

Abstract
Neurotropic alphaviruses, which include western equine encephalitis virus (WEEV) and Fort Morgan virus, are mosquito-borne pathogens that infect the central nervous system causing acute and potentially fatal encephalitis. We previously reported a novel series of indole-2-carboxamides as alphavirus replication inhibitors, one of which conferred protection against neuroadapted Sindbis virus infection in mice. We describe here further development of this series resulting in 10-fold improvement in potency in a WEEV replicon assay and up to 40-fold increases in half-lives in mouse liver microsomes. Using a rhodamine123 uptake assay in MDR1-MDCKII cells we were able to identify structural modifications that markedly reduce recognition by P-glycoprotein, the key efflux transporter at the blood brain barrier. In a preliminary mouse PK study we were able to demonstrate that two new analogs could achieve higher and/or longer plasma drug exposures than our previous lead, and that one compound achieved measurable drug levels in the brain.
Keywords: antiviral, alphavirus, indole, central nervous system, encephalitis, RNA replication inhibitor, metabolic stability, P-glycoprotein
Introduction
Alphaviruses are mosquito-borne pathogens that cause disease outbreaks in humans and animals worldwide.1 The neurotropic alphaviruses, which include western equine encephalitis virus (WEEV), infect the central nervous system (CNS) causing acute and potentially fatal encephalitis. In addition to natural insectborne disease transmission,2 these pathogens could be aerosolized and released into a population center as potential bioterrorism agents3, 4. As a result, the neurotropic alphaviruses are considered Category B Priority Pathogens by the National Institute of Allergy and Infectious Diseases (NIAID).5 There are no FDA-approved vaccines or antiviral drugs active against neurotropic alphaviruses, and thus there remains a pressing need for novel therapies to combat either naturally occurring or intentional outbreaks from these highly virulent pathogens.
Alphaviruses such as WEEV contain a single-stranded, positive polarity RNA genome that serves as a direct template for translation and replication.1 The genome encodes both non-structural proteins having RNA polymerase, protease, helicase, and methyltransferase activities, and structural capsid and envelope proteins, which are translated from a subgenomic RNA that is produced during viral RNA replication. However, alphavirus structural proteins are dispensable for RNA replication, and can be replaced with easily measured reporter genes to generate non-infectious replicons that facilitate drug discovery and development under reduced biosafety conditions. We previously generated WEEV replicons containing a firefly luciferase (fLUC) reporter gene, developed a cell-based assay amenable to high-throughput screening (HTS), and identified a novel series of thienopyrrole derivatives (represented by 1 in Figure 1) active against WEEV and related alphaviruses.6
Figure. 1. Original HTS hit 1 and initial key replicon SAR.
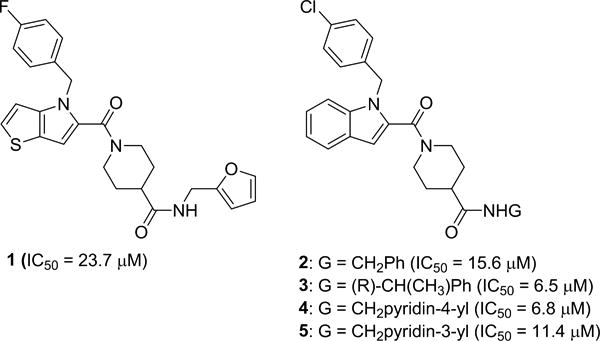
We subsequently undertook the synthesis of indole analogs of 1, designed to improve potency and metabolic stability.7 Initial structure-activity relationship (SAR) development of the terminal amide led to the discovery of enantiomer 3 (CCG-203926, Fig. 1). Although 3 achieved only a modest improvement in potency compared to 2, it proved to be significantly more stable to oxidative metabolism by liver micorosmes. Compound 3 was subsequently advanced to preclinical efficacy studies in mice, where it conferred protection against infection caused by a related alphavirus, neuroadapted Sindbis virus.7 During the course of our investigation, we noted that 4-pyridylmethyl amide 4 possessed increased potency relative to simple benzyl amide 2 (Fig. 1) and that simply moving the nitrogen to the 3-position (5) resulted in a 2-fold loss in activity. These results and the enantiospecific activity of 3 strongly suggested that the amide moiety was making close contact with the unknown molecular target.
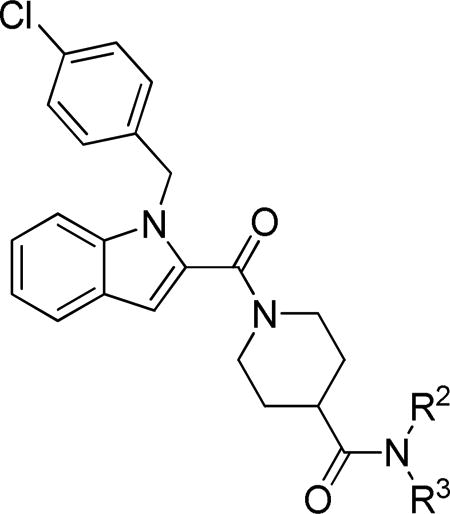
In the present work we therefore expanded our investigation of the benzylamide moiety in an effort to further enhance antiviral potency and also initiated an investigation of the N-benzyl position. To improve the potential for achieving in vivo activity, we also explored modifications of the indole template that reduced molecular weight and/or lipophilicity to improve aqueous solubility. Finally, we introduced an assay to estimate affinity of new compounds for P-glycoprotein (Pgp), the major efflux transporter at the blood brain barrier (BBB),8 in order to identify compounds with the greatest potential for CNS penetration in future in vivo studies.
Results
Chemistry
Indole carboxamide analogs 27 were generated as shown in Scheme 1 via standard peptide coupling conditions using our previously synthesized carboxylic acid intermediate 7 and commercially available amines, except for 3-(pyridin-4-yl)propan-1-amine used to prepare 27j, which was synthesized as previously reported.9, 10 The various indole carboxamide analogs synthesized are specified in Table 1.
Scheme 1. Preparation of Terminal Carboxamide Analogs 27a.

aReagents and conditions: (a) R1R2NH, EDCI, HOBt, DCM, DIEA, RT, overnight
Table 1. WEEV Replicon and In Vitro ADME Data for Carboxamide Analogs.
| |||||||
|---|---|---|---|---|---|---|---|
| No. | NR2R3 | IC50 (μM)a | CC50 (μM)b | BBB-PAMPA (log Peff)c | MLM T1/2 (min)d | MDR1 Recognition (increase in Rho123 uptake)e | Sol (μM)f |
| 3 | (R)-PhCH(CH3)NH | 6.5 ± 1.5 | >100 | -6.52 ± 3.14 | 2.1 | 24.4 ± 3.7 | 8-16 |
| 4 | 4-PyrCH2NH | 6.8 ± 1.7 | >100 | -4.16 ± 0.12 | 25.9 ± 2.6 | 31-63 | |
| 27a | 4-PyrCH(CH3)NH | 2.4 ± 0.8 | 90.1 | -3.99 ± 0.01 | 43.1 ± 5.9 | 31-63 | |
| 27b | (R)-4-PyrCH(CH3)NH | 0.6 ± 0.3 | 92.3 | -4.02 ± 0.03 | 23.4 | 35.9 ± 1.8 | |
| 27c | 4-Me-piperazine | 31.4 ± 16.7 | 75.6 | ||||
| 27d | 1-Me-piperidin-4-ylCH2NH | >50 | >50 | ||||
| 27e | 1-Me-pyrazol-3-ylCH2NH | 76.9 ± 30.8 | >100 | ||||
| 27f | 1-Me-imidazol-4-ylCH2NH | >50 | >50 | ||||
| 27g | 4-PyrCH2CH2NH | 0.5 ± 0.2 | 49.9 | -4.18 ± 0.06 | 11.5 | 30.9 ± 1.0 | 31-63 |
| 27h | 3-PyrCH2CH2NH | 5.3 ± 1.1 | 39.4 | 30.1 ± 2.3 | |||
| 27i | 2-PyrCH2CH2NH | 22.4 ± 10.0 | 86.2 | 21.9 ± 2.1 | |||
| 27j | 4-PyrCH2CH2CH2NH | 31.9 ± 37.9 | 71.5 | -4.11 ± 0.03 | 35.2 ± 4.3 | 16-32 | |
| 27k | 4-PyrCH(CH3)CH2NH | 1.7 ± 1.2 | 98.2 | 25.4 ± 0.9 | |||
| 27l | 4-PyrCH2C(CH3)2NH | 19.3 ± 21.0 | 43.6 | -4.19 ± 0.13 | |||
| 27m | 4-PyrCH2CH2NMe | 1.6 ± 0.8 | 47.9 | -4.21 ± 0.13 | 72.7 ± 5.9 | ||
| 27n |
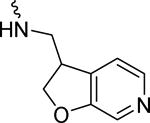
|
3.6 ± 1.7 | 75.8 | 47.0 ± 4.8 | |||
| 27o | Imidazol-1 -ylCH2CH2NH | 4.9 ± 2.6 | 99.6 | -5.76 ± 2.83 | 14.3 | 15.1 ± 0.7 | 31-63 |
| 27p | 3-MeO-PhCH2CH2NH | 11.1 ± 3.8 | >100 | ||||
| 27q | 4-F-PhCH2CH2NH | 21.4 ± 18.8 | 18.6 | ||||
| 27r | 4-MeO-PhCH2CH2NH | 15.5 ± 14.6 | 11.3 | ||||
| 27s | 4-Cl-PhCH2CH2NH | 8.1 ± 1.1 | 40.4 | ||||
| 27t | 4-iPr-PhCH2CH2NH | 10.8 ± 3.3 | 27.2 | ||||
| 27u | (S)-prolinol | 30.6 ± 4.5 | 98.9 | ||||
| 27v | Me2N | 10.5 ± 2.1 | >100 | -4.19 ± 0.03 | 23.2 ± 1.9 | 62-125 | |
| 27w | Pyrrolidine | 22.2 ± 4.3 | 65.6 | ||||
| 27x | morpholine | 12.8 ± 4.6 | >100 | ||||
Inhibition of luciferase expression in WEEV replicon assay. Ribavirin as positive control has an IC50 in the assay of 16 μM. Values are mean of at least n=3 independent experiments ± SE.
Cell viability determined by inhibition of cellular reduction of MTT. Values are mean of at least n=3 independent experiments.
Log of effective permeability (cm/s) determined using PAMPA Explorer (pION) with BBB lipid mixture measured at pH = 7.4.
Half-life in mouse liver microsome incubations. Values are mean of ≥ 2 independent incubations.
Rhodamine 123 uptake was measured in MDR1-MDCKII cells utilizing Glomax Multi Detection System (Promega). ‘MDR1 recognition’ was assessed by measuring uptake in the presence of MDR inhibitor, tariquidar (5 μM), and either 30 μM of antiviral or vehicle, and calculating: (Cav − Cveh)*100/(Ctar-Cveh), where Cav = concentration of rhodamine 123 in the presence of anti-viral, Cveh = concentration in the presence of vehicle, Ctar = concentration of rhodamine 123 in the presence of tariquidar. In the presence of tariquidar, rhodamine 123 uptake was 1123 ± 54% of vehicle controls (n=44).
Kinetic solubility measured using the same assay media as WEEV replicon assay, except with 10% fetal bovine serum. See Exp Section for methods and detailed synthetic procedures.
Analogs 28 varying the indole N-substituent were prepared as in Scheme 2. All contain a terminal N-methylbenzyl carboxamide (Table 2). We found that attempts to alkylate intermediate 10 lacking the amide N-methyl group under basic conditions resulted in decomposition, double alkylation, or no reaction. Our initial SAR established that methylation of the amide did not significantly compromise activity (compare 28a in Table 2 with 2 in Fig. 1), so we maintained the N-Me benzylamide through the course of this small SAR series. Hydrolysis of ethyl indole-2-carboxylate 8 followed by an EDCI-mediated coupling with ethyl isonipecotate generated intermediate 9 (Scheme 2). Subsequent hydrolysis followed by amide formation with N-methylbenzylamine afforded the desired key advanced common intermediate 10. Final compounds were then obtained through base-mediated N-alkylations of the indole.
Scheme 2. Preparation of N-alkyl Indole Analogsa.
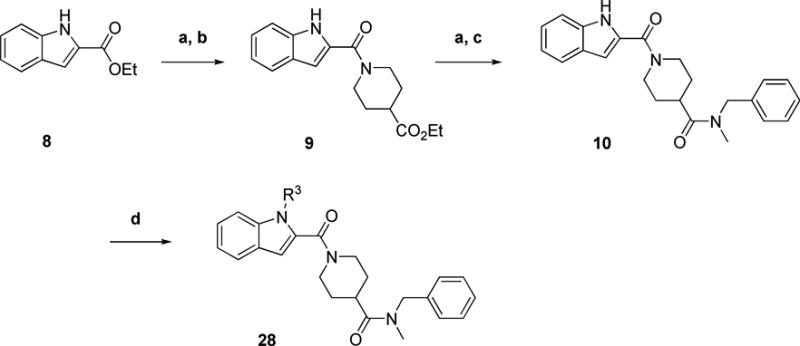
aReagents and conditions: (a) LiOH, THF, H2O (b) ethyl piperidine-4-carboxylate, EDCI, HOBt, DIEA, THF, RT, overnight (c) N-methylbenzylamine, EDCI, HOBt, DCM, DIEA, DCM, RT, overnight (d) X-R3, base, DMF, overnight, RT, X = I, Br, Cl, OMs.
Table 2. WEEV Replicon and PAMPA Data for N-Alkyl Indole Analogsa.
| ||||
|---|---|---|---|---|
| R1 | IC50 (uM) | CC50 (uM) | BBB-PAMPA (log Peff) | |
| 28a | 4-Cl-PhCH2 | 16.9 ± 4.4 | 87.0 | |
| 28b | 4-NO2-PhCH2 | 61.3 ± 35.5 | >100 | |
| 28c | 4-CN-PhCH2 | 95.9 ± 7.0 | >100 | -4.27 ± 0.03 |
| 28d | 4-MeO-PhCH2 | 16.5 ± 0.8 | 99.2 | |
| 28e | Ac | >50 | >50 | -5.01 ± 0.09 |
| 28f | MeOCH2CH2 | >50 | >50 | -4.39 ± 0.05 |
| 28g | i-Bu | 27.9 ± 3.6 | 58.2 | |
| 28h | PhCH2 | 17.0 ± 1.1 | 64.8 | |
| 28i | 4-Pyr-CH2 | 24.2 ± 3.6 | 80.3 | |
| 28j | 4-CF3-PhCH2 | 35.1 ± 43.5 | 90.3 | |
See Table 1 for assay definitions. See Exp Section for methods and detailed synthetic procedures.
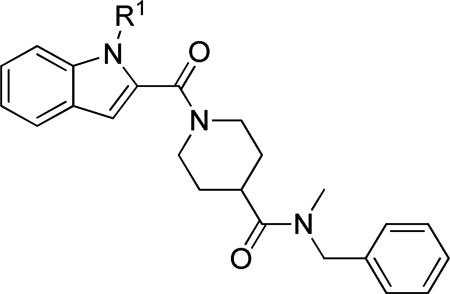
Pyrrole analog 29a was prepared as shown in Scheme 3. Methyl 1H-pyrrole-2-carboxylate 11 was alkylated with 4-chlorobenzyl chloride in the presence of potassium carbonate. Saponification resulted in acid 12. Amide bond formation with ethyl isonipecotate and subsequent hydrolysis generated carboxylic acid 13. A final peptide coupling resulted in 29a.
Scheme 3. Preparation of Pyrrole Analog 29aa.
aReagents and conditions: a) 1-chloro-4-(chloromethyl)benzene, cat. NaI, K2CO3, DMF, 90 °C; (b) 10% NaOH (aq), EtOH, RT to 50 °C, overnight; c) ethyl isonipecotate, EDCI, HOBT, DIPEA, DCM, RT, 24 h; d) LiOH, H2O, EtOH, RT, 24 h; e) 2-(4-pyridyl)ethylamine, EDCI, HOBT, TEA, DCM.
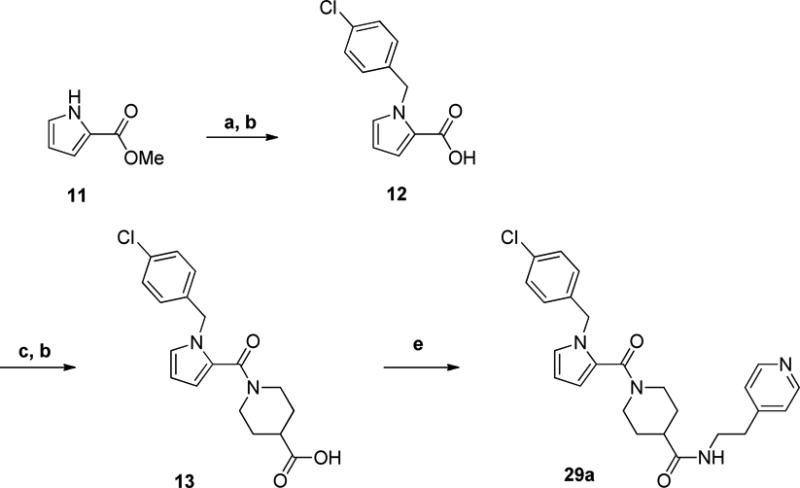
A convenient synthon for the synthesis of several indole replacement analogs is amine 16 (Scheme 4). Its synthesis began with the Boc protection of ethyl isonipecotate 14 using di-tert-butyl dicarbonate. The Bocprotected ester was hydrolyzed to acid 15 and subsequently coupled with 2-(4-pyridyl)ethylamine to afford piperidine amide 16.
Scheme 4. Preparation of Isonipecotamide Intermediate 16a.
aReagents and conditions: a) Boc2O, TEA, DCM, overnight b) 10% NaOH (aq), EtOH, 4 h c) 2-(4-pyridyl)ethylamine, EDCI, HOBt, DIEA, DCM d) 4N HCl in 1,4-Dioxane, Et2O
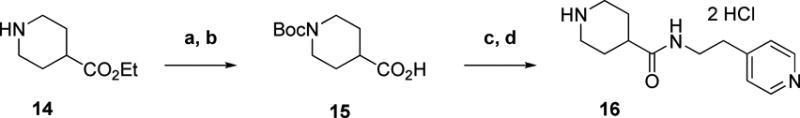
The synthesis of imidazole analog 29b (Scheme 5) commenced with the alkylation of ethyl 1H-imidazole-2-carboxylate (17b) with 1-chloro-4-(chloromethyl)benzene. Ester hydrolysis then gave 18b. The final step proceeded through a peptide coupling with amine 16. The synthesis of fluoropyrrole 29k proceeded through similar steps; however, it utilized methyl 4-fluoro-1H-pyrrole-2-carboxylate 17k which was synthesized according to a previously described procedure.11, 12
Scheme 5. Preparation of Monocyclic Template Analogsa.
aReagents and conditions: a) 1-chloro-4-(chloromethyl)benzene, Na2CO3, DMF, RT, 24 h b) 10% NaOH (aq), EtOH, RT, 15 h c) 16, EDCI, HOBt, DIEA, DCM, RT, 24 h
Urea analog 29d was prepared as shown in Scheme 6. Addition of ethyl isonipecotate to 1-chloro-4-(2-isocyanatoethyl)benzene resulted in urea 20. The ester was saponified followed by peptide bond formation with 2-(4-pyridyl)ethylamine to generate 29d.
Scheme 6. Preparation of Urea Analog 29da.
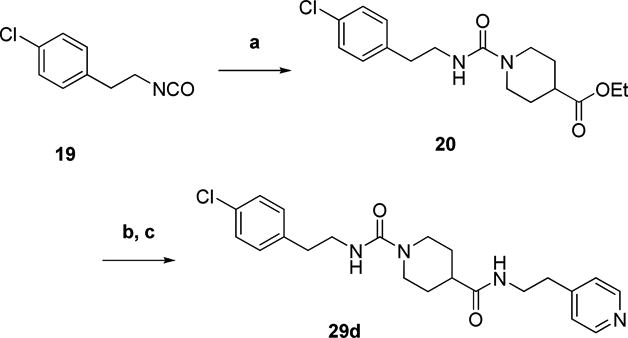
aReagents and conditions: a) ethyl isonipecotate, DCM, RT, 1 h b) 10% NaOH (aq), EtOH, 50 °C, 2 h c) 2-(4-pyridyl)ethylamine, EDCI, HOBt, TEA, DCM, RT, overnight
The synthesis of indole-modified analogs 29c and 29h proceeded through similar synthetic steps (Scheme 7). It began with a coupling reaction between benzimidazole or 6-fluoroindole carboxylates (21c or 21h, respectively) and ethyl isonipecotate. Alkylation with 1-chloro-4-(chloromethyl)benzene resulted in 22c or 22h. Subsequent hydrolysis and peptide coupling with 2-(4-pyridyl)ethylamine resulted in the final analogs.
Scheme 7. Preparation of Indole-modified Analogsa.
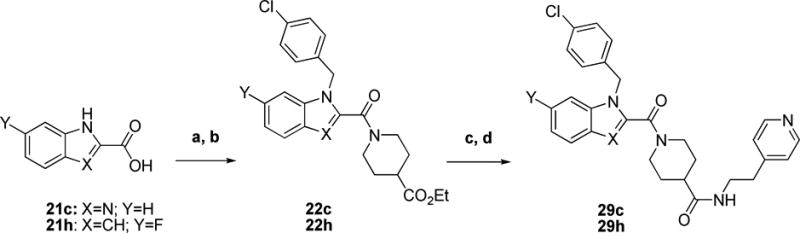
aReagents and conditions: a) ethyl isonipecotate, EDCI, HOBt, DIEA, DCM b) 1-chloro-4-(chloromethyl)benzene, Cs2CO3, DMF, 80 °C c) LiOH, THF/H2O d) EDCI, HOBt or DMAP, DIEA, 2-(4-pyridyl)ethylamine, DCM
Scheme 8 summarizes the preparation of three more indole-modified analogs. Compounds 29f and 29g utilized ethyl indole-2-carboxylate 8 for their indole starting material while 29j utilized azaindole ester 23j synthesized according to procedures described in the literature.13, 14 Each were N-alkylated under basic conditions and saponified to give intermediate acids 24f, 24g, and 24j. Final coupling reactions with piperidine 16 provided the final compounds.
Scheme 8. Preparation of Indole-modified Analogsa.
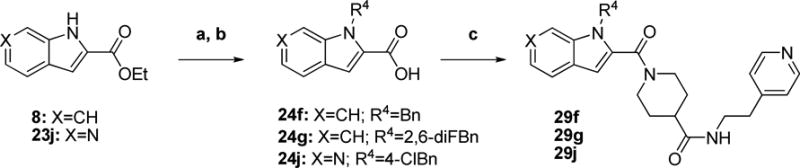
aReagents and conditions: a) Base, R4 -X (see Exp Section), DMF b) 10% NaOH (aq), THF or EtOH, RT, 24 h c) 16, EDCI, HOBt or DMAP, DIEA or TEA, THF or DCM, RT, overnight
Finally, the preparation of indoles 29e and 29i started with 8 and 25i, respectively (Scheme 9). Each was N-alkylated, saponified and coupled with ethyl isonipecotate to give amides 26e and 26i. After ester hydrolysis, the ethyl pyridine motif was appended using an amide bond coupling to generate the desired compounds.
Scheme 9. Preparation of Indole-modified Analogsa.
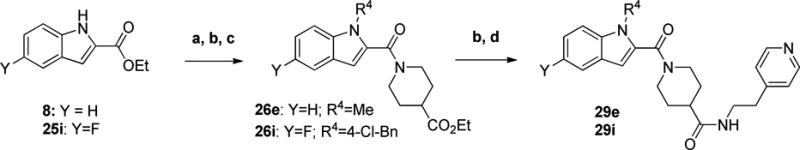
aReagents and conditions: a) R4 -X, K2CO3 or Cs2CO3, DMF, 80 °C b) LiOH, THF/H2O c) ethyl isonipecotate, EDCI, HOBt or DMAP, DIEA, DCM d) EDC, HOBt or DMAP, DIEA, 2-(4-pyridyl)ethylamine, DCM
WEEV replicon SAR
All new analogs were tested in the WEEV replicon assay as previously described.7 Specific modifications to the terminal amide are summarized in Table 1. Combining the two favorable modifications from our previous SAR noted in Figure 1 (4-pyridyl amide and alpha-methyl group) improved activity by about 3-fold in the racemate 27a. As anticipated, the (R)-enantiomer 27b proved to be more active than the racemate, consistent with what we observed previously with 3.7 Replacement of the pyridine ring of 4 with basic amines (27c and 27d) or isosteric heterocycles (27e and 27f) both greatly reduced potency.
The optimal distance between the pyridine and the carboxamide was established by generating homologs 27g and 27j. Relative to analog 4, the 10-fold increase in potency with addition of one methylene (27g) and 5-fold decrease with addition of a second methylene (27j) clearly indicates that the ethylene linker of 27g is optimal. Very interestingly, there is a nearly 40-fold decrease in potency as the nitrogen in the pyridine ring is migrated from para to ortho (27g-i), demonstrating the key importance of the nitrogen being in the 4-position of the pyridylethyl amide. N-methylation of the amide of 27g (27m) diminished potency by 3-fold and introduced some cytotoxicity, contrary to what we had earlier observed with 28a. Additional conformationally biased analogs (27k, 27l, and 27n) decreased potency compared to 27g. Replacement of the pyridine of 27g with an imidazole, in an attempt to introduce greater hydrogen-bonding potential (27o), was not productive. Various substituted phenethyl amides were also explored, ranging from hydrogen bonding (27p, 27r) to lipophilic (27q, 27s, 27t), but none matched the potency of pyridine 27g. Finally, amides 27u − 27x were prepared to improve solubility or reduce molecular weight, but all caused unacceptable potency reductions in the WEEV replicon assay.
In addition to the variations in the amide group, substitution at the N1 position of the indole was explored (Table 2). Replacing the 4-chloro group of the benzyl motif in 28a with other aromatic substituents or hydrogen did not improve activity (28b-d, 28h, 28j). Overall, the activity seemed to be more dependent on size than electronegativity, with H and OMe having the best activity among the new analogs. Aliphatic substitution (28f, 28g) or acetylation (28e) resulted in less active or inactive analogs. Replacement of the phenyl with 4-pyridine slightly diminished potency (28i).
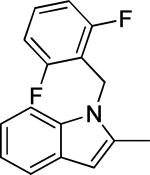
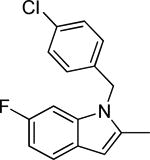
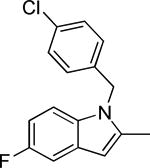
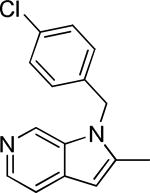
Based on the results outlined in Tables 1 and 2, the optimal 4-pyridylethyl amide and N-4-chlorobenzyl moieties were retained for an investigation of the indole template SAR (Table 3). Replacement with a pyrrole (29a) to reduce molecular weight maintained potency and actually diminished cytotoxicity compared to 27g, indicating a pyrrole may be a viable substitute for the indole. Decreasing lipophilicity with an imidazole (29b), a benzoimidazole (29c), or an azaindole (29j) scaffold decreased potency. Removal of the aromatic ring altogether (29d) resulted in nearly complete loss of activity, demonstrating the importance of an aromatic ring or a rigid scaffold for antiviral activity. Compounds 29h and 29i were synthesized to attenuate the potential for CYP450-mediated metabolism of the indole scaffold by decreasing the electron density of the indole. These analogs possessed activity and cytotoxicity similar to 27g. However, a similar attempt to increase metabolic stability of pyrrole 29a with a fluoro analog (29k) resulted in a significant increase in toxicity. Finally, a few modifications of the N1-indole position of 27g were investigated to improve solubility and/or metabolic stability. Replacing the benzyl motif with a methyl group (29e) eliminated activity, but removing the 4-chloro group was tolerated with only a small reduction in activity (29f). Insertion of ortho fluoro groups (29g) also did not overly diminish activity, but did increase cytotoxicity as evidenced by a decline in the CC50/IC50 ratio below our target of 50.
Table 3. WEEV Replicon and In Vitro ADME Data for Template Analogsa.
| |||||||
|---|---|---|---|---|---|---|---|
| G | IC50 (uM) | CC50 (uM) | BBB-PAMPA (log Peff) | MLM T1/2 (min) | MDR1 Recognition (increase in Rho123 uptake) | Sol (μM) | |
| 27g |
|
0.5 ± 0.2 | 49.9 | -4.18 ± 0.06 | 11.5 | 30.9 ± 1.0 | 31-63 |
| 29a |
|
0.7 ± 0.1 | >100 | -5.01 ± 0.03 | 2.9 | -1.2 ± 0.6 | 125-250 |
| 29b |
|
>50 | >50 | -5.06 ± 0.04 | 0.13 ± 0.37 | >500 | |
| 29c |
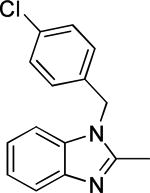
|
4.1 ± 1.0 | 74.6 | -4.50 ± 0.03 | 43.0 | 6.9 ± 0.4 | 63-125 |
| 29d |
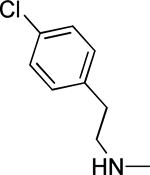
|
40.9 ± 7.8 | >100 | -4.85 ± 0.06 | 250-500 | ||
| 29e |
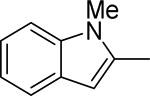
|
>50 | >50 | ||||
| 29f |
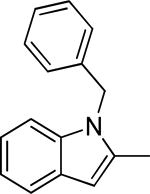
|
1.6 ± 0.5 | 45.6 | 77.9 | 11.7 ± 1.5 | 31-63 | |
| 29g |
|
1.3 ± 0.8 | 34.5 | 28.5 ± 0.9 | |||
| 29h |
|
0.6 ± 0.7 | 43.1 | 30.8 ± 2.7 | |||
| 29i |
|
0.9 ± 0.8 | 52.5 | -4.11 ± 0.03 | 42.6 ± 6.5 | 31-63 | |
| 29j |
|
4.4 ± 0.8 | 77.1 | -5.52 ± 0.03 | 9.1 | 1.2 ± 1.8 | 125-250 |
| 29k |
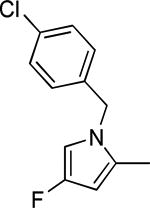
|
0.9 ± 0.3 | 8.0 | -4.90 ± 0.06 | 125-250 | ||
See Table 1 for assay definitions. See Exp Section for methods and detailed synthetic procedures.
Solubility
Aqueous solubility was an important physicochemical property targeted during our synthetic efforts. Poor solubility can result in low bioavailability after oral or intraperitoneal (IP) dosing.15 The kinetic solubility of selected compounds in our assay media was measured using a simple precipitation assay.15, 16 As expected, pyridyl amides (e.g. 4 and 27g) were about 4-fold more soluble than previous lead compound 3. Extending the pyridyl alkyl amide chain (27j) reduced solubility by half. Decreasing the molecular weight resulted in an increase in solubility (e.g. 27v) which was particularly dramatic when transitioning from a bicyclic aromatic core to a monocyclic core. This is evident when comparing 27g vs. 29a, 29b vs. 29c, and 29i vs. 29k. Finally, increasing polarity (e.g. benzimidazole 29c and azaindole 29j vs indole 27g) was effective at improving solubility.
BBB Permeability
Because our candidate inhibitors need to exert antiviral activity within the brain, compounds with good blood-brain barrier (BBB) permeability are a priority. An important physical property for BBB permeability is high passive permeability. Selected compounds were tested using the PAMPA Explorer™ program from pION with the PAMPA-BBB lipid mixture.17, 18 Due to the low solubility of our compounds, we utilized a co-solvent system containing 20% acetonitrile in donor wells.19 Data are shown in Tables 1-3. In general, compounds with logPeff > -4.7 are categorized as having high permeability while those exhibiting logPeff ≤ -6 are considered to have low permeability.17 Most of our compounds fell between these standards, indicating that our compounds would be predicted to exhibit moderate passive permeability through the BBB. Somewhat surprisingly, our data indicate no direct correlation between PAMPA-BBB permeability and either molecular weight or lipophilicity (r = -0.03 and r = 0.03, respectively) within this class of compounds.
The BBB expresses high levels of the efflux transporter, P-glycoprotein (Pgp/MDR1).8 Pgp facilitates the efflux of xenobiotics from the CNS. For our inhibitors to have the greatest potential for antiviral activity within the CNS, interactions with Pgp should be minimized. The degree to which Pgp interacts with our compounds was measured using a rhodamine 123 uptake assay in MDCK cells transfected with human Pgp (MDR1-MDCKII). Rhodamine 123 is a known Pgp substrate that is actively effluxed from MDR1-MDCKII cells20. In this assay, cellular uptake of rhodamine 123 is measured in the absence or presence of a test compound. If the test compound interacts with Pgp, it will impede efflux of rhodamine 123, thereby increasing its intracellular concentration. This fluorescent assay is similar to the [3H]vinblastine assay we have used previously in these cells.21 Tariquidar, a known inhibitor of Pgp, is included as a control. Results are included in Tables 1 and 3 as “MDR1 Recognition” where the increase in rhodamine 123 uptake in the presence of a test compound is reported as a percent of the increased rhodamine 123 uptake in the presence of control inhibitor tariquidar in the same assay. Higher values indicate greater recognition by Pgp and thus higher likelihood for efflux at the BBB. Our data indicates a small direct correlation between lipophilicity and Pgp recognition (r = 0.5). A similar trend can be seen for the relationship between molecular weight and Pgp interaction (r = 0.6). Interestingly, topological polar surface area (TPSA) actually has a small inverse relationship to Pgp recognition in our series (r = -0.3), contrary to the prevailing concept that TPSA contributes to recognition by Pgp.22 Some direct SAR comparisons are worth noting. N-methylation of the amide of 27g (27m) actually increased Pgp recognition suggesting that conformational effects are more important than lowering the TPSA, which normally reduces recognition. Similarly, conformational restriction through cyclization (27n vs 27g) increased Pgp recognition. The most effective modifications to attenuate Pgp recognition were replacing the indole template with a monocyclic template (e.g. 29a and 29b) and decreasing the lipophilicity of the indole (29c and 29j).
Antiviral Activity
Selected compounds were advanced for testing of their capacity to inhibit the replication of infectious virus directly in cells. We used two parallel assays to measure activity against infectious virus: reduction in cytopathic effect (CPE) and extracellular virus titers. For initial experiments we used infectious WEEV, which requires Biosafety Level-3 containment, and focused studies on initial analogs 27g, 29a, and 27a. For subsequent experiments we used Fort Morgan virus (FMV), a WEEV-serogroup alphavirus that can be safely handled under Biosafety Level-2 conditions.23 There was excellent correlation between results with WEEV and FMV using both CPE reduction (R=0.96, p < 0.005) and virus titer (R=0.92, p <0.01) assays. Of the eight novel compounds examined, all but 29j had activity in viral titer assays equivalent to or superior than our previous lead 3, and all analogs had superior activity in CPE reduction assays (Table 4). Analogs 27g, 27a and 29h were particularly effective, reducing viral titers by approximately ten-fold more than 3.
Table 4. Antiviral Data for Selected Analogsa.
| WEEV | FMV | |||
|---|---|---|---|---|
| Compound (25 μM) | Titer (x106 pfu/mL) | Viability (% uninfected control) | Titer (x106 pfu/mL) | Viability (% uninfected control) |
| DMSO | 25.7 ± 5.2 | 22.7 ± 3.1 | 47.1 ± 10.5 | 39.2 ± 1.9 |
| Ribavirin | 9.3 ± 3.4 | 35.3 ± 1.0 | 53.5 ± 6.6 | 32.6 ± 1.7 |
| 3 | 8.8 ± 1.0* | 36.7 ± 1.4** | 39.3 ± 13.4 | 47.4 ± 8.0 |
| 27g | 0.5 ± 0.2** | 60.7 ± 4.7** | 4.9 ± 1.9* | 71.9 ± 1.9** |
| 29a | 4.9 ± 2.3* | 59.1 ± 6.1** | 23.8 ± 2.9 | 63.0 ± 5.9* |
| 27a | 0.7 ± 0.4** | 63.2 ± 2.6** | 3.7 ± 1.1** | 68.2 ± 2.4** |
| 27b | ND | ND | 6.4 ± 3.2* | 65.5 ± 11.9* |
| 27k | ND | ND | 13.3 ± 4.2* | 74.0 ± 7.7** |
| 27o | ND | ND | 15.8 ± 9.8 | 70.3 ± 9.2* |
| 29c | ND | ND | 16.4 ± 4.6* | 69.7 ± 13.7 |
| 29f | ND | ND | 6.8 ± 1.5** | 79.4 ± 6.6** |
| 29h | ND | ND | 1.6 ± 0.3** | 69.2 ± 12.1* |
| 29j | ND | ND | 69.4 ± 43.7 | 66.1 ± 4.9** |
Assays utilized the alphaviruses, western equine encephalitis virus (WEEV) and Fort Morgan virus (FMV). Infections were done in cultured human BE(2)-C neuronal cells. Viability was measured using MTT assay and viral titers were measured using plaque reduction assays at 25 μM. Values are mean ± SEM of n=4 independent experiments.
p-value <0.05* or 0.005** compared to DMSO control.
Results for 3 antiviral activity against WEEV have been previously published,7 and are reproduced here for convenience to compare results with FMV and between 3 and newly generated analogs.
ND = not determined.
Pharmacokinetics
Selected compounds were assessed for stability to oxidative metabolism by mouse liver microsomes (MLMs), and results are included in Tables 1 and 3. Our previous lead compound 3 was included for comparison and to control for a change in the lot of microsomes from that used in our previous studies. In Table 1, it can be seen that transitioning to a heterocyclic carboxamide aryl group (27g and 27o) significantly improved metabolic stability, perhaps due to reduced lipophilicity. Branching of the alkyl chain (27b) improved stability further, consistent with what we observed in our earlier work7 and suggesting that oxidation of the alkyl chain is one metabolic pathway. In Table 3 it can be seen that replacement of the indole ring of 27g with pyrrole (29a) eroded metabolic stability despite lowering lipophilicity, suggesting that the central aromatic ring is a major site of metabolism. This is consistent with the improved stability realized by replacement of the indole with the more electron-deficient benzimidazole ring of 29c. Remarkably, simply removing the chlorine of 27g dramatically improved metabolic stability (29f), probably due to reduced overall lipophilicity and strongly suggesting that oxidation of the N-benzyl aromatic ring is not a major metabolic pathway.
Two new compounds (27g and 29a) were selected for preliminary in vivo PK studies in comparison to compound 3 that was previously shown to have some in vivo efficacy.7 Compound 27g was one of our most potent analogs and displayed significantly improved in vitro metabolic stability compared to compound 3. Compound 29a also possessed excellent potency, combined with a very low potential for Pgp recognition as determined by the rhodamine 123 uptake assay. C57BL/6 mice were injected IP with compounds 3, 27g, or 29a. Brain and plasma samples were collected from duplicate mice at multiple time points and drug levels quantified via LC/MS/MS (Table 5). Compound 29a achieved the highest plasma concentration and demonstrated measurable brain exposure at thirty minutes, but was cleared rapidly from both compartments. Analog 27g had the highest plasma concentration at two hours and was the only compound with measurable plasma levels at twelve hours; however, no drug was consistently detected in brain. Nonetheless, it was reassuring to learn that the analog with the greatest in vivo stability (27g) was also the one with the best in vitro stability, and the only analog that achieved measurable levels in the brain (29a) was the one with the lowest apparent Pgp recognition in the rhodamine 123 uptake assay. These findings provide significant validation of our in vitro approach to selecting compounds for progression to in vivo studies. Although neither 27g nor 29a have ideal PK properties, both appear superior to our previous in vivo active 3 in important ways. 29a achieved measurable levels in the brain, while 27g exhibited higher drug levels at all time points.
Table 5. In vivo Exposure Following IP Administration to Micea.
| Compound (dose) | Plasma (ng/mL) | Brain (ng/g) | ||||
|---|---|---|---|---|---|---|
| 30 min | 2 hr | 12 hr | 30 min | 2 hr | 12 hr | |
| 3 (30 mg/kg) | 10 | 30 | BLQ | BLQ | BLQ | BLQ |
| 27g (30 mg/kg) | 59 | 79 | 23 | BLQ | BLQ | BLQ |
| 29a (20 mg/kg) | 124 | 5 | BLQ | 19 | BLQ | BLQ |
Six-week old female C57BL/6 mice were injected IP with a single indicated dose. The data shown are mean values from 2 mice at each time point. BLQ: below the level of quantification.
Discussion And Conclusion
In this work our objectives were to develop analogs of our novel anti-alphaviral indole carboxamide 3 possessing greater potency and enhanced potential for in vivo efficacy against alphavirus infection. Although protection against infection by neuroadapted Sindbis virus in mice was achieved with 3 in our previous work, the in vitro ADME properties of this early lead compound were clearly not optimal, especially in light of its modest potency.
To improve potency, we focused on expanding our investigation of the terminal carboxamide, since the enantiospecific activity of 3 suggested that the amide was making intimate contact with the unknown molecular target. Six compounds were identified with submicromolar activity against our WEEV replicon, four of which achieved 10-fold improvements over 3. The SAR leading to the optimal terminal 4-pyridylethyl amide (e.g. 27g-i) is consistent with our hypothesis that this substituent is making intimate contact with the binding site, probably via hydrogen bonding through the pyridyl nitrogen. We also initiated an exploration of the SAR of the indole N-substituent. This was less fruitful; neither alteration of the aromatic substitution nor replacement with alkyl groups improved activity. Antiviral activity of replicon actives was confirmed with studies of WEEV- or FMV-infected neuronal cells. In all cases, cell viability was improved relative to lead 3 at equal concentrations, and in all but one case, viral titers were reduced below that achieved by 3. Two compounds reduced titers by over a log relative to 3 (27a and 29h).
With regard to improving potential for in vivo activity, we focused on both physical properties (aqueous solubility, passive lipid bilayer permeability) and in vitro predictors of pharmacokinetics (stability to Phase I metabolism by mouse liver mirosomes, recognition by the BBB efflux transporter Pgp). Increases in solubility were realized, as expected, by reducing lipophilicity or molecular weight (e.g. 29a-d,29j). Significant improvements in stability to metabolism by mouse liver microsomes were achieved through reductions in overall lipophilicity and/or electron density of the central indole ring. Two of the most stable compounds (29c and 29f) achieved 20-35-fold increases in half-lives relative to lead compound 3. Collectively, our structure-metabolism relationship data suggest that two sites for metabolism of this class of compounds are the alkyl chain of the terminal amide (27b vs 27g), and the pyrrole ring of the indole (27g vs 29c). Oxidation of the indole N-benzyl group, on the other hand, is unlikely based on the greater stability of 29f vs 27g. Finally, we demonstrated that apparent recognition by MDR1 (Pgp), a potential significant barrier to penetration of the BBB, could be attenuated by specific modifications to the indole ring. In particular, replacement of the bicyclic indole with monocyclic templates (e.g. 29a and 29b) or incorporation of a nitrogen into the indole (29c and 29j) strongly diminished or virtually completely eliminated recognition by Pgp as determined by our assay.
A preliminary PK study comparing the plasma and brain levels of three diverse compounds after a single IP dose was undertaken to “validate” our in vitro approach to selecting compounds for future in vivo infection studies. The results correlated well with our predictive in vitro assays for Pgp efflux and metabolism, with measurable brain levels being observed only with the compound with the lowest value in our MDR1 recognition assay (29a, CCG-20638124) and the highest plasma levels being achieved by the compound with the longest half-life in mouse liver microsome incubations (27g, CCG-20543224). Both of these compounds achieved higher plasma and/or brain levels than our previous lead 3 which has already successfully protected mice from alphaviral infection. We therefore intend to advance both compounds into in vivo efficacy studies in mice with WEEV encephalitis, the results of which will be reported in due course.
Experimental Section
General synthetic procedures
All reagents were used as received from commercial sources unless otherwise noted. 1H and 13C spectra were obtained in DMSO-d6 or CDCl3 at room temperature, unless otherwise noted, on a Varian Inova 400 MHz, Varian Inova 500 MHz or Bruker Avance DRX 500 instruments. Chemical shifts for the 1H NMR and 13C NMR spectra were recorded in parts per million (ppm) on the δ scale from an internal standard of residual tetramethylsilane (0 ppm). Rotamers are described as a ratio of rotamer A and B if possible. Otherwise, if the rotamers cannot be distinguished, the NMR peaks are described as multiplets. Mass spectroscopy data were obtained on a Waters Corporation LCT. HPLC retention times were recorded in minutes (min) using an Agilent 1100 Series with an Agilent Zorbax Eclipse Plus − C18 column with the gradient 10% ACN/water (1 min), 10-90% ACN/water (6 min), and 90% ACN/water (2 min). Solvent abbreviations used: MeOH (methanol), DCM (dichloromethane), EtOAc (ethyl acetate), Hex (hexanes), DMSO (dimethylsulfoxide), DMF (dimethylformamide), H2O (water), THF (tetrahydrofuran), ACN (acetonitrile). Reagent abbreviations used: HOBt (1-hydroxy-1,2,3-benzotriazole), EDCI (N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride), DMAP (4-Dimethylaminopyridine), DIEA (diisopropylethylamine), TEA (triethylamine), PPh3 (triphenylphosphine), MgSO4 (magnesium sulfate), Na2SO4 (sodium sulfate), NaHCO3 (sodium bicarbonate), Na2CO3 (sodium carbonate), Cs2CO3 (sodium carbonate), NH4Cl (ammonium chloride), K2CO3 (potassium carbonate), KOH (potassium hydroxide), HCl (hydrogen chloride). Assay abbreviations: LUC (luciferase), MTT ((3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide. All anhydrous reactions were run under an atmosphere of dry nitrogen.
Ethyl 1-(1H-indole-2-carbonyl)piperidine-4-carboxylate (9)
Ethyl 1H-indole-2-carboxylate (8, 3.0 g, 16 mmol) and lithium hydroxide, H2O (3.3 g, 79 mmol) were dissolved in 2/1 water/THF (30 mL). The reaction was stirred overnight. The reaction was diluted with water and diethyl ether. The water layer was washed with diethyl ether twice. The aqueous layer was acidified to pH 2 using 2N HCl. The suspension was extracted using ethyl acetate. The organic layer was washed with saturated sodium chloride solution, dried over magnesium sulfate, filtered and concentrated to obtain 1H-Indole-2-carboxylic acid as a white solid. Yield: 2.39 g, 94%. 1H NMR (400 MHz, DMSO- d6) δ 12.95 (s, 1H), 11.74 (s, 1H), 7.67 − 7.60 (m, 1H), 7.47 − 7.39 (m, 1H), 7.28 − 7.19 (m, 1H), 7.11 − 7.01 (m, 2H). 1H-indole-2-carboxylic acid (2.49 g, 15.45 mmol), EDCI (4.44 g, 23.18 mmol), and HOBt (3.55 g, 23.18 mmol) were dissolved in THF (30 mL). The reaction was allowed to stir for 15 minutes before DIEA (4.05 ml, 23.18 mmol) and ethyl piperidine-4-carboxylate (3.57 mL, 23.18 mmol) were added to the reaction. The reaction was diluted with water and ethyl acetate. The organic layer was washed with saturated sodium bicarbonate, 1M HCl, and saturated sodium chloride. The organic layer was then dried over magnesium sulfate, filtered and concentrated. The resulting solid was triturated with ethyl acetate to obtain the product as a white solid. Yield: 3.27 g, 71%. 1H NMR (400 MHz, DMSO- d6) δ 11.55 (s, 1H), 7.60 (d, J = 8.1 Hz, 1H), 7.41 (d, J = 8.1 Hz, 1H), 7.41 (d, J = 8.2 Hz, 1H), 7.22 − 7.13 (m, 1H), 7.08 − 7.00 (m, 1H), 6.80 − 6.74 (m, 1H), 4.34 (dt, J = 13.4, 3.9 Hz, 2H), 4.09 (q, J = 7.1 Hz, 2H), 3.18 (bs, 2H), 2.76 − 2.64 (m, 1H), 1.99 − 1.88 (m, 2H), 1.65 − 1.50 (m, 2H), 1.20 (t, J = 7.1 Hz, 3H).
N-Benzyl-1-(1H-indole-2-carbonyl)-N-methylpiperidine-4-carboxamide (10)
Ester 9 (3.0 g, 10 mmol) and lithium hydroxide, H2O (4.19 g, 100 mmol)) were dissolved inTHF (Ratio: 1, Volume: 20 ml)and Water (Ratio: 2, Volume: 40.0 ml) and allowed to stir overnight at room temperature. The reaction was diluted with water, and extracted with diethyl ether. The aqueous layer was acidified using 2N HCl to pH ∼2. A fine, white suspension resulted. The aqueous layer was extracted with ethyl acetate twice. The combined ethyl acetate layers were dried over magnesium sulfate, filtered and concentrated to afford the 1-(1H-indole-2-carbonyl)piperidine-4-carboxylic acid as a white solid. Yield: 678 mg, 25%. 1H NMR (400 MHz, DMSO- d6) δ 12.33 (s, 1H), 11.54 (s, 1H), 7.60 (d, J = 8.0 Hz, 1H), 7.41 (d, J = 7.5 Hz, 1H), 7.22 − 7.13 (m, 1H), 7.08 − 6.99 (m, 1H), 6.79 − 6.74 (m, 1H), 4.32 (d, J = 13.3 Hz, 2H), 3.17 (bs, 1H), 2.65 − 2.54 (m, 1H), 1.96 − 1.87 (m, 2H), 1.63 − 1.49 (m, 2H). HPLC ret. time = 5.31 min; purity > 95%. 1-(1H-indole-2-carbonyl)piperidine-4-carboxylic acid (1.00g, 3.67 mmol), EDCI (1.408 g, 7.34 mmol), and HOBt hydrate (1.125 g, 7.34 mmol) were dissolved in DCM (10 ml). The reaction was allowed to stir for 10 minutes before adding DIEA (1.283 ml, 7.34 mmol) and N-methyl-1-phenylmethanamine (0.836 ml, 7.34 mmol). The reaction was allowed to stir overnight at room temperature. The reaction was diluted with water and ethyl acetate. The organic phase was washed with saturated sodium bicarbonate, 1N HCl, and saturated sodium chloride solution. The organic layer was dried over magnesium sulfate, filtered and concentrated. The crude material was purified by silica gel chromatography using 30% to 60% ethyl acetate: hexanes to obtain product as a white solid. Yield: 1.03 g, 75% 1H NMR (400 MHz, DMSO- d6) 1.7:1 mixture of rotamers δ 11.59 − 11.53 (m, 1H), 7.64 − 7.56 (m, 1H), 7.45 − 7.13 (m, 7H), 7.09 − 6.99 (m, 1H), 6.78 (rotamer A, s), 6.75 (rotamer B, s, 1H), 4.69 (rotamer A, s, 1H), 4.54 − 4.37 (m, 3H), 3.28 − 2.92 (m, 5H), 2.79 (rotamer B, s, 1H), 1.86 − 1.77 (m, 1H), 1.75 − 1.55 (m, 3H). TOF ES+MS: 376.2 (M+H), 398.1 (M+Na). HPLC ret. time = 6.10 min; purity = 93%.
1-(4-Chlorobenzyl)-1H-pyrrole-2-carboxylic acid (12)
Methyl 1H-pyrrole-2-carboxylate 11 (2.77 g, 22.14 mmol) was dissolved in anhydrous DMF (15 mL). Granular potassium carbonate (1.94 g, 26.6 mmol) was added, followed by the addition of 4-chlorobenzylchloride (1.80 mL, 26.6 mmol). The reaction was permitted to stir at 60°C for 24 h, then again 90°C for 48 h. Cesium carbonate (21.6 g, 66.4 mmol) was then added and the reaction was allowed to stir at 90°C for 24 h, then sodium iodide (332 mg, 2.214 mmol) was added and stirring continued at 90°C for 24 h. At this time the reaction was allowed to cool to RT, diluted with ethyl acetate, and washed with brine (3X). The organic phase was then dried (magnesium sulfate), concentrated in vacuo, and purified by flash chromatography (150 g silica, 5% ethyl acetate/hexanes) to provide methyl 1-(4-chlorobenzyl)-1H-pyrrole-2-carboxylate as colorless crystals. Yield: 4.91 g (89%). 1H NMR (400 MHz, CDCl3) δ 7.32 (d, J = 4.7 Hz, 1H), 7.26 (d, J = 3.5 Hz, 1H), 7.05 − 6.98 (m, 3H), 6.92 − 6.86 (m, 1H), 6.20 (dd, J = 3.9, 2.6 Hz, 1H), 5.52 (s, 2H), 3.76 (s, 3H).
To methyl 1-(4-chlorobenzyl)-1H-pyrrole-2-carboxylate (4.5 g, 18.02 mmol) was added to EtOH (50 mL) and 10% aq. NaOH (20 mL) and stirred overnight at RT, then again overnight at 50°C to give completion. The ethanol was stripped by rotary evaporation until material began to precipitate, at which point the aqueous mixture was cooled in an ice bath and acidified with concentrated HCl, which elicited further precipitation. The precipitate was collected over a filter and washed with a small amount of cold 1M HCl and hexanes. The material was then dried under high vacuum to provide the title compound as a white powder. Yield: 4.20 g, (99%). 1H NMR (400 MHz, DMSO-d6) δ 7.34 (d, J = 8.2 Hz, 2H), 7.19 (s, 1H), 7.06 (d, J = 8.2 Hz, 2H), 6.82 (s, 1H), 6.16 − 6.08 (m, 1H), 5.53 (s, 2H).
1-(1-(4-Chlorobenzyl)-1H-pyrrole-2-carbonyl)piperidine-4-carboxylic acid (13)
The following were added sequentially to anhydrous DMF (20 mL): 12 (2.0 g mg, 8.5 mmol), DIEA (4.45 mL, 25.5 mmol), EDCI (1.79 g, 9.34 mmol), and HOBt (1.43 g, 9.34 mmol), and this solution was allowed to stir at RT for 30 min. Finally, ethyl isonipecotate (1.44 mL, 9.34 mmol) was added and the reaction was permitted to stir at RT for 24 h. At this time, DCM was removed in vacuo and the residue was dissolved in ethyl acetate, which was subsequently washed with 1M HCl (3X), water (2X), 10% aq. sodium carbonate (2X), and lastly brine (1X). The organic layer was then dried (magnesium sulfate) and concentrated in vacuo. The residue was crystallized from ethyl acetate/hexanes via the slow evaporation of solvent at RT to give ethyl 1-(1-(4-chlorobenzyl)-1H-pyrrole-2-carbonyl)piperidine-4-carboxylate as large translucent crystals. Yield: 2.16 g (68%). 1H NMR (400 MHz, CDCl3) δ 7.24 (d, J = 8.4 Hz, 2H), 7.03 (d, J = 8.4 Hz, 2H), 6.84 − 6.74 (m, 1H), 6.31 (dd, J = 3.7, 1.5 Hz, 1H), 6.18 − 6.06 (m, 1H), 5.27 (s, 2H), 4.21 (d, J = 11.5 Hz, 2H), 4.14 (q, J = 7.1 Hz, 2H), 2.94 (t, J = 11.5 Hz, 2H), 2.46 (tt, J = 10.7, 4.0 Hz, 1H), 1.79 (d, J = 12.7 Hz, 2H), 1.56 − 1.30 (m, 2H), 1.25 (t, J = 7.1 Hz, 3H).
Ethyl 1-(1-(4-chlorobenzyl)-1H-pyrrole-2-carbonyl)piperidine-4-carboxylate (700 mg, 1.867 mmol) was dissolved in EtOH (60 mL) and water (20 mL). Granular LiOH (134 mg, 5.60 mmol) was added to this, and the reaction was allowed to stir at RT for 24 h. After this time, most of the solvent was removed in vacuo, and the remaining aqueous solution was chilled to 0°C and acidified by concentrated HCl, which elicited a thick, sticky precipitate that adhered to the flask. The aqueous solution was decanted off, a small amount of 1 N HCl was added, and the material sonicated. The aqueous solution was again decanted, and a small amount of ice-cold water was added, mixed, and decanted. Remaining water and HCl was then removed by rotary evaporation, followed by high-vacuum drying to afford the title compound as a white powder. Yield: 620 mg (96%). 1H NMR (400 MHz, DMSO-d6) δ 7.33 (dd, J = 9.0, 2.7 Hz, 2H), 7.14 − 6.97 (m, 2H), 6.27 (dd, J = 3.6, 1.5 Hz, 1H), 6.10 − 5.94 (m, 1H), 5.25 (s, 2H), 3.98 (d, J = 11.0 Hz, 2H), 3.03 − 2.80 (m, 2H), 2.35 − 2.19 (m, 1H), 1.64 (d, J = 11.1 Hz, 2H), 1.24 (d, J = 11.5 Hz, 2H).
1-(tert-Butoxycarbonyl)piperidine-4-carboxylic acid (15)
Ethyl piperidine-4-carboxylate 14 was converted to 1-tert-butyl 4-ethyl piperidine-1,4-dicarboxylate as previously described.25 The ester (3.00 g, 11.66 mmol) was dissolved in ethanol (20 mL) and 10% aqueous sodium hydroxide (10 mL) and stirred at room temperature for 4 h. At this time, as much ethanol as possible was stripped off in vacuo, and the remaining mostly aqueous solution was cooled in an ice bath and acidified with concentrated HCl. The resulting precipitate was collected over a filter and washed with cold water, dried over the filter, then finally dried further under high vacuum to afford the desired carboxylic acid as a white powder. Yield: 2.43 g, 91%. 1H NMR (400 MHz, DMSO-d6) δ 12.25 (s, 1H), 3.81 (d, J = 13.3 Hz, 2H), 2.78 (s, 2H), 2.38 (tt, J = 11.1, 3.9 Hz, 1H), 1.76 (dd, J = 13.3, 3.1 Hz, 2H), 1.47 − 1.24 (m, 10H).
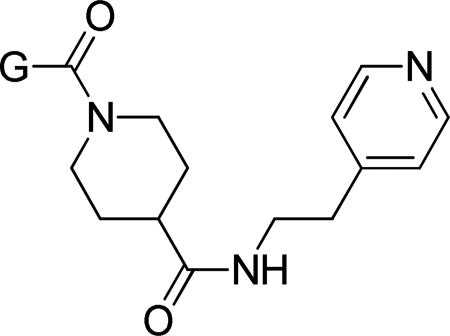
N-(2-(Pyridin-4-yl)ethyl)piperidine-4-carboxamide dihydrochloride (16)
The following was added to DCM: 15 (600 mg, 2.62 mmol), DIEA (1.37 mL, 7.85 mmol), EDCI (552 mg, 2.88 mL), HOBt (441 mg, 2.88 mmol), and 2-(4-pyridylethyl)amine (0.34 mL, 2.88 mL). The solution was stirred at rt for 17 h, at which time the DCM was stripped off and 10% aq. sodium carbonate was added. Material was extracted out with EtOAc (3X). The organic extractions were pooled, dried over magnesium sulfate, and concentrated in vacuo. The residue was taken up in a small amount of EtOAc and diethyl ether was added. The precipitate was collected over a filter and washed with diethyl ether to give tert-butyl 4-((2-(pyridin-4-yl)ethyl)carbamoyl)piperidine-1-carboxylate as an off-white solid. Yield: 634 mg (73%). 1H NMR (400 MHz, CDCl3) δ 8.49 (d, J = 5.9 Hz, 2H), 7.09 (d, J = 5.9 Hz, 2H), 5.59 (t, J = 5.3 Hz, 1H), 4.22 − 3.95 (m, 2H), 3.52 (q, J = 6.8 Hz, 2H), 2.81 (t, J = 6.9 Hz, 2H), 2.68 (t, J = 12.5 Hz, 2H), 2.14 (tt, J = 11.6, 3.8 Hz, 1H), 1.77 − 1.65 (m, 2H), 1.56 (qd, J = 12.1, 4.3 Hz, 2H), 1.43 (s, 9H).
tert-Butyl 4-((2-(pyridin-4-yl)ethyl)carbamoyl)piperidine-1-carboxylate (575 mg, 1.72 mmol) was suspended in diethyl ether at rt, and 4M HCl in dioxane (6 mL, 24 mmol) was added. The mixture was stirred at rt for 30 min, at which time the organic solution was decanted off and the solid material collected and dried in vacuo. The title compound was thus obtained as a tan powder. Yield: 448 mg (97%). 1H NMR (400 MHz, DMSO-d6) δ 9.22 (bs, 1H), 8.91 (bs, 1H), 8.79 (d, J = 6.4 Hz, 2H), 8.18 (t, J = 5.6 Hz, 1H), 7.89 (d, J = 6.3 Hz, 2H), 3.40 (q, J = 6.2 Hz, 2H), 3.15 (d, J = 12.6 Hz, 2H), 3.00 (t, J = 6.4 Hz, 2H), 2.83 − 2.69 (m, 2H), 2.39 − 2.26 (m, 1H), 1.80 − 1.59 (m, 4H). TOF ES+ MS: (M+H) 234.2, (M+Na) 256.1.
1-(4-Chlorobenzyl)-1H-imidazole-2-carboxylic acid (18b)
Ethyl 1H-imidazole-2-carboxylate 17b (4.00 g, 28.5 mmol), p-chlorobenzylchloride (4.38 mL, 34.3 mmol), and sodium carbonate (3.63 g, 34.3 mmol), was dissolved in DMF (8 mL). The solution was stirred at rt for 24 h, at which time water was added and material was extracted with EtOAc. The organic phase was collected, dried over magnesium sulfate, and decanted. Purification accomplished via silica gel flash chromatography (150 g silica, 10% EtOAc/Hexanes to 80% EtOAc/Hexanes). Ethyl 1-(4-chlorobenzyl)-1H-imidazole-2-carboxylate was obtained as a clear, yellowtinted oil. Yield: 7.28 g (96%). 1H NMR (400 MHz, CDCl3) δ 7.29 (d, J = 8.4 Hz, 2H), 7.18 (d, J = 0.9 Hz, 1H), 7.09 (d, J = 8.4 Hz, 2H), 7.06 (d, J = 0.9 Hz, 1H), 5.59 (s, 2H), 4.37 (q, J = 7.2 Hz, 2H), 1.39 (t, J = 7.1 Hz, 3H).
Ethyl 1-(4-chlorobenzyl)-1H-imidazole-2-carboxylate (7.28 g, 27.5 mmol), was dissolved in EtOH (10 mL) and 10% aq. NaOH (20 mL) and stirred at rt for 15 h. The solvent was then stripped off, water was added, and the solution acidified with HCl. The resulting precipitate was collected over a filter and washed with 1M HCl and dried to afford the title compound as a white powder. This material was taken directly into the next step. Yield: 6.506 g (100%).
1-(4-Chlorobenzyl)-4-fluoro-1H-pyrrole-2-carboxylic acid (18k)
Methyl 4-fluoro-1H-pyrrole-2-carboxylate 17k (130 mg, 0.91 mmol) was dissolved in anhydrous DMF at room temperature, followed by the addition of potassium carbonate (151 mg, 1.09 mmol) and 1-chloro-4-(chloromethyl)benzene (0.14 mL, 1.09 mmol). The reaction was then stirred for 30 h at 60 °C, after which time it was allowed to cool to room temperature. The reaction was diluted with a 1:1 solution of ethyl acetate/diethyl ether, washed with water (3X), brine (1X), dried with magnesium sulfate, and concentrated in vacuo. The resulting brown residue was purified via flash chromatography (60 g silica, 10% ethyl acetate/hexanes) to afford methyl 1-(4-chlorobenzyl)-4-fluoro-1H-pyrrole-2-carboxylate a light yellow oil. Yield: 203 mg (83%). 1H NMR (400 MHz, CDCl3) δ 7.27 (d, J = 8.3 Hz, 2H), 7.03 (d, J = 8.3 Hz, 2H), 6.69 − 6.59 (m, 2H), 5.44 (s, 2H), 3.75 (s, 3H).
Methyl 1-(4-chlorobenzyl)-4-fluoro-1H-pyrrole-2-carboxylate (150 mg, 0.56 mmol) was dissolved in ethanol (10 mL) at room temperature. 10% aqueous sodium hydroxide (2 mL) was added, and the reaction was stirred for 18 h at room temperature. At this time, solvent was stripped off in vacuo until material began to precipitate. Additional water was added (2 mL) and the solution was cooled in an ice bath, then acidified with concentrated HCl. The resulting precipitate was collected over a filter and washed with cold 1 M HCl, then dried under high vacuum to afford the desired carboxylic acid as a white powder. Yield: 111 mg (78%). 1H NMR (400 MHz, CDCl3) δ 7.29 (d, J = 8.3 Hz, 2H), 7.04 (d, J = 8.3 Hz, 2H), 6.79 (d, J = 2.0 Hz, 1H), 6.71 − 6.65 (m, 1H), 5.43 (s, 2H).
Ethyl 1-((4-chlorophenethyl)carbamoyl)piperidine-4-carboxylate (20)
1-chloro-4-(2-isocyanatoethyl)benzene 19 (0.45 mL, 2.94 mmol) was dissolved in DCM (10 mL) at RT. Ethyl isonipecotate was then added dropwise, which elicited precipitation. The slurry was allowed to stir at RT for 1 h, at which time the precipitate was collected and dried over a filter to afford ethyl 1-((4-chlorophenethyl)carbamoyl)piperidine-4-carboxylate as an off-white granular solid. This material was taken into the next reaction in this crude form. Yield: 916 mg (92%).
Ethyl 1-(1-(4-chlorobenzyl)-1H-benzo[d]imidazole-2-carbonyl)piperidine-4-carboxylate (22c)
1H-benzo[d]imidazole-2-carboxylic acid 21c, H2O (500 mg, 2.78 mmol), EDCI (1170 mg, 6.11 mmol), and HOBt (825 mg, 6.11 mmol) were dissolved in DCM. The reaction was allowed to stir for 10 minutes before DIEA (1.066 mL, 6.11 mmol) and ethyl piperidine-4-carboxylate (0.941 mL, 6.11 mmol) were added. The reaction was allowed to stir overnight at room temperature. The reaction was diluted with water and ethyl acetate. The organic phase was washed with 1N HCl, saturated sodium bicarbonate, and saturated sodium chloride solution. The ethyl acetate layer was dried over magnesium sulfate, filtered and concentrated. The resulting crude material was triturated with ethyl acetate to obtain ethyl 1-(1H-benzo[d]imidazole-2-carbonyl)piperidine-4-carboxylate as a white solid. Yield: 113 mg (14%). 1H NMR (400 MHz, DMSO- d6) δ 13.09 (s, 1H), 7.63 (dd, J = 84.6, 8.0 Hz, 2H), 7.28 (dt, 2H), 5.30 (d, J = 13.5 Hz, 1H), 4.42 (d, J = 13.0 Hz, 1H), 4.09 (q, J = 14.2, 7.0 Hz, 2H), 3.48 (t, J = 11.7 Hz, 1H), 3.05 (t, J = 11.3 Hz, 1H), 2.77 − 2.65 (m, 1H), 1.97 (d, J = 13.7 Hz, 2H), 1.69 − 1.52 (m, 2H), 1.19 (t, J = 7.1 Hz, 3H). TOF ES+ MS: 302.1 (M+H), 324.1 (M+Na). HPLC ret. time = 5.44 min; purity > 95%.
Ethyl 1-(1H-benzo[d]imidazole-2-carbonyl)piperidine-4-carboxylate (80 mg, 0.265 mmol) and cesium carbonate (130 mg, 0.398 mmol) were dissolved in DMF (Volume: 2.0 ml). 1-Chloro-4-(chloromethyl)benzene (0.051 ml, 0.40 mmol) was added and the reaction was heated at 80 °C overnight. After 18 hours, the reaction was cooled to room temperature and diluted with water and ethyl acetate. The organic phase was washed with saturated sodium chloride four times before it was dried over magnesium sulfate, filtered and concentrated. The isolated beige solid was taken directly to the next step without purification. Yield: 85 mg (75%). 1H NMR (400 MHz, DMSO- d6) δ 7.74 (d, J = 7.7 Hz, 1H), 7.66 (d, J = 7.7 Hz, 1H), 7.49 − 7.27 (m, 6H), 5.63 − 5.48 (m, 1H), 5.15 (s, 1H), 4.42 − 4.32 (m, 1H), 4.08 (q, J = 8 Hz, 8Hz, 2H), 3.97 − 3.86 (m, 1H), 3.22 − 3.10 (m, 1H), 3.05 − 2.93 (m, 1H), 2.70 − 2.55 (m, 1H), 1.97 − 1.88 (m, 1H), 1.76 − 1.67 (m, 1H), 1.53 − 1.38 (m, 1H), 1.21 (t, J = 8 Hz, 3H). TOF ES+ MS: 426.0 (M+H), 448.0 (M+Na).
Ethyl 1-(1-(4-chlorobenzyl)-6-fluoro-1H-indole-2-carbonyl)piperidine-4-carboxylate (22h)
6-fluoro-1H-indole-2-carboxylic acid 21h (500 mg, 2.79 mmol), EDCI (1070 mg, 5.58 mmol), and HOBt (754 mg, 5.58 mmol) were dissolved in DCM (14 mL). The reaction was allowed to stir for 20 minutes before adding ethyl piperidine-4-carboxylate (0.860 mL, 5.58 mmol) and DIEA (0.975 mL, 5.58 mmol). The reaction was stirred at room temperature overnight. After 18 hours, it was diluted with water and ethyl acetate. The organic phase was washed with 1N HCl, saturated sodium carbonate, and saturated sodium chloride. It was dried over sodium sulfate, filtered and concentrated. No further purification was done on the material. Yield: 140 mg (16%). 1H NMR (500 MHz, DMSO- d6) δ 11.64 (s, 1H), 7.62 (dd, J = 8.7, 5.5 Hz, 1H), 7.13 (dd, J = 10.0, 2.5 Hz, 1H), 6.96 − 6.88 (m, 1H), 6.81 (dd, J = 2.3, 0.9 Hz, 1H), 4.33 (dt, J = 13.3, 3.2 Hz, 2H), 4.09 (q, J = 7.1 Hz, 2H), 3.19 − 3.08 (m, 2H), 2.75 − 2.65 (m, 1H), 1.97 − 1.89 (m, 2H), 1.57 (q, J = 11.1 Hz, 2H), 1.20 (t, J = 7.1 Hz, 3H). TOF ES+ MS: 319.0 (M+H), 341.0 (M+Na). HPLC ret. time = 6.76 min; purity > 95%
Ethyl 1-(6-fluoro-1H-indole-2-carbonyl)piperidine-4-carboxylate (140 mg, 0.440 mmol) and Cs2CO3 (287 mg, 0.880 mmol) were dissolved in DMF (5.0 mL). 1-chloro-4-(chloromethyl)benzene (0.617 ml, 4.83 mmol) was added as a liquid. The reaction was heated at 60 °C overnight. The reaction was cooled and diluted with water and ethyl acetate. The aqueous layer was washed with another aliquot of ethyl acetate. The combined organic phases were washed with saturated sodium chloride solution four times. It was dried over sodium sulfate, filtered and concentrated. The crude oil was purified using flash chromatography (0-10% ethyl acetate/hexanes). The product was isolated as a white solid. Yield: 170 mg (87%). 1H NMR (500 MHz, DMSO- d6) δ 7.64 (dd, J = 8.7, 5.5 Hz, 1H), 7.54 (dd, J = 10.5, 2.3 Hz, 1H), 7.36 − 7.29 (m, 2H), 7.10 − 7.04 (m, 2H), 7.02 − 6.94 (m, 1H), 6.75 (d, J = 0.8 Hz, 1H), 5.50 − 5.46 (m, 2H), 4.30 (bs, 1H), 4.07 (q, J = 7.1 Hz, 2H), 3.88 (bs, 1H), 2.99 (m, 2H), 2.62 − 2.52 (m, 2H), 2.01 − 1.54 (m, 4H), 1.18 (t, J = 7.1 Hz, 3H). TOF ES+ MS: 443.1 (M+H), 465.0 (M+Na). HPLC ret. time = 8.39 min; purity >95%.
1-Benzyl-1H-indole-2-carboxylic acid (24f)
Ester 8 (1.0 g, 5.3 mmol), potassium carbonate (1.461 g, 10.57 mmol) were dissolved in DMF (15 ml) and heated to 60 °C. The reaction was stirred for 20 minutes before the addition of (bromomethyl)benzene (0.942 ml, 7.93 mmol). The reaction was stirred overnight at 60 °C. After cooling to room temperature, it was diluted with water and an ethyl acetate/diethyl ether mixture. The aqueous phase was washed with another aliquot of the same organic mixture. The organic phases were combined and washed with saturated sodium chloride (2x), dried over sodium sulfate, filtered and concentrated. The resulting crude material was purified via SP1 biotage (25 g silica cartridge) with 0-40% ethyl acetate/hexanes gradient. Ethyl 1-benzyl-1H-indole-2-carboxylate was obtained as an off-white solid. Yield: 396 mg, 27%. . 1H NMR (500 MHz, DMSO- d6) δ 7.72 (dt, J = 8.0, 1.0 Hz, 1H), 7.57 (dd, J = 8.5, 0.9 Hz, 1H), 7.38 (d, J = 0.9 Hz, 1H), 7.35 − 7.11 (m, 5H), 7.05 − 6.99 (m, 2H), 5.86 (s, 2H), 4.28 (q, J = 7.1 Hz, 2H), 1.28 (t, J = 7.1 Hz, 3H). HPLC ret. time = 8.58 min; purity > 95%.
The ester from above (390 mg, 1.396 mmol) was dissolved in THF (1 ml) and 2N aqueous sodium hydroxide (3.49 ml, 6.98 mmol). The reaction was stirred for 3 hours before it was concentrated under vacuum until a white precipitate formed. The resulting suspension was cooled by an ice bath and diluted with water until a stir bar was able to stir. 2N HCl was added dropwise until pH 2 was reached. The acidic aqueous suspension was extracted with ethyl acetate. The aqueous layer was washed with another aliquot of ethyl acetate. The organic phases were combined and washed with saturated sodium chloride, filtered and concentrated. No further purification of 24f was necessary. Yield: 339 mg, 27% 1H NMR (500 MHz, DMSO- d6) δ 12.97 (s, 1H), 7.70 (dt, J = 8.1, 0.9 Hz, 1H), 7.54 (dd, J = 8.5, 1.0 Hz, 1H), 7.35 − 7.17 (m, 5H), 7.16 − 7.09 (m, 1H), 7.05 − 7.00 (m, 2H), 5.88 (s, 2H). TOF ES+ MS: 252.1(M+H), 274.1(M+Na). HPLC ret. time = 7.13 min; purity > 95%.
1-(2,6-Difluorobenzyl)-1H-indole-2-carboxylic acid (24g)
60 wt% sodium hydride (127 mg, 3.17 mmol) was suspended in DMF (8 ml) and cooled with an ice bath. The reaction was stirred at for twenty minutes before a DMF (2 ml) solution of 8 (500 mg, 2.64 mmol) was added dropwise. The reaction was allowed to stir for 20 minutes before 2-(bromomethyl)-1,3-difluorobenzene (821 mg, 3.96 mmol) was added dropwise as a solution in DMF (3 ml). It was allowed to stir at room temperature overnight. The reaction was diluted with saturated ammonium chloride and a mixture of ethyl acetate/diethyl ether. The aquoeous phase was washed with another aliquout of ethyl acetate/diethyl ether. The organic phases were combined and washed with saturate sodium chloride, dried over sodium sulfate, filtered and concentrated. The resulting crude material was purified via SP1 Biotage (25 g silica gel column) using a gradient of 100% hexanes to 30% ethyl acetate/hexanes. The product ethyl 1-(2,6-difluorobenzyl)-1H-indole-2-carboxylate was isolated as a white solid. Yield: 586 mg (70%). 1H NMR (500 MHz, DMSO- d6) δ 7.68 (dt, J = 8.0, 1.0 Hz, 1H), 7.50 (dd, J = 8.4, 1.0 Hz, 1H), 7.39 − 7.27 (m, 3H), 7.16 − 7.09 (m, 1H), 7.08 − 7.01 (m, 2H), 5.99 (s, 2H), 4.32 (q, J = 7.1 Hz, 2H), 1.31 (t, J = 7.1 Hz, 3H). TOF ES+ MS: 316.1(M+H), 338.0 (M+Na). HPLC ret. time = 8.53 min; purity >95%.
The ester from above (500 mg, 1.59 mmol) was dissolved in THF (3 ml) and aqueous sodium hydroxide (3.96 ml, 7.93 mmol). The reaction was stirred for 3 hours before it was concentrated under vacuum until white precipitate formed. The resulting suspension was cooled using an ice bath and diluted with water until a stir bar was spinning freely. 2N HCl was added dropwise until the aqueous solution reached pH 2. It was diluted with ethyl acetate (2x). The organic phases were combined and washed with saturated sodium chloride, dried over sodium sulfate, filtered and concentrated. The resulting solid 24g was taken to the next step without purification. Yield: 396 mg, 87% 1H NMR (500 MHz, DMSO- d6) δ 12.99 (s, 1H), 7.67 (d, J = 7.9 Hz, 1H), 7.46 (d, J = 8.4 Hz, 1H), 7.40 − 7.23 (m, 3H), 7.14 − 6.99 (m, 3H), 6.02 (s, 2H). TOF ES+ MS: 288.1 (M+H), 310.1 (M+Na). HPLC ret. time = 7.10 min; purity > 95%.
1-(4-Chlorobenzyl)-1H-pyrrolo[2,3-c]pyridine-2-carboxylic acid (24j)
Ethyl 6-azaindole-2-carboxylate 23j (440 mg, 2.31 mmol) was dissolved in anhydrous DMF (15 mL) at RT under nitrogen in ovendried glassware. Granular potassium tert-butoxide (390 mg, 3.47 mmol) was added, which elicited a red color. This solution was allowed to stir for 30 min at RT under nitrogen. After this time, 4-chlorobenzylchloride (325 μL, 2.54 mmol) was added and the reaction stirred for 14 hours. The reaction was then taken up in ethyl acetate/diethyl ether (1:1) and washed with water (4X) and brine (1X), then dried (magnesium sulfate) and concentrated in vacuo. The resulting residue was purified by flash chromatography (150 g silica, 20% ethyl acetate/hexanes eluent) to give ethyl 1-(4-chlorobenzyl)-1H-pyrrolo[2,3-c]pyridine-2-carboxylate as off-white needles. Yield: 475 mg (65%). 1H NMR (400 MHz, CDCl3) δ 8.83 (s, 1H), 8.31 (d, J = 5.5 Hz, 1H), 7.58 (d, J = 5.5 Hz, 1H), 7.32 (s, 1H), 7.22 (d, J = 8.2 Hz, 2H), 7.00 (d, J = 8.1 Hz, 2H), 5.85 (s, 2H), 4.35 (q, J = 7.1 Hz, 2H), 1.37 (t, J = 7.1 Hz, 3H).
Ethyl 1-(4-chlorobenzyl)-1H-pyrrolo[2,3-c]pyridine-2-carboxylate (300 mg, 0.953 mmol) was dissolved into ethanol (8 mL) at RT, followed by the addition of 10% aqueous NaOH (8 mL), which initially caused precipitation, but homogeneity was achieved over time. The reaction was allowed to stir at RT for 12 h, at which time the solvent was removed in vacuo. The residue was taken up in a small amount of water (5 mL), the pH was adjusted to ∼4, and material was extracted with ethyl acetate (8X). The extracts were combined and the solvent removed again in vacuo to provide the title compound as a fine white powder. Yield: 226 mg (83%). 1H NMR (400 MHz, DMSO-d6) δ 9.64 (s, 1H), 8.40 (d, J = 6.3 Hz, 1H), 8.26 (d, J = 6.3 Hz, 1H), 7.61 (s, 1H), 7.35 (d, J = 8.5 Hz, 2H), 7.10 (d, J = 8.5 Hz, 2H), 6.06 (s, 2H).
Ethyl 1-(1-methyl-1H-indole-2-carbonyl)piperidine-4-carboxylate (26e)
Ethyl 1H-indole-2-carboxylate 8 (1.00 g, 5.29 mmol) and K2CO3 (1.46 g, 10.57 mmol) were dissolved in DMF (10 ml). Iodomethane (0.99 ml, 16 mmol) was added and the reaction was stirred at 60 °C overnight. The reaction was dissolved in water and diethyl ether. The water layer was washed with diethyl ether twice. The organic layers were combined and washed with brine twice. It was then dried over magnesium sulfate, filtered, and concentrated. The crude product ethyl 1-methyl-1H-indole-2-carboxylate was taken directly into the next step. 1H NMR (400 MHz, DMSO- d6) δ 7.69 (d, J = 8.0 Hz, 1H), 7.58 (d, J = 8.6 Hz, 1H), 7.35 (t, J = 7.8 Hz, 1H), 7.27 (s, 1H), 7.14 (t, J = 7.5 Hz, 1H), 4.32 (q, J = 7.1 Hz, 2H), 4.03 (s, 3H), 1.34 (t, J = 7.1 Hz, 3H). TOF ES+ MS: 204.1 (M+H). HPLC ret. time = 7.85 min; purity > 95%.
Ethyl 1-methyl-1H-indole-2-carboxylate (1.0 g, 4.92 mmol) and lithium hydroxide (1.178 g, 49.2 mmol) were dissolved in 2/1 water/THF (6 ml). The reaction was allowed to stir overnight. The reaction was diluted with water and washed with diethyl ether. The water layer was then acidified with 1N HCl to pH 2. The resulting suspension was extracted with ethyl acetate. The organic layer was washed with saturated sodium chloride, dried over magnesium sulfate, filtered and concentrated to obtain pure 1-methyl-1H-indole-2- carboxylic acid as a white solid. Yield: 560 mg (65%). 1H NMR (400 MHz, DMSO- d6) δ 12.91 (s, 1H), 7.67 (d, J = 8.0 Hz, 1H), 7.57 (d, J = 8.5 Hz, 1H), 7.37 − 7.29 (m, 1H), 7.22 (s, 1H), 7.16 − 7.08 (m, 1H), 4.02 (s, 3H). TOF ES+ MS: 176.1 (M+H). HPLC ret. time = 6.06 min; purity > 95%.
1-Methyl-1H-indole-2-carboxylic acid (200 mg, 1.14 mmol), HOBt (231 mg, 1.712 mmol), and EDCI (328 mg, 1.712 mmol) were dissolved in DCM (Volume: 4 ml). The reaction was allowed to stir for 10 minutes before adding DIEA (0.299 ml, 1.71 mmol) and ethyl piperidine-4-carboxylate (0.264 ml, 1.71 mmol). The reaction was stirred overnight. The reaction was diluted with water and extracted with ethyl acetate. The organic layer was washed with saturated sodium bicarbonate and 1N HCl. This was followed by a wash with saturated sodium chloride. The organic phase was dried over magnesium sulfate, filtered and concentrated to obtain 26e as a white solid. No further purification was required. Yield: 214 mg, 60%. 1H NMR (400 MHz, DMSO- d6) δ 7.57 (d, J = 8.0 Hz, 1H), 7.48 (d, J = 9.0 Hz, 1H), 7.21 (d, J = 15.3 Hz, 1H), 7.06 (t, J = 7.5 Hz, 1H), 6.61 (s, 1H), 4.37 − 3.83 (m, 4H), 3.71 (s, 3H), 3.10 (bs, 2H), 2.70 − 2.59 (m, 1H), 1.87 (bs, 2H), 1.61 − 1.46 (m, 2H), 1.16 (t, J = 7.1 Hz, 3H). TOF ES+ MS: 315.1 (M+H), 337.1 (M+Na). HPLC ret. time = 6.99 min; purity > 95%.
Ethyl 1-(1-(4-chlorobenzyl)-5-fluoro-1H-indole-2-carbonyl)piperidine-4-carboxylate (26i)
Ethyl 5-fluoro-1H-indole-2-carboxylate 25i (0.500 g, 2.413 mmol) and Cs2CO3 (1.179 g, 3.62 mmol) were dissolved in DMF (15 ml). 1-chloro-4-(chloromethyl)benzene (0.617 ml, 4.83 mmol) was added as a liquid. The reaction was heated at 60 °C overnight. The reaction was cooled and diluted with water and ethyl acetate. The aqueous layer was washed with another aliquot of ethyl acetate. The combined organic phases were washed with saturated sodium chloride solution four times. It was dried over sodium sulfate, filtered and concentrated. The crude oil was purified by flash chromatography using 0-10% ethyl acetate/hexanes. The product ethyl 1-(4-chlorobenzyl)-5-fluoro-1H-indole-2-carboxylate was isolated as a clear oil. Yield: 600 mg (75 %). 1H NMR (500 MHz, DMSO- d6) δ 7.63 (dd, J = 9.0, 5.3 Hz, 1H), 7.51 (dd, J = 9.4, 2.4 Hz, 1H), 7.47 − 7.41 (m, 1H), 7.37 − 7.33 (m, 2H), 7.21 (td, J = 9.3, 2.5 Hz, 1H), 7.03 (d, J = 8.5 Hz, 2H), 5.85 (s, 2H), 4.28 (q, J = 7.1 Hz, 2H), 1.28 (t, J = 7.1 Hz, 3H). HPLC ret. time = 9.06 min; purity > 95%.
Ethyl 1-(4-chlorobenzyl)-5-fluoro-1H-indole-2-carboxylate (465 mg, 1.402 mmol) and lithium hydroxide, H2O (588 mg, 14.0 mmol) were dissolved in 2/1 water/THF (12 ml). The reaction was allowed to stir overnight at room temperature. The reaction was diluted with water and diethyl ether. The organic phase was removed and the aqueous phase was acidified using 2N HCl to pH ∼2. The resulting suspension was extracted with ethyl acetate. The aqueous phase was washed with another aliquot of ethyl acetate. The organic phases were combined, washed with saturated sodium chloride, filtered and concentrated to obtain the product 1-(4-chlorobenzyl)-5-fluoro-1H-indole-2-carboxylic acid. No further purification was performed. Yield: 130 mg (31%). 1H NMR (500 MHz, DMSO- d6) δ 13.14 (s, 1H), 7.58 (dd, J = 9.2, 4.4 Hz, 1H), 7.49 (dd, J = 9.4, 2.6 Hz, 1H), 7.37 − 7.28 (m, 3H), 7.18 (td, J = 9.2, 2.6 Hz, 1H), 7.06 − 7.00 (m, 2H), 5.86 (s, 2H). HPLC ret. time = 7.55 min; purity > 95%.
1-(4-Chlorobenzyl)-5-fluoro-1H-indole-2-carboxylic acid (130 mg, 0.428 mmol), EDCI (164 mg, 0.856 mmol), and DMAP (105 mg, 0.856 mmol) were dissolved in DCM (Volume: 5 ml). The reaction was allowed to stir for 20 minutes before adding ethyl piperidine-4-carboxylate (0.132 ml, 0.856 mmol) and DIEA (0.150 ml, 0.856 mmol). The reaction was stirred at room temperature overnight. After 18 hours, it was diluted with water and ethyl acetate. The organic phase was washed with 1N HCl, saturated sodium carbonate, and saturated sodium chloride. It was dried over sodium sulfate, filtered and concentrated. The crude mixture was triturated with diethyl ether and ethyl acetate. The product 26i was isolated as a pale yellow oil solid. Yield: 90 mg (100%). 1H NMR (500 MHz, DMSO- d6) δ 7.62 (dd, J = 9.1, 4.4 Hz, 1H), 7.40 (dd, J = 9.5, 2.6 Hz, 1H), 7.35 − 7.30 (m, 6H), 7.11 − 7.05 (m, 9H), 6.71 (s, 1H), 5.49 (s, 2H), 4.31 (bs, 1H), 4.07 (q, J = 7.1, 7.1 Hz, 2H), 3.83 (bs, 1H), 3.08 (bs, 1H), 2.92 (bs, 1H), 2.62 − 2.53 (m, 1H), 1.86 (bs, 1H), 1.66 (bs, 1H), 1.40 (bs, 1H), 1.18 (t, J = 7.1 Hz, 3H), 1.05 (bs, 1H). TOF ES+ MS: 443.0 (M+H), 465.0 (M+Na). HPLC ret. time = 8.28 min; purity >95%.
Representative Procedure for Generating Analogs 27 from 7
1-(1-(4-Chlorobenzyl)-1H-indole-2-carbonyl)-N-(1-(pyridin-4-yl)ethyl)piperidine-4-carboxamide (27a)
Acid 77 (100 mg, 0.25 mmol), EDCI (121 mg, 0.63 mmol), and HOBt (85 mg, 0.63 mmol) were dissolved in 3.0 mL of DCM. The reaction was stirred at room temperature for ten minutes before DIEA (0.110 mL, 0.63 mmol) and 1-(pyridin-4-yl)ethanamine (77 mg, 0.63 mmol) as a 1.0 mL DCM solution were added. The reaction was allowed to stir at room temperature overnight. The reaction was diluted with water and extracted 3X with ethyl acetate. The organic phase was washed with saturated sodium bicarbonate (twice), and saturated sodium chloride. The organic phase was dried over magnesium sulfate, filtered and concentrated. The crude material was triturated with ether and ethyl acetate and filtered to obtain 27a as a white solid. Yield: 37.9 mg, 30% 1H NMR (400 MHz, DMSO-d6) δ 8.52 − 8.45 (m, 2H), 8.35 (d, J = 7.7 Hz, 1H), 7.62 (d, J = 7.8 Hz, 1H), 7.53 (d, J = 8.8 Hz, 1H), 7.37 − 7.17 (m, 5H), 7.14 − 7.05 (m, 3H), 6.72 (s, 1H), 5.48 (s, 2H), 4.87 (p, J = 7.1 Hz, 1H), 4.03 (m, 2H), 2.95 (bs, 2H), 2.49 − 2.42 (m, 1H), 1.79 − 1.28 (m, 7H). TOF ES+ MS: 501.2 (M+H). HPLC ret. time = 5.73 min; purity >95%.
(R)-1-(1-(4-Chlorobenzyl)-1H-indole-2-carbonyl)-N-(1-(pyridin-4-yl)ethyl)piperidine-4-carboxamide (27b)
Synthesized from 7 and (R)-1-(pyridin-4-yl)ethanamine as described for 27a. The crude solid was recrystallized using diethyl ether/ethyl acetate. The product was obtained as a white solid after filtration. Yield: 55%. 1H NMR (500 MHz, DMSO- d6) δ 8.51 − 8.46 (m, 2H), 8.37 (d, J = 7.7 Hz, 1H), 7.62 (d, J = 7.9 Hz, 1H), 7.54 (d, J = 8.3 Hz, 1H), 7.36 − 7.25 (m, 4H), 7.25 − 7.18 (m, 1H), 7.13 − 7.06 (m, 3H), 6.73 (s, 1H), 5.48 (s, 2H), 4.88 (p, J = 7.2 Hz, 1H), 4.44 (bs, 1H), 4.02 (bs, 1H), 3.13 − 2.70 (m, 2H), 2.50 − 2.44 (m, 1H), 1.85 − 1.30 (m, 7H). TOF ES+ MS: 501.2 (M+H). HPLC ret. time = 5.62 min; purity >95%.
(1-(4-Chlorobenzyl)-1H-indol-2-yl)(4-(4-methylpiperazine-1-carbonyl)piperidin-1-yl)methanone (27c)
Synthesized from 7 and 4-methylpiperazine as described for 27a. The crude material was purified by medium pressure chromatography using 100% DCM to 5% 7M ammonia in MeOH/95% DCM. The product was obtained as a white solid. Yield: 57%. 1H NMR (400 MHz, DMSO- d6) δ 7.62 (d, J = 7.9 Hz, 1H), 7.54 (d, J = 8.7 Hz, 1H), 7.37 − 7.29 (m, 2H), 7.26 − 7.17 (m, 1H), 7.14 − 7.05 (m, 3H), 6.73 (s, 1H), 5.49 (s, 2H), 4.52 − 3.90 (m, 2H), 3.57 − 3.39 (m, 4H), 3.19 − 2.78 (m, 3H), 2.39 − 2.11 (m, 7H), 1.77 − 1.20 (m, 4H). TOF ES+ MS: 479.1 (M+H). HPLC ret. time = 5.37 min; purity = 92%.
1-(1-(4-Chlorobenzyl)-1H-indole-2-carbonyl)-N-((1-methylpiperidin-4-yl)methyl)piperidine-4-carboxamide (27d)
Synthesized from 7 and (1-methylpiperidin-4-yl)methanamine as described for 27a. The crude product was triturated with diethyl ether/ethyl acetate to afford the product as a white solid. Yield: 65%. 1H NMR (400 MHz, DMSO- d6) δ 7.79 (t, J = 5.8 Hz, 1H), 7.62 (d, J = 7.9 Hz, 1H), 7.54 (d, J = 8.0 Hz, 1H), 7.38 − 7.29 (m, 2H), 7.26 − 7.17 (m, 1H), 7.15 − 7.05 (m, 3H), 6.72 (s, 1H), 5.49 (s, 2H), 4.61 − 3.82 (m, 2H), 2.99 − 2.73 (m, 4H), 2.44 − 2.30 (m, 1H), 2.19 (s, 3H), 1.96 − 1.05 (m, 13H). TOF ES+ MS: 507.1 (M+H). HPLC ret. time = 5.41 min; purity > 95%.
1-(1-(4-Chlorobenzyl)-1H-indole-2-carbonyl)-N-((1-methyl-1H-pyrazol-3-yl)methyl)piperidine-4-carboxamide (27e)
Synthesized from 7 and (1-methyl-1H-pyrazol-3-yl)methanamine as described for 27a. The crude product was triturated with ethyl acetate to obtain white solid as the product. Yield: 18%. 1H NMR (400 MHz, DMSO- d6) δ 8.17 (t, J = 5.7 Hz, 1H), 7.66 − 7.50 (m, 3H), 7.38 − 7.29 (m, 2H), 7.25 − 7.16 (m, 1H), 7.15 − 7.05 (m, 3H), 6.72 (s, 1H), 6.04 (d, J = 2.2 Hz, 1H), 5.49 (s, 2H), 4.17 (m, 4H), 3.76 (s, 3H), 2.92 (bs, 2H), 2.47 − 2.37 (m, 1H), 1.81 − 1.23 (m, 4H). TOF ES+ MS: 490.1 (M+H). HPLC ret. time = 6.62 min; purity = 94%.
1-(1-(4-Chlorobenzyl)-1H-indole-2-carbonyl)-N-((1-methyl-1H-imidazol-4-yl)methyl)piperidine-4-carboxamide (27f)
Synthesized from 7 and (1-methyl-1H-imidazol-4-yl)methanamine as described for 27a. The crude material was triturated with ether and ethyl acetate and filtered to obtain white solid as a product. Yield: 23%. 1H NMR (400 MHz, DMSO- d6) δ 8.07 (t, J = 5.5 Hz, 1H), 7.62 (d, J = 7.9 Hz, 1H), 7.53 (d, J = 8.3 Hz, 1H), 7.46 (s, 1H), 7.38 − 7.30 (m, 2H), 7.26 − 7.16 (m, 1H), 7.15 − 7.05 (m, 3H), 6.87 (s, 1H), 6.73 (s, 1H), 5.49 (s, 2H), 4.49 − 3.92 (m, 4H), 3.32 (s, 3H), 2.90 (bs, 2H), 2.48 − 2.36 (m, 1H), 1.78 − 1.33 (m, 4H). TOF ES+ MS: 490.1 (M+H). HPLC ret. time = 5.48 min; purity > 95%.
1-(1-(4-Chlorobenzyl)-1H-indole-2-carbonyl)-N-(2-(pyridin-4-yl)ethyl)piperidine-4-carboxamide (27g)
Synthesized from 7 and 2-(pyridin-4-yl)ethanamine as described for 27a. The crude material was triturated with ethyl acetate to afford the product as a white solid. Yield: 328 mg, 80%. 1H NMR (500 MHz, DMSO- d6) δ 8.48 − 8.42 (m, 2H), 7.89 (t, J = 5.5 Hz, 1H), 7.63 (d, J = 7.8 Hz, 1H), 7.54 (d, J = 8.5 Hz, 1H), 7.37 − 7.30 (m, 2H), 7.25 − 7.18 (m, 3H), 7.13 − 7.07 (m, 3H), 6.71 (s, 1H), 5.48 (s, 2H), 4.53 − 3.82 (m, 2H), 3.32 − 3.29 (m, 2H), 3.08-2.67 (m, 4H), 2.36 − 2.27 (m, 1H), 1.75 − 1.18 (m, 4H). 13C NMR (500 MHz, DMSO-d6) 226.99, 183.03, 173.53, 161.90, 149.39, 148.39, 137.21, 136.81, 131.91, 131.80, 128.80, 128.49, 126.16, 124.27, 123.15, 121.41, 120.27, 110.67, 103.40, 46.13, 41.42, 38.79, 34.20, 14.12, 14.09; Anal. Calcd for C29H29ClN4O2: C, 69.52%; H, 5.83%; N, 11.18%. Found: C, 69.28%; H, 5.88%; N, 11.23%. TOF ES+ MS: 501.0 (M+H). HPLC ret. time = 5.50 min; purity >95%.
1-(1-(4-Chlorobenzyl)-1H-indole-2-carbonyl)-N-(2-(pyridin-3-yl)ethyl)piperidine-4-carboxamide (27h)
Synthesized from 7 and 2-(pyridin-3-yl)ethanamine-HCl as described for 27a. The reaction was diluted with water and ethyl acetate. The organic layer was washed with saturated sodium bicarbonate, 10% aqeuous citric acid solution, followed by saturated sodium chloride solution. The organic layer was dried over magnesium sulfate, filtered and concentrated. The resulting solid was triturated with ethyl acetate to obtain the product as a white solid. Yield: 20%. 1H NMR (400 MHz, DMSO- d6) δ 8.44 − 8.37 (m, 2H), 7.88 (t, J = 5.7 Hz, 1H), 7.66 − 7.50 (m, 3H), 7.38 − 7.26 (m, 3H), 7.26 − 7.17 (m, 1H), 7.14 − 7.06 (m, 3H), 6.71 (s, 1H), 5.48 (s, 2H), 4.63 − 3.80 (m, 2H), 3.34 − 3.25 (m, 2H), 2.89 (bs, 2H), 2.73 (t, J = 7.0 Hz, 2H), 2.38 − 2.27 (m, 1H), 1.74 − 1.21 (m, 4H). TOF ES+ MS: 501.0 (M+H), 523.1 (M+Na). HPLC ret. time = 5.46 min; purity >95%.
1-(1-(4-Chlorobenzyl)-1H-indole-2-carbonyl)-N-(2-(pyridin-2-yl)ethyl)piperidine-4-carboxamide (27i)
Synthesized from 7 and 2-(pyridin-2-yl)ethanamine as described for 27a. The crude material was triturated with ether and ethyl acetate and filtered to obtain the product as a white solid. Yield: 10%. 1H NMR (400 MHz, DMSO- d6) δ 8.52 − 8.45 (m, 1H), 7.87 (t, J = 5.6 Hz, 1H), 7.72 − 7.66 (m, 1H), 7.63 (d, J = 7.9 Hz, 1H), 7.54 (d, J = 8.3 Hz, 1H), 7.38 − 7.30 (m, 2H), 7.25 − 7.17 (m, 3H), 7.14 − 7.05 (m, 3H), 6.72 (s, 1H), 5.49 (s, 2H), 4.52 − 3.78 (m, 2H), 3.40 (q, J = 6.9 Hz, 2H), 3.08 − 2.76 (m, 4H), 2.38 − 2.29 (m, 1H), 1.74 − 1.23 (m, 4H). TOF ES+ MS: 501.1 (M+H), 523.1 (M+Na). HPLC ret. time = 5.51 min; purity >95%.
1-(1-(4-Chlorobenzyl)-1H-indole-2-carbonyl)-N-(3-(pyridin-4-yl)propyl)piperidine-4-carboxamide (27j)
Synthesized from 7 and 3-(pyridin-4-yl)propan-1-amine as described for 27a. The crude solid was triturated with ethyl acetate, giving a white solid. Yield: 40 mg, 31%. 1H NMR (400 MHz, DMSO- d6) δ 8.48 − 8.41 (m, 2H), 7.85 (t, J = 5.6 Hz, 1H), 7.62 (d, J = 7.9 Hz, 1H), 7.54 (d, J = 8.4 Hz, 1H), 7.37 − 7.30 (m, 2H), 7.26 − 7.17 (m, 3H), 7.14 − 7.05 (m, 3H), 6.72 (s, 1H), 5.49 (s, 2H), 4.41 (bs, 2H), 4.14 − 3.99 (m, 4H), 3.10 − 2.75 (m, 4H), 2.62 − 2.50 (m, 2H), 2.38 − 2.30 (m, 1H), 1.77 − 1.64 (m, 4H), 1.49 − 1.25 (m, 2H). TOF ES+ MS: 514.9 (M+H). HPLC ret. time = 5.58 min; purity > 95%
1-(1-(4-Chlorobenzyl)-1H-indole-2-carbonyl)-N-(2-(pyridin-4-yl)propyl)piperidine-4-carboxamide (27k)
Synthesized from 7 and 2-(pyridin-4-yl)propan-1-amine as described for 27a. The crude material was triturated in ethyl acetate to obtain product. Yield: 40%. 1H NMR (500 MHz, CDCl3) δ 8.58 − 8.52 (m, 2H), 7.68 − 7.61 (m, 1H), 7.37 − 7.31 (m, 1H), 7.31 − 7.10 (m, 5H), 7.06 − 6.99 (m, 2H), 6.62 (s, 1H), 5.46 (s, 2H), 5.31 (t, J = 5.1 Hz, 1H), 4.68 − 3.98 (m, 3H), 3.64 − 3.54 (m, 1H), 3.37 − 3.26 (m, 1H), 3.04 − 2.96 (m, 1H), 2.88 − 2.81 (m, 1H), 2.22 − 2.13 (m, 1H), 1.85 − 1.20 (m, 6H). TOF ES+ MS: 515.1 (M+H). HPLC ret. time = 5.80 min; purity = 85%
1-(1-(4-Chlorobenzyl)-1H-indole-2-carbonyl)-N-(2-methyl-2-(pyridin-4-yl)propyl)piperidine-4-carboxamide (27l)
Synthesized from 7 and 2-methyl-2-(pyridin-4-yl)propan-1-amine as described for 27a. The crude material was triturated in ethyl acetate. The white solid was filtered to obtain product. Yield: 23%. 1H NMR (500 MHz, DMSO- d6) δ 8.49 − 8.45 (m, 2H), 7.69 − 7.59 (m, 2H), 7.53 (d, J = 8.5 Hz, 1H), 7.36 − 7.31 (m, 4H), 7.25 − 7.17 (m, 1H), 7.14 − 7.06 (m, 3H), 6.71 (s, 1H), 5.48 (s, 2H), 4.55 − 3.79 (m, 2H), 3.28 (d, J = 6.2 Hz, 2H), 2.88 (bs, 2H), 2.42 − 2.34 (m, 1H), 1.67 − 1.19 (m, 10H). TOF ES+ MS: 529.3 (M+H). HPLC ret. time = 6.01 min; purity > 95%.
1-(1-(4-Chlorobenzyl)-1H-indole-2-carbonyl)-N-methyl-N-(2-(pyridin-4-yl)ethyl)piperidine-4-carboxamide (27m)
Synthesized from 7 and N-methyl-2-(pyridin-4-yl)ethanamine as described for 27a. The resulting crude material was purified using SP1 Biotage chromatography with a 25 g silica cartridge and a gradient of 20% ethyl acetate/hexanes to 100% ethyl acetate. The product was obtained as a white solid. Yield: 32%. 1H NMR (400 MHz, DMSO- d6) 1.3:1 mixture of rotamers δ 8.49 − 8.45 (m, 2H), 7.64 − 7.61 (m, 1H), 7.54 (t, J = 10Hz, 1H), 7.35 − 7.30 (m, 3H), 7.24-7.19 (m, 2H), 7.12 − 7.08 (m, 3H), 6.72 (rotamer A, s), 6.70 (rotamer B, s, 1H), 4.50 − 4.75 (m, 2H), 4.41 (bs, 1H), 3.97 (bs, 1H), 3.63 (t, J = 6.8 Hz, 1H), 3.53 (t, J = 7.3 Hz, 1H), 3.09 - 2.69 (m, 8H), 2.84- 2.50 (m, 1H), 1.17 − 1.10 (m, 4H). TOF ES+ MS: 515.3 (M+H). HPLC ret. time = 5.84 min; purity > 95%.
1-(1-(4-Chlorobenzyl)-1H-indole-2-carbonyl)-N-((2,3-dihydrofuro[2,3-c]pyridin-3-yl)methyl)piperidine-4-carboxamide (27n)
Synthesized from 7 and (2,3-dihydrofuro[2,3-c]pyridin-3-yl)methanamine as described for 27a. The crude material was triturated in ethyl acetate, filtered and concentrated to obtain a white solid. Yield: 50%. 1H NMR (500 MHz, DMSO- d6) δ 8.14 − 8.06 (m, 3H), 7.63 (d, J = 7.9 Hz, 1H), 7.55 (d, J = 8.3 Hz, 1H), 7.37 − 7.28 (m, 4H), 7.25 − 7.18 (m, 1H), 7.14 − 7.06 (m, 2H), 6.72 (s, 1H), 5.49 (s, 2H), 4.63 − 4.55 (m, 1H), 4.39 − 4.32 (m, 2H), 4.14 − 3.87 (m, 2H), 3.73 − 3.63 (m, 1H), 3.17 (d, J = 5.3 Hz, 1H), 2.86 − 2.82 (m, 2H), 2.40 − 2.34 (m, 1H), 1.76 − 1.15 (m, 4H). TOF ES+ MS: 529.1 (M+H). HPLC ret. time = 5.78 min; purity > 95%.
N-(2-(1H-Imidazol-1-yl)ethyl)-1-(1-(4-chlorobenzyl)-1H-indole-2-carbonyl)piperidine-4-carboxamide (27o)
Synthesized from 7 and 2-(1H-imidazol-1-yl)ethanamine-HCl as described for 27a. The resulting crude solid was triturated with ethyl acetate to give a white solid. Yield: 18%. 1H NMR (500 MHz, DMSO- d6) δ 7.94 (t, J = 5.6 Hz, 1H), 7.63 (d, J = 7.9 Hz, 1H), 7.57 − 7.51 (m, 2H), 7.37 − 7.31 (m, 2H), 7.25 − 7.18 (m, 1H), 7.13 − 7.06 (m, 4H), 6.87 (s, 1H), 6.71 (s, 1H), 5.49 (s, 2H), 4.39 (s, 1H), 4.07 − 3.99 (m, 3H), 3.39 − 3.31 (m, 2H), 2.93 (bs, 2H), 2.39 − 2.29 (m, 1H), 1.79 − 1.20 (m, 4H). TOF ES+ MS: 490.1 (M+H). HPLC ret. time = 5.69 min; purity = 90%.
1-(1-(4-Chlorobenzyl)-1H-indole-2-carbonyl)-N-(3-methoxyphenethyl)piperidine-4-carboxamide (27p)
Synthesized from 7 and 2-(3-methoxyphenyl)ethanamine as described for 27a. The crude material was triturated with ethyl acetate to obtain product as a white solid. Yield: 67%. 1H NMR (400 MHz, DMSO- d6) δ 7.85 (t, J = 5.6 Hz, 1H), 7.63 (d, J = 7.8 Hz, 1H), 7.54 (d, J = 8.4 Hz, 1H), 7.37 − 7.30 (m, 2H), 7.26 − 7.14 (m, 2H), 7.14 − 7.06 (m, 3H), 6.78 − 6.73 (m, 3H), 6.71 (s, 1H), 5.49 (s, 2H), 4.51 − 3.86 (m, 2H), 3.72 (s, 3H), 3.26 (q, J = 6.7 Hz, 2H), 2.94 (s, 2H), 2.67 (t, J = 7.2 Hz, 2H), 2.37 − 2.30 (m, 1H), 1.62 (s, 2H), 1.37 (s, 2H). TOF ES+ MS: 530.1 (M+H), 552.1 (M+Na). HPLC ret. time = 7.86 min; purity > 95%.
1-(1-(4-Chlorobenzyl)-1H-indole-2-carbonyl)-N-(4-fluorophenethyl)piperidine-4-carboxamide (27q)
Synthesized from 7 and 2-(4-fluorophenyl)ethanamine as described for 27a. The crude material was triturated with ethyl acetate to obtain product as a white solid. Yield: 85%. 1H NMR (400 MHz, DMSO- d6) δ 7.84 (t, J = 5.7 Hz, 1H), 7.62 (d, J = 7.9 Hz, 1H), 7.54 (d, J = 8.4 Hz, 1H), 7.37 − 7.30 (m, 2H), 7.26 − 7.17 (m, 3H), 7.14 − 7.05 (m, 5H), 6.71 (s, 1H), 5.49 (s, 2H), 4.52 − 3.78 (m, 2H), 3.25 (q, J = 6.9 Hz, 2H), 2.91 (bs, 2H), 2.69 (t, J = 7.2 Hz, 2H), 2.38 − 2.28 (m, 1H), 1.62 (bs, 2H), 1.36 (bs, 2H). TOF ES+ MS: 518.1 (M+H), 540.1 (M+Na). HPLC ret. time = 7.91 min; purity > 95%.
1-(1-(4-Chlorobenzyl)-1H-indole-2-carbonyl)-N-(4-methoxyphenethyl)piperidine-4-carboxamide (27r)
Synthesized from 7 and 2-(4-methoxyphenyl)ethanamine as described for 27a. The crude material was triturated with ethyl acetate to obtain product as a white solid. Yield: 63%. 1H NMR (400 MHz, DMSO- d6) δ 7.83 (t, J = 5.6 Hz, 1H), 7.63 (d, J = 7.8 Hz, 1H), 7.54 (d, J = 8.4 Hz, 1H), 7.37 − 7.30 (m, 2H), 7.26 − 7.17 (m, 1H), 7.14 − 7.05 (m, 6H), 6.88 − 6.81 (m, 2H), 6.71 (s, 1H), 5.49 (s, 2H), 4.49 − 3.84 (m, 2H), 3.71 (s, 3H), 3.22 (q, J = 6.8 Hz, 2H), 2.91 (bs, 2H), 2.63 (t, J = 7.3 Hz, 2H), 2.39 − 2.28 (m, 1H), 1.62 (bs, 2H), 1.37 (bs, 2H). TOF ES+ MS: 530.1 (M+H), 552.1 (M+Na). HPLC ret. time = 7.81 min; purity > 95%.
1-(1-(4-Chlorobenzyl)-1H-indole-2-carbonyl)-N-(4-chlorophenethyl)piperidine-4-carboxamide (27s)
Synthesized from 7 and 2-(4-chlorophenyl)ethanamine as described for 27a. The crude material was triturated in ethyl acetate to give the product as a white solid. Yield: 45%. 1H NMR (400 MHz, DMSO- d6) δ 7.85 (t, J = 5.7 Hz, 1H), 7.63 (d, J = 7.8 Hz, 1H), 7.54 (d, J = 8.4 Hz, 1H), 7.37 − 7.30 (m, 4H), 7.26 − 7.17 (m, 3H), 7.14 − 7.06 (m, 3H), 6.71 (s, 1H), 5.49 (s, 2H), 4.54 − 3.86 (m, 2H), 3.26 (q, J = 6.7 Hz, 2H), 2.90 (bs, 2H), 2.69 (t, J = 7.1 Hz, 2H), 2.36 − 2.30 (m, 1H), 1.76 − 1.22 (m, 4H). TOF ES+ MS: 534.1 (M+H), 556.1 (M+Na). HPLC ret. time = 8.25 min; purity > 95%.
1-(1-(4-Chlorobenzyl)-1H-indole-2-carbonyl)-N-(4-isopropylphenethyl)piperidine-4-carboxamide (27t)
Synthesized from 7 and 2-(4-isopropylphenyl)ethanamine as described for 27a. The crude material purified using medium pressure silica gel chromatography with a gradient of 0-60% ethyl acetate/hexanes to obtain product as a white solid. Yield: 37%. 1H NMR (500 MHz, DMSO- d6) δ 7.89 (t, J = 5.7 Hz, 1H), 7.62 (d, J = 7.9 Hz, 1H), 7.55 (d, J = 8.3 Hz, 1H), 7.37 − 7.31 (m, 2H), 7.26 − 7.18 (m, 1H), 7.17 − 7.07 (m, 7H), 6.71 (s, 1H), 5.49 (s, 2H), 4.52 − 3.83 (m, 2H), 3.24 (q, J = 6.9 Hz, 2H), 3.10 − 2.75 (m, 3H), 2.66 (t, J = 7.4 Hz, 2H), 2.39 − 2.31 (m, 1H), 1.85 − 1.10 (m, 10H). TOF ES+ MS: 542.2 (M+H), 564.1 (M+Na). HPLC ret. time = 8.71 min; purity > 95%.
(S)-(1-(4-Chlorobenzyl)-1H-indol-2-yl)(4-(2-(hydroxymethyl)pyrrolidine-1-carbonyl)piperidin-1-yl)methanone (27u)
Synthesized from 7 and L-prolinol as described for 27a. The crude material was purified using flash chromatography with a gradient of 100% DCM to 5% 7M ammonia in methanol diluted with 95% DCM. The product was obtained as a white solid. Yield: 98%. 1H NMR (400 MHz, DMSO- d6) δ 7.62 (d, J = 8.1 Hz, 1H), 7.58 − 7.50 (m, 1H), 7.39 − 7.30 (m, 2H), 7.26 − 7.17 (m, 1H), 7.17 − 7.05 (m, 3H), 6.73 (s, 1H), 5.50 (s, 2H), 4.60 − 3.87 (m, 3H), 3.53 − 3.43 (m, 2H), 3.29 − 3.16 (m, 1H), 3.14 − 2.77 (m, 2H), 2.72 − 2.64 (m, 1H), 1.98 − 1.19 (m, 8H). TOF ES+ MS: 480.1 (M+H). HPLC ret. time = 6.82 min; purity = 94%.
1-(1-(4-Chlorobenzyl)-1H-indole-2-carbonyl)-N,N-dimethylpiperidine-4-carboxamide (27v)
Synthesized from 7 and dimethylamine as described for 27a. Purified by flash chromatography with a gradient of 100% DCM to 5% 7M ammonia in methanol diluted with 95% DCM. The product was obtained as a white solid. Yield: 68%. 1H NMR (400 MHz, DMSO- d6) δ 7.63 (d, J = 7.8 Hz, 1H), 7.54 (d, J = 8.4 Hz, 1H), 7.38 − 7.27 (m, 2H), 7.26 − 7.17 (m, 1H), 7.13 − 7.08 (m, 3H), 6.73 (s, 1H), 5.50 (s, 2H), 4.44 (bs, 1H), 3.99 (bs, 1H), 3.19 − 2.84 (m, 6H), 2.81 (s, 3H), 1.62 (bs, 2H), 1.36 (bs, 2H). TOF ES+ MS: 424.0 (M+H). HPLC ret. time = 7.09 min; purity > 95%.
(1-(4-Chlorobenzyl)-1H-indol-2-yl)(4-(pyrrolidine-1-carbonyl)piperidin-1-yl)methanone (27w)
Synthesized from 7 and pyrrolidine as described for 27a. Purified by flash chromatography with a gradient of 100% DCM to 5% 7M ammonia in methanol diluted with 95% DCM. The product was obtained as a white solid. Yield: 73%. 1H NMR (400 MHz, DMSO- d6) δ 7.63 (d, J = 7.8 Hz, 1H), 7.54 (d, J = 8.1 Hz, 1H), 7.38 − 7.30 (m, 2H), 7.26 − 7.17 (m, 1H), 7.16 − 7.05 (m, 3H), 6.73 (s, 1H), 5.50 (s, 2H), 4.63 − 3.86 (m, 2H), 3.47 (t, J = 6.7 Hz, 2H), 3.27 (t, J = 6.9 Hz, 2H), 3.13 − 2.80 (m, 2H), 2.77 − 2.64 (m, 1H), 1.87 (p, J = 6.7 Hz, 2H), 1.81 − 1.27 (m, 6H). TOF ES+ MS: 450.0 (M+H). HPLC ret. time = 7.35 min; purity > 95%.
(1-(4-Chlorobenzyl)-1H-indol-2-yl)(4-(morpholine-4-carbonyl)piperidin-1-yl)methanone (27x)
Synthesized from 7 and morpholine as described for 27a. Purified by flash chromatography with a gradient of 100% DCM to 5% 7M ammonia in methanol diluted with 95% DCM. The product was obtained as a white solid. Yield: 73%. 1H NMR (400 MHz, DMSO- d6) δ 7.63 (d, J = 7.8 Hz, 1H), 7.54 (d, J = 8.4 Hz, 1H), 7.37 − 7.29 (m, 2H), 7.26 − 7.17 (m, 1H), 7.14 − 7.05 (m, 3H), 6.73 (s, 1H), 5.49 (s, 2H), 4.43 (bs, 1H), 4.01 (bs, 1H), 3.58 − 3.38 (m, 8H), 3.11 − 2.85 (m, 3H), 1.77 − 1.20 (m, 4H). TOF ES+ MS: 466.1 (M+H). HPLC ret. time = 7.00 min; purity > 95%.
N-Benzyl-1-(1-(4-chlorobenzyl)-1H-indole-2-carbonyl)-N-methylpiperidine-4-carboxamide (28a)
Compound 77 (150 mg, 0.378 mmol), HOBt (102 mg, 0.756 mmol), and EDCI (145 mg, 0.756 mmol) were dissolved in 1.5 mL of DCM. The reaction was allowed to stir for ten minutes before N-methyl-1-phenylmethanamine (45.8 mg, 0.378 mmol) and DIEA (0.132 ml, 0.756 mmol) were added. The reaction was stirred for two days before it was diluted with water and ethyl acetate. The layers were separated and the organic layer was washed with the following saturated aqueous solutions: citric acid, sodium bicarbonate, and sodium chloride. The organic layer was dried over magnesium sulfate, filtered and concentrated. The crude mixture was purified using 50% Ethyl Acetate/Hexanes. The product was obtained as a white solid. Yield: 64 mg (34%). 1H NMR (400 MHz, DMSO- d6) 1.5:1 mixture of rotamers δ 7.67 − 7.59 (m, 1H), 7.58 − 7.50 (m, 1H), 7.41 − 7.17 (m, 8H), 7.14 − 7.07 (m, 3H), 6.75 (rotamer A, s), 6.71 (rotamer B, s, 1H), 5.53 − 5.47 (m, 2H), 4.65 (rotamer A, s, 1H), 4.53 − 4.33 (m, 2H), 4.01 (bs, 1H), 3.20 − 2.89 (m, 5H), 2.78 (rotamer B, s, 1H), 1.52 (bm, 4H). TOF ES+ MS: 500 (M+H). HPLC ret. time = 8.23 min; purity = 94%
N-Benzyl-N-methyl-1-(1-(4-nitrobenzyl)-1H-indole-2-carbonyl)piperidine-4-carboxamide (28b)
Compound 10 (50 mg, 0.133 mmol) was added to a suspension of sodium hydride (60 wt% in oil, 8.0 mg, 0.20 mmol) in DMF (2 ml). The reaction was allowed to stir for 10 minutes at 60 °C. 1-(chloromethyl)-4-nitrobenzene (45.7 mg, 0.266 mmol) and potassium iodide (44.2 mg, 0.266 mmol) were added and the reaction was stirred overnight at 60 °C. The reaction was diluted with water and ethyl acetate. The organic layer was washed with water and saturated sodium chloride solution. It was dried over magnesium sulfate, filtered and concentrated. The crude material was purified using 40-100% ethyl acetate: hexanes to afford the product as a white solid. Yield: 11 mg, 16%. 1H NMR (400 MHz, DMSO- d6) 1.7:1 mixture of rotamers δ 8.20 − 8.11 (m, 2H), 7.70 − 7.62 (m, 1H), 7.55 (d, J = 8.4 Hz, 1H), 7.40 − 7.10 (m, 9H), 6.80 (rotamer A, s), 6.77 (rotamer B, s, 1H), 5.70 − 5.63 (m, 2H), 4.62 (rotamer A, s, 1H), 4.49 − 4.25 (m, 2H), 4.08 (bs, 1H), 3.18 − 2.87 (m, 5H), 2.74 (rotamer B, s, 1H), 1.39 (bm, 4H). TOF ES+ MS: 511.1 (M+H). HPLC ret. time = 7.56 min; purity > 95%.
N-Benzyl-1-(1-(4-cyanobenzyl)-1H-indole-2-carbonyl)-N-methylpiperidine-4-carboxamide (28c)
Sodium iodide (99 mg, 0.660 mmol) and 4-(chloromethyl)benzonitrile (100 mg, 0.660 mmol) were dissolved in acetone (2.2 mL) and allowed to stir at room temperature for 4 hours. The resulting solid was filtered and the filtrate concentrated to obtain crude 4-(iodomethyl)benzonitrile (130 mg, 0.535 mmol, 81% yield) as an orange oil. It was taken directly to the next step without characterization and purification. Compound 10 (100 mg, 0.266 mmol) was dissolved in 1.0 mL of DMF and added to a suspension of sodium hydride (12.78 mg, 0.320 mmol) in 2.0 of DMF. The reaction was stirred at 80 °C for 10 minutes before adding 4-(iodomethyl)benzonitrile (129 mg, 0.533 mmol) dissolved in 2.0 mL of DMF. The reaction was allowed to stir overnight. After 18 hours, the reaction was cooled to room temperature before diluting with water and ethyl acetate. The organic phase was washed with water followed by saturated sodium chloride, dried over magnesium sulfate, filtered and concentrated. The crude material was purified via flash column chromatography using a gradient of 50% ethyl acetate/hexanes to 100% ethyl acetate. The purification provided product as a white solid. Yield: 50 mg, 38%. 1H NMR (400 MHz, DMSO- d6) 1.7:1 mixture of rotamers δ 7.78 − 7.70 (m, 2H), 7.69 − 7.61 (m, 1H), 7.52 (d, J = 8.4 Hz, 1H), 7.43 − 7.07 (m, 9H), 6.78 (rotamer A, s,), 6.75 (rotamer B, s, 1H), 5.64 − 5.57 (m, 2H), 4.64 (rotamer A, s, 1H), 4.52 − 4.25 (m, 2H), 4.04 (bs, 1H), 3.18 - 2.84 (m, 5H), 2.78 (rotamer B, s, 1H), 1.78 − 1.17 (m, 4H). TOF ES+ MS: 491.1 (M+H). HPLC ret. time = 7.54 min; purity = 94%.
N-Benzyl-1-(1-(4-methoxybenzyl)-1H-indole-2-carbonyl)-N-methylpiperidine-4-carboxamide (28d)
Compound 10 (100 mg, 0.266 mmol) was added to a suspension of sodium hydride (12.8 mg, 0.32 mmol) in THF (1.5 mL). The reaction was stirred for 10 minutes before 1-(bromomethyl)-4-methoxybenzene (0.039 mL, 0.27 mmol) was added. The reaction was allowed to stir at room temperature overnight. The reaction was diluted with water and extracted with ethyl acetate. The organic layer was washed with water and saturated sodium chloride solution. It was dried over magnesium sulfate, filtered and concentrated. The crude material was purified by flash chromatography using a gradient of 50% - 100% ethyl acetate in hexanes to obtain the product as a white solid. Yield: 42 mg (32%). 1H NMR (400 MHz, DMSO- d6) 1.7:1 mixture of rotamers δ 7.66 − 7.57 (m, 2H), 7.40 − 7.04 (m, 8H), 6.85-6.81(m, 3H), 6.69 (rotamer A, s), 6.65 (rotamer B, s, 1H), 5.46 − 5.39 (m, 2H), 4.64 (rotamer A, s, 1H), 4.53-4.20 (m, 2H), 3.97 (bs, 1H), 3.69-3.66(m, 3H), 3.19 − 2.83 (m, 5H), 2.78 (rotamer B, s, 1H), 1.82 − 1.24 (m, 4H). TOF ES+ MS: 496.1 (M+H), 518.1 (M+Na). HPLC ret. time = 7.83 min; purity = 92%.
1-(1-Acetyl-1H-indole-2-carbonyl)-N-benzyl-N-methylpiperidine-4-carboxamide (28e)
A solution of compound 10 (100 mg, 0.27 mmol) in 1 mL of DMF was added to a suspension of sodium hydride (11.7 mg, 0.29 mmol) in 2.0 mL DMF. The reaction was stirred at 80 °C for 10 minutes before adding acetic anhydride (0.050 ml, 0.53 mmol). The reaction was allowed to stir for 5 hours before it was cooled to room temperature and diluted with ammonium chloride and ethyl acetate. The organic phase was washed with saturated ammonium chloride twice, followed by saturated sodium chloride, dried over magnesium sulfate, filtered and concentrated. The crude material was purified via flash chromatography using a gradient of 50% ethyl acetate/hexanes to 100% ethyl acetate. Purification afforded the product as a white. Yield: 13 mg (12%). 1H NMR (400 MHz, DMSO- d6) 1.6:1 mixture of rotamers δ 8.15 − 8.07 (m, 1H), 7.69 − 7.61 (m, 1H), 7.44 − 7.15 (m, 6H), 6.91 (rotamer A, s,), 6.88 (rotamer B, s, 1H), 4.67 (rotamer A, s, 1H), 4.56 − 4.33 (m, 2H), 4.00 − 3.84 (m, 1H), 3.21 − 2.85 (m, 5H), 2.79 (s, 1H), 2.61 (rotamer A, s, 2H), 2.59 (rotamer B, s, 1H), 1.91 − 1.48 (m, 4H). TOF ES+ MS: 418.1 (M+H), 440.1 (M+Na). HPLC ret. time = 6.52 min; purity > 95%.
N-Benzyl-1-(1-(2-methoxyethyl)-1H-indole-2-carbonyl)-N-methylpiperidine-4-carboxamide (28f)
Compound 10 (50 mg, 0.13 mmol) dissolved in 0.5 mL of DMF was added to a suspension of sodium hydride (7.99 mg, 0.200 mmol) in 1.0 mL of DMF. The reaction was heated at 70 °C for ten minutes before adding 2-methoxyethyl 4-methylbenzenesulfonate (0.144 mL, 0.266 mmol). The reaction was allowed to stir overnight. After 18 hours, the reaction was cooled to room temperature before diluting with water and ethyl acetate. The organic phase was washed with water followed by saturated sodium chloride, dried over magnesium sulfate, filtered and concentrated. The crude material was purified via flash column chromatography using a gradient of 50-80% ethyl acetate/hexanes. The purification provided the product as a white solid. Yield: 20 mg (35%). 1H NMR (400 MHz, DMSO- d6) 1.7:1 mixture of rotamers δ 7.63 − 7.50 (m, 2H), 7.42 − 7.15 (m, 6H), 7.13 − 7.03 (m, 1H), 6.64 (rotamer A, s), 6.60 (rotamer B, s, 1H), 4.68 (s, 1H), 4.61 − 4.37 (m, 4H), 4.18 (bs, 1H), 3.62 − 3.44 (m, 2H), 3.19 − 3.14 (m, 3H), 3.11 − 2.92 (m, 5H), 2.79 (s, 1H), 1.89 − 1.48 (m, 4H). TOF ES+ MS: 434.2 (M+H), 456.1 (M+Na). HPLC ret. time = 7.03 min; purity > 95%.
N-Benzyl-1-(1-isobutyl-1H-indole-2-carbonyl)-N-methylpiperidine-4-carboxamide (28g)
Compound 10 (100 mg, 0.27 mmol) was dissolved in 1.0 mL of DMF and added to a suspension of sodium hydride (12.8 mg, 0.32 mmol) in 2.0 mL of DMF. The reaction was stirred at 80 °C for 10 minutes before adding 1-iodo-2-methylpropane (0.061 mL, 0.53 mmol). The reaction was allowed to stir overnight. After 18 hours, the reaction was cooled to room temperature before diluting with water and ethyl acetate. The organic phase was washed with water followed by saturated sodium chloride, dried over magnesium sulfate, filtered and concentrated. The crude material was purified via column chromatography using 50% ethyl acetate/hexanes to 100% ethyl acetate. The purification provided the product as a white solid. Yield: 28 mg (25%). 1H NMR (400 MHz, DMSO- d6) δ 7.64 − 7.50 (m, 2H), 7.42 − 7.15 (m, 6H), 7.12 − 7.03 (m, 1H), 6.67 (rotamer A, s), 6.64 (rotamer B, s, 1H), 4.67 (rotamer A, s, 1H), 4.60-4.26 (s, 3H), 4.24-3.98 (m, 2H), 3.00 (s, 5H), 2.79 (s, 1H), 2.06 − 1.92 (m, 1H), 1.78 (bs, 1H), 1.72 − 1.49 (m, 3H), 0.84 − 0.71 (m, 6H). TOF ES+ MS: 432.2 (M+H), 454.2 (M+Na). HPLC ret. time = 7.80 min; purity = 94%.
N-Benzyl-1-(1-benzyl-1H-indole-2-carbonyl)-N-methylpiperidine-4-carboxamide (28h)
Compound 10 (50 mg, 0.133 mmol) was dissolved in 0.5 mL of DMF and cooled to 0 °C. Lithium bis(trimethylsilyl)amide (0.146 ml, 0.146 mmol) was added dropwise and the reaction was allowed to stir for 10 minutes. Benzyl bromide (0.024 ml, 0.20 mmol) was added to the reaction, and it was allowed to stir overnight at room temperature. The reaction was quenched with water. It was then extracted with ethyl acetate. The organic extracts were washed with saturated sodium chloride solution three times and dried over magnesium sulfate, filtered and concentrated. The crude material was purified through flash silica gel chromatography using 25% EtOAc:DCM to give the product as a white solid. Yield: 41 mg (66%). 1H NMR (400 MHz, DMSO- d6) 1.6:1 mixture of rotamers δ 7.68 − 7.52 (m, 2H), 7.45 − 6.98 (m, 12H), 6.72 (rotamer A, s), 6.69 (rotamer B, s, 1H), 5.54-5.41 (m, 2H), 4.64 (s, 1H), 4.52 − 4.23 (m, 2H), 4.01 (bs, 1H), 3.18 − 2.85 (m, 5H), 2.78 (rotamer B, s, 1H), 1.73 − 1.23 (m, 4H). TOF ES+ MS: 466.2 (M+H), 488.2 (M+Na). HPLC ret. time = 7.86 min; purity > 95%.
N-Benzyl-N-methyl-1-(1-(pyridin-4-ylmethyl)-1H-indole-2-carbonyl)piperidine-4-carboxamide (28i)
Pyridin-4-ylmethanol (180 mg, 1.65 mmol) and TEA (0.22 mL, 1.6 mmol) were dissolved in THF (7 mL) and cooled to 0 °C. Methanesulfonyl chloride (0.125 mL, 1.6 mmol) was added dropwise and the reaction was allowed to warm to room temperature and stirred overnight. To a suspension of 60 wt% sodium hydride (21.5 mg, 0.538 mmol) in 3.5 mL of DMF, compound 10 (200 mg, 0.533 mmol) was added as a 1.0 mL DMF solution. The reaction was allowed to stir for 10 minutes before a 0.5 mL DMF solution of mesylated alcohol was added. The reaction was stirred at room temperature overnight. After 15 hours, the reaction was diluted with water and ethyl acetate. The aqueous phase was washed with another aliquot of ethyl acetate. The combined organic phases were washed with saturated sodium chloride, dried over magnesium sulfate, filtered and concentrated. The crude black oil was purified via SP1 Biotage using a gradient of 100% ethyl acetate to 10% methanol: ethyl acetate. The product isolated as a white solid. Yield: 25 mg (10%). 1H NMR (500 MHz, DMSO- d6) 1.8:1 mixture of rotamers δ 8.48 − 8.42 (m, 2H), 7.69 − 7.62 (m, 1H), 7.51 − 7.45 (m, 1H), 7.41 − 7.08 (m, 7H), 7.02 − 6.96 (m, 2H), 6.80 (rotamer A, s), 6.77 (rotamer B, s, 1H), 5.59 − 5.53 (m, 2H), 4.65 (rotamer A, s, 1H), 4.52 − 4.48 (m, 2H), 4.11 (bs, 1H), 3.17 (d, J = 5.3 Hz, 2H), 3.10-2.87 (m, H), 2.77 (rotamer B, s, 1H), 1.86-1.33 (bm, 4H). TOF ES+ MS: 467.0(M+H). HPLC ret. time = 5.52 min; purity > 95%.
N-Benzyl-N-methyl-1-(1-(4-(trifluoromethyl)benzyl)-1H-indole-2-carbonyl)piperidine-4-carboxamide (28j)
Compound 10 (200 mg, 0.533 mmol), Cs2CO3 (347 mg, 1.065 mmol), and 1-(bromomethyl)-4-(trifluoromethyl)benzene (0.124 mL, 0.799 mmol) were dissolved in DMF (5.0 mL) and heated to 70 °C overnight. The reaction was diluted with water and ethyl acetate and ether (1:1). The aqueous layer was back extracted with ethyl acetate. The organic phase was washed with saturated sodium chloride (3x), dried over sodium sulfate, filtered and concentrated. The resulting crude material was purified via SP1 Biotage with a gradient of 30% ethyl acetate/hexanes to 100% ethyl acetate. The product was isolated as a white solid. Yield: 90 mg (32%). 1H NMR (500 MHz, DMSO- d6) 2:1 mixture of rotamers δ 7.68 − 7.61 (m, 3H), 7.55 − 7.49 (m, 1H), 7.41 − 7.07 (m, 9H), 6.79 (rotamer A, s), 6.75 (rotamer B, s, 1H), 5.64 − 5.59 (m, 2H), 4.64 (s, 1H), 4.51 − 4.25 (m, 1H), 4.09 (bs, 1H), 2.96 (s, 5H), 2.77 (s, 1H), 1.86 − 1.22 (m, 4H). TOF ES+ MS: 534.0 (M+H), 556.0 (M+Na). HPLC ret. time = 8.25 min; purity = 92%.
1-(1-(4-Chlorobenzyl)-1H-pyrrole-2-carbonyl)-N-(2-(pyridin-4-yl)ethyl)piperidine-4-carboxamide (29a)
The following were added sequentially to DCM: 13 (600 mg, 1.73 mmol), TEA (0.725 mL, 5.19 mmol), EDCI (365 mg, 1.903 mmol), and HOBt (291 mg, 1.903 mmol). This was allowed to stir at rt for 30 min, at which time pyridylethylamine (0.227 mL, 1.90 mmol) was added. Stirring continued for 18 h. At this time, the DCM and TEA were stripped off, and the residue was taken up in EtOAc and washed with 10% aqueous sodium carbonate (3X). The organic phase was collected, dried over magnesium sulfate, and concentrated in vacuo. The resulting solid/oil mixture was recrystallized from EtOAc to afford the title compound as small, white crystals. Yield: 586 mg (75%). 1H NMR (400 MHz, CDCl3) δ 8.50 (d, J = 5.7 Hz, 2H), 7.21 (d, J = 8.3 Hz, 2H), 7.09 (d, J = 5.6 Hz, 2H), 7.02 (d, J = 8.2 Hz, 2H), 6.78 (s, 1H), 6.30 (dd, J = 3.6, 1.2 Hz, 1H), 6.13 − 6.09 (m, 1H), 5.48 (t, J = 5.4 Hz, 1H), 5.26 (s, 2H), 4.32 (d, J = 12.7 Hz, 2H), 3.53 (q, J = 6.8 Hz, 2H), 2.82 (t, J = 7.0 Hz, 4H), 2.19 (tt, J = 11.2, 3.6 Hz, 1H), 1.68 (d, J = 14.4 Hz, 2H), 1.35 (q, J = 11.0, 9.8 Hz, 2H); Anal. Calcd for C25H27ClN4O2: C, 66.58%; H, 6.04%; N, 12.42%. Found: C, 66.68%; H, 6.32%; N, 12.38%. TOF ES+ MS: (M+H) 451.2, (M+Na) 473.2; HPLC ret. time = 5.03 min, >95% purity.
1-(1-(4-Chlorobenzyl)-1H-imidazole-2-carbonyl)-N-(2-(pyridin-4-yl)ethyl)piperidine-4-carboxamide (29b)
The following were added sequentially to DCM (5 mL): 18b (73 mg, 0.309 mmol), 16 (100 mg, 0.371 mmol), DIEA (162 μL, 0.927 mmol), EDCI (65 mg, 0.340 mmol), and HOBt (52 mg, 0.340 mmol). The solution was allowed to stir at RT for 24 h, after which time the DCM was stripped off in vacuo and the resulting residue was taken up on 10% aq. sodium carbonate. Material was then extracted with ethyl acetate. The extract was dried (magnesium sulfate) and concentrated in vacuo. The residue was then triturated with ethyl acetate/hexanes and collected over a filter. The impure yellow powder was then recrystallized from ethyl acetate to provide the title compound as off-white crystals. Yield: 89 mg (64%). 1H NMR (400 MHz, CDCl3) δ 8.50 (d, J = 5.2 Hz, 2H), 7.28 (d, J = 8.3 Hz, 2H), 7.15 − 7.07 (m, 4H), 7.05 (s, 1H), 6.95 (s, 1H), 5.59 (t, J = 5.7 Hz, 1H), 5.35 (d, J = 8.9 Hz, 2H), 4.58 (d, J = 13.4 Hz, 2H), 3.53 (q, J = 6.7 Hz, 2H), 3.10 (t, J = 13.7 Hz, 1H), 2.87 − 2.71 (m, 3H), 2.29 (tt, J = 11.0, 3.4 Hz, 1H), 2.00 − 1.41 (m, 4H). TOF ES+ MS: (M+H) 452.2, (M+Na) 474.2; HPLC ret. time = 4.11 min, >90% purity.
1-(1-(4-Chlorobenzyl)-1H-benzo[d]imidazole-2-carbonyl)-N-(2-(pyridin-4-yl)ethyl)piperidine-4-carboxamide (29c)
22c (60 mg, 0.141 mmol) and lithium hydroxide, H2O (23.6 mg, 0.564 mmol) were dissolved in 2/1 water/THF (0.75 ml). The reaction was stirred for two hours before it was diluted with water and diethyl ether. The aqueous phase was washed with diethyl ether twice. It was then acidified to pH ∼ 2 and extracted with ethyl acetate. The organic phase was washed with saturated sodium chloride, dried over magnesium sulfate, filtered and concentrated to obtain crude acid as a white solid. No further purification was performed. Yield: 54 mg, 96%. 1H NMR (400 MHz, DMSO- d6) δ 12.31 (s, 1H), 7.74 (d, J = 7.8 Hz, 1H), 7.63 (d, J = 7.7 Hz, 2H), 7.41 − 7.20 (m, 6H), 5.55 (s, 2H), 4.39 − 4.30 (m, 1H), 3.95 − 3.86 (m, 1H), 3.21 − 3.09 (m, 1H), 3.06 − 2.95 (m, 1H), 1.98 − 1.85 (m, 1H), 1.76 − 1.68 (m, 1H), 1.50 − 1.42 (m, 1H), 1.30 − 1.22 (m, 1H). TOF ES+ MS: 398.1 (M+H), 420.1 (M+Na). HPLC ret. time = 6.01 min; purity > 95%.
The crude acid from above (50 mg, 0.13 mmol), EDCI (48.2 mg, 0.251 mmol), and HOBt (34.0 mg, 0.251 mmol) were dissolved in DCM (2.0 mL). The reaction was allowed to stir for 10 minutes before the addition of DIEA (0.044 mL, 0.251 mmol) and 2-(pyridin-4-yl)ethanamine (0.030 mL, 0.251 mmol). The reaction was allowed to stir overnight. The reaction was diluted with water and ethyl acetate. The organic layer was washed with saturated sodium bicarbonate and saturated sodium chloride. The ethyl acetate layer was dried over magnesium sulfate, filtered and concentrated. The crude material was triturated in diethyl ether/ethyl acetate to obtain a white solid. Yield: 19 mg (30%). 1H NMR (500 MHz, DMSO- d6) δ 8.48 − 8.43 (m, 2H), 7.92 (t, J = 5.7 Hz, 1H), 7.74 (d, J = 8.8 Hz, 1H), 7.61 (d, J = 8.1 Hz, 1H), 7.42 − 7.19 (m, 8H), 5.61 − 5.49 (m, 2H), 4.45 (d, J = 13.3 Hz, 1H), 3.98 (d, J = 13.6 Hz, 1H), 3.35 − 3.28 (m, 2H), 3.10 − 3.00 (m, 1H), 2.92 − 2.83 (m, 1H), 2.73 (t, J = 7.0 Hz, 2H), 2.41 − 2.31 (m, 1H), 1.79 − 1.31 (m, 4H). TOF ES+ MS: 502.1 (M+H), 524.1 (M+Na). HPLC ret. time = 5.20 min; purity > 95%.
N1-(4-Chlorophenethyl)-N4-(2-(pyridin-4-yl)ethyl)piperidine-1,4-dicarboxamide (29d)
Ethyl 1-((4-chlorophenethyl)carbamoyl)piperidine-4-carboxylate 20 (807 mg, 2.382 mmol) was dissolved in EtOH (20 mL) and 10% aq. NaOH (15 mL) was added. The resulting mixture was warmed to 50°C and stirred for 2 h. The now homogenous solution was then concentrated in vacuo, a small amount of water was added back, and the solution chilled in an ice bath. The solution was then acidified with concentrated HCl and the resulting precipitate was collected over a filter, washed with 1M HCl and ice-cold water, filter-dried, then further dried under high vacuum to afford 1-((4-chlorophenethyl)carbamoyl)piperidine-4-carboxylic acid as a white powder. Yield: 631 mg (85%). 1H NMR (400 MHz, DMSO-d6) δ 12.21 (s, 1H), 7.30 (d, J = 8.4 Hz, 2H), 7.18 (d, J = 8.4 Hz, 2H), 6.54 (t, J = 5.4 Hz, 1H), 3.80 (d, J = 13.3 Hz, 2H), 3.18 (q, J = 7.0 Hz, 2H), 2.76 − 2.62 (m, 4H), 2.36 (ddt, J = 11.0, 7.6, 3.9 Hz, 1H), 1.75 − 1.64 (m, 2H), 1.40 − 1.25 (m, 2H).
1-((4-Chlorophenethyl)carbamoyl)piperidine-4-carboxylic acid (60 mg, 0.193 mmol), TEA (81 μL, 0.579 mmol), EDCI (41 mg, 0.212 mmol), HOBt (33 mg, 0.212 mmol), and 4-(2-aminoethyl)pyridine (25 μL, 0.212 mmol) were all added sequentially to DCM (5 mL) at RT. This solution was allowed to stir for 16 h at RT, after which time the reaction was diluted with ethyl acetate and washed with water (3X), 10% aq. sodium carbonate (3X), and brine (1X), then dried (magnesium sulfate) and concentrated in vacuo. The resulting residue was then crystallized from ethyl acetate and washed with diethyl ether to provide the title compound as a tan solid. Yield: 46 mg (57%). 1H NMR (400 MHz, CDCl3) δ 8.61 − 8.26 (m, 2H), 7.30 − 7.19 (m, 2H), 7.17 − 7.01 (m, 4H), 5.91 − 5.66 (m, 1H), 4.59 (dd, J = 13.4, 5.6 Hz, 1H), 3.86 (d, J = 13.2 Hz, 2H), 3.53 (q, J = 6.7 Hz, 2H), 3.42 (q, J = 6.8 Hz, 2H), 2.82 (t, J = 6.8 Hz, 2H), 2.79 − 2.61 (m, 4H), 2.17 (t, J = 9.6 Hz, 1H), 1.73 (d, J = 12.9 Hz, 2H), 1.57 (qd, J = 12.9, 12.5, 3.8 Hz, 2H). TOF ES+ MS: (M+H) 415.2, (M+Na) 437.2; HPLC ret. time = 4.70 min, >95% purity.
1-(1-Methyl-1H-indole-2-carbonyl)-N-(2-(pyridin-4-yl)ethyl)piperidine-4-carboxamide (29e)
26e (210 mg, 0.668 mmol) and lithium hydroxide, H2O (280 mg, 6.68 mmol) were dissolved in 2/1 water/THF (2.3 ml). The reaction was stirred for 6 hours before it was diluted with water and diethyl ether. The aqueous layer was acidified using 2N HCl to pH ∼ 2. The suspension was extracted with ethyl acetate. The organic phase was washed with saturated sodium chloride, dried over magnesium sulfate, filtered and concentrated to obtain product as a white solid. No purification was performed on the material. The crude acid was taken directly to the next step. TOF ES+ MS: 287.1(M+H), 309.1(M+Na). HPLC ret. time = 5.60 min; purity = 94%.
1-(1-Methyl-1H-indole-2-carbonyl)piperidine-4-carboxylic acid (100 mg, 0.349 mmol), EDCI (100 mg, 0.524 mmol), and HOBt (70.8 mg, 0.524 mmol) were dissolved in DCM (Volume: 3.0 mL). The reaction was stirred for 10 minutes before 2-(pyridin-4-yl)ethanamine (0.059 mL, 0.524 mmol) and DIEA (0.091 mL, 0.524 mmol). The reaction was stirred overnight at room temperature. The reaction was diluted with water and ethyl acetate. The organic layer was washed with saturated sodium bicarbonate solution, and finally saturated sodium chloride. the organic layer was then dried over magnesium sulfate, filtered and concentrated. The resulting solid was triturated in ethyl acetate to give 29e. Yield: 50 mg (38%). 1H NMR (400 MHz, DMSO- d6) δ 8.49 − 8.42 (m, 2H), 7.94 (t, J = 5.6 Hz, 1H), 7.60 (d, J = 7.8 Hz, 1H), 7.50 (d, J = 8.0 Hz, 1H), 7.29 − 7.19 (m, 3H), 7.09 (t, J = 7.4 Hz, 1H), 6.62 (s, 1H), 4.40 (bs, 1H), 4.02 (bs, 1H), 3.74 (s, 3H), 3.32 − 3.29 (m, 2H), 3.04 (bs, 2H), 2.73 (t, J = 7.0 Hz, 2H), 2.41 − 2.36 (m, 1H), 1.71 − 1.66 (m, 2H), 1.57 − 1.46 (m, 2H). TOF ES+ MS: 391.1 (M+H). HPLC ret. time = 4.68 min; purity > 95%.
1-(1-Benzyl-1H-indole-2-carbonyl)-N-(2-(pyridin-4-yl)ethyl)piperidine-4-carboxamide (29f)
24f (48.5 mg, 0.193 mmol), DMAP (23.56 mg, 0.193 mmol), and EDCI (37.0 mg, 0.193 mmol) were dissolved in THF (3.0 ml). The reaction remained a suspension after 30 minutes. Amine 16 (30 mg, 0.129 mmol) was added followed by DIEA (0.034 ml, 0.193 mmol). The reaction was stirred overnight at room temperature. The reaction was diluted with water and ethyl acetate. The organic phase was washed with water and saturated sodium chloride, then it was dried over sodium sulfate. The resulting crude material was purified using SP1 Biotage using a gradient of 30% ethyl acetate: hexanes to 100% ethyl acetate. The product was isolated as a white solid. Yield: 20 mg (33%). 1H NMR (500 MHz, DMSO- d6) δ 8.48 − 8.43 (m, 2H), 7.88 (t, J = 5.6 Hz, 1H), 7.62 (dt, J = 7.8 Hz, 1H), 7.58 (dd, J = 8.4, 0.9 Hz, 1H), 7.29 − 7.17 (m, 7H), 7.13 − 7.05 (m, 4H), 6.69 (s, 1H), 5.49 (s, 2H), 4.40 (bs, 1H), 3.92 (bs, 1H), 3.30 (m, 2H), 2.89 (bs, 2H), 2.73 (t, J = 6.9 Hz, 2H), 2.35 − 2.25 (m, 1H), 1.74 − 1.21 (m, 4H). TOF ES+ MS: 467.2 (M+H), 489.2 (M+Na). HPLC ret. time = 5.30 min; purity >95%.
1-(1-(2,6-Difluorobenzyl)-1H-indole-2-carbonyl)-N-(2-(pyridin-4-yl)ethyl)piperidine-4-carboxamide (29g)
24g (104 mg, 0.361 mmol), HOBt (56.3 mg, 0.417 mmol), and EDCI (80 mg, 0.417 mmol) were dissolved in THF (Volume: 3.0 ml). The reaction was stirred for 30 minutes. Solid 16 (30 mg, 0.13 mmol) was added followed by DIEA (0.073 ml, 0.417 mmol). The reaction was stirred overnight at room temperature. The reaction was diluted with water and ethyl acetate. The organic phase was washed with water and saturated sodium chloride; it was dried over sodium sulfate. The resulting crude material was purified using SP1 Biotage using a gradient of 30% ethyl acetate: hexanes to 100% ethyl acetate. The product was isolated as a white solid. Yield: 68 mg (49%). 1H NMR (500 MHz, DMSO- d6) δ 8.49 − 8.44 (m, 2H), 7.91 (t, J = 5.6 Hz, 1H), 7.61 − 7.55 (m, 2H), 7.41 − 7.31 (m, 1H), 7.28 − 7.20 (m, 3H), 7.12 − 7.01 (m, 3H), 6.67 (s, 1H), 5.63 (s, 4H), 4.42 (bs, 1H), 3.97 (bs, 1H), 3.37 − 3.29 (m, 3H), 3.03 − 2.69 (m, 4H), 2.37 − 2.27 (m, 1H), 1.73 − 1.31 (m, 4H). TOF ES+ MS: 503.2 (M+H) HPLC ret. time = 5.37 min; purity = 92%.
1-(1-(4-Chlorobenzyl)-6-fluoro-1H-indole-2-carbonyl)-N-(2-(pyridin-4-yl)ethyl)piperidine-4-carboxamide (29h)
22h (140 mg, 0.316 mmol) and lithium hydroxide, H2O (133 mg, 3.16 mmol) were dissolved in 1/1 THF/water (4 ml). The reaction was stirred overnight at room temperature. After 16 hours, the reaction was concentrated until a suspension formed. It was cooled by an ice bath and diluted with minimal amount of water to allow a stir bar to move freely. 2N HCl was added dropwise until pH 2 was reached. The resulting suspension was diluted with ethyl acetate. The aqueous phase was washed with another aliquot of ethyl acetate. The organic phases were combined and washed with saturated sodium chloride, filtered, and concentrated. No further purification was performed. The product was isolated as a white solid. Yield: 106 mg, 81% 1H NMR (400 MHz, DMSO- d6) δ 12.28 (s, 1H), 7.64 (dd, J = 8.8, 5.5 Hz, 1H), 7.51 (dd, J = 10.5, 2.3 Hz, 1H), 7.37 − 7.29 (m, 2H), 7.12 − 7.05 (m, 2H), 7.03 − 6.93 (m, 1H), 6.75 (s, 1H), 5.47 (s, 2H), 4.26 (s, 1H), 3.87 (s, 1H), 3.00 (bs, 2H), 2.48 − 2.43 (m, 1H), 1.95 − 1.21 (m, 4H). TOF ES+ MS: 415.0 (M+H), 437.0 (M+Na). HPLC ret. time = 7.02 min; purity > 95%.
The crude acid from above (106 mg, 0.256 mmol), EDCI (98 mg, 0.511 mmol), and DMAP (62.4 mg, 0.511 mmol) were suspended in DCM (Volume: 5.0 mL). The reaction was stirred for 10 minutes before 2-(pyridin-4-yl)ethanamine (0.061 mL, 0.511 mmol) and DIEA (0.089 mL, 0.511 mmol) were added. The reaction was stirred overnight. The reaction was diluted with water and ethyl acetate. The organic phase was washed with water, saturated sodium carbonate, and saturated sodium chloride. It was dried over sodium sulfate, filtered and concentrated. The crude material was triturated using ethyl acetate. The white solid was filtered to obtain the desired product 29h. Yield: 90 mg (68%). 1H NMR (500 MHz, DMSO- d6) δ 8.48 − 8.43 (m, 2H), 7.89 (t, J = 5.7 Hz, 1H), 7.64 (dd, J = 8.7, 5.5 Hz, 1H), 7.49 (dd, J = 10.4, 2.3 Hz, 1H), 7.38 − 7.32 (m, 2H), 7.24 − 7.18 (m, 2H), 7.13 − 7.08 (m, 2H), 7.01 − 6.93 (m, 1H), 6.74 (s, 1H), 5.47 (s, 2H), 4.37 (bs, 1H), 3.93 (bs, 1H), 3.34 − 3.28 (m, 2H), 3.04 − 2.70 (m, 4H), 2.36 − 2.26 (m, 1H), 1.71 − 1.18 (m, 4H). TOF ES+ MS: 519.0 (M+H). HPLC ret. time = 5.83 min; purity > 95%.
1-(1-(4-Chlorobenzyl)-5-fluoro-1H-indole-2-carbonyl)-N-(2-(pyridin-4-yl)ethyl)piperidine-4-carboxamide (29i)
26i (190 mg, 0.429 mmol) and lithium hydroxide, H2O (180 mg, 4.29 mmol) were dissolved in 2/1 water/THF (4.5 ml). The reaction was stirred overnight at room temperature. After 16 hours, the reaction was concentrated until a suspension formed. It was cooled by an ice bath and diluted with minimal amount of water to allow a stir bar to move freely. 2N HCl was added dropwise until pH 2 was reached. The resulting suspension was diluted with ethyl acetate. The aqueous phase was washed with another aliquot of ethyl acetate. The organic phases were combined and washed with saturated sodium chloride, filtered, and concentrated. No further purification was performed. The product 1-(1-(4-chlorobenzyl)-5-fluoro-1H-indole-2-carbonyl)piperidine-4-carboxylic acid was isolated as a white solid. Yield: 129 mg, 73%. 1H NMR (400 MHz, DMSO- d6) δ 12.26 (s, 1H), 7.59 (dd, J = 9.0, 4.4 Hz, 1H), 7.40 (dd, J = 9.6, 2.6 Hz, 1H), 7.35 − 7.30 (m, 2H), 7.12 − 7.05 (m, 3H), 6.71 (s, 1H), 5.48 (s, 2H), 4.28 (bs, 1H), 3.84 (bs, 1H), 3.00 (bs, 2H), 2.48 − 2.41 (m, 1H), 1.53 (bm, 4H). HPLC ret. time = 5.51 min; purity > 95%.
1-(1-(4-Chlorobenzyl)-5-fluoro-1H-indole-2-carbonyl)piperidine-4-carboxylic acid (106 mg, 0.256 mmol), DMAP (62.4 mg, 0.51 mmol), and EDCI (98 mg, 0.51 mmol) were dissolved in DCM (5.0 mL). The reaction was stirred for 10 minutes before adding 2-(pyridin-4-yl)ethanamine (0.061 mL, 0.51 mmol) and DIEA (0.089 mL, 0.51 mmol). The reaction was stirred overnight at room temperature. It was diluted with water and ethyl acetate. The organic phase was washed with water, saturated sodium carbonate, and saturated sodium chloride. It was filtered and concentrated. The resulting crude material was triturated with ethyl acetate. The resulting white solid was filtered to obtain the desired product. Yield: 80 mg, 60%. 1H NMR (500 MHz, DMSO- d6) δ 8.48 − 8.43 (m, 2H), 7.90 (t, J = 5.7 Hz, 1H), 7.57 (dd, J = 9.1, 4.4 Hz, 1H), 7.40 (dd, J = 9.5, 2.5 Hz, 1H), 7.36 − 7.33 (m, 2H), 7.23 − 7.20 (m, 2H), 7.13 − 7.07 (m, 3H), 6.70 (s, 1H), 5.48 (s, 2H), 4.40 (bs, 1H), 3.90 (bs, 1H), 3.34 − 3.30 (m, 22H), 3.04 − 2.70 (m, 4H), 2.36 − 2.27 (m, 1H), 1.67 − 1.18 (m, 4H). TOF ES+ MS: 519.1 (M+H), 541.1 (M+Na). HPLC ret. time = 5.51 min; purity > 95%.
1-(1-(4-Chlorobenzyl)-1H-pyrrolo[2,3-c]pyridine-2-carbonyl)-N-(2-(pyridin-4-yl)ethyl)piperidine-4-carboxamide (29j)
24j (50 mg, 0.174 mmol) was dissolved into DCM (2 mL) at RT, followed by 16 (59 mg, 0.192 mmol) and TEA (122 μL, 0.872 mmol). Once all material was in solution, EDCI (37 mg, 0.192 mmol) and HOBt (29 mg, 0.192 mmol) were added and the reaction was allowed to stir for 18 h at RT. At this time, the reaction was diluted with a solution of ethyl acetate/diethyl ether (1:1) and washed with water (3X), 10% aq. sodium carbonate (3X), and brine (1X), then dried (magnesium sulfate) and concentrated in vacuo. The resulting residue was then purified by flash chromatography (50 g silica, 5% 7.0 M methanolic ammonia/ 95% ethyl acetate) to give the title compound as white solid. Yield: 53 mg (61%). 1H NMR (400 MHz, CDCl3) δ 8.78 (s, 1H), 8.49 (d, J = 5.4 Hz, 2H), 8.27 (d, J = 5.5 Hz, 1H), 7.51 (d, J = 5.5 Hz, 1H), 7.21 (d, J = 8.2 Hz, 2H), 7.09 (d, J = 5.3 Hz, 2H), 7.05 (d, J = 8.2 Hz, 2H), 6.56 (s, 1H), 5.67 (t, J = 5.6 Hz, 1H), 5.48 (s, 2H), 4.57 (s, 1H), 3.91 (s, 1H), 3.53 (q, J = 6.6 Hz, 2H), 2.96 − 2.69 (m, 4H), 2.21 (tt, J = 11.1, 7.5, 3.7 Hz, 1H), 1.88 − 1.69 (m, 1H), 1.67 − 1.46 (m, 2H), 1.33 − 1.22 (m, 1H). TOF ES+ MS: (M+H) 502.2, (M+Na) 524.2; HPLC ret. time = 4.01 min, >95% purity.
1-(1-(4-Chlorobenzyl)-4-fluoro-1H-pyrrole-2-carbonyl)-N-(2-(pyridin-4-yl)ethyl)piperidine-4-carboxamide (29k)
The following were added sequentially to DCM (3 mL): 18k (50 mg, 0.20 mmol), TEA (0.11 mL, 0.79 mmol), EDCI (45 mg, 0.24 mmol), HOBt (36 mg, 0.24 mmol), then 16 (64 mg, 0.24 mmol). The reaction was allowed to stir at room temperature for 14 h, at which time it was diluted with ethyl acetate, washed with water (3X), 10% aq. sodium carbonate (3X), and brine (1X), dried with magnesium sulfate, and concentrated in vacuo. The residue was the purified by flash chromatography (10 g silica, 1% 7 M methanolic ammonia/ethyl acetate) to provide the desired material as a white powder. Yield: 48 mg (52%). 1H NMR (400 MHz, CDCl3) δ 8.54 (d, J = 4.3 Hz, 2H), 7.25 − 7.20 (m, 2H), 7.14 (d, J = 4.4 Hz, 2H), 7.06 (d, J = 6.9 Hz, 2H), 6.58 − 6.54 (m, 1H), 6.05 (s, 1H), 5.46 − 5.38 (m, 1H), 5.17 (s, 2H), 4.29 (d, J = 6.7 Hz, 2H), 3.55 (q, J = 7.3 Hz, 2H), 2.83 (dt, J = 17.9, 9.0 Hz, 4H), 2.27 − 2.15 (m, 1H), 1.77 − 1.63 (m, 2H), 1.38 (d, J = 13.4 Hz, 2H). TOF ES+ MS: (M+H) 469.2, (M+Na) 491.2; HPLC ret time = 5.18 min, >95% purity.
Kinetic Solubility Assay
The experimental media contained 1 mM L-glutamine, 10 U/ml penicillin, 10 ug/ml streptomycin, 0.1 mM non-essential amino acids, and 110 ug/ml sodium pyruvate, 5% fetal bovine sera in high-glucose DMEM solution. Compounds were dissolved in DMSO to generate 25 mM solutions. 20 μL of each stock solution was serially diluted with DMSO through twelve 50% dilutions. 1 μL of each working solution was added to one well of a clear, round-bottom 96-well plate containing 199 μL of media. This resulted in 0.12 μM to 250 μM testing concentrations. The plate was placed in a Molecular Devices SpectraMax Plus UV/Vis spectrophotometer and shaken for 15 seconds. Measurement of the OD600 resulted in a series of curves. The concentration at which the average (n = 3 for each concentration and test compound) OD600 reading rose above background was noted, and the aqueous solubility is reported as the mean concentration between this point and the previous point.
Parallel Artificial Membrane Permeability Assay (PAMPA)
Compounds were dissolved in DMSO to generate 10 mM compound solution. The experiment was performed using the Double Sink protocol provided by pION, Inc. with the PAMPA explorer system. A cosolvent system with 20% ACN and 5 μL of BBB-1 lipid solution were used for the experiment.
Mouse Liver Microsomal (MLM T1/2) Stability Assay
Half-lives for compounds incubated with Balb-C mouse liver microsomes were determined as previously described.7
In Vitro Antiviral and Cytotoxicity Assays
WEEV replicon, MTT and cultured human BE(2)-C neuronal cell infection assays were performed as previously described.7
Rhodamine 123 Uptake Assay for Prediction of MDR1 Recognition
MDCKII cells overexpressing human MDR1 (MDR1-MDCKII; Netherlands Cancer Institute) were grown to confluence on 12 well plates in DMEM + 10% FBS media. The media was then replaced with fresh DMEM containing 10 μM rhodamine 123 (Sigma) and uptake measured at 1 or 240 min at 37°C. At those times, uptake was stopped by washing 3x with ice-cold PBS and cell fluorescence (excitation 525 nm, emission 560-640 nm) measured using a Glomax Multi Detection System (Promega). Rhodamine 123 uptake was measured in presence of vehicle, a MDR inhibitor tariquidar (5 μM), or different concentrations (1-30 μM) of potential anti-viral agents. In these studies, rhodamine 123 uptake from 1 to 240 min was 11.4+/-0.6 fold higher in the presence of tariquidar than in vehicle-treated cells (n=39). The impact of the anti-viral agents on MDR activity was assessed as a percentage of the tariquidar effect measured on the same day:
where, Cav is the concentration of rhodamine 123 in the presence of anti-viral, Cveh is the concentration in the presence of vehicle and Ctar is the concentration of rhodamine 123 in the presence of tariquidar. All experiments were performed in triplicate on at least three occasions.
In vivo Pharmacokinetic Study
5 week old C57Bl6 mice were injected intraperitoneally with 100 μL of test compound in 2% sterile DMSO and PBS. Blood was collected in EDTA coated tubes by cardiac puncture at 7 time points (30 min, 1hr, 2hr, 4hr, 8hr, 12hr and 24hr) and plasma isolated by centrifugation at 2000xg for 15 minutes. Mice were then perfused with 10mL PBS prior to harvest of the cerebrum, which was flash frozen on dry ice. Samples were analyzed for drug level by LC/MS/MS.
Acknowledgments
This work was supported by an NIH Partnerships for Biodefense Viral Pathogens grant (R01 AI089417, Principal Investigator DJM) and a UM Rackham Merit Scholarship for SJB. The VMCC is grateful for ongoing support from the Ella and Hans Vahlteich Fund and Beverly Vahlteich Delaney. The authors wish to thank Dr. Brian Shay of the UM Biomedical Mass Spec Facility for assistance with the LC/MS/MS analysis of samples from the MLM stability assays. The authors also wish to acknowledge the UM PK Core for LC/MS/MS analyses of the plasma and brain samples from the mouse PK study.
Abbreviations List
- CNS
central nervous system
- CPE
cytopathic effect
- EDC
N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NSV
neuroadapted Sindbis virus
- VEEV
Venezuelan equine encephalitis virus
- WEEV
western equine encephalitis virus
- PAMPA
parallel artificial membrane permeability assay
- MLM
mouse liver microsome
- Pgp
P-glycoprotein
- MDR1
multiple drug resistance protein 1
- rho123
rhodamine123
- FMV
Fort Morgan virus
- ADME
absorption, distribution, metabolism, excretion
References
- 1.Griffin DE. Alphaviruses. In: Knipe DM, H PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SS, editors. Fields Virology. Fourth. Lippincott Williams & Wilkins; Philadelphia: 2001. pp. 917–962. [Google Scholar]
- 2.Gubler DJ. The global emergence/resurgence of arboviral diseases as public health problems. Arch Med Res. 2002;33:330–342. doi: 10.1016/s0188-4409(02)00378-8. [DOI] [PubMed] [Google Scholar]
- 3.Bronze MS, Huycke MM, Machado LJ, Voskuhl GW, Greenfield RA. Viral agents as biological weapons and agents of bioterrorism. Am J Med Sci. 2002;323:316–325. doi: 10.1097/00000441-200206000-00004. [DOI] [PubMed] [Google Scholar]
- 4.Sidwell RW, Smee DF. Viruses of the Bunya- and Togaviridae families: potential as bioterrorism agents and means of control. Antivir Res. 2003;57:101–111. doi: 10.1016/s0166-3542(02)00203-6. [DOI] [PubMed] [Google Scholar]
- 5.NIH/NIAID Category A, B, & C Priority Pathogens. http://www.niaid.nih.gov/topics/BiodefenseRelated/Biodefense/research/Pages/CatA.aspx.
- 6.Peng W, Peltier DC, Larsen MJ, Kirchhoff PD, Larsen SD, Neubig RR, Miller DJ. Identification of thieno[3,2-b]pyrrole derivatives as novel small molecule inhibitors of neurotropic alphaviruses. J Infect Dis. 2009;199:950–957. doi: 10.1086/597275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sindac JA, Yestrepsky BD, Barraza SJ, Bolduc KL, Blakely PK, Keep RF, Irani DN, Miller DJ, Larsen SD. Novel Inhibitors of Neurotropic Alphavirus Replication That Improve Host Survival in a Mouse Model of Acute Viral Encephalitis. J Med Chem. 2012;55:3535–3545. doi: 10.1021/jm300214e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Begley DJ. ABC transporters and the blood-brain barrier. Curr Pharm Des. 2004;10:1295–1312. doi: 10.2174/1381612043384844. [DOI] [PubMed] [Google Scholar]
- 9.Frost JM, Dart MJ, Tietje KR, Garrison TR, Grayson GK, Daza AV, El-Kouhen OF, Yao BB, Hsieh GC, Pai M, Zhu CZ, Chandran P, Meyer MD. Indol-3-ylcycloalkyl Ketones: Effects of N1 Substituted Indole Side Chain Variations on CB2 Cannabinoid Receptor Activity. J Med Chem. 2010;53:295–315. doi: 10.1021/jm901214q. [DOI] [PubMed] [Google Scholar]
- 10.Beyer TA, Chambers RJ, Li M, Morrell AI, Thompson DD. Pyrido [2,3-d]pyrimidine-2,4-diamines as PDE 2 inhibitors. WO 2005/061497 A1. 2005
- 11.Kamal A, Rajender, Reddy DR, Reddy MK, Balakishan G, Shaik TB, Chourasia M, Sastry GN. Remarkable enhancement in the DNA-binding ability of C2-fluoro substituted pyrrolo[2,1-c][1,4]benzodiazepines and their anticancer potential. Bioorg Med Chem. 2009;17:1557–1572. doi: 10.1016/j.bmc.2008.12.068. [DOI] [PubMed] [Google Scholar]
- 12.Leroy J, Porhiel E, Bondon A. Synthesis and characterization of partially β-fluorinated 5,10,15,20-tetraphenylporphyrins and some derivatives. Tetrahedron. 2002;58:6713–6722. [Google Scholar]
- 13.Katritzky AR, Scriven EFV, Majumder S, Akhmedova RG, Vakulenko AV, Akhmedov NG, Murugan R, Abboud KA. Preparation of nitropyridines by nitration of pyridines with nitric acid. Org Biomol Chem. 2005;3:538–541. doi: 10.1039/b413285h. [DOI] [PubMed] [Google Scholar]
- 14.Guandalini L, Martini E, Gualtieri F, Romanelli MN, Varani K. Design, synthesis and preliminary pharmacological evaluation of rigid analogues of the nicotinic agonist 1,1-dimethyl-4-phenylpiperazinium iodide (DMPP) Arkivoc. 2004:286–300. [Google Scholar]
- 15.Kerns EH, Di L. Drug-like Properties: Concepts, Structure Design and Methods. Academic Press; San Diego: 2008. Chapter 7 - Solubility; pp. 56–85. [Google Scholar]
- 16.Bevan CD, Lloyd RS. A high-throughput screening method for the determination of aqueous drug solubility using laser nephelometry in microtiter plates. Anal Chem. 2000;72:1781–1787. doi: 10.1021/ac9912247. [DOI] [PubMed] [Google Scholar]
- 17.Di L, Kerns EH, Fan K, McConnell OJ, Carter GT. High throughput artificial membrane permeability assay for blood-brain barrier. Eur J Med Chem. 2003;38:223–232. doi: 10.1016/s0223-5234(03)00012-6. [DOI] [PubMed] [Google Scholar]
- 18.Avdeef A, Bendels S, Di L, Faller B, Kansy M, Sugano K, Yamauchi Y. PAMPA--critical factors for better predictions of absorption. J Pharm Sci. 2007;96:2893–2909. doi: 10.1002/jps.21068. [DOI] [PubMed] [Google Scholar]
- 19.Ruell JA, Tsinman O, Avdeef A. Acid-base cosolvent method for determining aqueous permeability of amiodarone, itraconazole, tamoxifen, terfenadine and other very insoluble molecules. Chem Pharm Bull. 2004;52:561–565. doi: 10.1248/cpb.52.561. [DOI] [PubMed] [Google Scholar]
- 20.Forster S, Thumser AE, Hood SR, Plant N. Characterization of Rhodamine-123 as a Tracer Dye for Use In In vitro Drug Transport Assays. PLoS One. 2012;3:e33252. doi: 10.1371/journal.pone.0033253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Larsen SD, Wilson MW, Abe A, Shu L, George CH, Kirchhoff P, Showalter HD, Xiang J, Keep RF, Shayman JA. Property-based design of a glucosylceramide synthase inhibitor that reduces glucosylceramide in the brain. J Lipid Res. 2011;53:282–291. doi: 10.1194/jlr.M021261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hitchcock SA. Structural modifications that alter the P-glycoprotein efflux properties of compounds. J Med Chem. 2012;55:4877–4895. doi: 10.1021/jm201136z. [DOI] [PubMed] [Google Scholar]
- 23.Bianchi TI, Aviles G, Sabattini MS. Biological characteristics of an enzootic subtype of western equine encephalomyelitis virus from Argentina. Acta Virol. 1997;41:13–20. [PubMed] [Google Scholar]
- 24.Larsen SD, Sindac JA, Barazza S, Miller DJ. Preparation of pyrrolyl(or imidazolyl) piperidinyl methanone derivatives as arbovirus inhibitors. US 20120252807 A1. 2012 Oct 04; 2012.
- 25.Lee J, Kang SU, Lim JO, Choi HK, Jin MK, Toth A, Pearce LV, Tran R, Wang Y, Szabo T, Blumberg PM. N-[4-(methylsulfonylamino)benzyl]thiourea analogues as vanilloid receptor antagonists: analysis of structure-activity relationships for the “C-Region”. Bioorg Med Chem. 2004;12:371–385. doi: 10.1016/j.bmc.2003.10.047. [DOI] [PubMed] [Google Scholar]