

Abstract
Cyclooxygenase-2 (COX-2) and 5-Lipoxygenase (5-LOX) enzyme have been found to play a role in promoting growth in colon cancer cell lines. The di-tert-butyl phenol class of compounds has been found to inhibit both COX-2 and 5-LOX enzymes with proven effectiveness in arresting tumor growth. In the present study, the structural analogs of 2, 6 di-tert-butyl-p-benzoquinone (BQ) appended with hydrazide side chain were found to inhibit COX-2 and 5-LOX enzymes at micromolar concentrations. Molecular docking of the compounds into COX-2 and 5-LOX protein cavities indicated strong binding interactions supporting the observed cytototoxicities. The signaling interaction between endogenous hyaluronan and CD44 has been shown to regulate COX-2 activities through ErbB2 receptor tyrosine kinase (RTK) activation. In the present studies it has been observed for the first time, that three of our COX/5-LOX dual inhibitors inhibit proliferation upon hydrazide substitution and prevent the activity of pro-angiogenic factors in HCA-7, HT-29, Apc10.1 cells as well as the hyaluronan synthase-2 (Has2) enzyme over-expressed in colon cancer cells, through inhibition of the Hyaluronan/CD44v6 cell survival pathway. Since there is a substantial enhancement in the antiproliferative activities of these compounds upon hydrazide substitution, the present work opens up new opportunities for evolving novel active compounds of BQ series for inhibiting colon cancer.
Keywords: Cyclooxygenase, Lipoxygenase, colon cancer, dual COX-LOX inhibitor, Hyaluronan, CD44v6, cell proliferation and drug resistance
1. Introduction
Colorectal cancer (CRC) is the third most common cause of cancer related deaths in the United States with an estimated 103,107 new cases and 51,690 number of deaths in 2012 1. Surgery remains the only treatment with curative potential for both primary and metastatic CRC2. Adjuvant chemotherapy based on 5-fluorouracil (5-FU) combined with radiotherapy after the radical resection of primary CRC has been found to prolong the survival of patients3. In cases of metastases improvements have been observed by combining 5-FU with irinotecan and oxaliplatin. Attempts in designing novel drugs have led to discovery of potential chemopreventive agents that are effective at the pre-clinical and clinical levels 4,5.
The cyclooxygenase enzyme exists in two different isoforms, viz. COX-1, and COX-2 respectively which uses arachidonic acid (AA) as a substrate to produce prostaglandins (PGs). AA is also a substrate for another enzyme, viz. 5-lipoxygenase (5-LOX), which generates leukotrienes (LTs)6. The roles of both downstream products of COX and 5-LOX, viz. PGE2 and LTB4, in the inflammatory process have been extensively explored in the past6. It has been demonstrated that the metabolism of AA, by either the COX or LOX pathway, generates a host of pro-inflammatory metabolites called eicosanoids including PGs, thromboxanes and leukotrienes (LTs), respectively6. Consequently increased levels of COX and LOX enzymes have been implicated in adenomatous polyposis7, 8 and in experimentally induced colon tumors in rodents 9. The cancer growth-promoting effects of downstream products of 5-LOX pathway such as 5-hydroxyeicosatetranoic acid (5-HETE) and leukotriene B4 (LTB4) have been reported10 which forms the basis of the use of Non-Steroidal Anti-Inflammatory Drugs (NSAIDs), different COX inhibitors and 5-LOX inhibitors such as REV5901, Zileuton, and Minocyclines 11-13. Since these two arachidonic acid-metabolizing enzymes are so closely related in their substrates and mechanisms of action, it can be envisaged that blocking one enzymatic pathway may activate the other14. This phenomenon might explain, at least in part, the observed limited efficacy of COX-2 inhibitors as anticancer agents15, 16. At higher concentrations, the anti-carcinogenic effects of Celecoxib have been explained by both COX-2-dependent and - independent mechanisms16, 17. These inter-relationship of their biological functions suggest that blocking both COX-2 and 5-LOX pathways using designed synthetic molecules may provide a promising integrated approach for prevention and therapy of colon cancer.
A number of studies have aimed at identifying molecules that are specifically expressed by epithelial tumor cells which correlate with tumor growth and drug resistance. Among such molecules, hyaluronan (HA) is a major component in the extra-cellular matrix of most mammalian tissues and animal models18,19, while CD44 is a trans-membrane glycoprotein that communicates with the environment by binding to its major ligand HA20 and thereby regulating cell survival and motility21-23. In the process of growth and spread of tumors, CD44/HA signaling employs at least three different functions of CD44 24. Firstly, CD44 needs to stimulate tumor cell proliferation, motility, and/or invasiveness through recruitment and enabling the activity of membrane-associated matrix metalloproteinases 25, 26 or receptor tyrosine kinases27, 28. Secondly, the variants of CD44 may function as co-receptors for the activation of growth-promoting tumor receptor tyrosine kinases29. Finally, the variant isoform CD44v6 is over-expressed in colon tumors 30-39 and the endogenous binding of HA to CD44v6 is more avid than to CD4418, 40, 41. We have shown that CD44v6/HA interaction regulates COX-2-induced prostaglandins E2 (PGE2) which in turn controls HA synthesis and hence the HA/CD44v6 signaling 42-46. We and other groups have shown that both HA and CD44 are involved in chemotherapeutic drug resistance in many cancer types 47-49, and the reversal of HA/CD44v6 signaling has been shown to modulate the cancer phenotype and adenoma growth in Apc min/+ mice suggesting potential of HA/CD44v6 as targets for anti-cancer/chemoprevention drugs 18, 50, 51. Conceptually, agents that target COX/LOX or CD44v6 are noteworthy because of their clinical efficacy in the prevention of polyp formation 18, 52.
Surprisingly, only few dual COX-LOX inhibitors have been studied clinically for their efficacy as the anti-inflammatory agents. Amongst them Licofelone is in phase III of clinical trials, while Tenidap developed by Pfizer has been withdrawn because of its high hepatotoxicity53. The di-tert-butyl phenols represent a potent class of dual COX/5-LOX inhibitors54-56 which is represented by Darbufelone (1), CI987 (2), S2474 (3) and KME4 (4) (Table 1). Darbufelone was recently found to inhibit growth of non-small cell lung cancer cell lines, inducing cell cycle arrest at G0/G1 phase and apoptosis by activation of caspase-3 and caspase-8 respectively57. However, these compounds or their pro-drugs di-tert-butyl benzoquinones, have remained inadequately explored for the inhibition of colon cancer in spite of their low ulcerogenicity and potent COX-LOX inhibitory activity58. Hence, in the present study, we have synthesized and structurally characterized three di-tert-butyl benzoquinone COX/LOX inhibitors appended with hydrazinic side chain, viz. BQBH, BQNH and BQIH as shown in Scheme 1. The compounds were docked into COX-2 and 5-LOX protein cavities and evaluated for their cyclooxygenase-1, -2 (COX-1, -2) and 5-lipoxygenase (5-LOX) enzyme inhibitory activities followed by evaluation of their cytotoxic effects against colon cancer cell line. Further, we also examined whether the COX/5-LOX pathway and the CD44v6/HA interactions co-operate in promoting proliferative stimulus in human colon cancer cells HT29, HCA7 and pre-neoplastic Apc10.1 cells isolated from Apc Min/+ mice59. It was found that these compounds were able to inhibit cell proliferation dramatically and differentially by increasing apoptosis in HT29 and HCA7 cells and hyaluronan synthase-2 over-expressed Apc10.1 cells59. It was also observed that CD44v6shRNA sensitizes these cells to chemotherapeutic drugs like Celecoxib and Licofelone as well as present COX-LOX dual inhibitor, viz. BQNH. Finally, it is reasonable to speculate that COX-2/PGE2 may regulate CD44v6, and HA synthesis since HA/CD44v6 signaling regulates COX-2/PGE2 signaling42, 43 and COX-2 and 5-LOX have been closely related in mechanisms of action and their substrates 60, 61. Our results suggest that COX/5-LOX dual inhibitors with hydrazinic side chain represent a potent class of chemopreventive agents for colon cancer.
TABLE 1.
Examples of the dual COX-LOX inhibitors
|
2. Results and Discussion
2.1 Chemistry
The compounds BQBH, BQNH and BQIH were shades of yellow-orange. The IR spectrum of BQ exhibited a sharp intense band at 1654 cm−1 due to the quinone carbonyl group flanked by the butyl groups and shoulder absorption at 1599 cm−1 due to other free carbonyl respectively 68. Upon Schiff base formation the free carbonyl stretch disappeared accompanied by the appearance of the C=N stretch in the region 1633-1680 cm−1 which overlaps with the remaining quinone carbonyl and the hydrazinic carbonyl frequency resulting in broadening of the band. The hydrazinic N-N stretch occurred in the range 1252-1258 cm−1, while the aromatic C=C stretches can be observed in the region 1521-1546 cm−1 69. The electronic spectra of the ligands in DMSO showed intra-ligand transitions in the range 26300-27700 cm−1 respectively.
The H1-NMR spectra of the parent BQ compound showed sharp singlets at 1.286 and 6.512 ppm ascribed to di-tert-butyl protons and protons of the quinone moiety 68. In case of the hydrazonate ligands signals in the region of 7.097- 7.856 ppm were due to aromatic protons confirming the formation of expected Schiff bases. The sharp singlet in the region of 12.027 - 12.374 ppm was ascribed to the imine (-NH) proton. Two signals appearing as singlets due to tert-butyl groups in the region of 1.271-1.281 and 1.320-1.334 ppm suggest that hydrazide-substitution induced asymmetry in the hydrazonate ligands70. The C13-NMR showed two prominent signals at 187.16 and 189.03 ascribed to two quinone carbonyls in the parent BQ compound. In the hydrazonate ligands one of these carbonyl signals (189.03 ppm) disappeared with emergence of a new signal in the region 151.79- 152.09 ppm corresponding to the imino carbon. The carbons of the tert-butyl groups 71, 72 in BQ with signals at 26.36 and 35.55 ppm undergo downfield shift to 29.59-29.89 and 35.15-36.41 ppm respectively upon Schiff base formation, while their aromatic carbon atoms can be observed in the region of 119.47-152.86 ppm respectively.
2.2 Molecular Docking Study
In order to identify the possible binding sites of the hydrazonate ligands, the compounds were docked into the pre-defined binding site of COX-2 and 5-LOX proteins. All presently synthesized compounds as well as standard dual COX-LOX inhibitor Darbufelone compound, exhibited extensive hydrogen bonding interactions in the COX-2 cavity. The binding energies calculated for the new analogs were in the range of −6.8 to −7.2 Kcal/mol compared to Darbufelone’s binding energy of −6.8 Kcal/mol (Table 2 and Figure 1: Panel 1). The best binding energy was exhibited by BQNH ligand. Darbufelone exhibited three hydrogen bonding interactions with residues VAL228 (3.4 A0), ASN537 (2.1A0) and GLY533 (2.0 A0) respectively, while the most potent compound BQNH showed interactions with ASN87 (3.3 A0) and HIS90 (2.0 A0) respectively. The Van der Waals interactions between nitrogen of the pyridine ring and oxygen of the nicotylhydrazide moiety were also found to be involved in the stabilization of ligand-enzyme complexes.
Table 2.
Docking Results and Consensus Scores of 2, 6 Di-Tert-Butyl Benzoquinone-4-Hydrazones in COX-2 and 5-LOX Protein Cavity
| Molecule | COX-2 | 5-LOX | |||||||
|---|---|---|---|---|---|---|---|---|---|
| LogP | B. E. (Kcal/mol) |
H bond | H bonding residues |
Distance (A0) |
B. E. (Kcal/mol) |
H bond | H bonding residues |
Distance (A0) |
|
| BQBH | 5.47 | −6.8 | 3 | HIS351, GLN192 |
3.3 2.2 and 3.4 |
−7.0 | 1 | ASP170 | 2.3 |
| BQNH | 3.34 | −7.2 | 2 | ASN87, HIS90 |
3.3, 2.0 |
−8.4 | 2 | GLN549, ARG370 |
2.9 2.2 |
| BQIH | 3.28 | −7.0 | 1 | GLN192 | 2.2 | −8.2 | 2 | ARG47 ARG401 |
2.3, 2.6 |
| Darbufelone | 4.98 | −6.7 | 3 | VAL228, ASN537, GLY533 |
3.4, 2.1, 2.0 |
−7.3 | 4 | ASP285, GLU287, ILE283, LEU244 |
2.5, 2.6, 3.3, 3.5 |
COX-2 = Cyclo-oxygenase-2, 5-LOX= 5-Lipoxygenase, B. E. = Binding Energy, H bond = Hydrogen Bond, H bonding residues = Hydrogen bonding residues
Figure 1.

Docking Features of 2, 6 Di-Tert-Butyl Benzoquinone-4-Hydrazones in COX-2 (Panel 1) and 5-LOX (Panel 2)
In 2011 Gilbert and coworkers (64) have reported on the crystal structure of human 5-LOX. The amino acid residues like HIS367, LEU368, HIS372, LEU373, ILE406, LEU414, HIS550, ASN554, LEU607 and ILE673 were identified in the active site of 5-LOX protein73. When docked into the protein cavity of 5-LOX, BQNH exhibited best binding energy of −8.4 Kcal/mol followed by compound BQIH, Darbufelone and BQBH with binding energies of −8.2, −7.3 and −7.0 Kcal/mol respectively indicating their tight binding in the protein cavity. Darbufelone was found to interact with ASP285 (2.5 A0), GLU287 (2.6 A0), ILE283 (3.3 A0), LEU244 (3.5 A0) respectively, while compound BQNH interacted with GLN549 (2.9 A0) and ARG370 (2.2 A0), suggesting that these interactions are involved in stabilizing these compounds in the protein cavity (Table 2 and Figure 1: Panel 2). Our docking study confirmed that the newly synthesized compounds can interact with COX-2 and 5-LOX protein cavities wherein the heterocyclic hydrazide moiety containing nitrogen contributes towards enhancing interaction with amino acid residues. BQNH showed best binding energy in both COX-2 and 5-LOX protein cavities which correlate well with its superior antiproliferative activity in vitro against the colon cancer cell lines employed in the present study.
2.3 Biological Activity
2.3.1 In vitro COX-LOX Assay
The potential of all synthesized compounds to inhibit COX-1, COX-2 isoforms and 5-LOX was determined on pure enzymes. Amongst the new compounds BQBH and BQIH exhibited most potent 5-LOX and COX-2 inhibitory activities. These results indicated that the hydrazonate pharmacophore in the side chain plays an important role in deciding the preferences for these enzyme sites. The present work thus confirms that 2, 6 di-tert-butyl-p-benzoquinone hydrazone ligands are effective COX/LOX dual inhibitors which can serve as promising therapeutic agents against colon cancers.
2.3.2 COX-2 and 5-LOX expression in colon tumor cells and Has2-overexpressing Apc10.1 cells
COX-2 and 5-LOX have been shown to over-express during the process of colonic adenoma formation and have been implicated as promoter for tumor development 61. In our earlier studies, we have established that constitutive hyaluronan-CD44 interaction stimulates a signaling pathway involving ErbB2, phosphoinositide-3-kinase/AKT, β-catenin, and 43 cyclooxygenase-2/prostaglandin E(2) in HCA7 colon carcinoma cells 43. Consequently in the present study, we sought to determine whether hyaluronan also regulates 5-LOX expression employing two human CRC HT29, HCA-7 cells, and Has2 over-expressing Apc10.1 cells with different basal COX-2 protein expression (Figure 2A). In our studies 5-LOX expression was detected in all three cell lines (Figure 2A), while Has2 over-expression in Apc 10.1-Has2 cells exhibited high levels of COX-2 compared to moderate levels expressed by HT29 and HCA-7 cells (Figure 2A, lane 2 vs. lane 1). In agreement with our previous study, Apc10.1 cells expressed low levels of COX-2 and 5-LOX protein, suggesting that COX-2 as well as 5-LOX is both regulated by hyaluronan. In further studies we used HT29 cells wherein our results indicated (Figure 2B) that very low levels of COX-2 and 5-LOX are present in the CD44v6shRNA transfected HT29 cells with a substantial inhibition by BQBH, BQNH and BQIH as well as Licofelone (standard COX/LOX inhibitor) and Celecoxib (COX-2 inhibitor). Subsequently, we determined that HA over-expression induces the recruitment of a significant amount of COX-2 and 5-LOX into the HA signaling cascade. This led us to conclude that just as HA/CD44-linked COX-2 activation is correlated with tumorigenic behavior, the 5-LOX expression is also regulated by HA and CD44v6 (Figure 2B, lane 8 compared to lane 2) and present COX-LOX dual inhibitors are able to target the surface adhesion molecules like CD44v6, (Figure 2B, lanes 3, 4, 5 and 7 vs. lane 2).
Figure 2.

Analyses of HA-induced CD44V6 associated COX-2 and 5-LOX expression in colorectal carcinoma (CRC) cell lines
2.3.3 Inhibition of COX-2, and COX-2/5-LOX attenuate HA-stimulated cell survival in colorectal cells
Although the role of the COX pathway in colonic cancer development has been widely studied, investigation of the role of the LOX pathway in colon cancer has been limited. We have previously shown that COX-2 induced PGE2 regulates HA production, and hence HA/CD44 induced signaling. The question is whether COX/LOX pathways and HA/CD44 pathways are co-regulated or act independently in the tumor specific behavior. Experiments in Figure 2 show that there exists a relationship between HA/CD44v6 and COX-2 and 5-LOX. Therefore, we investigated the anti-carcinogenic effect of dual inhibition of COX-2 and 5-LOX in the process of cell proliferation. The functional effect of 5-LOX and COX-2 on CRC cell proliferation and survival was analyzed by exposing cells to either colon cancer chemotherapeutic drug Celecoxib (inhibits COX-2), or Licofelone (inhibits 5-LOX), or the three COX-2/5-LOX inhibitors along with control shRNA or CD44v6shRNA transfection at various doses. Results in Figure 3 indicate that these antagonist drugs dose-dependently inhibited colon cancer cell proliferation. They also indicate that 5-LOX and COX-2 in addition to HA/CD44v6 could be involved in the mechanisms of CRC cell carcinogenesis because our new COX-LOX dual inhibitors (Scheme 1) decrease CRC proliferation similar to CD44v6shRNA, with the effect of the COX-2 inhibitor Celecoxib showing somewhat stronger inhibitory effect compared with the 5-LOX inhibitor Licofelone.
Figure 3.

Effect of COX/5-LOX, and CD44v6 inhibition of proliferation on colorectal carcinoma (CRC) cell lines.
Scheme 1.

Synthesis of 2, 6-Di-Tert-Butyl-Benzoquinone-4-Hydrazone Ligands
To further confirm whether the chemotherapeutic drug responses of Apc10.1, HT29, and HCA7 cells might be regulated by the HA/CD44v6 interaction, cell growth assays were done using colon cancer chemotherapeutic drugs Celecoxib and Licofelone along with BQBH, BQNH, BQIH, and CD44v6shRNA in the presence or absence of Has2 over expression (Table 4). In the absence of Has2, HT29, HCA-7 and Apc10.1 cells treated with BQBH or BQIH displayed a low level of tumor cell survival, with IC50 values of 12-20 μM (Tables 4). HT29, HCA-7 and Apc10.1 cells treated with BQNH exhibited a relatively low level of tumor cell survival, with IC50 values between 6.5-11 μM (Table 4). The cytotoxic activity of parent building blocks of the new inhibitors was also tested against HT29 cells which revealed higher IC50 values in the range 26.5±3.3 to 175.0±28.3 μM respectively. This suggests that the cytotoxic activity was due to intact compounds inside the cell. Celecoxib-treated cells exhibited a very low level of IC50 values between 2.0-4.2 μM, indicating that COX-2 may have a greater role in cell survival. Licofelone-treated cells showed higher IC50 values between 45-75 μM indicating that 5-LOX may have a greater role in apoptotic resistance. CD44v6shRNA-transfected cells exhibited the highest inhibition of cell survival (Tables 4), indicating that CD44v6 is necessary for cell survival. However, the over-expression of Has2 enhances cell survival in untreated controls (i.e. without chemotherapeutic drugs) and decreases the ability of BQBH, or BQNH, or BQIH, or the standard inhibitors to induce tumor cell death (Tables 4). These observations strongly suggest that HA causes an increase in tumor cell survival, leading to enhancement of chemo-resistance (i.e., resistance to cell death) to all the drug/inhibitors treatments. Furthermore, pre-treatment of these CRC cells with CD44v6shRNA followed by Has2 over-expression significantly reduces the HA-mediated resistance to cell death (Table 4). Thus, inhibition of HA/CD44v6 interaction, or inhibition of COX-2/5-Lox appears to be functionally linked to anti-apoptotic effects in CRC cells. These results indicate that HA/CD44v6 interaction promotes resistance to apoptosis (anti-apoptosis) in the presence of novel dual Cox-Lox inhibitors (BQBH, BQNH and BQIH) and chemotherapeutic drugs, Celecoxib and Licofelone.
Table-4.
IC50 analyses of drugs (COX-2/5-LOX inhibitor BQBH, BQNH, BQIH, Licofelone, COX-2 inhibitor Celecoxib and HA/CD44v6 antagonist CD44v6shRNA) in cell proliferation of HT29, HCA7, and Has2 over-expressed Apc10.1 cells
|
Cell
lines |
BQBH
IC50 (μM) |
BQNH
IC50 (μM) |
BQIH
IC50 (μM) |
Celecoxib
IC50 (μM) |
Licofelone
IC50 (μM) |
CD44v6shRNA
IC50 (pmole) |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Has2 | − | + | − | + | − | + | − | + | + | − | + | |
| HT29 | 15 ± 1.5 | 38.5 ± 3.6 |
11 ±1.3 | 28.6±3.2 | 15.2±1.9 | 5.4±5.7 | 3.0±0.8 | 18.3± 2.3 |
75±8.3 | 158±9.3 | 80.0 ±7 | 240±18 |
| HCA-7 | 12.5±0.9 | 26.4 ±1.9 |
6.5±1.2 | 34.5±4.3 | 12±1.5 | 38.6±3.9 | 2.0±0.9 | 17.5± 2.5 |
48±5.2 | 186±11.9 | 110.0±11 | 280±19 |
| Apc10.1 | 20 ± 3.7 | 31±4.2 | 7.5±2.2 | 12.2±1.8 | 8.9±1.2 | 18.3±2.0 | 4.2±1.2 | 6.0±1.5 | 45±7.2 | 86±7.2 | 180±17.2 | 100±9.5 |
IC50 values are presented as the means ± S.D (n = three replicates of 4 independent analyses) IC50 is designated as the concentration (μM) of these drugs that causes 50% inhibition of colon tumor cell proliferations.
2.3.4 The growth inhibitory effects of BQNH at different test concentrations by MTT assay
In order to further confirm the cytotoxic activity of the most potent compound BQNH, we have carried out another set of growth inhibition measurement using MTT assay (Figure 4). Since the tumor cells, such as HT29, HCA7 and Apc-Has2 express COX-2, 5-LOX, and CD44v6 19 (and Figure 2), the compounds active in this assay are indicated to be selective toward the tumor cells, and their cell growth-inhibiting activity is attributed to CD44v6- COX-LOX axis. In addition, the proliferation assay results of Figure 3 go parallel with MTT assay for the HT29, HCA7 and Apc-Has2 tumor cells. Importantly, this assay also showed that the new inhibitors have no effect on normal intestinal epithelial cells IEC6, which do not exhibit COX-2 61a, LOX and CD44v6 expression. Since pre-neoplastic Apc10.1 cells59 express both COX and LOX, the compound BQNH has reasonable cytotoxic activity. These results further confirm the potential of CD44v6- COX-LOX as targets in colon cancer therapy and prevention using our newly synthesized COX-LOX dual inhibitor compounds.
Figure 4.
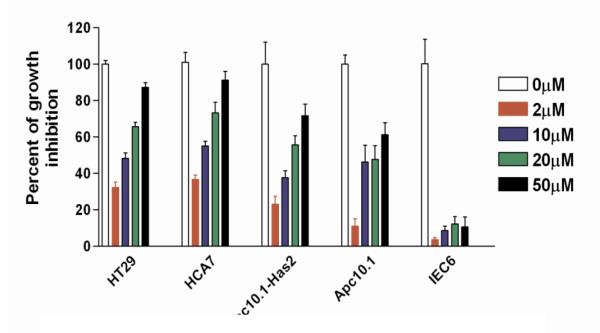
The inhibition curves of BQNH at different test concentrations using MTT assay
3. Conclusions
Thus, our present studies indicate that dual COX-LOX inhibitors are potent inhibitors of colon cancer growth and their consequent effect on drug resistance and CRC growth are regulated by HA, and CD44v6. The probable mechanism of action involves targeting COX-2/ 5-LOX proteins and abrogating their interaction with CD44v6 receptors thereby interfering with HA/CD44v6 signaling through autocrine /paracrine mechanism which offers a novel alternative therapeutic strategy for rational design of anticancer agents for colon cancer.
4. Material and Methods
All reagents used for synthesis were of analytical grade and were used without further purification. Solvents employed were purified by standard procedures prior to use. The reaction progress was monitored through thin-layer chromatography (TLC) on pre-coated silica gel on aluminum plates (Merck), while the reaction products were separated by column chromatography on silica gel 60 (Merck 60–120 mesh).
4.1 General Procedure for Preparation of compounds (2-4)
The compounds were synthesized as shown in Scheme 1 by condensation of equimolar amount of 2, 6-di-tert-1,4-benzoquinone (BQ) and respective isonicotyl (BQIH), nicotyl (BQNH) and benzoyl (BQBH) hydrazide in ethanol in presence of catalytic amount of conc. HCl with continuous stirring at temperature of 60-65°C. The reaction mixture was poured on crushed ice and then purified by silica gel column chromatography using chloroform: methanol as solvent system.
BQ: 2, 6-di-tert-butylcyclohexa-2,5-diene-1,4-dione
1H NMR (400 MHz, DMSO-D6): 1.286 (s, 18H), 6.512 (s, 2H). 13C-NMR (100 MHz, DMSO-D6): 26.36, 35.55, 130.13, 157.84, 187.16, and 189.03. IR (KBr, cm−1): 1654 (ν −C=O, adjacent to di-tert-butyl groups), 1654 (ν −C=O, free)
BQBH: N’- (3,5-di-tert-butyl-4-oxocyclohexa-2,5-dienylidene) benzohydrazide
Yield: 73%; m.p.: 160°C; 1H NMR (400 MHz, DMSO-D6): 1.281-1.329 (s, 18H), 7.108 (s, 1H), 7.505 (t, 2H, J= 7.64 Hz), 7.584 (t, 1H, J= 7.36 Hz), 7.66 (d, 1H, J= 11.3 Hz), 7.856 (d, 2H, J= 7.6 Hz), 12.027 (s, 1H, −NH), 13C-NMR (125.75 MHz, DMSO-D6): 29.614, 29.892, 35.151, 36.369, 119.523, 128.818, 129.273, 132.513, 133.849, 134.101, 149.196, 151.407, 159.958, 187.048. IR (KBr, cm−1): 1651(ν −C=O, 1535 (ν −C=N); UV-Vis: λmax(nm, DMSO): 369; Anal. Calcd. for C21H26N2O2: C, 74.55; H, 7.69; N, 8.28. Found: C, 73.87; H, 7.09; N, 7.73. ESIMS (m/z): Calc 338.44, found 339 (M++H) in accordance with MF C20H25N3O2.
BQNH: N’- (3,5-di-tert-butyl-4-oxocyclohexa-2,5-dienylidene) nicotinohydrazide
Yield: 65%; m.p.: 197°C; 1H NMR (400 MHz, DMSO-D6): 1.278-1.334 (s, 18H), 7.141 (s, 1H), 7.489 (t, 1H, J= 6.8 Hz), 7.618 (d, 1H, J=15.84 Hz), 8.24 (s, 1H), 8.7709 (s, 1H), 9.107 (s, 1H), 12.178 (s, 1H, −NH). 13C-NMR (125.75 MHz, DMSO-D6): 29.592, 29.890, 35.160, 36.416, 119.478, 123.904, 129.802, 133.906, 137.251, 149.452, 149.977, 151.728, 152.865, 187.024. IR (KBr, cm−1): 1666(ν C=O), 1539 (ν −C=N); UV-Vis: λmax(nm, DMSO): 366; Anal. Calcd. for C20H25N3O2: C, 70.79; H, 7.37; N, 12.38. Found: C, 70.07; H, 7.37; N, 12.09. ESIMS (m/z): Calc 339.43, found 340 (M++H) in accordance with MF C21H26N2O2.
BQIH: N’- (3,5-di-tert-butyl-4-oxocyclohexa-2,5-dienylidene) isonicotinohydrazide
Yield: 77%; m.p.: 170°C; 1H NMR (400 MHz, DMSO-D6): 1.271-1.320 (s, 18H), 7.097 (d, 1H, J=13Hz), 7.658-7.943 (m, 3H), 8.777 (s, 2H), 12.374 (s, 1H, −NH). 13C-NMR (125.75 MHz, DMSO-D6): 29.587, 29.879, 35.178, 36.419, 119.486, 122.968, 133.887, 149.612, 150.519, 151.820, 187.032. IR (KBr, cm−1): 1666(ν C=O), 1541 (ν −C=N); UV-Vis: λmax(nm, DMSO): 363; Anal. Calcd. for C20H25N3O2: C, 70.79; H, 7.37; N, 12.38. Found: C, 68.87; H, 7.09; N, 11.973%. ESIMS (m/z): Calc 339.43, found 339 (M+) in accordance with MF C20H25N3O2.
4.2 Molecular Docking Study
All calculations were performed using AutoDock-Vina software 62. The crystal structure of COX-2 protein was obtained from PDB ID (6COX) 63. The active site of the enzyme was defined to include residues of the COX-2 within the grid size of 50 A° X 50 A° X 50 A° to any of the inhibitory atoms. The AutoDock-Vina program which is an automated docking program was used to dock all three 2, 6 di-tert-butyl-p-benzoquinone hydrazone analogs as well as Darbufelone molecule in the active site of COX-2 enzyme. For each compound the most stable docking model was selected based upon conformation of best scored predicted by Auto Dock scoring function. The compounds were energy minimized with MMFF94 force field. From the histogram relevant parameters such as binding energy, total number of hydrogen bond formed and hydrogen bonding pattern were determined.
The 3D crystal structure of stable 5-lipoxygenase was obtained from Protein Data Bank (PDB ID: 3O8Y) 64. The ligands were energy minimized and partial charges were added using PRODRG algorithms. Each ligand was then docked individually into the active site of 5-LOX using AutoDock-Vina. The AutoDockTools graphical user interface was used to add polar hydrogens and partial charges, using Kollman United charges, to 5-LOX. Atomic solvation parameters and fragment volumes were assigned using the ADDSOL sub-routine. The grid map was calculated using the auxiliary program Autogrid3. Grid maps of 50 A° X 50 A° X 50 A° points centered on the active site of the ligand were calculated for each atom types found on the adducts. Lamarckian Genetic Algorithm (LGA) was selected for ligand conformational search. The Genetic Algorithm (GA) population size was set to 150, the maximum number of GA energy evaluations as 2500000, GA mutation rate as 0.02, GA crossover rate as 0.8 and GA docking runs was set as 100. The resulting docking conformations were clustered into families of similar conformation, with root mean square deviation (RMSD) clustering tolerance as 1.0 A°. The lowest docking energy conformations were included as a rule in the largest cluster. Flexible torsion in the ligands was assigned with Autotors, an auxiliary module for AutoDockTools. Each ligand was docked individually with 5-LOX to obtain the best binding conformation.
4.3 In vitro Cyclooxygenase (COX) Inhibition Assay
The ability of the synthesized compounds to inhibit the conversion of arachidonic acid (AA) to prostaglandin H2 by ram seminal vesicle COX-1 and sheep placental COX-2 was determined using a COX-2/COX-2 inhibitor screening assay kit (No. 560101; Cayman Chemical, Ann Arbor, MI). Cyclooxygenase catalyzes the first step in the biosynthesis of AA to PGH2. PGF2α produced from PGH2 by reduction with stannous chloride was measured by enzyme immunoassay. This assay is based on the competition between PGs and a PG-acetyl cholinesterase conjugate (PG tracer) for limited amount of PG antiserum. The amount of PG tracer that is able to bind to the PG antiserum is inversely proportional to the concentration of PGs in the wells since the concentration of PG tracer is held constant while that of PGs varies. This antibody-PG complex binds to a mouse anti-rabbit monoclonal antibody that has been previously attached to the well. The plate is washed to remove any unbound reagents and the Ellman’s Reagent is added to the well, which contains the substrate to Acetyl cholinesterase the product of this enzymatic reaction produces a distinct yellow color that absorbs at 405 nm. The intensity of this color is determined spectrophotometrically and is proportional to the amount of the well that in turn is inversely proportional to the amount of PGs present in the well during the incubation. Percent inhibition was calculated by comparison of compound treated to various control incubations. The concentration of the test compound causing 50% inhibition (IC50, μM) was calculated from the concentration-inhibition response curve 65, 66.
4.4 In vitro Lipoxygenase (5-LOX) Inhibition Assay
The ability of the test compounds to inhibit potato 5-LOX (catalog number 60401, Cayman Chemical, Ann Arbor, MI) was determined using an enzyme immunoassay (EIA) kit (catalog number 760700, Cayman Chemical, Ann Arbor, MI) according to the manufacturer’s instructions. The Cayman Chemical Lipoxygenase inhibitor screening assay detects and measures the hydroperoxides produced in the lip oxygenation reaction using a purified lipoxygenases. Stock solutions of test compounds were dissolved in a minimum volume of DMSO and were diluted using the supplied buffer solution. The lipoxygenases reaction was initiated by the addition of 10 μL arachidonic acid. After maintaining the 96-well plate on a shaker for 5 min, 100 μL of chromogen was added and the plate was retained on a shaker for 5 min. The lipoxygenases activity was determined after measuring absorbance at a wavelength of 500 nm. Percent inhibition was calculated by the comparison of compound-treated to the control incubations. The concentration of the test compound causing 50% inhibition (IC50, μM) was calculated from the concentration-inhibition response curve (duplicate determinations) 67.
4.5 Cell Proliferation Assay
Cell proliferation was measured by CellTiter 96® AQueous Assay (Promega). The reagent consists of solutions of a novel tetrazolium compound (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt; MTS) and an electron coupling reagent (phenazine methosulfate; PMS). MTS is reduced by cells into a soluble formazan product in tissue culture medium. The absorbance of the formazan is measured.. Growing cells were harvested, counted, and seeded at the 20 × 103 cells (200-μL volume) into 96-well microplates. After 18 h, culture medium was replaced by medium containing experimental agents (200 μL) as indicated in Figure 2. MTS/PMS solution was freshly prepared at 2/0.92 mg/mL in DPBS. Ten micro liters of MTS/PMS reagent (excess MTS/PMS) were added to each well and then incubated at 5% CO2 atmosphere. Absorbance at 490 nm was recorded at 90 min. A blank experiment detecting cell-free background absorbance was also performed in parallel. Absorbance shown in the figures was obtained by subtracting the absorbance of cell-free equivalents. Trypan blue exclusion showed less than 1% cell death both before and after the assays.
4.6 The MTT assay
The MTT viability assay was performed with slight modifications as previously described74. A stock solution of MTT was first prepared as a 5 mg/ml in phosphate buffer (PBS, pH 7.2) and filtered. At the end of the treatment period of our tested compounds (24 h), with four different concentrations in triplicate, 25 μl of MTT solution was added to each well. After incubation for 4 h at 37°C, 100 μl of solubilizing buffer (10% sodium dodecyl sulfate dissolved in 0.01 N HCl) was added to each well. After incubation, the 96-well plate was read by an enzyme-linked immunosorbent assay (ELISA) reader at 570 nm for absorbance density values to determine the cell viability. The viable cells produced a dark blue formazan product, whereas no such staining was formed in the dead cells.
Table 3.
COX-LOX Inhibitory Potential of 2, 6-Di-Tert-Butyl-Benzoquinone-4-Hydrazone compounds
| Compound | IC50 COX-2 μM |
IC50 COX-1 μM |
IC50 COX-2/IC50 COX-1 |
IC50 5-LOX μM |
|---|---|---|---|---|
| BQBH | 9.5 | 14.0 | 0.68 | 5.1 |
| BQNH | 9.3 | 48.6 | 0.19 | 7.4 |
| BQIH | 7.6 | 55.7 | 0.14 | 20 |
| Darbufelone | 0.48 | 35.00 | 0.014 | 0.77 |
Acknowledgments
We thank Dr. Vincent C. Hascall (Cleveland Clinic, Cleveland, Ohio), and Dr. Roger R. Markwald (Medical University of South Carolina, Charleston, SC) for their support and academic discussions in this work. We also thank Dr. Carla De Giovanni (University of Bologna, Bologna, Italy) for Apc 10.1 cells. This work was supported, in whole or in part, by National Institutes of Health Grants P20RR021949 (to S. G.), P20RR016434 (to S. M., S. G., and R. R. M.), P20RR16461-05 (to S.G., and R. R. M), HL033756-24 and 1 P30AR050953 (to V. C. H.). This work was also supported by Mitral-07 CVD 04 (to R. R. M.), Medical University of South Carolina University Research Project 2204000-24330 (to S. M.) and 2204000-24329 (to S. G.). P.D. acknowledges CSIR, New Delhi, INDIA for providing Senior Research Fellowship. SP acknowledges Mr. P. A. Inamdar and Dr. E. M. Khan for lab facilities for this project.
Abbreviations
- COX-2
cyclooxygenase
- CD44v6
variant exon 6 of CD44
- CD44s
CD44 standard form
- HA
hyaluronan
- Has2
hyaluronan synthase-2
- RTK
receptor tyrosine kinase
- PGE2
prostaglandin E2
- LOX
lipoxygenase
- AA
arachidonic acid
- Darbufelone
5-((Z)-3,5-Di-tert-butyl-4-hydroxybenzylidene)-2-imino-4-thiazolidinone
- Licofelone
[6-(4-chlorophenyl)-2,2-dimethyl-7-phenyl-2,3-dihydro-1H-pyrrolizin-5-yl]acetic acid
- CRC
colon rectal cancer
- 5-FU
5-fluorouracil
- LTB4
leukotriene B4
- 5-HETE
5-hydroxyeicosatetranoic acid
- NSAID
Non-Steroidal Anti-Inflammatory Drugs
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest: Author declare no conflicts of interest
REFERENCES
- 1.Siegel R, Ward E, Brawley O, Jemal A. CA Cancer J Clin. 2011;61:212–236. doi: 10.3322/caac.20121. [DOI] [PubMed] [Google Scholar]
- 2.Heinemann V. Oncology. 2001;60:8–18. doi: 10.1159/000055290. [DOI] [PubMed] [Google Scholar]
- 3.Sarkar FH, Banerjee S, Li Y. Toxicol Appl Pharmacol. 2007;224:326–336. doi: 10.1016/j.taap.2006.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reddy BS, Rao CV. J Environ Pathol Toxicol Onco l. 2002;21:155–164. [PubMed] [Google Scholar]
- 5.Reddy BS, Hirose Y, Lubet R, Steele V, Kelloff G, Paulson S, Seibert K, Rao CV. Cancer Res. 2000;60:293–297. [PubMed] [Google Scholar]
- 6.Kulkarni SK, Singh VP. Curr Top Med Chem. 2007;7:251–263. doi: 10.2174/156802607779941305. [DOI] [PubMed] [Google Scholar]
- 7.Rioux N, Castonguay A. Carcinogenesis. 1998;19:1393–1400. doi: 10.1093/carcin/19.8.1393. [DOI] [PubMed] [Google Scholar]
- 8.Ding XZ, Hennig R, Adrian TE. Mol Cancer. 2003;2:10. doi: 10.1186/1476-4598-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shureiqi I, Lippman SM. Cancer Res. 2001;61:6307–6312. [PubMed] [Google Scholar]
- 10.Steele VE, Holmes CA, Hawk ET, Kopelovich L, Lubet RA, Crowell JA, Sigman CC, Kelloff GJ. Cancer Epidemiol Biomarkers Prev. 1999;8:467–483. [PubMed] [Google Scholar]
- 11.Song Y, Wei EQ, Zhang WP, Zhang L, Liu JR, Chen Z. Neuroreport. 2004;15:2181–2184. doi: 10.1097/00001756-200410050-00007. [DOI] [PubMed] [Google Scholar]
- 12.Melstrom LG, Bentrem DJ, Salabat MR, Kennedy TJ, Ding XZ, Strouch M, Rao SM, Witt RC, Ternent CA, Talamonti MS, et al. Clin Cancer Res. 2008;14:6525–6530. doi: 10.1158/1078-0432.CCR-07-4631. [DOI] [PubMed] [Google Scholar]
- 13.Lu P, Schrag ML, Slaughter DE, Raab CE, Shou M, Rodrigues AD. Drug Metab Dispos. 2003;31:1352–1360. doi: 10.1124/dmd.31.11.1352. [DOI] [PubMed] [Google Scholar]
- 14.Duffield-Lillico AJ, Boyle JO, Zhou XK, Ghosh A, Butala GS, Subbaramaiah K, Newman RA, Morrow JD, Milne GL, Dannenberg AJ. Cancer Prev Res (Phila) 2009;2:322–329. doi: 10.1158/1940-6207.CAPR-09-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamazaki R, Kusunoki N, Matsuzaki T, Hashimoto S, Kawai S. FEBS Lett. 2002;531:278–284. doi: 10.1016/s0014-5793(02)03535-4. [DOI] [PubMed] [Google Scholar]
- 16.Maier TJ, Schilling K, Schmidt R, Geisslinger G, Grosch S. Biochem Pharmacol. 2004;67:1469–1478. doi: 10.1016/j.bcp.2003.12.014. [DOI] [PubMed] [Google Scholar]
- 17.Hawk ET, Viner JL, Dannenberg A, DuBois RN. J Natl Cancer Inst. 2002;94:545–546. doi: 10.1093/jnci/94.8.545. [DOI] [PubMed] [Google Scholar]
- 18.Bourguignon LY. J Mammary Gland Biol Neoplasia. 2001;6:287–297. doi: 10.1023/a:1011371523994. [DOI] [PubMed] [Google Scholar]
- 19.Misra S, Hascall VC, De Giovanni C, Markwald RR, Ghatak S. J Biol Chem. 2009;284:12432–12446. doi: 10.1074/jbc.M806772200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aruffo A, Stamenkovic I, Melnick M, Underhill CB, Seed B. Cell. 1990;61:1303–1313. doi: 10.1016/0092-8674(90)90694-a. [DOI] [PubMed] [Google Scholar]
- 21.Lesley J, Hascall VC, Tammi M, Hyman R. J Biol Chem. 2000;275:26967–26975. doi: 10.1074/jbc.M002527200. [DOI] [PubMed] [Google Scholar]
- 22.Lesley J, Hyman R. Eur J Immunol. 1992;22:2719–2723. doi: 10.1002/eji.1830221036. [DOI] [PubMed] [Google Scholar]
- 23.Lesley J, English N, Charles C, Hyman R. Eur J Immunol. 2000;30:245–253. doi: 10.1002/1521-4141(200001)30:1<245::AID-IMMU245>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 24.Ponta H, Sherman L, Herrlich PA. Nat Rev Mol Cell Biol. 2003;4:33–45. doi: 10.1038/nrm1004. [DOI] [PubMed] [Google Scholar]
- 25.Yu Q, Stamenkovic I. Genes Dev. 2000;14:163–176. [PMC free article] [PubMed] [Google Scholar]
- 26.Yu Q, Stamenkovic I. Genes Dev. 1999;13:35–48. doi: 10.1101/gad.13.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Misra S, Toole BP, Ghatak S. J Biol Chem. 2006;281:34936–34941. doi: 10.1074/jbc.C600138200. [DOI] [PubMed] [Google Scholar]
- 28.Bourguignon LY, Gilad E, Peyrollier K. J Biol Chem. 2007;282:19426–19441. doi: 10.1074/jbc.M610054200. [DOI] [PubMed] [Google Scholar]
- 29.Orian-Rousseau V, Chen L, Sleeman JP, Herrlich P, Ponta H. Genes Dev. 2002;16:3074–3086. doi: 10.1101/gad.242602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ishida T. Surg Today. 2000;30:28–32. doi: 10.1007/PL00010042. [DOI] [PubMed] [Google Scholar]
- 31.Heider KH, Hofmann M, Hors E, van den Berg F, Ponta H, Herrlich P, Pals ST. J Cell Biol. 1993;120:227–233. doi: 10.1083/jcb.120.1.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Neumayer R, Rosen HR, Reiner A, Sebesta C, Schmid A, Tuchler H, Schiessel R. Dis Colon Rectum. 1999;42:50–55. doi: 10.1007/BF02235182. [DOI] [PubMed] [Google Scholar]
- 33.Wielenga VJ, Heider KH, Offerhaus GJ, Adolf GR, van den Berg FM, Ponta H, Herrlich P, Pals ST. Cancer Res. 1993;53:4754–4756. [PubMed] [Google Scholar]
- 34.Wielenga VJ, van der Neut R, Offerhaus GJ, Pals ST. Adv Cancer Res. 2000;77:169–187. doi: 10.1016/s0065-230x(08)60787-3. [DOI] [PubMed] [Google Scholar]
- 35.Wielenga VJ, van der Voort R, Mulder JW, Kruyt PM, Weidema WF, Oosting J, Seldenrijk CA, van Krimpen C, Offerhaus GJ, Pals ST. Scand J Gastroenterol. 1998;33:82–87. doi: 10.1080/00365529850166257. [DOI] [PubMed] [Google Scholar]
- 36.Ghatak S, Hascall VC, Karamanos NK, Markwald RR, Misra S. DeGruyter; Berlin, Germany: 2012. [Google Scholar]
- 37.Misra S, Hascall V, Karamanos N, Markwald Ra, Ghatak S. De Gruyter; 2012. [Google Scholar]
- 38.Misra S, Hascall VC, Karamanos NK, Markwald RR, Ghatak S. DeGruyter; Berlin, Germany: 2012. [Google Scholar]
- 39.Misra S, Heldin P, Hascall VC, Karamanos NK, Skandalis SS, Markwald RR, Ghatak S. Febs J. 2011;278:1429–1443. doi: 10.1111/j.1742-4658.2011.08071.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Naor D, Nedvetzki S, Golan I, Melnik L, Faitelson Y. Crit Rev Clin Lab Sci. 2002;39:527–579. doi: 10.1080/10408360290795574. [DOI] [PubMed] [Google Scholar]
- 41.Yamada Y, Itano N, Narimatsu H, Kudo T, Morozumi K, Hirohashi S, Ochiai A, Ueda M, Kimata K. Clin Exp Metastasis. 2004;21:57–63. doi: 10.1023/b:clin.0000017203.71293.e0. [DOI] [PubMed] [Google Scholar]
- 42.Misra S, Hascall VC, Berger FG, Markwald RR, Ghatak S. Connect Tissue Res. 2008;49:219–224. doi: 10.1080/03008200802143356. [DOI] [PubMed] [Google Scholar]
- 43.Misra S, Obeid LM, Hannun YA, Minamisawa S, Berger FG, Markwald RR, Toole BP, Ghatak SJ. Biol Chem. 2008;283:14335–14344. doi: 10.1074/jbc.M703811200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang SJ, Bourguignon LY. Arch Otolaryngol Head Neck Surg. 2006;132:19–24. doi: 10.1001/archotol.132.1.19. [DOI] [PubMed] [Google Scholar]
- 45.Wang SJ, Bourguignon LY. Arch Otolaryngol Head Neck Surg. 2006;132:771–778. doi: 10.1001/archotol.132.7.771. [DOI] [PubMed] [Google Scholar]
- 46.Ohashi R, Takahashi F, Cui R, Yoshioka M, Gu T, Sasaki S, Tominaga S, Nishio K, Tanabe KK, Takahashi K. Cancer Lett. 2007;252:225–234. doi: 10.1016/j.canlet.2006.12.025. [DOI] [PubMed] [Google Scholar]
- 47.Misra S, Ghatak S, Zoltan-Jones A, Toole BP. J Biol Chem. 2003;278:25285–25288. doi: 10.1074/jbc.C300173200. [DOI] [PubMed] [Google Scholar]
- 48.Misra S, Ghatak S, Toole BP. J Biol Chem. 2005;280:20310–20315. doi: 10.1074/jbc.M500737200. [DOI] [PubMed] [Google Scholar]
- 49.Bourguignon LY, Spevak CC, Wong G, Xia W, Gilad E. J Biol Chem. 2009;284:26533–26546. doi: 10.1074/jbc.M109.027466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ghatak S, Hascall VC, Markwald RR, Misra SJ. Biol Chem. 2010;285:19821–19832. doi: 10.1074/jbc.M110.104273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ghatak S, Hascall VC, Berger FG, Penas MM, Davis C, Jabari E, He X, Norris JS, Dang Y, Markwald RR, Misra S. Connect Tissue Res. 2008;49:265–269. doi: 10.1080/03008200802147845. [DOI] [PubMed] [Google Scholar]
- 52.Bertagnolli MM, Eagle CJ, Zauber AG, Redston M, Breazna A, Kim K, Tang J, Rosenstein RB, Umar A, Bagheri D, et al. Cancer Prev Res (Phila) 2009;2:310–321. doi: 10.1158/1940-6207.CAPR-08-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang B, Wang CL, Zhao WH, Lv M, Wang CY, Zhong WX, Zhou WY, Yu WS, Zhang Y, Li S. World J Gastroenterol. 2008;14:2494–2500. doi: 10.3748/wjg.14.2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Song Y, Connor DT, Doubleday R, Sorenson RJ, Sercel AD, Unangst PC, Roth BD, Gilbertsen RB, Chan K, Schrier DJ, et al. J Med Chem. 1999;42:1151–1160. doi: 10.1021/jm9805081. [DOI] [PubMed] [Google Scholar]
- 55.Song Y, Connor DT, Sercel AD, Sorenson RJ, Doubleday R, Unangst PC, Roth BD, Beylin VG, Gilbertsen RB, Chan K, et al. J Med Chem. 1999;42:1161–1169. doi: 10.1021/jm980570y. [DOI] [PubMed] [Google Scholar]
- 56.Inagaki M, Tsuri T, Jyoyama H, Ono T, Yamada K, Kobayashi M, Hori Y, Arimura A, Yasui K, Ohno K, et al. J Med Chem. 2000;43:2040–2048. doi: 10.1021/jm9906015. [DOI] [PubMed] [Google Scholar]
- 57.Ye X, Zhou W, Li Y, Sun Y, Zhang Y, Ji H, Lai Y. Cancer Chemother Pharmacol. 2010;66:277–285. doi: 10.1007/s00280-009-1161-z. [DOI] [PubMed] [Google Scholar]
- 58. http://www.pslgroup.com/dg/14878E.htm.
- 59.De Giovanni C, Landuzzi L, Nicoletti G, Astolfi A, Croci S, Micaroni M, Nanni P, Lollini PL. Int J Cancer. 2004;109:200–206. doi: 10.1002/ijc.11690. [DOI] [PubMed] [Google Scholar]
- 60.Ohd JF, Nielsen CK, Campbell J, Landberg G, Lofberg H, Sjolander A. Gastroenterology. 2003;124:57–70. doi: 10.1053/gast.2003.50011. [DOI] [PubMed] [Google Scholar]
- 61.Ye YN, Wu WK, Shin VY, Bruce IC, Wong BC, Cho CH. Carcinogenesis. 2005;26:827–834. doi: 10.1093/carcin/bgi012. [DOI] [PubMed] [Google Scholar]; (a) Sasaki T, Yoshida K, Shimura H, Ichiba M, Sasahira T, Shimomoto T, Denda A, Kuniyasu H. Int J Cancer. 2006;118:593–9. doi: 10.1002/ijc.21393. [DOI] [PubMed] [Google Scholar]
- 62.Trott O, Olson AJ. Journal of Computational Chemistry. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rowlinson SW, Kiefer JR, Prusakiewicz JJ, Pawlitz JL, Kozak KR, Kalgutkar AS, Stallings WC, Kurumbail RG, Marnett LJ. J Biol Chem. 2003;278:45763–45769. doi: 10.1074/jbc.M305481200. [DOI] [PubMed] [Google Scholar]
- 64.Gilbert NC, Bartlett SG, Waight MT, Neau DB, Boeglin WE, Brash AR, Newcomer ME. Science. 2011;331:217–219. doi: 10.1126/science.1197203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Habeeb AG, Praveen Rao PN, Knaus EE. Drug Dev Res. 2000;51:273–286. [Google Scholar]
- 66.Penning TD, Talleym JJ, Bertenshaw SR, Carter JS, Collins PW, Docter S, Graneto MJ, Lee LF, Malecha JW, Miyashiro JM, Rogers RS, Rogier DJ, Yu SS, Anderson GD, Burton EG, Cogburn JN, Gregory SA, Koboldt CM, Perkins WE, Seibert K, Veenhuizen AW, Zhang YY, Isakson PC. J Med Chem. 1997;40:1347–65. doi: 10.1021/jm960803q. [DOI] [PubMed] [Google Scholar]
- 67.Rao PN, Chen QH, Knaus EE. J Med Chem. 2006;49:1668–83. doi: 10.1021/jm0510474. [DOI] [PubMed] [Google Scholar]
- 68.Guidote AM, Jr, Ando K, Terada K, Kurusu Y, Nagao H, Masuyama Y. Inorg Chim Acta. 2001;324:203–211. [Google Scholar]
- 69.Coates J. In: Encyclopedia of Analytical Chemistry. Meyers RA, editor. John Wiley & Sons Ltd; Chichester: 2000. pp. 10815–10837. [Google Scholar]
- 70.Figueras J, Scullard PW, Mack AR. J Org Chem. 1971;36:3497–3501. [Google Scholar]
- 71.Milaeva ER, Gerasimova OA, Jingwei Z, Shpakovsky DB, Syrbu SA, Semeykin AS, Koifman OI, Kireeva EG, Shevtsova EF, Bachurin SO, Zefirov NS. J Inorg Biochem. 2008;102:1348–1358. doi: 10.1016/j.jinorgbio.2008.01.022. [DOI] [PubMed] [Google Scholar]
- 72.Kilic A, Tas E, Deveci B, Yilmaz I. Polyhedron. 2007;26:4009–4018. [Google Scholar]
- 73.Laskowski RA. Nucleic Acids Res. 2009;37:D355–359. doi: 10.1093/nar/gkn860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mosmann T. J Immunol Meth. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]