

Abstract
Rationale
Muscarinic cholinergic M1 and M4 receptors may participate in schizophrenia's etiology, and have been proposed as targets for antipsychotic medications.
Objective
Here we investigated the involvement of these receptors in behavioral measures pertinent to schizophrenia using knockout mice lacking M1 receptors (M1−/−), M4 receptors (M4−/−) or both (M1−/−M4−/−).
Methods
We measured prepulse inhibition of startle (PPI) without drugs, and after treatment with scopolamine (0.32–1.8 mg/kg), xanomeline (3.2 mg/kg) oxotremorine (0.032–0.1 mg/kg), clozapine (1.0–5.6 mg/kg), or haloperidol (0.32–3.2 mg/kg).
Results
In female (but not male) mice, combined deletion of both M1 and M4 receptors decreased PPI relative to wild-type mice, while knockout of either receptor alone had no significant effect. Scopolamine disrupted PPI in wild-type and M4−/− mice, but not in female M1−/−M4−/− or female M1−/− mice. When administered before scopolamine, xanomeline restored PPI in wild-type mice and M1−/− mice, but not in M4−/− mice. In contrast, pretreatment with oxotremorine increased PPI regardless of genotype. Effects of clozapine and haloperidol on PPI were not hindered by either mutation.
Conclusions
Deletion of both M1 and M4 receptors can disrupt PPI, suggesting that (at least partially redundant) M1 and M4 receptor-dependent functions are involved in sensorimotor gating mechanisms. PPI-disrupting effects of muscarinic antagonists appeared dependent upon M1 receptor blockade. Our data also suggest that xanomeline exerts antipsychotic-like effects mainly through M4 receptor stimulation, while stimulation of non-M1/M4 subtypes may also have antipsychotic potential. Finally, our results do not support a role of M1/M4 receptors in mediating antipsychotic-like effects of clozapine.
Keywords: prepulse inhibition, muscarinic, acetylcholine receptor, antipsychotic, knockout mice, M1, M4
Introduction
Despite an abundance of available antipsychotic drugs, there is still a need for highly efficacious schizophrenia medications with favorable side-effect profiles. In particular, cognitive deficits are often debilitating symptoms of schizophrenia that are typically poorly improved by current medications (Buchanan et al. 2007). Evidence suggests cholinergic dysregulation is part of schizophrenia's etiology, including abnormal densities and/or function of the muscarinic M1 and M4 receptor subtypes, which have therefore been proposed as novel antipsychotic drug targets (Bymaster et al. 2002, 2003; Sarter et al. 2005; Vakalopoulos 2006; Raedler et al. 2007). These cholinergic hypotheses likely extend, rather than contradict, the “dopamine hypothesis” of schizophrenia, as the two systems are intricately interconnected. Muscarinic antagonists induce striatal dopamine efflux in rodents and humans, can produce psychotic-like effects similar to dopamine agonists and psychostimulants, and worsen positive symptoms in schizophrenic patients (Tandon et al. 1991; Dewey et al. 1993; Chapman et al. 1997; Perry and Perry 1995; Halpern 2004). Conversely, muscarinic agonists have been shown to produce functional dopamine antagonism, which may account, at least in part, for their antipsychotic-like effects in various preclinical assays (Bymaster et al. 1998; Fink-Jensen et al. 1998; Shannon et al. 1999,2000; Stanhope et al. 2001; Andersen et al. 2003; Jones et al. 2005).
Muscarinic systems are also critically involved in memory and cognition (Power et al. 2003; Hasselmo 2006). Muscarinic antagonists disrupt memory and cognitive performance in laboratory animals as well as in healthy individuals, and worsen cognitive impairment in schizophrenic patients (Sitaram et al. 1978; Johnstone et al. 1983; Minzenber et al. 2004; Ellis et al. 2006). Conversely, muscarinic agonists can improve cognitive performance in laboratory animals and humans (Davis et al. 1978; Sitaram et al. 1978; Bartus 1979; Harries et al. 1998; Hodges et al. 1999). Muscarinic agonists such as the M1/M4-preferring xanomeline are being evaluated and shown some promise as cognitive enhancers in schizophrenia as well as other disorders, e.g., Alzheimer's disease (Bodick et al. 1997; Friedman 2004; Langmead et al. 2008b; Shekhar et al. 2008).
There are five cloned muscarinic receptor subtypes (M1–M5), and little is known about which of these mediate potential therapeutic effects, and whether different subtypes mediate undesirable effects. While the usefulness of nonselective muscarinic agonists and acetylcholinesterase inhibitors are limited by side effects, the recent emergence of highly subtype-selective allosteric ligands has intensified the interest in effects mediated through specific subtypes (Chan et al. 2008; Jones et al. 2008; Langmead et al. 2008a; Shirey et al. 2008; Conn et al. 2009). M1 and M4 receptors are expressed in striatal areas and in areas relevant to cognitive function, e.g., cingulate cortex, prefrontal cortex and hippocampus (Levey et al. 1991). In contrast, the M2 and M3 receptors are sparser in the brain but represent the major subtypes mediating (undesirable) peripheral effects such as alterations in heart rate, gastrointestinal contractility, and exocrine gland secretion, causing symptoms like nausea, diarrhea, and salivation (Bymaster et al. 2002; Wess 2004). M1 and M4 are the most abundant muscarinic receptor subtypes on striatal neurons, with M4 receptors and D1 receptors exerting directly opposing effects on cyclic AMP synthesis, while M1 receptors oppose the effects of D2 receptors (Di Chiara et al. 1994; McGinty 1999; Onali and Olianas 2002). Functional dopamine antagonism in striatal pathways therefore seems most likely mediated through M1 and/or M4 receptors. Accordingly the M1 and M4 receptors are generally thought to mediate cognition-enhancing and antipsychotic-like effects of muscarinic agonists (Bymaster et al. 2002; Langmead et al. 2008b; Conn et al. 2009).
One of the most validated preclinical assays for predicting antipsychotic drug action in humans is prepulse inhibition (PPI) of the startle response, in which the reflex elicited by a startling stimulus, such as a loud noise, is reduced when it is immediately preceded by a low-intensity “prepulse” stimulus (Geyer et al. 2001,2002; Swerdlow et al. 2008). PPI is on average reduced in the schizophrenic population, and is believed to reflect sensorimotor gating, or the ability of the brain to “filter out” irrelevant stimuli (Braff et al. 2001; Swerdlow et al. 2008). PPI can be disrupted pharmacologically in both rodents and healthy humans by drugs that can induce psychotic-like symptoms – including muscarinic receptor antagonists (Wu et al. 1993; Jones and Shannon 2000; Geyer et al. 2001; Kumari et al. 2001). Such disruptions in PPI can be reversed by administration of dopamine receptor antagonists or antipsychotic drugs (Geyer et al. 2001; Swerdlow et al. 2008). In the present study, we tested the hypothesis that the M1 and/or M4 receptors play a role in sensorimotor gating by evaluating PPI in knockout mice lacking either or both receptor subtypes. We also tested the hypothesis that M1 and/or M4 receptors are involved in the responsiveness to muscarinic ligands or antipsychotic drugs. Specifically, we evaluated PPI in M1−/−, M4−/−, M1−/−M4−/− and wild-type mice after treatment with the non-selective muscarinic antagonist scopolamine, with and without xanomeline (M1/M4-preferring agonist) or oxotremorine (non-selective muscarinic agonist) pretreatments. We also evaluated the effects of the atypical antipsychotic clozapine, which has measurable affinity for muscarinic receptors including M1 and M4 subtypes, and the typical antipsychotic haloperidol, which does not (Olianas et al. 1999; Weiner et al. 2004; Davies et al. 2005).
Materials and methods
Animals and housing
M1−/− and M4−/− mice were generated as described previously (Gomeza et al. 1999; Miyakawa et al. 2001) and backcrossed for 11 generations to C57BL/6NTac females. Double knockout mice were bred by intercrossing the single knockout lines and then maintained as a separate line, due to the low yield of M1−/−M4−/− mice if bred by heterozygous intermating. Age- and sex-matched C57BL/6NTac were therefore used as wild-type controls. All mice were bred at Taconic Farms (Germantown, NY). Male and female double knockout mice were tested first, together with wild-type mice, in two sequential batches of N = 5–7/sex/genotype. A second batch was tested because initial results indicated an effect of sex, therefore additional mice were obtained both to duplicate the findings, and to increase sample size to allow for the detection of sex effects. Results obtained from both batches were comparable, and all data are therefore reported as both batches combined. Subsequently to the double knockout mice, female M1−/− mice and female M4−/− mice (each N=12) were tested simultaneously with an additional group of female wild-type mice (N=9), in a single batch.
Animals were kept in a 12-h light/dark cycle (lights on at 07:00) at 22°C and 55% humidity. Tap water and standard rodent chow (rodent diet 5001, PMI Feeds, Inc., St. Louis, MO) were accessible ad libitum. For enrichment purposes rodent “treats” were given once or twice weekly, and nesting material (cotton), hiding/nesting devices and exercise devices were provided. Fresh litter (pine wood shavings) was provided twice weekly. The air in each cage was actively circulated. Animals were group housed up to 4 per cage, and were left to acclimate to the housing facilities at least 7 days before experiments began. All testing was conducted in animals at least 8 weeks of age and during the light phase of the circadian cycle (the latter, to match relevant previous behavioral studies). Animal health was evaluated daily. All procedures were carried out in accordance with the NIH Guide for the Care and Use of Laboratory Animals and the Principles of Laboratory Animal Care.
Drugs
Scopolamine hydrobromide, oxotremorine sesquifumarate and xanomeline were dissolved in 0.9% saline. Clozapine and haloperidol were dissolved in a small volume of sterile water acidified to pH≈1 with HCl, then diluted to the desired concentrations with saline and water, respectively. Final pH was adjusted to 5–5.5 with NaOH. For clozapine and haloperidol vehicles, equivalent amounts of HCl and NaOH were added to saline or water (for comparable pH). Xanomeline was synthesized at the McLean Hospital following published methods (Sauerberg et al. 1992; Kane et al. 2008). Other drugs were purchased from Sigma-Aldrich (St Louis, MO). Doses refer to weights of the drug salts.
Startle and prepulse inhibition of startle
Apparatus and session
Startle reactivity was assessed using four startle chambers (SR-LAB, San Diego Instruments, San Diego, CA). The apparatus and procedures have been described in detail elsewhere (Thomsen et al., 2007). Each 20-min session consisted of startle trials (“pulse alone”; a 40 msec 120 dB broadband pulse), prepulse trials (“prepulse+pulse”; a 20 ms prepulse followed by a 100 ms interstimulus interval and a 40 msec 120 dB pulse), and no-stimulus trials (“nostim”; background noise alone, 65 dB). Prepulse intensities were 6, 12 and 16 dB above background (i.e., 71, 77 and 81 dB). A session consisted of a 5 min acclimation period with background noise, five startle (pulse alone) trials, then 10 presentations of each trial type (pulse alone, prepulse+pulse of each prepulse intensity and nostim) in pseudorandom order, then five startle trials. Intertrial intervals averaged 15 sec (range: 12–18 sec). Each animal was assigned one apparatus, balanced for sex and genotype. Chambers were cleaned with a paper towel and water after each session. The amount of PPI for each prepulse intensity was calculated as %PPI = 100 – [[(ASR for prepulse+pulse)/(ASR for pulse alone)] × 100], using the average of the ten presentations of each trial type. Startle amplitude (ASR for pulse alone) was calculated as the average of the 10 mid-session pulse alone trials.
Spontaneous and drug-modulated PPI
Drug- and experimentally naïve mice were used. Dose-effect functions were assessed for saline and scopolamine (0.32, 1.0 and 1.8 mg/kg), and the saline data were analyzed for baseline startle and PPI differences. Following scopolamine experiments, female mice only were studied further. The M1/M4 preferring agonist xanomeline (3.2 mg/kg) was tested as a pretreatment to 1.0 mg/kg scopolamine. The non-selective muscarinic agonist oxotremorine was tested as pretreatment to 1.0 mg/kg scopolamine (0.032, 0.1 mg/kg) and alone (0.01, 0.032, 0.1 mg/kg). Clozapine was tested at doses of 1.0, 1.8, 3.2 and 5.6 mg/kg, and in the first batch of double knockout mice only, haloperidol at doses of 0.3, 1.0 and 3.2 mg/kg. Drug doses plus vehicle were tested according to a Latin square design within-subjects, with the exception of 5.6 mg/kg clozapine which was tested last. Animals were allowed at least three days between tests, and at least one week between each drug. Scopolamine, clozapine and haloperidol were injected i.p. (10 ml/kg), oxotremorine and xanomeline s.c. (10 ml/kg). Scopolamine was injected immediately before placing the animal in the test chamber (i.e., 5 min before the first startle stimulus), clozapine and haloperidol 30 min before placing the animal in the chamber, oxotremorine and xanomeline 15 min before placing the animal in the chamber. Drug doses were chosen based on previous studies (Thomsen et al., 2007) and pilot experiments in wild-type mice (scopolamine, xanomeline, oxotremorine).
Statistical analysis
Statistical comparisons were performed by mixed model ANOVA with genotype and sex as between-subjects factors and drug dose as a within-subjects factor (repeated measures), unless otherwise indicated. Significant effects of dose were analyzed post-hoc by Dunnett's multiple comparisons test vs. vehicle, and significant effects of genotype by Bonferroni-corrected two-sided unpaired t-test vs. wild-type. Genotype comparisons were designed to examine effects of each mutation relative to their respective group of wild-type controls, and no comparisons were made either between the various knockout lines or between the two groups of wild-type controls. The level of significance was set at p ≤ 0.05. All t-tests reported are two-sided. For PPI, prepulse intensity was always included as a factor in an initial analysis, and was always highly significant (p < 0.0001). For brevity, %PPI was collapsed across prepulse intensity for graphic representations of the data and for analyses (with the exception of baseline PPI). In cases where drug effects were detected for startle amplitude, possible correlations between startle and PPI in individual mice were investigated (in each genotype) by linear regression.
Results
Baseline effects
Baseline PPI
PPI was assessed in male and female M1−/−M4−/− and wild-type mice, then in female M1−/−, M4−/− and wild-type mice. An ANOVA using genotype and sex as between-subjects factors and prepulse intensity as a within-subjects factor showed a significant effect of genotype [F1,43 = 6.10, p < 0.05], prepulse intensity [F2,86 = 77.09, p < 0.0001], and a sex by prepulse intensity interaction [F2,86 = 3.31, p < 0.05]. Because of the significant interaction, the data were re-analyzed for female and male mice separately. As expected, %PPI increased with prepulse intensity in both female mice [F2,44 = 20.28, p < 0.0001] and male mice [F2,42 = 75.21, p < 0.0001]. There was a significant effect of genotype only in the female mice [F1,22 = 8.96, p < 0.01]. Figure 1A–D shows %PPI as a function of prepulse intensity in male (A) and female (B) M1−/−M4−/− and wild-type mice. It can be seen that PPI was reduced in the female M1−/−M4−/− mice relative to wild-type at all prepulse intensities (p < 0.05 to p < 0.01), while there was no effect of genotype in the male mice.
Figure 1. PPI was decreased in female mice lacking both M1 and M4 receptors.

Abscissae: prepulse intensity (dB above background noise; A–D), genotype (E–H). Ordinates: %PPI (A–D), startle amplitude (arbitrary units; E–H). N=9–13. Data are group means, bars represent one s.e.m. †p<0.05, ††p<0.01 vs. wild-type, unpaired-sample t-test.
Because there was an effect of the double mutation in female mice, female single knockout mice lacking either M1 or M4 receptors were subsequently tested, with an additional cohort of female wild-type mice. Figure 1C–D shows %PPI in the M1−/− mice and M4−/− mice, which demonstrated only a small and non-significant decrease in PPI relative to wild-type mice. Separate ANOVAs were performed comparing each knockout to wild-type, with genotype as between-subjects factor and prepulse intensity as within-subjects factor. For the M1−/− mice, %PPI was related to prepulse intensity [F2,38 = 52.84, p < 0.0001], but there was no significant effect of M1 genotype or genotype by intensity interaction (Fig. 1C). Similarly for the M4−/− mice, %PPI was related to prepulse intensity [F2,38 = 20.99, p < 0.0001], with no significant effect of M4 genotype or interaction (Fig. 1D). It is noteworthy that PPI levels were very similar between cohorts of wild-type mice.
Baseline startle reactivity
Baseline startle amplitudes (i.e., following saline administration) are shown in Figure 1E–H. Startle amplitudes did not differ significantly between wild-type and M1−/−M4−/− mice, but were higher in male mice than in female mice [F1,44 = 7.55, p < 0.01]. There was no significant genotype by sex interaction. In the single knockout experiment, baseline startle reactivity was not significantly altered by the M1 mutation, but was increased in the M4−/− mice (p < 0.05; Fig. 1H).
Scopolamine effects
Effect of scopolamine on PPI
Figure 2A–D shows PPI dose-effect functions for scopolamine in double and single knockout mice. For the double knockout experiment, because of the differential effect of the mutation on baseline PPI between sexes, the data were analyzed separately in male and female mice. In male mice, scopolamine decreased PPI dose-dependently [F3,66 = 9.13, p < 0.0001], but there was no effect of genotype or dose by scopolamine interaction (see Fig. 2A for the significance of specific scopolamine doses). In female mice, PPI was related to scopolamine dose [F3,66 = 9.25, p < 0.0001] with a genotype by scopolamine interaction [F3,66 = 3.84, p < 0.05], but no main effect of genotype. Further analysis showed the effect of scopolamine was significant only in wild-type mice (p < 0.0001; Fig. 2B). In contrast, scopolamine did not affect PPI significantly in the female M1−/−M4−/− mice. Also, PPI was significantly lower in female M1−/−M4−/− mice relative to wild-type in the saline condition only (p < 0.05).
Figure 2. The PPI-suppressing effect of scopolamine was absent in female mice lacking M1 receptors or both M1 and M4 receptors.

In contrast, scopolamine increased startle reactivity in all genotypes. Abscissae: scopolamine dose [mg/kg]. Ordinates: Total %PPI (A–D), startle amplitude (E–H). N=9–13. Data are group means, bars represent one s.e.m. *p<0.05, **p<0.01 vs. vehicle, Dunnett's multiple comparisons test; †p<0.05, ††p<0.01 vs. wild-type, Bonferroni-corrected unpaired-sample t-test.
In the M1−/− single knockout experiment, PPI was related to scopolamine dose [F3,57 = 9.18, p < 0.0001], with a significant genotype by scopolamine interaction [F3,57 = 3.69, p < 0.05], but no main effect of genotype. Further analysis showed scopolamine decreased PPI in wild-type mice [F3,24 = 8.09, p < 0.001], but had no significant effect in M1−/− mice (Fig. 2C). In the M4−/− single knockout experiment, PPI was related to scopolamine dose [F3,57 = 14.53, p < 0.0001], with a trend for genotype [F1,19 = 3.81, p = 0.07], with no significant interaction. Scopolamine decreased PPI in both wild-type mice and M4−/− mice (both p < 0.001; Fig. 2D). Thus with respect to scopolamine's effects, the deletion of M1 receptors alone produced a phenotype comparable to the double mutation while the deletion of M4 receptors alone had little or no effect.
Effect of scopolamine on startle amplitude
Figure 2E–H shows the effect of scopolamine on startle amplitude. In the double knockout experiment, ANOVA showed a significant effect of genotype [F1,43 = 9.11, p < 0.01], sex [F1,43 = 12.20, p < 0.01], and scopolamine dose [F3,129 = 39.89, p < 0.0001], with no significant interactions (Fig. 2E–F). Data were analyzed in each sex, showing significant effects of genotype in both male mice [F1,21 = 4.47, p < 0.05] and female mice [F1,22 = 4.85, p < 0.05], significant effects of scopolamine dose in both male mice [F3,63 = 16.67, p < 0.0001] and female mice [F3,66 = 30.28, p < 0.0001], and a genotype by scopolamine interaction in the female mice only [F3,66 = 5.03, p < 0.01]. Further analysis in each genotype confirmed that scopolamine increased startle reactivity in all four groups (all p < 0.001). There was no significant correlation between scopolamine's effect on startle and PPI in any group. The M1−/−M4−/− mice appeared more sensitive to scopolamine-induced increases in startle, although all three doses of scopolamine reached significance in all groups. In the female mice, startle amplitudes were significantly higher in M1−/−M4−/− mice than in wild-type mice at the two highest scopolamine doses (p < 0.05). No doses reached significance in the male mice.
In the single knockout experiment, for the M1 mutation, startle amplitude was increased by scopolamine [F3,57 = 14.65, p < 0.0001], with no significant effect of M1 genotype or interaction (Fig. 2G). In contrast for the M4 mutation, startle amplitude was related to genotype [F1,19 = 29.21, p < 0.0001] and scopolamine dose [F3,57 = 9.87, p < 0.0001], with no significant interaction. Startle amplitude and PPI were negatively correlated in the M4−/− mice (r2 = 0.19, p < 0.01), with no correlation in the other genotypes. While scopolamine increased startle reactivity in both M4−/− mice [F3,33 = 7.29, p < 0.001] and wild-type mice [F3,24 = 6.45, p < 0.01], only the lowest dose scopolamine reached significance relative to saline in the wild-type mice, while all three doses did so in the M4−/− mice, and startle was significantly higher in M4−/− mice relative to wild-type at the two highest doses (p < 0.01; Fig. 2H). Thus, in contrast to the PPI modulation effects, the effect of the double mutation on startle reactivity was mimicked by the single M4 knockout, but not the M1 knockout.
Xanomeline effects
Effect of xanomeline on PPI
Following baseline and scopolamine tests, only the female mice were selected for further evaluation in the double knockout experiment. Figure 3A–C shows the effect of xanomeline pretreatment (3.2 mg/kg) in female double and single knockout mice treated with 1.0 mg/kg scopolamine. Vehicle and scopolamine-alone data are the same as in Figure 2, shown to aid visual comparisons. Pre-planned t-tests were done to evaluate the effect of xanomeline pretreatment vs. scopolamine alone in each genotype, as well as between-genotype comparisons in the xanomeline-treated state. In wild-type mice from both the double and single knockout experiments, the scopolamine-induced decrease in PPI was completely reversed by xanomeline (p < 0.001, p < 0.05). In contrast, female M1−/−M4−/− mice showed no significant effect of either scopolamine or xanomeline. Similar to the vehicle condition, the M1−/−M4−/− mice had lower PPI than wild-type mice after xanomeline + scopolamine treatment (p < 0.01).
Figure 3.

Xanomeline increased scopolamine-suppressed PPI in wild-type mice and in mice lacking M1 receptors, but not in mice lacking M4 or both receptors. Abscissae: treatment, Veh. = vehicle, Scop. = scopolamine 1.0 mg/kg, +Xano = scopolamine 1.0 mg/kg with xanomeline 3.2 mg/kg pretreatment. Ordinates: Total %PPI (A–D), startle amplitude (E–H). N=9–12. Data are group means, bars represent one s.e.m. *p<0.05, **p<0.01, ***p<0.001 vs. vehicle; #p≤0.05, ###p<0.001 vs. scopolamine alone, paired-sample t-test; †p<0.05, ††p<0.01, †††p<0.001vs. wild-type, unpaired-sample t-test.
In the M1−/− single knockout mice, xanomeline increased PPI (p = 0.050). As in the vehicle condition, PPI levels were comparable between M1−/− and wild-type mice after xanomeline + scopolamine treatment. In contrast, in the M4−/− single knockout mice, xanomeline failed to reverse the effect of scopolamine on PPI. PPI levels after xanomeline + scopolamine treatment thus appeared reduced in the M4−/− mice relative to the wild-type mice, an effect which approached significance (p = 0.051). In summary, the ability of xanomeline to block a scopolamine-induced PPI deficit was absent in the M1−/−M4−/− mice and in the M4−/− mice, but appeared intact in the M1−/− mice.
Effect of xanomeline on startle amplitude
As described above, 1.0 mg/kg scopolamine increased startle reactivity in all genotypes (Fig. 3D–F). Pre-planned t-tests were done as for PPI Xanomeline pretreatment did not significantly alter startle amplitudes relative to scopolamine alone in any genotype. The exaggerated effect of scopolamine in the female M1−/−M4−/− mice relative to wild-type mice was not significant after xanomeline pretreatment. Comparable to the baseline and scopolamine-alone tests, no difference was found between M1−/− mice and wild-type mice in startle reactivity, while startle was increased in M4−/− mice relative to wild-type regardless of xanomeline treatment (Fig. 3F).
Oxotremorine effects
Effect of oxotremorine on scopolamine-modulated PPI
The ability of the non-selective muscarinic agonist oxotremorine to reverse scopolamine-induced PPI deficits was then evaluated. Figure 4A–C shows the effect of 1.0 mg/kg scopolamine alone and after pretreatment with 0.032 or 0.1 mg/kg oxotremorine in female double and single knockout mice. Vehicle and scopolamine-alone data are the same as in Figure 2, shown to aid visual comparisons. ANOVAs were performed for each mutation with genotype as between-subjects factor and dose oxotremorine as within-subjects factor (relative to scopolamine alone). In the double-knockout experiment, oxotremorine increased PPI [F2,40 = 31.02, p < 0.0001], with no significant effect of genotype and no interaction. In both genotypes, oxotremorine increased PPI at both doses (p <0.01 vs. scopolamine). Similarly for both the M1 and the M4 single knockout mutations, there was a main effect of oxotremorine dose [F2,38 = 9.26, p < 0.001; F2,38 = 10.95, p < 0.001], but no effect of genotype and no interaction. In all three genotypes, oxotremorine increased PPI significantly from the scopolamine level for at least one dose (p <0.05 or p < 0.01).
Figure 4. Oxotremorine increased scopolamine-suppressed PPI regardless of M1 or M4 genotype.

Abscissae: treatment, Veh. = vehicle, Scop = scopolamine 1.0 mg/kg, +Ox = scopolamine 1.0 mg/kg with oxotremorine pretreatment, 0.032 and 0.1 mg/kg. Ordinates: Total %PPI (A–D), startle amplitude (E–H). N=9–12. Data are group means, bars represent one s.e.m. *p<0.05, **p<0.01. ***p<0.001 vs. vehicle, paired-sample t-test; †p<0.05, ††p<0.01, †††p<0.001 vs. wild-type, unpaired-sample t-test; #p<0.05, ##p<0.01 vs. scopolamine alone, Dunnett's multiple comparisons test.
Effect of oxotremorine on scopolamine-modulated startle amplitude
As described above, 1.0 mg/kg scopolamine increased startle reactivity in all genotypes (Fig. 4 D–F). ANOVAs were performed for startle amplitude the same way as for PPI. In the double-knockout experiment, startle was higher in M1−/−M4−/− mice than wild-type [F1,40 = 9.15, p < 0.01], significant for scopolamine alone (p < 0.05, Fig. 4D). There was a main effect of oxotremorine [F2,40 = 6.35, p < 0.01], which was significant only in the wild-type mice [F2,22 = 3.49, p < 0.05], but neither dose reached significance vs. scopolamine post-hoc. There was also a main effect of oxotremorine dose for both the M1 and the M4 single knockout comparisons [F2,38 = 7.46, p < 0.01; F2,38 = 8.05, p < 0.01], which was significant in M1−/− mice [F1,22 = 7.95, p < 0.01] and M4−/− mice [F1,22 = 9.00, p < 0.01], but not wild-type mice. Oxotremorine 0.1 mg/kg reached significance post-hoc in the M4−/− mice only (p < 0.01); in the other genotypes, no dose oxotremorine reached significance vs. scopolamine. There was an effect of the M4 genotype [F1,38 = 24.81, p < 0.0001] but not M1, consistent with the scopolamine alone data. There was also a M4 genotype by oxotremorine interaction [F2,38 = 3.13, p < 0.05]. Startle was significantly higher in the M4−/− mice relative to wild-type for scopolamine alone and scopolamine+0.032 mg/kg oxotremorine (p < 0.01, p < 0.001, Fig 4F). Thus there was a general trend for oxotremorine to reverse scopolamine's effects on startle. Effects on startle and PPI were not correlated in any genotype.
Effect of oxotremorine alone on PPI and startle amplitude
Oxotremorine alone was also tested, in M1−/−M4−/− mice and wild-type mice (Fig. 5A–B). PPI was related to oxotremorine dose [F3,60 = 7.71, p < 0.001], with a genotype by oxotremorine interaction [F3,60 = 16.57, p < 0.0001], but no main effect of genotype. When analyzed in each genotype, the oxotremorine effect was significant only in wild-type mice [F3,30 = 30.69, p < 0.0001], in which 0.1 mg/kg oxotremorine produced a decrease in PPI (p < 0.01 vs. vehicle). Genotype effects were explored due to the interaction, again showing lower PPI in M1−/−M4−/− mice than wild-type after vehicle administration (p < 0.05). In contrast PPI was higher in the M1−/−M4−/− mice relative to wild-type at the highest oxotremorine dose (p < 0.0001). Oxotremorine also decreased startle amplitude [F3,60 = 34.37, p < 0.001], with a significant effect of genotype [F1,60 = 9.90, p < 0.01] but no interaction. Oxotremorine decreased startle amplitude in both M1−/−M4−/− mice [F3,30 = 2.26, p < 0.05] and wild-type mice [F3,30 = 4.16, p < 0.01], at doses of 0.032 and/or 0.1 mg/kg (p < 0.05 to p < 0.01, see Fig. 5B). Startle amplitude and PPI were positively correlated in the wild-type mice (r2 = 0.55, p < 0.0001), but not correlated in the M1−/−M4−/− mice. Further analysis of the genotype effect showed that startle amplitude was higher in M1−/−M4−/− mice than in wild-type mice under vehicle conditions (p < 0.05, in contrast to previous evaluations), and at the highest dose oxotremorine (p < 0.01).
Figure 5. Oxotremorine alone did not increase PPI.
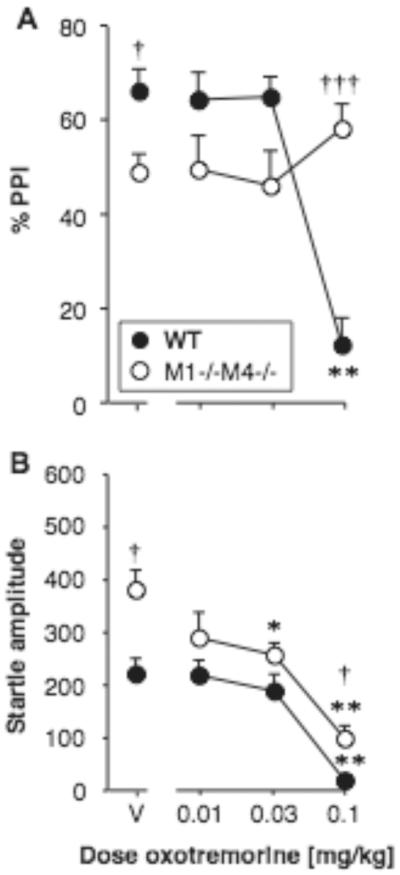
Abscissae: dose oxotremorine [mg/kg]. Ordinates: Total %PPI (A), startle amplitude (B). N=11. Data are group means, bars represent one s.e.m. *p<0.05, **p<0.01 vs. vehicle, Dunnett's multiple comparisons test; †p<0.05, †††p<0.001 vs. wild-type, Bonferroni-corrected unpaired-sample t-test.
Clozapine and haloperidol effects
Effect of clozapine on PPI
Figure 6A–C shows the effect of clozapine on PPI in female M1−/−M4−/− mice, M1−/− mice, M4−/− mice, and their wild-type controls. In the double knockout experiment, PPI was related to clozapine dose [F4,88 = 5.10, p = 0.001], with a genotype by clozapine interaction [F4,88 = 7.77, p < 0.0001], while the overall effect of genotype was not significant. Further analysis showed a significant effect of clozapine in both M1−/−M4−/− mice [F4,44 = 3.51, p < 0.05] and wild-type mice [F4,44 = 7.58, p < 0.0001]. Clozapine increased PPI above vehicle level at 1.8, 3.2 and 5.6 mg/kg (p < 0.05, p < 0.01) in the M1−/−M4−/− mice, while increases in the wild-type mice did not reach significance. At the highest dose clozapine decreased PPI in the wild-type mice (p < 0.01), making PPI significantly higher in the M1−/−M4−/− mice relative to wild-type (p < 0.05) - likely related to a profound suppression of startle amplitude (see Fig. 6D and next section of results).
Figure 6. Clozapine ameliorated the PPI deficit in the female mice lacking M1 and M4 receptors, while startle-suppressing effects of high doses were absent in mice lacking M4 or both receptors.

Abscissae: dose clozapine [mg/kg]. Ordinates: Total %PPI (A–C), startle amplitude (D–F). N=9–12. Data are group means, bars represent one s.e.m. *p<0.05, **p<0.01 vs. vehicle, Dunnett's multiple comparisons test; †p<0.05, ††p<0.01 vs. wild-type, Bonferroni-corrected unpaired-sample t-test.
For the single M1 mutation, PPI was related to clozapine dose [F4,79 = 35.21, p < 0.0001], with no genotype effect or interaction. PPI was decreased at 5.6 mg/kg clozapine in both genotypes (p < 0.01), likely related to the profound reduction in startle amplitude (see Fig. 6E and next section). In contrast, for the single M4 mutation, there was an effect of genotype [F1,19 = 13.61, p < 0.01] and clozapine dose [F4,76 = 13.97, p < 0.0001], with a significant interaction [F4,76 = 7.78, p < 0.0001]. Further analysis showed a significant effect of clozapine in the wild-type mice [F4,32 = 14.24, p < 0.0001], but not in the M4−/− mice. Because clozapine decreased PPI in the wild-type mice, PPI was significantly higher in the M4−/− mice relative to wild-type at 3.2 and 5.6 mg/kg (p < 0.01, p < 0.05).
Effect of clozapine on startle amplitude
Figure 6D–F shows the effect of clozapine on startle amplitude in female M1−/−M4−/− mice, M1−/− mice, M4−/− mice, and wild-type controls. In the double knockout experiment, startle was related to genotype [F1,22 = 16.86, p < 0.001] and clozapine dose [F4,88 = 11.58, p < 0.0001], with a significant interaction [F4,88 = 4.51, p < 0.01]. Further exploration of the interaction showed a significant effect of clozapine in both M1−/−M4−/− mice [F4,44 = 2.81, p < 0.05] and wild-type mice [F4,44 = 20.92, p < 0.0001]. Startle amplitude and PPI were positively correlated in the wild-type mice (r2 = 0.38, p < 0.0001), but were not correlated in the M1−/−M4−/− mice. Clozapine increased startle amplitude above vehicle level at 1.8 mg/kg (p < 0.05) in the M1−/−M4−/− mice, and decreased startle amplitude at 3.2 and 5.6 mg/kg in the wild-type mice (p < 0.01).
In the single M1 mutation, startle amplitude was related to clozapine dose [F4,76 = 53.07, p < 0.0001], with no effect of M1 genotype and no interaction. For both genotypes 3.2 and 5.6 mg/kg clozapine significantly suppressed startle (p <0.01 vs. vehicle). Startle amplitude and PPI were positively correlated in both genotypes (r2 = 0.48 and r2 = 0.37, both p < 0.0001). For the single M4 mutation, startle was related to genotype [F1,19 = 10.86, p < 0.01] and clozapine dose [F4,76 = 21.06, p < 0.0001], with a significant interaction [F4,76 = 2.57, p < 0.05]. There was an effect of clozapine in both M4−/− mice [F4,44 = 5.86, p < 0.001] and wild-type mice [F4,32 = 23.72, p < 0.0001], with significant decreases in startle amplitude at 5.6 mg/kg in the M4−/− mice (p < 0.05) and at 3.2 and 5.6 mg/kg in the wild-type mice (p < 0.01). Startle amplitude and PPI were not correlated in the M4−/− mice.
Effect of haloperidol on PPI and startle amplitude
Haloperidol was tested in the first batch of female M1−/−M4−/− mice and their wild-type controls. Haloperidol increased PPI in both genotypes (Fig. 7A). PPI was related to genotype [F1,10 = 9.20, p < 0.05] and haloperidol dose [F3,30 = 4.50, p < 0.05], with no significant interaction. Because of the small sample size and the lack of interaction, dose effects are reported for both genotypes combined. Figure 7B shows startle amplitudes, which were not significantly related to genotype or haloperidol dose, and with no interaction. Because the effect of haloperidol did not differ between genotypes, it was not tested further.
Figure 7. Haloperidol increased PPI regardless of M1/M4 genotype.
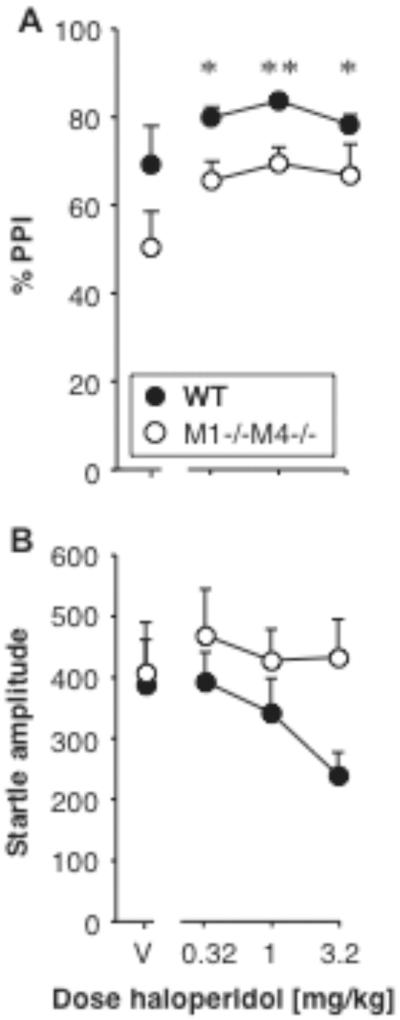
Abscissae: dose haloperidol [mg/kg]. Ordinates: Total %PPI (A), startle amplitude (B). N=6. Data are group means, bars represent one s.e.m. *p<0.05, **p<0.01 vs. vehicle, Dunnett's multiple comparisons test (genotypes combined).
Discussion
We evaluated PPI and pharmacological modulation of PPI in mice constitutively lacking muscarinic cholinergic M1 or M4 receptors or both receptor subtypes. PPI was decreased in female mice lacking both M1 and M4 receptors, while the deletion of either subtype alone had no significant effect on baseline PPI. Thus a lack of M1 and M4 receptors can generate a phenotype indicative of reduced sensorimotor gating. While a constitutive inactivation of a gene in mice cannot “model” a complex human brain disorder, this finding supports a possible involvement of muscarinic M1 and M4 receptors in the etiology of schizophrenia, suggested by numerous studies reporting decreased densities of muscarinic M1 and/or M4 receptors in the brains of schizophrenic patients (Crook et al. 1999; Dean et al. 2002; Mancama et al. 2003; Raedler et al. 2003; Zavitsanou et al. 2004; Deng and Huang 2005; Newell et al. 2007, Scarr et al. 2007; and see Vakalopoulos 2006). In an intriguing article Borda et al. (2002) demonstrated the presence of anti-M1 antibodies in the serum of schizophrenic patients, but not of healthy controls, and suggested an autoimmune etiology to schizophrenia (see also Stefansson et al. 2009). Our results further suggest overlapping functions of M1 and M4 receptors, at least to the extent that constitutive deletion of either subtype can be compensated for in mice to maintain largely normal function. Similar to our findings, a previous study reported unaltered baseline PPI in M1−/− mice, although only male mice were tested (Miyakawa et al. 2001). But M1−/− mice have also shown some phenotypes reminiscent of schizophrenia-like symptoms, such as striatal hyperdopaminergia, hyperactivity and impaired performance in some cognitive tests (Gerber et al. 2001; Miyakawa et al. 2001; Anagnostaras et al. 2003). M4−/− mice showed normal passive avoidance and only inconsistent locomotor hyperactivity at baseline, but did show striatal hyperdopaminergia and exaggerated responses to direct and indirect dopamine agonists or phencyclidine (Gomeza et al. 1999; Felder et al. 2001; Tzavara et al. 2003, 2004). Electrophysiological recordings in striatal slices suggested altered synaptic plasticity in M4−/− mice (Bonsi et al. 2008). Thus deletion of either M1 or M4 receptors may cause less overt impairments which when combined produce a phenotype including reduced PPI. It should be noted however that in earlier studies sexes were combined, unspecified, or male only, while we detected significant effects of the M1/M4 deletion only in female mice.
In the initial experiments of this investigation we tested both male and female mice. We found that in female mice the double mutation affected both baseline PPI and responses to scopolamine, while this phenotype was not observed in male mice. The only statistically significant difference between wild-type and M1−/−M4−/− male mice was increased startle reactivity after scopolamine administration, similarly observed in the female mice. As previous studies in M1−/− or M4−/− mice typically used only male mice or reported results as sexes combined, we cannot draw parallels to potential sex effects in those investigations. More generally, in rodents most cholinergic neurons express estrogen receptors and cholinergic systems show many sex differences, including rates of maturation and aging, cell size and enzyme activity, and sensitivity to pharmacological manipulations or lesions (Fragkouli et al. 2006). Sex differences in brain muscarinic binding sites were also reported in wild-type mice (Fragkouli et al. 2006). Thus it may be that background sex differences in cholinergic systems resulted in male and female mice developing compensatory mechanisms differently, the male mice being perhaps better able to surmount effects of the mutation as measured in this investigation. There are numerous examples of sex differences in the expression of gene deletions in mice, including of muscarinic receptors. We previously saw PPI deficits in M5−/− mice that were more pronounced in female than in male mice (Thomsen et al. 2007). In other studies, female M5−/− mice were protected from the reduction in cerebral blood flow seen in male mice, and ovariectomy uncovered this phenotype in the female mice (Kitamura et al. 2009). Male and female M1−/− or M2−/− mice showed differential phenotypes in stress responses, with the clearest phenotypes in female versus male M1−/− mice, but the converse in M2−/− mice (Rhodes et al. 2005, 2008). In humans, reports of reduced muscarinic receptor levels in schizophrenic patients typically reported sexes combined, and were also observed in an all-male sample (Deng and Huang 2005). Thus the differences between sexes we observed may be more related to general sex differences in mouse cholinergic systems development, rather than being predictive of related sex differences in human schizophrenic patients, although the latter cannot be excluded. While sex differences in human cholinergic systems are not well-characterized, recent evidence that estrogen therapy in post-menopausal women protected against age-related loss of brain M1/M4 receptor density suggests some interaction between brain muscarinic systems and sex hormones in humans (Norbury et al. 2007). Further studies, e.g., using overiectomized mice, are warranted to explore interactions between sex, muscarinic receptor-related phenotypes, and drug effects.
In addition to affecting baseline PPI, the M1 and M4 receptor mutations affected the pharmacological modulation of startle and PPI. The non-subtype selective muscarinic antagonist scopolamine decreased PPI in male and female wild-type mice, consistent with previous reports in rats and mice (Wu et al. 1993; Jones and Shannon 2000; Sipos et al. 2001; Stanhope et al. 2001; Ukai et al. 2004; Jones et al. 2005). We found that in female mice lacking M1 receptors alone or both M1 and M4 receptors, scopolamine failed to significantly suppress PPI. Mice lacking M4 receptors alone showed decreases in PPI comparable to wild-type mice. While it cannot be excluded that the lack of effect in the M1−/−M4−/− mice reflects a “floor effect” due to the low baseline PPI, this is unlikely to be the case for the M1−/− mice, which showed baseline PPI comparable to wild-type mice. Based on pharmacological studies, some authors have suggested an involvement of M3 or M4 receptors in muscarinic antagonist-induced PPI disruption (Ukai et al. 2004). Others demonstrated the difficulty in obtaining conclusive evidence to implicate one receptor subtype based on available, moderately selective, muscarinic antagonists (Jones and Shannon 2000). While not excluding the possible contribution of other subtypes, our results support a role of M1, but not M4 receptors in the disruption of PPI by scopolamine. Scopolamine also increased startle reactivity in all genotypes, in agreement with a previous study in rats (Sipos et al. 2001), although in many investigations scopolamine did not increase startle reactivity significantly (Jones and Shannon 2000; Ouagazzal et al. 2001; Stanhope et al. 2001). In contrast to PPI, the deletion of M4 receptors or of both M1 and M4 receptors seemed to exacerbate the effect of scopolamine on startle but also resulted in increased baseline startle reactivity, while the M1 mutation had little effect. This dissociation between startle and PPI, along with a general lack of correlation between startle and PPI, makes it unlikely that scopolamine-induced decreases in PPI were attributable to direct effects on startle reactivity. In summary our data are consistent with a role of both M1 and M4 receptors in scopolamine's behavioral effects in mice, but most clearly supports a role of M1 receptors in its PPI-disrupting effect.
The M1/M4-preferring agonist xanomeline has been suggested as a novel antipsychotic agent, showing antipsychotic-like effects in both preclinical and clinical studies (Shannon et al. 2000; Stanhope et al. 2001; Andersen et al. 2003; Bodick et al. 1997; Shekhar et al. 2008). In the present study xanomeline pretreatment reversed scopolamine-induced disruption of PPI in wild-type mice, in agreement with previous studies in rats showing amelioration of apomorphine- or scopolamine-induced PPI disruption (Stanhope et al. 2001; Jones et al. 2005). Xanomeline has moderate functional selectivity for M1/M4 receptors over other muscarinic subtypes, and may also elicit effects via non-muscarinic receptors, such as 5-HT receptors (Shannon et al. 1994; Watson et al. 1998; Langmead et al. 2008a). It is not well established which receptors mediate antipsychotic-like effects of xanomeline. Here we found that the PPI-ameliorating effect of xanomeline was absent in the M4−/− and M1−/−M4−/− mice, but was largely intact in the M1−/− mice, suggesting a critical role of M4 but not M1 receptors in xanomeline's antipsychotic efficacy. This finding is consistent with the hypothesis advanced by Bymaster and coworkers, who proposed that M4 agonists may have antipsychotic effects while M1 agonists may be useful primarily as cognitive enhancers (Bymaster et al. 2002). Similarly a recent report indicated that xanomeline's ability to reverse amphetamine-induced hyperactivity was absent in M4−/− mice, with a trend for attenuation in M1−/− mice (Woolley et al. 2009). Also, recently developed M4-selective positive allosteric modulators showed antipsychotic-like effects in preclinical assays in rats, including PPI (Brady et al. 2008; Chan et al. 2008). Taken together these observations suggest xanomeline exerts antipsychotic-like effects primarily through M4 receptor stimulation.
We further assessed whether a non-selective muscarinic agonist would reverse scopolamine-induced PPI disruption and whether those effects would similarly be lacking in the M1−/−M4−/− and M4−/− mice. We found that oxotremorine ameliorated PPI regardless of genotype. The same doses of oxotremorine per se failed to increase PPI, with an apparent but non-significant increase in the M1−/−M4−/− mice only. The highest doses decreased startle amplitude dramatically, ruling out that too low doses were tested to observe an effect. Previous investigations similarly showed oxotremorine ameliorated PPI disrupted by apomorphine, methamphetamine or ketamine in rats, and increased PPI only in a mouse strain with low baseline PPI (Jones et al. 2005; Maehara et al. 2008). Therefore the lack of increase in PPI (without scopolamine challenge) in the present study likely reflects a “ceiling effect” rather than a scopolamine/oxotremorine interaction. Because M1 and M4 receptor deletions did not diminish oxotremorine's effects in the present investigation, our findings indicate that M2, M3 or M5 receptor stimulation is sufficient to ameliorate PPI, at least in the knockout mice. The extent to which compensatory changes in the knockout mice may have altered contributions of other receptors cannot be fully ascertained. However the targets that were investigated revealed no changes: no changes in muscarinic M2, dopamine D1-like or D2-like receptor expression were detected in M4−/− mice, and no changes in striatal M2-M5 receptor expression were detected in M1−/− mice (Gomeza et al. 1999; Miyakawa et al. 2001). We previously reported decreased PPI in M5−/− mice, suggesting a role of M5 receptors (Thomsen et al. 2007). While M2−/− mice showed normal PPI, they have shown profound deficits in neuronal plasticity as well as impaired performance in cognitive tests (Felder et al. 2001; Bymaster et al. 2002; Tzavara et al. 2003; Seeger et al. 2004). In addition the M2/M4 partial agonist M1 antagonist BuTAC reversed apomorphine-suppressed PPI in rats (Jones et al. 2005). Thus M2 and M5 receptor stimulation both appear plausible additional mechanisms of PPI amelioration, at least in mice lacking M1 and/or M4 receptors.
Finally we evaluated the ability of the atypical antipsychotic clozapine and of the typical antipsychotic haloperidol to ameliorate the PPI deficit seen in the female M1−/−M4−/− mice. Clozapine and its metabolites have high affinity for muscarinic receptors, and M1 stimulation has been speculated to contribute to clozapine's antipsychotic effects (Olianas et al. 1999; Sur et al. 2003; Weiner et al. 2004; Davies et al. 2005). We found that clozapine increased PPI in the female M1−/−M4−/− mice, reversing the PPI deficit to wild-type levels. Thus neither M1 nor M4 receptors likely played a significant role in the antipsychotic-like effects of clozapine in the present experiments. Our results are consistent with a previous report showing comparable attenuation of locomotor activity by clozapine in wild-type and M1−/− mice (Gerber et al. 2001). Clozapine appears to be only a weak partial agonist at M1 receptors in vivo, and behavioral data are consistent with general muscarinic antagonist, rather than agonist, actions of clozapine (Bymaster et al. 2003; Prus et al. 2004). Taken together these observations do not support a significant role of M1 (or M4) receptor stimulation in mediating antipsychotic-like effects of clozapine. At high doses, we found that clozapine strongly suppressed startle reactivity and produced parallel decreases in PPI in wild-type mice. This is in agreement with similar effects on startle in rats and mice (Ouagazzal et al. 2001; Maehara et al. 2008). In contrast to PPI this effect of clozapine was absent in the M1−/−M4−/− mice and dramatically reduced in the M4−/− mice, but intact in the M1−/− mice. Thus it is possible that M4 receptors play a role in adverse effects of clozapine, although the doses at which these effects were overt in the present study may not be clinically relevant. Finally, haloperidol also increased PPI in the M1−/−M4−/− mice, comparable to its effect in wild-type mice. Thus the PPI deficit seen in M1−/−M4−/− mice could be reverted by clozapine or haloperidol administration, with no indication that M1 or M4 receptors were essential to the antipsychotic-like effects of either drug.
In conclusion, we evaluated PPI and pharmacological modulation of PPI in mice lacking muscarinic cholinergic M1 or M4 receptors or both receptor subtypes. We found that constitutive deletion of either subtype did not affect baseline PPI but that combined deletion of both subtypes decreased PPI in female mice, lending support to the theory that muscarinic dysfunction with decreased M1/M4 receptor densities is involved in the etiology of schizophrenia. The reason why the phenotype was apparent only in female animals remains unclear and deserves further investigation. Our data also suggested that suppression of PPI by the muscarinic antagonist scopolamine was dependent upon M1 receptors and that amelioration of PPI by the potential antipsychotic drug xanomeline was dependent upon M4 receptors. The latter finding supports the concept that the M4 receptor, rather than the M1 receptor, represents a key target for developing potential novel antipsychotic drugs. Finally, clozapine and haloperidol both ameliorated PPI in the double knockout mice, failing to support the notion that M1 stimulation is critical to clozapine's antipsychotic efficacy.
Acknowledgements
This research was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Disorders (J.W). M.T. was supported in part by a NARSAD Young Investigator Award and a Eleanor and Miles Shore / Harvard Medical School Fellowship during this work. All procedures were carried out in accordance with the Guidelines for the Care and Use of Mammals in Neuroscience and Behavioral Research (National Research Council 2003). We thank Joon Y. Boon and Kate Woodard for technical assistance.
References
- Anagnostaras SG, Murphy GG, Hamilton SE, Mitchell SL, Rahnama NP, Nathanson NM, Silva AJ. Selective cognitive dysfunction in acetylcholine M1 muscarinic receptor mutant mice. Nat Neurosci. 2003;6:51–8. doi: 10.1038/nn992. [DOI] [PubMed] [Google Scholar]
- Andersen MB, Fink-Jensen A, Peacock L, Gerlach J, Bymaster F, Lundbaek JA, Werge T. The muscarinic M1/M4 receptor agonist xanomeline exhibits antipsychotic-like activity in Cebus apella monkeys. Neuropsychopharmacology. 2003;28:1168–75. doi: 10.1038/sj.npp.1300151. [DOI] [PubMed] [Google Scholar]
- Bartus RT. Physostigmine and recent memory: effects in young and aged nonhuman primates. Science. 1979;206:1087–9. doi: 10.1126/science.227061. [DOI] [PubMed] [Google Scholar]
- Bodick NC, Offen WW, Levey AI, Cutler NR, Gauthier SG, Satlin A, Shannon HE, Tollefson GD, Rasmussen K, Bymaster FP, Hurley DJ, Potter WZ, Paul SM. Effects of xanomeline, a selective muscarinic receptor agonist, on cognitive function and behavioral symptoms in Alzheimer disease. Arch Neurol. 1997;54:465–73. doi: 10.1001/archneur.1997.00550160091022. [DOI] [PubMed] [Google Scholar]
- Bonsi P, Martella G, Cuomo D, Platania P, Sciamanna G, Bernardi G, Wess J, Pisani A. Loss of muscarinic autoreceptor function impairs long-term depression but not long-term potentiation in the striatum. J Neurosci. 2008;28:6258–63. doi: 10.1523/JNEUROSCI.1678-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borda T, Perez Rivera R, Joensen L, Gomez RM, Sterin-Borda L. Antibodies against cerebral M1 cholinergic muscarinic receptor from schizophrenic patients: molecular interaction. J Immunol. 2002;168:3667–74. doi: 10.4049/jimmunol.168.7.3667. [DOI] [PubMed] [Google Scholar]
- Brady AE, Jones CK, Bridges TM, Kennedy JP, Thompson AD, Heiman JU, Breininger ML, Gentry PR, Yin H, Jadhav SB, Shirey JK, Conn PJ, Lindsley CW. Centrally active allosteric potentiators of the M4 muscarinic acetylcholine receptor reverse amphetamine-induced hyperlocomotor activity in rats. J Pharmacol Exp Ther. 2008;327:941–53. doi: 10.1124/jpet.108.140350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braff DL, Geyer MA, Swerdlow NR. Human studies of prepulse inhibition of startle: normal subjects, patient groups, and pharmacological studies. Psychopharmacology (Berl) 2001;156:234–58. doi: 10.1007/s002130100810. [DOI] [PubMed] [Google Scholar]
- Buchanan RW, Freedman R, Javitt DC, Abi-Dargham A, Lieberman JA. Recent advances in the development of novel pharmacological agents for the treatment of cognitive impairments in schizophrenia. Schizophr Bull. 2007;33:1120–30. doi: 10.1093/schbul/sbm083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bymaster FP, Felder C, Ahmed S, McKinzie D. Muscarinic receptors as a target for drugs treating schizophrenia. Curr Drug Targets CNS Neurol Disord. 2002;1:163–81. doi: 10.2174/1568007024606249. [DOI] [PubMed] [Google Scholar]
- Bymaster FP, Felder CC, Tzavara E, Nomikos GG, Calligaro DO, McKinzie DL. Muscarinic mechanisms of antipsychotic atypicality. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:1125–43. doi: 10.1016/j.pnpbp.2003.09.008. [DOI] [PubMed] [Google Scholar]
- Bymaster FP, Shannon HE, Rasmussen K, Delapp NW, Mitch CH, Ward JS, Calligaro DO, Ludvigsen TS, Sheardown MJ, Olesen PH, Swedberg MD, Sauerberg P, Fink-Jensen A. Unexpected antipsychotic-like activity with the muscarinic receptor ligand (5R,6R)6-(3-propylthio-1,2,5-thiadiazol-4-yl)-1-azabicyclo[3.2.1]octane. Eur J Pharmacol. 1998;356:109–19. doi: 10.1016/s0014-2999(98)00487-7. [DOI] [PubMed] [Google Scholar]
- Chan WY, McKinzie DL, Bose S, Mitchell SN, Witkin JM, Thompson RC, Christopoulos A, Lazareno S, Birdsall NJ, Bymaster FP, Felder CC. Allosteric modulation of the muscarinic M4 receptor as an approach to treating schizophrenia. Proc Natl Acad Sci U S A. 2008;105:10978–83. doi: 10.1073/pnas.0800567105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman CA, Yeomans JS, Blaha CD, Blackburn JR. Increased striatal dopamine efflux follows scopolamine administered systemically or to the tegmental pedunculopontine nucleus. Neuroscience. 1997;76:177–86. doi: 10.1016/s0306-4522(96)00358-2. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Jones CK, Lindsley CW. Subtype-selective allosteric modulators of muscarinic receptors for the treatment of CNS disorders. Trends Pharmacol Sci. 2009;30:148–55. doi: 10.1016/j.tips.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crook JM, Dean B, Pavey G, Copolov D. The binding of [3H]AF-DX 384 is reduced in the caudate-putamen of subjects with schizophrenia. Life Sci. 1999;64:1761–71. doi: 10.1016/s0024-3205(99)00114-9. [DOI] [PubMed] [Google Scholar]
- Davies MA, Compton-Toth BA, Hufeisen SJ, Meltzer HY, Roth BL. The highly efficacious actions of N-desmethylclozapine at muscarinic receptors are unique and not a common property of either typical or atypical antipsychotic drugs: is M1 agonism a pre-requisite for mimicking clozapine's actions? Psychopharmacology (Berl) 2005;178:451–60. doi: 10.1007/s00213-004-2017-1. [DOI] [PubMed] [Google Scholar]
- Davis KL, Mohs RC, Tinklenberg JR, Pfefferbaum A, Hollister LE, Kopell BS. Physostigmine: improvement of long-term memory processes in normal humans. Science. 1978;201:272–4. doi: 10.1126/science.351807. [DOI] [PubMed] [Google Scholar]
- Dean B, McLeod M, Keriakous D, McKenzie J, Scarr E. Decreased muscarinic1 receptors in the dorsolateral prefrontal cortex of subjects with schizophrenia. Mol Psychiatry. 2002;7:1083–91. doi: 10.1038/sj.mp.4001199. [DOI] [PubMed] [Google Scholar]
- Deng C, Huang XF. Decreased density of muscarinic receptors in the superior temporal gyrusin schizophrenia. J Neurosci Res. 2005;81:883–90. doi: 10.1002/jnr.20600. [DOI] [PubMed] [Google Scholar]
- Dewey SL, Smith GS, Logan J, Brodie JD, Simkowitz P, MacGregor RR, Fowler JS, Volkow ND, Wolf AP. Effects of central cholinergic blockade on striatal dopamine release measured with positron emission tomography in normal human subjects. Proc Natl Acad Sci U S A. 1993;90:11816–20. doi: 10.1073/pnas.90.24.11816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Chiara G, Morelli M, Consolo S. Modulatory functions of neurotransmitters in the striatum: ACh/dopamine/NMDA interactions. Trends Neurosci. 1994;17:228–33. doi: 10.1016/0166-2236(94)90005-1. [DOI] [PubMed] [Google Scholar]
- Ellis JR, Ellis KA, Bartholomeusz CF, Harrison BJ, Wesnes KA, Erskine FF, Vitetta L, Nathan PJ. Muscarinic and nicotinic receptors synergistically modulate working memory and attention in humans. Int J Neuropsychopharmacol. 2006;9:175–89. doi: 10.1017/S1461145705005407. [DOI] [PubMed] [Google Scholar]
- Felder CC, Porter AC, Skillman TL, Zhang L, Bymaster FP, Nathanson NM, Hamilton SE, Gomeza J, Wess J, McKinzie DL. Elucidating the role of muscarinic receptors in psychosis. Life Sci. 2001;68:2605–13. doi: 10.1016/s0024-3205(01)01059-1. [DOI] [PubMed] [Google Scholar]
- Fink-Jensen A, Kristensen P, Shannon HE, Calligaro DO, Delapp NW, Whitesitt C, Ward JS, Thomsen C, Rasmusseen T, Sheardown MJ, Jeppesen L, Sauerberg P, Bymaster FP. Muscarinic agonists exhibit functional dopamine antagonism in unilaterally 6-OHDA lesioned rats. Neuroreport. 1998;9:3481–6. doi: 10.1097/00001756-199810260-00027. [DOI] [PubMed] [Google Scholar]
- Fragkouli A, Stamatakis A, Zographos E, Pachnis V, Stylianopoulou F. Sexually dimorphic effects of the Lhx7 null mutation on forebrain cholinergic function. Neuroscience. 2006;137:1153–64. doi: 10.1016/j.neuroscience.2005.10.037. [DOI] [PubMed] [Google Scholar]
- Friedman JI. Cholinergic targets for cognitive enhancement in schizophrenia: focus on cholinesterase inhibitors and muscarinic agonists. Psychopharmacology (Berl) 2004;174:45–53. doi: 10.1007/s00213-004-1794-x. [DOI] [PubMed] [Google Scholar]
- Gerber DJ, Sotnikova TD, Gainetdinov RR, Huang SY, Caron MG, Tonegawa S. Hyperactivity, elevated dopaminergic transmission, and response to amphetamine in M1 muscarinic acetylcholine receptor-deficient mice. Proc Natl Acad Sci U S A. 2001;98:15312–7. doi: 10.1073/pnas.261583798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyer MA, Krebs-Thomson K, Braff DL, Swerdlow NR. Pharmacological studies of prepulse inhibition models of sensorimotor gating deficits in schizophrenia: a decade in review. Psychopharmacology (Berl) 2001;156:117–54. doi: 10.1007/s002130100811. [DOI] [PubMed] [Google Scholar]
- Geyer MA, McIlwain KL, Paylor R. Mouse genetic models for prepulse inhibition: an early review. Mol Psychiatry. 2002;7:1039–53. doi: 10.1038/sj.mp.4001159. [DOI] [PubMed] [Google Scholar]
- Gomeza J, Zhang L, Kostenis E, Felder C, Bymaster F, Brodkin J, Shannon H, Xia B, Deng C, Wess J. Enhancement of D1 dopamine receptor-mediated locomotor stimulation in M(4) muscarinic acetylcholine receptor knockout mice. Proc Natl Acad Sci U S A. 1999;96:10483–8. doi: 10.1073/pnas.96.18.10483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpern JH. Hallucinogens and dissociative agents naturally growing in the United States. Pharmacol Ther. 2004;102:131–8. doi: 10.1016/j.pharmthera.2004.03.003. [DOI] [PubMed] [Google Scholar]
- Harries MH, Samson NA, Cilia J, Hunter AJ. The profile of sabcomeline (SB-202026), a functionally selective M1 receptor partial agonist, in the marmoset. Br J Pharmacol. 1998;124:409–15. doi: 10.1038/sj.bjp.0701844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselmo ME. The role of acetylcholine in learning and memory. Curr Opin Neurobiol. 2006;16:710–5. doi: 10.1016/j.conb.2006.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges H, Peters S, Gray JA, Hunter AJ. Counteractive effects of a partial (sabcomeline) and a full (RS86) muscarinic receptor agonist on deficits in radial maze performance induced by S-AMPA lesions of the basal forebrain and medial septal area. Behav Brain Res. 1999;99:81–92. doi: 10.1016/s0166-4328(98)00075-8. [DOI] [PubMed] [Google Scholar]
- Johnstone EC, Crow TJ, Ferrier IN, Frith CD, Owens DG, Bourne RC, Gamble SJ. Adverse effects of anticholinergic medication on positive schizophrenic symptoms. Psychol Med. 1983;13:513–27. doi: 10.1017/s0033291700047942. [DOI] [PubMed] [Google Scholar]
- Jones CK, Brady AE, Davis AA, Xiang Z, Bubser M, Tantawy MN, Kane AS, Bridges TM, Kennedy JP, Bradley SR, Peterson TE, Ansari MS, Baldwin RM, Kessler RM, Deutch AY, Lah JJ, Levey AI, Lindsley CW, Conn PJ. Novel selective allosteric activator of the M1 muscarinic acetylcholine receptor regulates amyloid processing and produces antipsychotic-like activity in rats. J Neurosci. 2008;28:10422–33. doi: 10.1523/JNEUROSCI.1850-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CK, Eberle EL, Shaw DB, McKinzie DL, Shannon HE. Pharmacologic interactions between the muscarinic cholinergic and dopaminergic systems in the modulation of prepulse inhibition in rats. J Pharmacol Exp Ther. 2005;312:1055–63. doi: 10.1124/jpet.104.075887. [DOI] [PubMed] [Google Scholar]
- Jones CK, Shannon HE. Muscarinic cholinergic modulation of prepulse inhibition of the acoustic startle reflex. J Pharmacol Exp Ther. 2000;294:1017–23. [PubMed] [Google Scholar]
- Kane BE, Grant MK, El-Fakahany EE, Ferguson DM. Synthesis and evaluation of xanomeline analogs--probing the wash-resistant phenomenon at the M1 muscarinic acetylcholine receptor. Bioorg Med Chem. 2008;16:1376–92. doi: 10.1016/j.bmc.2007.10.058. [DOI] [PubMed] [Google Scholar]
- Kitamura N, Araya R, Kudoh M, Kishida H, Kimura T, Murayama M, Takashima A, Sakamaki Y, Hashikawa T, Ito S, Ohtsuki S, Terasaki T, Wess J, Yamada M. Beneficial effects of estrogen in a mouse model of cerebrovascular insufficiency. PLoS One. 2009;4:e5159. doi: 10.1371/journal.pone.0005159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari V, Zachariah E, Galea A, Mehrotra R, Taylor D, Sharma T. Effects of procyclidine on prepulse inhibition of the acoustic startle response in healthy human volunteers. Psychopharmacology (Berl) 2001;154:221–9. doi: 10.1007/s002130000656. [DOI] [PubMed] [Google Scholar]
- Langmead CJ, Austin NE, Branch CL, Brown JT, Buchanan KA, Davies CH, Forbes IT, Fry VA, Hagan JJ, Herdon HJ, Jones GA, Jeggo R, Kew JN, Mazzali A, Melarange R, Patel N, Pardoe J, Randall AD, Roberts C, Roopun A, Starr KR, Teriakidis A, Wood MD, Whittington M, Wu Z, Watson J. Characterization of a CNS penetrant, selective M1 muscarinic receptor agonist, 77-LH-28-1. Br J Pharmacol. 2008a;154:1104–15. doi: 10.1038/bjp.2008.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead CJ, Watson J, Reavill C. Muscarinic acetylcholine receptors as CNS drug targets. Pharmacol Ther. 2008b;117:232–43. doi: 10.1016/j.pharmthera.2007.09.009. [DOI] [PubMed] [Google Scholar]
- Levey AI, Kitt CA, Simonds WF, Price DL, Brann MR. Identification and localization of muscarinic acetylcholine receptor proteins in brain with subtype-specific antibodies. J Neurosci. 1991;11:3218–26. doi: 10.1523/JNEUROSCI.11-10-03218.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maehara S, Hikichi H, Satow A, Okuda S, Ohta H. Antipsychotic property of a muscarinic receptor agonist in animal models for schizophrenia. Pharmacol Biochem Behav. 2008;91:140–9. doi: 10.1016/j.pbb.2008.06.023. [DOI] [PubMed] [Google Scholar]
- Mancama D, Arranz MJ, Landau S, Kerwin R. Reduced expression of the muscarinic 1 receptor cortical subtype in schizophrenia. Am J Med Genet B Neuropsychiatr Genet. 2003;119B:2–6. doi: 10.1002/ajmg.b.20020. [DOI] [PubMed] [Google Scholar]
- McGinty JF. Regulation of neurotransmitter interactions in the ventral striatum. Ann N Y Acad Sci. 1999;877:129–39. doi: 10.1111/j.1749-6632.1999.tb09265.x. [DOI] [PubMed] [Google Scholar]
- Minzenberg MJ, Poole JH, Benton C, Vinogradov S. Association of anticholinergic load with impairment of complex attention and memory in schizophrenia. Am J Psychiatry. 2004;161:116–24. doi: 10.1176/appi.ajp.161.1.116. [DOI] [PubMed] [Google Scholar]
- Miyakawa T, Yamada M, Duttaroy A, Wess J. Hyperactivity and intact hippocampus-dependent learning in mice lacking the M1 muscarinic acetylcholine receptor. J Neurosci. 2001;21:5239–50. doi: 10.1523/JNEUROSCI.21-14-05239.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newell KA, Zavitsanou K, Jew SK, Huang XF. Alterations of muscarinic and GABA receptor binding in the posterior cingulate cortex in schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31:225–33. doi: 10.1016/j.pnpbp.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Norbury R, Travis MJ, Erlandsson K, Waddington W, Ell PJ, Murphy DG. Estrogen therapy and brain muscarinic receptor density in healthy females: a SPET study. Horm Behav. 2007;51:249–57. doi: 10.1016/j.yhbeh.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Olianas MC, Maullu C, Onali P. Mixed agonist-antagonist properties of clozapine at different human cloned muscarinic receptor subtypes expressed in Chinese hamster ovary cells. Neuropsychopharmacology. 1999;20:263–70. doi: 10.1016/S0893-133X(98)00048-7. [DOI] [PubMed] [Google Scholar]
- Onali P, Olianas MC. Muscarinic M4 receptor inhibition of dopamine D1-like receptor signalling in rat nucleus accumbens. Eur J Pharmacol. 2002;448:105–11. doi: 10.1016/s0014-2999(02)01910-6. [DOI] [PubMed] [Google Scholar]
- Ouagazzal AM, Jenck F, Moreau JL. Drug-induced potentiation of prepulse inhibition of acoustic startle reflex in mice: a model for detecting antipsychotic activity? Psychopharmacology (Berl) 2001;156:273–83. doi: 10.1007/s002130100763. [DOI] [PubMed] [Google Scholar]
- Perry EK, Perry RH. Acetylcholine and hallucinations: disease-related compared to drug-induced alterations in human consciousness. Brain Cogn. 1995;28:240–58. doi: 10.1006/brcg.1995.1255. [DOI] [PubMed] [Google Scholar]
- Power AE, Vazdarjanova A, McGaugh JL. Muscarinic cholinergic influences in memory consolidation. Neurobiol Learn Mem. 2003;80:178–93. doi: 10.1016/s1074-7427(03)00086-8. [DOI] [PubMed] [Google Scholar]
- Prus AJ, Baker LE, Meltzer HY. Discriminative stimulus properties of 1.25 and 5.0 mg/kg doses of clozapine in rats: examination of the role of dopamine, serotonin, and muscarinic receptor mechanisms. Pharmacol Biochem Behav. 2004;77:199–208. doi: 10.1016/j.pbb.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Raedler TJ, Bymaster FP, Tandon R, Copolov D, Dean B. Towards a muscarinic hypothesis of schizophrenia. Mol Psychiatry. 2007;12:232–46. doi: 10.1038/sj.mp.4001924. [DOI] [PubMed] [Google Scholar]
- Raedler TJ, Knable MB, Jones DW, Urbina RA, Gorey JG, Lee KS, Egan MF, Coppola R, Weinberger DR. In vivo determination of muscarinic acetylcholine receptor availability in schizophrenia. Am J Psychiatry. 2003;160:118–27. doi: 10.1176/appi.ajp.160.1.118. [DOI] [PubMed] [Google Scholar]
- Rhodes ME, Billings TE, Czambel RK, Rubin RT. Pituitary-adrenal responses to cholinergic stimulation and acute mild stress are differentially elevated in male and female M(2) muscarinic receptor knockout mice. J Neuroendocrinol. 2005;17:817–26. doi: 10.1111/j.1365-2826.2005.01376.x. [DOI] [PubMed] [Google Scholar]
- Rhodes ME, Rubin RT, McKlveen JM, Karwoski TE, Fulton BA, Czambel RK. Pituitary-adrenal responses to oxotremorine and acute stress in male and female M1 muscarinic receptor knockout mice: comparisons to M2 muscarinic receptor knockout mice. J Neuroendocrinol. 2008;20:617–25. doi: 10.1111/j.1365-2826.2008.01700.x. [DOI] [PubMed] [Google Scholar]
- Sarter M, Nelson CL, Bruno JP. Cortical cholinergic transmission and cortical information processing in schizophrenia. Schizophr Bull. 2005;31:117–38. doi: 10.1093/schbul/sbi006. [DOI] [PubMed] [Google Scholar]
- Sauerberg P, Olesen PH, Nielsen S, Treppendahl S, Sheardown MJ, Honore T, Mitch CH, Ward JS, Pike AJ, Bymaster FP, et al. Novel functional M1 selective muscarinic agonists. Synthesis and structure-activity relationships of 3-(1,2,5-thiadiazolyl)-1,2,5,6-tetrahydro-1-methylpyridines. J Med Chem. 1992;35:2274–83. doi: 10.1021/jm00090a019. [DOI] [PubMed] [Google Scholar]
- Scarr E, Sundram S, Keriakous D, Dean B. Altered hippocampal muscarinic M4, but not M1, receptor expression from subjects with schizophrenia. Biol Psychiatry. 2007;61:1161–70. doi: 10.1016/j.biopsych.2006.08.050. [DOI] [PubMed] [Google Scholar]
- Seeger T, Fedorova I, Zheng F, Miyakawa T, Koustova E, Gomeza J, Basile AS, Alzheimer C, Wess J. M2 muscarinic acetylcholine receptor knock-out mice show deficits in behavioral flexibility, working memory, and hippocampal plasticity. J Neurosci. 2004;24:10117–27. doi: 10.1523/JNEUROSCI.3581-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon HE, Bymaster FP, Calligaro DO, Greenwood B, Mitch CH, Sawyer BD, Ward JS, Wong DT, Olesen PH, Sheardown MJ, et al. Xanomeline: a novel muscarinic receptor agonist with functional selectivity for M1 receptors. J Pharmacol Exp Ther. 1994;269:271–81. [PubMed] [Google Scholar]
- Shannon HE, Hart JC, Bymaster FP, Calligaro DO, DeLapp NW, Mitch CH, Ward JS, Fink-Jensen A, Sauerberg P, Jeppesen L, Sheardown MJ, Swedberg MD. Muscarinic receptor agonists, like dopamine receptor antagonist antipsychotics, inhibit conditioned avoidance response in rats. J Pharmacol Exp Ther. 1999;290:901–7. [PubMed] [Google Scholar]
- Shannon HE, Rasmussen K, Bymaster FP, Hart JC, Peters SC, Swedberg MD, Jeppesen L, Sheardown MJ, Sauerberg P, Fink-Jensen A. Xanomeline, an M(1)/M(4) preferring muscarinic cholinergic receptor agonist, produces antipsychotic-like activity in rats and mice. Schizophr Res. 2000;42:249–59. doi: 10.1016/s0920-9964(99)00138-3. [DOI] [PubMed] [Google Scholar]
- Shekhar A, Potter WZ, Lightfoot J, Lienemann J, Dube S, Mallinckrodt C, Bymaster FP, McKinzie DL, Felder CC. Selective muscarinic receptor agonist xanomeline as a novel treatment approach for schizophrenia. Am J Psychiatry. 2008;165:1033–9. doi: 10.1176/appi.ajp.2008.06091591. [DOI] [PubMed] [Google Scholar]
- Shirey JK, Xiang Z, Orton D, Brady AE, Johnson KA, Williams R, Ayala JE, Rodriguez AL, Wess J, Weaver D, Niswender CM, Conn PJ. An allosteric potentiator of M4 mAChR modulates hippocampal synaptic transmission. Nat Chem Biol. 2008;4:42–50. doi: 10.1038/nchembio.2007.55. [DOI] [PubMed] [Google Scholar]
- Sipos ML, Burchnell V, Galbicka G. Effects of selected anticholinergics on acoustic startle response in rats. J Appl Toxicol. 2001;21(Suppl 1):S95–101. doi: 10.1002/jat.821. [DOI] [PubMed] [Google Scholar]
- Sitaram N, Weingartner H, Gillin JC. Human serial learning: enhancement with arecholine and choline impairment with scopolamine. Science. 1978;201:274–6. doi: 10.1126/science.351808. [DOI] [PubMed] [Google Scholar]
- Stanhope KJ, Mirza NR, Bickerdike MJ, Bright JL, Harrington NR, Hesselink MB, Kennett GA, Lightowler S, Sheardown MJ, Syed R, Upton RL, Wadsworth G, Weiss SM, Wyatt A. The muscarinic receptor agonist xanomeline has an antipsychotic-like profile in the rat. J Pharmacol Exp Ther. 2001;299:782–92. [PubMed] [Google Scholar]
- Stefansson H, Ophoff RA, Steinberg S, Andreassen OA, Cichon S, Rujescu D, Werge T, Pietilainen OP, Mors O, Mortensen PB, Sigurdsson E, Gustafsson O, Nyegaard M, Tuulio-Henriksson A, Ingason A, Hansen T, Suvisaari J, Lonnqvist J, Paunio T, Borglum AD, Hartmann A, Fink-Jensen A, Nordentoft M, Hougaard D, Norgaard-Pedersen B, Bottcher Y, Olesen J, Breuer R, Moller HJ, Giegling I, Rasmussen HB, Timm S, Mattheisen M, Bitter I, Rethelyi JM, Magnusdottir BB, Sigmundsson T, Olason P, Masson G, Gulcher JR, Haraldsson M, Fossdal R, Thorgeirsson TE, Thorsteinsdottir U, Ruggeri M, Tosato S, Franke B, Strengman E, Kiemeney LA, Melle I, Djurovic S, Abramova L, Kaleda V, Sanjuan J, de Frutos R, Bramon E, Vassos E, Fraser G, Ettinger U, Picchioni M, Walker N, Toulopoulou T, Need AC, Ge D, Yoon JL, Shianna KV, Freimer NB, Cantor RM, Murray R, Kong A, Golimbet V, Carracedo A, Arango C, Costas J, Jonsson EG, Terenius L, Agartz I, Petursson H, Nothen MM, Rietschel M, Matthews PM, Muglia P, Peltonen L, St Clair D, Goldstein DB, Stefansson K, Collier DA. Common variants conferring risk of schizophrenia. Nature. 2009;460:744–7. doi: 10.1038/nature08186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sur C, Mallorga PJ, Wittmann M, Jacobson MA, Pascarella D, Williams JB, Brandish PE, Pettibone DJ, Scolnick EM, Conn PJ. N-desmethylclozapine, an allosteric agonist at muscarinic 1 receptor, potentiates N-methyl-D-aspartate receptor activity. Proc Natl Acad Sci USA. 2003;100:13674–9. doi: 10.1073/pnas.1835612100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, Weber M, Qu Y, Light GA, Braff DL. Realistic expectations of prepulse inhibition in translational models for schizophrenia research. Psychopharmacology (Berl) 2008;199:331–88. doi: 10.1007/s00213-008-1072-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tandon R, Shipley JE, Greden JF, Mann NA, Eisner WH, Goodson JA. Muscarinic cholinergic hyperactivity in schizophrenia. Relationship to positive and negative symptoms. Schizophr Res. 1991;4:23–30. doi: 10.1016/0920-9964(91)90006-d. [DOI] [PubMed] [Google Scholar]
- Thomsen M, Wortwein G, Fink-Jensen A, Woldbye DP, Wess J, Caine SB. Decreased prepulse inhibition and increased sensitivity to muscarinic, but not dopaminergic drugs in M5 muscarinic acetylcholine receptor knockout mice. Psychopharmacology (Berl) 2007;192:97–110. doi: 10.1007/s00213-006-0682-y. [DOI] [PubMed] [Google Scholar]
- Tzavara ET, Bymaster FP, Davis RJ, Wade MR, Perry KW, Wess J, McKinzie DL, Felder C, Nomikos GG. M4 muscarinic receptors regulate the dynamics of cholinergic and dopaminergic neurotransmission: relevance to the pathophysiology and treatment of related CNS pathologies. Faseb J. 2004;18:1410–2. doi: 10.1096/fj.04-1575fje. [DOI] [PubMed] [Google Scholar]
- Tzavara ET, Bymaster FP, Felder CC, Wade M, Gomeza J, Wess J, McKinzie DL, Nomikos GG. Dysregulated hippocampal acetylcholine neurotransmission and impaired cognition in M2, M4 and M2/M4 muscarinic receptor knockout mice. Mol Psychiatry. 2003;8:673–9. doi: 10.1038/sj.mp.4001270. [DOI] [PubMed] [Google Scholar]
- Ukai M, Okuda A, Mamiya T. Effects of anticholinergic drugs selective for muscarinic receptor subtypes on prepulse inhibition in mice. Eur J Pharmacol. 2004;492:183–7. doi: 10.1016/j.ejphar.2004.03.066. [DOI] [PubMed] [Google Scholar]
- Vakalopoulos C. Neuropharmacology of cognition and memory: a unifying theory of neuromodulator imbalance in psychiatry and amnesia. Med Hypotheses. 2006;66:394–431. doi: 10.1016/j.mehy.2005.09.037. [DOI] [PubMed] [Google Scholar]
- Watson J, Brough S, Coldwell MC, Gager T, Ho M, Hunter AJ, Jerman J, Middlemiss DN, Riley GJ, Brown AM. Functional effects of the muscarinic receptor agonist, xanomeline, at 5-HT1 and 5-HT2 receptors. Br J Pharmacol. 1998;125:1413–20. doi: 10.1038/sj.bjp.0702201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner DM, Meltzer HY, Veinbergs I, Donohue EM, Spalding TA, Smith TT, Mohell N, Harvey SC, Lameh J, Nash N, Vanover KE, Olsson R, Jayathilake K, Lee M, Levey AI, Hacksell U, Burstein ES, Davis RE, Brann MR. The role of M1 muscarinic receptor agonism of N-desmethylclozapine in the unique clinical effects of clozapine. Psychopharmacology (Berl) 2004;177:207–16. doi: 10.1007/s00213-004-1940-5. [DOI] [PubMed] [Google Scholar]
- Wess J. Muscarinic acetylcholine receptor knockout mice: novel phenotypes and clinical implications. Annu Rev Pharmacol Toxicol. 2004;44:423–50. doi: 10.1146/annurev.pharmtox.44.101802.121622. [DOI] [PubMed] [Google Scholar]
- Woolley ML, Carter HJ, Gartlon JE, Watson JM, Dawson LA. Attenuation of amphetamine-induced activity by the non-selective muscarinic receptor agonist, xanomeline, is absent in muscarinic M4 receptor knockout mice and attenuated in muscarinic M1 receptor knockout mice. Eur J Pharmacol. 2009;603:147–9. doi: 10.1016/j.ejphar.2008.12.020. [DOI] [PubMed] [Google Scholar]
- Wu MF, Jenden DJ, Fairchild MD, Siegel JM. Cholinergic mechanisms in startle and prepulse inhibition: effects of the false cholinergic precursor N-aminodeanol. Behav Neurosci. 1993;107:306–16. doi: 10.1037//0735-7044.107.2.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavitsanou K, Katsifis A, Mattner F, Huang XF. Investigation of m1/m4 muscarinic receptors in the anterior cingulate cortex in schizophrenia, bipolar disorder, and major depression disorder. Neuropsychopharmacology. 2004;29:619–25. doi: 10.1038/sj.npp.1300367. [DOI] [PubMed] [Google Scholar]