

Abstract
A fluorescence-based, high-resolution imaging approach was used to visualize longitudinally the cellular events unfolding during T cell-mediated tumor destruction. The dynamic interplay of T cells, cancer cells, cancer antigen loss variants, and stromal cells—all color-coded in vivo—was analyzed in established, solid tumors that had developed behind windows implanted on the backs of mice. Events could be followed repeatedly within precisely the same tumor region—before, during and after adoptive T cell therapy—thereby enabling for the first time a longitudinal in vivo evaluation of protracted events, an analysis not possible with terminal imaging of surgically exposed tumors. T cell infiltration, stromal interactions, and vessel destruction, as well as the functional consequences thereof, including the elimination of cancer cells and cancer cell variants were studied. Minimal perivascular T cell infiltrates initiated vascular destruction inside the tumor mass eventually leading to macroscopic central tumor necrosis. Prolonged engagement of T cells with tumor antigen-crosspresenting stromal cells correlated with high IFNγ cytokine release and bystander elimination of antigen-negative cancer cells. The high-resolution, longitudinal, in vivo imaging approach described here will help to further a better mechanistic understanding of tumor eradication by T cells and other anti-cancer therapies.
Keywords: imaging, cancer, tumor microenvironment, tumor immunology, CD8 T cell, stroma
Introduction
Solid tumors consist of cancer cells embedded in a network of non-malignant cells and extracellular matrix, referred to as tumor stroma. Cancer cell-intrinsic alterations, including antigen loss, antigen-processing, and presentation defects, and upregulation of negative co-stimulatory molecules, are major obstacles to anti-cancer immunotherapy.1 In addition, cancer cells secrete chemokines and cytokines to establish paracrine stimulatory loops with stromal cells, which allow antigenic cancer cells to escape immune destruction.2-4 Pre-clinical and clinical data have revealed that such paracrine stimulatory loops between the cancer cell and the microenvironment induce tumorigenic processes including angiogenesis, invasion, and metastasis.5 Although tumor stroma contributes to immune-suppressive networks, which restrict the effectiveness of certain cancer therapies,6-8 stromal cells are non-malignant and generally genetically stable and therefore less likely to escape chemo-, radiation-, and/or immunotherapy,9 thus making tumor stroma an attractive target for new anti-cancer therapeutic strategies.10
Cancer cells in solid tumors sensitize the surrounding stroma to T cell recognition through the constant release of antigens. Stromal cells can pick up cancer antigens, present antigenic epitopes on their surface MHC molecules, and become targets for antigen-specific T cells.11-13 The source of tumor antigen for stromal loading can be apoptotic or necrotic cancer cells, microvesicles or nanometer-sized exosomes secreted by fully viable cancer cells,14-16 or peptides transferred directly via gap junctions.17,18 Previously, we showed that T-cell mediated eradication of solid tumors, including antigen-loss variants (ALV), is successful when stroma cross-presents tumor-antigen11,19. Although we previously demonstrated that sensitized stroma could be lysed by T cells ex vivo, the question of whether and how the recognition of stroma by T cells in vivo promotes the death of cancer cells and/or their variants remained unanswered.
In this study, we set out to capture the kinetics of T cell-mediated tumor destruction and to dissect the dynamic events and cellular interactions between cancer cells, T cells and stromal cells in solid, established tumors in vivo. Technological advances in optical imaging have allowed the observation of some intratumoral immune events, but focused mainly on the visualization of T cell-cancer cell interactions.20-27 Furthermore, surgically exposed tumors were imaged in previous studies that depended on reconstructing a putative sequence of events from observations of serially sacrificed mice. Caution is needed in interpreting these results because of the acute effects of surgery on blood flow, and leukocyte infiltration.28-30 Ideally one would like to “revisit” the exact same site in a tumor at different time points to better understand the interaction of T cells with cancer and stromal cells over hours and tissue morphology responses over days or even weeks. Thus, in this study our objective was to follow in vivo the response of solid tumor tissue to adoptive T cell therapy over short and long time frames in single cell resolution. For this, we established an experimental system combining the implantable tumor window technology31 with a custom-made precision holder and an optimized multi-color reporter system to stably color-code T cells, cancer cells, including cancer antigenic variants, and the non-malignant stromal microenvironment in mice for the longitudinal observation. Here we report the longitudinal sequence of events unfolding in precise locations within solid tumors following T cell transfer and provide novel insights into T-cell mediated tumor destruction.
Results
Cancer cells, tumor stroma, and adoptively transferred CD8+ T cells express distinct fluorescent reporter proteins for in vivo visualization
For our in vivo imaging studies, we used the well-characterized 2C TCR-transgenic mouse model. 2C CD8+ T cells recognize the synthetic SIYRYYGL (SIY) peptide in association with the MHC class I molecule Kb. SIY antigen can be directly presented by cancer cells expressing SIY or cross-presented by antigen-presenting cells (APC) expressing Kb (Fig. 1A). In addition to recognizing the SIY antigen, 2C T cells also recognize Ld, which is an allogeneic MHC class I molecule that presents the peptide p2Ca, LSPFPFDL, derived from the house-keeping gene α-ketoglutarate dehydrogenase.32,33 p2Ca cannot be cross-presented because 2C T cells only recognize it in the context of Ld and not Kb. Thus p2Ca can only be presented by cancer cells directly as p2Ca/Ld (Fig. 1A).

Figure 1. Tumor model for in vivo longitudinal imaging of solid established tumors growing behind skin-fold window chambers. (A) Model of T cell-recognized tumor antigens presented directly and/or indirectly to 2C transgenic CD8+ T cells. 2C T cells recognize the self-peptide p2Ca presented on MHC class I Ld which can only be presented directly. 2C T cells also recognize the synthetic SIYRYYGL (SIY) peptide in context of MHC class I Kb. SIY can be directly presented by Kb-positive cancer cells as well as cross-presented by antigen-presenting cells (APC) expressing Kb. (B) Color-coded T cells, cancer cells and host stromal cells; T cells express EYFP as transgene, cancer cells are transduced to express Cerulean, and host cells, including tumor stromal cells, express DsRed. Numbers indicate excitation/emission wavelengths for fluorescent proteins. (C) Fluorescence intensity of T cells from 2C TCR × EYFP transgenic F1 mice. EYFP expression of CD8+ and Vß8.1 double-positive cells was analyzed by flow cytometry; gray line represents the background fluorescence of T cells from 2C TCR transgenic mice lacking the fluorescent transgene. (D) CD11b+ cells isolated from a spleen of DsRed Rag1−/− mice express high level of DsRed. CD11b+ splenocytes from a litter mate that does not express the transgene DsRed are used as control (gray line); DsRed expression might potentially vary in different types of stromal cells. (E) The MC57 cell line was transduced to express the model tumor antigens SIY-Cerulean or Ld and Cerulean. Pro4L was transduced to express SIY-Cerulean. All transduced tumor lines were FACS-sorted for high expression of Cerulean. The inset numbers indicate the mean fluorescence intensity. Untransduced tumor cells are included for comparison (gray lines). (F) Tumor growth and T cell-mediated eradication of large, established tumors developing behind the dorsal window. A dorsal window and MC57-SIY-Cerulean cancer cells were implanted onto the back skin fold of C57Bl/6 Rag−/− mice as described in Methods and Figure S2. By day 21, large established tumors developed behind the window. After adoptive transfer of activated, antigen-specific 2C T cells tumors regressed and were eliminated (day 32). (G) Fourteen hours after window implantation and MC57-SIY-Cerulean cancer cell transplantation, cancer cells were imaged by confocal microscopy (top picture); to visualize pre-established vasculature, 2.000.000 MW fluorescein-conjugated dextran was injected i.v. into the mouse (lower picture); data are representative of more than 15 displays of different injection areas, n = 3 mice. Z-stack images were recorded and x-y plane, xz and yz projections are shown to the right (z = 220μm). (H) Eighteen days later, the same tumor, now large and established, was imaged. The existence of irregular micro-vasculature typical for established tumors was visualized by dextran-FITC injection; data are representative of at least 2 mice.
To stably color-code cancer cells, T cells and tumor stroma, we used fluorescent reporter proteins that (1) differ significantly in their peak absorption and emission wavelengths, (2) do not affect the functional properties of the cells, (3) are bright and stable, and (4) have spectral profiles that are unaltered by environmental effects such as pH.34,35 We used the fluorescent reporters EYFP (yellow/green) to visualize adoptively transferred transgenic 2C T cells, Cerulean (blue) to image cancer cells, and DsRed (red) to visualize non-malignant host stromal cells (Fig. 1B).36-38 By combining these fluorescent proteins we could readily distinguish each cellular component in the confocal microscopy mode using standard filter sets. Compared with single laser multiphoton excitation, the spectral separation of multiple fluorophores benefited from excitation multiplexing, and the spatial resolution was excellent. As shown in Figure 1C and 2C CD8+ T cells isolated from 2C × EYFP F1 transgenic TCR mice expressed high levels of the EYFP protein. To highlight the tumor microenvironment, we crossed DsRed transgenic mice into the recombination activating gene 1 (Rag1) knockout background, and DsRed Rag1−/− mice (> 8 weeks old) were used as host mice for all our tumor transplantation studies, unless otherwise stated. Host cells, including antigen-presenting cells such as CD11b+ cells isolated from spleens of DsRed Rag1−/− mice showed high expression levels of DsRed (Fig. 1D). For the visualization of cancer cells, a retroviral vector pMFG was constructed encoding the trimeric peptide (SIYRYYGL-AAY)3 fused to the Cerulean-coding sequence. MC57 is a C57BL/6-derived fibrosarcoma and the Pro4L tumor cell line was derived from a UV-induced tumor in a C3H mouse; both cell lines have been extensively used in our previously described studies.39 MC57 and Pro4L were transduced to express the tumor antigen SIY-Cerulean (MC57-SIY-Cerulean, Pro4L-SIY-Cerulean). In addition, MC57 was transduced to co-express Ld and Cerulean (MC57-Ld-Cerulean) (Fig. 1E). In combination with 2C CTL, this tumor model allowed to study 3 different scenarios of T cell mediated tumor and/or stromal cell interactions: (1) MC57-SIY-Cerulean, in which both cancer and stroma are T cell targets, (2) Pro4L-SIY-Cerulean, in which only stromal cells cross-presenting SIY antigen are targeted, and (3) MC57-Ld-Cerulean, in which only cancer cells are recognized by 2C CTL (Fig. S1).
Tumor growth and T cell-mediated eradication of large, established tumors using a modified dorsal window
In order to longitudinally visualize the microscopic events occurring in tumors before, during and after T cell transfer, we needed to generate an optical imaging setting that allows cancer cells to grow naturally and form large tumors. Therefore, we used the modified dorsal window chamber model as previously described.31,40,41 We implanted customized titanium window frames onto the backs of mice as to sandwich an extended double layer of skin. The front epidermal layer of skin was removed in a circular area of approximately 1cm in diameter, leaving the rear skin layer with its dermis and fascia intact (for technical details, see Methods and Fig. S2A). MC57-SIY-Cerulean cancer cells were injected between the fascia and dermis of the rear skin layer and the area was then covered with a circular glass slide and stabilized by a C-ring (Fig. 1F; Fig. S2A). Fourteen hours later, the mouse was placed onto a custom-made adaptor for a motorized microscope stage and the injection sites within the window area were imaged using confocal laser scanning microscopy at constant ambient temperatures. Deposits of round, cerulean-blue cancer cells were visible through the window moments after the placement (Fig. 1G, upper picture). To image the pre-existing vasculature in the window at the injection sites, the mouse was injected intravenously with a solution of high molecular weight fluorescein-labeled dextran. Three-dimensional (3D) reconstruction analysis revealed that cerulean-blue cancer cells were in very close proximity to the preexisting blood vessels of fascia and dermis of the back skin fold (Fig. 1G, lower picture). The cancer cells continued to grow, and within 3 weeks, a large established tumor had formed behind the window (Fig. 1F, left picture). The tumor grew with similar growth kinetics as subcutaneous tumors (Fig. S2B) and displayed the irregular vascularization with abnormal branching patterns typical of established solid tumors (Fig. 1H). In vitro activated 2C T cells were adoptively transferred into the window-tumor-bearing mouse on day 21. The MC57-SIY-Cerulean solid tumor regressed and was eliminated within 12 d (Fig. 1F, right picture). Thus, the modified dorsal window model not only provided an environment for cancer cells to form established tumors unimpeded by the dimensions of the window chamber, but also allowed T cell-mediated destruction with similar kinetics compared with window-less MC57-SIY cancers growing subcutaneously12 (Fig. S2B).
Visualization of tumor stroma
To visualize the formation of tumor stroma during tumor development, windows were implanted onto the backs of DsRed Rag1−/− mice and MC57-SIY-Cerulean cancer cells were placed as described above. Fourteen hours post implantation we imaged the site of cancer cell transplantation through the window. In addition to Cerulean-blue cancer cells, high numbers of rapidly moving “amoeboid,” round, red host cells were present (Fig. 2A; Video 1). These cells, possibly granulocytes and/or monocytes/macrophages, infiltrated the site in response to pro-inflammatory effects of the window and cancer cell implantation. Over the next 4–5 d, the amoeboid host cells diminished and non-migratory fibroblastoid (stellate or spindle-shaped) red host cells accumulated forming a cohesive network (Fig. 2A, corresponding to Video 2). A similar ratio of mobile and sessile cellular stromal elements was present at later time points (day 8) (Video 2), and by day 21 stroma and cancer cells together formed a dense network including vasculature (Fig. S3A). Window implantation into DsRed Rag1−/− mice without the injection of cancer cells (only PBS solution) also caused the rapid infiltration of amoeboid motile host cells; however, no fibroblastoid cells were visible at later time points (Fig. S3B), suggesting that malignant cells recruited and promoted the accumulation of fibroblastoid cells. Similar observations regarding the stromal composition were made with Pro4L-SIY-Cerulean tumors (Fig. S3C).
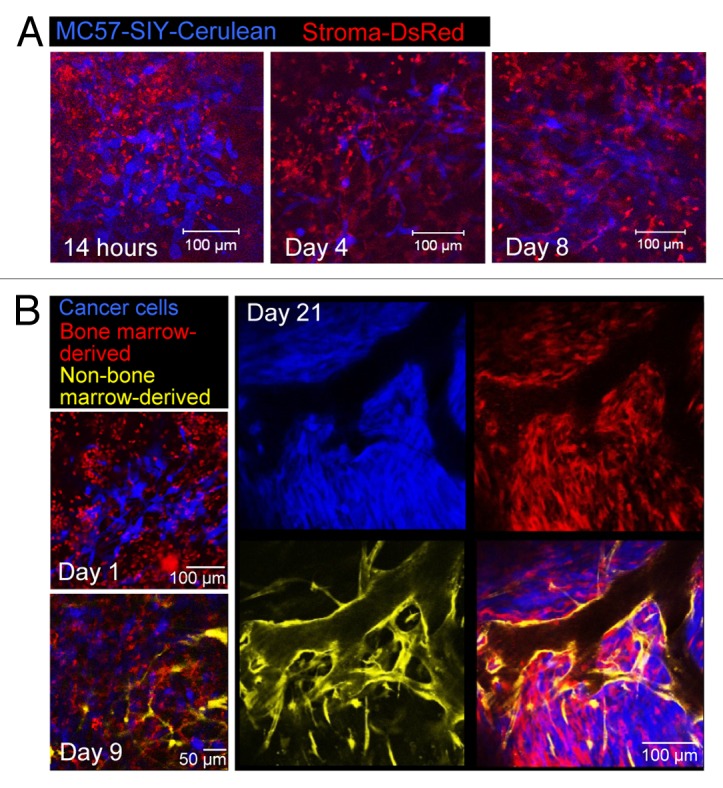
Figure 2. Longitudinal visualization of tumor stroma formation. (A) MC57-SIY-Cerulean cells were injected into a DsRed Rag1−/− mouse. Images were taken 14h, 4d, and 8d post window and cancer cell implantation. While 14h after window/cancer cell implantation, the site is densely infiltrated with round, rapidly moving stromal cells (image corresponds to Video 1), 4–8d later the number of these cells is greatly reduced and many stromal cells acquire a stellate and spindle-like shape (image corresponds to Video 2). (B) Tumor growth in color-coded bone-marrow (BM) chimeric mice with cerulean-blue tumor cells, DsRed BM-derived host cells, and EYFP-yellow non-BM-derived host cells. Cancer cells mobilize BM-cells and non-BM cells for the formation of tumor stroma. Non-BM-derived cells account mainly for tumor vasculature. Data are representative of 3 independent experiments.
Tumor stroma consists of heterogeneous bone marrow (BM) and non-BM derived cell populations and targeting both populations is crucial for the complete elimination of tumors, including cancer variants.11,19 Such stromal cells include immature CD11b+ myeloid-derived cells,42,43 tumor-associated macrophages,44 cancer-associated fibroblasts45-47, and tumor vasculature.48 To define the origin and composition of tumor stroma and to elucidate whether and when cells are recruited for stroma formation from systemic BM and/or nearby local sources, we developed color-coded BM-chimeras (Fig. 2B). Longitudinal imaging for 21 d revealed that early during tumor formation mainly BM-derived stromal cells were recruited and that these cells began to acquire a fibroblastoid shape by day 9. Previously, we demonstrated that the majority (> 90%) of CD45+ cells in established MC57 tumors are CD11b+F4/80+ macrophages,12 and < 10% of CD11b+ cells are Gr1hi granulocytic cells. Non-BM-cells delineated the vessels with a few of these cells interspersed throughout the stroma (Fig. 2B, day 21). These cells included endothelial cells,49 and type I collagen-producing fibroblasts (unpublished data). These results highlight the capacity of cancer cells to mobilize different sources of cells, at different times, from different compartments for the formation of stroma.
Entry and dissemination of adoptively transferred T cells in solid tumors
To characterize the course of tumor destruction following adoptive T cell therapy, we asked where, when, and how adoptively transferred tumor-specific CD8+ T cells arrived and distributed within the tumor tissue. In vitro activated 2C EYFP CD8+ T cells were injected intravenously into MC57-SIY-Cerulean tumor-bearing DsRed Rag1−/− mice. It was not until 2–3 d post transfer that EYFP+ 2C T cells extravasated preferentially at a few discrete sites (Fig. 3A and B; Video 3). When the same tumor was re-imaged 24 h later, many T cells had now infiltrated the tumor mass (Fig. 3C and D). However, T cell distribution and action within the tumor was not homogenous; at 96 h post-T cell transfer, some blue tumor areas contained hardly any T cells (Fig. 3C and D lower half, and 3E), whereas adjacent areas showed massive T cell infiltration and red “stroma-only-areas” (Fig. 3D, upper half, and 3E).

Figure 3. Entry, distribution, cytolytic activity, and motility of tumor-specific T cells in tumors after adoptive transfer. (A) and (B) T cells enter the tumor as early as 2–3 d after transfer. In vitro activated, tumor-specific 2C EYFP T cells adoptively transferred into a DsRed Rag1−/− mouse bearing a MC57-SIY-Cerulean tumor extravasate and enter into the tumor tissue at few distinct sites (data are representative of more than 3 independent experiments). (C) and (D) 24 h later, T cells do not uniformly distribute within the tumor, but heterogeneously. (E) Fluorescence intensity profiles for Cerulean, EYFP, and DsRed were acquired throughout total fields of view (FOV) of Figure 3D. Increased fluorescence intensity indicates accumulation of cell populations. (F) and (G) Cell-cell interactions during T cell-mediated tumor destruction. 2C EYFP T cells engage with MC57-SIY-Cerulean cancer cells; apoptotic membrane blebbing of cancer cells (yellow arrows); enlarged, 3-dimensional, rotated display of the T cell-cancer cell engagement (right). T cells also engage with stromal cells [(F), yellow arrows]; image corresponds to Video 4. (H) Perforin is not needed for the rejection of established MC57-SIY tumors. In vitro activated 2C Prf−/− or 2C WT T cells were adoptively transferred into MC57-SIY tumor bearing mice (when tumors reached a size of about 500 mm3 [between days 13 and 17 as indicated by the horizontal bars (┣┫)]. The number of rejected tumors per total number of tumors is indicated. Data are pooled from 5 independent experiments. P = 0.026 (2C Prf−/− and 2C WT). (I) 1–2 d after cancer cell elimination T cells remain in microenvironment and stably engage with stromal cells; image corresponds to Video 6. Data are representative of 3 independent experiments (n = 4 mice). (J) Comparison of diffusion coefficient and average velocities of T cells in Video 6 (T cells remain arrested) and Video 7 (T cells regain motility ~4 d post cancer cell elimination). Each dot represents an individual T cell; red lines indicate mean; black bars indicate the time-domain standard deviation (td-StDev). Data are representative of 2 independent experiments (**P = 0.0064; ***P < 0.0001).
Visualization of cell-cell interactions during T cell-mediated tumor destruction
We next performed high-resolution imaging to visualize cell-cell interactions at the onset of tumor death in order to obtain mechanistic insights into T-cell mediated cancer elimination and to elucidate the role of stroma during this process. As shown in Figures 3F and G and Fig. 2C EYFP T cells engaged with blue MC57-SIY-Cerulean cancer cells, forming T cell-cancer cell conjugates (Fig. 3F, yellow arrows). Membrane blebbing, one of the common features of cells undergoing programmed cell death was observed during CTL-mediated tumor destruction (Video 4), and subsequently, apoptotic blue tumor material was engulfed by red host stromal cells (Video 4). However, cell-cell contact dependent CTL-mediated cancer cell apoptosis and membrane blebbing typically associated with perforin-mediated lysis50 was an infrequent event. Thus, to evaluate the role of perforin in T cell-mediated tumor destruction we generated 2C T cells deficient in perforin (2C Prf-−/−) and evaluated anti-tumor activity in vivo. Interestingly, perforin was not needed for the rejection of large MC57-SIY tumors, as 2C Prf−/− T cells eliminated MC57-SIY tumors as efficiently as 2C WT T cells (Fig. 3H).
Cross-presentation of SIY antigen by CD11b+ tumor stromal cells has been demonstrated by the use of high-(nM) affinity TCR tetramers ex vivo12,13 and we hypothesized that cross-presenting stromal cells might be a direct target for 2C T cells in vivo and eventually being killed. Thus far, the physical interaction between T cells and stromal cells and direct T-cell mediated killing of stromal cells within the tumor microenvironment in vivo has never been demonstrated. We observed that initially rapidly migrating 2C T cells arrested upon encounter with stromal cells, resulting in stable interactions (Fig. 3F and G; Video 5). However, we did not observe apoptosis of stromal cells coincident with a T cell attachment. Even after MC57-SIY-Cerulean cancer cells were eliminated, round stromal cells persisted and T cells engaged with stromal cells forming stable interactions (Fig. 3I and J; Video 6). At later time points post cancer elimination, T cells displayed faster migration patterns associated with lower arrest coefficients, and higher mean velocities and diffusion coefficients (Fig. 3J; Video 7).
T cells form long-lasting, stable, and cognate antigen-dependent interactions with stromal cells resulting in the production and release of IFNγ
Because, contrary to our expectation, we observed T cell-stromal interactions rather than killing of the stromal cells by tumor-specific CD8+ T cells, we next focused our efforts on elucidating the functional consequences of CTL-stromal interactions and engagements for tumor destruction. First, we confirmed antigen-dependent stromal T cell-stromal cell interactions in a second tumor model Pro4L-SIY-Cerulean. Again, T cells formed long-lasting interactions with stromal cells without subsequent apoptosis (Fig. 4A corresponding to Video 8); this T cell behavior was dependent on cognate antigen, since 2C T cells in antigen-negative Pro4L control tumors had a significantly lower arrest coefficient (0.69 vs 0.98) and significantly higher mean velocity (1.61 vs 0.48 μm/min) (Fig. 4B-D; Video 9). Antigen-dependent stromal engagements were stable and long-lived as revealed by longitudinal imaging over the course of several days (Fig. S4A and B). Similarly, antigen-dependent T cell arrest was observed in MC57-SIY-Cerulean tumors (Fig. S4C). Importantly, imaging procedures (over 2 h long) did not induce phototoxicity and did not decrease or damage T cell motilities (Fig. S4D).

Figure 4. Antigen-dependent stromal engagement of 2C-EYFP CD8+ T cells in Pro4L-SIY-Cerulean tumors (A–D). T cells interact with DsRed-positive stromal cells (yellow arrows); scale bar = 75μm. Image corresponds to Video 8. (B) Left panel. Stromal engagement of T cells is antigen-dependent. Percentage of T cell/stromal cell engagements in Pro4L-SIY-Cerulean (Ag-positive) and Pro4L-Cerulean (Ag-negative) tumors. See Methods for further details. Right panel, Arrest coefficient, (C) average velocities, and (D) representative displacement tracks for 2C T cells in antigen-positive Pro4L-SIY and antigen-negative Pro4L control tumors are graphed (scale bar = 50μm). Red lines indicate mean; black bars indicate td-StDev. Data are representative of 3 experiments and mice, 2 with Pro4L-SIY tumors and 1 with Pro4L. A total of 12 movies (Pro4L-SIY) and 3 movies (Pro4L) were analyzed, 30–150 min long. In B and C, each dot represents an individual T cell. P values were obtained with Mann–Whitney analysis for (B–D); ***P < 0.0001. (E) Left panel: IFNγ secretion by 2C T cells stimulated with tumor-derived stromal cells or cancer cells. CD11b+ stromal cells were isolated from established antigen-positive MC57-SIY tumors (or antigen-negative MC57-hgp100 tumors as control) by magnetic separation and co-cultured with peptide-activated 2C T cells. Stromal cells were compared with equal numbers of cultured cancer cells expressing the same antigen. Supernatants were harvested after 24 h of co-culture and IFNγ was measured by ELISA. Non-stimulated T cells served as negative control (none) and a Data are representative for 4 independent experiments. Right panel: High in vivo IFNγ levels in solid tumors after adoptive T cell transfer only when cross-presentation is enabled. Rag−/−mice with established MC57, MC57-Ld, or MC57-SIY tumors were treated with activated 2C T cells. Three and 6 d after T cell transfer, in vivo IFNγ levels were determined in homogenized tissues of individual tumors by ELISA as per manufacturer’s instructions (eBioscience). Bars represent IFNγ levels per gram of tumor tissue. Data are representative for 2 experiments with 3 to 5 tumors per group.
To elucidate the effects of stromal cross-presentation on CD8+ T cells, we next isolated CD11b+ stromal cells from large established antigen-positive (Ag+) MC57-SIY, or antigen-negative (Ag−) MC57 control tumors and co-cultured them together with effector 2C T cells for 24 h ex vivo. 2C T cells released high amounts of IFNγ when re-stimulated with stromal cells from Ag+ tumors, but not from Ag− tumors, and cross-presenting stromal cells from Ag+ MC57-SIY tumors induced significantly higher levels of IFNγ release than Ag+ cancer cells (Fig. 4E, left panel). Consistent with these data, high levels of IFNγ within tumor tissue was only detected in the cross-presentation-enabled MC57-SIY tumors, but not in the antigen-expressing but cross-presentation-disabled MC57-Ld tumors, the latter inducing only marginally higher IFNγ levels than control tumors (Fig. 4E, right panel). In conclusion, our imaging technology allowed us to demonstrate that CTL engage with stromal cells (Fig. 4B) without killing them, which activates CTL to release IFNγ (Fig. 4E).
Minimal, perivascular infiltration of antigen-specific T cells rapidly initiates vessel destruction
We next sought to visualize with our longitudinal imaging approach the kinetics of T cell mediated effects on tumor vasculature. We imaged every 12 h the same tumor regions over several days after T cell transfer, and quantified 3D volume and integrity of tumor vasculature by developing a novel vessel perfusion measurement approach (Fig. 5; Fig. S5): 1,1-dioctadecyl-3,3,3,3-tetramethylindodicarbocyanine perchlorate (DiD)-labeled red blood cells (RBC) were injected into mice with established MC57-SIY tumors or Ag− MC57 control tumors, and tumors were imaged longitudinally in 3D by acquiring z-stack images, every 12 h over several days. Systemic injection of DiD-labeled RBCs is a useful and validated technique to measure blood flow (or absence thereof) and vascular networks, and ideal for long-term studies due to the long half-life of labeled RBC.51 While we never observed T cells directly killing tumor vasculature and/or endothelial cells, we observed that minimal, perivascular infiltration of antigen-specific T cells resulted in vessel leakiness indicating vessel damage in Ag+ MC57-SIY tumors (Fig. 5A; Fig. S5A; corresponding Videos 10,11, and 3D reconstruction images Video 12), but not in Ag- MC57 tumors (Fig. S5B; corresponding Video 13). For all time points, maximal projections of z-stacks were generated (Fig. 5B), vessel volumes were calculated, and a novel algorithm was generated to render a “vessel perfusion index” as described in Materials and Methods (Fig. 5B–E). While perfusion indexes and 3D vessel volume remained constant over time in Ag− tumors, both vessel perfusion and volume dramatically decreased in Ag+ MC57-SIY tumors (Fig. 5D and E). Thus, vessel regression began and coincided with the earliest time points of T cell entry in Ag+ tumors but not in Ag− tumors (Fig. 5F). However, while immediate vessel regression was observed upon T cell entry, cancer regression was observed at later time points (˃ 3 d post T cell transfer) (Figs. 3D, 5A and F; Fig. S5). These kinetics and observations were solely dependent on antigen-expression by the tumor, as Ag− control tumors did not reveal any signs of vessel regression even at later time points (Fig. 5F).

Figure 5. Longitudinal imaging and quantification of vessel integrity, vessel perfusion, and 3D vessel volume in solid tumors after adoptive T cell transfer. (A) Activated 2C T cells were adoptively transferred into a DsRed host mice with a 21-d-old established solid tumor (95% MC57-SIY EGFP + 5% MC57 Cerulean). At indicated time points post T cell transfer same tumor area was imaged (scale bar = 50μm). (B–F) DsRed Rag1−/− mice with established MC57-Cerulean or MC57 SIY-Cerulean tumors growing behind windows were injected with activated 2C EYFP CD8 T cells at day 18 of tumor growth. DiD-labeled red blood cells were injected intravenously into mice on the day of T cell transfer and tumors were imaged in 3D and longitudinally, every 12 h, at indicated time points by acquiring numerous sets of z-stack volumes. (B) Bright field images (top) and maximal projections of z-stacks (bottom) of DiD-labeled red blood cells (purple), 2C EYFP T cells (yellow), MC57-Cerulean (right), and MC57 SIY-Cerulean (left) cancer cells (cyan blue) at indicated time points post T cell transfer. (C) Representation of vessel perfusion indexes (VPI) generated from time-domain standard deviations (td-StDev) of DiD-intensities throughout the entire set of z-stacks. The higher the td-StDev values in the DiD channel per pixel, the higher the assigned color intensity values in the pseudo-colored image (option Fire LUT; Fiji). A detailed description of the generation of VPI is provided in Materials and Methods. VPI is an indirect measure of vessel integrity: high td-StDev in the DiD channel indicates fast blood flow and consequently good vessel integrity; low td-StDev results from static DiD-RBC reflecting impaired vessel integrity. Vessel leakiness/damage is observed in antigen-positive MC57 SIY tumors, but not in antigen-negative MC57 tumors after T cell transfer. (D) Vessel Perfusion Indexes, and (E) 3D volumetric vessel analysis for MC57 and MC57 SIY tumors measured over several days post T cell transfer. Calculations and methods used to quantify vessel density in 3D images are described in Materials and Methods. (D) and (E) show averages and StDev from several tumor regions followed longitudinally. N of regions at each time point is as follows (day, N); for antigen-positive tumor: (d1,6;d1.5,7;d2,9;d2.5,9;d3,4); for control tumor (d1,6;d1.5,5;d2.5,5;d3,4;d3.5,3). A second, independent longitudinal experiment with another mouse showed similar results. (F) The area occupied by cancer cells, T cells and vessels was measured for images displayed in Figure 5B. The maximum value of area fraction for each channel was assigned the value of 100% and all subsequent values for the specific channel were plotted as percent of maximum.
Longitudinal real-time in vivo imaging of cancer variants elimination
The outgrowth of variant cancer cells is a frequent cause for the failure of cancer treatments in humans, including T cell therapy.52-54 Our previous work has demonstrated that cancer cells that have lost antigen expression, or antigen-loss variants (ALV), are eliminated if the stroma is sensitized by antigen released from the cancer cells that have not lost the antigen. However, the mechanism by which CD8+ T cells eliminate ALV has remained elusive. Two scenarios have been proposed: (1) CD8+ T cells become indiscriminate killers and eliminate ALV by direct cell-cell engagement, or (2) CD8+ T cells target sensitized stromal cells resulting in indirect, bystander killing of ALV. Interestingly, it was recently demonstrated that IFNγ not only acts synaptically (i.e., toward antigenic target cells) but multidirectionally affecting non-antigenic bystanders in vitro.55 Obviously, the kinetics of elimination in these 2 scenarios would be very different. In the first, Ag+ cancer cells and ALV would die simultaneously, whereas in the second, Ag+ bulk tumor would be killed by T cells first, causing sensitization of the stroma, and subsequently bystander death of ALV. Therefore, we set out to visualize and determine the kinetics and mechanism(s) of ALV elimination. Parental MC57 cancer cells were transduced to express DsRed (Fig. 6A). MC57-DsRed cells are not recognized by 2C T cells because they do not express the SIY or Ld antigen and thus mimic ALV in our model. DsRed-ALV were mixed in a 1:20 ratio with either Ag+ MC57-SIY-Cerulean or Ag+ MC57-Ld-Cerulean blue cancer cells. The difference between these 2 models is that only MC57-SIY but not MC57-Ld can sensitize stroma. The cancer cell mixtures were inoculated into Rag1−/− window-bearing mice (Fig. 6B). As shown in Figure 6C and D (panel 1), DsRed+ ALV grew and were homogeneously embedded in the blue, Ag+ bulk tumor tissues of both, MC57-SIY-Cerulean or MC57-Ld-Cerulean (quantified in Figure 6E, left panel). In vitro activated 2C EYFP T cells were adoptively transferred. T cells arrived at the tumor site (Fig. 6C and D, panel 2), and again infiltrated the tumor heterogeneously (Fig. 6C and D, panel 3). While T cells eliminated Ag+ MC57-SIY-Cerulean and MC57-Ld-Cerulean cancer cells, red ALVs were not eliminated; 5 d after T cell transfer, ALV persisted and several “red-only” areas emerged [Figure 6C (panel 3, lower part) and D (panel 3, upper part)]. However, T cells remained in the microenvironment: while T cells were arrested in MC57-SIY-tumors, T cells in the microenvironment of MC57 Ld tumors remained motile, showing lower arrest coefficient, higher average velocity and higher diffusion coefficient (Fig. 6F and G; and Videos 14 and 15). Interestingly, while MC57-DsRed ALV eventually disappeared in MC57-SIY tumors, ALV in MC57-Ld tumors persisted [Figure 6C, D (panel 4), and E (right panel)]. To confirm that T cells indeed interacted with stromal cells in an antigen-dependent fashion, we developed a 4-color model to simultaneously visualize Ag+ cancer cells, ALV, stromal cells, as well as T cells (Fig. S6A). Again, T cells in MC57-SIY tumors engaged with stromal cells, in contrast to T cells in MC57-Ld tumors, and showed a significantly decreased velocity and higher arrest coefficient (Fig. S6D-G; Videos 16 and 17). Most importantly, ALV in MC57-Ld tumors grew, while ALV in MC57-SIY tumors eventually died (Fig. S6B and S6C). Since ALV in both models were completely eliminated in sensitized stroma of Kb/SIY tumors but not in Ld-tumors, we hypothesize that in this tumor model T cell-stromal engagements are required for the elimination and prevention of re-growth of cancer variants.

Figure 6. Visualization of bystander elimination of antigen-loss variants (ALV). (A) To visualize ALV, parental MC57 cells were transduced to express DsRed. The inset numbers indicate the mean fluorescence intensity by flow analysis. Parental MC57 is used as control (gray line). (B) Scheme. Model 1: antigen-positive MC57-SIY-Cerulean cancer cells are mixed with 5% of MC57-DsRed ALV and transplanted into C57Bl/6 Rag1−/− mice (n = 3). Model 2: antigen-positive MC57-Ld-Cerulean cancer cells are mixed with 5% of MC57-DsRed ALV and transplanted into C57Bl/6 Rag1−/− mice (n = 2). (C–D) Both cell mixtures grow with red ALV homogeneously embedded in the blue, antigen-positive tumor tissue [image 1]. Adoptively transferred 2C EYFP T cells arrive in the tumors [image 2; 3–4 d post T cell transfer]. While T cells eliminate the blue antigen-positive MC57-SIY-Cerulean and MC57-Ld-Cerulean cancer cells, the red ALV are not killed and “red-only areas” emerge [image 3; 4 d post T cell transfer]. While red ALV in the MC57-SIY-Cerulean microenvironment disappear over time (model 1 [image 4; 6 d post T cell transfer]), ALV in the MC57-Ld Cerulean microenvironment persist and continue to grow (model 2 [image 4; 5 d post T cell transfer]). (E) Percent of image occupied by DsRed ALV and blue SIY- or Ld-expressing cancer cells before (left panel; corresponding to Figure 6C and D (image 2) or during/after (right panel; corresponding to Figures 6C and D (images 4)) T cell mediated destruction. While the presence of ALV in the SIY model significantly decreases, ALV in the Ld-tumor persist and continue to grow. Images were acquired using different magnifications and subsequently adjusted digitally to be in the same scale. (F) Arrest coefficient, diffusion coefficient and average velocities of 2C T cells in the microenvironment of MC57-SIY or MC57-Ld tumors are graphed. Red lines indicate mean, black bars indicate td-StDev; ***P < 0.0001; *P = 0.0122. (G) Representative displacement tracks from 2C T cells in MC57-SIY or MC57-Ld tumors (scale bar = 40μm).
Discussion
We developed an in vivo window imaging model to longitudinally monitor the complex interplay of color-coded cancer cells, ALV, BM- and non-BM-derived stromal cells, tumor-specific CD8 T cells, as well as tumor vasculature in solid cancers. By overcoming the limitations of previous approaches using terminal intravital imaging of surgically exposed tumors, we captured for the first time in real-time and high resolution, the dynamic, immune-mediated sequence of events within a given tumor over hours, days, and even weeks after adoptive T cell transfer. The powerful, technological advance is the ability to longitudinally monitor the same area, and to image how events unfold within precisely the same region over time. In contrast, non-longitudinal intravital imaging can only capture single end points and does not allow revisiting the same area at later time points. Even when large numbers of animals for each different time point are being used to compensate for the variability of tumor regions chosen for analysis, any resulting sequences of events are reconstructed. Here, we found that vessel regression begins and coincides with the earliest time points of T cell entry from the blood stream into the tumor (Fig. 5). This may explain the enigma why solid tumors “melt” from the inside following successful T cell attack, although only few T cells deeply infiltrate into the tumor early after T cell transfer.56 Importantly, the use of long-term tumor window implant device eliminated the necessity to surgically expose the tumor tissue just prior imaging, thereby eliminating potential confounding influences due to acute tissue injury and blood vessel destruction, and associated immediate inflammatory effects.28-30
Our imaging approach also allowed us to develop quantitative contextual analyses of T cell interactions with cancer cells and stroma, and design novel algorithms for the quantitative assessment of vessel integrity and perfusion over time after T cell therapy. These analyses revealed that perforin-mediated direct CTL-killing was, contrary to our expectation, not the primary mechanism leading to cancer destruction (Fig. 3H). Furthermore, stromal cells cross-presenting tumor-antigen are not a target for perforin-mediated destruction but serve as a prolonged and highly effective stimulus for antigen-specific T cells resulting in the production and release of high amounts of cytokines (IFNγ) within tumors, which has been shown to be required in some models for the elimination of tumors, including antigen loss variants.11,19,57,58 Tumor relapse of cancer variants often occurs after initial T cell-mediated tumor destruction11,12,59 and usually resumes from the tumor margin surviving after central necrosis. Even in the face of a potential “field effect” of vessel destruction, causing some bystander cancer cell death, including ALV, we found that when the tumor bulk was being destroyed, ALV may survive and remain within the microenvironment. Tumor-specific CD8+ T cells in Kb/SIY tumors interacted with antigen-sensitized stromal cells. Subsequently, this interaction triggered the production and release of high levels of IFNγ (Fig. 4E). In agreement with these results, we found higher amounts of IFNγ in vivo in stroma of Kb/SIY tumors than in the stroma of Ld- or control tumors (Fig. 4E). While the cytokine release is clearly dependent on and mediated by the formation of conjugates between T cells and cells presenting the antigen, the effect of the released cytokines (cytokine-mediated killing of target cells) does not require conjugate formation. While perforin, not needed in our model (Fig. 3H), depends on effector-target conjugates for achieving cell death, IFNγ- and TNFα-dependent killing can be mediated without direct target contact,60 and can act multidirectionally affecting non-antigenic bystanders.55 Since ALV were eliminated in sensitized stroma of Kb/SIY tumors but not in Ld-tumors, we propose that stromal cross-presentation, T cell-stromal engagements, and subsequent high cytokine release in this model are helping the elimination of ALV and prevention of re-growth of cancer variants. Identifying precisely how cytokines such as IFNγ and TNF released by T cells contribute to ALV elimination, prevention of ALV re-growth (e.g., cytokine-mediated cytotoxicity, IFNγ-induced Fas/FasL-dependent apoptosis,59,61 or IFNγ-mediated prevention of tumor vasculature regeneration), and vessel destruction, or if other factors could potentially override the requirement of stromal cross-presentation for tumor elimination and/or cancer variants, will be the focus of future studies.
In conclusion, the ability to visualize distinct immune, stroma and cancer cell subsets in distinct compartments of tumors and to longitudinally follow their fate in vivo will help to elucidate the underlying mechanism(s) responsible for success or failure of anti-cancer immunotherapies or other anti-cancer therapeutic approaches.
Materials and Methods
Mice and Cell lines
C57Bl/6 Rag1−/− mice were purchased from The Jackson Laboratory; Tg(ACTB-DsRed*MST)1Nagy/J mice were purchased from The Jackson Laboratory and crossed to C57BL/6 Rag-1 KO mice to obtain DsRed Rag-1 KO mice. 2C TCR Rag KO mice were provided by J Chen (Massachusetts Institute of Technology). To generate 2CEYFP mice, 2C TCR mice were crossed to 129-Tg(ACTB-EYFP)7AC5Nagy/J, obtained from The Jackson Laboratory. 2C × EYFP mice were backcrossed to C57BL/6 for 7 generations. 2C × EGFP mice were generated by crossing 2C TCR to EGFP transgenic mice (obtained from Jackson Laboratory). Over 95% of CD8-EYFP T cells expressed the Vβ8.1 8.2-chain. All mice were bred and maintained in a specific pathogen-free barrier facility at The University of Chicago according to the Institutional Animal Care and Use Committee (IACUC) guidelines. To generate colored bone-marrow chimeras, EYFP Rag1−/− mice were lethally irradiated and reconstituted 24 h later with 5 × 106 bone-marrow cells from DsRed Rag1−/− mice. Imaging experiments were initially performed at the Vanderbilt University Cell Imaging Shared Resource (CISR) and subsequently at The University of Chicago Light Microscopy Core Facility. For experiments done at Vanderbilt University, mice were shipped via special airplane carriers from University of Chicago and maintained in a special mouse facility at Vanderbilt University. All animal experiments were approved by the IACUC of Vanderbilt University and The University of Chicago. MC57G was provided by P Ohashi (University of Toronto), with permission of H Hengartner (University Hospital Zurich). The murine fibrosarcoma cell line Pro4L was derived from the C3H/HeN mouse undifferentiated spindle cell cancer 1591-PRO4L and has been described preciously.6
Antibodies, plasmids, and retroviral vectors
PE-anti-mouse H-2Kb (AF6–88.5), PE-anti-mouse Vß8.1, 8.2 TCR (MR5–2), and anti-mouse CD8a (53–6.7) antibodies used for flow cytometry were obtained from BD PharMingen. The anti-Ld antibody (30–5-7) was obtained from D Sachs (Massachusetts General Hospital). The retroviral vector pMFG was obtained from RC Mulligan and retroviral infections were performed as described.62 The plasmid pDsRedT1-N1 was obtained from Clontech. The plasmid mCerulean-C1 was obtained from DW Piston, Vanderbilt University and used for subsequent cloning to generate the retroviral vectors pMFG-Cerulean and pMFG (SIY)3-Cerulean.
Generation of pMFG Cerulean and pMFG (SIY)3-Cerulean vectors
To generate pMFG-Cerulean, Cerulean was amplified from mCerulean-C1 using the primers 5′ - GCG CCA TGG TGA GCA AGG GCG AGG AGC - 3′ and 5′ - GCG GAT CCT TAC TTG TAC AGC TCG TCC ATG CCG - 3′, digested with NcoI and BamHI (New England Biolabs), and ligated into pMFG digested with NcoI/BamHI. To generate pMFG (SIY)3-Cerulean, the minigene SIYRYYGL-AAY trimer was cut from pLEGFP-SIY with NcoI and ligated into pMFG- Cerulean.
Generation of tumor cell lines
MC57, MC57-Ld, and Pro4L were retrovirally infected to express the fluorescent proteins Cerulean or (SIY)3-Cerulean respectively as described 39. After infection, cell lines MC57-(SIY)3-Cerulean, Pro4L-(SIY)3-Cerulean, MC57-Ld-Cerulean were sorted multiple times by FACS for high expression of Cerulean. To obtain the cell line MC57-DsRed, MC57 was transfected with pDsRedT1-N1 (Clontech) using Ca-phosphate transfection. MC57-DsRed cells were FACS-sorted for high DsRed protein expression.
In vitro T cell activation and adoptive transfer of 2C T cells
For the adoptive transfer of activated 2C-EYFP or 2C-EGFP CD8+ T cells, (NH4)Cl–treated splenocytes from 2C × EYFP or 2C × EGFP mice were stimulated in vitro with IL-2 (8U/ml) (in some experiments without IL-2) and SIYRYYGL peptide (7.5 μg/ml) for 3–4 d at a concentration of 4 × 106 cells/ml. Approximately 5 × 106 activated T cells were injected intravenously into the retro-orbital plexus. H Auer and S Meredith (University of Chicago) synthesized the 2C-recognized peptide SIYRYYGL.
Flow cytometric analysis
Flow cytometric analysis was performed using FACSCalibur, FACScan, and DakoCytomation CyanADP and analyzed with FlowJo software (BD Biosciences). Tumor cell lines were sorted using DakoCytomation MoFlo HTS and BD FACS Aria.
Window implantation and cancer cell injection
The in vivo tumor window model was performed as the window chamber model previously described41,63 with some modifications. In brief, before surgery, hair was removed from the back of the mouse and skin was surface treated with iodine solution. For initial experiments, mice were anesthetized by administering intraperitoneally a mixture of ketamin and xylazine, later inhaled isoflurane was used exclusively. A circular hole of 1 cm diameter was dissected in one side of the skin surface of the dorsal skin flap by removing skin and fascial plane, leaving the opposite skin layer and its fascial plane with associated vasculature. The window frame was then implanted as previously described.41 Cancer cells (re-suspended in 20–30 μl 1 × PBS) were injected at 3–5 different sites in between the fascia and dermis of the rear skin layer. The window frame was sealed with only 1 glass coverslip and clamped to the window frame with a C-ring. Window frames were tightened via 3 screws and sutured additionally. Titanium window frames were purchased from and specially modified by APJ Trading Co. Inc. All procedures were performed using sterile material and equipments.
Confocal microscopy
Window chamber mice were anesthetized by administering a mixture of ketamine and xylazine intraperitoneally; isoflurane was used in later experiments. The window was fastened to the main stage of the microscope using a custom-made holder. (For experiments performed at University of Chicago: a stage insert was custom-built by the engineering team at the University of Chicago Engineering Center (UCEC) to hold the anesthetized animal in the same position on the microscope. The stage insert fixed mouse and implanted window always in the same position by indexing the 3 screws (asymmetric triangle) that are used to hold the window frame plates together. Mechanical clamps were holding the imaging window frame to the stage insert. A motorized microscope XY scanning stage and Leica LAS-AF software allowed recording individual 3-dimensional positions per field-of-view and returning to them later with high precision (stated accuracy +/− 3μm; reproducibility < 1.0μm). Using blood vessels as “landmarks,” same vessels could be located within 50μm when returning on the same day, or within 100μm on the next day). Body temperature was maintained by placing every 10 min a warm (37 °C), water filled glove on the mouse’s body. Confocal images were captured with a Zeiss LSM 510 Confocal Microscope System (Vanderbilt), or a Leica SP5 II TCS Tandem scanner 2-photon spectral confocal with 4 × and 20 × /0.45 LWD IR objectives from Olympus (University of Chicago). A X-Y motorized stage was used for multiple point visiting during imaging experiments. For visualization of the tumor vasculature, mice were injected with 2.000.000 MW anionic, fluorescein-conjugated dextran (Molecular Probes), or 1,1-dioctadecyl-3,3,3,3-tetramethylindodicarbocyanine perchlorate (DiD)-labeled red blood cells (RBC).64 Briefly, RBC were obtained from the peripheral blood of C57Bl/6 Rag1−/− mice and stained with DiD (Invitrogen) for 30 min at 37 °C. After 3 washes with PBS, DiD-labeled RBC were injected intravenously. Absorption and fluorescence emission maxima of DiD are 644nm and 665nm respectively. The optical penetration of tissue ranged between 90–500μm with the average of 120–150μm.
Data analysis
Initial digital image processing was performed using Zeiss LSM browser and Leica LAS-AF Lite and selected images were further analyzed using ImagePro 6.3 (MediaCybernetics), Fiji (NIH), and SlideBook 4 software (Intelligent imaging Innovations). Time-lapse recordings were corrected for drift based on landmark features and T cell motility was tracked and quantified using Imaris 7.5 software (Bitplane). T cell diffusion coefficient (D) was calculated as D = L2/t, with L being the cell track displacement length (μm) and t the track duration in minutes. Arrest coefficient was calculated as the fraction of time that T cell velocity was less than 3 μm/min. The long-term arrest was defined as when a cell centroid did not displace further than 1 half of the cell diameter (5 μm) for at least 15 min; in shorter movies, the entire length of the movie was considered.. For Figure 5F: images displayed in Figure 5B were analyzed using Fiji to determine the area occupied by cerulean or EYFP expressing cells, as well as by vessels in maximum intensity projections of z-stacks .
Contextual analysis of T cell interaction targets
T cell interactions were analyzed using the Leica Application Suite version 1.7.0 build 1240 (Leica Microsystems), Imaris 7.5 (Bitplane Inc.) and Slidebook software (Intelligent Imaging Innovations Inc.). An interaction was defined as cell immobilization for at least 15 min. To detect the sites of T cell immobilization in an unbiased manner, raw 3-D time-lapse recordings were first corrected for drift, if any, then threshold and binarized. Next, the binarized time-lapse data sets were processed to generate 2 types of time projection images: the average intensity time projection (AITP) and the maximum intensity time projection (MITP). The AITP image has the important property that pixel intensities represent the duration of cell persistence in the area, and the MITP image represents all places visited by cells during intravital motility recording. To detect the sites of cell persistence for 15 min or longer in a 60 min long recording, the AITP image was thresholded at the corresponding pixel intensity value. The thresholded AITP image was then merged with the MITP image into a single 2-color image such that the areas of overlap represented the areas of 15 min long T cell persistence in relation to all T cell-visited areas. The image representing T cell persistence sites was then overlaid over the images of other fluorescence channels that were co-visualized during the intravital recording, and the cell persistence sites were categorized based on the type of immediately adjacent neighboring cells.
Analysis of vessel volume and vessel perfusion index
The index of vessel perfusion was designed based on the observation that movements of brightly-labeled RBCs during collection of the 3D time series data become blurred due to the blood flow, resulting in a low frequency components in the XYZ Fourier space and high frequency components in the time domain. In contrast, the lack of blood flow results in stationary, discernible RBCs giving rise to higher frequency components in the XYZ Fourier space but decreasing the high frequency components in the time domain, which is quantifiable as low values of time-domain standard deviation (td-StDev). Maximal projections of z-stacks of DiD-labeled RBC were generated to document vessel morphology, and the functional vessel volume within those 3-D stacks was calculated as the space occupied by DiD-RBC using the ImageJ “3D Object Counter” plugin. When vessels showed compromised/impaired integrity (as evidenced by lack of blood flow in the 3D stacks viewed as a temporal series), a value of 0 was manually assigned. Subsequently, an algorithm was designed by using a statistical quantification method to render a “vessel perfusion index” (VPI) from the td-StDev of DiD-intensities throughout the entire set of z-stacks. An ImageJ macro was created that automatically calculated pairwise td-StDev of per-pixel intensity changes in 3D stack images. Pairwise comparisons limited the intensity changes to RBC movements and lessened contributions of changes in background structure. The mean td-StDev was normalized to the number of frames and corrected by subtracting the image median to reveal RBC intensity changes. The resulting value is what we called VPI. VPI is an indirect measure of vessel integrity: high td-StDev in the DiD channel indicates fast blood flow and consequently good vessel integrity; low td-StDev results from static DiD-RBC reflecting impaired vessel integrity. 3D vessel reconstruction/rotation videos were done using Imaris.
Statistical Analyses
Statistical analysis was performed using GraphPad Prism 5.03, typically using the Mann Whithey test. Measurement differences resulting in P values of 0.05 or less were considered as statistically significant and are referred to as such in the text. All error bars represent standard deviations, unless indicated as standard errors.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Ann Schue for excellent help in arranging the mouse transfers from University of Chicago to Vanderbilt University; Laura DeBusk, Kimberly Boelte, and CarolAnn Bonner for help and technical assistance, Anita Chong, Alexander Chervonsky, Maki Motobu, Karin Schreiber, Heather Booras, David Binder, and Christian Idel for technical advice and helpful discussions, and at the University of Texas MD Anderson Cancer Center, Anna Zal and Felix Nwajei for help with the contextual analysis of cell motility. We thank Mary Philip for critical reading of the manuscript and valuable suggestions. We thank the University of Chicago Cancer Research Center Core facilities, especially R Duggan, D Leclerc and M Olson for expert assistance with cell sorting and flow cytometric analysis, the Vanderbilt Cell Imaging Shared Resource, and Christine Labno, University of Chicago Light Microscopy Core, for technical assistance. This work was supported by National Institute of Health, Activities to Promote Research Collaborations (APRC) program from the NCI, and grants, R01-CA22677, R01-CA37516 and P01-CA97296 to H.S, as well as R01-CA137059 to T.Z.
Citation: Schietinger A, Arina A, Liu RB, Wells S, Huang J, Engels B, Bindokas V, Bartkowiak T, Lee D, Herrmann A, et al. Longitudinal confocal microscopy imaging of tumor eradication following adoptive T-cell transfer. OncoImmunology 2013; 2:e26677; 10.4161/onci.26677
Footnotes
Previously published online: www.landesbioscience.com/journals/oncoimmunology/article/26677
References
- 1.Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267–96. doi: 10.1146/annurev.immunol.25.022106.141609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pekarek LA, Starr BA, Toledano AY, Schreiber H. Inhibition of tumor growth by elimination of granulocytes. J Exp Med. 1995;181:435–40. doi: 10.1084/jem.181.1.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seung LP, Rowley DA, Dubey P, Schreiber H. Synergy between T-cell immunity and inhibition of paracrine stimulation causes tumor rejection. Proc Natl Acad Sci U S A. 1995;92:6254–8. doi: 10.1073/pnas.92.14.6254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seung LP, Seung SK, Schreiber H. Antigenic cancer cells that escape immune destruction are stimulated by host cells. Cancer Res. 1995;55:5094–100. [PubMed] [Google Scholar]
- 5.Wyckoff J, Wang W, Lin EY, Wang Y, Pixley F, Stanley ER, Graf T, Pollard JW, Segall J, Condeelis J. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 2004;64:7022–9. doi: 10.1158/0008-5472.CAN-04-1449. [DOI] [PubMed] [Google Scholar]
- 6.Singh S, Ross SR, Acena M, Rowley DA, Schreiber H. Stroma is critical for preventing or permitting immunological destruction of antigenic cancer cells. J Exp Med. 1992;175:139–46. doi: 10.1084/jem.175.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spiotto MT, Yu P, Rowley DA, Nishimura MI, Meredith SC, Gajewski TF, Fu YX, Schreiber H. Increasing tumor antigen expression overcomes “ignorance” to solid tumors via crosspresentation by bone marrow-derived stromal cells. Immunity. 2002;17:737–47. doi: 10.1016/S1074-7613(02)00480-6. [DOI] [PubMed] [Google Scholar]
- 8.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–74. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qiu W, Hu M, Sridhar A, Opeskin K, Fox S, Shipitsin M, Trivett M, Thompson ER, Ramakrishna M, Gorringe KL, et al. No evidence of clonal somatic genetic alterations in cancer-associated fibroblasts from human breast and ovarian carcinomas. Nat Genet. 2008;40:650–5. doi: 10.1038/ng.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chometon G, Jendrossek V. Targeting the tumour stroma to increase efficacy of chemo- and radiotherapy. Clin Transl Oncol. 2009;11:75–81. doi: 10.1007/s12094-009-0317-y. [DOI] [PubMed] [Google Scholar]
- 11.Spiotto MT, Rowley DA, Schreiber H. Bystander elimination of antigen loss variants in established tumors. Nat Med. 2004;10:294–8. doi: 10.1038/nm999. [DOI] [PubMed] [Google Scholar]
- 12.Zhang B, Bowerman NA, Salama JK, Schmidt H, Spiotto MT, Schietinger A, Yu P, Fu YX, Weichselbaum RR, Rowley DA, et al. Induced sensitization of tumor stroma leads to eradication of established cancer by T cells. J Exp Med. 2007;204:49–55. doi: 10.1084/jem.20062056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang B, Zhang Y, Bowerman NA, Schietinger A, Fu YX, Kranz DM, Rowley DA, Schreiber H. Equilibrium between host and cancer caused by effector T cells killing tumor stroma. Cancer Res. 2008;68:1563–71. doi: 10.1158/0008-5472.CAN-07-5324. [DOI] [PubMed] [Google Scholar]
- 14.Cocucci E, Racchetti G, Meldolesi J. Shedding microvesicles: artefacts no more. Trends Cell Biol. 2009;19:43–51. doi: 10.1016/j.tcb.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 15.Théry C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009;9:581–93. doi: 10.1038/nri2567. [DOI] [PubMed] [Google Scholar]
- 16.Andre F, Schartz NE, Movassagh M, Flament C, Pautier P, Morice P, Pomel C, Lhomme C, Escudier B, Le Chevalier T, et al. Malignant effusions and immunogenic tumour-derived exosomes. Lancet. 2002;360:295–305. doi: 10.1016/S0140-6736(02)09552-1. [DOI] [PubMed] [Google Scholar]
- 17.Neijssen J, Herberts C, Drijfhout JW, Reits E, Janssen L, Neefjes J. Cross-presentation by intercellular peptide transfer through gap junctions. Nature. 2005;434:83–8. doi: 10.1038/nature03290. [DOI] [PubMed] [Google Scholar]
- 18.Groothuis TA, Neefjes J. The many roads to cross-presentation. J Exp Med. 2005;202:1313–8. doi: 10.1084/jem.20051379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang B, Karrison T, Rowley DA, Schreiber H. IFN-gamma- and TNF-dependent bystander eradication of antigen-loss variants in established mouse cancers. J Clin Invest. 2008;118:1398–404. doi: 10.1172/JCI33522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boissonnas A, Fetler L, Zeelenberg IS, Hugues S, Amigorena S. In vivo imaging of cytotoxic T cell infiltration and elimination of a solid tumor. J Exp Med. 2007;204:345–56. doi: 10.1084/jem.20061890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boissonnas A, Scholer-Dahire A, Fetler L, Amigorena S. Multiphoton imaging of cytotoxic T lymphocyte-mediated antitumor immune responses. Curr Top Microbiol Immunol. 2009;334:265–87. doi: 10.1007/978-3-540-93864-4_11. [DOI] [PubMed] [Google Scholar]
- 22.Breart B, Lemaître F, Celli S, Bousso P. Two-photon imaging of intratumoral CD8+ T cell cytotoxic activity during adoptive T cell therapy in mice. J Clin Invest. 2008;118:1390–7. doi: 10.1172/JCI34388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pittet MJ, Grimm J, Berger CR, Tamura T, Wojtkiewicz G, Nahrendorf M, Romero P, Swirski FK, Weissleder R. In vivo imaging of T cell delivery to tumors after adoptive transfer therapy. Proc Natl Acad Sci U S A. 2007;104:12457–61. doi: 10.1073/pnas.0704460104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mrass P, Takano H, Ng LG, Daxini S, Lasaro MO, Iparraguirre A, Cavanagh LL, von Andrian UH, Ertl HC, Haydon PG, et al. Random migration precedes stable target cell interactions of tumor-infiltrating T cells. J Exp Med. 2006;203:2749–61. doi: 10.1084/jem.20060710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pittet MJ. Behavior of immune players in the tumor microenvironment. Curr Opin Oncol. 2009;21:53–9. doi: 10.1097/CCO.0b013e32831bc38a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lohela M, Werb Z. Intravital imaging of stromal cell dynamics in tumors. Curr Opin Genet Dev. 2010;20:72–8. doi: 10.1016/j.gde.2009.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zal T, Chodaczek G. Intravital imaging of anti-tumor immune response and the tumor microenvironment. Semin Immunopathol. 2010;32:305–17. doi: 10.1007/s00281-010-0217-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Germain RN, Miller MJ, Dustin ML, Nussenzweig MC. Dynamic imaging of the immune system: progress, pitfalls and promise. Nat Rev Immunol. 2006;6:497–507. doi: 10.1038/nri1884. [DOI] [PubMed] [Google Scholar]
- 29.Ryan GB, Majno G. Acute inflammation. A review. Am J Pathol. 1977;86:183–276. [PMC free article] [PubMed] [Google Scholar]
- 30.Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol. 2006;6:173–82. doi: 10.1038/nri1785. [DOI] [PubMed] [Google Scholar]
- 31.Algire GH. An adaptation of the transparent chamber technique to the mouse. J Natl Cancer Inst. 1943;4:1–11. [Google Scholar]
- 32.Sha WC, Nelson CA, Newberry RD, Kranz DM, Russell JH, Loh DY. Selective expression of an antigen receptor on CD8-bearing T lymphocytes in transgenic mice. Nature. 1988;335:271–4. doi: 10.1038/335271a0. [DOI] [PubMed] [Google Scholar]
- 33.Udaka K, Wiesmüller KH, Kienle S, Jung G, Walden P. Self-MHC-restricted peptides recognized by an alloreactive T lymphocyte clone. J Immunol. 1996;157:670–8. [PubMed] [Google Scholar]
- 34.Vaupel P, Kallinowski F, Okunieff P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: a review. Cancer Res. 1989;49:6449–65. [PubMed] [Google Scholar]
- 35.Patterson G, Day RN, Piston D. Fluorescent protein spectra. J Cell Sci. 2001;114:837–8. doi: 10.1242/jcs.114.5.837. [DOI] [PubMed] [Google Scholar]
- 36.Hadjantonakis AK, Nagy A. The color of mice: in the light of GFP-variant reporters. Histochem Cell Biol. 2001;115:49–58. doi: 10.1007/s004180000233. [DOI] [PubMed] [Google Scholar]
- 37.Rizzo MA, Springer GH, Granada B, Piston DW. An improved cyan fluorescent protein variant useful for FRET. Nat Biotechnol. 2004;22:445–9. doi: 10.1038/nbt945. [DOI] [PubMed] [Google Scholar]
- 38.Vintersten K, Monetti C, Gertsenstein M, Zhang P, Laszlo L, Biechele S, Nagy A. Mouse in red: red fluorescent protein expression in mouse ES cells, embryos, and adult animals. Genesis. 2004;40:241–6. doi: 10.1002/gene.20095. [DOI] [PubMed] [Google Scholar]
- 39.Engels B, Engelhard VH, Sidney J, Sette A, Binder DC, Liu RB, Kranz DM, Meredith SC, Rowley DA, Schreiber H. Relapse or eradication of cancer is predicted by peptide-major histocompatibility complex affinity. Cancer Cell. 2013;23:516–26. doi: 10.1016/j.ccr.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Papenfuss HD, Gross JF, Intaglietta M, Treese FA. A transparent access chamber for the rat dorsal skin fold. Microvasc Res. 1979;18:311–8. doi: 10.1016/0026-2862(79)90039-6. [DOI] [PubMed] [Google Scholar]
- 41.Huang Q, Shan S, Braun RD, Lanzen J, Anyrhambatla G, Kong G, Borelli M, Corry P, Dewhirst MW, Li CY. Noninvasive visualization of tumors in rodent dorsal skin window chambers. Nat Biotechnol. 1999;17:1033–5. doi: 10.1038/13736. [DOI] [PubMed] [Google Scholar]
- 42.Marigo I, Dolcetti L, Serafini P, Zanovello P, Bronte V. Tumor-induced tolerance and immune suppression by myeloid derived suppressor cells. Immunol Rev. 2008;222:162–79. doi: 10.1111/j.1600-065X.2008.00602.x. [DOI] [PubMed] [Google Scholar]
- 43.Serafini P, Borrello I, Bronte V. Myeloid suppressor cells in cancer: recruitment, phenotype, properties, and mechanisms of immune suppression. Semin Cancer Biol. 2006;16:53–65. doi: 10.1016/j.semcancer.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 44.Sica A, Schioppa T, Mantovani A, Allavena P. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: potential targets of anti-cancer therapy. Eur J Cancer. 2006;42:717–27. doi: 10.1016/j.ejca.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 45.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 46.Orimo A, Weinberg RA. Stromal fibroblasts in cancer: a novel tumor-promoting cell type. Cell Cycle. 2006;5:1597–601. doi: 10.4161/cc.5.15.3112. [DOI] [PubMed] [Google Scholar]
- 47.Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature. 2004;432:332–7. doi: 10.1038/nature03096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Noonan DM, De Lerma Barbaro A, Vannini N, Mortara L, Albini A. Inflammation, inflammatory cells and angiogenesis: decisions and indecisions. Cancer Metastasis Rev. 2008;27:31–40. doi: 10.1007/s10555-007-9108-5. [DOI] [PubMed] [Google Scholar]
- 49.Purhonen S, Palm J, Rossi D, Kaskenpää N, Rajantie I, Ylä-Herttuala S, Alitalo K, Weissman IL, Salven P. Bone marrow-derived circulating endothelial precursors do not contribute to vascular endothelium and are not needed for tumor growth. Proc Natl Acad Sci U S A. 2008;105:6620–5. doi: 10.1073/pnas.0710516105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lieberman J. Cell-Mediated Cytotoxicity in Fundamental Immunology., W. Paul, Editor. 2013, Paul, W., Lippincott-Williams & Wilkins: Philadelphia, PA. p. 891-909. [Google Scholar]
- 51.Kamoun WS, Chae SS, Lacorre DA, Tyrrell JA, Mitre M, Gillissen MA, Fukumura D, Jain RK, Munn LL. Simultaneous measurement of RBC velocity, flux, hematocrit and shear rate in vascular networks. Nat Methods. 2010;7:655–60. doi: 10.1038/nmeth.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Khong HT, Wang QJ, Rosenberg SA. Identification of multiple antigens recognized by tumor-infiltrating lymphocytes from a single patient: tumor escape by antigen loss and loss of MHC expression. J Immunother. 2004;27:184–90. doi: 10.1097/00002371-200405000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lehmann F, Marchand M, Hainaut P, Pouillart P, Sastre X, Ikeda H, Boon T, Coulie PG. Differences in the antigens recognized by cytolytic T cells on two successive metastases of a melanoma patient are consistent with immune selection. Eur J Immunol. 1995;25:340–7. doi: 10.1002/eji.1830250206. [DOI] [PubMed] [Google Scholar]
- 54.Yee C, Thompson JA, Byrd D, Riddell SR, Roche P, Celis E, Greenberg PD. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci U S A. 2002;99:16168–73. doi: 10.1073/pnas.242600099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sanderson NS, Puntel M, Kroeger KM, Bondale NS, Swerdlow M, Iranmanesh N, Yagita H, Ibrahim A, Castro MG, Lowenstein PR. Cytotoxic immunological synapses do not restrict the action of interferon-γ to antigenic target cells. Proc Natl Acad Sci U S A. 2012;109:7835–40. doi: 10.1073/pnas.1116058109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Blohm U, Potthoff D, van der Kogel AJ, Pircher H. Solid tumors “melt” from the inside after successful CD8 T cell attack. Eur J Immunol. 2006;36:468–77. doi: 10.1002/eji.200526175. [DOI] [PubMed] [Google Scholar]
- 57.Gerbitz A, Sukumar M, Helm F, Wilke A, Friese C, Fahrenwaldt C, Lehmann FM, Loddenkemper C, Kammertoens T, Mautner J, et al. Stromal interferon-γ signaling and cross-presentation are required to eliminate antigen-loss variants of B cell lymphomas in mice. PLoS One. 2012;7:e34552. doi: 10.1371/journal.pone.0034552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Briesemeister D, Sommermeyer D, Loddenkemper C, Loew R, Uckert W, Blankenstein T, Kammertoens T. Tumor rejection by local interferon gamma induction in established tumors is associated with blood vessel destruction and necrosis. Int J Cancer. 2011;128:371–8. doi: 10.1002/ijc.25350. [DOI] [PubMed] [Google Scholar]
- 59.Listopad JJ, Kammertoens T, Anders K, Silkenstedt B, Willimsky G, Schmidt K, Kuehl AA, Loddenkemper C, Blankenstein T. Fas expression by tumor stroma is required for cancer eradication. Proc Natl Acad Sci U S A. 2013;110:2276–81. doi: 10.1073/pnas.1218295110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Urban JL, Shepard HM, Rothstein JL, Sugarman BJ, Schreiber H. Tumor necrosis factor: a potent effector molecule for tumor cell killing by activated macrophages. Proc Natl Acad Sci U S A. 1986;83:5233–7. doi: 10.1073/pnas.83.14.5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Thilenius AR, Sabelko-Downes KA, Russell JH. The role of the antigen-presenting cell in Fas-mediated direct and bystander killing: potential in vivo function of Fas in experimental allergic encephalomyelitis. J Immunol. 1999;162:643–50. [PubMed] [Google Scholar]
- 62.Pear WS, Nolan GP, Scott ML, Baltimore D. Production of high-titer helper-free retroviruses by transient transfection. Proc Natl Acad Sci U S A. 1993;90:8392–6. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Algire GH, Legallais FY. Recent developments in the transparent-chamber technique as adapted to the mouse. J Natl Cancer Inst. 1949;10:225–53, 8. [PubMed] [Google Scholar]
- 64.Li Y, Song Y, Zhao L, Gaidosh G, Laties AM, Wen R. Direct labeling and visualization of blood vessels with lipophilic carbocyanine dye DiI. Nat Protoc. 2008;3:1703–8. doi: 10.1038/nprot.2008.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.