

Abstract
Overexpression of suprachiasmatic nucleus circadian oscillatory protein (SCOP), a negative ERK regulator, blocks long-term memory encoding. Inhibition of calpain-mediated SCOP degradation also prevents the formation of long-term memory, suggesting rapid SCOP breakdown is necessary for memory encoding. However, whether SCOP levels also control the magnitude of long-term synaptic plasticity is unknown. Here we show that following synaptic activity-induced SCOP degradation, SCOP is rapidly replaced via mTOR-mediated protein synthesis. We further show that early SCOP degradation is specifically catalyzed by µ–calpain while late SCOP re-synthesis is mediated by m-calpain. We propose that µ–calpain promotes long-term potentiation induction by degrading SCOP and activating ERK, while m-calpain activation limits the magnitude of potentiation by terminating the ERK response via enhanced SCOP synthesis. This unique braking mechanism could account for the advantages of spaced vs. massed training in the formation of long-term memory.
Keywords: SCOP, calpain, LTP, induction, consolidation, learning and memory
Long-term potentiation (LTP) of glutamatergic transmission induced by brief, patterned stimulation of afferents fulfills several requirements for a high-capacity memory mechanism, including rapid induction, extreme persistence and synapse specificity1. Accordingly, a large amount of work has led to a detailed description of the induction, expression, and stabilization phases of LTP and the identification of key signaling cascades involved in each of these steps. In contrast, the factors that control the magnitude of LTP are still poorly understood. The present study introduces a novel set of signaling events that impose a limit on the degree of lasting potentiation produced by naturalistic induction events.
Transient ERK activation has been repeatedly shown to be necessary for LTP induction2–4 and for triggering mTOR-dependent local protein synthesis3. SCOP (suprachiasmatic nucleus [SCN] circadian oscillatory protein) inhibits ERK1/2 by binding and trapping its activator Ras in the inactive form5. In cultured neurons, BDNF or NMDA receptor activation can remove this inhibition through calpain-mediated SCOP degradation. Calpain regulation of SCOP has important functional implications, as SCOP overexpression blocked novel object recognition memory6, while calpain inhibition blocked learning-induced decrease in SCOP levels in hippocampus. We had earlier proposed that calpain activation leads to modifications of the dendritic spine cytoskeleton and a redistribution of glutamate receptors, resulting in a long-lasting changes in synapse structure and function7,8. Several lines of evidence support this hypothesis, including reports from different groups that calpain inhibitors applied before and during inducing stimulation block the subsequent development of LTP9–11. More recently we found that BDNF, whose release after LTP induction12 is required for stable potentiation, activates m-calpain, likely via ERK-mediated phosphorylation at serine 5013, resulting in mTOR-stimulated local protein synthesis via PTEN truncation14. This raises the possibility that mTOR activation might serve to replenish SCOP pools, thereby terminating TBS-induced ERK activation.
Our studies were directed at testing the idea that both degradation and synthesis of SCOP are important for LTP process, and at understanding the mechanism as well as the functional significance of the rapid return of ERK inhibition. Combined with earlier findings, the results strongly suggest that µ-calpain-mediated SCOP degradation is involved in LTP induction while m-calpain-mediated stimulation of mTOR-dependent SCOP synthesis restricts the magnitude of potentiation during early consolidation. Thus, rapid activity-dependent changes in SCOP levels resulting from the dual and opposite effects of µ- and m-calpain control the formation and extent of LTP in hippocampus. The existence of this molecular mechanism controlling the magnitude of potentiation has clear implications for our understanding of learning and memory
Results
Reciprocal changes in SCOP and ERK following BDNF treatment
Shimizu et al.6 showed that BDNF causes a rapid decrease in SCOP levels associated with increase in ERK phosphorylation/activation in cultured neurons. We extended this result to acute hippocampal slices, and found, as was observed in cultured neurons, that both effects returned to baseline levels within 40 min despite the continued presence of the neurotrophin (Fig. 1a–c). Pretreatment with calpain inhibitor III (10 µM) completely eliminated BDNF-induced decrease in SCOP levels and the concomitant increase in phosphorylated (p-)ERK levels (Fig. 1d–f). Collectively, these results suggest that BDNF-driven calpain activation results in degradation of a negative modulator (SCOP) of ERK, as described in previous work, followed by a surprisingly rapid recovery of the modulator associated with termination of ERK activation.
Figure 1. Effects of calpain inhibitor III on BDNF-induced changes in levels of SCOP and p-ERK.

a, d, g. Experimental protocols: hippocampal slices were prepared from adult rats and were treated with BDNF (100 ng/ml) for the indicated periods of time in the absence or presence of calpain inhibitor III (10 µM), as indicated. b, e, h. Representative western blots for SCOP, Akt, p-ERK and ERK under various experimental conditions. c, f, i. Quantitative analysis of the levels of SCOP (normalized to the values of Akt) and p-ERK/ERK ratios under various experimental conditions. In all cases, results are means ± S.E.M. of 3 experiments. * p < 0.05, as compared to time point 0, one-way ANOVA followed by Bonferroni test.
We next asked if calpain was involved in SCOP recovery by applying its inhibitor starting 20 minutes after the onset of BDNF treatment. The results were remarkable: the neurotrophin produced the expected decrease in SCOP levels with associated increase in ERK activation but these effects continued throughout the remainder of the 1 hour incubation period, and ERK recovery was eliminated (Fig. 1g–i).
We recently reported that calpain degrades PTEN, an inhibitor of mTOR-mediated protein synthesis14, raising the possibility that BDNF-induced activation of the protease results in accelerated synthesis of replacement copies of SCOP. In support of this idea, when BDNF treatment of hippocampal slices was performed in the presence of cycloheximide, a general protein synthesis inhibitor, or rapamycin, an inhibitor of mTOR-mediated protein synthesis, the recovery of SCOP levels was prevented and p-ERK levels remained elevated above control levels (Fig. 2a–f). To further confirm that BDNF promotes de novo SCOP synthesis, we performed pulse-chase experiments in cortical synaptoneurosomes using a modified aminoacid, L-azidohomoalanine, which is incorporated into newly synthesized proteins and can later be labeled with biotin. Total SCOP was immunoprecipitated after metabolic labeling of nascent proteins. Newly synthesized (biotin-labeled) and total SCOP were then detected, using infrared dye conjugated Streptavidin and anti-SCOP antibody, respectively. Treatment of synaptoneurosomes with BDNF for 60 min significantly increased the levels of newly synthesized SCOP as compared to control, an effect that was absent in synaptoneurosomes pre-treated with rapamycin (Fig. 2g–i). This result further confirms the hypothesis proposed by Shimizu et al.6 that balance between degradation and synthesis is responsible for the dual effect of BDNF on SCOP levels and ERK activation.
Figure 2. Effects of cycloheximide and rapamycin on BDNF-induced changes in levels of SCOP and p-ERK.

a, d, Experimental design: hippocampal slices were treated with BDNF (100 ng/ml) for the indicated periods of time in the presence of cycloheximide (25 µM) or rapamycin (1 µM). b, e, Representative western blots for SCOP, Akt, p-ERK and ERK under various experimental conditions. c, f. Quantitative analysis of the levels of SCOP (normalized to the values of Akt) and p-ERK/ERK ratios under various experimental conditions. In all cases, results are means ± S.E.M. of 3 experiments. * p < 0.05, as compared to time point 0, one-way ANOVA followed by Bonferroni test. g, Representative blot for all newly synthesized (biotin-labeled) proteins larger than 150 kDa from synaptoneurosomes before immunoprecipitation (input) as detected by IRDye® 800CW Streptavidin. Note that the second lane (BDNF-treated sample) shows denser labeling than the other lanes. NL (not-labeled) line is from naïve synaptoneurosomes without metabolic labeling (negative control). h, Representative western blots for newly synthesized (biotin-labeled) and total SCOP, after treatment of cortical synaptoneurosomes with BDNF (100 ng/ml) in the absence or presence of rapamycin (1 µM) and subsequent immunoprecipitation with SCOP antibody. i, Quantitative analysis of the levels of newly synthesized SCOP (normalized to total SCOP). Data are means ± S.E.M. of 4 experiments. * p < 0.05, as compared to control, two-way ANOVA followed by Bonferroni test.
SCOP decrease mediates transient ERK activation in LTP
To directly test whether calpain regulates SCOP levels and subsequently ERK activity during LTP, we repeated the above experiments using TBS stimulation of hippocampal CA1 mini-slices from adult rats. SCOP levels, as assessed with immunoblots, were significantly reduced 8 min after TBS stimulation in CA1 mini-slices, but returned to control levels 50 min after TBS. As in BDNF-treated hippocampal slices, p-ERK levels followed a reciprocal pattern: a marked elevation when SCOP levels were decreased and a delayed return to baseline as SCOP subsequently returned to control values (Fig. 3a–c). Pretreatment of slices with calpain inhibitor III eliminated TBS-induced decrease in SCOP levels and the associated increase in ERK activation (Fig. 3d–f). However, when calpain inhibitor III (10 µM) was applied after TBS application, SCOP levels remained decreased, and correspondingly p-ERK levels remained elevated (Fig. 3g–j). Similar results were obtained when using tetraethylammonium (TEA)-induced chemical LTP15–17 in rat acute hippocampal slices (Supplementary Fig. 1). These findings suggested that SCOP is rapidly synthesized following its degradation stimulated by increased neuronal activity. Under basal conditions, cycloheximide treatment of cortical synaptoneurosomes decreased SCOP levels with a t1/2 of about 90 min, suggesting that SCOP turns over rapidly and is replenished by protein synthesis (Supplementary Fig. 2a). Consistently, ERK phosphorylation gradually increased over time after treatment with cycloheximide (Supplementary Fig. 2b). The results obtained with BDNF, TBS or TEA lead to the conclusion that LTP is associated with rapid degradation and recovery of SCOP, resulting in a pronounced but transient stimulation of ERK, consistent with previous study showing that TBS stimulation causes a rapid, short-lived increase in ERK phosphorylation in CA3-CA1 synapses of hippocampal slices18. Calpain appears to be involved in both the loss and recovery of SCOP, and thus in the initiation and then the termination of ERK activation.
Figure 3. Effects of calpain inhibition on TBS-induced changes in adult hippocampal CA1 mini-slices.

a, d, g. Experimental protocol: hippocampal CA1 mini-slices were prepared from adult rats and were stimulated by TBS in the absence or presence of calpain inhibitor III (10 µM), as indicated. b, e, h. Representative western blots for SCOP, Akt, p-ERK and ERK under various experimental conditions. c, f, i. Quantitative analysis of the levels of SCOP (normalized to the values of Akt) and p-ERK/ERK ratios under various experimental conditions. Results are means ± S.E.M. of 3–8 experiments. * p < 0.05, as compared to time point 0, one-way ANOVA followed by Bonferroni test.
We then used immunohistochemistry to verify whether calpain inhibition before TBS could block ERK activation, while the same treatment applied after TBS could prolong ERK phosphorylation in dendritic spines. A single train of 10 theta bursts was applied to the Schaffer-commissural pathway in field CA1b of hippocampus and slices were collected at various times post-TBS. Sectioning and immunostaining with antibodies selective for p-ERK were performed as described in Methods. A marked increase in p-ERK labeling of spines and dendrites was observed at five minutes post-TBS in the apical dendritic field adjacent to the stimulation site, an effect that was altogether absent in zones outside the Schaffer-commissural termination field. The increased labeling was, as expected from the literature, absent in slices collected 50 min after TBS (Fig. 4). Calpain inhibitor III applied before TBS partially but significantly blocked ERK activation at 5 min after TBS (Fig. 4). In contrast, when slices were treated with calpain inhibitor III starting 10 min after TBS, a large number of dendrites and spines in the vicinity of the stimulating electrode were still p-ERK-immunopositive at 50 min post-TBS (Fig. 4), an effect that was highly significant relative to TBS alone.
Figure 4. TBS-induced ERK activation is differentially affected by pre- or post-TBS calpain inhibitor III treatment.
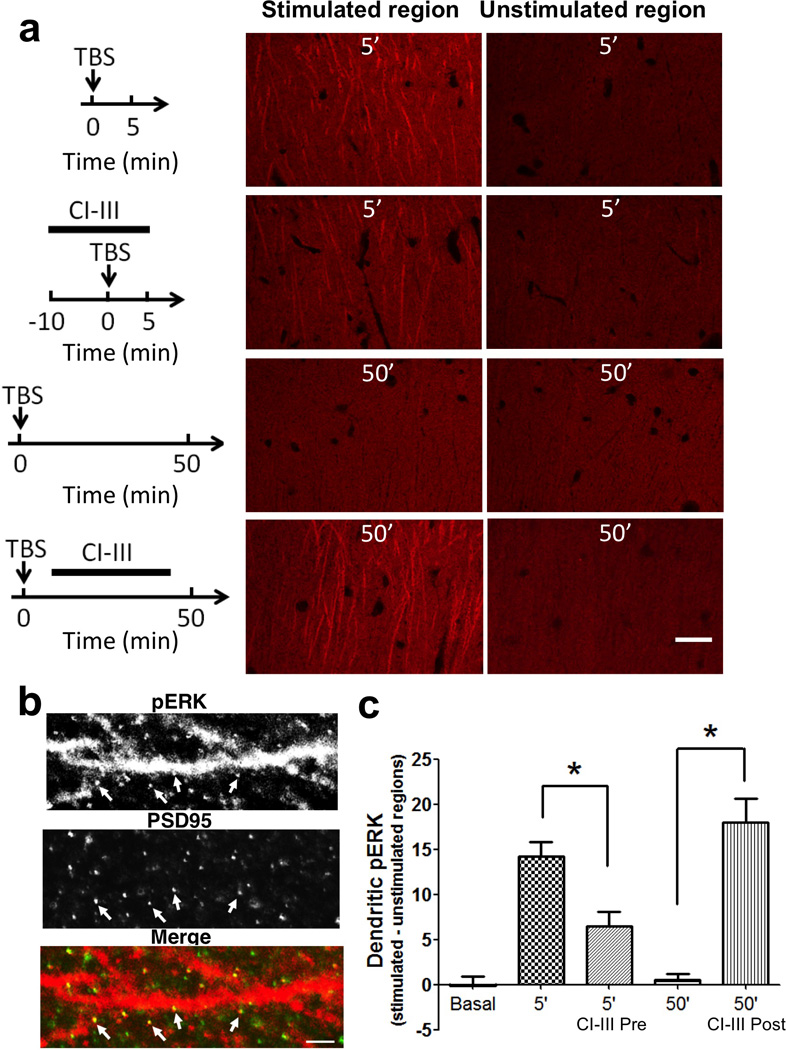
Hippocampal slices were prepared from adult rats and subjected to various experimental conditions. At the indicated times, slices were fixed, re-sectioned and processed for immunohistochemistry for p-ERK. Images of stimulated and unstimulated regions from two most representative sections of each slice were chosen for analysis. a. Representative images. Top: p-ERK immunoreactivity at 5 min after TBS (10 bursts of 4 pulses (100 Hz) delivered at 5 Hz) in slices treated with vehicle or calpain inhibitor III (10 µM) for 10 min before and continued for 5 min after TBS. Bottom: p-ERK immunoreactivity at 50 min after TBS in slices treated with vehicle or calpain inhibitor III (10 µM) for 30 min starting 10 min after TBS. Scale bar: 20 µm. b. High magnification images of dendrites in stimulated region double-stained with p-ERK and PSD-95 antibodies, showing the co-localization of p-ERK and PSD-95 (arrows). Scale bar: 2 µm. c. Quantitative analysis of p-ERK immunostaining. The average p-ERK intensity was determined in a 120×60 µm2 area in the dendritic field of CA1. Image intensities from stimulated regions were subtracted from those of unstimulated regions of the same slice to correct for slice-to-slice variation in staining intensity, as described in38. Shown are means ± S.E.M. of n = 8. * p < 0.01, one-way ANOVA followed by Bonferroni test.
Calpain activity after TBS restricts the magnitude of LTP
What, if any, are the physiological consequences of blocking SCOP recovery and prolonging ERK activation following LTP induction? We began answering this question by first repeating prior work on the effects of calpain inhibition on the induction, expression, and rapid consolidation of LTP. Infusion of calpain inhibitor III (10 µM) starting 10 min before TBS had no evident effects on baseline or on the initial expression of LTP but, in agreement with previous reports10,11, completely blocked the subsequent stabilization of the potentiation effect (Fig. 5a). Notably, it also caused no detectable changes in the burst responses during TBS (Supplementary Fig. 3), indicating that calpain inhibition did not interfere with frequency facilitation or NMDA receptor function. We also reproduced a previous finding that application of calpain inhibitor III 1 h after TBS did not modify established LTP (Fig. 5a)10. Remarkably, infusion of calpain inhibitor III starting 10 min after TBS produced an unexpected result: the slope of the already potentiated responses rapidly increased, and over the next 30 min, reached values more than twice as large as those normally found 1 h after a single train of theta bursts (Fig. 5a,b). Similar results were obtained with calpain inhibitor IV (Supplementary Fig. 4), which is reported to have a different pattern of inhibition for cysteine proteases than calpain inhibitor III19
Figure 5. Effects of calpain inhibition at various times before or after LTP induction.

a. Time-dependent effects of calpain inhibition on LTP. Hippocampal slices were prepared from adult rats and LTP was induced in field CA1 by TBS (black arrow). Vehicle or calpain inhibitor III (10 µM) was applied for 10 min before and continued 5 min after TBS (cyan). Alternatively, calpain inhibitor III (10 µM) was applied for 30 min starting 10 min (blue) or 60 min (magenta) post-TBS. Slopes of field EPSPs (fEPSPs) are expressed as percent of the average values recorded during the 10 min baseline (means ± S.E.M. of 5–10 slices from 3–5 animals). Basal synaptic transmission did not change during the recording period (red). b. Representative traces for fEPSPs recorded at the indicated time-points (1, 2, and 3), under different experimental conditions. Calibration: 0.5 mV/5 msec. c. Post-TBS calpain inhibition blocks TBS2-induced further potentiation. TBS was applied as indicated by arrows. Calpain inhibitor III (10 µM) was applied during the time indicated by the horizontal lines. A second TBS was delivered 60 min after the first TBS and fEPSP was recorded for an additional 40 min (means ± S.E.M of 6–8 slices from 3–4 animals). d. Top: Calpain inhibitor III (10 µM) was applied for 30 min starting 10 min after TBS1 (arrow). At the end of calpain inhibitor treatment, stimulation intensity was decreased to obtain a response equivalent to the pre-TBS value. After 20 min, TBS2 was delivered and responses recorded for an additional 40 min (representative experiment). Bottom: Calpain inhibitor III (10 µM) was applied to naïve slices for 30 min. After 20 min, TBS was applied and responses recorded for an additional 40 min (representative experiment). e. Comparison of the responses highlighted in d (means ± S.E.M. of 6–8 slices).
Recent work showed that delivery of a second TBS bout (‘TBS2’) after a 1 h delay more than doubles the magnitude of LTP produced by TBS120–22 and that the enhancement is likely due to recruitment of additional synapses into the potentiated state22. We confirmed the ‘LTP2’ effect in the present studies and found that its magnitude is about the same as that produced by delayed application of calpain inhibitor III (Fig. 5c, open circles). These observations raise the possibility that post-TBS infusion of a calpain inhibitor, acting via increase in p-ERK, unmasks a latent potentiation produced by TBS1 in a large population of synapses. If so, then the novel enhancement phenomenon described here should occlude the LTP2 effect. To test this hypothesis, we first confirmed that the enhanced LTP remained in place following a 20 min washout of the calpain inhibitor (Fig. 5c) and that normal LTP could be induced in a ‘naive’ pathway at this time point (Fig. 5d, top). TBS2 delivered after washout, and 1 h after TBS1, had no further effect on the magnitude of LTP (Fig. 5c, closed circles). To confirm that the absence of additional LTP was due to the saturation of synaptic potentiation but not to a ceiling effect on evoked synaptic responses, we decreased the intensity of stimulation to generate responses of the same magnitude as during baseline, before delivering the second TBS. Again, no evidence for LTP2 was found (Fig. 5d, bottom; also see Fig 5e).
To further test the selectivity of the effect of calpain inhibitor on stimulated synapses, we used a second stimulation electrode (S2), which was placed on the opposite side of the recording electrode from the first stimulation electrode (S1) (Supplementary Fig. 5a). In the S1 pathway TBS induced LTP, and calpain inhibitor III (10 µM) applied 10 min post-TBS elicited further potentiation. However, responses in the control pathway (S2) remained unchanged in the absence or presence of calpain inhibitor III (Supplementary Fig. 5b). These data indicated that calpain inhibitor III only had effects on TBS-stimulated synapses.
ERK inactivation after TBS is necessary for LTP restriction
We then tested the hypothesis that SCOP degradation and the resulting ERK activation were involved in the LTP enhancement effect of post-TBS calpain inhibition by applying two different inhibitors of MAP kinase (MEK), U0126 (5 µM) or PD98029 (5 µM), alone or in the presence of calpain inhibitor III (10 µM) 10 min after TBS. Both inhibitors completely prevented the enhancing effect of calpain inhibitor III on LTP (Fig. 6a,b). Application of the MEK inhibitors alone at this concentration had no effect on LTP consolidation, a result consistent with a previous report that ERK activation is not required for LTP consolidation23. Quantitative analysis of the results indicated that the effects of calpain inhibitor III on LTP and the blocking effects of both U0126 and PD98209 were highly statistically significant, as compared to TBS alone (Fig. 6d). As our results indicated that SCOP synthesis was responsible for the late phase of ERK inhibition induced by BDNF treatment or TBS stimulation, we also tested the effects of rapamycin applied 10 min after TBS. Interestingly, application of rapamycin shortly after TBS caused a progressive enhancement of LTP, similar to that produced by post-TBS application of calpain inhibitor III (Fig. 6c,d). Like rapamycin, a 10 min post-TBS application of cycloheximide, a general protein synthesis inhibitor, further enhanced the magnitude of LTP (Supplementary Fig. 6).
Figure 6. ERK inhibition prevents and mTOR inhibition mimics calpain inhibition-induced LTP enhancement.

Hippocampal slices were prepared from adult rats and LTP was induced by TBS as under Fig. 1. The MEK inhibitors PD98059 (5 µM) (a) or U0126 (5 µM) (b) were applied together with calpain inhibitor III (10 µM) for 30 min. The data for the grey symbols in a and b, which were used as the reference, are from Fig.5a (blue symbol). In all cases, the slopes of the field EPSPs are expressed as percent of the average values recorded during the 10 min baseline, and are means ± S.E.M. of 5–10 slices prepared from 3–5 animals. c. Effects of rapamycin (1 µM) applied for 30 min (horizontal bar) starting at 10 min after TBS on hippocampal LTP. d. Summary of the amplitude of LTP measured within the gray area corresponding to 49–50 min after TBS (Note that the data for TBS and TBS + CI–III are from Fig. 5a). * p < 0.01 as compared to TBS alone; § p < 0.01 as compared to TBS + Calpain inhibitor III (one-way ANOVA followed by Bonferroni test). The slopes of the fEPSPs are expressed as percent of the average values recorded during the 10 min baseline, and are means ± S.E.M. of 5–10 slices prepared from 3–5 animals.
m-calpain activation limits the magnitude of LTP
Two major isoforms of calpain are present in the brain, µ- and m-calpain, which differ by their calcium requirements for activation in in vitro assays, with µ-calpain requiring micromolar and m-calpain requiring millimolar calcium concentration for activation. As we previously showed that BDNF stimulated m-calpain through ERK-mediated phosphorylation13, and that m-calpain-dependent PTEN degradation was involved in BDNF-mediated mTOR stimulation and stimulation of local protein synthesis14, we postulated that m-calpain activation could be responsible for the changes in SCOP metabolism following TBS. We tested the effects of a dipeptide ketoamide, Z-Leu-Abu-CONH-CH2-C6H3(3,5-(OMe)2) (mCalp-I), which has been reported to have a Ki of 22 nM against m-calpain vs a Ki of 2.3 µM against µ-calpain24. We first verified that mCalp-I was indeed more selective for m- than for µ-calpain, by determining its IC50 for purified m-calpain- or µ-calpain-induced degradation of Succinyl-Leu-Tyr-7-amino-4-methylcoumarin (Suc-Leu-Tyr-AMC) (Supplementary Fig. 7). From the IC50s, we used the Cheng-Prusoff equation to determine the Kis of mCalp-I for m- and µ-calpain; in our hand, the Ki for m-calpain was 25 nM and for µ-calpain 0.94 µM, in close agreement with the reported values. We then tested the effects of mCalp-I on TEA-mediated SCOP and PTEN degradation (Fig. 7a–f). Interestingly, mCalp-I (200 nM) did not prevent TEA-mediated SCOP degradation when added before TEA but it prevented its recovery during prolonged incubation (Fig. 7d–f). On the other hand, mCalp-I did prevent TEA-mediated PTEN degradation, further suggesting that m-calpain activation is responsible for TEA-mediated PTEN degradation and stimulation of SCOP synthesis. Application of mCalp-I before TBS did not affect LTP induction (Fig. 7g–h); however, it did produce the same enhancement of LTP starting about 15 min after TBS (Fig. 7g–h). It also enhanced LTP when applied 20 min after TBS (Fig. 7g–h).
Figure 7. Effects of an m-calpain specific inhibitor on TEA-mediated SCOP and PTEN degradation and on LTP.

a–f: Hippocampal slices were prepared from adult rats and were treated with TEA (20 mM) for 10 min, and collected at the indicated times. TEA and mCalp-I (200 nM) were applied as shown in a and d. b and e: Representative western blots for SCOP, PTEN and Akt at the indicated times. c and f: Quantitative analysis of the levels of SCOP and PTEN (normalized to the values of Akt). In all cases, results are means ± S.E.M. of 3 experiments. * p < 0.05, as compared to time point 0, one-way ANOVA followed by Bonferroni test. g–h. Effects of mCalp-I on TBS-induced LTP. mCalp-I (200 nM) was applied either before TBS (vertical arrow) (blue circles) or 20 min after TBS (red circles). mCalp-I alone had no effect on baseline responses (blue triangles).
h. Summary of the amplitude of LTP measured within the gray area corresponding to 49–50 min after TBS. * p < 0.01 as compared to TBS alone.
Discussion
The results described here constitute the first evidence that an active process regulates the magnitude of LTP in the minutes to hour following the inducing stimulus. Not surprisingly, the pertinent mechanisms incorporate many novel features, including a time of action that continues for a considerable period after induction of the potentiation effect. It thus appears that theta burst stimulation both enhances synaptic responses and triggers previously undetected processes that set limits on the degree of potentiation. The present findings point to the serial activation of two isoforms of the protease calpain, both targeting a negative regulator of ERK, as being critically involved in the LTP induction/capping sequence. This collection of new phenomenology is logically related to recently reported LTP timing rules and thereby to particular aspects of learning. Below, we consider the complex cellular events uncovered in our studies before turning to these functional implications.
Our results indicate that the metabolism of SCOP, a protein exhibiting a circadian rhythm in the SCN, is subjected to rapid, activity-dependent changes, and that these lead to a critical role in regulating the timing and extent of synaptic plasticity in hippocampus. As previously reported6, SCOP is rapidly degraded by calpain following BDNF treatment of hippocampal slices; it is also rapidly degraded following TBS- or TEA-induced LTP in CA1. Under all these conditions, SCOP levels rapidly recover through mTOR-mediated protein synthesis, an event that is blocked by delayed treatment with a calpain inhibitor. Our results indicate that this is due to de novo SCOP synthesis, and not to some indirect effect producing masking/unmasking epitopes of SCOP. A previous study has identified SCOP mRNA among the numerous mRNAs present in the neuropil of hippocampal CA1 region25, indicating that SCOP is likely synthesized locally as a result of mTOR activation. In accord with this, we recently showed that stimulation of the mTOR pathway and local protein synthesis by BDNF is due, at least in part, to m-calpain-mediated PTEN truncation14. Calpain activation at different time points can thus account for both the loss and recovery of SCOP following BDNF treatment, TBS- or TEA-induced potentiation. SCOP suppresses ERK and, as expected from this, changes in activation/phosphorylation of the kinase, which plays a critical role in LTP, occurred in parallel with the above-described fluctuations in levels of the negative regulator. Thus, the present results indicate that SCOP is one of the growing list of plasticity-related proteins (e.g., Arc, CamKII, and possibly various glutamate receptor subunits26) that are rapidly degraded and synthesized in dendrites as a result of increased neuronal activity.
Studies using various inhibitors have indicated that calpain activation is a necessary step in LTP induction. Moreover, down-regulation of calpain activity through knock-out of calpain-4, the small calpain subunit common to both µ-calpain and m-calpain, similarly impairs LTP27. As ERK activation is a necessary early step for LTP induction2–4,23, calpain could exert its effects on LTP induction via SCOP degradation. We confirmed the necessary prediction from this hypothesis that pretreatment of slices with a calpain inhibitor prevented SCOP degradation and the ERK activation that normally occurs with induction of LTP by theta bursts. The question then arose of what, if any, role calpain-mediated recovery of SCOP plays following the induction and initial expression of LTP. We investigated this by infusing a calpain inhibitor minutes after delivering a train of theta bursts. This led to a marked increase in the magnitude of synaptic potentiation. We therefore propose that the suppression of ERK activity by calpain-mediated increase in SCOP synthesis in the post-induction period serves to prevent the further growth of LTP. The dual role of calpain in SCOP metabolism raised the question of whether the two major isoforms of the protease (‘µ’ and ‘m’) are differentially involved in degradation vs. enhanced synthesis of SCOP. Results obtained with a specific inhibitor implicated m-calpain in mTOR-dependent SCOP synthesis following induction and strongly suggested that µ-calpain is involved in SCOP degradation during the initial phase of LTP production. The differential effects of µ-and m-calpain on SCOP could be due to a different subcellular localization of the proteases. In particular, a fraction of SCOP has been shown to be present in membrane rafts, and this fraction could be degraded following NMDA receptor channel opening and the resulting µ-calpain activation28. On the other hand, m-calpain might be present in a different subcellular compartment at the base of dendritic spines where local dendritic protein synthesis is taking place. Thus, the biphasic effect of TBS on ERK activation, initial activation followed by delayed prevention of activation could reflect the sequential activation of µ- and m-calpain following TBS, as well as their opposite effects on SCOP levels and ERK activation (Fig. 8). An alternative hypothesis is suggested by the recent discovery that BDNF promotes m-calpain activation via phosphorylation. Release of the neurotrophin during theta burst stimulation, as evidenced by activation of its TrkB receptor within synapses, is known to facilitate the cytoskeletal changes required for LTP consolidation. But its effects on m-calpain suggest that it also engages ERK suppression machinery shown here to place limits on the magnitude of the LTP produced by those changes.
Figure 8. Schematic representation of the functions of µ- and m-calpain in LTP induction and consolidation.

a. µ-calpain activation is necessary for synaptic potentiation during LTP induction, and its inhibition prevents LTP (middle panel). m-calpain activation during consolidation limits the extent of synaptic potentiation and its inhibition results in enhanced LTP (right panel). µ-calpain activation is indicated by yellow triangles and m-calpain by red squares. Calpain inhibitor III (CI–III) application is indicated by red arrows. Note that we also postulate that µ-calpain and m-calpain are differentially localized in synapses. b. Signaling pathways downstream of µ- and m- calpain in LTP induction and consolidation. In LTP induction, µ-calpain activation, possibly resulting from Ca2+ influx through the NMDA receptors, results in SCOP truncation followed by ERK activation. In the consolidation period, m-calpain activation, possibly resulting from BDNF-mediated ERK activation13 stimulates mTOR-dependent protein synthesis through calpain-mediated PTEN degradation14, and in particular SCOP synthesis, which would restore normal SCOP levels, thereby preventing ERK activation.
Our results also provide interesting information regarding the role of local protein synthesis in LTP. While there is much debate regarding this issue with contradictory reports supporting or negating the requirement for protein synthesis29,30, our results indicate that, at least during a short window of time following TBS, protein synthesis inhibition can enhance and not prevent LTP consolidation. This paradoxical effect of protein synthesis inhibition might have been easily missed, as the prevalent hypothesis is that protein synthesis is necessary for consolidation of LTP and memory. In particular, Cammalleri et al.31 did not observe changes in LTP when they applied rapamycin after high frequency stimulation. However, their stimulation protocol was very different from the one we used and could have produced LTP saturation. Our result also underscores the complexity of the function of local protein synthesis in LTP and in learning and memory and the critical role of the spatio-temporal sequence of events that are activated by TBS or by a learning experience. Protein homeostasis through synthesis and proteasome-mediated degradation has recently been proposed to play an important role in synaptic plasticity26. Our results indicate that calpain-mediated degradation is also an important contributor of protein homeostasis, as it provides a direct link between various extracellular signals, including glutamate and BDNF, with the local protein synthesis machinery regulated by the mTOR pathway.
Previous studies had indicated that calpain-mediated SCOP degradation played a role in novel object recognition learning6, although the lack of effect of SCOP overexpression on fear conditioning suggested the possibility that different forms of learning and memory might use different types of signaling. Our results extend these findings and indicate that calpain-mediated SCOP regulation is more complex than previously reported, as calpain promotes both the degradation and synthesis of SCOP. The latter part of this duality provides a plausible explanation for recently discovered timing rules that obviate the normal limits on the magnitude of LTP. In particular, it has been reported that a second bout of theta burst stimulation does not produce additional LTP unless delivered after a delay of about 1 h, at which point it doubles the magnitude of potentiation21. It thus appears that induction and expression are followed by an extended refractory period. Additional work suggested that adult hippocampus contains a large population of spine synapses with high plasticity threshold and that these are not driven into the potentiated state by a single episode of theta pattern stimulation22. Notably, the timing of the dual role of calpain in regulating SCOP metabolism matches that for the refractory period during which the high threshold synapses do not respond to a second train of theta bursts. If the two phenomena are closely related, then removing the calpain-mediated brake on LTP size should occlude the LTP2 effect. We confirmed this prediction by showing that a delayed theta train has no effect on the enhanced potentiation produced by a brief, post-TBS1 infusion of a calpain inhibitor.
Based on the above, we propose that m-calpain-mediated increase of SCOP synthesis in the minutes following LTP depresses ERK activity at high threshold synapses and thereby prevents them from transitioning into the potentiated state. The protease in this scenario serves to restrict single trial encoding of new information to a subset of synapses and imposes a delay before the initial representation can be expanded. It will be noted that this mechanism relates naturally to the distributed practice effect, a fundamental aspect of learning that has received little attention from neuroscientists. Specifically, training trials spaced apart by sizable delays produce much stronger memory than does a single, massed learning session32. The molecular brake described here may thus contribute importantly to the optimization of learning. It will be interesting to test whether m-calpain inhibitors enhance learning but do so at the cost of interfering with the spaced trials effect.
In any event, our results provide a new description of the succession of events that take place following TBS in dendritic spines of field CA1 of the hippocampus (Fig. 8). They place calpain activation at a central node of the various cascades of biochemical reactions that have been proposed to participate in the induction and consolidation stages of LTP33,34, in particular through the regulation of protein degradation and local protein synthesis of rapidly turning-over proteins, such as SCOP. Through this network, µ-calpain participates in LTP induction, while m-calpain limits the extent of synaptic enhancement, possibly by restricting the number of potentiated synapses to a subset of activated synapses. Superimposed on these cascades, calpain activation could also play important roles in the restructuring of the actin cytoskeleton and modification of the shape of dendritic spines, as we had proposed in our original hypothesis7. Thus, our findings provide a new framework to assemble the pieces of the complex events that are required to transform patterns of presynaptic activity in long-lasting changes in synaptic structure and function, and to explain certain temporal features of learning and memory.
Material and Methods
Acute hippocampal slice preparation
Animal use in all experiments followed NIH guidelines and all protocols were approved by the Institution Animal Care and Use Committee of Western University of Health Sciences.
Adult male Sprague-Dawley rats (3–4-month-old) were anesthetized with halothane and decapitated. Brains were quickly removed and transferred to oxygenated, ice-cold cutting medium (in mM): 124 NaCl, 26 NaHCO3, 10 glucose, 3 KCl, 1.25 KH2PO4, 5 MgSO4, and 3.4 CaCl2. Hippocampal transversal slices (400 µm-thick) were prepared using a McIlwain-type tissue chopper and transferred to either an interface recording chamber and exposed to a warm, humidified atmosphere of 95%O2/5%CO2 and continuously perfused with oxygenated and preheated (33 ± 0.5 °C) artificial cerebrospinal fluid (aCSF) (in mM) [110 NaCl, 5 KCl, 2.5 CaCl2, 1.5 MgSO4, 1.24 KH2PO4, 10 D-glucose, 27.4 NaHCO3], perfused at 1.4 ml/min (electrophysiology); or a recovery chamber with a modified aCSF medium, containing (in mM): 124 NaCl, 2.5 KCl, 2.5 CaCl2, 1.5 MgSO4, 1.25 NaH2PO4, 24 NaHCO3, 10 D-glucose, and saturated with 95%O2 /5%CO2 for 1 h at 37 °C (biochemical assays). CA1 mini-slices were obtained by dissecting out the CA1 region before transferring them in the recording chamber.
Electrophysiology
After 1.5 h incubation at 33 ± 0.5 °C in the recording chamber, a single glass pipette filled with 2 M NaCl was used to record field EPSPs (fEPSPs) elicited by stimulation of the Schaffer collateral pathway with twisted nichrome wires (single bare wire diameter, 50 µm) placed in CA1 stratum radiatum. Responses were recorded through a differential amplifier (DAM 50, World Precision Instruments, USA) with a 10 kHz high-pass and 0.1 Hz low-pass filter. Before each experiment, the input/output (I/O) relation was examined by varying the intensity of the stimulation. Data were collected and digitized by Clampex, and the slope of fEPSP was analyzed. All Data are expressed as means ± SEM, and statistical significance of differences between means was calculated with appropriate statistical tests as indicated in figure legends.
Treatments of acute hippocampal slices
After 1 h recovery, hippocampal slices were then transferred into screw-cap microfuge tubes containing 2 ml of freshly oxygenated aCSF medium with various drugs and were incubated at 37 °C for the indicated periods of times.
For induction of chemical LTP, slices were transferred to a modified aCSF containing (in mM): NaCl (124), KCl (5), CaCl2 (5), MgCl2 (0.1), KH2PO4 (1.25), NaHCO3 (24), D-glucose (10) and tetraethylammonium (25) for 10 min, and transferred back to normal aCSF. For calpain-inhibitor pretreatment, 10 µM calpain inhibitor III (CI–III) (Calbiochem) was applied 15 min before changing the medium to the modified aCSF and during the incubation in modified aCSF. For calpain inhibitor post-treatment, CI-III was applied 5 min after changing the medium back to normal aCSF.
For BDNF treatment, slices were incubated in normal aCSF containing 100 ng/ml BDNF (Millipore) for the indicated time periods. For calpain-inhibitor pretreatment, CI-III was applied 15 min before application of BDNF. For calpain inhibitor post-treatment, CI-III was applied 20 min after the application of BDNF. To inhibit protein synthesis, 25 µM cycloheximide (Tocris Bioscience) or 1 µM rapamycin (Cell Signaling) was applied 20 min before application of BDNF. At the indicated time points, 2–3 slices were rapidly frozen on dry ice. Slices were then lysed and protein concentrations were measured using BCA protein assay kit (Thermo). Equivalent amounts of proteins were processed for SDS-PAGE and western blot. The primary antibodies used were SCOP (1:1000, 07-1341, Millipore), actin (1:10000, MAB1501, Millipore), phospho-ERK (1:2000, 9101, Cell Signaling), ERK (1:3000, 9107), and Akt (1:1500, 2920). Full gel scans of all western blots in this paper can be found in Supplementary Fig. 8.
Calpain assay
The m-calpain specific inhibitor, Z-Leu-Abu-CONH-CH2-C6H3(3,5-(OMe)2) (mCalp-I), was a generous gift from Dr. James Power (Georgia Tech University). The hydrolysis of the fluorogenic substrate Suc-Leu-Tyr-AMC by µ-calpain and m-calpain was performed as previously described35, with minor modifications. Briefly, purified µ-calpain from porcine erythrocytes (8 µg, Millipore) or rat recombinant m-calpain (8 µg, a generous gift by Dr. Peter Davis from Queen’s University, Ontario, CA) was incubated with Suc-Leu-Tyr-AMC (0.5 mM) in 60 mM imidazole-HCl buffer, pH 7.3, containing 5 mM CaCl2, 5 mM cysteine, 2.5 mM β-mercaptoethanol, and different concentrations of mCalp-I (ranging from 0 to 20 µM). The reaction was initiated by adding the enzyme and continued at 30 °C for 15 min, while the fluorescence of 7-amino-4-methylcoumarin (Ex 380 nm/Em 450 nm) was monitored every 30 sec in a POLARstar Omega fluorescence polarization microplate reader (BMG Labtech). The rate of hydrolysis (increase in fluorescence/min) was determined from the linear portion of the curve. The IC50 values were obtained by adjusting data from each experiment into a sigmoidal dose-response curve. The Ki values for mCalp-I were calculated from the average of the IC50 values and from a single substrate concentration by using a Ki calculator tool for fluorescence-based competitive binding assays (http://sw16.im.med.umich.edu/software/calc_ki/). The Km values of Suc-Leu-Tyr-AMC for µ-calpain and m-calpain are 4.74 mM and 2.21 mM, respectively35. The final concentrations for µ-calpain and m-calpain were 357 and 367 nM.
Immunohistochemistry
At the indicated time points after TBS, hippocampal slices were fixed in 4% paraformaldehyde for 1 h and cryoprotected in 30% sucrose for 1 h at 4 °C, and sectioned on a freezing microtome at 20 µm. Sections were blocked in 0.1 M PBS containing 10% goat serum and 0.4% Triton X-100, and then incubated in primary antibody mixture including rabbit anti-pERK (1:500, 4370, Cell Signaling) and mouse anti-PSD95 (1:500, MA1-045, Thermo) in 0.1 M PBS containing 5% goat serum and 0.4% Triton X-100 overnight at 4 °C. Sections were washed 3 times (10 min each) in PBS and incubated in Alexa Fluor 594 goat anti-rabbit IgG (A-11037, Life Technologies) and Alexa Fluor 488 goat anti-mouse IgG (A-11001) for 2 h at room temperature36.
Cortical synaptoneurosome preparation
Synaptoneurosomes were prepared from 3–4 month-old male Sprague-Dawley rats, as previously described37, with minor modifications. Briefly, cortices were rapidly dissected and placed into chilled modified Krebs solution (mKrebs) containing (in mM): NaCl (118.5), KCl (4.7), KH2PO4 (1.18), NaHCO3 (24.9); MgSO4 (1.18), CaCl2 (2.5), D-glucose (10), HEPES (1), pH 7.4, and equilibrated with 95%O2/5%CO2. Cortices were homogenized in a 7-mL Kontes tissue Dounce homogenizer with ten strokes. Homogenized tissue was filtered through a 100-µm nylon mesh filter (BD Falcon) and the resulting suspension was filtered again through a 5-µm pore size Acrodisc syringe filter with a Supor membrane (Pall Life Sciences). The filtrate was then centrifuged at 1000 g for 15 min at 4 °C, washed once and centrifuged again. The pellet was resuspended in mKrebs and drugs were directly added to the pre-warmed (5 min, 37 °C) synaptoneurosome suspension for the indicated periods of time.
Metabolic labeling of synaptoneurosomes and immunoprecipitation
Detection of de novo protein synthesis was determined by using metabolic labeling with Click-iT® L-azidohomoalanine (AHA, Molecular probes). Cortical synaptoneurosomes were pre-incubated with 500 µM AHA in the absence and presence of 1 µM rapamycin for 20 min, and then treated with 100 ng/ml of BDNF for 60 min. After washing out the treatments, samples were incubated with phosphine-PEG3-Biotin (Thermo) for 4 h at 37 °C, to conjugate biotin to AHA containing proteins. Samples were then eluted on Zeba Spin desalting columns (7K MWCO, Thermo) to remove the excess of phosphine-PEG3-Biotin. Equal amount of proteins (~ 5 mg/ml) were incubated with SCOP antibody (1:100, Bethyl Laboratories) over night at 4 °C. Subsequently, 50 µl of protein A-sepharose beads (1:1 slurry, Sigma) were added to each sample and incubated for 1 h at 4 °C with gentle rocking. After three washes, samples were processed for SDS-PAGE and western blot. IRDye® 800CW Streptavidin (1:2000, LI-COR Biosciences) was used to detect biotin-conjugated (newly synthesized) SCOP. Anti-SCOP antibody (1:1000, Millipore) was used to detect total immunoprecipitated SCOP.
Statistical analyses
For LTP experiments where only two groups were compared, we used the two-tail t-test for determining statistical significance. When more than 2 groups were compared, we used one-way ANOVA followed by Bonferroni test to determine statistical significance. For the analyses of time-dependent change of SCOP, PTEN and pERK after various treatments, the ratios of SCOP to Akt (loading control), PTEN to Akt and phospho-ERK 1/2 to total ERK 1/2 were compared at different time points. All values were divided by the average value at time point 0 for normalization. Comparison to time point 0 was performed by one-way ANOVA followed by Bonferroni test.
Supplementary Material
Acknowledgements
This work was supported by grant P01NS045260-01 from NINDS (PI: Dr. C.M. Gall), and grant R01NS057128 from NINDS to MB. XB is also supported by funds from the Daljit and Elaine Sarkaria Chair.
Footnotes
Author Contributions
YW and GZ performed most of the experiments, and prepared the figures.
VB performed some of the experiments.
Y-TH performed some preliminary experiments regarding calpain-mediated SCOP truncation.
XB provided technical assistance, worked on the figures and wrote the manuscript.
MB directed the research and wrote the manuscript.
The authors declare that they have no competing financial interests
References
- 1.Morris RG, et al. Elements of a neurobiological theory of the hippocampus: the role of activity-dependent synaptic plasticity in memory. Philos Trans R Soc Lond B Biol Sci. 2003;358:773–786. doi: 10.1098/rstb.2002.1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jin SX, Feig LA. Long-term potentiation in the CA1 hippocampus induced by NR2A subunit-containing NMDA glutamate receptors is mediated by Ras-GRF2/Erk map kinase signaling. PLoS One. 2010;5:e11732. doi: 10.1371/journal.pone.0011732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kelleher RJ, 3rd, Govindarajan A, Jung HY, Kang H, Tonegawa S. Translational control by MAPK signaling in long-term synaptic plasticity and memory. Cell. 2004;116:467–479. doi: 10.1016/s0092-8674(04)00115-1. [DOI] [PubMed] [Google Scholar]
- 4.Winder DG, et al. ERK plays a regulatory role in induction of LTP by theta frequency stimulation and its modulation by beta-adrenergic receptors. Neuron. 1999;24:715–726. doi: 10.1016/s0896-6273(00)81124-1. [DOI] [PubMed] [Google Scholar]
- 5.Shimizu K, Okada M, Nagai K, Fukada Y. Suprachiasmatic nucleus circadian oscillatory protein, a novel binding partner of K-Ras in the membrane rafts, negatively regulates MAPK pathway. J Biol Chem. 2003;278:14920–14925. doi: 10.1074/jbc.M213214200. [DOI] [PubMed] [Google Scholar]
- 6.Shimizu K, Phan T, Mansuy IM, Storm DR. Proteolytic degradation of SCOP in the hippocampus contributes to activation of MAP kinase and memory. Cell. 2007;128:1219–1229. doi: 10.1016/j.cell.2006.12.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lynch G, Baudry M. The biochemistry of memory: a new and specific hypothesis. Science. 1984;224:1057–1063. doi: 10.1126/science.6144182. [DOI] [PubMed] [Google Scholar]
- 8.Lynch G, Baudry M. Brain spectrin, calpain and long-term changes in synaptic efficacy. Brain Res Bull. 1987;18:809–815. doi: 10.1016/0361-9230(87)90220-6. [DOI] [PubMed] [Google Scholar]
- 9.Staubli U, Larson J, Thibault O, Baudry M, Lynch G. Chronic administration of a thiol-proteinase inhibitor blocks long-term potentiation of synaptic responses. Brasin Res. 1988;444:153–158. doi: 10.1016/0006-8993(88)90922-5. [DOI] [PubMed] [Google Scholar]
- 10.Denny JB, Polan-Curtain J, Ghuman A, Wayner MJ, Armstrong DL. Calpain inhibitors block long-term potentiation. Brain Res. 1990;534:317–320. doi: 10.1016/0006-8993(90)90148-5. [DOI] [PubMed] [Google Scholar]
- 11.Oliver MW, Baudry M, Lynch G. The protease inhibitor leupeptin interferes with the development of LTP in hippocampal slices. Brain Res. 1989;505:233–238. doi: 10.1016/0006-8993(89)91448-0. [DOI] [PubMed] [Google Scholar]
- 12.Gartner A, Staiger V. Neurotrophin secretion from hippocampal neurons evoked by long-term-potentiation-inducing electrical stimulation patterns. Proc Natl Acad Sci U S A. 2002;99:6386–6391. doi: 10.1073/pnas.092129699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zadran S, Jourdi H, Rostamiani K, Qin Q, Bi X, Baudry M. Brain-derived neurotrophic factor and epidermal growth factor activate neuronal m-calpain via mitogen-activated protein kinase-dependent phosphorylation. J Neurosci. 2010;30:1086–1095. doi: 10.1523/JNEUROSCI.5120-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Briz V, Hsu YT, Li Y, Lee E, Bi X, Baudry M. Calpain-2-Mediated PTEN Degradation Contributes to BDNF-Induced Stimulation of Dendritic Protein Synthesis. J Neurosci. 2013;33:4317–4328. doi: 10.1523/JNEUROSCI.4907-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hanse E, Gustafsson B. TEA elicits two distinct potentiations of synaptic transmission in the CA1 region of the hippocampal slice. J Neurosci. 1994;14:5028–5034. doi: 10.1523/JNEUROSCI.14-08-05028.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang YY, Malenka RC. Examination of TEA-induced synaptic enhancement in area CA1 of the hippocampus: the role of voltage-dependent Ca2+ channels in the induction of LTP. J Neurosci. 1993;13:568–576. doi: 10.1523/JNEUROSCI.13-02-00568.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aniksztejn L, Ben-Ari Y. Novel form of long-term potentiation produced by a K+ channel blocker in the hippocampus. Nature. 1991;349:67–69. doi: 10.1038/349067a0. [DOI] [PubMed] [Google Scholar]
- 18.Harvey CD, et al. A genetically encoded fluorescent sensor of ERK activity. Proc Natl Acad Sci U S A. 2008;105:19264–19269. doi: 10.1073/pnas.0804598105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Angliker H, Anagli J, Shaw E. Inactivation of calpain by peptidyl fluoromethyl ketones. J Med Chem. 1992;35:216–220. doi: 10.1021/jm00080a003. [DOI] [PubMed] [Google Scholar]
- 20.Babayan AH, et al. Integrin dynamics produce a delayed stage of long-term potentiation and memory consolidation. J Neurosci. 2012;32:12854–12861. doi: 10.1523/JNEUROSCI.2024-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kramar EA, et al. Synaptic evidence for the efficacy of spaced learning. Proc Natl Acad Sci U S A. 2012;109:5121–5126. doi: 10.1073/pnas.1120700109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lynch G, Kramar EA, Babayan AH, Rumbaugh G, Gall CM. Differences between synaptic plasticity thresholds result in new timing rules for maximizing long-term potentiation. Neuropharmacology. 2013;64:27–36. doi: 10.1016/j.neuropharm.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.English JD, Sweatt JD. A requirement for the mitogen-activated protein kinase cascade in hippocampal long term potentiation. J Biol Chem. 1997;272:19103–19106. doi: 10.1074/jbc.272.31.19103. [DOI] [PubMed] [Google Scholar]
- 24.Li Z, et al. Novel peptidyl alpha-keto amide inhibitors of calpains and other cysteine proteases. J Med Chem. 1996;39:4089–4098. doi: 10.1021/jm950541c. [DOI] [PubMed] [Google Scholar]
- 25.Cajigas IJ, Tushev G, Will TJ, tom Dieck S, Fuerst N, Schuman EM. The local transcriptome in the synaptic neuropil revealed by deep sequencing and high-resolution imaging. Neuron. 2012;74:453–466. doi: 10.1016/j.neuron.2012.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cajigas IJ, Will T, Schuman EM. Protein homeostasis and synaptic plasticity. EMBO J. 2010;29:2746–2752. doi: 10.1038/emboj.2010.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Amini M, et al. Conditional Disruption of Calpain in the CNS Alters Dendrite Morphology, Impairs LTP, Promotes Neuronal Survival following Injury. J Neurosci. 2013;33:5773–5784. doi: 10.1523/JNEUROSCI.4247-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vanderklish PW, Krushel LA, Holst BH, Gally JA, Crossin KL, Edelman GM. Marking synaptic activity in dendritic spines with a calpain substrate exhibiting fluorescence resonance energy transfer. Proc Natl Acad Sci U S A. 2000;97:2253–2258. doi: 10.1073/pnas.040565597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bramham CR. Local protein synthesis, actin dynamics, and LTP consolidation. Curr Opin Neurobiol. 2008;18:524–531. doi: 10.1016/j.conb.2008.09.013. [DOI] [PubMed] [Google Scholar]
- 30.Villers A, Godaux E, Ris L. Long-lasting LTP requires neither repeated trains for its induction nor protein synthesis for its development. PLoS One. 2012;7:e40823. doi: 10.1371/journal.pone.0040823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cammalleri M, et al. Time-restricted role for dendritic activation of the mTOR-p70S6K pathway in the induction of late-phase long-term potentiation in the CA1. Proc Natl Acad Sci U S A. 2003;100:14368–14373. doi: 10.1073/pnas.2336098100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Benjamin AS, Tullis J. What makes distributed practice effective? Cogn Psychol. 2010;61:228–247. doi: 10.1016/j.cogpsych.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frey S, Frey JU. 'Synaptic tagging' and 'cross-tagging' and related associative reinforcement processes of functional plasticity as the cellular basis for memory formation. Prog Brain Res. 2008;169:117–143. doi: 10.1016/S0079-6123(07)00007-6. [DOI] [PubMed] [Google Scholar]
- 34.Kennedy MB, Beale HC, Carlisle HJ, Washburn LR. Integration of biochemical signalling in spines. Nat Rev Neurosci. 2005;6:423–434. doi: 10.1038/nrn1685. [DOI] [PubMed] [Google Scholar]
- 35.Sasaki T, Kikuchi T, Yumoto N, Yoshimura N, Murachi T. Comparative specificity and kinetic studies on porcine calpain I and calpain II with naturally occurring peptides and synthetic fluorogenic substrates. J Biol Chem. 1984;259:12489–12494. [PubMed] [Google Scholar]
- 36.Chen LY, Rex CS, Casale MS, Gall CM, Lynch G. Changes in synaptic morphology accompany actin signaling during LTP. J Neurosci. 2007;27:5363–5372. doi: 10.1523/JNEUROSCI.0164-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dominguez R, Liu R, Baudry M. 17-Beta-estradiol-mediated activation of extracellular-signal regulated kinase, phosphatidylinositol 3-kinase/protein kinase B-Akt and N-methyl-D-aspartate receptor phosphorylation in cortical synaptoneurosomes. J Neurochem. 2007;101:232–240. doi: 10.1111/j.1471-4159.2006.04360.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dudek SM, Fields RD. Somatic action potentials are sufficient for late-phase LTP-related cell signaling. Proc Natl Acad Sci U S A. 2002;99:3962–3967. doi: 10.1073/pnas.062510599. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.