

Abstract
Advances in molecular biology have significantly increased the understanding of the biology of different diseases. However, these discoveries have not yet been fully translated into improved treatments for patients with diseases such as cancers. One of the factors limiting the translation of knowledge from preclinical studies to the clinic has been the limitations of in vivo diseases models. In this brief review, we will discuss the advantages and disadvantages of rodent models that have been developed to simulate human pathologies, focusing in models that employ xenografts and genetic modification. Within the framework of genetically engineered mouse (GEM) models, we will review some of the current genetic strategies for modeling diseases in the mouse and the preclinical studies that have already been undertaken. We will also discuss how recent improvements in imaging technologies may increase the information derived from using these GEMs during early assessments of potential therapeutic pathways. Furthermore, it is interesting to note that one of the values of using a mouse model is the very rapid turnover rate of the animal, going through the process of birth to death in a very short timeframe relative to that of larger mammalian species.
KEY WORDS: Cancer, genetically modified animals, human diseases, rodents, transgenic
Animal experiments remain essential to understand the fundamental mechanisms underlying the onset of malignancies and to discover improved methods to prevent, diagnose and treat diseases. The current excellence of animal care standards are consistent with the experimental conditions needed when conducting cancer research.[1]
For scientists to understand how diseases develop and spread throughout the body and to discover new and more effective ways to diagnose and treat these diseases, it is necessary to conduct live animal research. Although animal studies (over 95% of which are conducted in mice) are essential for gaining the necessary understanding of complex disease processes, they should be performed only after exhausting information that can be derived from other tools such as disease cell lines. Furthermore, animal studies are also required by regulatory authorities before any trials of new drugs can be tested in humans. Indeed, preclinical animal studies are required for toxicological assessments and to support human drug approvals. Humane considerations are emphasized in an effort to minimize adverse effects. Nevertheless, since we cannot replace all animal experiments in the immediate future, the highest standards of welfare are upheld.[1]
In terms of oncology drug products, the preclinical studies were previously limited to the transplantation of murine or human cancers into mice. Most recently, some investigators have explored alternatives to these transplantation models, such as the study of animals that naturally develop cancers which are similar to that in humans. An example of this is seen in the parallel assessments underway for canine and human osteosarcoma.[2] Alternatively, animals can be genetically engineered to develop cancers with features relevant to the human disease.[3] Experiments with these animals produce valuable information about the role of specific genes in biological processes and diseases.
The first transgenic mouse was created in 1974 at the Massachusetts Institute of Technology. It was created through an in vitro micro-injection of Simian virus 40 into mouse blastocysts and early embryonic infection with retrovirus. The technology to create transgenic animals broke new ground in the scientific community and enabled scientists to seek new ways of treating diseases and developing new drugs. The ability to introduce new genetic information into the germ line of complex organisms has completely changed and enhanced the study of all aspects of biologic processes. Transgenic mouse models in toxicology have primarily been used to screen drugs for carcinogenicity and to understand the mechanisms of toxicity. These mouse models can reliably predict the carcinogenic potential of compounds and significantly reduce the risk of using these drugs in clinics to treat human diseases.
Use of short-term experiments on transgenic mice in combination with 2-year chronic studies on rats could increase the overall accuracy of detecting carcinogens and non-carcinogens. Testing new drugs using different species also reduces false results. Additional advantages of using transgenic assays include reduced duration of studies, conservative use of animals and lower cost relative to a traditional 2-year rodent chronic toxicity study (http://www.ruro.com/blog/3752, viewed 27th January 2013).
Xenografts
Xenografts (xenos-from the Greek meaning “foreign”), is a graft obtained from a member of one species and transplanted to a member of another species, genus, or family. The transplantation can consist of living cells, tissues or organs from one species to another. Xenografts are used to answer key questions in the field of cancer research when it is necessary to rely upon the use of animal model systems that closely resemble tumor progression in the human patient. Human xenografts growing in immunodeficient mice are a well-established and useful model for studying human tumor biology in a system that better mimics the primary tumor as compared to cells grown in vitro. Often times, xenograft studies use highly passaged cell lines that have been genetically modified and artificially cultured, leading to a clonal selection that may not be observed in patients. Ideal primary tumor tissue xenografts result from patient-derived explants, established as models, at low passage numbers (<10 passes removed from patient). Furthermore, these cell lines are not grown in plastic or propagated as cell cultures.[4,5] Establishing xenograft tumor models from patient-derived tumor tissue at low passage is believed to conserve original tumor characteristics such as heterogeneous histology, clinical biomolecular signature, malignant phenotypes and genotypes, tumor architecture and tumor vasculature. Based on this prevalent hypothesis, primary tumor xenografts are believed to offer relevant predictive insights into clinical outcomes when evaluating the efficacy of novel cancer therapies.
Subcutaneous xenograft models are the standard technique for determining the efficacy of new cancer agents in vivo and the most commonly performed. It is also the fastest and lowest cost model. In these subcutaneous models, the tumor is grown under the skin of a nude or severe combined immunodeficiency (SCID) mouse and the tumor growth is monitored by measuring the size of the tumor with calipers. In xenograft studies, cultured cells are generally injected subcutaneously into immunodeficient mice, which are then treated with the compound of interest for 2-6 weeks during which time subcutaneous tumors develop. The study is “positive” if the compound of interest reduces the rate of growth of new tumors.
A large variety of human and murine cell lines derived from both, solid tumors or leukemias, covering a wide range of tumor geno-and phenotypes, have been adapted to grow in a murine host and thus allow testing of a compound in the appropriate tumor model [Table 1].
Table 1.
Subcutaneous in vivo tumor cell lines models
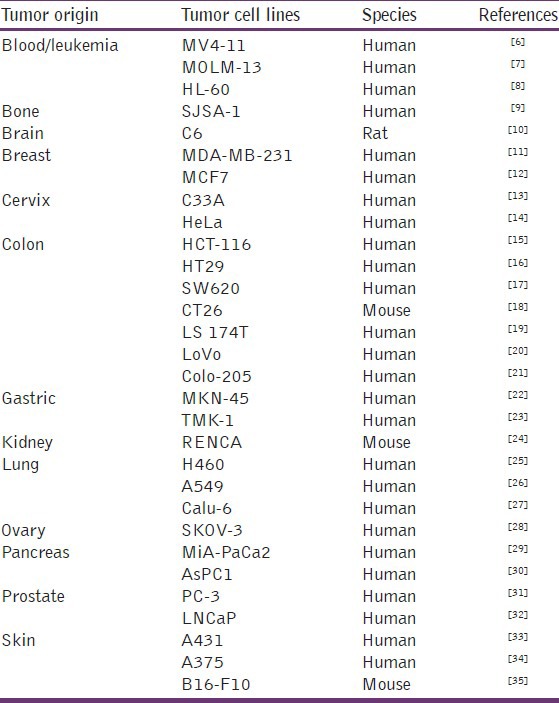
The earliest xenograft models in which human tumor cells were grown in immunosuppressed mice involved subcutaneous implantation of human cell lines. Subcutaneous xenograft models have been popular because they are easy to establish, easy to manage and lend themselves to ready quantitation of the tumor burden. More recently, orthotopic xenograft models, in which the tumor cells are implanted in the tumor site of origin, have been used with greater frequency in animal studies of cancers. Orthotopic xenograft models are advantageous for their ability to mimic local tumor growth and recapitulate the pathways of metastasis seen in human cancers. In addition, recent innovations in cell labeling techniques and small-animal imaging have enabled investigators to monitor the metastatic process and quantitate the growth and spread of orthotopically implanted tumors.[36]
Progress in oncology drug development has been hampered by a lack of preclinical models that reliably predict clinical activity of novel compounds in cancer patients. In an effort to address these shortcomings, there has been a recent increase in the use of patient-derived tumor xenografts (PDTX), engrafted into immune-compromised rodents such as athymic nude or non-obese diabetic/SCID mice, for use as preclinical models. Numerous tumor-specific PDTX models have been established and importantly, they are biologically stable when passaged in mice in terms of global gene-expression patterns, mutational status, metastatic potential, drug responsiveness and tumor architecture. These characteristics might provide significant improvements over standard cell-line xenograft models.[5]
Genetically Engineered Animal Models
Because the genetics and histology of xenografts do not recapitulate the genetics and histology of human tumors, genetically engineered animal models were developed.
The use of genetically modified mice for carcinogenicity evaluation began more than 20 years ago, when researchers found that different strains of genetically engineered mice demonstrated that cancer incidence is increased and tumor latency is decreased in mice whose germ line, the Ha-ras oncogene, has been inserted. Previously, the selection of mouse oncology models was based simply on availability of a mouse strain and a known compatible tumor. This frequently resulted in the use of tumor models that while long on history were short on homology and quality control. For these reasons, preclinical efficacy testing for anti-tumor therapies should progress through a series of models of increasing sophistication that includes incorporation of genetically engineered animals and orthotopic and combination therapy models.
Genetically modified animals are organisms in which specific genes have been altered (added or ablated) to create models for human and animal diseases. A transgenic animal is defined as an animal that carries one or more foreign genes, deliberately introduced through insertion into the animal's genome. The foreign gene is constructed using recombinant deoxyribonucleic acid (DNA) technologies. The introduction of a gene can also generate therapeutic medicinal products. Standard genetically modified animals include laboratory flies, fish, worms, rodents and (for agricultural or production purposes) pigs, sheep and cows. In no case is genetic modification of man implied.[37,38]
In a modern GEM models, oncogenes are activated and/or tumor-suppressor genes (TSGs) are inactivated somatically, generally through temporally controlled and tissue-specific expression of CRE recombinase (tyrosine recombinase enzyme derived from the P1 Bacteriophage). Animals then develop tumors in the tissue of interest (in this case the lung). Tumor-bearing genetically engineered mice are then treated with the compound of interest and serially assessed for response.[39]
The benefits of engineered animal models are numerous. Examples include[40,41]
Development and testing of safe and effective products for human application (e.g., human antibodies).
The production of recombinant products (anti-coagulant; therapeutic antibodies).
A means to study disease mechanisms in a complex organism (diabetes).
Understanding the mechanistic causes and pathways underlying human disease, to permit the development of efficient and targeted treatments (e.g., leukemia, hypertension and obesity).
The major applications for genetically modified animals in agriculture are
To generate animal with desired breeding traits (e.g., lower phosphorous in the dung).
To induce resistance against disease (e.g., fish farming) (Basel Declaration, 2010).
One of the goals in using transgenic animals is that it allows for a reduction in the use of larger animals. The use of GEMs also provides an opportunity to improve our understanding of the mechanisms of action for potential therapeutic compounds. At present, the European Commission consider that mice appear to be the most common genetically engineered animal models to study new drug compounds for different diseases for the following reasons[42]
The mouse is the most common model organism for preclinical studies even though it has not proven particularly reliable at predicting the outcome of studies in humans.
Mice have many advantages over other model organisms: Their genome is similar to the human genome (99%), a good genetic/molecular toolbox is available and the animal's small size facilitates large scale/high throughput studies making it a cost-efficient model. Therefore, it's potential for making medical research and in particular drug development more efficient could be increased by solving a range of identified bottlenecks.
Mouse models have been successfully used to validate drug targets and to determine efficacious and safe dosage schemes for combination treatments in humans. These cases have one factor in common: They do not aim to fully model a disease or disease mechanisms, but rather set out to obtain specific functional information.
Genetically modified mice need to be validated, reproducible, robust and cost-effective to be considered optimal by the pharmaceutical industry.
Transgenic humanized mice could provide good preclinical screening and safety testing models for use in lead identification and optimization.
The large-scale phenotyping of genetically engineered mice can provide valuable information on gene function which is relevant for human health and disease.
The use of mice in clinical studies has proven effective in a number of cases. Dosage regimes of new treatments or treatment combinations can rapidly be optimized by co-clinical trials with mice, allowing fast application in humans with greater patient safety. This provides increasing opportunities in particular for the rapid development of treatments for very rare diseases where low patient numbers otherwise hamper the creation of clinical trials.
One of the key bottlenecks that needs to be solved concerns the aggressive patenting strategy (including the patenting of mouse genes and broad based methods patents) and overly restrictive licensing terms in material transfer agreements, which hamper the construction, sharing, use and proper exploitation of mouse models. Representatives from the European Medicines Agency and the Food and Drug Administration confirmed that data stemming from mouse models will be taken into account wherever the relevance has been clearly proven within the given context.
The efficient development and use of these mouse models necessitates the collaboration of experts within academia, clinicians and industry. In this regard, there is a need for specialist training programs that provide venue whereby insights can be shared from mouse pathologists, human pathologists and clinicians in order to better define the opportunities and limitations associated with use of these mouse models.
The Use of Mouse Models to Support Therapeutic Drug Development
Animal models are being increasingly used in the study of disease progression and for safety assessments of new compounds. They are a powerful tool for developing a more detailed understanding of the role specific genes play in biological pathways. At the present time, the ability to introduce new genetic information into the germ line of complex organisms has completely changed and enhanced the study of all aspects of biologic processes.
Cancer
Mouse models of human cancer are valuable tools for cancer research. Although xenografts and GEMs possess limitations as well as advantages, each system plays a significant role in preclinical testing.
Tumor xenografts are easy to use, relatively inexpensive and reproducible. The main drawback of xenografts is that the genetics and histology of the tumors are frequently not representative of the respective human tumor and thus far, these models have not been as predictive of therapeutic success as one would like.[43] The use of xenografts to design novel specific anti-tumor therapies for enhanced and targeted drug delivery such as anti-angiogenesis (inhibitors and enhanced permeation), immunotherapy (vaccines, monoclonal antibodies, toxin conjugates, prodrug activators, cytokine antagonists), small molecules (inhibitors of growth, matrix and adhesion), apoptosis (enhancers, inducers, proteasome inhibitors, reverse DNA methylation), anti-sense and gene therapy (TSGs) and cell cycle alterations (inhibitors) are not sufficient to support their use as the sole drug screen in an anti-tumor evaluation. Therefore, to support the development of cancer therapeutics, there needs to be a series of models used, including GEMs and orthotopic and combination therapy models.
In contrast to studies involving xenografts, GEMs are histologically and genetically accurate models of human cancer but have disadvantages of heterogeneity with regard to frequency, latency and growth. These disadvantages are reminiscent of the variable behavior of actual human tumors. Recently, these shortcomings have been partly overcome with the development of anatomic and molecular in vivo imaging techniques such as magnetic resonance imaging (MRI) and bioluminescence imaging. These new technologies will hopefully support the use of GEMs in preclinical trials and help determine if trials in GEMs are more predicative than xenografts of human responses.[43]
The selection of murine cancer models is often based simply on the availability of a mouse strain and on a known compatible tumor. Frequently this information results in use of tumor models long on history, but short on homology and quality control. Other factors need to be considered, including genetics, sex, immunological status, method and site of tumor implantation, technical competence and biological activity of the tumor, protocol sequence and timing and selection of endpoints. Each of these variables can influence the data derived from use of these tumor models.
It is frequently observed that murine tumor models error towards false positive therapeutic results, curing the cancer in mice but not in humans. This perceived inadequacy of classical transplantation models for the development of anti-tumor therapies is currently a pivotal factor in driving the development and use of GEMs. The underlying limitations of tumor models also serve to reinforce the need for careful attention to design (applying correct models to the question), conduct (using multiple models) and interpretation (recognizing limitations and applying stringent criteria to outcomes) of efficacy studies for tumor modulation. Thus, while animal models can provide a form of “high throughput” in the selection of potential drug gable candidates, the use of these results to predict human clinical outcomes is premature. Therefore, new strategies, techniques and continued improvements in stringency and consistency of criteria used for evaluating outcomes are necessary to ensure that tumor models in mice remain a useful tool for development of anticancer agents and devices.[44] Moreover, transitions from in vitro to preclinical and then to clinical testing for tumor modulation remain difficult with a low rate of clinical entry for most therapeutic classes. Increased understanding of mechanisms of neoplasia through macromolecular biology, genomics and bioinformatics is helping to address treatment bottlenecks. These bottlenecks include a lack of specificity, low efficacy, toxicity and drug resistance and the need to identify critical targets for clinical exploitation.
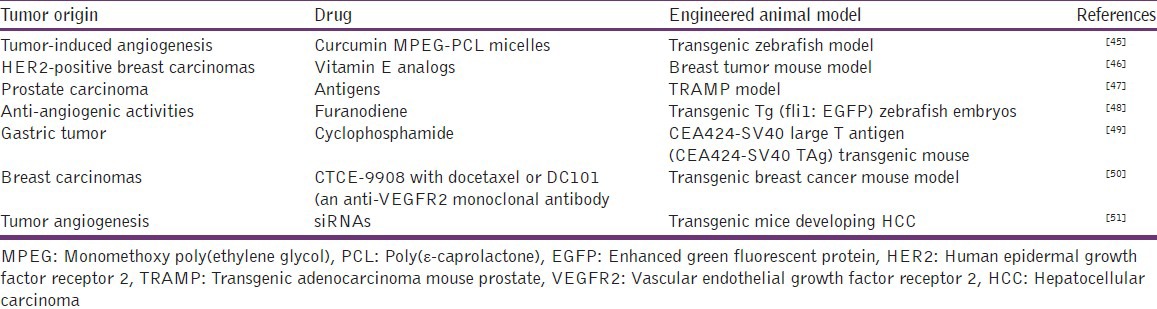
The numerous studies evaluating new antitumor drugs use engineered animal models [Table 2].
Table 2.
Engineered animal models for anti-tumor evaluations
Other applications
Intestinal inflammation
Rennick and Fort used interleukin (IL)-10-/-mice, which spontaneously develop this pathological condition that is characterized by discontinuous transmural lesions affecting the small and large intestine. This inflammatory response also includes the dysregulated production of proinflammatory cytokines.[52] The uncontrolled generation of interferon-gamma-producing CD41 T-cells (Th1 type) has been shown to play a causal role in the development of enterocolitis affecting these mutants. the study of IL-102/2 mice have been applied to an evaluation of the role of enteric organisms in triggering intestinal disease, the mediators responsible for initiating and maintaining intestinal disease, the role IL-10 plays in the generation and/or function of regulatory cells and the results of IL-10 therapy in experimental animal models of inflammatory bowel disease (IBD) and human patients with IBD.
Hepatic steatosis and/or insulin resistance
Although rodent models do not always perfectly reproduce the human pathology of non-alcoholic fatty liver disease (NAFLD), Postic and Girard concluded that the use of transgenic, knockout and knockdown mouse models have helped over the past years to better the understanding of the molecular determinants of NAFLD. Key enzymes of fatty acid synthesis, such as acetyl-CoA carboxylase, elongation of long-chain fatty acids family member 6, stearoyl-Coenzyme A desaturase 1 (SCD1), glycerol-3-phosphate acyltransferase, or diacylglycerol acyltransferase, have been shown, when knocked down, to reverse many of the metabolic defects associated with hepatic steatosis and/or insulin resistance, indicating that decreased TG synthesis in liver is a potential and interesting target for the treatment of NAFLD.[53] Therefore, a better understanding of the function and/or regulation of the transcription factors that control the activity of the enzymes modulating fatty acid synthesis in liver, namely carbohydrate response element binding protein, sterol regulatory element binding protein-1c and liver X receptors, may in the future help the development of potential therapeutic approaches for this disease
Patho-physiology of transglutaminase
The human TG family consists of a structural protein, protein 4.2, that lacks catalytic activity and eight zymogens/enzymes, designated factor XIII-A (FXIII-A) and TG1-7, that catalyze three types of posttranslational modification reactions: Transamidation, esterification and hydrolysis. These reactions are essential for biological processes such as blood coagulation, skin barrier formation and extracellular matrix assembly. However, they can also contribute to the pathophysiology of various inflammatory, autoimmune and degenerative conditions. Some members of the TG family, for example, TG2, can participate in biological processes through actions unrelated to transamidase catalytic activity. Iismaa et al.[54] in their study reviewed recent insights into the physiology and pathophysiology of TG family members that have come from studies of GEM models and/or inherited disorders. The review focused on FXIII-A, TG1, TG2, TG5 and protein 4.2, as mice deficient in TG3, TG4, TG6, or TG7 and they underlined the necessity to use these engineered animals to study these proteins linked to human disease.
Nicotine addiction
Recently, Jean-Pierre Changeux underlined the interest in using transgenic animals to study nicotine addiction and nicotinic receptors. Indeed, in an interesting review, he mentions that recent studies in mice involving deletion and replacement of nicotinic acetylcholine receptor subunits have begun to identify the molecular mechanisms underlying nicotine addiction and might offer new therapeutic strategies to treat this addiction.[55]
Arachidonic acid metabolism and central nervous system pathology
Bosetti used genetic mouse models to investigate the physiological and pathological roles of Arachidonic acid (AA) release and metabolism in brain.[56] She concluded that although data obtained in genetically altered mouse models should be interpreted with care because of possible compensatory changes, knockout and transgenic mice provide a useful approach to identify the roles of specific enzymes of the AA cascade in physiological and pathological models. Therefore, these genetic animal models combined with the use of conditional knockouts, in vivo ribonucleic acid interference and specific pharmacological inhibitors, may help to develop novel therapeutic strategies for diseases involving altered brain AA metabolism.
AA is freed from a phospholipid molecule by the enzyme phospholipase A2 (PLA2), which cleaves off the fatty acid, but can also be generated from diacylglycerol by diacylglycerol lipase. AA is generated for signaling purposes appears to be derived by the action of a phosphatidylcholine-specific cytosolic PLA2 (85 kDa), whereas inflammatory AA is generated by the action of a low molecular-weight secretory PLA2 (14-18 kDa). AA is a precursor in the production of eicosanoids: (i) The enzymes cyclooxygenase and peroxidase lead to prostaglandin H2, which in turn is used to produce the prostaglandins, prostacyclin and thromboxanes; (ii) the enzyme 5-lipoxygenase leads to 5-HPETE, which in turn is used to produce the leukotrienes; (iii) AA is also used in the biosynthesis of anandamide; (iiii) some AA is converted into hydroxyeicosatetraenoic acids and epoxyeicosatrienoic acids by epoxygenase. The production of these derivatives and their action in the body are collectively known as the “AA cascade”.
Ovarian endometrioid adenocarcinoma
Using the combination of an OEA GEM and GEM of OEA of molecular-imaging technologies, Wang et al. studied the activation of the AKT serine/threonine kinase in response to long-term cisplatin therapy.[57] The authors showed that the treatment of cells in culture and tumor-bearing animals with cisplatin resulted in activation of AKT, a key mediator of cell survival. On the basis of these results, they investigated the therapeutic use of AKT inhibition in combination with cisplatin, which resulted in enhanced and prolonged induction of apoptosis and in significantly improved tumor control when compared with either agent alone. They concluded that their results provide an impetus for clinical trials using combination therapy. To facilitate these trials, they also showed the use of diffusion-weighted MRI as an imaging biomarker for evaluation of therapeutic efficacy in OEA.
Pancreatic cancer
The lack of early detection and the use of ineffective interventions are major factors contributing to the poor prognosis and dismal survival rates of pancreatic cancer patients for more than 60 years. Detection of pancreatic cancer at an early stage might permit life-saving intervention. Clinical and preclinical diagnosis and evaluation of pancreatic cancers involve several imaging technologies MRI, positron emission tomography, computed tomography, ultrasound, bioluminescent imaging and single photon emission computed tomography. The advent of genetically engineered animal models that recapitulate the cellular and molecular pathology of human pancreatic intraepithelial neoplasia and pancreatic ductal adenocarcinoma (PDAC) has not yet yielded translational implications. Although the use of tumor xenografts to predict drug efficacy in patients has been disappointing, use of novel transgenic mice models should permit improved early detection and development of drug regimens through integration of appropriate imaging modalities. In a very descriptive review, Mohammed et al.[58] consider issues that are unique to working with transgenic mouse models, such as the biology of GEM models, stage-tumor-specific detection using imaging technologies, use of monoclonal antibodies, nanoparticles and biomarkers and development of chemopreventive and chemotherapeutic drugs for PDAC.
Conclusion
The absence of effective in vivo systems that accurately predict clinical efficacy has hindered drug development in therapeutics. Indeed, deficiencies in the standard preclinical methods for evaluating potential bioactive drugs, such as xenograft mouse models, have been highlighted as a key obstacle in the translation of the major advances in basic therapeutic research into meaningful clinical benefits. At present, uses of xenograft mouse models for different drug developments appear to be limited. In contrast, opportunities for applying GEM models more faithfully mimic biological evolution of different human diseases. Greater use of such GEMs for preclinical studies in target validation, assessment of disease response, investigation of pharmacodynamic markers of drug action, the modeling of drug resistance and for understanding drug toxicity can each potentially improve the likelihood of successful drug development. Incorporating a 6-month transgenic mouse model into safety testing strategies for new drugs/chemicals makes valid scientific, ethical and sound business sense, since these assays are shorter in duration, use fewer animals and the cost is well below the traditional 2-year mouse bioassay.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
References
- 1.Workman P, Aboagye EO, Balkwill F, Balmain A, Bruder G, Chaplin DJ, et al. Guidelines for the welfare and use of animals in cancer research. Br J Cancer. 2010;102:1555–77. doi: 10.1038/sj.bjc.6605642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Angstadt AY, Thayanithy V, Subramanian S, Modiano JF, Breen M. A genome-wide approach to comparative oncology: High-resolution oligonucleotide aCGH of canine and human osteosarcoma pinpoints shared microaberrations. Cancer Genet. 2012;205:572–87. doi: 10.1016/j.cancergen.2012.09.005. [DOI] [PubMed] [Google Scholar]
- 3.Hansen K, Khanna C. Spontaneous and genetically engineered animal models; use in preclinical cancer drug development. Eur J Cancer. 2004;40:858–80. doi: 10.1016/j.ejca.2003.11.031. [DOI] [PubMed] [Google Scholar]
- 4.Cree IA, Glaysher S, Harvey AL. Efficacy of anti-cancer agents in cell lines versus human primary tumour tissue. Curr Opin Pharmacol. 2010;10:375–9. doi: 10.1016/j.coph.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 5.Tentler JJ, Tan AC, Weekes CD, Jimeno A, Leong S, Pitts TM, et al. Patient-derived tumour xenografts as models for oncology drug development. Nat Rev Clin Oncol. 2012;9:338–50. doi: 10.1038/nrclinonc.2012.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Batra V, Maris JM, Kang MH, Reynolds CP, Houghton PJ, Alexander D, et al. Initial testing (stage 1) of SGI-1776, a PIM1 kinase inhibitor, by the pediatric preclinical testing program. Pediatr Blood Cancer. 2012;59:749–52. doi: 10.1002/pbc.23364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McCormack E, Haaland I, Venås G, Forthun RB, Huseby S, Gausdal G, et al. Synergistic induction of p53 mediated apoptosis by valproic acid and nutlin-3 in acute myeloid leukemia. Leukemia. 2012;26:910–7. doi: 10.1038/leu.2011.315. [DOI] [PubMed] [Google Scholar]
- 8.Yun SH, Park ES, Shin SW, Na YW, Han JY, Jeong JS, et al. Stichoposide C induces apoptosis through the generation of ceramide in leukemia and colorectal cancer cells and shows in vivo antitumor activity. Clin Cancer Res. 2012;18:5934–48. doi: 10.1158/1078-0432.CCR-12-0655. [DOI] [PubMed] [Google Scholar]
- 9.Beltran PJ, Chung YA, Moody G, Mitchell P, Cajulis E, Vonderfecht S, et al. Efficacy of ganitumab (AMG 479), alone and in combination with rapamycin, in Ewing's and osteogenic sarcoma models. J Pharmacol Exp Ther. 2011;337:644–54. doi: 10.1124/jpet.110.178400. [DOI] [PubMed] [Google Scholar]
- 10.Yu Y, Gao S, Li Q, Wang C, Lai X, Chen X, et al. The FGF2-binding peptide P7 inhibits melanoma growth in vitro and in vivo. J Cancer Res Clin Oncol. 2012;138:1321–8. doi: 10.1007/s00432-012-1201-7. [DOI] [PubMed] [Google Scholar]
- 11.Chughtai K, Jiang L, Greenwood TR, Glunde K, Heeren RM. Mass spectrometry images acylcarnitines, phosphatidylcholines, and sphingomyelin in MDA-MB-231 breast tumor models. J Lipid Res. 2013;54:333–44. doi: 10.1194/jlr.M027961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martin LA, Pancholi S, Farmer I, Guest S, Ribas R, Weigel MT, et al. Effectiveness and molecular interactions of the clinically active mTORC1 inhibitor everolimus in combination with tamoxifen or letrozole in vitro and in vivo. Breast Cancer Res. 2012;14:R132. doi: 10.1186/bcr3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chang JT, Kuo TF, Chen YJ, Chiu CC, Lu YC, Li HF, et al. Highly potent and specific siRNAs against E6 or E7 genes of HPV16- or HPV18-infected cervical cancers. Cancer Gene Ther. 2010;17:827–36. doi: 10.1038/cgt.2010.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Y, Ma J, Wang F, Hu J, Cui A, Wei C, et al. Amygdalin induces apoptosis in human cervical cancer cell line HeLa cells. Immunopharmacol Immunotoxicol. 2013;35:43–51. doi: 10.3109/08923973.2012.738688. [DOI] [PubMed] [Google Scholar]
- 15.Tu Z, Ma Y, Tian J, Li H, Akers W, Achilefu S, et al. Estrogen receptor β potentiates the antiproliferative effect of raloxifene and affects the cell migration and invasion in HCT-116 colon cancer cells. J Cancer Res Clin Oncol. 2012;138:1091–103. doi: 10.1007/s00432-011-1145-3. [DOI] [PubMed] [Google Scholar]
- 16.Lin J, Yu Y, Shigdar S, Fang DZ, Du JR, Wei MQ, et al. Enhanced antitumor efficacy and reduced systemic toxicity of sulfatide-containing nanoliposomal doxorubicin in a xenograft model of colorectal cancer. PLoS One. 2012;7:e49277. doi: 10.1371/journal.pone.0049277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yin P, Wang Y, Qiu Y, Hou L, Liu X, Qin J, et al. Bufalin-loaded mPEG-PLGA-PLL-cRGD nanoparticles: Preparation, cellular uptake, tissue distribution, and anticancer activity. Int J Nanomedicine. 2012;7:3961–9. doi: 10.2147/IJN.S32063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang H, Zhang S, He H, Zhao W, Chen J, Shao RG. GAP161 targets and downregulates G3BP to suppress cell growth and potentiate cisplaitin-mediated cytotoxicity to colon carcinoma HCT116 cells. Cancer Sci. 2012;103:1848–56. doi: 10.1111/j.1349-7006.2012.02361.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hsu CW, Chang YJ, Chang CH, Chen LC, Lan KL, Ting G, et al. Comparative therapeutic efficacy of rhenium-188 radiolabeled-liposome and 5-fluorouracil in LS-174T human colon carcinoma solid tumor xenografts. Cancer Biother Radiopharm. 2012;27:481–9. doi: 10.1089/cbr.2011.1158. [DOI] [PubMed] [Google Scholar]
- 20.Burden RE, Gormley JA, Kuehn D, Ward C, Kwok HF, Gazdoiu M, et al. Inhibition of Cathepsin S by Fsn0503 enhances the efficacy of chemotherapy in colorectal carcinomas. Biochimie. 2012;94:487–93. doi: 10.1016/j.biochi.2011.08.017. [DOI] [PubMed] [Google Scholar]
- 21.James J, Ruggeri B, Armstrong RC, Rowbottom MW, Jones-Bolin S, Gunawardane RN, et al. CEP-32496: A novel orally active BRAF (V600E) inhibitor with selective cellular and in vivo antitumor activity. Mol Cancer Ther. 2012;11:930–41. doi: 10.1158/1535-7163.MCT-11-0645. [DOI] [PubMed] [Google Scholar]
- 22.Jagoda EM, Lang L, Bhadrasetty V, Histed S, Williams M, Kramer-Marek G, et al. Immuno-PET of the hepatocyte growth factor receptor Met using the 1-armed antibody onartuzumab. J Nucl Med. 2012;53:1592–600. doi: 10.2967/jnumed.111.102293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wada Y, Yoshida K, Suzuki T, Mizuiri H, Konishi K, Ukon K, et al. Synergistic effects of docetaxel and S-1 by modulating the expression of metabolic enzymes of 5-fluorouracil in human gastric cancer cell lines. Int J Cancer. 2006;119:783–91. doi: 10.1002/ijc.21879. [DOI] [PubMed] [Google Scholar]
- 24.Ellis L, Shah P, Hammers H, Lehet K, Sotomayor P, Azabdaftari G, et al. Vascular disruption in combination with mTOR inhibition in renal cell carcinoma. Mol Cancer Ther. 2012;11:383–92. doi: 10.1158/1535-7163.MCT-11-0748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Y, Su HH, Yang Y, Hu Y, Zhang L, Blancafort P, et al. Systemic delivery of modified mRNA encoding herpes simplex virus 1 thymidine kinase for targeted cancer gene therapy. Mol Ther. 2013;21:358–67. doi: 10.1038/mt.2012.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xie H, Lee MH, Zhu F, Reddy K, Peng C, Li Y, et al. Identification of an Aurora kinase inhibitor specific for the Aurora B isoform. Cancer Res. 2013;73:716–24. doi: 10.1158/0008-5472.CAN-12-2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gerber DE, Gupta P, Dellinger MT, Toombs JE, Peyton M, Duignan I, et al. Stromal platelet-derived growth factor receptor α (PDGFRα) provides a therapeutic target independent of tumor cell PDGFRα expression in lung cancer xenografts. Mol Cancer Ther. 2012;11:2473–82. doi: 10.1158/1535-7163.MCT-12-0431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heyerdahl H, Abbas N, Brevik EM, Mollatt C, Dahle J. Fractionated therapy of HER2-expressing breast and ovarian cancer xenografts in mice with targeted alpha emitting 227Th-DOTA-p-benzyl-trastuzumab. PLoS One. 2012;7:e42345. doi: 10.1371/journal.pone.0042345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang H, Galbán S, Wu R, Bowman BM, Witte A, Vetter K, et al. Molecular imaging reveals a role for AKT in resistance to cisplatin for ovarian endometrioid adenocarcinoma. Clin Cancer Res. 2013;19:158–69. doi: 10.1158/1078-0432.CCR-12-2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosa R, Melisi D, Damiano V, Bianco R, Garofalo S, Gelardi T, et al. Toll-like receptor 9 agonist IMO cooperates with cetuximab in K-ras mutant colorectal and pancreatic cancers. Clin Cancer Res. 2011;17:6531–41. doi: 10.1158/1078-0432.CCR-10-3376. [DOI] [PubMed] [Google Scholar]
- 31.Choi ES, Jung JY, Lee JS, Park JH, Cho NP, Cho SD. Myeloid cell leukemia-1 is a key molecular target for mithramycin A-induced apoptosis in androgen-independent prostate cancer cells and a tumor xenograft animal model. Cancer Lett. 2013;328:65–72. doi: 10.1016/j.canlet.2012.09.009. [DOI] [PubMed] [Google Scholar]
- 32.Authier S, Tremblay S, Dumulon V, Dubuc C, Ouellet R, Lecomte R, et al. [11C] acetoacetate utilization by breast and prostate tumors: A PET and biodistribution study in mice. Mol Imaging Biol. 2008;10:217–23. doi: 10.1007/s11307-008-0143-6. [DOI] [PubMed] [Google Scholar]
- 33.Cheng KW, Mattheolabakis G, Wong CC, Ouyang N, Huang L, Constantinides PP, et al. Topical phospho-sulindac (OXT-328) is effective in the treatment of non-melanoma skin cancer. Int J Oncol. 2012;41:1199–203. doi: 10.3892/ijo.2012.1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kamran MZ, Gude RP. Preclinical evaluation of the antimetastatic efficacy of Pentoxifylline on A375 human melanoma cell line. Biomed Pharmacother. 2012;66:617–26. doi: 10.1016/j.biopha.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 35.Yu K, Chen Z, Pan X, Yang Y, Tian S, Zhang J, et al. Tetramethylpyrazine-mediated suppression of C6 gliomas involves inhibition of chemokine receptor CXCR4 expression. Oncol Rep. 2012;28:955–60. doi: 10.3892/or.2012.1866. [DOI] [PubMed] [Google Scholar]
- 36.Sano D, Myers JN. Xenograft models of head and neck cancers. Head Neck Oncol. 2009;1:32. doi: 10.1186/1758-3284-1-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blandini F, Armentero MT. Animal models of Parkinson's disease. FEBS J. 2012;279:1156–66. doi: 10.1111/j.1742-4658.2012.08491.x. [DOI] [PubMed] [Google Scholar]
- 38.Forabosco F, Löhmus M, Rydhmer L, Sundström LF. Genetically modified farm animals and fish in agriculture: A review. Livestock Sci. 2013;153:1–9. [Google Scholar]
- 39.Sharpless NE, Depinho RA. The mighty mouse: Genetically engineered mouse models in cancer drug development. Nat Rev Drug Discov. 2006;5:741–54. doi: 10.1038/nrd2110. [DOI] [PubMed] [Google Scholar]
- 40.Jones TS, Holland EC. Animal models for glioma drug discovery. Expert Opin Drug Discov. 2011;6:1271–83. doi: 10.1517/17460441.2011.632628. [DOI] [PubMed] [Google Scholar]
- 41.Flisikowska T, Kind A, Schnieke A. The new pig on the block: Modelling cancer in pigs. Transgenic Res. 2013;22:673–80. doi: 10.1007/s11248-013-9720-9. [DOI] [PubMed] [Google Scholar]
- 42.European Commission. Of mice and men – Are mice relevant models for human disease? Outcomes of the European Commission workshop “Are mice relevant models for human disease?” held in London, UK, on May. 2010 [Google Scholar]
- 43.Becher OJ, Holland EC. Genetically engineered models have advantages over xenografts for preclinical studies. Cancer Res. 2006;66:3355–8. doi: 10.1158/0008-5472.CAN-05-3827. 3358. [DOI] [PubMed] [Google Scholar]
- 44.Schuh JC. Trials, tribulations, and trends in tumor modeling in mice. Toxicol Pathol. 2004;32(Suppl 1):53–66. doi: 10.1080/01926230490424770. [DOI] [PubMed] [Google Scholar]
- 45.Gong C, Deng S, Wu Q, Xiang M, Wei X, Li L, et al. Improving antiangiogenesis and anti-tumor activity of curcumin by biodegradable polymeric micelles. Biomaterials. 2013;34:1413–32. doi: 10.1016/j.biomaterials.2012.10.068. [DOI] [PubMed] [Google Scholar]
- 46.Dong LF, Grant G, Massa H, Zobalova R, Akporiaye E, Neuzil J. α-Tocopheryloxyacetic acid is superior to α-tocopheryl succinate in suppressing HER2-high breast carcinomas due to its higher stability. Int J Cancer. 2012;131:1052–8. doi: 10.1002/ijc.26489. [DOI] [PubMed] [Google Scholar]
- 47.Mueller M, Reichardt W, Koerner J, Groettrup M. Coencapsulation of tumor lysate and CpG-ODN in PLGA-microspheres enables successful immunotherapy of prostate carcinoma in TRAMP mice. J Control Release. 2012;162:159–66. doi: 10.1016/j.jconrel.2012.06.015. [DOI] [PubMed] [Google Scholar]
- 48.Zhong ZF, Hoi PM, Wu GS, Xu ZT, Tan W, Chen XP, et al. Anti-angiogenic effect of furanodiene on HUVECs in vitro and on zebrafish in vivo. J Ethnopharmacol. 2012;141:721–7. doi: 10.1016/j.jep.2011.08.052. [DOI] [PubMed] [Google Scholar]
- 49.van den Engel NK, Rüttinger D, Rusan M, Kammerer R, Zimmermann W, Hatz RA, et al. Combination immunotherapy and active-specific tumor cell vaccination augments anti-cancer immunity in a mouse model of gastric cancer. J Transl Med. 2011;9:140. doi: 10.1186/1479-5876-9-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hassan S, Buchanan M, Jahan K, Aguilar-Mahecha A, Gaboury L, Muller WJ, et al. CXCR4 peptide antagonist inhibits primary breast tumor growth, metastasis and enhances the efficacy of anti-VEGF treatment or docetaxel in a transgenic mouse model. Int J Cancer. 2011;129:225–32. doi: 10.1002/ijc.25665. [DOI] [PubMed] [Google Scholar]
- 51.Bergé M, Bonnin P, Sulpice E, Vilar J, Allanic D, Silvestre JS, et al. Small interfering RNAs induce target-independent inhibition of tumor growth and vasculature remodeling in a mouse model of hepatocellular carcinoma. Am J Pathol. 2010;177:3192–201. doi: 10.2353/ajpath.2010.100157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rennick DM, Fort MM. Lessons from genetically engineered animal models. XII. IL-10-deficient (IL-10(-/-) mice and intestinal inflammation. Am J Physiol Gastrointest Liver Physiol. 2000;278:G829–33. doi: 10.1152/ajpgi.2000.278.6.G829. [DOI] [PubMed] [Google Scholar]
- 53.Postic C, Girard J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: Lessons from genetically engineered mice. J Clin Invest. 2008;118:829–38. doi: 10.1172/JCI34275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Iismaa SE, Mearns BM, Lorand L, Graham RM. Transglutaminases and disease: Lessons from genetically engineered mouse models and inherited disorders. Physiol Rev. 2009;89:991–1023. doi: 10.1152/physrev.00044.2008. [DOI] [PubMed] [Google Scholar]
- 55.Changeux JP. Nicotine addiction and nicotinic receptors: Lessons from genetically modified mice. Nat Rev Neurosci. 2010;11:389–401. doi: 10.1038/nrn2849. [DOI] [PubMed] [Google Scholar]
- 56.Bosetti F. Arachidonic acid metabolism in brain physiology and pathology: Lessons from genetically altered mouse models. J Neurochem. 2007;102:577–86. doi: 10.1111/j.1471-4159.2007.04558.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang X, Deng Y, Mao Z, Ma X, Fan X, Cui L, et al. CCN1 promotes tumorigenicity through Rac1/Akt/NF-κB signaling pathway in pancreatic cancer. Tumour Biol. 2012;33:1745–58. doi: 10.1007/s13277-012-0434-z. [DOI] [PubMed] [Google Scholar]
- 58.Mohammed A, Janakiram NB, Lightfoot S, Gali H, Vibhudutta A, Rao CV. Early detection and prevention of pancreatic cancer: Use of genetically engineered mouse models and advanced imaging technologies. Curr Med Chem. 2012;19:3701–13. doi: 10.2174/092986712801661095. [DOI] [PubMed] [Google Scholar]