

Abstract
The incidence of metabolic disease, including type 2 diabetes and obesity, has increased to epidemic levels in recent years. A growing body of evidence suggests that the intrauterine environment plays a key role in the development of metabolic disease in offspring. Among other perturbations in early life, alteration in the provision of nutrients has profound and lasting effects on the long term health and well being of offspring. Rodent and non-human primate models provide a means to understand the underlying mechanisms of this programming effect. These different models demonstrate converging effects of a maternal high fat diet on insulin and glucose metabolism, energy balance, cardiovascular function and adiposity in offspring. Furthermore, evidence suggests that the early life environment can result in epigenetic changes that set the stage for alterations in key pathways of metabolism that lead to type 2 diabetes or obesity. Identifying and understanding the causal factors responsible for this metabolic dysregulation is vital to curtailing these epidemics.
Introduction
The thrifty phenotype hypothesis of Hales and Barker proposes that poor nutrition in early life results in poor fetal growth and increased susceptibility to type 2 diabetes and the metabolic syndrome [1, 2]. Evidence from epidemiological studies as well as animal models suggest that the intrauterine environment plays a role in the development of obesity and metabolic disorders. These studies demonstrate that suboptimal maternal nutrition, whether under-nutrition or over-nutrition, has negative effects on the offspring [3-9]. Fetal development is dependent on maternal supply of nutrients. Thus, alterations in maternal metabolism would expose the fetus to a perturbed intrauterine milieu that may predispose offspring to metabolic disease in later life. This concept, known as developmental origins of health and disease (DOHaD) states that a stimulus or insult at a critical period of developmental plasticity in early life causes disruptions in normal growth and development, as a result one genotype can give rise to different phenotypes in response to different environmental conditions during development [10-12]. Furthermore, a mismatch between the pre- and postnatal environment may result in inappropriate adaptations and subsequent metabolic disease, also known as the predictive adaptive response (PAR) hypothesis [13]. In animal models there is a commonality in the phenotypic outcomes of offspring from mothers exposed to either under- or over-nutrition. This phenotype is usually associated with poor fetal growth, mediated by the insulin growth factor axis, followed by the development of metabolic disease later in life [14-16].
Although initial studies on the thrifty phenotype hypothesis focused predominantly on the effects of maternal under-nutrition such as a diet low in protein or calories [17-19], recent studies have sought to investigate the effects of maternal over-nutrition. A maternal high fat diet (HFD) is more reflective of the dietary habits in western society. A maternal HFD can alter the development of various organs and thus the offspring become more susceptible to disease in later life. It has been shown that not only are dietary exposures during pregnancy important in mediating this disease susceptibility but also the timing of this exposure during development [20, 21]. Thus, since the fetus or neonate is undergoing rapid cell division, tissues may be affected differently by the same exposure depending on the timing of the insult.
Animal models provide an invaluable tool to study the underlying mechanisms of developmental programming. Rodent and non-human primates (NHP) are mammalian systems with similar embryology, anatomy and physiology to humans. Utilization of these systems has led to a greater understanding of the underlying mechanisms of DOHaD. Rodents are advantageous to study due for practical and economic reasons and the effect of manipulation of offspring can be seen within a short time. In contrast, NHPs are non-litter bearing animals with similarity in development to humans, however, they have the disadvantage of requiring considerable resources to maintain and have a lengthy gestation and lifespan.
The phenotypic outcome of the offspring exposed to a maternal HFD during development varies based on the species, diet composition, timing and length of HFD consumption. In addition, maternal factors such as obesity, the presence of glucose intolerance, diabetes and insulin resistance may contribute independently of the diet to the phenotype observed in the offspring. Thus, most of the methods used to develop animals models of developmental programming can be confounded by the presence of more than one insult. Similarly, other dietary components such as protein, carbohydrates and micronutrients have to be considered. To be able to maintain the same caloric content, changes in one component may result in the increase or reduction of another [22].
Prior DOHaD reviews [23] have either focused on maternal over-nutrition (combining studies on HFD, high sugar or junk food diet) [24], maternal obesity [25, 26] or “only” one outcome i.e., how maternal HFD causes epigenetic changes [27]. This review focuses on animal models that demonstrate offspring effects resulting from exposure to a maternal HFD irrespective of maternal obesity. The PubMed database was searched for articles published between 1995 and 2013 using the key terms high fat in utero and maternal high fat programming. Articles obtained from this search are discussed in this review. A brief summary of all the known metabolic and molecular changes that have been identified from animal models of a HFD in utero are summarized in Table 1.
Table 1.
Effect of a maternal high fat diet on offspring phenotype.
| Animal | Duration of Maternal HFD | Composition | Effect (offspring) | Organ affected (offspring) |
Ref |
|---|---|---|---|---|---|
| Mouse | 2 weeks prior to mating, during G and L |
36% of calories from fat |
Decreased fetal growth and birth weight, increased expression of hepatic lipogenic genes | Liver | 28 |
| 4, 12 or 23 weeks prior to mating |
60% of calories from fat |
No effect on hepatic inflammation or expression of genes involved in lipid metabolism | - | 36 | |
| 8 weeks prior to mating, during G and L |
32% of calories from fat |
Increased fetal weight and placental GLUT1 and SNAT2 mRNA | Placenta | 29 | |
| 8 weeks prior to mating, during G |
60% of calories from fat |
Type 2 diabetes in F1 and F2, higher β-cell mass in F1, lower β-cell mass in F2 | Pancreas | 51 | |
| During P and L until 3 months |
49% of calories from fat |
Insulin resistance, increased hepatic triglycerides, hepatic steatosis | Liver | 41 | |
| 1 week prior to mating, during G and L |
35% of calories from fat |
Metabolic syndrome, hepatic lipid accumulation, increased hepatic JNK phosphorylation | Liver | 30 | |
| 4 weeks prior to conception during G and L |
45% of calories from fat |
Reduced mitochondrial function, increased hepatic lipogenesis, oxidative stress, changes the expression of inflammatory genes |
Liver | 47 | |
| During G, L, G and L | 49% of calories from fat |
Insulin resistance, increased expression of hepatic lipogenic genes | Liver | 37 | |
| 6 weeks prior to mating, during G and L |
53% of calories from fat |
Increased birth weight, increased protein levels in fatty acid oxidation, altered hepatic microRNA |
Liver | 48, 49 | |
| 6 weeks prior to mating, during G and L |
18% fat (w/w) | Hypertension, hepatic steatosis | Liver | 33 | |
| During G and L | 18% fat (w/w) | Effects on bone quantity and quality | Bone | 52 | |
| 3 weeks prior to mating, during G and L |
60% of calories from fat |
Increased anxiety behavior, molecular changes in the hippocampus | Brain | 53 | |
| During G and L | 20% fat (PUFA or SFA) |
SFA increased total plasma cholesterol and low density lipoprotein cholesterol | - | 42 | |
| During G and L | 20% w/w (PUFA or SFA) |
Reduced hepatic and heart DHA content | Liver, Heart | 43 | |
| 4 weeks prior to mating | 62% of calories from fat |
Increased blood pressure and body weight, altered histone modification of ADIPOQ promoter |
Adipose | 95 | |
| 12 weeks prior to mating, during G and L |
60% of calories from fat |
Decrease in global methylation in brain | Brain | 96 | |
| During G | 45% of calories from fat |
Increased birth weight and glucose level, altered histone modification of PCK1 gene | Liver | 98 | |
| 2 weeks prior to mating, during G and L |
21% fat (w/w) | Altered vascular function and CpG methylation of FADS2 promoter | Liver, aorta | 99, 100 | |
| 4 weeks prior to mating, during G and L |
45% of calories from fat |
Reduced insulin sensitivity, altered growth hormone GH axis, increased body length, altered expression of paternally imprinted genes in F3 |
Liver, Brain | 31, 103 | |
| Rat | During G and L | 40% of calories from fat |
No difference in birth weight | - | 38 |
| From postnatal day 24, during G and L |
60% of calories from fat |
Increased body weight, glucose and insulin levels, and insulin secretion | Pancreas | 54 | |
| From pregnancy to postnatal day 10 |
21% fat (w/w) | Increased serum insulin and leptin, increased blood pressure and left ventricular wall thickness |
Left ventricle | 62 | |
| 4 weeks prior to mating, during G and L |
60% of calories from fat |
Increased adiposity and serum leptin | - | 57 | |
| 10 days prior to mating, during G and L |
33% fat (w/w) | Reduced serum DHA | - | 64 | |
| 15 days prior to mating, during G and L |
40% (w/w) | Decreased hepatic mtDNA copy number and PGC1α mRNA expression | Liver | 59 | |
| 6 weeks prior to mating, during G |
35% of calories from fat |
Increased serum leptin, body weight and alterations in food motivated behavior | - | 60 | |
| 10 days prior to mating, during G and L |
26% of calories from fat |
Increased blood pressure, altered aortic structure and renal function, insulin resistance, lower heart rate, endothelial dysfunction, and altered vascular fatty acid content |
Aorta |
58, 65- 70 |
|
| 6 weeks prior to mating until the end of weaning |
40% of calories from fat |
Increased body weight, serum leptin and insulin, and reduced hypothalamic leptin- dependent STAT3 phosphorylation |
Brain | 71 | |
| 11 weeks prior to mating (from postnatal 23), G |
60% of calories from fat |
Increased serum leptin and insulin, and expression of hypothalamic appetite-regulating genes |
Brain | 72 | |
| Started embryonic day 6 | 50% of calories from fat |
Precocious puberty, increased proliferation of neuroepithelial and neuronal precursor cells | Brain | 73 | |
| 8 weeks prior to mating, during G and L |
29% of calories from fat |
Increased body weight, blood glucose and leptin, reduced phospho-STAT3 and SOCS3 expression in the arcuate nucleus |
Brain | 55 | |
| From gestational day 2 | 60% of calories from fat |
Increased body weight and serum leptin, and reduced phosphorylation of STAT3 in the arcuate nucleus |
Brain | 56 | |
| Defined periods of G and L | 40% of calories from fat |
Increased serum glucose and reduced pancreatic glucokinase mRNA | Pancreas | 74 | |
| During G, L or G and L | 40% of calories from fat |
Increased serum glucose, increased serum leptin, and pancreatic β-cell hyperplasia and hypertrophy |
Pancreas | 61, 75 | |
| During the different period of G |
40% of calories from fat |
Hyperglycemic, impaired insulin release, changes β-cell volume and number, reduced PDX-1 and GK expression in β-cells |
Pancreas | 76, 77 | |
| 2 weeks prior to mating, during G and L |
18% fat (w/w) | Reduced birth weight and weight gain on unsaturated fatty acids diet, and fewer large islets on saturated fatty acids diet |
Pancreas | 78 | |
| 4 weeks prior to mating, during G |
59% of calories from fat |
Increased serum insulin and altered expression of insulin signaling proteins in skeletal muscle |
Skeletal muscle |
63 | |
| 10 weeks prior to mating | 40% of calories from fat |
Increased abdominal adiposity, serum leptin and insulin | - | 108 | |
|
NHP |
Up to 4 years prior to mating |
32% of calories from fat |
Altered fetal metabolite profile, fetal thyroid axis, expression and methylation of hypothalamic gene, decreased fetal HDAC activity, SIRT1 gene expression and activity, decreased fetal hepatic apoptosis, increased placental inflammatory cytokines, placental dysfunction, early lower body weight and decreased lean mass, increased serum leptin, early onset obesity, increased proinflammatory cytokine levels, modulated serotogenic system, increased hepatic expression of the gluconeogenic enzymes and transcription factor mRNA |
Liver |
81, 82, 84-89, 93, 94 |
| Up to 4 years prior to mating | 36% of calories from fat |
Increased serum insulin | Vasucular abnormality |
83 |
Abbreviations: (G: gestation, L: lactation)
Mouse
Using mouse as a model, several studies have demonstrated adverse metabolic outcomes in offspring exposed to a maternal HFD. Included are models of poor fetal growth and adverse metabolic events including liver dysfunction, high blood pressure, alterations in bone structure and effects on behavior and or emotional health.
In the mother, HFD has been shown to alter maternal food intake, substrate utilization, body composition, glucose and lipid metabolism, leptin levels and placenta nutrient transport [26, 28-31]. Hartil et al. has demonstrated that different metabolic outcomes in offspring such as catch up growth, increased adiposity and impaired glucose tolerance and insulin sensitivity was associated with maternal metabolic dysfunction illustrated by increased serum levels of nonesterified fatty acids (NEFA) and lactate accompanied by increased expression of peroxisome proliferator activated receptor γ coactivator-1β (PGC1β) mRNA in liver [28]. In contrast, glucose transporter 4 heterozygous (GLUT4+/−) females (a model of insulin resistance) consuming the same diet display enhanced lipid clearance and no significant alterations in expression of genes of glucose or lipid metabolism in their liver, suggesting that the mother’s ability to adequately utilize different diets may play a role on the programming effects seen in the offspring.
In addition to acute alteration in maternal substrate utilization, chronic high fat feeding leading to maternal obesity is another factor that can have adverse effects on the offspring. This is cause for concern because of the significant increase of obesity in women of child-bearing age [32]. Animal models that have investigated the effect of maternal obesity on the offspring have reported hyperphagia, adiposity, hypertension and insulin resistance in the offspring [33-35] dyslipidemia and hepatic steatosis [33]. Krasnow et. al. has shown that switching obese mothers to a low fat diet (LFD) for a subsequent pregnancy can minimize the metabolic effects seen by the HFD/maternal obesity in C57BL/6 mice [36]. Similar findings have been observed by introducing the HFD shortly before mating, without changing maternal body composition [26, 28, 37, 38]. These data suggest a maternal HFD has adverse effects on the offspring, whether or not maternal obesity is present and that it is possible to reverse these effects by switching the diet.
HFD diet during pregnancy has been shown to alter the placenta leading to either small or large fetuses [29, 39]. While some studies report HFD offspring being born smaller than controls [40], others report the opposite. The differences may be due to different dietary components, fat source or the strain of mice used. Despite being born small, some studies report catch up growth in the HFD growth restricted offspring, this in itself is also associated with the development of metabolic disease [28]. Pregnancies associated with large fetuses are accompanied by upregulation of placental nutrient transport (glucose and neutral amino acids) compared to offspring whose mothers consumed a control diet [29]. HFD increased glucose transporter 1 (GLUT1) and sodium coupled neutral amino acid transporter 2 (SNAT2) protein expression in trophoblast microvillus plasma membranes isolated from the placentas of HFD animals at embryonic day 18.5 [29]. These data suggest nutrients are the most important factors during development.
Decreased fetal body weight has been associated with early catch up growth. Using dams that were WT or heterozygous for GLUT4 (G4+/−) expression, a decrease in birth weight was observed in offspring born to both genotypic mothers that consumed the HFD irrespective of genotype. At embryonic day 18.5 these offspring had decreased fetal weight and crown rump length [40]. Only the WT male offspring born to WT mothers consuming a HFD demonstrated early life catch up growth (CUG) and increased adiposity accompanied by impaired glucose tolerance and insulin sensitivity despite their being weaned to a LFD. Furthermore, these offspring had increased serum glucose and PAI-1 and decreased adiponectin [40]. Conversely, WT male offspring born to GLUT4+/− mothers that consumed a HFD were protected from precocious manifestation of these characteristics of the Metabolic Syndrome [28]. These data suggest that although offspring exposed to a maternal HFD are born small, this may not always translate to a negative metabolic outcome. Early life CUG appears to have a negative impact on the offspring. Furthermore, the mother’s ability to utilize the HFD appears to be a determining factor in whether or not the offspring is susceptible to metabolic disease.
Metabolic Syndrome, a cluster of disorders that increase the risk for diabetes and cardiovascular disease, is the common phenotype seen in models of DOHaD. Offspring exposed to a maternal HFD in utero and/or postnatally developed manifestations of the Metabolic Syndrome, such as insulin resistance, increased liver mass and triglyceride content, hepatic steatosis, increased visceral fat mass and adipocyte hypertrophy but not central leptin resistance [30, 41]. These changes are accompanied by increased serum concentrations of tumor necrosis factor α (TNFα) and interleukin 1β (IL-1β) [30]. These data suggest serum factors may play a role in the mechanisms involved in the development of the Metabolic Syndrome in HFD exposed offspring.
Changes in blood pressure (increase in systolic blood pressure), another feature of the Metabolic Syndrome, were seen in offspring exposed to a maternal HFD [33]. Similarly, exposing G4+/− mice to a HFD in utero was associated with a premature increase in systolic blood pressure as early as 13 wks of age [40]. These data demonstrate that a HFD during pregnancy as well as a heterozygous deletion of GLUT4 increases offspring susceptibility to increased systolic blood pressure.
Similar to changes in blood pressure, alterations in the offspring plasma lipids are seen in response to a maternal HFD. In a study by Elahi et al., plasma lipids were increased in a sexually dimorphic manner. In female offspring exposed to either maternal control (C) or HFD and weaned to a HFD have increased cholesterol levels whereas only males exposed to a maternal HFD in utero have increased cholesterol [33]. Changes in offspring lipid levels have also been seen after the exposure to different dietary fats, specifically exposure to saturated fatty acids (SFA). The group in which the mothers and offspring were exposed to SFA had the highest plasma concentrations of total cholesterol and low-density lipoprotein (LDL) cholesterol while the polyunsaturated fatty acid exposed group had the lowest levels of total cholesterol and LDL cholesterol compared to the other groups. The plasma high-density lipoprotein (HDL) cholesterol concentrations were significantly higher in the animals exposed to polyunsaturated fatty acids (PUFA) in utero and weaned to SFA [42]. This demonstrates a maternal HFD affects lipid metabolism of the offspring and alters their lipid profile, but the effect differs depending on the quality of fat as well as the sex of the offspring. Furthermore, a PUFA or SFA diet throughout gestation and lactation was associated with reduced docosahexaenoic acid (DHA) in both liver and heart in comparison to control [43].
Non-alcoholic fatty liver disease (NAFLD) encompasses a spectrum of liver disorders from steatosis to nonalcoholic steatohepatitis (NASH) associated with insulin resistance and the metabolic syndrome [44-46]. In the offspring, maternal HFD exposure has been associated with increased triglyceride content, increased phosphoenolpyruvate carboxykinase 1 (PCK1) gene expression and increased c-jun N-terminal kinase (JNK) and IκB kinase (IKK) phosphorylation which is consistent with development of NAFLD. In addition, maternal HFD exposure has been shown to reduce hepatic insulin signaling and basal acetyl-CoA carboxylase (ACC) phosphorylation in the offspring [30]. Similar changes are observed even when the offspring are weaned to a non-HFD. Interestingly, exposure to HFD in utero has a more detrimental effect on the liver of the offspring than does HFD after weaning [41]. Moderate steatosis and NASH has been shown to be associated with increased expression of genes of lipogenesis, oxidative stress and inflammation, in addition to alterations in activity of enzymes of the electron transport chain [37, 47]. Further supporting that maternal HFD predisposes the offspring to NAFLD, offspring exposed to a HFD developed hepatic steatosis; which was more pronounced in the offspring exposed to HFD during gestation, lactation and postweaning. HFD exposed offspring also had increased expression of sterol regulatory element binding protein -1c (SREBP-1c) suggesting an effect on hepatic lipogenesis [37]. Offspring that are weaned to the HFD appear to have a more pronounced effect suggesting that there may be a “two hit ” effect. a maternal HFD enhances offspring susceptibility to liver dysfunction and NAFLD. Interestingly, in another study there was no effect of maternal diet on hepatic inflammation or expression of lipid metabolism genes [36]. It is plausible that the negative outcomes reported in this study could be due to the young age of the offspring.
In the fetus, a maternal HFD was associated with increased hepatic expression of genes involved in glycolysis, gluconeogenesis, inflammation and oxidative stress [40]. This provides insight into the mechanism(s) underlying insulin resistance and metabolic disease. The upregulation of genes involved in glycolysis and gluconeogenesis suggest that the offspring is being programmed for hepatic insulin resistance. Additionally, inflammation and oxidative stress has been implicated in the development of metabolic disease.
Consistent with prior studies, female sex may have a protective effect on the programming of the offspring liver. In female offspring, exposure to a HFD in utero resulted in a reduction in hepatic triglyceride content and increased hepatic protein levels for cluster of differentiation 36 (CD36), carnitine palmitoyltransferase-1 (CPT-1) and peroxisomal proliferator activated receptor α (PPARα) in an IGF2 dependent upon manner [48]. These changes can lead to upregulation of fatty acid oxidation in the liver, which is different from that observed in male offspring. HFD exposure in utero was also associated with altered expression of miRNA in the HFD exposed female offspring [48]. These data suggest liver is a major target for programming during development [30, 37, 41, 44-49].
Maternal HFD in utero has been shown to enhance offspring susceptibility to type 2 diabetes [50]. One potential mechanism for this involves alterations in pancreatic beta cell mass which could potentially be transmitted to subsequent generations. Despite changes in diet, type 2 diabetes was seen in F1 mice exposed to the HFD during fetal or neonatal life and F2 mice whose mothers were exposed to the HFD during fetal life. In the F1 generation, pancreatic beta cell mass, replication, and neogenesis were increased in animals exposed to HFD. In contrast, F2 mice had lower beta cell mass. In the F1 generation, the exposure to HFD in utero had an even more dramatic effect when the HFD was also provided during the neonatal period [51]. These data suggest exposure to a HFD during fetal life alters beta cell structure and function thereby resulting in type 2 diabetes and this is transmissible to their offspring (F2 generation) in the absence of an additional insult.
HF exposure in utero results in remodeling of the bone structure of the offspring in a sexually dimorphic manner. HFD has been shown to affect bone structure and length [52]. Bone marrow adiposity was increased and femur length decreased in male and female offspring. Female offspring also demonstrated alterations in trabecular structure [52]. These data provide insight into the effect of a maternal HFD on the skeletal structure of the offspring and how these alterations may lead to the development of future disease such as osteoporosis.
Little is known about the effect of HFD exposure in utero on animal behavior [53]. HFD exposed offspring displayed increased anxiety (reduced time in open arms of elevated plus maze as well as increased inhibition to consume novel food) associated with molecular changes in the hippocampus (increased expression of bone-derived neurotrophic factor (BDNF and GABAA alpha2 receptor subunit (GABAA α2R) and 5-hydroxytryptamine 1A (5HT1A) [53]. These data suggest a maternal HFD may increase anxiety behaviors in the offspring via alterations in the GABAergic and neurotrophin systems in early life.
In conclusion, in HFD mouse models of DOHaD, it is clear that there may be critical periods of vulnerability to the suboptimal environment. In utero exposure seems to have a more dramatic effect offspring programming. It is also clear that offspring have a sexually dimorphic response to HFD exposure. In the mother, intake of a HFD resulted in alterations in substrate utilization and/or changes on body composition. Maternal HFD exposure does not have to be accompanied by maternal obesity to impart metabolic changes to their offspring. In the offspring, in utero exposure to HFD resulted in placenta dysfunction associated with alterations in fetal growth, obesity, insulin resistance, hypertension, NAFLD, and altered pancreatic beta cell mass. These phenotypic outcomes are associated with changes in organ structure as well as gene expression in metabolically important organs (e.g., liver). Further emphasizing the relevance of these early life perturbations with respect to the rise in the metabolic disease incidence, these changes may be passed on to subsequent generations without additional exposure to the HFD.
Rat
The interaction of pregnancy and dietary fat on the pregnancy outcome as well as the metabolism in the offspring exposed to a maternal HFD has also been investigated using rat models. In the field of DOHaD, Sprague-Dawley and Wistar rats are the two most frequently used rat strains. When compared to mouse models, rats present several advantages including that they are bigger and easier to handle and they could potentially be more resistant to insults. The pregnancy duration is longer and the fetuses are larger. In contrast to many commonly used inbred strains of mice, Sprague-Dawley and Wistar rats are outbred strains. Similar to mouse models, maternal HFD exposure in rats produces similar effects on the metabolic phenotype of the offspring as well as organ specific effects including the liver and pancreas as well as hypothalamic changes.
The Sprague-Dawley dams exposed to a HFD during pregnancy experienced increased body weight and elevated plasma insulin, glucose and triglycerides levels [54], these effects are similar to what is reported in the mouse [29-31]. In addition, HF exposure has been shown to alter the quality of breast milk. HFD breast milk had increased protein, cholesterol, triglycerides [55] and leptin concentration [56, 57]. These data suggest alterations in maternal milk composition may contribute to offspring obesity.
In the rat, a maternal HFD has been shown to reduce fetal growth and to alter the placental structure by reducing the placental junctional zone, without altering the placental labyrinth zone [39]. These changes were not accompanied by alterations in gene expression of perosixome proliferator-activated receptor gamma (PPARγ) and vascular endothelial growth factor A (VEGFa) which are markers of placental vascular development. This study suggests mechanisms responsible for fetal growth restriction may include effects on placental growth.
Similar to a HFD during pregnancy, maternal obesity secondary to prolonged HF feeding was associated with an increase in offspring adiposity and increased serum leptin as well as reduced insulin tolerance [57]. Consistent with mouse models, HFD in utero is sex dependent and seems to be more detrimental that exposure to HFD during lactation. Cross-fostering studies showed that in males in utero HFD exposure results in an increase in adiposity independent of the diet exposure during lactation. In contrast, in females, significant differences in adiposity were associated with exposure to HFD in utero and during lactation [58, 59]. The post-weaning diet also appears to affect offspring body composition. When exposed to a HFD in utero and weaned to a “junk food” diet, female offspring from borderline hypertensive fathers are hyperphagic and have greater fat pad mass compared to controls [60]. They also had increased fasting serum leptin and insulin levels. This suggests there is enhanced susceptibility metabolic disease when offspring exposed in utero to a HFD are weaned to a junk food diet.
Similar to mouse models, HFD rat fetuses had enhanced susceptibility to the Metabolic Syndrome, HFD fetuses display hyperinsulinemia and altered insulin secretory response to insulin secretagogues [54]. As adults, HFD offspring had increased adiposity, increased liver weight and hepatic liver lipid content, increased blood glucose [61], triglycerides [38] and plasma corticosterone levels, increased adipocyte area and left ventricular wall thickness [62]. In addition, HFD offspring were glucose intolerant, and their pancreatic islets, secreted more insulin in response to low glucose stimuli. When weaned to a high sucrose diet HF offspring displayed an impaired metabolic profile (increased body weight, impaired glucose tolerance and increased FFA compared to those weaned to LFD [54]. These data further support the negative effects of a maternal HFD and its contribution to metabolic disease in the offspring. Furthermore, if the offspring are also weaned to an adverse diet, this worsens their metabolic profile.
Similar to mouse models the diet composition plays a major role in the offspring phenotype. A diet rich in omega-6 polyunsaturated fat programs offspring for Metabolic Syndome. Specifically, early life exposure to a diet rich on omega-6 PUFA diet has been shown to increase total and abdominal adiposity. Hyperinsulinemia and altered hepatic triglyceride content, in association with changes in expression of several insulin signaling genes in the skeletal muscle, suggest insulin resistance is also present [63]. These data demonstrate that adverse maternal nutrition results in multiple negative outcomes that program offspring for metabolic disease in later life.
This susceptibility to the Metabolic Syndrome, is further enhanced by alterations in the hepatic mitochondria content in response to the HFD exposure. Wistar rats exposed to HFD during pregnancy had a decreased copy number of mitochondrial DNA in liver regardless of offspring sex or weaning diet. There was a male specific reduction of hepatic PGC1α mRNA which had a significant effect on peripheral insulin resistance (higher HOMA-IR) with the opposite effect being observed in the female offspring [59]. Furthermore, exposure to a HFD in utero resulted in a reduction in DHA in liver lipids in the offspring [64]. This suggests a maternal HFD results in alterations in the fatty acid profile of the liver and a decrease in the levels of an essential lipid.
As observed in mice, several rat models have also demonstrated altered cardiovascular function following exposure to a maternal HFD. Studies have reported increases in diastolic, systolic and mean arterial blood pressure in older female offspring exposed to a maternal HFD [65]. In contrast, another study using female offspring from borderline hypertensive fathers exposed to a HFD in utero and weaned to either control or a “junk food” diet reported lower mean arterial and diastolic blood pressure in HFD exposed offspring in comparison to controls. Whereas there was no difference in blood pressure in offspring placed on different postweaning diets [60]. While the results from both studies are not consistent, the effect of the borderline hypertensive father used in one mating strategy cannot be ignored. Furthermore, these studies clearly demonstrate that a HFD in utero results in cardiovascular dysfunction. Additionally, cross-fostering studies showed that in utero HFD exposed male offspring suckled by a control dam compared with HFD offspring suckled by the same dam had no difference in blood pressure. However, female offspring of fat fed dams suckled by a control dam and control offspring suckled by a fat fed dam were hypertensive [58]. There appears to be a sex-dependent susceptibility to these cardiovascular outcomes, with the female offspring being more susceptible.
All offspring exposed to the HFD during gestation or during suckling demonstrated endothelial dysfunction seen as a blunting of acetylcholine (Ach) induced relaxation in mesenteric arteries [58]. HFD exposure was associated with a reduction in the maximal endothelial-dependent relaxation to Ach in small mesenteric arteries in comparison to control animals [65]. Furthermore, there was a reduction in aortic endothelial cell layer volume and smooth muscle number, increased aortic stiffness and reduced endothelium-dependent relaxation in comparison to control. In the kidney, glomerular number and volume was altered depending on the maternal diet. Additionally, renin and Na+, K+-ATPase activity was reduced in offspring exposed to a maternal HFD [66]. This suggests that a maternal HFD programs hypertension via alterations in the vascular and renal structure of the offspring. Follow up studies in the HFD adult female offspring demonstrated a decrease in mtDNA content in kidney and alterations in the expression of genes such as AJ223355 (mitochondrial dicarboxylate carrier) and D12770 (mitochondrial adenine nucleotide transporter) in aorta that suggested mitochondrial dysfunction [67].
Consistent with vascular dysfunction the femoral arteries of offspring of HFD diabetic mothers fed a HFD during pregnancy, had an enhanced sensitivity to norephinephrine with a decreased or blunted arterial relaxation in response to Ach [68, 69]. These alterations, which enhance offspring susceptibility to cardiovascular dysfunction, were accompanied by changes in the fatty acid composition of the aortas including decreased arachidonic acid and DHA content, which are cardioprotective fatty acids [69]. Diabetes was induced via streptozocin on day 1 or 2 of pregnancy. In this case there are two possible mechanisms for the programming effects seen in the offspring; the maternal HFD and the maternal diabetes.
The predictive adaptive response (PAR) was investigated in a Sprague-Dawley rat model [70]. The outcomes demonstrate that the PAR to a maternal HFD prevents endothelial dysfunction but not hypertension in the adult offspring of Sprague-Dawley rats. There was a partial protection from the cardiovascular effects of maternal HFD exposure displayed by the offspring when re-exposed to a HFD in later life displayed. However, the female offspring were hypertensive when maintained on the same HFD diet as their mothers [70]. This study suggests the adverse effects of maternal HFD diet on blood pressure may be irreversible.
The impact of maternal HFD on hypothalamic leptin sensitivity has been investigated in HFD rat models. Male and female offspring exposed to a HFD in utero and weaned to a HFD had altered feed efficiency [71]. In addition, in offspring exposed to HFD in utero, STAT3 phosphorylation in response to leptin was abolished suggesting hypothalamic leptin resistance [71]. Similarly, decreased STAT3 and IRS-2 protein expression was detected in the hypothalamus of HFD exposed offspring [72]. These changes were accompanied by an increase in mRNA levels of insulin receptor β (IRβ) and leptin long receptor (OB-Rb). Alterations in gene expression were also detected in the hypothalamus. Specifically, increased mRNA levels of neuropeptide Y (NPY), Agouti-related peptide (AGRP) pro-opiatemelanocortin (POMC), and melanocortin receptor-4 (MC4R) were reported. This outcome is consistent with hypothalamic dysregulation after exposure to a HFD in utero being associated with upregulation of the orexigenic (appetite stimulating) neuropeptides during early development. In addition, HFD in utero stimulated the proliferation of neuroepithelial and neuronal precursor cells of the embryonic hypothalamic third ventricle, as well as stimulated proliferation and differentiation of neurons and their migration toward hypothalamic areas [73]. HFD increased expression of orexigenic peptides, galanin, enkephalin and dynorphin in the paraventricular nucleus (PVN) and orexin and melanin-concentrating hormone in the perifornical lateral hypothalamus (PFLH) of the offspring [73].. In addition to changes in the appetite centers, females developed precocious puberty (day 29-32 in comparison to day 33-36 in BD offspring). This data suggests the mechanisms involved in hyperphagia and increased adiposity seen in offspring exposed to HFD in utero may include increased neurogenesis as well as stimulation of hypothalamic peptides.
Several studies have investigated the effects of a HFD at different periods within gestation and lactation. A HFD exposure during critical periods of development alters pancreatic morphology and function. Changes in glucokinase (GK) and pancreatic and duodenal homeobox 1 (PDX-1) immunoreactivity expression has been observed in offspring exposed to a HFD in utero. When HFD diet exposure was limited to the gestation period PDX-1 and GK mRNA expression and immunoreactivity were unaffected and the impairment in glucose tolerance was less pronounced [74]. HFD exposure during development was also associated with pancreatic alpha and beta cell hyperplasia, increased pancreatic acinar cell proliferation and changes in beta and alpha cell size [75, 76] as well as impaired glucose stimulated insulin release [77]. This suggests an adverse programming effect on beta cell function may predispose offspring to beta cell failure.
The effect of fatty acid composition of the maternal diet on fetal and postnatal growth, and pancreas morphology was examined [78]. A HFD rich in unsaturated fat (UFA: menhaden fish oil) was associated with an increase in pancreatic islet numbers. A diet rich in saturated fat (SFA: coconut oil) was associated with a reduction in the number of large islets and a faster and more robust insulin response to a glucose load [78]. The programming effect on the pancreas alters the structure and function of islets associated with a predisposition towards glucose intolerance and diabetes. These data demonstrate the fatty acid composition of the maternal diet is important as the diets rich in saturated fatty acids and unsaturated fatty acids had opposite effects on pancreatic islet development and insulin response of offspring.
In conclusion, despite the size and breed differences between the mouse and rats, in rodent models, no major differences have been observed in the phenotype of their respective offspring exposed to a maternal HFD. In both models, the effects were seen in a sex-dependent manner during critical stages of development. Similar to the mouse model, a maternal HFD in rats affects offspring body composition, glucose and lipid metabolism, endothelial and vascular function and blood pressure, liver function, pancreatic morphology, pancreatic function and hypothalamic signaling. Hypothalamic reprogramming of offspring exposed to a maternal HFD results in altered expression of hypothalamic peptides that regulate food intake and weight gain that, in the long term, may affect offspring behavior and physiology.
Summarized in Figure 1 are the effects of a maternal HFD on various organ systems of exposed offspring derived from several animal models. These effects vary based on the animal model, timing and duration of the HFD exposure, as well as the gender of the offspring. The programming effects are further enhanced if the exposure is extended to the post-weaning period. Although not implicated in these studies, future work in the field could point to the gut microbiota as a potential source of programming [79, 80].
Figure 1.

Organ-specific pathologies associated with exposure to a maternal HF diet.
Non-human primates
The effect of a maternal HFD on the offspring has also been investigated in non-human primates (NHP). Similar to the rodent models, NHPs exposed to a maternal HFD is associated with predisposition to metabolic disease associated with effects in multiple organ systems (including liver and placenta), as well as circulating factors in the mother and her offspring. Additionally, several of these studies have focused on epigenetic factors that may explain the increased susceptibility to metabolic disease in response to early life exposure to HFD. Many of these changes are observed during fetal life (prior to birth), which suggests that the intrauterine environment is a period vulnerable to nutrient perturbations.
At 5-7 years of age, female Japanese macaques were fed a C or a HFD for 2-4 years. A subset of these NHPs were switched from HFD to normal chow 1–3 months before becoming pregnant in the fifth year of the study [81]. Animals fed the HFD were divided into two groups based on their weight gain and insulin resistance and were considered either sensitive to the HFD (HFD-S) or resistant to the HFD (HFD-R). Approximately 60% of the NHPs were categorized as HFD-S. The fetal (e130) offspring of HFD (O-HFD) NHPs had lower body weight compared to CD offspring, which was likely due to decreased lean body mass. These data suggest, regardless of the mother’s insulin status, a HFD in utero results in the offspring being born smaller than control.
The consumption of a HFD during pregnancy was shown to alter fetal placental hemodynamics [82]. Uterine hemodynamic parameters were examined in early third-trimester pregnancies (gestational day 120). Uterine artery blood flow volume was decreased in both HFD-R (38% reduction) and HFD-S (56% reduction). A maternal HFD resulted in expression of increased placental inflammatory cytokines (IL-1β and Monocyte Chemotactic Protein-1 (MCP-1) and increased expression of toll-like receptor 4 (TLR4R) associated with an increase in the frequency of placental infarctions and stillbirth. These data suggest that maternal obesity and insulin resistance associated with chronic consumption of a HFD exacerbates placental dysfunction and results in an increased frequency of stillbirth seen in HFD pregnancies in general [82]. In agreement to what is seen in rodents, a maternal HFD results in placental alterations. In the rodent, studies reported alterations in placenta structure and nutrient transport, whereas placental inflammation was investigated in NHPs. The potential role of placenta in offspring programming is very important as maternal factors can cross the placenta and affect the fetus. Any placenta dysfunction can affect fetal growth and development. In the NHP inflammation of the placenta resulted in adverse effects including increased stillbirth.
The impact of a maternal HFD on cardiovascular disease was examined in 13 month old offspring by investigating endothelial function [83]. The mothers were fed the same diet discussed in previous studies and after weaning the offspring were either maintained on the same diet (CD/CD, HFD/HFD), or switched to the opposite diet (CD/HFD, HFD/CD). HFD/HFD juveniles showed increased plasma insulin level and glucose-stimulated insulin secretion compared to CD/CD. In abdominal aorta, HFD/HFD juveniles showed decreased Ach-induced vasorelaxation compared to CD/CD. HFD/HFD animals also showed a thicker intima wall and an abnormal vascular morphology. mRNA expression of proinflammatory factors such as vascular endothelial growth factor A (VEGFA), TNFα and Intercellular adhesion molecule 1 (ICAM-1) were increased in HFD/HFD compared to CD/CD in the abdominal aorta tissue. Post-weaning diet reversal (HFD/CD) did not reverse the expression of VEGFA or TNFα, however, ICAM-1 tended to be lower in the HFD/CD animals compared to HFD/HFD. These data suggest a maternal HFD exposure, as well as a post-weaning HFD exposure, affects endothelial function and this is partially reversed by switching to the CD [83].
Using NHP it has been demonstrated that a maternal HFD is associated with decreased plasma n–3 fatty acids (FA) and fetal hepatic apoptosis [84]. Lipid analysis of fetal and maternal plasma revealed total n-3 FA and DHA levels were significantly lower in the HFD compared to the CD group. Also, the n-6:n-3 ratio was increased and correlated with the maternal ratio in the HFD group. Furthermore, the number of apoptotic cells was increased in the HFD fetal liver compared to CD and switching the diet normalized the fetal DHA, n-3 FAs and hepatic apoptosis to CD levels. The HFD also had an effect on the breast milk composition such that mothers consuming a HFD displayed lower levels of eicosopentanoic acid and DHA [84]. Alterations in the composition of milk have also been reported in the rodent. These compositional changes can be a potential mechanism underlying offspring phenotypic outcomes.
Fetal O-HFD had increased triglycerides and increased evidence of hepatic oxidative stress that are early signs of NAFLD, similar to the what is reported for rodent models. Increased triglyceride levels and fatty liver is a feature of maternal HFD programming that occurs in rodent and NHP models. Furthermore, when the offspring are also weaned to a HFD they have a more adverse effect [81]. Exposure to a maternal HFD resulted in increased liver triglycerides and histologic evidence of NAFLD. These hepatic changes were associated with significant hyperacetylation at H3K14 and a trend towards increased H3K9 and H3K18 acetylation in fetal HFD liver [85]. Fetal HDAC1 mRNA and protein expression were significantly decreased, as was in vitro HDAC activity. Fetal hepatic AcH3K14 ‘reprogrammed’ genes (Glutamic pyruvate transaminase 2 (GPT2), DnaJ homolog subfamily A member 2 (DNAJA2) and Retinol dehydrogenase 12 (RDH12)) were increased by exposure to a maternal HFD [85]. These data suggest epigenetic modifications involving alterations to chromatin structure may be the molecular basis of the outcomes seen in offspring exposed to a maternal HFD.
Glycerol levels were higher in O-HFD NHPs compared to CD fetal offspring. Fetal OHFD exhibited elevated hepatic expression of the gluconeogenic enzymes and transcription factors, glucose 6-phosphatase (G6P), fructose-1,6-bisphosphatase 1 (FBP1), PCK1, PGC1α, and hepatocyte nuclear factor 4α (HNF4α) protein was significantly elevated compared to CD liver. Maternal diet reversal improved fetal hepatic triglyceride levels and partially normalized the mRNA expression of the gluconeogenic genes. Furthermore, the offspring had normal total triglyceride and glycerol levels [81]. Metabolomic analysis of the fetal serum revealed significant alterations in the HFD exposed offspring [86]. Twenty two metabolites had statistical significance over the entire study and eight metabolites (2-hydroxybutyrate, ascorbic acid, α-tocopherol, cholesterol, 3-hydroxybutyrate, un(1462_100), un(1574_304) and un(1952_334) were differentially present in serum of HFD-exposed compared CD offspring [86]. These data suggest alterations in the fetal metabolite profile occur in response to a maternal HFD and may play a role in the development of metabolic disease.
Since sirtuin 1 (SIRT1) is a lysine deacetylase and has been shown to play a role in cellular metabolism, it was hypothesized that SIRT1 may be involved in the altered H3K14ac seen in liver of HFD offspring. Fetal hepatic SIRT1 gene expression, protein level, and activity were all found to be dysregulated following exposure to a maternal HFD [87]. In addition, the SIRT1-associated genes, peroxisome proliferator-activated receptor gamma (PPARγ), PPARα, sterol regulatory element-binding transcription factor 1 (SREBF1), cholesterol 7 alpha-hydroxylase (CYP7A1), fatty acid synthase (FAS) and stearoyl-CoA desaturase (SCD) were increased in the fetal macaque liver with HFD exposure. These findings suggest that SIRT1 likely plays a key role in remodeling the fetal hepatic epigenome and metabolome in a HFD milieu [87]. These are all key genes involved in fat and cholesterol metabolism and may correlate fetal lipid oxidation and NAFLD [87].
Using the same experimental paradigm the effects of fetal exposure to HFD on the development of the melanocortin system in the hypothalamus was investigated [88]. Third-trimester fetuses from mothers on HFD showed increased POMC and decreased AGRP mRNA expression in the hypothalamus. A cytokine array showed eight inflammatory factors and receptors, namely IL-1β, Interleukin-1 receptor type I (IL-1R1), Interleukin-1 family member 6 (IL-1F6), Interleukin-1 family member 7 (IL-1F7), Chemokine (C-C motif) ligand 26 (CCL26), Chemokine (C-C motif) receptor 3 (CCR3), Chemokine (C-C motif) ligand 19 (CCL19) and Chemokine (C-C motif) ligand 2 (CCL2) were increased in the hypothalamus of fetus exposed to a maternal HFD. Reversing the diet normalized fetal POMC and AGRP mRNA expression as well as circulating cortisol and cytokine levels. These results suggest chronic consumption of a HFD during pregnancy leads to widespread activation of proinflammatory cytokines that affect development of the melanocortin system in a way that can be reversed by a healthy maternal diet [88].
The effect of exposure to a maternal HFD on the peripheral circadian system has also been examined in this NHP model [89]. Exposure to a maternal HFD resulted in alteration in fetal hepatic neuronal PAS domain protein 2 (NPAS2), a paralog of the clock transcription factor, gene expression and switching to a LF maternal diet normalized this effect. Expression of other circadian genes was also affected by exposure to a maternal HFD. In particular, period circadian protein homolog 1 (PER1) and nuclear receptor subfamily 1, group D, member 1(Reverb-α) mRNAs were up-regulated in fetal liver of HFD compared to CD NHPs. The NPAS2 promoter did not display alterations in DNA methylation at any CpG dinucleotide between CD and HFD exposed animals suggesting another epigenetic modification may be responsible for alteration in NPAS2 gene expression. To address the hypothesis that the change in NPAS2 gene expression may be due to alterations in histone modifications in the functional promoter, an antibody against H3K14 was used. There was significant enrichment for the Rev-erb-α/ROR response element promoter region, which drives NPAS2 transcription, in NHPs exposed in utero to a maternal HFD compared to CD as well as significant increase in enrichment of promoter occupancy with the gene activating mark, H3K14ac [89]. These changes in expression of circadian genes, and the observed epigenetic modifications, may be important in programming metabolic disease since disruption of circadian rhythmicity is associated with obesity and cardiovascular disease [90-92].
Thyroid hormone homeostasis was investigated in the maternal HFD NHP model because of its effects on development and energy balance [93]. Fetal free T3 was not changed but free T4 was significantly decreased in the maternal HFD exposed offspring compared to CD offspring. Since thyroid hormones are important regulators of fetal development and energy balance, alterations in the thyroid hormone homeostasis could be an underlying mechanism for the development of metabolic disease in later life. Analysis of expression of genes involved in thyroid function (thyrotropin releasing hormone (TRH), thyroid stimulating hormone receptor (TSHR), thyroglobulin (TRHR), thyroid peroxidase (TPO), and sodium iodide symporter/solute carrier family 5 member 5 (SLC5A5)) showed a decrease in the hypothalamus and thyroid gland. Additionally, transcription of the deiodinase iodothyroine (DIO) gene that helps to maintain thyroid homeostasis was dysregulated in the fetal liver, thyroid and hypothalamus. Chromatin immunoprecipitation (ChIP) showed that the thyroid response element (TRE) of the thyroid hormone receptor beta (THRB) is associated with an increase in deacetylation of histone H3 at lysines 9 and 14 (H3K9,14ac) in liver of NHPs exposed to a maternal HFD. This data suggests a maternal HFD modifies the histone code and thus alters the fetal thyroid axis and thyroid gene expression [93].
The serotonergic system, which is involved in behavioral disorders, was also investigated in NHP fetuses on gestational day 130 (G130; early 3rd trimester; full term is 175 days) from CD and HFD mothers [94]. Maternal HFD consumption was associated with perturbations in the serotonergic system of fetal offspring. Female, but not male, HFD offspring exhibited increased latency to touch a potentially threatening novel object as compared to CD offspring while none of the CD offspring or male HFD offspring exhibited increased latency. HFD female offspring also took longer than CD female offspring to touch a threatening object, while male CD and HFD offspring were not different. These results show that a maternal HFD increases anxiety-like behavior in a sexually dimorphic manner [94]. Similarly, in mice [53], increased anxiety and alterations in brain circuitry suggest a maternal HFD increases the risk of developing behavioral disorders.
In conclusion, these studies demonstrate that NHP offspring exposed to a maternal HFD have altered glucose metabolism, liver and thyroid functions, circadian rhythm, hepatic gene expression and histone modifications. Thus, the same the phenotypic changes observed in rodents in response to a maternal HFD are manifested in this NPH model that is much closer to humans. Interestingly, in the NHP models many of the negative effects induced by the maternal HFD were reversible when the diet was switched postnatally or prior to pregnancy. Studies using rodent models demonstrate that exposure to a HFD in utero has a more profound effect than exposure during lactation only. While switching the diets did not completely reverse the effects, improved metabolic outcomes were observed in the offspring.
Epigenetic Effects
Epigenetics is the study of heritable changes in gene expression without changes in the underlying DNA sequence. Epigenetic effects have also been examined in rodent models exposed of maternal HFD exposure. As described above in the NHP, many of the phenotypic changes observed in the HFD exposed offspring could be linked to alterations of the epigenome [85, 87, 89, 93]. Histone modifications at the adiponectin (ADIPOQ) and leptin (LEP) genes were examined in mice exposed to a maternal HFD [95]. Female 8 wk old ICR mice were consumed either a CD (12% fat) or HFD (62% fat) for 4 weeks and males were fed CD for 4 weeks before mating. All offspring were weaned to CD at postnatal day 21. OH mice (offspring from mothers exposed to an HFD during pregnancy) were heavier than OC (offspring from mothers exposed to an CD during pregnancy) mice. Systolic blood pressure in OH mice was elevated, and they had impaired glucose tolerance compared to OC mice. Serum leptin and triglyceride were higher and total triglyceride lower compared to OC. This was in association with increased LEP and ADIPOQ mRNA expression in white adipose tissue of OH. The acetyl H3K9 level in the ADIPOQ promoter region was lower and the dimethyl H3K9 was increased in adipose tissue in OH mice compared to OC mice. On the other hand, the monomethyl H4K20 level was significantly increased in the LEP promoter region of OH compared to OC mice [95]. This study demonstrates that maternal HFD induced metabolic syndrome leads to epigenetic modifications of adiponectin and leptin and their gene expression.
Vucetic et al. have demonstrated that there are alterations in the gene expression and methylation of the hypothalamic dopamine genes using the C57BL/6J mouse model of maternal HFD [96]. Females were maintained on either CD (12% fat) or HFD (60% fat) for 3 months before breeding with DBA/2J males. HFD was maintained through lactation and offspring were weaned to the CD. Expression of the preproenkephalin (PENK) and opioid receptor mu 1 (MOR) genes were increased nucleus accumbens, prefrontal cortex and hypothalamus. This was correlated with hypomethylation of the promoter region of these genes in the nucleus accumbens (NAc) and hypothalamus. Similarly, dopamine reuptake transporter (SLC6A3) mRNA was increased in the NAc and this was also associated with hypomethylation of the SLC6A3 promoter region [96]. These results show that alterations in CpG methylation of DNA is associated with altered expression genes in brain regions and with energy balance and motivated feeding behavior present in offspring exposed to a maternal HFD and they demonstrated a preference for palatable foods. This is a plausible mechanism for epigenetic transfer of the phenotype as the maternal HFD was able to change the methylation marks of the offspring. Additionally, these results are largely consistent with what is known about effects of CpG methylation of DNA in a promoter on gene expression [97].
Fetal gluconeogenic gene expression and potential regulatory mechanisms related to histone modifications were examined in an obesity resistant strain of rats (Crl:OR(CD)) [98]. Females consumed a CD (16% fat) or a HFD (45% fat) throughout gestation. Hepatic mRNA expression of PCK1 was higher in HFD compared to CD offspring. ChIP analysis of the hepatic PCK1 promoter showed the decreasing of H3Ac, H3K4Me2, H3K9Me3 and H3K27Me3 [98], though the analysis of PCK1 coding region and upstream showed the increasing of H4Ac and H3K4Me2. These results suggest that maternal HFD leads to certain histone modifications that are associated with transcriptional activation of a main control point for the regulation of gluconeogenesis that could result in elevated glucose production and reduced insulin sensitivity.
To address the question if the variation in the type and amount of maternal HFD leads to epigenetic regulation of hepatic fatty acid desaturase 2 (FADS2) gene expression, female Wistar rats were fed either 3.5% (LFD), 7% [adequate fat (AFD)] or 21% (HFD) butter or fish oil (FO) from 14 days before mating and throughout pregnancy and lactation [99]. Offspring were weaned to a diet containing 4% fat (soybean oil). Increasing the amount of fat in the maternal diet decreased FADS2 mRNA expression in liver of offspring. The methylation status of the proximal promoter of FADS2 was increased with the amount of fat in the maternal diet, while the level of methylation tended to be greater in offspring of mothers fed FO compared to those fed butter fat. Methylation of CpG −394 showed a strong negative correlation with FADS2 mRNA expression in offspring, while CpGs – 623, −84 and −76 correlated weakly with FADS2 mRNA expression. Methylation of CpG −394, but not CpGs −623, −84 and – 76 correlated negatively with the proportions of 20:4n-6 and 22:6n-3 in liver PC and PE of the offspring [99]. Further studies have revealed that there were significant single factor effects of the amount of maternal dietary fat on the methylation of CpG -394 of the FADS2 gene [100]. These data demonstrate that alterations in the amount and type of maternal dietary fat may result in epigenetic modifications in the offspring.
All of these studies show that exposure to a maternal HFD is correlated with epigenetic changes in the offspring correlated with in alterations in gene expression and dysregulation of various key metabolic pathways. The mechanism(s) by which metabolic disease develops is quite complex and involves multiple factors, however alteration of the epigenome appears to play a significant role in this process.
Figure 2 summarizes the pathways by which exposure to a maternal HF diet may alter offspring development and manifestation of metabolic disease. Specifically, exposure to a maternal HF diet may reprogram offspring development through epigenetic modifications that impart a molecular memory of an adverse early life milieu. Maternal HF diet is associated with altered fetal growth and a precocious increase in adiposity and insulin resistance. Also, pathologic effects upon serum metabolites and cytokines are observed in exposed offspring as well as alterations in gene expression and tissue morphology in major organs. In sum, these changes dramatically increase offspring risk for development of metabolic disease. This highlights the importance of the maternal environment and its impact on the life long health of the offspring.
Figure 2.
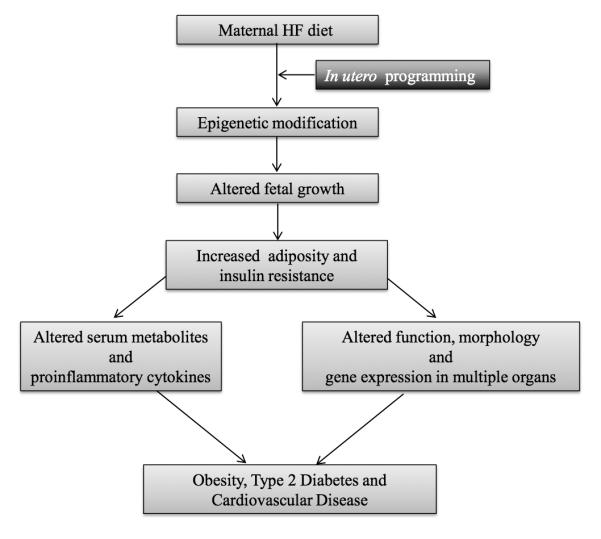
Overview of events that occur in response to exposure to a maternal HF diet and increased susceptibility to metabolic disease.
Paternal Effect
While this article focuses on the programming effects of a maternal HFD, the contribution of the father cannot be ignored [101]. In an article by Ng et al., it was shown using Sprague–Dawley rats that chronic paternal consumption of a HFD had profound effects on the pancreatic beta-cell function of female offspring. In this study, male Sprague–Dawley founder rats fed either CD or HFD (43% fat) were mated with females consuming a CD. The female offspring from HFD fed fathers had increased blood glucose and reduced insulin secretion. A paternal HFD reduced relative islet area and beta-cell area in female offspring that is typically associated with impaired beta-cell replication. Pancreatic gene expression revealed differentially expressed genes involved in calcium signaling pathway mitogen activated protein kinase (MAPK) pathway and wingless type MMTV integration site (Wnt) signaling pathways, apoptosis, and the cell cycle [102]. Dunn and Bale also reported that the effect of a HFD was passed through the paternal lineage in C57Bl/6:129 hybrid mice [31, 103]. Several other studies also reported that HFD induced paternal obesity is associated with impaired embryo development and diminished reproductive health of their offspring [104-107]. This impaired development coincided with embryonic genome activation suggesting paternal obesity affects sperm, possibly at the genetic or epigenetic level. These data demonstrate that paternal HFD has a negative effect on the offspring and that female offspring may be more susceptible to this perturbation.
In C57Bl/6:129 hybrid mice, a maternal HFD (45% fat) resulted in increased body length and decreased insulin sensitivity in the offspring for two generations and that these outcomes were transmitted via the maternal or the paternal lineage. Mothers were fed HFD for 4 wks prior to mating and throughout gestation and lactation and control mice were fed a chow diet (12% fat) [31]. Both E17 embryos and male and female HFD exposed adult offspring had increased body length in comparison to control offspring. In the F2 generation the effect of in utero HFD exposure to fathers was investigated and the effects on the offspring were enhanced if both maternal and paternal lines were exposed to HFD in utero versus only one or the other. Offspring exposed to maternal or paternal HFD showed decreased insulin sensitivity in the F1 and F2 generations. F1 and F2 females exposed to HFD in utero had increased plasma levels of IGF-I and increased hypothalamic expression of Growth hormone secretagogue receptor (GHSR). Methylation studies of GHSR on micropunches of the arcuate nucleus showed decreased methylation at the GHSR promoter in both maternal and paternal lineages [31]. This study was extended to the F3 generation since the primordial germ cells of the first generation mice may have been affected by the gestational exposure to HFD. The effect of HFD on weight and length only occurred in females and was only passed through the paternal lineage [103]. Imprinted genes showed a trend in paternally expressed genes to have a “greater volatility of expression” (genes that show greater than 50% deviation from baseline) in comparison to maternally expressed genes [103]. These data clearly demonstrate both maternal and paternal HFD can influence offspring susceptibility to metabolic disease. The mechanisms underlying this susceptibility may be explained via epigenetic modifications perhaps at the level of imprinted genes.
In a study using Sprague-Dawley rats exposed to a HFD (from both mother and father) increased body fat accumulation was observed in male offspring. Male and female rats were fed a HFD (40% fat) or LFD (5% fat) and mated to the same diet group [108]. The offspring were nurtured by their own mothers and weaned to a HFD or LFD (experiment 1), by an F344 chow fed foster mother (experiment 2), or 2 days after mating, the fertilized eggs were collected from HFD or LFD mothers and transplanted to the oviducts of a sham pregnant foster mother fed a 12% fat diet (experiment 3) and subsequently weaned to a HFD only for 12-17 weeks (experiments 2,3). The total body weight and abdominal tissue weight was significantly greater in male offspring of HFD parents regardless of weaning diet and plasma leptin concentrations were increased in comparison to control. Both HFD-LFD and HFD-HFD offspring had increased food intake and food efficiency however, weaning to the HFD enhanced this effect [108]. Although the maternal and paternal contributions were not separated in this study the various parental contributions to programmed metabolic disturbances in offspring should not be ignored.
These studies demonstrate that, as with the mother, paternal environmental exposures can also influence the health of their offspring; though maternal and paternal programming effects vary in their potency.
Sexual dimorphism
The studies discussed above predominantly focus on the male offspring. However, studies that investigate the effects of a maternal HFD on both sexes demonstrate sex-specific responses to nutrient perturbation during development [33, 49, 51, 52]. Female hormones may play a protective role in the development of insulin resistance and associated disorders and may be the basis of some of the results seen. However, other studies have shown that the females are more susceptible, especially when the insult is from the paternal lineage [102, 103]. Not only are these changes seen after birth, several studies reported sex specific differences in the placenta during fetal life. Mao et al. examined the impact of diet and fetal sex on placental gene expression in NIH Swiss female mice fed CD, LFD or HFD [109]. At day 12.5 of pregnancy, the placenta gene expression patterns of male offspring clearly clustered separately from the placenta of females and the female placenta showed more striking changes in gene expression in response to maternal diet than male placenta [110]. Gallou-Kabani et al. also reported the effect of HFD on sexual dimorphism in the placenta. Twenty imprinted genes were examined and sexual dimorphism and sensitivity to diet were observed for nine of these genes. Additionally, exposure to a HFD during gestation triggered sex-specific epigenetic alterations within CpG and throughout the genome [111]. Another study in mice also identified differential expression at imprinted genes and suggested that these may be at least partially responsible for the sex-specific differences between offspring [103].
These studies suggest the mechanism for developing metabolic disease may be sex-dependent, with one sex being more sensitive/susceptible than the other to a particular perturbation. The mechanism for this susceptibility may involve sex-dependent epigenetic modifications. This emphasizes the importance of studying the response of both sexes to dietary interventions.
Transgenerational effect
As discussed above, maternal HFD leads to obesity and metabolic syndrome in the first-generation (F1) offspring. Furthermore, recent studies show that the F1 can transmit a similar phenotype to the second (F2) or third generation (F3). Using C57BL/6:129 hybrid mice, Dunn and Bale reported that maternal HFD exposure results in the increasing of body length in the F2 generation and also found that the heritable feature of reduced insulin sensitivity could be seen for at least two generations [31]. Moreover, they found the increase in body size was transmitted to F3 females [103]. The imprinted gene array detected a potential dynamic pattern of paternally expressed genes from the paternal lineage that was not noted in the maternal lineage.
These studies demonstrate that either the maternal or the paternal environment can affect programming events at imprinted loci associated with developmental regulation of growth and body size in future generations.
Concluding Remarks
It is well known that the maternal nutritional environment plays an important role in determining offspring susceptibility to metabolic diseases. The studies reviewed here demonstrate that maternal HFD exposure during pregnancy and/or lactation increases offspring risk for the development of metabolic diseases such as type 2 diabetes and obesity. Direct evidence linking specific HFD exposure and metabolic disease in humans is limited, however animal models, such as those described here, provide significant insight into the molecular changes associated with this condition, while implicating an epigenetic basis for these diseases. Interestingly, independent of the animal model studied, all models showed similar phenotypic as well as structural changes in metabolically relevant organs as well as molecular changes when exposed to a HFD in utero. The changes were sex specific, happened during a critical period of development, were transmitted by the mother as well as the father, and were seen in subsequent generations. In addition, these symptoms were accompanied by changes in expression of key genes involved in metabolic and inflammatory pathways and were sometimes associated with changes in DNA methylation or histone post-translational modifications. Thus, even though the mechanism by which a HFD influences the health of the offspring is not clear, disruption of normal epigenetic regulation is likely involved.
Unfortunately, the precise functional role of the epigenetic changes related to a HFD exposure in utero and the increase in disease susceptibility is not well understood and requires further investigation. Part of the issue is limited not simply to practical issues concerning study design (type of diet, timing, sex) and the confounding effects of genetic variation (animal model and background) but also to the difficulty associated with interpreting the epigenetic outcomes.
Epigenetic modifications associated thus far with in utero programming associated with a maternal HFD include DNA methylation and histone modifications. Other epigenetic changes, such as chromatin structural patterns, gene expression changes controlled by RNAi, hydroxymethylation [100], or O-linked N-acetylglucosamine transferase changes [101-103], although central to the topic of epigenetics, have not been studied widely in the context of HFD exposure and DOHaD. Thus, future studies aimed at elucidating involvement of these processes in the context of metabolic disease pathogenesis associated with early life exposure to HFD may shed new light on molecular mechanisms underlying this epidemic.
Although these animal models have extended our knowledge of the epigenetic mechanisms associated with metabolic disease, particularly DNA methylation changes, several questions still remain. One such question is whether additional pathologies linked to a HFD in utero can be correlated with epigenetic changes other than DNA methylation and what animal model is best suited to studies on fetal programming of adult disease. It also remains to be determined whether similar outcomes are encountered in human populations. If so, dietary changes during a critical period of development should be identified with the aim to develop future guidelines for a healthy pregnancy. Meanwhile, the growing body of knowledge derived from animal models suggests cautionary measures should be taken regarding dietary habits during pregnancy and postnatal development to minimize offspring risk for enhanced susceptibility to develop metabolic disease as adults.
Acknowledgements
The authors wish to thank Ms. Lovisa Heyman for critical reading of this review and current and past members of the Charron laboratory for fruitful discussions on the subject of this review. Generous support of this work was obtained from the following agencies: National Institutes of Health (R21 DK081194 to MJC, KO8 HD042172 to PMV, F31 DK093332 to LW), Diabetes Research and Training Center P60 DK020541, Epigenomics, Liver, O’Brien Kidney, and Comprehensive Cancer Centers of Albert Einstein College of Medicine), Diabetes Action Foundation and American Diabetes Association.
References
- [1].Hales CN, Barker DJ. Type 2 (non-insulin-dependent) diabetes mellitus: the thrifty phenotype hypothesis. Diabetologia. 1992;35:595–601. doi: 10.1007/BF00400248. [DOI] [PubMed] [Google Scholar]
- [2].Hales CN, Barker DJ. The thrifty phenotype hypothesis. Br. Med. Bull. 2001;60:5–20. doi: 10.1093/bmb/60.1.5. [DOI] [PubMed] [Google Scholar]
- [3].Yang Z, Huffman SL. Nutrition in pregnancy and early childhood and associations with obesity in developing countries. Matern. Child Nutr. 2013;9(Suppl 1):105–119. doi: 10.1111/mcn.12010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Muhlhausler BS, Ong ZY. The fetal origins of obesity: early origins of altered food intake. Endocr. Metab. Immune Disord. Drug Targets. 2011;11:189–197. doi: 10.2174/187153011796429835. [DOI] [PubMed] [Google Scholar]
- [5].Rooney K, Ozanne SE. Maternal over-nutrition and offspring obesity predisposition: targets for preventative interventions. Int. J. Obes. 2011;35:883–890. doi: 10.1038/ijo.2011.96. [DOI] [PubMed] [Google Scholar]
- [6].Tamashiro KL, Moran TH. Perinatal environment and its influences on metabolic programming of offspring. Physiol. Behav. 2010;100:560–566. doi: 10.1016/j.physbeh.2010.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Le Clair C, Abbi T, Sandhu H, Tappia PS. Impact of maternal undernutrition on diabetes and cardiovascular disease risk in adult offspring. Can. J Physiol. Pharmacol. 2009;87:161–179. doi: 10.1139/y09-006. [DOI] [PubMed] [Google Scholar]
- [8].Metges CC. Early nutrition and later obesity: animal models provide insights into mechanisms. Adv. Exp. Med. Biol. 2009;646:105–112. doi: 10.1007/978-1-4020-9173-5_11. [DOI] [PubMed] [Google Scholar]
- [9].Cerf ME. High fat programming of beta-cell failure. Adv. Exp. Med. Biol. 2010;654:77–89. doi: 10.1007/978-90-481-3271-3_5. [DOI] [PubMed] [Google Scholar]
- [10].Taylor PD, Poston L. Developmental programming of obesity in mammals. Exp. Physiol. 2007;92:287–298. doi: 10.1113/expphysiol.2005.032854. [DOI] [PubMed] [Google Scholar]
- [11].Barker DJ. The developmental origins of adult disease. J. Am. Coll. Nutr. 2004;23:588S–595S. doi: 10.1080/07315724.2004.10719428. [DOI] [PubMed] [Google Scholar]
- [12].Langley-Evans SC, McMullen S. Developmental origins of adult disease. Med. Princ. Prac. 2010;19:87–98. doi: 10.1159/000273066. [DOI] [PubMed] [Google Scholar]
- [13].Gluckman PD, Hanson MA. The developmental origins of the metabolic syndrome. Trends Endocrinol. Metab. 2004;15:183–187. doi: 10.1016/j.tem.2004.03.002. [DOI] [PubMed] [Google Scholar]
- [14].Larnkjaer A, Molgaard C, Michaelsen KF. Early nutrition impact on the insulin-like growth factor axis and later health consequences. Curr. Opin. Clin. Nutr. Metab. Care. 2012;15:285–292. doi: 10.1097/MCO.0b013e328351c472. [DOI] [PubMed] [Google Scholar]
- [15].Setia S, Sridhar MG. Changes in GH/IGF-1 axis in intrauterine growth retardation: consequences of fetal programming? Horm. Metab. Res. 2009;41:791–798. doi: 10.1055/s-0029-1231026. [DOI] [PubMed] [Google Scholar]
- [16].Chard T. Insulin-like growth factors and their binding proteins in normal and abnormal human fetal growth. Growth Regul. 1994;4:91–100. [PubMed] [Google Scholar]
- [17].Brawley L, Poston L, Hanson MA. Mechanisms underlying the programming of small artery dysfunction: review of the model using low protein diet in pregnancy in the rat. Arch. Physiol. Biochem. 2003;111:23–35. doi: 10.1076/apab.111.1.23.15138. [DOI] [PubMed] [Google Scholar]
- [18].Hoet JJ, Ozanne S, Reusens B. Influences of pre- and postnatal nutritional exposures on vascular/endocrine systems in animals. Environ. Health Perspec. 2000;108(Suppl 3):563–568. doi: 10.1289/ehp.00108s3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Langley-Evans SC. Fetal programming of cardiovascular function through exposure to maternal undernutrition. Proc. Nutr. Soc. 2001;60:505–513. doi: 10.1079/pns2001111. [DOI] [PubMed] [Google Scholar]
- [20].Lucas A. Programming by early nutrition: an experimental approach. J. Nutr. 1998;128:401S–406S. doi: 10.1093/jn/128.2.401S. [DOI] [PubMed] [Google Scholar]
- [21].Symonds ME, Stephenson T, Gardner DS, Budge H. Long-term effects of nutritional programming of the embryo and fetus: mechanisms and critical windows. Reprod. Fertil. Dev. 2007;19:53–63. doi: 10.1071/rd06130. [DOI] [PubMed] [Google Scholar]
- [22].Warden CH, Fisler JS. Comparisons of diets used in animal models of high-fat feeding. Cell Metab. 2008;7:277–277. doi: 10.1016/j.cmet.2008.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Symonds ME, Budge H. Nutritional models of the developmental programming of adult health and disease. Proc. Nutr. Soc. 2009;68:173–178. doi: 10.1017/S0029665109001049. [DOI] [PubMed] [Google Scholar]
- [24].Alfaradhi MZ, Ozanne SE. Developmental programming in response to maternal overnutrition. Front. Genet. 2011;2:27. doi: 10.3389/fgene.2011.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Nathanielsz PW, Poston L, Taylor PD. In utero exposure to maternal obesity and diabetes: animal models that identify and characterize implications for future health. Clin. Perinatol. 2007;34:515–526. v. doi: 10.1016/j.clp.2007.09.005. [DOI] [PubMed] [Google Scholar]
- [26].Li M, Sloboda DM, Vickers MH. Maternal obesity and developmental programming of metabolic disorders in offspring: evidence from animal models. Exp. Diabetes Res. 2011;2011:592408. doi: 10.1155/2011/592408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Seki Y, Williams L, Vuguin PM, Charron MJ. Minireview: Epigenetic programming of diabetes and obesity: animal models. Endocrinology. 2012;153:1031–1038. doi: 10.1210/en.2011-1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hartil K, Vuguin PM, Kruse M, Schmuel E, Fiallo A, Vargas C, Warner MJ, Durand JL, Jelicks LA, Charron MJ. Maternal Substrate Utilization Programs the Development of the Metabolic Syndrome in Male Mice Exposed to High Fat In Utero. Pediatr. Res. 2009;66:368–373. doi: 10.1203/PDR.0b013e3181b33375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Jones HN, Woollett LA, Barbour N, Prasad PD, Powell TL, Jansson T. High-fat diet before and during pregnancy causes marked up-regulation of placental nutrient transport and fetal overgrowth in C57/BL6 mice. FASEB J. 2009;23:271–278. doi: 10.1096/fj.08-116889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ashino NG, Saito KN, Souza FD, Nakutz FS, Roman EA, Velloso LA, Torsoni AS, Torsoni MA. Maternal high-fat feeding through pregnancy and lactation predisposes mouse offspring to molecular insulin resistance and fatty liver. J. Nutr. Biochem. 2012;23:341–348. doi: 10.1016/j.jnutbio.2010.12.011. [DOI] [PubMed] [Google Scholar]
- [31].Dunn GA, Bale TL. Maternal high-fat diet promotes body length increases and insulin insensitivity in second-generation mice. Endocrinology. 2009;150:4999–5009. doi: 10.1210/en.2009-0500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Dong M, Zheng Q, Ford SP, Nathanielsz PW, Ren J. Maternal obesity, lipotoxicity and cardiovascular diseases in offspring. J. Mol. Cell. Cardiol. 2013;55:111–116. doi: 10.1016/j.yjmcc.2012.08.023. [DOI] [PubMed] [Google Scholar]
- [33].Elahi MM, Cagampang FR, Mukhtar D, Anthony FW, Ohri SK, Hanson MA. Long-term maternal high-fat feeding from weaning through pregnancy and lactation predisposes offspring to hypertension, raised plasma lipids and fatty liver in mice. Br. J. Nutr. 2009;102:514–519. doi: 10.1017/S000711450820749X. [DOI] [PubMed] [Google Scholar]
- [34].Samuelsson AM, Matthews PA, Argenton M, Christie MR, McConnell JM, Jansen EH, Piersma AH, Ozanne SE, Twinn DF, Remacle C, Rowlerson A, Poston L, Taylor PD. Diet-induced obesity in female mice leads to offspring hyperphagia, adiposity, hypertension, and insulin resistance: a novel murine model of developmental programming. Hypertension. 2008;51:383–392. doi: 10.1161/HYPERTENSIONAHA.107.101477. [DOI] [PubMed] [Google Scholar]
- [35].Nivoit P, Morens C, Van Assche FA, Jansen E, Poston L, Remacle C, Reusens B. Established diet-induced obesity in female rats leads to offspring hyperphagia, adiposity and insulin resistance. Diabetologia. 2009;52:1133–1142. doi: 10.1007/s00125-009-1316-9. [DOI] [PubMed] [Google Scholar]
- [36].Krasnow SM, Nguyen ML, Marks DL. Increased maternal fat consumption during pregnancy alters body composition in neonatal mice. Am. J Physiol. Endocrinol Metab. 2011;301:E1243–1253. doi: 10.1152/ajpendo.00261.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gregorio BM, Souza-Mello V, Carvalho JJ, Mandarim-de-Lacerda CA, Aguila MB. Maternal high-fat intake predisposes nonalcoholic fatty liver disease in C57BL/6 offspring. Am. J Obstet. Gynecol. 2010;203:495, e1–8. doi: 10.1016/j.ajog.2010.06.042. [DOI] [PubMed] [Google Scholar]
- [38].Guo F, Jen KLC. High fat feeding during pregnancy and lactation affects offspring metabolism in rats. Physiol. Behav. 1995;57:681–686. doi: 10.1016/0031-9384(94)00342-4. [DOI] [PubMed] [Google Scholar]
- [39].Mark PJ, Sisala C, Connor K, Patel R, Lewis JL, Vickers MH, Waddell BJ, Sloboda DM. A maternal high-fat diet in rat pregnancy reduces growth of the fetus and the placental junctional zone, but not placental labyrinth zone growth. J. Dev. Orig. Hlth. Dis. 2011;2:63–70. [Google Scholar]
- [40].Vuguin PM, Hartil K, Kruse M, Kaur H, Lin CL, Fiallo A, Glenn AS, Patel A, Williams L, Seki Y, Katz EB, Charron MJ. Shared effects of genetic and intrauterine and perinatal environment on the development of metabolic syndrome. PloS One. 2013;8:e63021. doi: 10.1371/journal.pone.0063021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Volpato AM, Schultz A, Magalhaes-da-Costa E, Correia ML, Aguila MB, Mandarim-de-Lacerda CA. Maternal High-Fat Diet Programs for Metabolic Disturbances in Offspring despite Leptin Sensitivity. Neuroendocrinology. 2012;96:272–284. doi: 10.1159/000336377. [DOI] [PubMed] [Google Scholar]
- [42].Chechi K, Cheema SK. Maternal diet rich in saturated fats has deleterious effects on plasma lipids of mice. Exp. Clinic. Cardiol. 2006;11:129–135. [PMC free article] [PubMed] [Google Scholar]
- [43].Chechi K, Herzberg GR, Cheema SK. Maternal dietary fat intake during gestation and lactation alters tissue fatty acid composition in the adult offspring of C57Bl/6 mice. Prostaglandins Leukot. Essent. Fatty Acids. 2010;83:97–104. doi: 10.1016/j.plefa.2010.06.001. [DOI] [PubMed] [Google Scholar]
- [44].Varela-Rey M, Embade N, Ariz U, Lu SC, Mato JM, Martinez-Chantar ML. Non-alcoholic steatohepatitis and animal models: understanding the human disease. Int. J. Biochem. Cell Biol. 2009;41:969–976. doi: 10.1016/j.biocel.2008.10.027. [DOI] [PubMed] [Google Scholar]
- [45].Jou J, Choi SS, Diehl AM. Mechanisms of disease progression in nonalcoholic fatty liver disease. Semin. Liver Dis. 2008;28:370–379. doi: 10.1055/s-0028-1091981. [DOI] [PubMed] [Google Scholar]
- [46].Musso G, Gambino R, Cassader M. Recent insights into hepatic lipid metabolism in non-alcoholic fatty liver disease (NAFLD) Prog. Lipid Res. 2009;48:1–26. doi: 10.1016/j.plipres.2008.08.001. [DOI] [PubMed] [Google Scholar]
- [47].Bruce KD, Cagampang FR, Argenton M, Zhang J, Ethirajan PL, Burdge GC, Bateman AC, Clough GF, Poston L, Hanson MA, McConnell JM, Byrne CD. Maternal high-fat feeding primes steatohepatitis in adult mice offspring, involving mitochondrial dysfunction and altered lipogenesis gene expression. Hepatology. 2009;50:1796–1808. doi: 10.1002/hep.23205. [DOI] [PubMed] [Google Scholar]
- [48].Zhang J, Zhang F, Didelot X, Bruce KD, Cagampang FR, Vatish M, Hanson M, Lehnert H, Ceriello A, Byrne CD. Maternal high fat diet during pregnancy and lactation alters hepatic expression of insulin like growth factor-2 and key microRNAs in the adult offspring. BMC Genomics. 2009;10:478. doi: 10.1186/1471-2164-10-478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Zhang J, Wang C, Terroni PL, Cagampang FR, Hanson M, Byrne CD. High-unsaturated-fat, high-protein, and low-carbohydrate diet during pregnancy and lactation modulates hepatic lipid metabolism in female adult offspring. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005;288:R112–118. doi: 10.1152/ajpregu.00351.2004. [DOI] [PubMed] [Google Scholar]
- [50].Ainge H, Thompson C, Ozanne SE, Rooney KB. A systematic review on animal models of maternal high fat feeding and offspring glycaemic control. Int. J. Obes. 2011;35:325–335. doi: 10.1038/ijo.2010.149. [DOI] [PubMed] [Google Scholar]
- [51].Gniuli D, Calcagno A, Caristo ME, Mancuso A, Macchi V, Mingrone G, Vettor R. Effects of high-fat diet exposure during fetal life on type 2 diabetes development in the progeny. J. Lipid Res. 2008;49:1936–1945. doi: 10.1194/jlr.M800033-JLR200. [DOI] [PubMed] [Google Scholar]
- [52].Lanham SA, Roberts C, Hollingworth T, Sreekumar R, Elahi MM, Cagampang FR, Hanson MA, Oreffo RO. Maternal high-fat diet: effects on offspring bone structure. Osteoporos. Int. 2010;21:1703–1714. doi: 10.1007/s00198-009-1118-4. [DOI] [PubMed] [Google Scholar]
- [53].Peleg-Raibstein D, Luca E, Wolfrum C. Maternal high-fat diet in mice programs emotional behavior in adulthood. Behav. Brain Res. 2012;233:398–404. doi: 10.1016/j.bbr.2012.05.027. [DOI] [PubMed] [Google Scholar]
- [54].Srinivasan M, Katewa SD, Palaniyappan A, Pandya JD, Patel MS. Maternal high-fat diet consumption results in fetal malprogramming predisposing to the onset of metabolic syndrome-like phenotype in adulthood. Am. J. Physiol. Endocrinol. Metab. 2006;291:E792–799. doi: 10.1152/ajpendo.00078.2006. [DOI] [PubMed] [Google Scholar]
- [55].Franco JG, Fernandes TP, Rocha CP, Calvino C, Pazos-Moura CC, Lisboa PC, Moura EG, Trevenzoli IH. Maternal high-fat diet induces obesity and adrenal and thyroid dysfunction in male rat offspring at weaning. J. Physiol. 2012;590:5503–5518. doi: 10.1113/jphysiol.2012.240655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Sun B, Purcell RH, Terrillion CE, Yan J, Moran TH, Tamashiro KL. Maternal high-fat diet during gestation or suckling differentially affects offspring leptin sensitivity and obesity. Diabetes. 2012;61:2833–2841. doi: 10.2337/db11-0957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].White CL, Purpera MN, Morrison CD. Maternal obesity is necessary for programming effect of high-fat diet on offspring. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009;296:R1464–R1472. doi: 10.1152/ajpregu.91015.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Khan IY, Dekou V, Douglas G, Jensen R, Hanson MA, Poston L, Taylor PD. A high-fat diet during rat pregnancy or suckling induces cardiovascular dysfunction in adult offspring. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005;288:R127–133. doi: 10.1152/ajpregu.00354.2004. [DOI] [PubMed] [Google Scholar]
- [59].Burgueno AL, Cabrerizo R, Mansilla N. Gonzales, Sookoian S, Pirola CJ. Maternal high-fat intake during pregnancy programs metabolic-syndrome-related phenotypes through liver mitochondrial DNA copy number and transcriptional activity of liver PPARGC1A. J. Nutr. Biochem. 2013;24:6–13. doi: 10.1016/j.jnutbio.2011.12.008. [DOI] [PubMed] [Google Scholar]
- [60].Mitra A, Alvers KM, Crump EM, Rowland NE. Effect of high-fat diet during gestation, lactation, or postweaning on physiological and behavioral indexes in borderline hypertensive rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009;296:R20–28. doi: 10.1152/ajpregu.90553.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Cerf ME, Louw J. High fat programming induces glucose intolerance in weanling Wistar rats. Horm. Metab. Res. 2010;42:307–310. doi: 10.1055/s-0030-1248303. [DOI] [PubMed] [Google Scholar]
- [62].Parente LB, Aguila MB, Mandarim-de-Lacerda CA. Deleterious effects of high-fat diet on perinatal and postweaning periods in adult rat offspring. Clin. Nutr. 2008;27:623–634. doi: 10.1016/j.clnu.2008.05.005. [DOI] [PubMed] [Google Scholar]
- [63].Buckley AJ, Keseru B, Briody J, Thompson M, Ozanne SE, Thompson CH. Altered body composition and metabolism in the male offspring of high fat-fed rats. Metabolism. 2005;54:500–507. doi: 10.1016/j.metabol.2004.11.003. [DOI] [PubMed] [Google Scholar]
- [64].Ghebremeskel K, Bitsanis D, Koukkou E, Lowy C, Poston L, Crawford MA. Maternal diet high in fat reduces docosahexaenoic acid in liver lipids of newborn and sucking rat pups. Br. J. Nutr. 1999;81:395–404. [PubMed] [Google Scholar]
- [65].Khan Y, Taylor P, Dekou V, Seed P, Lakasing L, Graham L, Dominiczak A, Hanson M, Poston L. Gender-linked hypertension in offspring of lard-fed pregnant rats. Hypertension. 2003;41:168–175. doi: 10.1161/01.hyp.0000047511.97879.fc. [DOI] [PubMed] [Google Scholar]
- [66].Armitage JA, Lakasing L, Taylor PD, Balachandran AA, Jensen RI, Dekou V, Ashton N, Nyengaard JR, Poston L. Developmental programming of aortic and renal structure in offspring of rats fed fat-rich diets in pregnancy. J. Physiol. 2005;565:171–184. doi: 10.1113/jphysiol.2005.084947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Taylor P, McConnell J, Khan Y, Holemans K, Lawrence K, Asare-Anane H, Persaud S, Jones P, Petrie L, Hanson M, Poston L. Impaired glucose homeostasis and mitochondrial abnormalities in offspring of rats fed a fat-rich diet in pregnancy. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005;288:R134–139. doi: 10.1152/ajpregu.00355.2004. [DOI] [PubMed] [Google Scholar]
- [68].Koukkou E, Ghosh P, Lowy C, Poston L. Offspring of normal and diabetic rats fed saturated fat in pregnancy demonstrate vascular dysfunction. Circulation. 1998;98:2899–2904. doi: 10.1161/01.cir.98.25.2899. [DOI] [PubMed] [Google Scholar]
- [69].Ghosh P, Bitsanis D, Ghebremeskel K, Crawford MA, Poston L. Abnormal aortic fatty acid composition and small artery function in offspring of rats fed a high fat diet in pregnancy. J. Physiol. 2001;533:815–822. doi: 10.1111/j.1469-7793.2001.00815.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Khan I, Dekou V, Hanson M, Poston L, Taylor P. Predictive adaptive responses to maternal high-fat diet prevent endothelial dysfunction but not hypertension in adult rat offspring. Circulation. 2004;110:1097–1102. doi: 10.1161/01.CIR.0000139843.05436.A0. [DOI] [PubMed] [Google Scholar]
- [71].Ferezou-Viala J, Roy AF, Serougne C, Gripois D, Parquet M, Bailleux V, Gertler A, Delplanque B, Djiane J, Riottot M, Taouis M. Long-term consequences of maternal high-fat feeding on hypothalamic leptin sensitivity and diet-induced obesity in the offspring. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007;293:R1056–R1062. doi: 10.1152/ajpregu.00117.2007. [DOI] [PubMed] [Google Scholar]
- [72].Gupta A, Srinivasan M, Thamadilok S, Patel MS. Hypothalamic alterations in fetuses of high fat diet-fed obese female rats. J. Endocrinol. 2009;200:293–300. doi: 10.1677/JOE-08-0429. [DOI] [PubMed] [Google Scholar]
- [73].Chang GQ, Gaysinskaya V, Karatayev O, Leibowitz SF. Maternal high-fat diet and fetal programming: increased proliferation of hypothalamic peptide-producing neurons that increase risk for overeating and obesity. J. Neurosci. 2008;28:12107–12119. doi: 10.1523/JNEUROSCI.2642-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Cerf ME, Muller CJ, Toit D.F. Du, Louw J, Wolfe-Coote SA. Hyperglycaemia and reduced glucokinase expression in weanling offspring from dams maintained on a high-fat diet. Br.J. Nutr. 2006;95:391–396. doi: 10.1079/bjn20051632. [DOI] [PubMed] [Google Scholar]
- [75].Cerf ME, Chapman CS, Louw J. High-fat programming of hyperglycemia, hyperinsulinemia, insulin resistance, hyperleptinemia, and altered islet architecture in 3-month-old wistar rats. ISRN endocrinology. 2012;2012:627270. doi: 10.5402/2012/627270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Cerf ME, Williams K, Nkomo XI, Muller CJ, Du Toit DF, Louw J, Wolfe-Coote SA. Islet cell response in the neonatal rat after exposure to a high-fat diet during pregnancy. Am. J. Physiol.. Regul. Integr. Comp. Physiol. 2005;288:R1122–1128. doi: 10.1152/ajpregu.00335.2004. [DOI] [PubMed] [Google Scholar]
- [77].Cerf ME, Chapman CS, Muller CJ, Louw J. Gestational high-fat programming impairs insulin release and reduces Pdx-1 and glucokinase immunoreactivity in neonatal Wistar rats. Metabolism. 2009:1787–1792. doi: 10.1016/j.metabol.2009.06.007. [DOI] [PubMed] [Google Scholar]
- [78].Siemelink M, Verhoef A, Dormans JA, Span PN, Piersma AH. Dietary fatty acid composition during pregnancy and lactation in the rat programs growth and glucose metabolism in the offspring. Diabetologia. 2002;45:1397–1403. doi: 10.1007/s00125-002-0918-2. [DOI] [PubMed] [Google Scholar]
- [79].Everard A, Cani PD. Diabetes, obesity and gut microbiota. Best Pract. Res. Clin. Gastroenterol. 2013;27:73–83. doi: 10.1016/j.bpg.2013.03.007. [DOI] [PubMed] [Google Scholar]
- [80].Kovatcheva-Datchary P, Arora T. Nutrition, the gut microbiome and the metabolic syndrome. Best Practice Res. Clin. Gastroenterol. 2013;27:59–72. doi: 10.1016/j.bpg.2013.03.017. [DOI] [PubMed] [Google Scholar]
- [81].McCurdy CE, Bishop JM, Williams SM, Grayson BE, Smith MS, Friedman JE, Grove KL. Maternal high-fat diet triggers lipotoxicity in the fetal livers of nonhuman primates. J. Clin. Invest. 2009;119:323–335. doi: 10.1172/JCI32661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Frias AE, Morgan TK, Evans AE, Rasanen J, Oh KY, Thornburg KL, Grove KL. Maternal High-Fat Diet Disturbs Uteroplacental Hemodynamics and Increases the Frequency of Stillbirth in a Nonhuman Primate Model of Excess Nutrition. Endocrinology. 2011;152:2456–2464. doi: 10.1210/en.2010-1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Fan L, Lindsley SR, Comstock SM, Takahashi DL, Evans AE, He GW, Thornburg KL, Grove KL. Maternal high-fat diet impacts endothelial function in nonhuman primate offspring. Int. J. Obes. (Lond) 2013;37:254–262. doi: 10.1038/ijo.2012.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Grant WF, Gillingham MB, Batra AK, Fewkes NM, Comstock SM, Takahashi D, Braun TP, Grove KL, Friedman JE, Marks DL. Maternal high fat diet is associated with decreased plasma n-3 fatty acids and fetal hepatic apoptosis in nonhuman primates. PloS One. 2011;6:e17261. doi: 10.1371/journal.pone.0017261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Aagaard-Tillery KM, Grove K, Bishop J, Ke X, Fu Q, McKnight R, Lane RH. Developmental origins of disease and determinants of chromatin structure: maternal diet modifies the primate fetal epigenome. J. Mol. Endocrinol. 2008;41:91–102. doi: 10.1677/JME-08-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Cox J, Williams S, Grove K, Lane RH, Aagaard-Tillery KM. A maternal high-fat diet is accompanied by alterations in the fetal primate metabolome. Am. J. Obstet. Gynecol. 2009;201:281, e1–9. doi: 10.1016/j.ajog.2009.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Suter MA, Chen A, Burdine MS, Choudhury M, Harris RA, Lane RH, Friedman JE, Grove KL, Tackett AJ, Aagaard KM. A maternal high-fat diet modulates fetal SIRT1 histone and protein deacetylase activity in nonhuman primates. FASEB J. 2012;26:5106–5114. doi: 10.1096/fj.12-212878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Grayson BE, Levasseur PR, Williams SM, Smith MS, Marks DL, Grove KL. Changes in melanocortin expression and inflammatory pathways in fetal offspring of nonhuman primates fed a high-fat diet. Endocrinology. 2010;151:1622–1632. doi: 10.1210/en.2009-1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Suter M, Bocock P, Showalter L, Hu M, Shope C, McKnight R, Grove K, Lane R, Aagaard-Tillery K. Epigenomics: maternal high-fat diet exposure in utero disrupts peripheral circadian gene expression in nonhuman primates. FASEB J. 2011;25:714–726. doi: 10.1096/fj.10-172080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Li MD, Li CM, Wang Z. The role of circadian clocks in metabolic disease. Yale J. Biol. Med. 2012;85:387–401. [PMC free article] [PubMed] [Google Scholar]
- [91].Arble DM, Ramsey KM, Bass J, Turek FW. Circadian disruption and metabolic disease: findings from animal models. Best Pract. Res.. Clin. Endocrinol. Metab. 2010;24:785–800. doi: 10.1016/j.beem.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Maury E, Ramsey KM, Bass J. Circadian rhythms and metabolic syndrome: from experimental genetics to human disease. Circ. Res. 2010;106:447–462. doi: 10.1161/CIRCRESAHA.109.208355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Suter MA, Sangi-Haghpeykar H, Showalter L, Shope C, Hu M, Brown K, Williams S, Harris RA, Grove KL, Lane RH, Aagaard KM. Maternal high-fat diet modulates the fetal thyroid axis and thyroid gene expression in a nonhuman primate model. Mol. Endocrinol. 2012;26:2071–2080. doi: 10.1210/me.2012-1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Sullivan EL, Grayson B, Takahashi D, Robertson N, Maier A, Bethea CL, Smith MS, Coleman K, Grove KL. Chronic consumption of a high-fat diet during pregnancy causes perturbations in the serotonergic system and increased anxiety-like behavior in nonhuman primate offspring. J. Neurosci. 2010;30:3826–3830. doi: 10.1523/JNEUROSCI.5560-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Masuyama H, Hiramatsu Y. Effects of a high-fat diet exposure in utero on the metabolic syndrome-like phenomenon in mouse offspring through epigenetic changes in adipocytokine gene expression. Endocrinology. 2012;153:2823–2830. doi: 10.1210/en.2011-2161. [DOI] [PubMed] [Google Scholar]
- [96].Vucetic Z, Kimmel J, Totoki K, Hollenbeck E, Reyes TM. Maternal high-fat diet alters methylation and gene expression of dopamine and opioid-related genes. Endocrinology. 2010;151:4756–4764. doi: 10.1210/en.2010-0505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Vinson C, Chatterjee R. CG methylation. Epigenomics. 2012;4:655–663. doi: 10.2217/epi.12.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Strakovsky RS, Zhang X, Zhou D, Pan YX. Gestational high fat diet programs hepatic phosphoenolpyruvate carboxykinase gene expression and histone modification in neonatal offspring rats. J. Physiol. 2011;589:2707–2717. doi: 10.1113/jphysiol.2010.203950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Hoile SP, Irvine NA, Kelsall CJ, Sibbons C, Feunteun A, Collister A, Torrens C, Calder PC, Hanson MA, Lillycrop KA, Burdge GC. Maternal fat intake in rats alters 20:4n-6 and 22:6n-3 status and the epigenetic regulation of Fads2 in offspring liver. J. Nutr. Biochem. 2012:245–248. doi: 10.1016/j.jnutbio.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Kelsall CJ, Hoile SP, Irvine NA, Masoodi M, Torrens C, Lillycrop KA, Calder PC, Clough GF, Hanson MA, Burdge GC. Vascular dysfunction induced in offspring by maternal dietary fat involves altered arterial polyunsaturated fatty acid biosynthesis. PloS One. 2012;7:e34492. doi: 10.1371/journal.pone.0034492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Rando OJ. Daddy issues: paternal effects on phenotype. Cell. 2012;151:702–708. doi: 10.1016/j.cell.2012.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Ng SF, Lin RC, Laybutt DR, Barres R, Owens JA, Morris MJ. Chronic high-fat diet in fathers programs beta-cell dysfunction in female rat offspring. Nature. 2010;467:963–966. doi: 10.1038/nature09491. [DOI] [PubMed] [Google Scholar]
- [103].Dunn GA, Bale TL. Maternal High-Fat Diet Effects on Third-Generation Female Body Size via the Paternal Lineage. Endocrinology. 2011;152:2228–2236. doi: 10.1210/en.2010-1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Binder NK, Hannan NJ, Gardner DK. Paternal diet-induced obesity retards early mouse embryo development, mitochondrial activity and pregnancy health. PloS One. 2012;7:e52304. doi: 10.1371/journal.pone.0052304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Binder NK, Mitchell M, Gardner DK. Parental diet-induced obesity leads to retarded early mouse embryo development and altered carbohydrate utilisation by the blastocyst. Reprod. Fertil. Dev. 2012;24:804–812. doi: 10.1071/RD11256. [DOI] [PubMed] [Google Scholar]
- [106].Fullston T, Palmer NO, Owens JA, Mitchell M, Bakos HW, Lane M. Diet-induced paternal obesity in the absence of diabetes diminishes the reproductive health of two subsequent generations of mice. Hum. Reprod. 2012;27:1391–1400. doi: 10.1093/humrep/des030. [DOI] [PubMed] [Google Scholar]
- [107].Mitchell M, Bakos HW, Lane M. Paternal diet-induced obesity impairs embryo development and implantation in the mouse. Fertil. Steril. 2011;95:1349–1353. doi: 10.1016/j.fertnstert.2010.09.038. [DOI] [PubMed] [Google Scholar]
- [108].Wu Y.m. Qing, masahiko komiya, Tatsuhiro Matsuo, Masashige Suzuki. Body Fat Accumulation in the Male Offspring of Rats Fed High-Fat Diet. J. Clin. Biochem. Nutr. 1998;25:71–79. [Google Scholar]
- [109].Mao J, Zhang X, Sieli PT, Falduto MT, Torres KE, Rosenfeld CS. Contrasting effects of different maternal diets on sexually dimorphic gene expression in the murine placenta. Proc. Natl. Acad. Sci. USA. 2010;107:5557–5562. doi: 10.1073/pnas.1000440107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Gabory A, Ferry L, Fajardy I, Jouneau L, Gothie JD, Vige A, Fleur C, Mayeur S, Gallou-Kabani C, Gross MS, Attig L, Vambergue A, Lesage J, Reusens B, Vieau D, Remacle C, Jais JP, Junien C. Maternal diets trigger sex-specific divergent trajectories of gene expression and epigenetic systems in mouse placenta. PloS One. 2012;7:e47986. doi: 10.1371/journal.pone.0047986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Gallou-Kabani C, Gabory A, Tost J, Karimi M, Mayeur S, Lesage J, Boudadi E, Gross MS, Taurelle J, Vige A, Breton C, Reusens B, Remacle C, Vieau D, Ekstrom TJ, Jais JP, Junien C. Sex- and diet-specific changes of imprinted gene expression and DNA methylation in mouse placenta under a high-fat diet. PloS One. 2010;5:e14398. doi: 10.1371/journal.pone.0014398. [DOI] [PMC free article] [PubMed] [Google Scholar]