawagishi-Kobayashi et al. 1997; Dar and Sicheri 2002; Seo et al. 2008), variola virus C3L protein (Seo et al. 2008), swinepox virus C8L protein (Kawagishi-Kobayashi et al. 2000), and ranavirus vIF2α protein (Rothenburg et al. 2011) resemble the N-terminal OB-fold domain of eIF2α (Dar and Sicheri 2002; Dar et al. 2005). Binding of these proteins to activated PKR (Dar et al. 2005) is thought to prevent access to eIF2α and thus block eIF2α phosphorylation.
Whereas vaccinia virus mutants lacking K3L remain competent to infect cells as well as mice, vaccinia virus mutants lacking E3L are nonpathogenic in wild-type mice (Brandt and Jacobs 2001; Rice et al. 2011). Consistent with the sensitivity of pkr−/− mice to infection with ΔE3L virus (Rice et al. 2011), replication of the mutant virus in HeLa cells was rescued by knockdown of PKR expression by RNA interference (Zhang et al. 2008). It is noteworthy that the vaccinia virus E3L protein has been reported to inhibit PKR by sequestering dsRNA activators of the kinase and by directly binding to PKR and inhibiting kinase activation (Chang et al. 1992; Davies et al. 1993; Romano et al. 1998; Sharp et al. 1998). The E3L protein consists of two functional domains: an N-terminal domain (NTD) containing a Z-DNA/RNA binding domain (ZBD), and a C-terminal dsRBD (Fig. 2A, left). The dsRBD shares a dsRNA-binding motif (dsRBM) found in many proteins that interact with dsRNA, including PKR, Staufen, RNaseIII, and Xlrbpa (St Johnston et al. 1992; Ryter and Schultz 1998; Saunders and Barber 2003). The dsRBD is required for E3L protein binding to dsRNA in vitro (Chang and Jacobs 1993), and point mutations in the E3L dsRBD that severely impair its ability to bind dsRNA in vitro (Ho and Shuman 1996) not only abolish its ability to inhibit PKR in yeast (Romano et al. 1998), but also eliminate its ability to prevent dsRNA-, 5′-triphosphate single-stranded RNA-, and DNA-induced activation of the IFN-β gene in human cell lines (Marq et al. 2009). In addition to its role in nucleic acid–induced cellular responses, the dsRNA-binding activity of E3L is also critical for vaccinia virus pathogenesis in mouse as well as virus replication in cell culture (Chang et al. 1995; Shors et al. 1997; Brandt and Jacobs 2001).
In support of the dsRNA sequestration model for E3L inhibition of PKR, heterologous dsRNA-binding proteins such as reovirus σ3 (Beattie et al. 1995), rotavirus NSP3 (Langland et al. 1994), human cytomegalovirus TRS1 (Bierle et al. 2013), TAR RNA-binding protein TRBP (Park et al. 1994), and Escherichia coli RNase III (Shors and Jacobs 1997) were shown to functionally complement an E3L-deleted virus. However, E3L was also reported to directly bind to PKR. It was proposed that in the presence of dsRNA, the dsRBDs of PKR and E3L heterodimerize, and that this interaction enables the NTD of E3L to interact with the PKR kinase domain (Romano et al. 1998; Sharp et al. 1998). Furthermore, mutations that disrupt dsRNA binding to E3L were proposed to block PKR inhibition by preventing both dsRNA sequestration and PKR–E3L heterodimer formation (Chang and Jacobs 1993; Davies et al. 1993; Shors and Jacobs 1997; Romano et al. 1998). In contrast, mutations in the E3L NTD that do not impair dsRNA binding were proposed to weaken PKR inhibition by interfering with the interaction between the E3L NTD and the PKR kinase domain (Romano et al. 1998).
While the nucleic acid–binding activity of the E3L dsRBD plays an important role for the virus in sequestering dsRNA and in mediating dsRNA-dependent heterodimerization of E3L with PKR, the function of the N-terminal ZBD of E3L is not as well understood. Moreover, it is not clear whether the Z-DNA binding function of the ZBD is important for E3L inhibition of PKR. The E3L ZBD belongs to the functional Zα family of Z-DNA binding domains based on its sequence similarity with the first (Zα domain) of two ZBDs found in the enzyme adenosine deaminase acting on RNA1 (ADAR1). In addition to ADAR1, ZBDs are also found in DNA-dependent activator of interferon regulatory factor DAI (also known as DLM-1 or ZBP1) (Schwartz et al. 2001), and the protein kinase PKZ (Rothenburg et al. 2005). Interestingly, the ZBD of E3L can be functionally replaced by the heterologous ZBDs from ADAR1 (ZαADAR1) or DAI (ZαDAI) in mouse infection assays (Kim et al. 2003).
The crystal structures of ZBDs from human ADAR1 (Schwartz et al. 1999), mouse DLM-1 (Schwartz et al. 2001), the yatapoxvirus E3L ortholog (Ha et al. 2004), the cyprinid herpesvirus 3 ORF112 protein (Tome et al. 2013), human DAI (ZBP1) (Ha et al. 2008), and zebrafish PKZ (de Rosa et al. 2013) have been solved either free or in complex with Z-DNA. Consistent with amino acid sequence identities of ∼30%, the ZBDs fold into similar α/β structures with three α-helices bundled on one side of a triple-stranded β-sheet (Schwartz et al. 2001; Ha et al. 2004, 2008; de Rosa et al. 2013). Moreover, the solution structure of vaccinia virus E3L revealed a similar topology (Kahmann et al. 2004). Notably, five of the nine residues that contact Z-DNA are highly conserved across the family of E3L proteins and related ZBDs (Fig. 1; Kim et al. 2003; Ha et al. 2004). Mutation of these five residues substantially impaired or abolished the Z-DNA binding activity of the isolated first ZBD (Zα domain) from ADAR1 (Schade et al. 1999; Kim et al. 2003). Similarly, mutation of these same residues impaired the ability of an ADAR-E3L chimera, in which the vaccinia E3L ZBD was replaced by the Zα domain of ADAR1, to functionally substitute for E3L and maintain vaccinia virus pathogenicity in mice (Kim et al. 2003). Likewise, substitution of alanine in place of vaccinia E3L residues Y48 and P63 markedly reduced viral lethality following intracranial inoculation in mice (Kim et al. 2003). In contrast to these studies revealing the importance of the E3L Z-DNA binding activity, the ability of E3L to antagonize the innate immune response in plasmacytoid dendritic cells is dependent on the ZBD, but not its Z-DNA binding activity (Dai et al. 2011; Cao et al. 2012). Taken together, these data reveal important roles for the E3L ZBD- and its Z-DNA binding activity; however, the contribution of Z-DNA binding to E3L inhibition of PKR is not known.
FIGURE 1.

Multiple sequence alignment of the N-terminal domains of the indicated poxvirus E3L proteins and the first Z-DNA binding (ZBD or Zα) domains from mouse ADAR1 and mouse DLM1 (ZBD1). Sequences were aligned using the program PROMALS3D (Pei et al. 2008). (Red) Residues perfectly conserved among all of the ZBDs; (magenta) highly conserved residues. Indicated below the alignment are the conserved hydrophobic (h), polar (p), and aliphatic (l) residues, and the Z-DNA contact residues in the structure of the ADAR1 Zα domain bound to Z-DNA (*). Indicated above the alignment are the sites (red !) of the E3L-N44D,Y48A, and W66A mutations that impair Z-DNA binding and the sites (#) of the variola E3L L36R and L50P mutations (shown in reversed type) that impair PKR inhibitory activity, but not Z-DNA binding.
Here, we use yeast, mammalian cell, and in vitro assays to further examine the inhibition of PKR by a poxviral E3L protein. As previous studies revealed that PKR has evolved under selection by viral inhibitors (Elde et al. 2009; Rothenburg et al. 2009) and that various poxvirus E3L proteins differ in their ability to inhibit human PKR (Myskiw et al. 2011), we focused our studies on variola virus E3L and its natural target human PKR. Our selection of variola E3L was also spurred by the critical biodefense importance of variola virus. We show that variola virus E3L protein, hereafter referred to as E3L, readily suppresses PKR toxicity in yeast, PKR inhibition of reporter gene expression in mammalian cells, and PKR phosphorylation of eIF2α in vitro. Whereas deletion of the NTD abolishes E3L inhibition of PKR in yeast, in vitro, and in mammalian cells treated with dsRNA, point mutations that substantially reduce Z-DNA binding to E3L do not impair PKR inhibition. Thus, these results support a nucleic acid–binding-independent function of the E3L NTD required for inhibition of PKR.
RESULTS
NTD of E3L is required for inhibition of PKR in yeast and in vitro
Variola and vaccinia virus E3L proteins differ at 10 out of the 190 residues in these proteins. To test the ability of variola virus E3L to inhibit PKR, we generated a variola virus E3L expression vector by introducing the 10 required mutations (see Materials and Methods) in a vaccinia virus E3L expression vector. Yeast strain H2544, which expresses human PKR under the control of a galactose-inducible GAL–CYC1 hybrid promoter, was transformed with either empty vector or plasmids that express N-terminally HA-tagged vaccinia or variola E3L also under the control of the GAL–CYC1 promoter. High-level expression of PKR is toxic in yeast due to phosphorylation of eIF2α on Ser51 (Chong et al. 1992; Vazquez de Aldana et al. 1993; Kawagishi-Kobayashi et al. 1997). Consistently, the empty vector transformant of H2544 failed to grow when PKR expression was induced on galactose medium (Fig. 2B, right panel, row 1). While high-level expression of E3L was slightly toxic in yeast even in the absence of PKR (Fig. 2B, left panel, row 2 vs. 1), expression of variola E3L suppressed the lethal phenotype associated with high-level expression of PKR (Fig. 2B, row 2). Deletion of the NTD (residues 1–80) from E3L, generating E3L-Δ80, abolished its ability to suppress PKR toxicity in yeast (Fig. 2B, row 3). Previous studies have shown that a C-terminal truncated version of PKR, retaining residues 1–258 and consisting of the two dsRBDs (PKR-ΔKD), functioned in a dominant-negative manner and blocked PKR toxicity in yeast (Fig. 2B, row 4). This inhibitory function was attributed to formation of heterodimers between PKR-ΔKD and the full-length kinase (Romano et al. 1995). We generated a corresponding version of E3L in which the E3L dsRBD was substituted for the NTD, generating a protein with two dsRBDs (E3L-R:R) (Fig. 2B, row 5). In contrast to PKR-ΔKD, E3L-R:R failed to inhibit PKR toxicity in yeast (Fig. 2B, row 5). Although we cannot assess whether the dsRBDs are folding properly in E3L-R:R, this result suggests that simply linking two dsRBDs in a single protein, as found in E3L-R:R, is not sufficient to confer PKR inhibitory function. Western analyses demonstrated that both E3L-Δ80 and E3L-R:R were readily expressed in yeast, albeit at lower levels than E3L (Fig. 2C). While the reduced expression of the E3L mutants limits the strength of the conclusion that a second copy of the E3L dsRBD cannot functionally replace the NTD, the in vitro experiments described below support the conclusion that the NTD of E3L is critical for the inhibition of PKR.
To directly examine E3L inhibition of PKR, we carried out in vitro kinase assays. Flag-tagged PKR was purified from yeast and tested for the ability to phosphorylate recombinant His6-eIF2α (residues 1–200) purified from E. coli. As the PKR expressed in yeast is purified in a pre-activated form, already phosphorylated on Thr446 (Dey et al. 2005a,b), it was unnecessary to add dsRNA to activate the kinase. Moreover, inhibitors that simply sequester dsRNA or other activators are unlikely to inhibit this pre-activated kinase. The in vitro kinase assays were conducted in the presence or absence of GST, GST-E3L, or GST-E3L-Δ80, and eIF2α phosphorylation was monitored by immunoblot analysis using antibodies that specifically detect phospho-Ser51 on eIF2α. As shown in Figure 3A, PKR readily phosphorylated eIF2α on Ser51 (lane 1). Pre-incubation of PKR with a 10-fold molar excess of GST-E3L (lane 5), but not GST (lane 3), substantially reduced eIF2α phosphorylation. In contrast, pre-incubation of Flag-PKR with GST-E3L-Δ80 failed to inhibit eIF2α phosphorylation (Fig. 3A, lanes 6,7). Thus, deletion of the NTD abolished the ability of E3L to inhibit PKR both in yeast (Fig. 2B) and in vitro (Fig. 3A). Immunoblot analyses confirmed that equivalent amounts of PKR, eIF2α, and the various GST or GST-E3L fusion proteins were used in each assay (Fig. 3A). Moreover, unlike GST, which as shown in Figure 3B was largely localized in the unbound fraction, both GST-E3L and GST-E3L-Δ80 readily bound to an agarose affinity resin containing poly(I:C) (polyinosinic–polycytidylic acid, a double-stranded RNA homopolymer known to activate PKR) (Minks et al. 1979). Thus, the loss of PKR inhibitory activity by GST-E3L-Δ80 was not due to impaired dsRNA-binding ability. Taken together, these in vitro experiments and yeast studies demonstrate that the E3L NTD plays a critical role in PKR inhibition, and they indicate that E3L can inhibit PKR following kinase activation.
FIGURE 3.
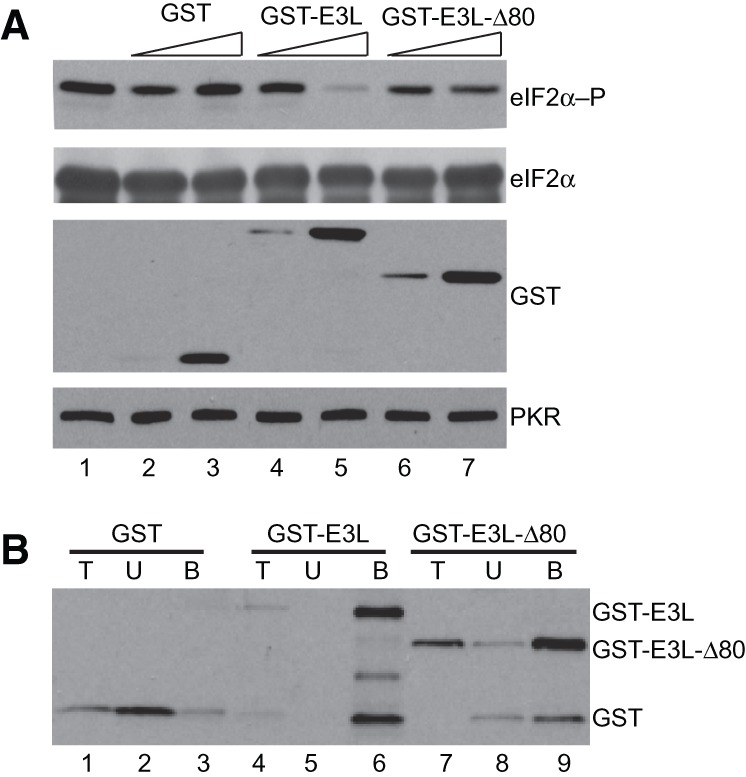
E3L ZBD is required for inhibition of PKR in vitro. (A) E3L inhibition of eIF2α phosphorylation; 2.5 nM purified Flag-His6-PKR was pre-incubated with 2.5 or 25 nM purified GST (lanes 2,3), GST-E3L (lanes 4,5), or GST-E3L-Δ80 (lanes 6,7) for 10 min at room temperature, then kinase reactions were initiated by addition of 0.2 µM His6-tagged eIF2α1–200 and 0.2 mM ATP. Reactions were resolved by SDS-PAGE and subjected to immunoblot analysis using phosphospecific antibodies against Ser51 in eIF2α (top panel, eIF2α-P). The membranes were then sequentially stripped and probed using polyclonal antiserum against yeast eIF2α (second panel, eIF2α), polyclonal anti-GST antiserum (third panel), and monoclonal anti-PKR antibody (bottom panel). (B) GST-E3L-Δ80 mutation does not impair dsRNA binding in vitro. Purified GST, GST-E3L, and GST-E3L-Δ80 were incubated with poly(I:C)-agarose for 1 h at 4°C. Bound proteins were collected by centrifugation and eluted by boiling in SDS sample buffer. Proteins were resolved by SDS-PAGE and subjected to immunoblot analysis using polyclonal anti-GST antiserum. (T) 5% of total protein used in the assay; (U) unbound (20%); (B) protein bound (20%) to the poly(I:C) agarose.
Abundance of dsRNA alters the requirement of the E3L NTD for PKR inhibition in mammalian cells
PKR knockdown HeLa cells (PKRkd), which express an shRNA that targets the PKR mRNA and efficiently abolishes endogenous PKR expression and blocks dsRNA-induced eIF2α phosphorylation (Zhang and Samuel 2007), were used to test the role of the E3L NTD for inhibition of PKR in mammalian cells. Previous studies demonstrated that PKR expression could be restored in the PKRkd cells using PKR expression constructs in which the shRNA target sequence was altered (Rothenburg et al. 2009). The PKRkd cells were transiently transfected with a luciferase reporter plasmid, the knockdown-resistant PKR construct, and various C-terminally HA-tagged E3L expression vectors. As expected, expression of PKR impaired luciferase expression (Fig. 4A) and increased eIF2α phosphorylation (Fig. 4C, lane 2 vs. 1). Cotransfection of E3L with PKR increased luciferase activity (Fig. 4A) and lowered eIF2α phosphorylation (Fig. 4C, lane 3), indicating that E3L was able to inhibit PKR in mammalian cells. Surprisingly, unlike the yeast and in vitro studies, E3L-Δ80 functioned like wild-type (WT) E3L in the mammalian cell transfection assays, increasing luciferase activity (Fig. 4A) and lowering eIF2α phosphorylation levels in the transfected cells (Fig. 4C, lane 4). Western analyses revealed that WT E3L, which, as previously reported (Watson et al. 1991; Chang et al. 1992; Yuwen et al. 1993), is produced as two separate species in mammalian cells, was expressed at higher levels than E3L-Δ80 in these experiments (Fig. 4C). Thus, the unexpected activity of E3L-Δ80 cannot be attributed to elevated levels of the protein, and contrary to the yeast and in vitro kinase assay results, these findings demonstrate that the NTD of E3L is dispensable for PKR inhibition in mammalian cells.
FIGURE 4.
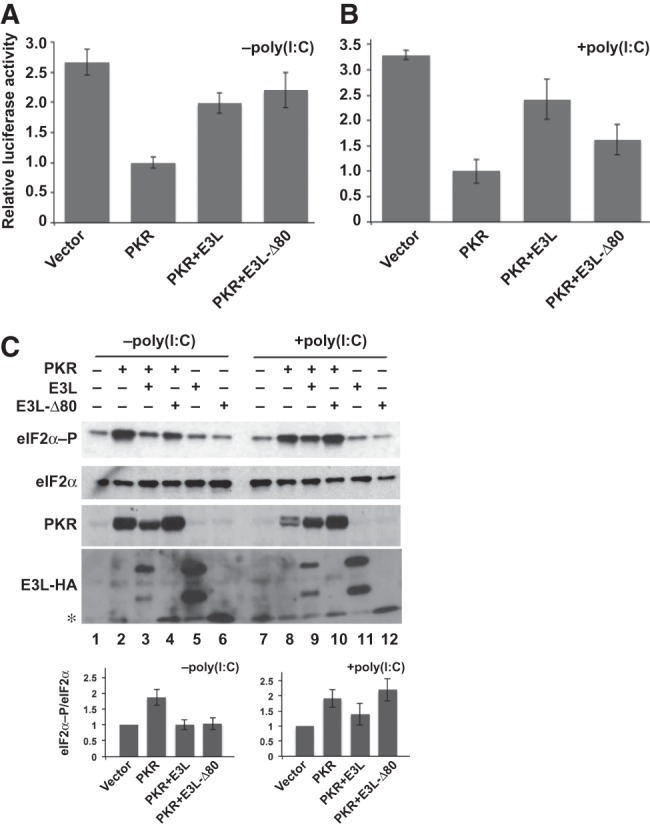
Abundance of dsRNA alters the requirement of the E3L ZBD for PKR inhibition in mammalian cells. (A) Human HeLa PKRkd cells were cotransfected with expression vectors for luciferase, an empty vector or knockdown-resistant human PKR (100 ng), and C-terminally HA-tagged variola E3L-HA or E3L-Δ80-HA (400 ng). After 40 h, cells were harvested, lysed, and extracts were assayed for luciferase activity. Luciferase activity was normalized to the transfections containing PKR but lacking E3L. Error bars indicate the standard deviation for three independent transfections. (B) Analysis of luciferase activity following cotransfection of PKRkd cells with poly(I:C) (500 ng) and the expression vectors for luciferase, vector or PKR (100 ng), and E3L-HA or E3L-Δ80-HA (400 ng) as described in panel A. (C) Immunoblot analyses of WCEs from cells transfected as described in panels A and B with or without poly(I:C) and the indicated expression vectors for PKR, E3L, or E3L-Δ80. Blots were probed with antibodies specific for phosphorylated Ser51 in eIF2α (top panel), polyclonal antiserum against human eIF2α (second panel), monoclonal human PKR antibody (third panel), and monoclonal anti-HA antibody to detect HA-tagged E3L (fourth panel). Note that E3L-HA resolves as two species on SDS-PAGE (see text). (*) E3L-Δ80 comigrates with a nonspecific protein that cross-reacts with the anti-HA antibody. (Bottom panel) Relative level of eIF2α-P to total eIF2α was determined for three independent experiments using quantitative densitometry and Image J software (NIH) and normalized to the vector transformant.
The contrasting observations in yeast and mammalian cells led us to propose that the requirement for the E3L NTD is dependent on the abundance of PKR activators. We hypothesized that in the presence of low levels of dsRNA, such as in mammalian cells, the inhibition of PKR by E3L is entirely through sequestration of the dsRNA activators. Thus, due to its intact dsRBD, E3L-Δ80, which lacks the NTD, is able to inhibit PKR in mammalian cells. However, when PKR activators are abundant or in the presence of the pre-activated PKR used in the in vitro assays, dsRNA sequestration by E3L is not sufficient to inhibit PKR. Instead, we propose that E3L must heterodimerize with PKR to inhibit kinase activity when PKR activators are abundant. The nature of the PKR activator in yeast cells is not known. The activator does not appear to be the dsRNA L-A or related killer viruses of yeast because PKR expression is toxic in yeast cured of all dsRNA viruses (TE Dever, unpubl.). However, as mutations that impair dsRNA binding reduce PKR toxicity in yeast (Romano et al. 1995; Zhang et al. 2001), we propose that dsRNA structures in yeast mRNAs or that other, perhaps non-RNA, activators are abundant in yeast cells leading to high PKR activity. Consistent with the notion that PKR activators are abundant in yeast, the kinase-dead variant PKR-K296R, which functions in a dominant-negative manner in mammalian cells (Barber et al. 1993), fails to inhibit PKR in yeast (Romano et al. 1995). It is presumed that PKR-K296R effectively sequesters the low-abundance activators in mammalian cells, but is unable to sequester the activators present in yeast cells.
To test the hypothesis that the E3L NTD is critical for inhibition of PKR when activators are abundant, we increased the levels of PKR activators in the PKRkd cells by cotransfecting poly(I:C) together with PKR and E3L. As shown in Figure 4B, the ability of E3L-Δ80 to suppress the inhibition of luciferase expression by PKR was diminished in the presence of added poly(I:C) dsRNA. E3L readily restored luciferase expression both in the presence (Fig. 4B) and absence (Fig. 4A) of added dsRNA, and likewise lowered eIF2α phosphorylation levels (Fig. 4C, lane 3 vs. 2, lane 9 vs. 8). In contrast, E3L-Δ80 only partially restored luciferase expression (Fig. 4B) and failed to lower eIF2α phosphorylation when poly(I:C) was cotransfected with PKR (lane 10). Thus, increasing the abundance of PKR activators in mammalian cells mimics the conditions in yeast, or in vitro with pre-activated PKR, and reveals the importance of the NTD for E3L inhibition of PKR.
ZBD functions independent of Z-DNA binding in PKR inhibition
As the best-characterized function of a ZBD is to bind Z-DNA/RNA, it was intriguing to determine whether this nucleic acid–binding activity is required for E3L inhibition of PKR. It is noteworthy that the dsRNA-binding activity of E3L is critical for inhibition of PKR in vitro (Chang and Jacobs 1993). Moreover, mutation of Lys167 in the E3L dsRBD impairs dsRNA binding (Chang and Jacobs 1993; Ho and Shuman 1996; Romano et al. 1998) and the ability of E3L to inhibit PKR toxicity in yeast (Romano et al. 1998), and to block PKR phosphorylation of eIF2α and inhibition of luciferase expression in mammalian cells (data not shown). Analysis of the sequence and structure of ZBDs from viral E3L proteins as well as from mouse ADAR1 and DLM-1 (Fig. 1) identified seven perfectly conserved residues including the variola E3L residues Asn44, Tyr48, and Pro63. These three residues are among the set of residues that directly contact Z-DNA in the cocrystal structure of the ADAR1 ZBD bound to Z-DNA (Schwartz et al. 1999), and mutation of the corresponding residues (N173D, Y177A, P192A) in the Zα domain of ADAR1 impaired Z-DNA binding (Schade et al. 1999; Kim et al. 2003). Furthermore, similar mutations significantly reduced the ability of a ZαADAR–RBDE3L chimera, consisting of the ADAR1 Zα domain fused to the E3L RBD to substitute for E3L in a viral pathogenesis assay in mice (Kim et al. 2003). Since these three Z-DNA binding residues are identical in E3L and ADAR1, we introduced the corresponding N44D, Y48A, and P63A mutations in the E3L ZBD and examined the ability of the mutant proteins to inhibit PKR in yeast. As shown in Figure 5A, expression of WT E3L or the N44D, Y48A, or P63A mutants of E3L suppressed the lethal effects associated with high-level expression of PKR in yeast. Combining these mutations to create double (N44D,Y48A [DM] and Y48A,P63A) or triple (N44D,Y48A,P63A) mutants failed to abolish the ability of E3L to suppress PKR toxicity in yeast (Fig. 5A). Consistent with their potent activity to inhibit PKR, the E3L mutants were all well expressed in yeast (Fig. 5B).
FIGURE 5.
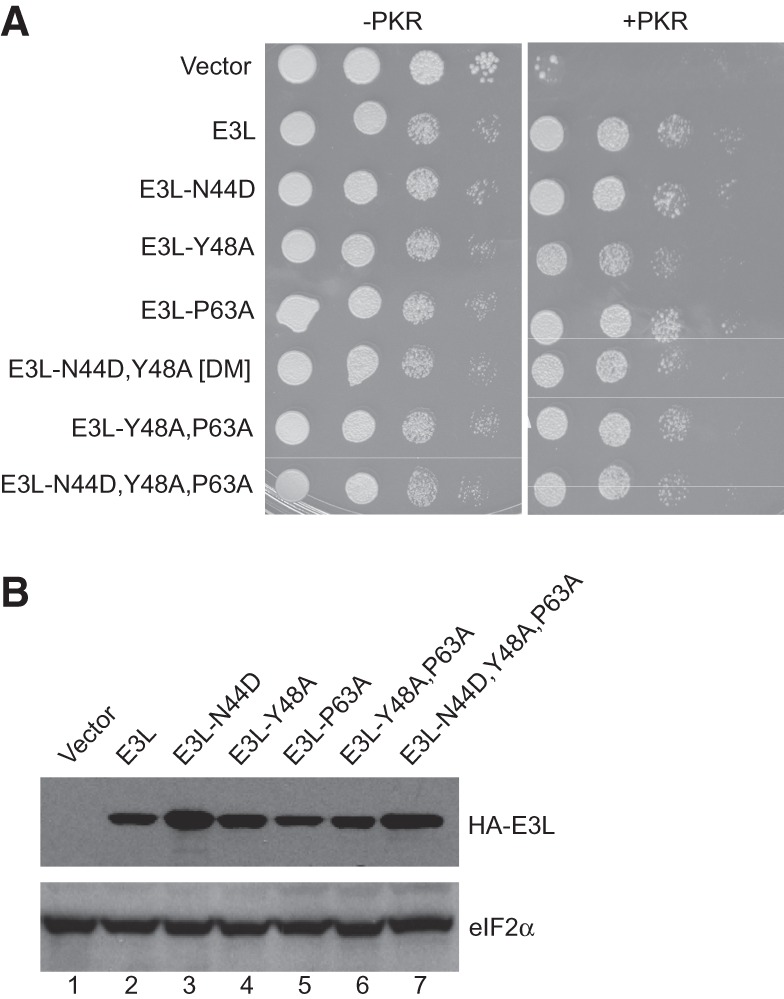
E3L inhibition of PKR in yeast is independent of Z-DNA binding activity. (A) Transformants of yeast strains J110 (−PKR) and H2544 (+PKR) bearing empty vector pEMBLyex4 or plasmids expressing the indicated N-terminally HA-tagged E3L derivatives under the control of the GAL–CYC1 promoter were grown to saturation, and 5 µL of serial dilutions (of OD600 = 1.0, 0.1, 0.01, and 0.001) was spotted on SCGal (10% galactose) medium and incubated for 3 d at 30°C. (B) Transformants of H2544 (+PKR) from panel A were grown in SD medium at 30°C until OD ∼ 0.6, then shifted to SCGal medium for 2 h. WCEs were fractionated by SDS-PAGE and subjected to immunoblot analysis using monoclonal antibodies against the HA epitope to detect HA-tagged E3L (upper panel) and polyclonal antisera against yeast eIF2α (lower panel).
To directly examine the impact of the ZBD mutations on the Z-DNA binding activity of E3L, recombinant GST-E3L fusion proteins were purified from E. coli and tested for Z-DNA binding ability. Electrophoretic gel mobility shift assays were used to monitor the binding of the E3L proteins with [32P]-labeled partially brominated synthetic poly(dG:dC), which was previously shown to be stable in the Z conformation (Zhang et al. 1992; Kim et al. 2006). As shown in Figure 6A, incubation of the poly(dG:dC) with increasing amounts of GST-E3L resulted in the appearance of multiple slower-migrating species consistent with the binding of GST-E3L to Z-DNA. The exact number of complexes observed varied in different experiments (see Fig. 6A,B), perhaps reflecting heterogeneity in the Z-DNA or the proteins. However, the appearance of the upper complex in Figure 6A, and to a lesser extent the lower complex, was reduced when unlabeled Z-DNA (lanes 6,7), but not unlabeled B-DNA (lanes 8,9), was added as a competitor in the binding reaction. These results demonstrate that E3L binds specifically to Z-DNA. Next, this assay was used to monitor the Z-DNA binding activity of the E3L-N44D,Y48A double mutant (E3L-DM). Note that the corresponding single mutations in the Zα domain of ADAR1 were shown to impair Z-DNA binding activity (Schade et al. 1999; Kim et al. 2003). Consistent with the results of these previous studies, the Z-DNA binding ability of GST-E3L (Fig. 6B, lane 1) was severely impaired, with reduced detection of all Z-DNA–protein complexes, following deletion of the ZBD (E3L-Δ80, lane 2) or by the N44D,Y48A double mutation (E3L-DM, lane 5). In contrast to these ZBD mutations, the K167A mutation in the E3L dsRBD, which abolishes dsRNA binding (Romano et al. 1998), did not impair Z-DNA binding (lane 3). These data demonstrate that the E3L-N44D,Y48A double mutant is impaired in binding to Z-DNA.
FIGURE 6.

E3L inhibition of PKR in vitro and in mammalian cells treated with dsRNA is independent of Z-DNA binding activity. (A) Electrophoretic mobility shift assay of GST-E3L binding to the Z-DNA substrate [32P]-labeled d(G:5BrC)20. Indicated concentrations of GST-E3L were incubated with labeled d(G:5BrC)20 in the presence of 30 nM or 300 nM unlabeled Z-DNA (lanes 6,7) or B-DNA (lanes 8,9). Positions of the GST-E3L–Z-DNA complex and free probe are indicated. (B) Z-DNA binding activity of E3L mutants. GST or the indicated GST-E3L derivative (5 µM) was incubated with the Z-DNA substrate and analyzed as described in A. (C) Z-DNA binding mutations do not impair E3L inhibition of PKR in vitro. The indicated GST or GST-E3L derivatives were incubated with purified Flag-His6-PKR and then mixed with His6-eIF2α1–200 and ATP. Reactions were resolved by SDS-PAGE and subjected to immunoblot analysis using antibodies specific for the Ser51-phosphorylated form of eIF2α (top panel) or using polyclonal antiserum against total yeast eIF2α (middle panel) or GST (bottom panel). (D) Human HeLa PKRkd cells were cotransfected with poly(I:C) (500 ng) and expression vectors for luciferase, knockdown-resistant human PKR (100 ng), and the indicated E3L-HA derivative (400 ng). After 40 h, cells were harvested, lysed, and extracts were assayed for luciferase activity. Luciferase activity was normalized to the transfections containing PKR but lacking E3L. Error bars indicate the standard deviation for three independent transfections. (E) Immunoblot analyses of WCEs from cells transfected as described in panel D. Blots were probed with antibodies specific for phosphorylated Ser51 in eIF2α (top panel), polyclonal antiserum against human eIF2α (second panel), monoclonal human PKR antibody (third panel), and monoclonal anti-HA antibody to detect HA-tagged E3L (bottom panel). The relative level of eIF2α-P to total eIF2α (2α-P/2α) was determined for three independent experiments using quantitative densitometry and Image J software (NIH) and normalized to the vector transformant.
Having shown that E3L-DM fails to bind Z-DNA, yet retains PKR inhibitory activity in yeast, we next examined the ability of this mutant to directly inhibit PKR using in vitro kinase assays. PKR purified from yeast was incubated with GST-E3L, GST-E3L-DM, or GST-E3L-K167A and then tested for eIF2α phosphorylation. As shown in Figure 6C, GST-E3L and GST-E3L-DM, but not GST-E3L-K167A, substantially reduced the ability of PKR to phosphorylate eIF2α (lanes 2–4). Thus, the Z-DNA binding ability of E3L, but not its dsRNA-binding ability, is dispensable for PKR inhibition in vitro.
Next, we examined the importance of Z-DNA binding for E3L inhibition of PKR in mammalian cells. Consistent with our previous data showing that deletion of the NTD did not impair PKR inhibition in this assay system (Fig. 4A), E3L-DM (N44D,Y48A) functioned like WT E3L and lowered eIF2α phosphorylation levels and restored luciferase expression in cells cotransfected with the PKR expression vector (data not shown). Importantly, unlike E3L-Δ80, but similar to WT E3L, E3L-DM retained the ability to suppress the PKR inhibition of luciferase reporter expression in cells treated with excess dsRNA (Fig. 6D). Likewise, both WT E3L and E3L-DM lowered the level of eIF2α phosphorylation in the cells treated with dsRNA (Fig. 6E, lanes 3,4 vs. 2), although for unknown reasons E3L-DM was expressed at higher levels than WT E3L in the transfected cells (Fig. 6E). While this enhanced expression of E3L-DM could contribute to its retention of PKR inhibitory activity, these data together with the yeast (Fig. 5A) and in vitro (Fig. 6C) data demonstrate that the PKR inhibitory activity of E3L requires the NTD but is independent of Z-DNA binding.
Identification of E3L ZBD mutations that impair PKR inhibition but not Z-DNA binding
Having found that the E3L NTD, but not its Z-DNA binding activity, is required for inhibition of PKR, we subjected the E3L NTD to random mutagenesis and screened for mutants that lost the ability to inhibit PKR in yeast. Error-prone PCR was used to generate a library of E3L mutants containing mutations in the NTD. The mutant library was introduced into the yeast strain expressing PKR under the control of the galactose-inducible promoter. Approximately 2000 transformants were patched on SD medium where PKR and E3L are not expressed and then replica-printed to galactose medium to induce PKR and E3L expression. The E3L plasmid was isolated from the colonies growing on SD medium that failed to grow on galactose medium. Following retesting and sequencing, only two of the mutants were found to contain single amino acid changes. These two mutants, E3L-L36R and E3L-L50P, were selected for further characterization. As shown in Figure 7A, E3L-L36R and E3L-L50P, unlike wild-type E3L, were unable to restore the growth of yeast coexpressing PKR. The mutant proteins were expressed at levels comparable to wild-type E3L in cells lacking PKR (Fig. 7B). The expression of E3L-L36R and E3L-L50P was slightly lower than E3L in cells expressing PKR, consistent with the notion that induction of PKR inhibited protein synthesis including expression of these nonfunctional E3L mutants. As expected, given their intact dsRBDs, the E3L mutants retained dsRNA-binding activity (Fig. 7C). The L50 residue is perfectly conserved among viral E3L proteins and the mouse ADAR1 and DLM-1 ZBDs (Fig. 1). Likewise, the L36 residue is conserved among the E3L proteins except for swinepox E3L, where Ile is substituted for Leu. However, neither residue was observed to directly contact Z-DNA in the structure of the ADAR1 Zα domain bound to Z-DNA (Schwartz et al. 1999; Kahmann et al. 2004). To determine whether mutation of these conserved residues affects Z-DNA binding, we produced fusion proteins consisting of GST linked to the ZBD of wild-type E3L or various E3L mutants. As shown in Figure 7D, GST-E3L-ZBD (lane 2) and the GST-E3L-ZBD-L36R (lane 4) and GST-E3L-ZBD-L50P (lane 7) mutants readily formed a complex with Z-DNA. In contrast, few or no DNA–protein complexes were detected in assays with GST, GST-E3L-ZBD-DM (N44D,Y48A), or GST-E3L-ZBD-W66A, which contains a previously described mutation that prevents PKR inhibition (Fig. 7D, lanes 3,5,6; Romano et al. 1998). Finally, the E3L-L50P mutant was tested in the mammalian cell transfection assay. In contrast to WT E3L, E3L-L50P was unable to lower eIF2α phosphorylation (Fig. 6E, lane 5) and restore luciferase expression in cells transfected with the PKR expression vector and dsRNA (Fig. 6D). Thus, the L50P mutation impairs E3L inhibition of PKR in both yeast and mammalian cell assays. The finding that the E3L-L36R and E3L-L50P mutants retain Z-DNA binding activity, yet fail to inhibit PKR, further demonstrates that the PKR inhibitory function of the E3L NTD is distinct from its nucleic acid–binding activity.
FIGURE 7.

Identification of point mutations in the ZBD of E3L that impair PKR inhibition, but not Z-DNA binding. (A) Growth phenotypes of E3L mutants identified through random mutagenesis. Transformants of yeast strains J110 (−PKR) or H2544 (+PKR) bearing empty vector pEMBLyex4 or plasmids that express the indicated HA-E3L derivatives under the control of a GAL–CYC1 promoter were grown to saturation, and 5 µL of serial dilutions (of OD600 = 1.0, 0.1, 0.01, and 0.001) was spotted on SCGal (10% galactose) medium and incubated for 3 d at 30°C. (B) Immunoblot analysis of WCEs from yeast strains in panel A. Transformants were grown in SD medium at 30°C to OD600 ∼ 0.6, and then shifted to SCGal medium for 2 h. WCEs (5 µg) were fractionated by SDS-PAGE and subjected to immunoblot analysis using monoclonal antibodies against the HA epitope to detect HA-E3L (upper panel) and polyclonal antisera against yeast eIF2α as loading control (lower panel). (C) dsRNA binding. Purified GST or the indicated GST-E3L derivative was incubated with poly(I:C)-agarose for 1 h at 4°C. Bound proteins were collected by centrifugation and eluted by boiling in SDS sample buffer. Proteins were resolved by SDS-PAGE and subjected to immunoblot analysis using polyclonal anti-GST antiserum. (T) 5% of total protein used in the assay; (U) unbound (20%); (B) protein bound (20%) to the poly(I:C) agarose. (D) Z-DNA binding activity of E3L ZBD derivatives. The indicated GST-E3L-ZBD (residues 1–80) derivatives (5 µM) were incubated with the Z-DNA substrate [32P]-labeled d(C:5BrG)20 and assayed for Z-DNA binding ability by electrophoretic mobility shift assay.
DISCUSSION
Successful viral replication is dependent on subversion of host cell defense mechanisms. It was previously shown that vaccinia virus expresses an inhibitor of the interferon-induced host cell kinase PKR, and this activity was eventually linked to the viral E3L protein (Rice and Kerr 1984; Whitaker-Dowling and Youngner 1984; Chang et al. 1992). As the E3L inhibition of PKR could be overcome by adding excess dsRNA (Davies et al. 1993; Jagus and Gray 1994), and as E3L was found to contain a dsRBD at its C terminus (Chang et al. 1992), it was concluded that E3L inhibits PKR by sequestering dsRNA activators of the kinase (Davies et al. 1993). Consistent with this idea, point mutations that blocked dsRNA binding to E3L likewise interfered with the ability of E3L to inhibit PKR (Shors et al. 1997). At odds with this simple model for E3L inhibition of PKR, truncation mutations that removed the NTD of E3L (E3L-Δ80), yet left the dsRBD intact, blocked the ability of E3L to suppress PKR toxicity in yeast (Romano et al. 1998). The inability of this truncated E3L mutant, which retains dsRNA-binding activity (Fig. 3B), to inhibit PKR indicates that sequestration of dsRNA activators is not sufficient for E3L inhibition of PKR. In accord with this latter idea, it was shown that E3L could bind directly to PKR, leading to the hypothesis that E3L inhibits PKR both by sequestering dsRNA activators of the kinase and by forming inactive heterodimers with PKR (Romano et al. 1998; Sharp et al. 1998). In this study, we further explored the role of the E3L NTD and found that the ZBD, but not its nucleic acid–binding function, is important for PKR inhibition. It is noteworthy that the E3L ZBD, but not its Z-DNA binding activity, is likewise required to block the induction of the innate immune response and cytokine production in poxvirus-infected plasmacytoid dendritic cells (Dai et al. 2011; Cao et al. 2012). In contrast, the Z-DNA binding activity of E3L was previously shown to be critical for viral pathogenicity (Kim et al. 2003). Taken together, we propose that the E3L ZBD performs a nucleic acid–binding independent function to inhibit PKR and perhaps other components of the innate immune response, whereas the Z-DNA binding activity of this domain is critical for E3L subversion of other cellular anti-viral defenses.
Our studies provide several insights into the mechanism of E3L inhibition of PKR. We show that the abundance of PKR activators determines the requirements for PKR inhibition. Whereas full-length E3L impairs the phosphorylation of eIF2α and the inhibition of reporter gene expression by PKR in either untreated mammalian cells or in cells treated with dsRNA (Fig. 4), the inhibition of PKR in mammalian cells by E3L-Δ80 (Fig. 4) or the Z-DNA binding mutant E3L-DM (Fig. 6) was reversed by increasing the abundance of dsRNA. We propose that under native conditions, where dsRNA activators are not abundant, the E3L RBD is sufficient to sequester the dsRNA activators and prevent PKR activation. However, under conditions where dsRNA activators are plentiful, E3L inhibition of PKR is dependent on both the RBD and the ZBD. The requirement for the E3L ZBD observed in mammalian cells treated with dsRNA mimics the E3L requirements for PKR inhibition in yeast, supporting the notion that PKR activators are abundant in yeast. While it could have been proposed that the ZBD enhances the nucleic acid–binding properties of E3L and thus enables the protein to effectively titrate the activators, the fact that the Z-DNA binding defective mutant E3L-DM effectively inhibits PKR both in yeast and in mammalian cells treated with excess dsRNA (Figs. 5, 6) suggests that the ZBD contributes to PKR inhibition in a manner independent of nucleic acid binding. Further supporting this notion, we identified the E3L-L36R and E3L-L50P mutants that readily bind dsRNA as well as Z-DNA but are unable to inhibit PKR in yeast or in mammalian cells treated with dsRNA. Taken together, our molecular dissection of E3L has revealed a PKR inhibitory function in the NTD that is distinct from its nucleic acid–binding activity.
We reasoned that the NTD could contribute to PKR inhibition by promoting heterodimer formation with PKR. Previously, the N-terminal half of vaccinia virus E3L was shown to interact with the PKR KD in yeast two-hybrid assays (Romano et al. 1998; Sharp et al. 1998). We found that full-length variola E3L interacted with full-length PKR in yeast two-hybrid assays (data not shown). Moreover, this interaction was not disrupted by the N44D and Y48A mutations that impair Z-DNA binding. However, as both full-length E3L and PKR contain dsRBDs, the two-hybrid interaction we detected could be mediated solely by the dsRBD in E3L. Unfortunately, we were unable to test the effect of the ZBD mutations on binding of the isolated E3L NTD to PKR. While the variola E3L ZBD interacted with full-length PKR in a yeast two-hybrid assay, thus eliminating the concern about dsRNA-mediated interactions, the ZBD mutations caused the DNA-binding domain fusion to promiscuously activate the two-hybrid reporter in the absence of a partner activation domain (data not shown). Moreover, and consistent with previous reports (Romano et al. 1998; Sharp et al. 1998), issues with nonspecific binding prevented the use of pull-down assays to monitor an interaction between the E3L ZBD and PKR. Thus, while we were unable to directly show that disruption of its Z-DNA binding activity does not affect the ability of the E3L NTD to bind PKR, our data are consistent with the notion that the E3L NTD contributes to PKR inhibition in a manner that is independent of its nucleic acid–binding function and is presumably mediated by direct protein–protein interactions with PKR.
The E3L gene is critical for vaccinia virus replication in mammalian cells (Chang et al. 1995) and for viral pathogenicity in mice (Brandt and Jacobs 2001). As knockdown of PKR expression in HeLa cells complemented the defect in virus yield and virus-induced apoptosis by E3L-deleted vaccinia virus (Zhang et al. 2008), it is clear that PKR is an important target of E3L. Consistently, the attenuated phenotype of E3L-deleted virus following intratracheal infection of wild-type mice was partially reversed upon infection of mice lacking PKR (Rice et al. 2011). The fact that deletion of PKR does not fully rescue the pathogenicity of the E3L-deleted virus (Rice and Kerr 1984; Xiang et al. 2002) indicates that E3L likely targets other components of the cellular anti-viral defense, such as the mitochondrial anti-viral signaling (MAVS) pathway for interferon induction (Deng et al. 2008). In support of this idea, the E3L-deleted virus was pathogenic in mice lacking both PKR and the anti-viral endoribonuclease RNAse L (Rice et al. 2011). However, the PKR and RNase L pathways are likely not the sole targets of E3L, as E3L-deleted virus failed to replicate to high titers in mice triply lacking PKR, RNase L, and the interferon-induced Mx1 protein (Xiang et al. 2002). Our results are also consistent with the notion that E3L inhibits anti-viral targets other than PKR. While our data show that the Z-DNA binding domain of E3L is not required for PKR inhibition in transfected mammalian cells (not treated with excess dsRNA) and that the Z-DNA binding activity is dispensable for PKR inhibition in yeast, in vitro, and in mammalian cells treated with dsRNA, mutations impairing the Z-DNA binding function of E3L were previously shown to reduce the pathogenicity of vaccinia virus in mice (Kim et al. 2003). Thus, while the E3L ZBD has a nucleic acid–binding independent function to inhibit PKR, the nucleic binding property of this domain is important for viral pathogenicity. Taken together, these results lead us to suggest that the Z-DNA binding properties of E3L are necessary for the inhibition of other components of the interferon-induced anti-viral defense, consistent with the recent report that the pathogenicity of ΔE3L vaccinia virus is fully rescued by knockout of the type I interferon receptor in mice (White and Jacobs 2012).
MATERIALS AND METHODS
Plasmids
Plasmids used in this study are listed in Table 1. Construction of the plasmid pEMBLyex4-E3L (p2245), which expresses hemagglutinin (HA) epitope-tagged vaccinia E3L under the control of a yeast GAL–CYC1 hybrid promoter, was described previously (Romano et al. 1998). A SacI–BamH1 fragment carrying HA-E3L was subcloned in pUC19 to generate pC2490. Site-directed mutagenesis was performed at 10 residues to convert vaccinia E3L to variola E3L (N11D, T21N, I24L, A27V, A30V, D78M, A79T, G115N V121I, and R130K), generating pUC19-varE3L (pC2491). The SacI/BamHI fragment from pC2491 was subcloned in pEMBLyex4 to generate pEMBLyex4-var.E3L (pC2492). Site-directed mutagenesis of pC2492 was performed to generate E3L single (K167A, W66A, N44D, Y48A, P63A, L36R, and L50P), double (N44D/Y48A; Y48A/P63A), and triple (N44D/Y48A/P63A) point mutants.
TABLE 1.
Plasmids used in this study
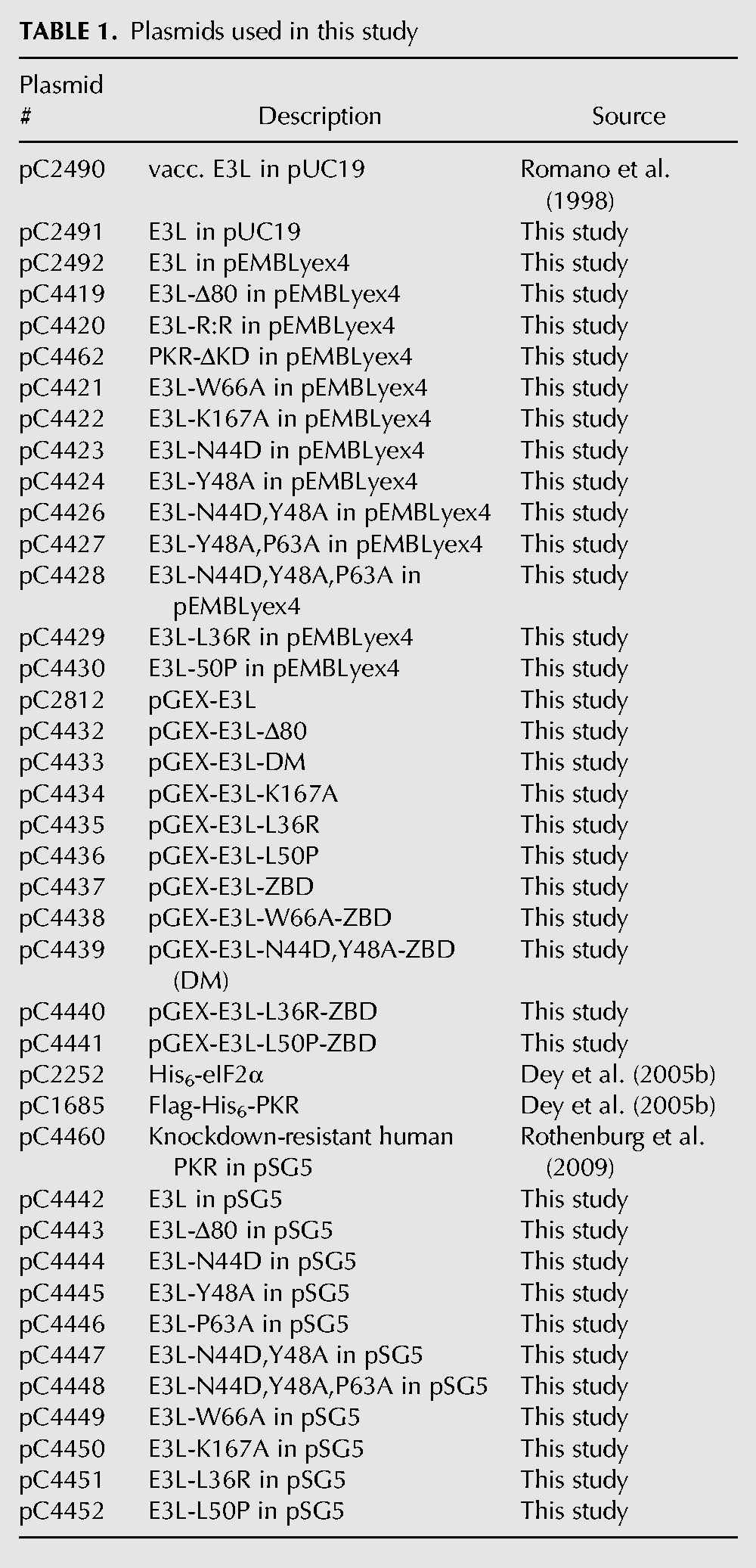
Plasmids encoding GST-E3L (pC2812), GST-E3L-Δ80 (pC4432) and GST-E3L-ZBD (E3L residues 1–80; pC4437) were constructed by inserting the relevant PCR products between the BamHI and EcoRI sites of the vector pGEX-5x-3. Point mutations in GST-E3L were generated by site-directed mutagenesis of pC2812.
Construction of knockdown-resistant human PKR (pSG5-PKR) was described previously (Rothenburg et al. 2009). pSG5-E3L-HA (pC4442) and pSG5-E3L-Δ80-HA (pC4443) were generated by PCR inserting a C-terminal HA-epitope and cloning between the EcoRI and BamHI sites of pSG5. Point mutants pSG5-E3L-N44D (pC4444), pSG5-E3L-Y48A (pC4445), pSG5-E3L-P63A (pC4446), pSG5-E3L-N44D,Y48A (pC4447), pSG5-E3L-N44D,Y48A,P63A (pC4448), pSG5-E3L-W66A (pC4449), and pSG5-E3L-K167A (pC4450) were generated by site-directed mutagenesis using pC4442 as a template.
Yeast strains
Construction of yeast strains H2544 (MATa ura3-52 leu2-3 leu2-112 trp1-Δ63 gcn2Δ <GAL-CYC1-PKR, LEU2>) and J110 (MATa ura3-52 leu2-3 leu2-112 trp1-Δ63 gcn2Δ <LEU2>) has been described previously (Romano et al. 1995; Seo et al. 2008). Strain H2766 (MATa ura3-52 leu2-3 leu2-112 trp1-Δ63 gcn2Δ gcd2Δ::hisG p[GCD2-K627T, TRP1]) was used for expression and purification of Flag- and His6-tagged PKR (Krishnamoorthy et al. 2001).
Immunoblot analysis
Yeast transformants were grown in SC-Ura medium (synthetic minimal medium with all amino acids) overnight at 30°C to saturation, diluted in fresh medium to OD600 ≈ 0.1, and grown at 30°C until OD600 ≈ 0.6. Cells were harvested and transferred to SCGal-Ura medium (SC-Ura except 10% galactose) for 2 h at 30°C to induce PKR and E3L expression. Cells were harvested, and whole cell extracts (WCEs) were prepared as described previously (Dey et al. 2005a). Five micrograms of WCEs was resolved by SDS-PAGE and subjected to immunoblot analysis using rabbit phosphospecific antibodies directed against phosphorylated Ser51 of eIF2α (BioSource International), rabbit polyclonal antiserum that recognizes yeast eIF2α irrespective of Ser51 phosphorylation, mouse monoclonal anti-HA (Roche), or mouse monoclonal anti-PKR antibodies (R&D Systems). Immune complexes were detected by enhanced chemiluminescence.
dsRNA-binding assay
Polyinosinic–polycytidylic acid bound to agarose [Poly(I:C) agarose, Pharmacia] was equilibrated in Buffer A (50 mM Tris-HCl at pH 8.0, 150 mM NaCl) and incubated with 2 µg of purified GST-E3L variants in 200 µL of Buffer A for 1 h at 4°C. Agarose beads were collected, washed five times with Buffer B (50 mM Tris-HCl at pH 8.0, 150 mM NaCl, 0.1% Triton X-100), resuspended in 30 µL of SDS sample buffer, and boiled for 5 min. Proteins were resolved by 10% SDS-PAGE, transferred to nitrocellulose membranes, and immunoblotted with anti-GST antibody (Santa Cruz Biotechnology). Immune complexes were detected by enhanced chemiluminescence.
Recombinant protein expression and purification
Flag- and His6-tagged PKR was expressed in yeast strain H2766. Yeast transformants were grown to saturation in 5 mL of SC-Ura medium at 30°C. Cells were harvested, transferred to 50 mL of SGR medium (SC medium containing 10% galactose and 2% raffinose), and incubated for 48 h at 30°C. Cells were then harvested, washed with ice-cold water, and resuspended in Flag Binding Buffer (20 mM Tris-HCl at pH 7.5, 500 mM NaCl, 0.1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride [PMSF], 10 mM NaF, 50 mM β-glycerophosphate, 125 μM Na3VO4, and complete protease inhibitor mixture [Roche]). Cells were broken by high speed mixing with glass beads on a vortex. WCEs were mixed with 100 μL of anti-Flag-M2 affinity gel (50% slurry; Sigma-Aldrich) pre-equilibrated with Flag Binding Buffer, and incubated with gentle rocking for 1 h at 4°C. Immune complexes were collected by centrifugation and washed three times with Flag Binding Buffer and once with 1× Kinase Buffer (20 mM Tris-HCl at pH 8.0, 50 mM KCl, 25 mM MgCl2, and 1 μM PMSF). Bound proteins were eluted with 25 μL Flag-peptide (500 μg/mL), and individual aliquots were used for immunoblot analysis and for protein kinase assays with recombinant yeast eIF2α as described previously (Dey et al. 2005a).
GST fusion proteins and His6-eIF2α1–200 were expressed in E. coli strain BL21(DE3) and purified using glutathione Sepharose 4B and Ni2+-NTA resin, respectively, according to the manufacturer's protocol.
In vitro kinase assay
Purified Flag-His6-PKR, GST-E3L fusion proteins, and His6-eIF2α were incubated with 0.5 mM ATP for 10 min at room temperature in Kinase Buffer (20 mM Tris-HCl at pH 8.0, 50 mM KCl, 25 mM MgCl2, and 1 mM PMSF). Reaction products were separated by SDS-PAGE, and eIF2α phosphorylation was monitored by immunoblot analysis using phospho-specific and total eIF2α antibodies.
Mammalian cell luciferase assay and immunoblot analysis
HeLa PKRkd cells (Zhang and Samuel 2007) were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (v/v) fetal bovine serum and 1 μg/mL puromycin (Sigma-Aldrich) at 37°C in 5% CO2. For luciferase assays, 3 × 104 cells were seeded in a 24-well plate, incubated for 24 h, and then transfected at 70%–80% confluence with 2 μL of GenJet (SignaGen Laboratories), 0.05 μg of pGL3 Promoter luciferase plasmid (Promega), 0.1 μg of PKR expression vector, and varying amounts of variola E3L plasmids using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's instructions. Transfections were performed in triplicate, and cells were harvested 40 h post-transfection using passive lysis buffer (Promega). Luciferase activity was determined using the Luciferase Assay System (Promega) and a luminometer (Centro XS Microplate Reader LB 960).
For Western blot analysis, 1 or 5 × 105 cells were seeded into 12-well or six-well plates, respectively, a day before transfection. Cells were cotransfected with GenJet reagent (SignaGen Laboratories), PKR expression vector, and E3L or mutant expression vectors. At 24 h post-transfection, WCEs were obtained by lysis in 50 mM Tris (pH 8.0), 150 mM NaCl, 1% Triton X-100, 100 mM sodium fluoride, 50 mM β-glycerophosphate, and 10 mM sodium pyrophosphate. Six micrograms of WCEs was subjected to SDS-PAGE, and immunoblot analysis was performed using monoclonal anti-HA (Roche) or anti-PKR (R&D Systems) antibodies, or with polyclonal antibodies against total eIF2α (Santa Cruz Biotechnology) or rabbit phosphospecific antibodies directed against Ser51 in eIF2α. Immune complexes were detected by enhanced chemiluminescence.
Z-DNA binding assay
The synthetic oligomer (5′-CG[5Br-dC]GCGCG[5Br-dC]GCGCG[5Br-dC]GCGCG[5Br-dC]GCGCG[5Br-dC]GCGCG[5Br-dC]GCGCG[5Br-dC]G), which is stable in the left-handed Z-DNA conformation (Schwartz et al. 1999; Kim et al. 2006), was annealed (95°C for 2 min, ramp-cooled to 25°C over a period of 45 min) and 5′-end-labeled using polynucleotide kinase and [γ-32P]ATP. The labeled DNA was incubated with GST-E3L proteins in Binding Buffer (10 mM Tris-HCl at pH 7.5, 10 mM MgCl2, 20 mM NaCl, 5 mM DTT, 0.1 mg/mL BSA, and 0.5 µg/µL calf thymus DNA) for 10 min in a final volume of 15 µL. The mixture was separated on a 6% TBE–Polyacrylamide Gel at 10 V/cm using 0.5× TBE as running buffer, and protein–DNA complexes were detected by autoradiography. Synthetic annealed B-DNA (TCCTTCTGTAGTTGGTCGCCCGGTTCATCTCGGTACTAT) was used as a competitor.
E3L random mutagenesis
An SmaI restriction site was engineered between amino acid residues 80 and 81 of the yeast E3L expression vector pC2492. Error-prone PCR (GeneMorph II Random Mutagenesis Kit, Agilent Technologies, Inc.) was used to amplify the SacI–SmaI fragment encoding E3L residues 1–80 under low-stringency conditions (one or two mutations per cycle), and the products were subcloned to pC2492. Sequencing of randomly selected clones from the mutagenesis library revealed that 70% of the plasmids contained mutations. The mutant library was transformed in the PKR strain H2544, and about 2000 independent yeast transformants were patched on glucose medium and then replica-printed to galactose medium to induce E3L and PKR expression. The E3L plasmid was isolated from the seven transformants that failed to grow on galactose medium and retested in H2544. Plasmids passing the retest were sequenced, and then E3L single mutants were generated by site-directed mutagenesis of pC2492 and tested in H2544.
ACKNOWLEDGMENTS
We thank members of the Dever and Hinnebusch laboratories for helpful discussions. This work was supported by the Intramural Research Program of the National Institutes of Health, NICHD.
REFERENCES
- Barber GN, Wambach M, Wong ML, Dever TE, Hinnebusch AG, Katze MG 1993. Translational regulation by the interferon-induced double-stranded-RNA-activated 68-kDa protein kinase. Proc Natl Acad Sci 90: 4621–4625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beattie E, Denzler KL, Tartaglia J, Perkus ME, Paoletti E, Jacobs BL 1995. Reversal of the interferon-sensitive phenotype of a vaccinia virus lacking E3L by expression of the reovirus S4 gene. J Virol 69: 499–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierle CJ, Semmens KM, Geballe AP 2013. Double-stranded RNA binding by the human cytomegalovirus PKR antagonist TRS1. Virology 442: 28–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt TA, Jacobs BL 2001. Both carboxy- and amino-terminal domains of the vaccinia virus interferon resistance gene, E3L, are required for pathogenesis in a mouse model. J Virol 75: 850–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bratke KA, McLysaght A, Rothenburg S 2013. A survey of host range genes in poxvirus genomes. Infect Genet Evol 14: 406–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao H, Dai P, Wang W, Li H, Yuan J, Wang F, Fang CM, Pitha PM, Liu J, Condit RC, et al. 2012. Innate immune response of human plasmacytoid dendritic cells to poxvirus infection is subverted by vaccinia E3 via its Z-DNA/RNA binding domain. PLoS ONE 7: e36823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll K, Elroy-Stein O, Moss B, Jagus R 1993. Recombinant vaccinia virus K3L gene product prevents activation of double-stranded RNA-dependent, initiation factor 2α-specific protein kinase. J Biol Chem 268: 12837–12842 [PubMed] [Google Scholar]
- Chang HW, Jacobs BL 1993. Identification of a conserved motif that is necessary for binding of the vaccinia virus E3L gene products to double-stranded RNA. Virology 194: 537–547 [DOI] [PubMed] [Google Scholar]
- Chang HW, Watson JC, Jacobs BL 1992. The E3L gene of vaccinia virus encodes an inhibitor of the interferon-induced, double-stranded RNA-dependent protein kinase. Proc Natl Acad Sci 89: 4825–4829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HW, Uribe LH, Jacobs BL 1995. Rescue of vaccinia virus lacking the E3L gene by mutants of E3L. J Virol 69: 6605–6608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong KL, Feng L, Schappert K, Meurs E, Donahue TF, Friesen JD, Hovanessian AG, Williams BR 1992. Human p68 kinase exhibits growth suppression in yeast and homology to the translational regulator GCN2. EMBO J 11: 1553–1562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai P, Cao H, Merghoub T, Avogadri F, Wang W, Parikh T, Fang CM, Pitha PM, Fitzgerald KA, Rahman MM, et al. 2011. Myxoma virus induces type I interferon production in murine plasmacytoid dendritic cells via a TLR9/MyD88-, IRF5/IRF7-, and IFNAR-dependent pathway. J Virol 85: 10814–10825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dar AC, Sicheri F 2002. X-ray crystal structure and functional analysis of vaccinia virus K3L reveals molecular determinants for PKR subversion and substrate recognition. Mol Cell 10: 295–305 [DOI] [PubMed] [Google Scholar]
- Dar AC, Dever TE, Sicheri F 2005. Higher-order substrate recognition of eIF2α by the RNA-dependent protein kinase PKR. Cell 122: 887–900 [DOI] [PubMed] [Google Scholar]
- Davies MV, Elroy-Stein O, Jagus R, Moss B, Kaufman RJ 1992. The vaccinia virus K3L gene product potentiates translation by inhibiting double-stranded-RNA-activated protein kinase and phosphorylation of the α subunit of eukaryotic initiation factor 2. J Virol 66: 1943–1950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies MV, Chang HW, Jacobs BL, Kaufman RJ 1993. The E3L and K3L vaccinia virus gene products stimulate translation through inhibition of the double-stranded RNA-dependent protein kinase by different mechanisms. J Virol 67: 1688–1692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rosa M, Zacarias S, Athanasiadis A 2013. Structural basis for Z-DNA binding and stabilization by the zebrafish Z-DNA dependent protein kinase PKZ. Nucleic Acids Res 41: 9924–9933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Dai P, Parikh T, Cao H, Bhoj V, Sun Q, Chen Z, Merghoub T, Houghton A, Shuman S 2008. Vaccinia virus subverts a mitochondrial antiviral signaling protein-dependent innate immune response in keratinocytes through its double-stranded RNA binding protein, E3. J Virol 82: 10735–10746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dever TE, Chen JJ, Barber GN, Cigan AM, Feng L, Donahue TF, London IM, Katze MG, Hinnebusch AG 1993. Mammalian eukaryotic initiation factor 2α kinases functionally substitute for GCN2 protein kinase in the GCN4 translational control mechanism of yeast. Proc Natl Acad Sci 90: 4616–4620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey M, Cao C, Dar AC, Tamura T, Ozato K, Sicheri F, Dever TE 2005a. Mechanistic link between PKR dimerization, autophosphorylation, and eIF2α substrate recognition. Cell 122: 901–913 [DOI] [PubMed] [Google Scholar]
- Dey M, Trieselmann B, Locke EG, Lu J, Cao C, Dar AC, Krishnamoorthy T, Dong J, Sicheri F, Dever TE 2005b. PKR and GCN2 kinases and guanine nucleotide exchange factor eukaryotic translation initiation factor 2B (eIF2B) recognize overlapping surfaces on eIF2α. Mol Cell Biol 25: 3063–3075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elde NC, Child SJ, Geballe AP, Malik HS 2009. Protein kinase R reveals an evolutionary model for defeating viral mimicry. Nature 457: 485–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia MA, Meurs EF, Esteban M 2007. The dsRNA protein kinase PKR: Virus and cell control. Biochimie 89: 799–811 [DOI] [PubMed] [Google Scholar]
- Ha SC, Lokanath NK, Van Quyen D, Wu CA, Lowenhaupt K, Rich A, Kim YG, Kim KK 2004. A poxvirus protein forms a complex with left-handed Z-DNA: Crystal structure of a Yatapoxvirus Zα bound to DNA. Proc Natl Acad Sci 101: 14367–14372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha SC, Kim D, Hwang HY, Rich A, Kim YG, Kim KK 2008. The crystal structure of the second Z-DNA binding domain of human DAI (ZBP1) in complex with Z-DNA reveals an unusual binding mode to Z-DNA. Proc Natl Acad Sci 105: 20671–20676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho CK, Shuman S 1996. Mutational analysis of the vaccinia virus E3 protein defines amino acid residues involved in E3 binding to double-stranded RNA. J Virol 70: 2611–2614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagus R, Gray MM 1994. Proteins that interact with PKR. Biochimie 76: 779–791 [DOI] [PubMed] [Google Scholar]
- Kahmann JD, Wecking DA, Putter V, Lowenhaupt K, Kim YG, Schmieder P, Oschkinat H, Rich A, Schade M 2004. The solution structure of the N-terminal domain of E3L shows a tyrosine conformation that may explain its reduced affinity to Z-DNA in vitro. Proc Natl Acad Sci 101: 2712–2717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawagishi-Kobayashi M, Silverman JB, Ung TL, Dever TE 1997. Regulation of the protein kinase PKR by the vaccinia virus pseudosubstrate inhibitor K3L is dependent on residues conserved between the K3L protein and the PKR substrate eIF2α. Mol Cell Biol 17: 4146–4158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawagishi-Kobayashi M, Cao C, Lu J, Ozato K, Dever TE 2000. Pseudosubstrate inhibition of protein kinase PKR by swine pox virus C8L gene product. Virology 276: 424–434 [DOI] [PubMed] [Google Scholar]
- Kim YG, Muralinath M, Brandt T, Pearcy M, Hauns K, Lowenhaupt K, Jacobs BL, Rich A 2003. A role for Z-DNA binding in vaccinia virus pathogenesis. Proc Natl Acad Sci 100: 6974–6979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YG, Park HJ, Kim KK, Lowenhaupt K, Rich A 2006. A peptide with alternating lysines can act as a highly specific Z-DNA binding domain. Nucleic Acids Res 34: 4937–4942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitajewski J, Schneider RJ, Safer B, Munemitsu SM, Samuel CE, Thimmappaya B, Shenk T 1986. Adenovirus VAI RNA antagonizes the antiviral action of interferon by preventing activation of the interferon-induced eIF-2α kinase. Cell 45: 195–200 [DOI] [PubMed] [Google Scholar]
- Krishnamoorthy T, Pavitt GD, Zhang F, Dever TE, Hinnebusch AG 2001. Tight binding of the phosphorylated α subunit of initiation factor 2 (eIF2α) to the regulatory subunits of guanine nucleotide exchange factor eIF2B is required for inhibition of translation initiation. Mol Cell Biol 21: 5018–5030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langland JO, Pettiford S, Jiang B, Jacobs BL 1994. Products of the porcine group C rotavirus NSP3 gene bind specifically to double-stranded RNA and inhibit activation of the interferon-induced protein kinase PKR. J Virol 68: 3821–3829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marq JB, Hausmann S, Luban J, Kolakofsky D, Garcin D 2009. The double-stranded RNA binding domain of the vaccinia virus E3L protein inhibits both RNA- and DNA-induced activation of interferon beta. J Biol Chem 284: 25471–25478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews MB, Shenk T 1991. Adenovirus virus-associated RNA and translation control. J Virol 65: 5657–5662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minks MA, West DK, Benvin S, Baglioni C 1979. Structural requirements of double-stranded RNA for the activation of 2′,5′-oligo(A) polymerase and protein kinase of interferon-treated HeLa cells. J Biol Chem 254: 10180–10183 [PubMed] [Google Scholar]
- Myskiw C, Arsenio J, Hammett C, van Bruggen R, Deschambault Y, Beausoleil N, Babiuk S, Cao J 2011. Comparative analysis of poxvirus orthologues of the vaccinia virus E3 protein: Modulation of protein kinase R activity, cytokine responses, and virus pathogenicity. J Virol 85: 12280–12291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Davies MV, Langland JO, Chang HW, Nam YS, Tartaglia J, Paoletti E, Jacobs BL, Kaufman RJ, Venkatesan S 1994. TAR RNA-binding protein is an inhibitor of the interferon-induced protein kinase PKR. Proc Natl Acad Sci 91: 4713–4717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei J, Kim BH, Grishin NV 2008. PROMALS3D: A tool for multiple protein sequence and structure alignments. Nucleic Acids Res 36: 2295–2300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice AP, Kerr IM 1984. Interferon-mediated, double-stranded RNA-dependent protein kinase is inhibited in extracts from vaccinia virus-infected cells. J Virol 50: 229–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice AD, Turner PC, Embury JE, Moldawer LL, Baker HV, Moyer RW 2011. Roles of vaccinia virus genes E3L and K3L and host genes PKR and RNase L during intratracheal infection of C57BL/6 mice. J Virol 85: 550–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano PR, Green SR, Barber GN, Mathews MB, Hinnebusch AG 1995. Structural requirements for double-stranded RNA binding, dimerization, and activation of the human eIF-2α kinase DAI in Saccharomyces cerevisiae. Mol Cell Biol 15: 365–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano PR, Zhang F, Tan SL, Garcia-Barrio MT, Katze MG, Dever TE, Hinnebusch AG 1998. Inhibition of double-stranded RNA-dependent protein kinase PKR by vaccinia virus E3: Role of complex formation and the E3 N-terminal domain. Mol Cell Biol 18: 7304–7316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenburg S, Deigendesch N, Dittmar K, Koch-Nolte F, Haag F, Lowenhaupt K, Rich A 2005. A PKR-like eukaryotic initiation factor 2α kinase from zebrafish contains Z-DNA binding domains instead of dsRNA binding domains. Proc Natl Acad Sci 102: 1602–1607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenburg S, Seo EJ, Gibbs JS, Dever TE, Dittmar K 2009. Rapid evolution of protein kinase PKR alters sensitivity to viral inhibitors. Nat Struct Mol Biol 16: 63–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenburg S, Chinchar VG, Dever TE 2011. Characterization of a ranavirus inhibitor of the antiviral protein kinase PKR. BMC Microbiol 11: 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryter JM, Schultz SC 1998. Molecular basis of double-stranded RNA-protein interactions: Structure of a dsRNA-binding domain complexed with dsRNA. EMBO J 17: 7505–7513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders LR, Barber GN 2003. The dsRNA binding protein family: Critical roles, diverse cellular functions. FASEB J 17: 961–983 [DOI] [PubMed] [Google Scholar]
- Schade M, Turner CJ, Lowenhaupt K, Rich A, Herbert A 1999. Structure–function analysis of the Z-DNA-binding domain Zα of dsRNA adenosine deaminase type I reveals similarity to the (α + β) family of helix–turn–helix proteins. EMBO J 18: 470–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz T, Rould MA, Lowenhaupt K, Herbert A, Rich A 1999. Crystal structure of the Zα domain of the human editing enzyme ADAR1 bound to left-handed Z-DNA. Science 284: 1841–1845 [DOI] [PubMed] [Google Scholar]
- Schwartz T, Behlke J, Lowenhaupt K, Heinemann U, Rich A 2001. Structure of the DLM-1–Z-DNA complex reveals a conserved family of Z-DNA-binding proteins. Nat Struct Biol 8: 761–765 [DOI] [PubMed] [Google Scholar]
- Seo EJ, Liu F, Kawagishi-Kobayashi M, Ung TL, Cao C, Dar AC, Sicheri F, Dever TE 2008. Protein kinase PKR mutants resistant to the poxvirus pseudosubstrate K3L protein. Proc Natl Acad Sci 105: 16894–16899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp TV, Schwemmle M, Jeffrey I, Laing K, Mellor H, Proud CG, Hilse K, Clemens MJ 1993. Comparative analysis of the regulation of the interferon-inducible protein kinase PKR by Epstein-Barr virus RNAs EBER-1 and EBER-2 and adenovirus VAI RNA. Nucleic Acids Res 21: 4483–4490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp TV, Moonan F, Romashko A, Joshi B, Barber GN, Jagus R 1998. The vaccinia virus E3L gene product interacts with both the regulatory and the substrate binding regions of PKR: Implications for PKR autoregulation. Virology 250: 302–315 [DOI] [PubMed] [Google Scholar]
- Shors T, Jacobs BL 1997. Complementation of deletion of the vaccinia virus E3L gene by the Escherichia coli RNase III gene. Virology 227: 77–87 [DOI] [PubMed] [Google Scholar]
- Shors T, Kibler KV, Perkins KB, Seidler-Wulff R, Banaszak MP, Jacobs BL 1997. Complementation of vaccinia virus deleted of the E3L gene by mutants of E3L. Virology 239: 269–276 [DOI] [PubMed] [Google Scholar]
- St Johnston D, Brown NH, Gall JG, Jantsch M 1992. A conserved double-stranded RNA-binding domain. Proc Natl Acad Sci 89: 10979–10983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tome AR, Kus K, Correia S, Paulo LM, Zacarias S, de Rosa M, Figueiredo D, Parkhouse RM, Athanasiadis A 2013. Crystal structure of a poxvirus-like Zα domain from cyprinid herpesvirus 3. J Virol 87: 3998–4004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez de Aldana CR, Dever TE, Hinnebusch AG 1993. Mutations in the α subunit of eukaryotic translation initiation factor 2 (eIF-2α) that overcome the inhibitory effect of eIF-2α phosphorylation on translation initiation. Proc Natl Acad Sci 90: 7215–7219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson JC, Chang HW, Jacobs BL 1991. Characterization of a vaccinia virus-encoded double-stranded RNA-binding protein that may be involved in inhibition of the double-stranded RNA-dependent protein kinase. Virology 185: 206–216 [DOI] [PubMed] [Google Scholar]
- Whitaker-Dowling P, Youngner JS 1984. Characterization of a specific kinase inhibitory factor produced by vaccinia virus which inhibits the interferon-induced protein kinase. Virology 137: 171–181 [DOI] [PubMed] [Google Scholar]
- White SD, Jacobs BL 2012. The amino terminus of the vaccinia virus E3 protein is necessary to inhibit the interferon response. J Virol 86: 5895–5904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y, Condit RC, Vijaysri S, Jacobs B, Williams BR, Silverman RH 2002. Blockade of interferon induction and action by the E3L double-stranded RNA binding proteins of vaccinia virus. J Virol 76: 5251–5259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Z, Shatkin AJ 1997. Double-stranded RNA-dependent protein kinase (PKR) is regulated by reovirus structural proteins. Virology 234: 364–371 [DOI] [PubMed] [Google Scholar]
- Yuwen H, Cox JH, Yewdell JW, Bennink JR, Moss B 1993. Nuclear localization of a double-stranded RNA-binding protein encoded by the vaccinia virus E3L gene. Virology 195: 732–744 [DOI] [PubMed] [Google Scholar]
- Zhang P, Samuel CE 2007. Protein kinase PKR plays a stimulus- and virus-dependent role in apoptotic death and virus multiplication in human cells. J Virol 81: 8192–8200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Lockshin C, Herbert A, Winter E, Rich A 1992. Zuotin, a putative Z-DNA binding protein in Saccharomyces cerevisiae. EMBO J 11: 3787–3796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Romano PR, Nagamura-Inoue T, Tian B, Dever TE, Mathews MB, Ozato K, Hinnebusch AG 2001. Binding of double-stranded RNA to protein kinase PKR is required for dimerization and promotes critical autophosphorylation events in the activation loop. J Biol Chem 276: 24946–24958 [DOI] [PubMed] [Google Scholar]
- Zhang P, Jacobs BL, Samuel CE 2008. Loss of protein kinase PKR expression in human HeLa cells complements the vaccinia virus E3L deletion mutant phenotype by restoration of viral protein synthesis. J Virol 82: 840–848 [DOI] [PMC free article] [PubMed] [Google Scholar]