

Abstract
The cannabinoid 1 receptor (CB1R) has a well-established role in appetite regulation. Central CB1R antagonists, notably rimonabant, induced weight loss and improved the metabolic profile in obese individuals, but were discontinued due to psychiatric side-effects. The CB1R is also expressed peripherally, where its effects include promotion of liver fat accumulation, which consumes ATP. Type 2 diabetes in obese subjects is linked to excess liver fat, whilst there is a negative correlation between hepatic ATP content and insulin resistance. A decreased hepatic ATP/AMP ratio increases food intake by signals via the vagus nerve to the brain. The hepatic cannabinoid system is highly upregulated in obesity, and the effects of hepatic CB1R activation include increased activity of lipogenic and gluconeogenic transcription factors. Thus, blockade of hepatic CB1Rs could contribute significantly to the weight-reducing and insulin-sensitizing effects of CB1R antagonists. Additionally, upregulation of the hepatic CB1R may contribute to chronic liver inflammation, fibrosis and cirrhosis from causes including obesity, alcoholism and viral hepatitis. Peripheral CB1R antagonists induce weight loss and metabolic improvements in obese rodents; however, as there is evidence that hepatic CB1Rs are predominately intracellular, due to high intrinsic clearance, many drugs may not effectively block these receptors and therefore have limited efficacy. Hepatoselective CB1R antagonists may be effective at reducing hepatic steatosis, insulin resistance and bodyweight in obese, diabetic patients, with far fewer side-effects than first-generation CB1R antagonists. Additionally, such compounds may be effective in treating inflammatory liver disease, such as non-alcoholic steatohepatitis, reducing the likelihood of disease progression to cirrhosis or cancer.
Keywords: cannabinoid 1 receptor, diabetes, fatty liver, hepatic energy state, non-alcoholic steatohepatitis, obesity
Hepatic energy state and its effects on food intake
The cellular energy state is defined by adenine nucleotide levels. Healthy cells maintain a ratio of ATP to ADP of the order of 10:1. Cellular concentrations of ADP typically remain constant, while ATP and AMP levels deviate in reciprocal directions [1].
Hepatic energy state influences food intake [2, 3]. For instance, infusion of various lipids and carbohydrates into the hepatic portal vein of rodents was found to suppress food intake more effectively than administration of the same nutrients into the jugular vein [2]. Supporting results were found when injecting the fructose analogue 2,5-anhydro-d-mannitol (2,5-AM) into the portal vein. In the liver, 2,5-AM is phosphorylated at the 1 and 6 positions, but not metabolized further, thus lowering the levels of free intracellular phosphates, which reduces the generation of ATP, whilst increasing its degradation by disinhibiting adenosine deaminase. The net result is a lower hepatocellular ATP concentration, which increases feeding [4]. Pretreatment with sodium phosphate prevents the decrease in liver ATP levels and the increase in feeding [5]. Administration of the amino-acid analogue l-ethionine, which reduces ATP production by trapping the adenosine moiety of ATP, also increased food intake [6]. Also, liver fructose-1,6-bisphosphatase, which is upregulated in obesity and affects hepatic gluconeogenesis, has recently been demonstrated to regulate appetite and adiposity in mice [7].
Increased fatty acid oxidation in the liver reduces food intake, whereas inhibition of fatty acid oxidation increases it. Ablation of the hepatic branches of the vagus nerve prevented these effects. The consequences for feeding are probably not due to fatty acid synthesis or oxidation per se, but rather due to the effects of fatty acid metabolism on liver energy production [2].
How changes in hepatocellular energy state lead to a signal that affects food intake has not been fully elucidated. However, in vitro studies suggest that reduced ATP production may affect sodium pumps, increasing the intracellular sodium concentration [8]. 2,5-Anhydro-d-mannitol also increases the calcium concentration in hepatocytes, which is a typical step in many signal transduction pathways [9]. Hence, intracellular sodium and/or calcium levels may play a role in signalling between hepatocytes and neurons.
The impact of the hepatic energy state on feeding has also been demonstrated in ruminants [3], suggesting that the liver influences feeding in many different species. Thus, from studies such as these, the liver has been recognized as an important organ in appetite and bodyweight regulation, reflecting the strong influence that hepatic energy state has on feeding behaviour, with signals being carried from the liver to the brain by the vagus nerve [2].
Hepatic energy metabolism is impaired in patients with type 2 diabetes. Due to a markedly lower ATP production, such patients were found to have 42% lower hepatic ATP turnover (i.e. nucleotide phosphorylation rate, measured with 31P magnetic resonance spectroscopy) than control subjects. There was a strong negative correlation of ATP turnover to hepatic fat content and insulin resistance [10]. These findings are consistent with earlier studies showing that demonstrated 40% reduced mitochondrial oxidative phosphorylation in elderly, insulin-resistant patients with fatty liver compared with young, healthy control subjects [11] and that individuals with type 2 diabetes have less hepatic ATP and inorganic phosphate (Pi) than control subjects, with ATP and Pi content being negatively related to insulin resistance [12].
Taken together, these findings suggest that increased fatty acid synthesis and decreased fatty acid breakdown in the liver cause fat accumulation and a lowered hepatic energy state, marked by a low ATP : AMP ratio. In response to the low energy state, the liver generates signals that are carried via the vagus nerve to the brain, leading to increased food intake. This may lead to fatty liver, obesity and/or type 2 diabetes. Hence, targeting a main mediator of fatty liver and decreased hepatic energy state would counteract this chain of events. Studies of recent years suggest that the hepatic cannabinoid 1 receptor (CB1R) may be such a target.
The role of cannabinoid receptors in fatty liver
Endocannabinoids are lipid ligands that bind to cannabinoid receptors. The best characterized of these receptors are the cannabinoid 1 receptor and the cannabinoid 2 receptor [13]. Although traditionally associated with the central nervous system (CNS), cannabinoid receptors in hepatocytes are increasingly being recognized as key mediators of fatty liver [14] and associated insulin resistance [15–17] caused by high-fat diet [18], viral hepatitis [19–21] and ethanol intake [22]. Additionally, cannabis smoking has been shown to be a risk factor for hepatic steatosis [23] and, in a separate study, patients with non-alcoholic fatty liver disease were found to have a 34.2 ± 9.7-fold increase in the amount of hepatic CB1R mRNA compared with patients without liver pathology [24].
One of the downstream effects of CB1R activation is to increase the expression of the transcription factor sterol regulatory element-binding protein 1c (SREBP-1c), which controls the expression of a number of key lipogenic genes [25]. Among the more important effects of SREBP-1c are the upregulation of acetyl-CoA carboxylase 1 and fatty acid synthase (key enzymes of fatty acid synthesis) and inhibition of carnitine palmitoyltransferase I, which controls the rate-limiting step of β-oxidation [26]. See reference [27] for a more detailed review of the steps leading from CB1R activation to steatosis and Figure 1 for a simplified representation of the key mediators of this process.
Figure 1.
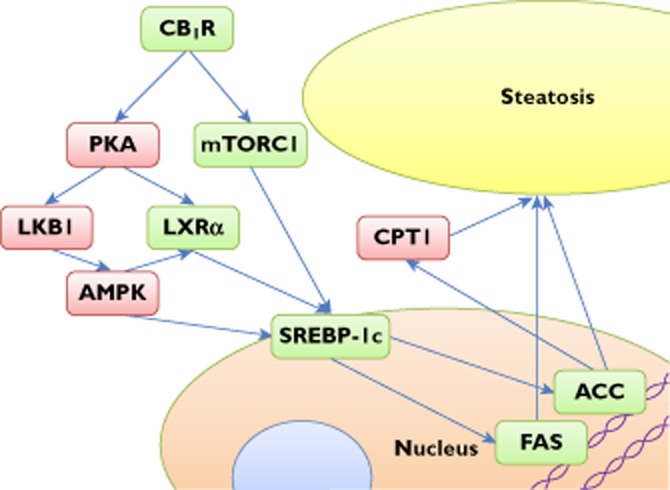
Central mediators of steatosis activated by CB1R. Green boxes indicate increased activity; red ones decreased activity. Abbreviations are as follows: ACC, acetyl-CoA carboxylase; AMPK, AMP-activated protein kinase; CB1R, cannabinoid 1 receptor; CPT1, carnitine palmitoyltransferase I; FAS, fatty acid synthase; LKB1, liver kinase B1; LXRα, Liver X receptor alpha; mTORC1, mammalian target of rapamycin complex 1; PKA, protein kinase A; SREBP-1c, sterol regulatory element-binding protein 1c
Activation of CB1Rs, causing fatty liver by a combination of increased lipogenesis and reduced fatty acid oxidation, depletes ATP, which decreases the cellular ATP : AMP ratio. Additionally, CB1R activation has been demonstrated to increase activation of the endoplasmic reticulum stress-associated liver-specific transcription factor cyclic AMP response element-binding protein H [16, 17, 24], which regulates expression of gluconeogenic genes and, at least in part, accounts for the CB1R-induced reduction in insulin sensitivity and raised glucose levels, whilst further exacerbating the reduced hepatic energy status.
In a healthy cell, the low ATP : AMP ratio would activate AMP-activated protein kinase (AMPK) [1], which then inhibits anabolic pathways and promotes catabolic ones, in part by reducing SREBP-1c activity and expression [28]. In contrast to this, AMPK is less sensitive to stimulation by AMP in the livers of rats with ethanol-induced fatty liver [29, 30]. Also, obese diabetic and nondiabetic individuals have decreased AMPK activity in skeletal muscle in response to exercise [31], as do mice made obese by a high-fat diet [32]. Therefore, in these diseases, the stimulatory effects of increased AMP on AMPK appear to be counteracted. In other studies, CB1R activation has been shown to counteract the effects of high AMP on AMPK [27], with steatosis as the net effect.
Liver-specific knockout of the CB1R made mice resistant to high-fat diet-induced steatosis, although overall adiposity and weight gain were not affected in this situation [18]. However, knockout of genes that control important metabolic regulators may lead to compensatory antenatal changes affecting appetite; thus, the knockout does not always have the same effects as inhibition of the corresponding metabolic regulators in adults. For example, deletion of the gene for acetyl CoA carboxylase 2 did not improve glucose homeostasis in db/db mice [33], but a small-molecule acetyl CoA carboxylase 2 inhibitor has been reported to do so [34]. Acetyl CoA carboxylase 2 is a key regulator of fatty acid oxidation, but adaptation during fetal development and/or early life may compensate for reduced hunger signalling to the brain in acetyl CoA carboxylase 2 null mice. The finding that liver-specific CB1R knockout mice did not become resistant to diet-induced obesity may have been due to similar compensatory mechanisms.
Nevertheless, studies with liver-specific CB1R knockout mice demonstrated that the hepatic CB1R contributes significantly to steatosis or metabolic disturbances that lead indirectly to steatosis. Also, treatment of cultured mouse liver explants with rimonabant increased fat oxidation [35], showing that hepatic CB1R inverse agonism reduced expression of genes for prolipogenic enzymes. Studies such as these demonstrate that the effects of CB1R inhibition on liver fat are mediated peripherally, independently of the nervous system.
Based upon evidence from the peripherally selective CB1R antagonist JD5037, it has recently been proposed that reversal of obesity-related leptin resistance accounts for the hypophagic effect of peripheral CB1R inverse agonism and that this is achieved by normalization of the hyperleptinaemia of diet-induced obese mice through the suppression of leptin secretion in adipose tissue and increased leptin clearance via the kidney [36]. However, whilst it seems likely that activation of the CB1R is an important regulator of diet-induced leptin resistance [36], which probably contributes to the development of obesity, this is due to the inability of the high levels of leptin, normally associated with obesity, to reduce appetite effectively, rather than to the high leptin levels themselves [37]. Thus, leptin resistance is an additional mechanism by which peripheral CB1R activation may exacerbate the development of obesity.
The efficacy of hepatic or peripheral CB1R inhibition is also suggested by data in a clinical trial with 20 mg rimonabant daily for the treatment of abdominal obesity and dyslipidaemia. According to regression analysis, 72% of the improvement in high-density lipoprotein-cholesterol could not be explained by the weight loss alone [38]. Hence, peripheral effects of rimonabant probably contribute to its metabolic benefits. However, although hepatic CB1Rs presumably contribute to this efficacy, it is not clear exactly how effectively receptors in the liver are blocked at doses used clinically. This is because, as will be discussed in the next section, hepatic CB1Rs, which are highly upregulated in obesity and a number of inflammatory liver conditions, appear to be mainly intracellular and, whilst functional activity of intrahepatic CB1Rs has not been confirmed, studies in other cell types have demonstrated intracellular receptors to be highly important for CB1R-mediated effects [39–43]. Thus, the efficacy of a CB1R antagonist drug may be influenced by how well it maintains exposure inside hepatocytes, in the vicinity of high levels of metabolizing enzymes.
Cytosolic localization, functional activity and hyperactivation of CB1R in hepatocytes
The main direct evidence for intracellular localization of CB1Rs in hepatocytes is from immunostaining experiments with rodent [19, 44–48] and human [19, 48, 49] liver tissue samples. Although this does not provide definitive proof for intracellular localization of upregulated hepatic CB1Rs, due to the absence of information from classical biochemical tests (such as preparing heavy organelle, light organelle and cytosolic fractions from cells by directed Förster resonance energy transfer assays on binding partners) or by ultrastructural analyses using immune-electron microscopy, the pattern of staining in the published images, particularly in articles by van der Poorten et al. [19] and Floreani et al. [44], nevertheless strongly indicates cytosolic localization.
These suppositions are also consistent with emerging evidence that the hepatic CB1R may become upregulated in response to elevated intracellular endocannabinoid levels as part of a hyperactivation of the cannabinoid system. For instance, of the two most-studied endocannabinoids, 2-arachodonylglycerol has been shown to mediate binding of the retinoic acid receptor subunit γ to the regulatory domain for the CB1R [50], whilst anandamide has also been demonstrated to increase CB1R mRNA [35] Furthermore, it has also been proposed that CB1R upregulation may in turn lead to increased endocannabinoid synthesis [48]. Elevated cellular endocannabinoid levels may be caused by faster synthesis or increased uptake from the systemic circulation or reduced activity of the intracellular metabolizing enzymes; in particular, monoacylglycerol lipase and fatty acid amide hydrolase (FAAH) are important for regulating levels of 2-AG and AEA, respectively. For instance, whilst FAAH is primarily expressed in the liver, human FAAH gene mutations are associated with increased bodyweight and obesity [19, 51], whilst FAAH–/– mice have altered energy homeostasis, demonstrated by decreased oxygen consumption and hyperinsulinaemia, with adipose, skeletal muscle and hepatic insulin resistance [52].
Whilst high cannabinoid concentrations can be maintained for hours insde hepatocytes, rapid fluid flow through hepatic vessels and the space of Disse washes away extracellular cannabinoids. It would therefore seem reasonable to assume that elevated endocannabinoid levels in the liver can be maintained only if they are intracellular. It thus seems likely that the resulting elevated endocannabinoid levels will have maximal effect if they act through intracellular receptors. These suppositions are consistent with observations made in other cell types; for instance, in retinal epithelial cells high glucose was shown to downregulate the expression of FAAH 1 mRNA and protein in ARPE-19 cells, whilst upregulating CB1R mRNA and protein. Furthermore, overexpression of FAAH 1 and treatment with the CB1R antagonist AM 251 blocked high-glucose-induced internalization of CB1R in HEK 293 cells and ARPE-19 cells, expression of CB1R in the cytosolic fraction, as well as generation of reactive oxygen species (ROS) and lipid peroxide formation [53].
Although the precise mechanism by which endocannabinoid levels are elevated in hepatocytes is not fully elucidated, there is evidence in other cell types implicating oxidative stress; for instance, in hepatic stellate cells, acetaldehyde, H2O2, as well as 2-arachodonylglycerol and tetrahydrocannabinol, alone or in combination with acetaldehyde, were shown to induce CB1 mRNA expression in vitro [47]. This may explain the pathogenesis of CB1R-mediated liver disease, because circulating ROS have been shown to be elevated due to hyperlipidaemia [54], high-fat [55] or carbohydrate [56] diets. However, given that elevated ROS may be caused by CB1R activation [57], it is necessary to distinguish between cause and effect before drawing firm conclusions about what stimulus induces upregulation of the cannabinoid system in hepatocytes. Nevertheless, the concept that elevated ROS leads to upregulation and activation of hepatic CB1R is intriguing, because this might imply a positive feedback loop whereby hepatic CB1R activation leads to increased food intake, which in turn leads to increased ROS production and further increased CB1R activation, but this remains to be proved.
In summary, we postulate that localized hyperactivation of the cannabinoid system occurs internally in various cell types, possibly in response to inflammatory stimuli, such as elevated ROS, to activate specific cellular processes. In this context, considerable evidence implicates the cannabinoid system as a key mediator of apoptosis in many cell types [58], including immune cells [59] and many forms of cancer cells [60, 61], whilst, in neuronal cells, intracellular CB1R activation has been shown to play a role in stabilizing lysosomes against amyloid toxicity, thereby improving cell viability and conferring neuroprotection [62]. Anandamide activation of the hepatic cannabinoid system does not induce apoptosis in hepatocytes [63], possibly due to elevated levels of glutathione, which protects against elevated ROS, and high levels of FAAH [64]. Moreover, in the light of a recent report implicating the hepatic CB1R as a regulator of transcription for enzymes involved in M-phase progression and as an important mediator in the early phase of liver regeneration [48], it may seem reasonable to assume that hepatic cannabinoid system upregulation has a physiological role as part of the mechanism inducing cell replication.
Understanding of the effects of alterations in the expression level of cannabinoid receptors in the pathogenesis of many chronic diseases is emerging [65], and the key role played by aberrant chronic activation of the hepatic CB1R in the development of inflammatory liver conditions, including non-alcoholic fatty liver disease [18], non-alcoholic steatohepatitis [66], alcoholic steatohepatitis [57] and fibrosis/cirrhosis [46, 67, 68], is well documented. Recent evidence links development of fibrosis to increased hepatocyte apoptosis [69] mediated by NADPH oxidase [70, 71] due to cell damage caused by chronic inflammation, which is, at least in part, attributable to CB1R activation. Thus, it is expected that, as well as reducing obesity, liver fat and insulin resistance, peripheral CB1R antagonists may additionally have significant clinical benefit in the treatment and prevention of fibrosis in many forms of inflammatory liver disease.
Pharmacological applications
Challenges in the development of peripheral CB1R antagonists
Rimonabant is a CB1R inverse agonist that was designed to target the central nervous system. In obese patients, the drug caused weight loss and improved several metabolic risk factors [72]. However, rimonabant was removed from the market because of its psychiatric side-effects, most significantly depression, anxiety and suicidal ideation [73]. To avoid these side-effects while retaining the benefits of the drug, some groups have sought peripherally restricted CB1R antagonists, hoping that drugs with an improved risk–benefit profile will be approved for the treatment of conditions believed to be mediated by peripheral CB1Rs, such as obesity, steatosis and insulin resistance [74]. Indeed, peripheral CB1R antagonists with promising efficacy have been reported, such as AM6545, currently in preclinical testing [75], and TM38837, in early clinical development [76].
A good peripheral CB1R antagonist should not reach concentrations in the CNS that cause adverse reactions at doses giving significant efficacy. The brain is separated from the systemic circulation by the blood–brain barrier, which consists of endothelial cells differing from those in the rest of the body by having extensive tight junctions, sparse pinocytotic transport and no fenestrations [77]. Free drug concentrations are equal on both sides of a physiological membrane at steady state. Thus, for drugs with normal permeability, the free concentration in the brain will resemble the free concentration in plasma during long-term treatment, unless active transport moves drug into or out of the brain, or the drug is metabolized in the endothelial cells of the blood–brain barrier. It is the free drug that exerts the physiological effects [78]; thus, a peripherally selective drug needs to be a substrate for an active (energy-consuming) process that limits long-term brain exposure. Ideally, the drug should not have high permeability, because this counteracts the active transport. Several methods are available to estimate the free drug concentration in the brain interstitial fluid and immediately near the target receptor [79].
ATP-binding cassette transporters are ATP-dependent integral membrane proteins that translocate solutes across cellular membranes [80]. These transporters are present in several tissues; those relevant to this article move solutes across the blood–brain barrier, the blood–cerebrospinal fluid barrier and the cell membrane of hepatocytes. Among these transporters, permeability glycoprotein 1 (P-gp) is particularly important for transporting xenobiotics out of the brain across the blood–brain barrier [81]. Permeability glycoprotein 1 is an efflux pump with broad substrate specificity, implicated in limiting the access of drugs to the brain parenchyma [80]. Permeability glycoprotein 1 also reduces absorption from the gastrointestinal tract and transports some drugs and metabolites from hepatocytes into the bile [82].
Taranabant is a CB1R inverse agonist that is not a P-gp substrate [83]; it caused psychiatric side-effects, even at low doses [84]. In contrast, AM6545 and JD5037 are CB1R inverse agonists that are actively transported by P-gp [85]. Thus, P-gp-mediated extrusion of AM6545 and JD5037 reduces their concentration in the brain, lessening the risk of psychiatric side-effects. JD5037 has high CB1R binding affinity (Ki = 0.35 nm) [36], which suggests that effective doses in humans would be low. JD5037 reduces appetite and weight in mice with diet-induced obesity and does so as effectively as a central CB1R inverse agonist, SLV319 [86]. Publications have not reported the ratio of free concentration of JD5037 in brain to free concentration in plasma and have not clarified whether effects on hepatic CB1Rs contribute in a relevant manner to the efficacy of JD5037.
Several peripheral CB1R antagonists are under development [76], with researchers reporting often extensive studies to demonstrate reduced CNS exposure compared with first-generation drugs, such as rimonabant. However, a compound similar to rimonabant but without CNS exposure might be expected to have reduced in vivo efficacy, in comparison to rimonabant, with limited anticipated clinical effectiveness. Such an argument assumes that, at doses used clinically, rimonabant achieves high levels of receptor occupancy, both centrally and peripherally; thus, close to maximal efficacy is observed. However, centrally mediated effects upon food intake may arise at very low occupancy [87, 88], and dose limitations, due to psychiatric adverse events, might mean that the full peripheral effect is not realized. Furthermore, the efficiency with which these compounds block receptors in the liver, which may vary considerably between compounds, has generally not been considered, and this may significantly affect the observed efficacy of a particular compound. The liver has a major role in regulating whole-body energy and lipid homeostasis, whilst CB1Rs in hepatocytes are believed strongly to influence enzymes controlling energy usage and storage. Thus, considering the cytosolic distribution of CB1Rs, we propose that the ability of CB1R antagonist drugs to maintain high unbound concentrations in hepatocytes, where they are vulnerable to metabolic clearance, may be a key determinant of in vivo and clinical efficacy, perhaps of greater importance than the degree of CNS exposure. Moreover, a strategy of specifically blocking CB1Rs in the liver of patients with metabolic syndrome may have been overlooked as a therapeutic mechanism. Such a strategy would negate adverse events from CNS exposure as well as reduce side-effects from blockade of other peripheral receptors (such as those in the gut), thus providing an opportunity for safe, effective drugs.
Strategies to increase hepatoselectivity
Although G protein-coupled receptors are generally transported to the cell membrane [89], as mentioned previously, several studies have demonstrated that most CB1Rs in various tissues are present and active in intracellular compartments, such as endosomes, lysosomes and mitochondria [39–44]. Thus, a hepatoselective CB1R antagonist should maintain a high unbound concentration in the cytosol of hepatocytes relative to the plasma and other tissues. In this context, it should be noted that a high apparent total (free and bound) concentration in the liver does not necessarily indicate good exposure. If the drug maintains an adequate unbound concentration in the interstitial space, lipophilic compounds (such as rimonabant and taranabant) will be strongly associated with cell membranes and thus may attain apparent high concentrations in whole liver even though cytosolic free concentrations are low.
Strategies that have been employed to achieve pharmacological hepatoselectivity include producing molecules with the following characteristics: (i) they are actively transported into liver cells by liver-specific transporters, such as organic anion transporting polypeptide (OATP) transporters or organic cation transporters; (ii) they are conjugates with liver-targeting substances, such as bile acids; and (iii) they undergo metabolic activation in the liver [90].
The OATPs are believed to play an important role in drug disposition [82]. The hepatic specificity of pravastatin, a hydrophilic HMG-CoA reductase inhibitor, largely depends on OATPs [91, 92]. Pfefferkorn et al. report designing a glucokinase activator optimized for active liver uptake via members of the OATP family, leading to a greater than 50-fold higher substrate distribution in the liver than in the pancreas [93]. Likewise, Oballa et al. have designed a stearoyl-CoA desaturase inhibitor that accumulates to a higher concentration in the liver than in the skin [94].
A hepatoselective drug should be designed to have low passive permeability to reduce diffusion out of hepatocytes. To maintain a high intrahepatic concentration, it should also be metabolically stable and not a substrate for transporters that move compounds into the bile. Rimonabant and other lipophilic inverse agonists of CB1R may not be particularly metabolically stable in hepatocytes, and little is known about how intrahepatic free concentrations vary with plasma concentrations in vivo.
Summary
Overall, the references of this review suggest that a CB1R inverse agonist that achieves sufficient selectivity for hepatocytes may provide efficacy with no or negligible psychiatric side-effects. In patients with overexpression of CB1R inside hepatocytes, as implicated in many aspects of the metabolic syndrome, a hepatoselective CB1R antagonist could improve ATP turnover and hepatic energy state, which may in turn reduce food intake. In these patients, the hepatoselective CB1R antagonist could ameliorate hepatic steatosis, insulin resistance, dyslipidaemia and inflammatory liver disease, as well as promote significant weight loss. We propose that long-term metabolic benefits could exceed those from rimonabant 20 mg daily in obese diabetic patients with hepatic steatosis, as well as in patients with various forms of inflammatory liver disease from other causes.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare: no support from any organization for the submitted work; no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years; no other relationships or activities that could appear to have influenced the submitted work.
References
- 1.Hardie DG, Hawley SA. AMP-activated protein kinase: the energy charge hypothesis revisited. Bioessays. 2001;23:1112–1119. doi: 10.1002/bies.10009. [DOI] [PubMed] [Google Scholar]
- 2.Friedman MI. Obesity and the hepatic control of feeding behavior. Drug News Perspect. 2007;20:573–578. doi: 10.1358/dnp.2007.20.9.1162243. [DOI] [PubMed] [Google Scholar]
- 3.Allen MS, Bradford BJ, Oba M. The hepatic oxidation theory of the control of feed intake and its application to ruminants. J Anim Sci. 2009;87:3317–3334. doi: 10.2527/jas.2009-1779. [DOI] [PubMed] [Google Scholar]
- 4.Koch JE, Ji H, Osbakken MD, Friedman MI. Temporal relationships between eating behavior and liver adenine nucleotides in rats treated with 2,5-AM. Am J Physiol. 1998;274:R610–617. doi: 10.1152/ajpregu.1998.274.3.R610. [DOI] [PubMed] [Google Scholar]
- 5.Rawson NE, Friedman MI. Phosphate loading prevents the decrease in ATP and increase in food intake produced by 2,5-anhydro-D-mannitol. Am J Physiol. 1994;266:R1792–1796. doi: 10.1152/ajpregu.1994.266.6.R1792. [DOI] [PubMed] [Google Scholar]
- 6.Rawson NE, Ulrich PM, Friedman MI. L-ethionine, an amino acid analogue, stimulates eating in rats. Am J Physiol. 1994;267:R612–615. doi: 10.1152/ajpregu.1994.267.2.R612. [DOI] [PubMed] [Google Scholar]
- 7.Visinoni S, Khalid NF, Joannides CN, Shulkes A, Yim M, Whitehead J, Tiganis T, Lamont BJ, Favaloro JM, Proietto J, Andrikopoulos S, Fam BC. The role of liver fructose-1,6-bisphosphatase in regulating appetite and adiposity. Diabetes. 2012;61:1122–1132. doi: 10.2337/db11-1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Friedman MI, Graczyk-Millbrandt G, Ji H, Rawson NE, Osbakken MD. 2,5-Anhydro-D-mannitol increases hepatocyte sodium: transduction of a hepatic hunger stimulus? Biochim Biophys Acta. 2003;1642:53–58. doi: 10.1016/s0167-4889(03)00098-3. [DOI] [PubMed] [Google Scholar]
- 9.Rawson NE, Ji H, Friedman MI. 2,5-Anhydro-D-mannitol increases hepatocyte calcium: implications for a hepatic hunger stimulus. Biochim Biophys Acta. 2003;1642:59–66. doi: 10.1016/s0167-4889(03)00099-5. [DOI] [PubMed] [Google Scholar]
- 10.Schmid AI, Szendroedi J, Chmelik M, Krssák M, Moser E, Roden M. Liver ATP synthesis is lower and relates to insulin sensitivity in patients with type 2 diabetes. Diabetes Care. 2011;34:448–453. doi: 10.2337/dc10-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Petersen KF, Befroy D, Dufour S, Dziura J, Ariyan C, Rothman DL, DiPietro L, Cline GW, Shulman GI. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science. 2003;300:1140–1142. doi: 10.1126/science.1082889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Szendroedi J, Chmelik M, Schmid AI, Nowotny P, Brehm A, Krssak M, Moser E, Roden M. Abnormal hepatic energy homeostasis in type 2 diabetes. Hepatology. 2009;50:1079–1086. doi: 10.1002/hep.23093. [DOI] [PubMed] [Google Scholar]
- 13.Cota D, Marsicano G, Tschöp M, Grübler Y, Flachskamm C, Schubert M, Auer D, Yassouridis A, Thöne-Reineke C, Ortmann S, Tomassoni F, Cervino C, Nisoli E, Linthorst AC, Pasquali R, Lutz B, Stalla GK, Pagotto U. The endogenous cannabinoid system affects energy balance via central orexigenic drive and peripheral lipogenesis. J Clin Invest. 2003;112:423–431. doi: 10.1172/JCI17725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Purohit V, Rapaka R, Shurtleff D. Role of cannabinoids in the development of fatty liver (steatosis) AAPS J. 2010;12:233–237. doi: 10.1208/s12248-010-9178-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gary-Bobo M, Elachouri G, Gallas JF, Janiak P, Marini P, Ravinet-Trillou C, Chabbert M, Cruccioli N, Pfersdorff C, Roque C, Arnone M, Croci T, Soubrié P, Oury-Donat F, Maffrand JP, Scatton B, Lacheretz F, Le Fur G, Herbert JM, Bensaid M. Rimonabant reduces obesity-associated hepatic steatosis and features of metabolic syndrome in obese Zucker fa/fa rats. Hepatology. 2007;46:122–129. doi: 10.1002/hep.21641. [DOI] [PubMed] [Google Scholar]
- 16.Chanda D, Kim DK, Li T, Kim YH, Koo SH, Lee CH, Chiang JY, Choi HS. Cannabinoid receptor type 1 (CB1R) signaling regulates hepatic gluconeogenesis via induction of endoplasmic reticulum-bound transcription factor cAMP-responsive element-binding protein H (CREBH) in primary hepatocytes. J Biol Chem. 2011;286:27971–27979. doi: 10.1074/jbc.M111.224352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chanda D, Kim YH, Kim DK, Lee MW, Lee SY, Park TS, Koo SH, Lee CH, Choi HS. Activation of cannabinoid receptor type 1 (Cb1r) disrupts hepatic insulin receptor signaling via cyclic AMP-response element-binding protein H (Crebh)-mediated induction of Lipin1 gene. J Biol Chem. 2012;287:38041–38049. doi: 10.1074/jbc.M112.377978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Osei-Hyiaman D, Liu J, Zhou L, Godlewski G, Harvey-White J, Jeong W, Bátkai S, Marsicano G, Lutz B, Buettner C, Kunos G. Hepatic CB1 receptor is required for development of diet-induced steatosis, dyslipidemia, and insulin and leptin resistance in mice. J Clin Invest. 2008;118:3160–3169. doi: 10.1172/JCI34827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van der Poorten D, Shahidi M, Tay E, Sesha J, Tran K, McLeod D, Milliken JS, Ho V, Hebbard LW, Douglas MW, George J. Hepatitis C virus induces the cannabinoid receptor 1. PLoS ONE. 2010;5:pii: e12841. doi: 10.1371/journal.pone.0012841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Enjoji M, Kohjima M, Kotoh K, Nakamuta M. Metabolic disorders and steatosis in patients with chronic hepatitis C: metabolic strategies for antiviral treatments. Int J Hepatol. 2012;2012:264017–264023. doi: 10.1155/2012/264017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Toyoda M, Kitaoka A, Machida K, Nishinakagawa T, Yada R, Kohjima M, Kato M, Kotoh K, Sakamoto N, Shiota G, Nakamuta M, Nakashima M, Enjoji M. Association between lipid accumulation and the cannabinoid system in Huh7 cells expressing HCV genes. Int J Mol Med. 2011;27:619–624. doi: 10.3892/ijmm.2011.622. [DOI] [PubMed] [Google Scholar]
- 22.Jeong WI, Osei-Hyiaman D, Park O, Liu J, Bátkai S, Mukhopadhyay P, Horiguchi N, Harvey-White J, Marsicano G, Lutz B, Gao B, Kunos G. Paracrine activation of hepatic CB1 receptors by stellate cell-derived endocannabinoids mediates alcoholic fatty liver. Cell Metab. 2008;7:227–235. doi: 10.1016/j.cmet.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 23.Hézode C, Zafrani ES, Roudot-Thoraval F, Costentin C, Hessami A, Bouvier-Alias M, Medkour F, Pawlostky JM, Lotersztajn S, Mallat A. Daily cannabis use: a novel risk factor of steatosis severity in patients with chronic hepatitis C. Gastroenterology. 2008;134:432–439. doi: 10.1053/j.gastro.2007.11.039. [DOI] [PubMed] [Google Scholar]
- 24.Liu J, Zhou L, Xiong K, Godlewski G, Mukhopadhyay B, Tam J, Yin S, Gao P, Shan X, Pickel J, Bataller R, O'Hare J, Scherer T, Buettner C, Kunos G. Hepatic cannabinoid receptor-1 mediates diet-induced insulin resistance via inhibition of insulin signaling and clearance in mice. Gastroenterology. 2012;142:1218–1228. doi: 10.1053/j.gastro.2012.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu HM, Yang YM, Kim SG. Rimonabant, a cannabinoid receptor type 1 inverse agonist, inhibits hepatocyte lipogenesis by activating liver kinase B1 and AMP-activated protein kinase axis downstream of Gα i/o inhibition. Mol Pharmacol. 2011;80:859–869. doi: 10.1124/mol.111.072769. [DOI] [PubMed] [Google Scholar]
- 26.Osei-Hyiaman D, DePetrillo M, Pacher P, Liu J, Radaeva S, Bátkai S, Harvey-White J, Mackie K, Offertáler L, Wang L, Kunos G. Endocannabinoid activation at hepatic CB1 receptors stimulates fatty acid synthesis and contributes to diet-induced obesity. J Clin Invest. 2005;115:1298–1305. doi: 10.1172/JCI23057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Regnell SE. Cannabinoid 1 receptor in fatty liver. Hepatol Res. 2013;43:131–138. doi: 10.1111/j.1872-034X.2012.01085.x. [DOI] [PubMed] [Google Scholar]
- 28.Li Y, Xu S, Mihaylova MM, Zheng B, Hou X, Jiang B, Park O, Luo Z, Lefai E, Shyy JY, Gao B, Wierzbicki M, Verbeuren TJ, Shaw RJ, Cohen RA, Zang M. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011;13:376–388. doi: 10.1016/j.cmet.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.García-Villafranca J, Guillén A, Castro J. Ethanol consumption impairs regulation of fatty acid metabolism by decreasing the activity of AMP-activated protein kinase in rat liver. Biochimie. 2008;90:460–466. doi: 10.1016/j.biochi.2007.09.019. [DOI] [PubMed] [Google Scholar]
- 30.You M, Matsumoto M, Pacold CM, Cho WK, Crabb DW. The role of AMP-activated protein kinase in the action of ethanol in the liver. Gastroenterology. 2004;127:1798–1808. doi: 10.1053/j.gastro.2004.09.049. [DOI] [PubMed] [Google Scholar]
- 31.Sriwijitkamol A, Coletta DK, Wajcberg E, Balbontin GB, Reyna SM, Barrientes J, Eagan PA, Jenkinson CP, Cersosimo E, DeFronzo RA, Sakamoto K, Musi N. Effect of acute exercise on AMPK signaling in skeletal muscle of subjects with type 2 diabetes: a time-course and dose-response study. Diabetes. 2007;56:836–848. doi: 10.2337/db06-1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee-Young RS, Ayala JE, Fueger PT, Mayes WH, Kang L, Wasserman DH. Obesity impairs skeletal muscle AMPK signaling during exercise: role of AMPKα2 in the regulation of exercise capacity in vivo. Int J Obes (Lond) 2011;35:982–989. doi: 10.1038/ijo.2010.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Olson DP, Pulinilkunnil T, Cline GW, Shulman GI, Lowell BB. Gene knockout of Acc2 has little effect on body weight, fat mass, or food intake. Proc Natl Acad Sci U S A. 2010;107:7598–7603. doi: 10.1073/pnas.0913492107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Glund S, Schoelch C, Thomas L, Niessen HG, Stiller D, Roth GJ, Neubauer H. Inhibition of acetyl-CoA carboxylase 2 enhances skeletal muscle fatty acid oxidation and improves whole-body glucose homeostasis in db/db mice. Diabetologia. 2012;55:2044–2053. doi: 10.1007/s00125-012-2554-9. [DOI] [PubMed] [Google Scholar]
- 35.Jourdan T, Demizieux L, Gresti J, Djaouti L, Gaba L, Vergès B, Degrace P. Antagonism of peripheral hepatic cannabinoid receptor-1 improves liver lipid metabolism in mice: evidence from cultured explants. Hepatology. 2012;55:790–799. doi: 10.1002/hep.24733. [DOI] [PubMed] [Google Scholar]
- 36.Tam J, Cinar R, Liu J, Godlewski G, Wesley D, Jourdan T, Szanda G, Mukhopadhyay B, Chedester L, Liow JS, Innis RB, Cheng K, Rice KC, Deschamps JR, Chorvat RJ, McElroy JF, Kunos G. Peripheral cannabinoid-1 receptor inverse agonism reduces obesity by reversing leptin resistance. Cell Metab. 2012;16:167–179. doi: 10.1016/j.cmet.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scarpace PJ, Zhang Y. Leptin resistance: a prediposing factor for diet-induced obesity. Am J Physiol Regul Integr Comp Physiol. 2009;296:R493–500. doi: 10.1152/ajpregu.90669.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Després JP, Ross R, Boka G, Alméras N, Lemieux I ADAGIO-Lipids Investigators. Effect of rimonabant on the high-triglyceride/ low-HDL-cholesterol dyslipidemia, intraabdominal adiposity, and liver fat: the ADAGIO-Lipids trial. Arterioscler Thromb Vasc Biol. 2009;29:416–423. doi: 10.1161/ATVBAHA.108.176362. [DOI] [PubMed] [Google Scholar]
- 39.Brailoiu GC, Oprea TI, Zhao P, Abood ME, Brailoiu E. Intracellular cannabinoid type 1 (CB1) receptors are activated by anandamide. J Biol Chem. 2011;286:29166–29174. doi: 10.1074/jbc.M110.217463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leterrier C, Lainé J, Darmon M, Boudin H, Rossier J, Lenkei Z. Constitutive activation drives compartment-selective endocytosis and axonal targeting of type 1 cannabinoid receptors. J Neurosci. 2006;26:3141–3153. doi: 10.1523/JNEUROSCI.5437-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leterrier C, Bonnard D, Carrel D, Rossier J, Lenkei Z. Constitutive endocytic cycle of the CB1 cannabinoid receptor. J Biol Chem. 2004;279:36013–36021. doi: 10.1074/jbc.M403990200. [DOI] [PubMed] [Google Scholar]
- 42.Rozenfeld R, Devi LA. Regulation of CB1 cannabinoid receptor trafficking by the adaptor protein AP-3. FASEB J. 2008;22:2311–2322. doi: 10.1096/fj.07-102731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bénard G, Massa F, Puente N, Lourenço J, Bellocchio L, Soria-Gómez E, Matias I, Delamarre A, Metna-Laurent M, Cannich A, Hebert-Chatelain E, Mulle C, Ortega-Gutiérrez S, Martín-Fontecha M, Klugmann M, Guggenhuber S, Lutz B, Gertsch J, Chaouloff F, López-Rodríguez ML, Grandes P, Rossignol R, Marsicano G. Mitochondrial CB(1) receptors regulate neuronal energy metabolism. Nat Neurosci. 2012;15:558–564. doi: 10.1038/nn.3053. [DOI] [PubMed] [Google Scholar]
- 44.Floreani A, Lazzari R, Macchi V, Porzionato A, Variola A, Colavito D, Leon A, Guido M, Baldo V, De Caro R, Bergasa NV. Hepatic expression of endocannabinoid receptors and their novel polymorphisms in primary biliary cirrhosis. J Gastroenterol. 2010;45:68–76. doi: 10.1007/s00535-009-0122-y. [DOI] [PubMed] [Google Scholar]
- 45.Giannone FA, Baldassarre M, Domenicali M, Zaccherini G, Trevisani F, Bernardi M, Caraceni P. Reversal of liver fibrosis by the antagonism of endocannabinoid CB1 receptor in a rat model of CCl(4)-induced advanced cirrhosis. Lab Invest. 2012;92:384–395. doi: 10.1038/labinvest.2011.191. [DOI] [PubMed] [Google Scholar]
- 46.Teixeira-Clerc F, Julien B, Grenard P, Tran Van Nhieu J, Deveaux V, Hezode C, Mallat A, Lotersztajn S. CB1 cannabinoid receptor antagonism: a new strategy for the treatment of liver fibrosis. Nat Med. 2006;12:671–676. doi: 10.1038/nm1421. [DOI] [PubMed] [Google Scholar]
- 47.Patsenker E, Stoll M, Millonig G, Agaimy A, Wissniowski T, Schneider V, Mueller S, Brenneisen R, Seitz HK, Ocker M, Stickel F. Cannabinoid receptor type I modulates alcohol-induced liver fibrosis. Mol Med. 2011;17:1285–1294. doi: 10.2119/molmed.2011.00149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mukhopadhyay B, Cinar R, Yin S, Liu J, Tam J, Godlewski G, Harvey-White J, Mordi I, Cravatt BF, Lotersztajn S, Gao B, Yuan Q, Schuebel K, Goldman D, Kunos G. Hyperactivation of anandamide synthesis and regulation of cell-cycle progression via cannabinoid type 1 (CB1) receptors in the regenerating liver. Proc Natl Acad Sci U S A. 2011;108:6323–6328. doi: 10.1073/pnas.1017689108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee TY, Lee KC, Chang HH. Modulation of the cannabinoid receptors by andrographolide attenuates hepatic apoptosis following bile duct ligation in rats with fibrosis. Apoptosis. 2010;15:904–914. doi: 10.1007/s10495-010-0502-z. [DOI] [PubMed] [Google Scholar]
- 50.Mukhopadhyay B, Liu J, Osei-Hyiaman D, Godlewski G, Mukhopadhyay P, Wang L, Jeong WI, Gao B, Duester G, Mackie K, Kojima S, Kunos G. Transcriptional regulation of cannabinoid receptor-1 expression in the liver by retinoic acid acting via retinoic acid receptor-gamma. J Biol Chem. 2010;285:19002–19011. doi: 10.1074/jbc.M109.068460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Müller TD, Brönner G, Wandolski M, Carrie J, Nguyen TT, Greene BH, Scherag A, Grallert H, Vogel CIG, Scherag S, Rief W, Wichmann HE Illig T, Schäfer H, Hebebrand J, Hinney A. Mutation screen and association studies for the fatty acid amide hydrolase (FAAH) gene and early onset and adult obesity. BMC Med Genet. 2010;11:2. doi: 10.1186/1471-2350-11-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vaitheesvaran B, Yang L, Hartil K, Glaser S, Yazulla S, Bruce JE, Kurland IJ. Peripheral effects of FAAH deficiency on fuel and energy homeostasis: role of dysregulated lysine acetylation. PLoS ONE. 2012;7:e33717. doi: 10.1371/journal.pone.0033717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lim SK, Park MJ, Lim JC, Kim JC, Han HJ, Kim GY, Cravatt BF, Woo CH, Ma SJ, Yoon KC, Park SH. Hyperglycemia induces apoptosis via CB1 activation through the decrease of FAAH 1 in retinal pigment epithelial cells. J Cell Physiol. 2012;227:569–577. doi: 10.1002/jcp.22756. [DOI] [PubMed] [Google Scholar]
- 54.Aliev G, Bodin P, Burnstock G. Free radical generators cause changes in endothelial and inducible nitric oxide synthases and endothelin-1 immunoreactivity in endothelial cells from hyperlipidemic rabbits. Mol Genet Metab. 1998;63:191–197. doi: 10.1006/mgme.1997.2664. [DOI] [PubMed] [Google Scholar]
- 55.Faure P, Rossini E, Lafond JL, Richard MJ, Favier A, Halimi S. Vitamin E improves the free radical defense system potential and insulin sensitivity of rats fed high fructose diets. J Nutr. 1997;127:103–107. doi: 10.1093/jn/127.1.103. [DOI] [PubMed] [Google Scholar]
- 56.Slim RM, Toborek M, Watkins BA, Boissonneault GA, Hennig B. Susceptibility to hepatic oxidative stress in rabbits fed different animal and plant fats. J Am Coll Nutr. 1996;15:289–294. doi: 10.1080/07315724.1996.10718600. [DOI] [PubMed] [Google Scholar]
- 57.Kim DK, Kim YH, Jang HH, Park J, Kim JR, Koh M, Jeong WI, Koo SH, Park TS, Yun CH, Park SB, Chiang JY, Lee CH, Choi HS. Estrogen-related receptor γ controls hepatic CB1 receptor-mediated CYP2E1 expression and oxidative liver injury by alcohol. Gut. 2012 doi: 10.1136/gutjnl-2012-303347. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Maccarrone M, Finazzi-Agró A. The endocannabinoid system, anandamide and the regulation of mammalian cell apoptosis. Cell Death Differ. 2003;10:946–955. doi: 10.1038/sj.cdd.4401284. [DOI] [PubMed] [Google Scholar]
- 59.Rieder SA, Chauhan A, Singh U, Nagarkatti M, Nagarkatti P. Cannabinoid-induced apoptosis in immune cells as a pathway to immunosuppression. Immunobiology. 2010;215:598–605. doi: 10.1016/j.imbio.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Guindon J, Hohmann AG. The endocannabinoid system and cancer: therapeutic implication. Br J Pharmacol. 2011;163:1447–1463. doi: 10.1111/j.1476-5381.2011.01327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fogli S, Breschi MC. The molecular bases of cannabinoid action in cancer. Cancer Ther. 2008;6:103–116. [Google Scholar]
- 62.Noonan J, Tanveer R, Klompas A, Gowran A, McKiernan J, Campbell VA. Endocannabinoids prevent β-amyloid-mediated lysosomal destabilization in cultured neurons. J Biol Chem. 2010;285:38543–38554. doi: 10.1074/jbc.M110.162040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Siegmund SV, Schwabe RF. Endocannabinoids and liver disease. II. Endocannabinoids in the pathogenesis and treatment of liver fibrosis. Am J Physiol Gastrointest Liver Physiol. 2008;294:G357–G362. doi: 10.1152/ajpgi.00456.2007. [DOI] [PubMed] [Google Scholar]
- 64.Siegmund SV, Seki E, Osawa Y, Uchinami H, Cravatt BF, Schwabe RF. Fatty acid amide hydrolase determines anandamide-induced cell death in the liver. J Biol Chem. 2006;281:10431–10438. doi: 10.1074/jbc.M509706200. [DOI] [PubMed] [Google Scholar]
- 65.Miller LK, Devi LA. The highs and lows of cannabinoid receptor expression in disease: mechanisms and their therapeutic implications. Pharmacol Rev. 2011;63:461–470. doi: 10.1124/pr.110.003491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mallat A, Lotersztajn S. Cannabinoid receptors as novel therapeutic targets for the management of non-alcoholic steatohepatitis. Diabetes Metab. 2008;34:680–684. doi: 10.1016/S1262-3636(08)74604-4. [DOI] [PubMed] [Google Scholar]
- 67.Tam J, Liu J, Mukhopadhyay B, Cinar R, Godlewski G, Kunos G. Endocannabinoids in liver disease. Hepatology. 2011;53:346–355. doi: 10.1002/hep.24077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Reichenbach V, Ros J, Jiménez W. [Endogenous cannabinoids in liver disease: many darts for a single target] Gastroenterol Hepatol. 2010;33:323–329. doi: 10.1016/j.gastrohep.2009.06.010. [DOI] [PubMed] [Google Scholar]
- 69.Chakraborty JB, Oakley F, Walsh MJ. Mechanisms and biomarkers of apoptosis in liver disease and fibrosis. Int J Hepatol. 2012;2012 doi: 10.1155/2012/648915. 648915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jiang F, Zhang Y, Dusting GJ. NADPH oxidase-mediated redox signaling: roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol Rev. 2011;63:218–242. doi: 10.1124/pr.110.002980. [DOI] [PubMed] [Google Scholar]
- 71.Jiang JX, Chen X, Serizawa N, Szyndralewiez C, Page P, Schröder K, Brandes RP, Devaraj S, Török NJ. Liver fibrosis and hepatocyte apoptosis are attenuated by GKT137831, a novel NOX4/NOX1 inhibitor in vivo. Free Radic Biol Med. 2012;53:289–296. doi: 10.1016/j.freeradbiomed.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Després JP, Golay A, Sjöström L. Effects of rimonabant on metabolic risk factors in overweight patients with dyslipidemia. N Engl J Med. 2005;353:2121–2134. doi: 10.1056/NEJMoa044537. [DOI] [PubMed] [Google Scholar]
- 73.Christensen R, Kristensen PK, Bartels EM, Bliddal H, Astrup A. Efficacy and safety of the weight-loss drug rimonabant: a meta-analysis of randomised trials. Lancet. 2007;370:1706–1713. doi: 10.1016/S0140-6736(07)61721-8. [DOI] [PubMed] [Google Scholar]
- 74.Kunos G, Tam J. The case for peripheral CB1 receptor blockade in the treatment of visceral obesity and its cardiometabolic complications. Br J Pharmacol. 2011;163:1423–1431. doi: 10.1111/j.1476-5381.2011.01352.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cluny NL, Vemuri VK, Chambers AP, Limebeer CL, Bedard H, Wood JT, Lutz B, Zimmer A, Parker LA, Makriyannis A, Sharkey KA. A novel peripherally restricted cannabinoid receptor antagonist, AM6545, reduces food intake and body weight, but does not cause malaise, in rodents. Br J Pharmacol. 2010;161:629–642. doi: 10.1111/j.1476-5381.2010.00908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kirilly E, Gonda X, Bagdy G. CB1 receptor antagonists: new discoveries leading to new perspectives. Acta Physiol (Oxf) 2012;205:41–60. doi: 10.1111/j.1748-1716.2012.02402.x. [DOI] [PubMed] [Google Scholar]
- 77.Liu X, Chen C, Smith BJ. Progress in brain penetration evaluation in drug discovery and development. J Pharmacol Exp Ther. 2008;325:349–356. doi: 10.1124/jpet.107.130294. [DOI] [PubMed] [Google Scholar]
- 78.Hammarlund-Udenaes M, Fridén M, Syvänen S, Gupta A. On the rate and extent of drug delivery to the brain. Pharm Res. 2008;25:1737–1750. doi: 10.1007/s11095-007-9502-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liu X, Van Natta K, Yeo H, Vilenski O, Weller PE, Worboys PD, Monshouwer M. Unbound drug concentration in brain homogenate and cerebral spinal fluid at steady state as a surrogate for unbound concentration in brain interstitial fluid. Drug Metab Dispos. 2009;37:787–793. doi: 10.1124/dmd.108.024125. [DOI] [PubMed] [Google Scholar]
- 80.Löscher W, Potschka H. Blood-brain barrier active efflux transporters: ATP-binding cassette gene family. NeuroRx. 2005;2:86–98. doi: 10.1602/neurorx.2.1.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schinkel AH. P-Glycoprotein, a gatekeeper in the blood-brain barrier. Adv Drug Deliv Rev. 1999;36:179–194. doi: 10.1016/s0169-409x(98)00085-4. [DOI] [PubMed] [Google Scholar]
- 82.Evers R, Chu XY. Role of the murine organic anion-transporting polypeptide 1b2 (Oatp1b2) in drug disposition and hepatotoxicity. Mol Pharmacol. 2008;74:309–311. doi: 10.1124/mol.108.048991. [DOI] [PubMed] [Google Scholar]
- 83.Addy C, Rothenberg P, Li S, Majumdar A, Agrawal N, Li H, Zhong L, Yuan J, Maes A, Dunbar S, Cote J, Rosko K, Van Dyck K, De Lepeleire I, de Hoon J, Van Hecken A, Depré M, Knops A, Gottesdiener K, Stoch A, Wagner J. Multiple-dose pharmacokinetics, pharmacodynamics, and safety of taranabant, a novel selective cannabinoid-1 receptor inverse agonist, in healthy male volunteers. J Clin Pharmacol. 2008;48:734–744. doi: 10.1177/0091270008317591. [DOI] [PubMed] [Google Scholar]
- 84.Proietto J, Rissanen A, Harp JB, Erondu N, Yu Q, Suryawanshi S, Jones ME, Johnson-Levonas AO, Heymsfield SB, Kaufman KD, Amatruda JM. A clinical trial assessing the safety and efficacy of the CB1R inverse agonist taranabant in obese and overweight patients: low-dose study. Int J Obes (Lond) 2010;34:1243–1254. doi: 10.1038/ijo.2010.38. [DOI] [PubMed] [Google Scholar]
- 85.Tam J, Vemuri VK, Liu J, Bátkai S, Mukhopadhyay B, Godlewski G, Osei-Hyiaman D, Ohnuma S, Ambudkar SV, Pickel J, Makriyannis A, Kunos G. Peripheral CB1 cannabinoid receptor blockade improves cardiometabolic risk in mouse models of obesity. J Clin Invest. 2010;120:2953–2966. doi: 10.1172/JCI42551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chorvat RJ, Berbaum J, Seriacki K, McElroy JF. JD-5006 and JD-5037: peripherally restricted (PR) cannabinoid-1 receptor blockers related to SLV-319 (Ibipinabant) as metabolic disorder therapeutics devoid of CNS liabilities. Bioorg Med Chem Lett. 2012;22:6173–6180. doi: 10.1016/j.bmcl.2012.08.004. [DOI] [PubMed] [Google Scholar]
- 87.Erkekoğlu P, Giray B, Şahin G. The toxicological evaluation of rimonabant, taranabant, surinabant and otenabant in the treatment of obesity: why the trials on endocannabinoid receptor antagonists and inverse agonists are suspended? FABAD J Pharm Sci. 2008;33:95–108. [Google Scholar]
- 88.Need AB, Davis RJ, Alexander-Chacko JT, Eastwood B, Chernet E, Phebus LA, Sindelar DK, Nomikos GG. The relationship of in vivo central CB1 receptor occupancy to changes in cortical monoamine release and feeding elicited by CB1 receptor antagonists in rats. Psychopharmacology (Berl) 2006;184:26–35. doi: 10.1007/s00213-005-0234-x. [DOI] [PubMed] [Google Scholar]
- 89.Neves SR, Ram PT, Iyengar R. G protein pathways. Science. 2002;296:1636–1639. doi: 10.1126/science.1071550. [DOI] [PubMed] [Google Scholar]
- 90.Pfefferkorn JA, Litchfield J, Hutchings R, Cheng XM, Larsen SD, Auerbach B, Bush MR, Lee C, Erasga N, Bowles DM, Boyles DC, Lu G, Sekerke C, Askew V, Hanselman JC, Dillon L, Lin Z, Robertson A, Olsen K, Boustany C, Atkinson K, Goosen TC, Sahasrabudhe V, Chupka J, Duignan DB, Feng B, Scialis R, Kimoto E, Bi YA, Lai Y, El-Kattan A, Bakker-Arkema R, Barclay P, Kindt E, Le V, Mandema JW, Milad M, Tait BD, Kennedy R, Trivedi BK, Kowala M. Discovery of novel hepatoselective HMG-CoA reductase inhibitors for treating hypercholesterolemia: a bench-to-bedside case study on tissue selective drug distribution. Bioorg Med Chem Lett. 2011;21:2725–2731. doi: 10.1016/j.bmcl.2010.11.103. [DOI] [PubMed] [Google Scholar]
- 91.Hsiang B, Zhu Y, Wang Z, Wu Y, Sasseville V, Yang WP, Kirchgessner TG. A novel human hepatic organic anion transporting polypeptide (OATP2). Identification of a liver-specific human organic anion transporting polypeptide and identification of rat and human hydroxymethylglutaryl-CoA reductase inhibitor transporters. J Biol Chem. 1999;274:37161–37168. doi: 10.1074/jbc.274.52.37161. [DOI] [PubMed] [Google Scholar]
- 92.Zaher H, Meyer zu Schwabedissen HE, Tirona RG, Cox ML, Obert LA, Agrawal N, Palandra J, Stock JL, Kim RB, Ware JA. Targeted disruption of murine organic anion-transporting polypeptide 1b2 (Oatp1b2/Slco1b2) significantly alters disposition of prototypical drug substrates pravastatin and rifampin. Mol Pharmacol. 2008;74:320–329. doi: 10.1124/mol.108.046458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pfefferkorn JA, Guzman-Perez A, Litchfield J, Aiello R, Treadway JL, Pettersen J, Minich ML, Filipski KJ, Jones CS, Tu M, Aspnes G, Risley H, Bian J, Stevens BD, Bourassa P, D'Aquila T, Baker L, Barucci N, Robertson AS, Bourbonais F, Derksen DR, Macdougall M, Cabrera O, Chen J, Lapworth AL, Landro JA, Zavadoski WJ, Atkinson K, Haddish-Berhane N, Tan B, Yao L, Kosa RE, Varma MV, Feng B, Duignan DB, El-Kattan A, Murdande S, Liu S, Ammirati M, Knafels J, Dasilva-Jardine P, Sweet L, Liras S, Rolph TP. Discovery of (S)-6-(3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanamido)nicotinic acid as a hepatoselective glucokinase activator clinical candidate for treating type 2 diabetes mellitus. J Med Chem. 2012;55:1318–1333. doi: 10.1021/jm2014887. [DOI] [PubMed] [Google Scholar]
- 94.Oballa RM, Belair L, Black WC, Bleasby K, Chan CC, Desroches C, Du X, Gordon R, Guay J, Guiral S, Hafey MJ, Hamelin E, Huang Z, Kennedy B, Lachance N, Landry F, Li CS, Mancini J, Normandin D, Pocai A, Powell DA, Ramtohul YK, Skorey K, Sørensen D, Sturkenboom W, Styhler A, Waddleton DM, Wang H, Wong S, Xu L, Zhang L. Development of a liver-targeted stearoyl-CoA desaturase (SCD) inhibitor (MK-8245) to establish a therapeutic window for the treatment of diabetes and dyslipidemia. J Med Chem. 2011;54:5082–5096. doi: 10.1021/jm200319u. [DOI] [PubMed] [Google Scholar]