

Abstract
Purpose
Hyperthermia enhances cytotoxic effects of chemotherapeutic agents such as cisplatin. However, the underlying molecular mechanisms remain unclear. We hypothesised that hyperthermia increases cisplatin accumulation and efficacy by modulating function of copper transport protein 1 (Ctr1), a major regulator of cellular cisplatin uptake. We examined the significance of Ctr1 in the synergistic interaction between hyperthermia and cisplatin. We assessed the importance of cisplatin- and hyperthermia-induced Ctr1 multimerisation in sensitising cells to cisplatin cytotoxicity.
Materials and methods
Ctr1 protein levels and cisplatin sensitivities were assessed in bladder cancer cell lines with immunoblotting and clonogenic survival assays. Using Myc-tagged-Ctr1 HEK293 cells, we assessed the effect of hyperthermia on Ctr1 multimerisation with immunoblotting. The effect of hyperthermia on cisplatin sensitivity and accumulation was assessed in wild-type (WT) and Ctr1 knockout (Ctr1−/−) mouse embryonic fibroblasts (MEFs) with clonogenic assays and inductively coupled plasma-mass spectrometry (ICP-MS).
Results
Increased Ctr1 protein expression was observed for the most cisplatin-sensitive bladder cancer cell lines and MEFs. Heat-induced increase in Ctr1 multimerisation with cisplatin was observed in Myc-tagged Ctr1 cells. Hyperthermia enhanced cisplatin-mediated cytotoxicity in WT more than Ctr1−/− cells (dose modifying factors 1.75 versus 1.4, respectively). WT cells accumulated more platinum versus Ctr1−/− cells; this was further increased by hyperthermia in WT cells.
Conclusions
Hyperthermia enhanced cisplatin uptake and cytotoxicity in WT cells. Heat increased Ctr1 activity by increasing multimerisation, enhancing drug cytotoxicity. Furthermore, Ctr1 protein profiles of bladder tumours, as well as other tumour types, may predict their response to cisplatin and overall efficacy of treatment.
Keywords: Bladder cancer, cisplatin, Ctr1, hyperthermia
Introduction
The chemotherapeutic agent cisplatin, first described by Rosenberg et al. in 1965, has been used clinically since the late 1970s for the treatment of multiple types of cancer, including testicular, ovarian, bladder, cervical, head and neck, and small cell lung cancers [1]. Although the ability of hyperthermia to synergistically enhance the cytotoxic effects of cisplatin has long been recognised [2–4], the mechanism underlying this process remains unclear. It is known that hyperthermic sensitisation is associated with increased drug accumulation and subsequent platinum-DNA adduct formation [3], but the underlying mechanisms for these responses remain incompletely unexplained. For example, previous studies have shown increased drug uptake, but the mechanism underlying the increase in uptake has not been elucidated. Additional mechanisms that have been identified include altered drug metabolism, increased drug reaction rates with DNA, and heat-induced inhibition in DNA repair, indicating that multiple mechanisms may be involved in the synergism at the cell surface and intracellularly [5]. Hyperthermia has also been shown to reverse cisplatin resistance in vitro [3,4]. Here, we investigated the role of the copper transport protein 1 (Ctr1) in this process. Under normal metabolic conditions, Ctr1 maintains a homeostatic balance between intra- and extracellular copper levels; however, Ctr1 can also function as a cisplatin transporter [6]. We hypothesised that the observed synergistic interaction between heat and cisplatin is due, in part, to the ability of heat to enhance Ctr1 function.
Ctr1 is an ATP-independent transporter in the cell membrane. Its high affinity for copper allows sufficient levels of copper to enter the cell for normal metabolic function. Ctr1 RNA has been detected in all organs and tissues examined with the greatest expression in the heart, liver, pancreas, prostate, colon, and intestine [7]. Ctr1 consists of three membrane-spanning segments with an extracellular amino terminus and a cytoplasmic carboxyl-terminal tail. Using electron crystallography, De Feo et al. demonstrated that Ctr1 trimers create a pore across the cell membrane [8]. The amino terminus contains methionine-rich motifs that are involved in Ctr1 multimerisation and subsequent copper transport [9].
Ctr1 expression is regulated at both the transcriptional and translational levels. Dancis et al. observed that copper deprivation induced Ctr1 gene expression, whereas excess copper resulted in decreased Ctr1p gene expression [10]. Early work done on yeast Ctr1 showed that overexpression of Ctr1p resulted in increased cellular copper uptake. At the translational level, Petris et al. demonstrated that exposure to elevated copper levels resulted in decreased surface Ctr1 levels, increased Ctr1 endocytosis, and degradation of the protein [11].
In addition to copper, Ctr1 also transports platinum-based compounds such as cisplatin. Previous studies have shown that knocking out Ctr1 in cells decreases cisplatin uptake and increases cisplatin resistance [6,12]. Song et al. exogenously overexpressed Ctr1 in a small cell lung carcinoma cell line and a cisplatin-resistant variant to enhance cisplatin uptake and sensitivity [13]. Expression of Ctr1 protein with C-terminus mutations showed normal function in the uptake and sensitivity to cisplatin, whereas N-terminus mutations rendered the protein inactive, suggesting the NH2-terminal amino acid sequence is necessary for cisplatin transport.
The precise mechanisms by which copper and cisplatin enter the cell via Ctr1 are distinct but have yet to be clearly elucidated [14]. Copper exposure results in Ctr1 internalisation, reducing surface Ctr1 levels due to both endocytosis and degradation of the transporter [11]. In contrast, cisplatin exposure results in stable multimer (trimer) formation and only basal levels of internalisation [11,15]. This group hypothesised that stabilisation of the Ctr1 multimer on the cell surface may create a pore to facilitate cisplatin uptake [15]. Guo et al. demonstrated that cisplatin exposure stabilised a multimer of Ctr1 [15]. Formation of this multimer was assessed in the presence of platinum chelators or in cells with mutant methionine-rich motifs (M1/M2) in the amino-terminal region of Ctr1, and the largest multimer complex was not formed following cisplatin exposure, again emphasising the importance of the amino-terminal region of Ctr1.
Three recent studies have also shown that Ctr1 mRNA expression levels are associated with platinum sensitivity in ovarian cancer patients [16–18]. Lee et al. assessed mRNA levels in ovarian carcinoma patients and found that higher Ctr1 expression was associated with sensitivity to platinum-based therapy and was a prognostic factor for improved survival [17]. Ishida et al. also assessed Ctr1 levels in advanced stage ovarian carcinoma patients and found that lower Ctr1 expression levels were associated with platinum resistance and decreased disease-free survival [16]. Finally, Liang et al. found an association between elevated Ctr1 mRNA levels and progression free and overall survival in ovarian cancer patients following treatment with platinum-based therapy [18]. These studies demonstrate the clinical importance of Ctr1 in cancer types commonly treated with cisplatin.
Due to the significant role Ctr1 plays in cisplatin uptake, we hypothesised that hyperthermia increases cisplatin accumulation and efficacy in part by modulating Ctr1 function. In this study, we examine the significance of Ctr1 function in the synergistic interaction between hyperthermia and cisplatin in the context of bladder cancer. Interestingly, Ctr1 has been shown to be predominantly localised in caveolin-enriched lipid rafts in vascular smooth muscle cells [19], and recent work by Mace et al. demonstrates enhanced clustering of ganglioside (GM1+) lipid microdomains, a predominant marker of lipid rafts [20], in CD8+ T cells following hyperthermia treatment (39.5 °C) [21,22]. Based on these studies, we hypothesise that hyperthermia may be enhancing cisplatin-induced Ctr1 multimerisation. Here, we assess the importance of cisplatin- and hyperthermia-induced Ctr1 multimerisation in sensitising cells to cisplatin cytotoxicity.
Materials and methods
Reagents and chemicals
Cisplatin was purchased from APP Pharmaceuticals (Schaumburg, IL, USA). Cycloheximide and hydrochloric acid (HCl, 1.0 N) were supplied by Sigma (St Louis, MO, USA). Nitric acid (Optima*) was obtained from Fisher Scientific (Pittsburgh, PA, USA). All cell culture reagents including media and supplementation were purchased from Gibco (Grand Island, NY, USA).
Cell lines
Wild-type (WT) and Ctr1 knockout (Ctr1−/−) mouse embryonic fibroblasts (MEFs) were kindly provided by Dennis Thiele. These cells were cultured in RPMI-1640 supplemented with 10% foetal bovine serum (FBS) and 1% penicillin/streptomycin. The bladder cancer cell lines J82, NBT2, MBT2, and MB49 were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS and 1% antimycotic/antibiotic. The human bladder cancer cell lines 5637 and RT4 were cultured in RPMI and McCoy's 5a, respectively, supplemented with 10% FBS and 1% antimycotic/antibiotic. The WT Myc-tagged Ctr1 and pcDNA3.1 vector control cell lines as well as the M1/M2 mutant Ctr1 plasmid were kindly provided by Michael J. Petris. The HEK293 cells were cultured in DMEM supplemented with 10% FBS, 1% antibiotic/antimycotic, and 0.1% 2-mercaptoethanol. All cell lines were maintained at 37 °C/20% O2/5% CO2.
Human bladder cancer samples and real-time PCR
RNA samples from human bladder cancer patients were kindly provided by Brant Inman, and a normal bladder tissue control sample was obtained from the Duke Cancer Institute Shared Resource Biorepository (Durham, NC). RNA was isolated from paraffin-embedded tissue using the AllPrep DNA/RNA FFPE kit according to the manufacturer's instructions (QIAGEN, Germantown, MD, USA). RNA concentrations were measured using a NanoDrop 2000 spectrophotometer (Thermo Scientific, Wilmington, DE, USA). The iScript™ cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA) was used to make cDNA in a 20-μL reaction with 1 μg RNA according to the manufacturer's instructions. Real-time polymerase chain reaction (PCR) was conducted on a Bio-Rad iCycler using iQ™ SYBR® Green Supermix (Bio-Rad). Ctr1 primers were used as described in Kitada et al. [23] (forward: ACAAGTCAGCATTCGCTACAATTC and reverse: TTGCAGGAGGTGAGGAAAGC). The reaction was performed at 95 °C for 10 min, followed by 40 cycles of 95 °C for 5 s and 60 °C for 34 s. The dissociation stage was initiated at 95 °C for 15 s, followed by 60 °C for 15 s and 95 °C for 15 s. The threshold cycles (CT) were used to quantify the PCR product, and relative expression levels were calculated by subtracting control gene (GAPDH, forward: GAAGGTGAAGGTCGGAGTC and reverse: GAAGATGGTGATGGGATTTC) CT from Ctr1 CT. All samples were run in triplicate.
A second set of genomic data from human bladder samples (normal and malignant) was generated using the NCBI Gene Expression Omibus (GEO) Dataset Browser. (NCBI: GSE3167; GDS1479) [24].
Transfections
HEK293 cells were transfected with either the M1/M2 mutant Myc-tagged plasmid or the empty pcDNA3.1 vector control, kindly provided by Michael Petris, Ph.D. Transfections were performed with Lipofectamine™ 2000 (Invitrogen, Grand Island) according to the manufacturer's protocol. Briefly, HEK293 cells were plated in 10-cm dishes and allowed to attach overnight. Approximately 4 μg of either the vector control or M1/M2 DNA was transfected into the cells. All experiments with these cells were performed 24 h post-transfection.
Clonogenic survival
Clonogenic survival following a 2-h exposure to cisplatin was assessed in all cell types (WT and Ctr1 −/− MEFs); bladder cancer cell lines 5637, J82, MB49, MBT2, NBT2, and RT4; WT and M1/M2 Myc-tagged Ctr1 HEK293 cells). Cells were plated in 6-well plates at known densities 18–24 h before drug treatment. Cell density varied from 100 to 3000 cells per well in the bladder lines and from 200 to 6000 per well in the MEFs, depending on the plating efficiency and cisplatin concentration (higher densities received higher drug concentrations). Due to the poor attachment of HEK293 cells, these cells were plated in BD BioCoat™ poly-L-lysine-coated 6-well plates (BD Biosciences, San Jose, CA) at a density of 500 cells per well. The day after plating, cells were treated with indicated cisplatin doses (2–12 μM cisplatin or the saline control) and allowed to incubate for 2 h. Hyperthermia treatment was conducted by sealing the plates with parafilm. The bottom of the plates were then placed in a water bath set at 37 °C (normothermia control) or 41 °C. Following 1 h of hyperthermia treatment, cells were returned to the incubator for 1 h more. After drug treatment, media was removed, cells were washed with phosphate buffered saline (PBS), and fresh media was added.
Once colonies were detectable by the naked eye (7–12 days), media was removed, and cells were washed with PBS and incubated for 10 min at room temperature in a fixation solution (10% methanol, 10% glacial acetic acid, and 80% water). The fixation solution was removed and colonies were stained with crystal violet (0.4% crystal violet solution in 20% ethanol and water) for 10 min at room temperature. Plates were then rinsed with water and allowed to dry for 24 h. Colonies were counted either by hand or with a ColCount™ (Oxford Optronix, Oxford, UK), and the plating efficiency and survival fraction were calculated for each cell line at each cisplatin concentration.
Multimerisation experiments
Cells were plated in 10-cm cell culture dishes. Once the cells reached 80% confluency, they were treated with either 0 (saline vehicle), 100, or 200 μM cisplatin. Approximately 30 min prior to cisplatin treatment, 100 μg/mL cycloheximide was added to the cells to inhibit protein synthesis during the experiment. Hyperthermia treatment was conducted by sealing the dish with parafilm and placing the bottom of the dishes in a water bath set at 37° (normothermia control), 39°, 40°, 41°, 42°, or 43 °C. Cells were collected at 0, 15, 30, 45, and 60 min during hyperthermia treatment. Following 1 h of hyperthermia treatment, cells were returned to the incubator for 1 h more and then harvested. All cells were washed with PBS and harvested for protein extraction.
Protein extraction
Cells were grown to 70–90% confluency. Cells were washed once with cold 1 × PBS, and all adherent cells were scraped off and suspended in cold PBS in 1.5 mL Eppendorf tubes. Tubes were centrifuged at 4 °C for 1 min at 8000 rpm. The supernatant was aspirated from the cell pellet, and Triton-X lysis buffer (1 M Tris (pH 7.5), 5 M sodium chloride, 100 mM EDTA, 100 mM EGTA, 10% Triton X-100, and 1% protease inhibitor cocktail) was added to the pellet. Cells were placed on a rocker at 4 °C for 45 min and then centrifuged for 10 min at 12,000 rpm at 4 °C. The supernatant was collected in a 1.5 mL Eppendorf tube, frozen and stored at −80 °C, and the pellet was discarded.
Western blotting
Protein concentration was measured with the DC Protein Assay (Bio-Rad) according to the manufacturer's instructions, and western blots were performed. Briefly, proteins were separated by SDS-PAGE at 20 mA on 4–20% precast polyacrylamide gels (Mini-PROTEAN TGX, Bio-Rad). Tris-HCl gels were transferred to a polyvinylidene fluoride membrane (Bio-Rad) and then blocked either for 1 h at room temperature in 5% non-fat, dry milk reconstituted in 0.1% TBST (1x Tris buffered saline with 0.1% Tween 20). Ctr1 rabbit anti-human IgG primary antibody (kindly provided by Dennis J. Thiele, Ph.D.) was used, diluted 1:1000 in 5% milk in 1% TBST (pH 8), and incubated at 4 °C overnight. When probing for the Myc-tag, 0.5 μg/mL of the human c-Myc antibody from the supernatant of 9E10 hybridoma media (Developmental Studies Hybridoma Bank, University of Iowa, IA, USA) was diluted in 5% milk. The membrane was washed three times for 10 min in 0.1% TBST. The secondary goat anti-rabbit and goat anti-mouse IgG horse-radish peroxidase-linked antibodies (Bio-Rad) were diluted 1:2000 in 5% milk and incubated at room temperature for 1 h. The membrane was washed three more times followed by a 5-min incubation with ECL western plus blotting detection system (GE Healthcare, Amersham, UK). Kodak film (Rochester, NY) was used to capture the luminescent protein bands, and the film was developed on the Kodak processor with Spectra-2 developer (Merry X-Ray, Mentor, OH, USA) for X-ray film processing. Membranes were stripped with Restore™ PLUS western blot stripping buffer (Thermo Scientific, Rockford, IL) for 15 min at room temperature and reprobed for the loading control actin for quantification purposes.
Platinum accumulation
Cells were plated in 10-cm cell culture dishes. Once the cells reached 80% confluency, they were treated with either 0 (saline vehicle), 2, 6, or 10 μM cisplatin. Hyperthermia treatment was conducted by sealing the dish with parafilm and placing the bottom of the dishes in a water bath set at 37° (normothermia control), 41°, or 42 °C. Following 1 h of hyperthermia treatment, cells were returned to the incubator for 1 h more. Cells were washed with PBS, trypsinised, and washed in PBS once more. A small aliquot of the resuspended cell pellet was collected from each sample for measuring protein levels to normalise the samples.
Platinum measurements
To assess platinum accumulation, nitric acid digestion was performed on the cell samples as described in Minami et al. [25]. Briefly, cell samples were dried overnight at 80 °C. Each sample was then digested in 500 mL nitric acid for 20 min at 100 °C. In order to prevent platinum from falling out of solution, 250 μL of HCl was added to each sample prior to analysis. Samples were measured using ICP-MS at North Carolina State University within the Department of Soil Science (Raleigh, NC).
Statistical analyses
Clonogenic data were analysed using R software and all other data were analysed with IBM(R) SPSS(R) Statistics (Version 19.0.0). Survival curves were fitted with a single exponential fit (Surviving fraction = exp(−b × Dose)) and tested for differences in the exponent between groups; lethal concentration to 50% of cells (LC50), LC90, and dose modifying factor were calculated based on the exponential fit. Bladder cancer cell LC50s were determined based on the exponential fit, and a Spearman correlation was used to test the significance between Ctr1 protein expression and cisplatin LC50. All other data were tested with a linear regression, ANOVAs to assess response to temperature and treatment, and Tukey's post-hoc test for multiple comparisons. All experiments were done in triplicate.
Results
Human bladder cancer Ctr1 expression
Ctr1 mRNA levels were measured in human bladder cancer RNA samples. Ctr1 expression levels were also assessed from a human bladder data set available through the NCBI GEO Dataset Browser. Expression levels were variable across patient samples, for the normal and malignant tissues (Figure 1A and 1B). A waterfall plot (Figure 1A) shows the range of Ctr1 expression levels across the patient samples. Staging of the bladder cancer samples were provided for these samples; however, significant differences between tumour stage were not observed (p>0.05; data not shown). Figure 1(B) shows Ctr1 expression levels across different stages of bladder cancer and in normal bladder tissue. These data demonstrate that Ctr1 mRNA expression levels vary considerably across human bladder samples, in both normal and malignant tissues. Because of the variability in expression, it may be of interest to determine whether these levels are associated with sensitivity to platinum-based therapy.
Figure 1.

Ctr1 mRNA expression is variable in human bladder cancer samples. (A) Waterfall plot of Ctr1 mRNA expression in human bladder cancer samples, showing the range of Ctr1 expression levels. (B) Genomic data from human bladder tissue samples (normal and malignant) generated using the NCBI Gene Expression Omibus (GEO) Dataset Browser. (NCBI: GSE3167; GDS1479) [24].
Bladder cancer cell Ctr1 expression and cisplatin sensitivity
Because of the variability of Ctr1 mRNA expression observed in the human bladder samples, we assessed cisplatin sensitivity (clonogenic survival) and Ctr1 protein expression levels in six different bladder cancer cell lines of human or rodent origin (Figure 2A and 2B). Basal Ctr1 expression levels varied, as did cisplatin sensitivity. Survival curves were all significantly different from each other (p<0.05) except for the differences between 5637 and MBT2 (the two most sensitive cell lines) and between MB49 and RT4 (p>0.05). As depicted in Figure 2, we observed increased cisplatin sensitivity in the cell lines with higher basal Ctr1 expression, specifically 5637 and MBT2. Cell lines with a more cisplatin-resistant phenotype (higher LC50) expressed lower levels of Ctr1. When LC50s were plotted against relative basal Ctr1 protein expression levels (Figure 2C), a negative correlation (R2=−0.71) was detected, suggesting cell lines expressing higher levels of Ctr1 have lower cisplatin LC50s; however, this was not statistically significant (p=0.111).
Figure 2.

Differential Ctr1 protein expression and cisplatin sensitivity in bladder cancer cell lines. (A) Basal Ctr1 protein levels were measured in six different bladder cancer cell lines of human or rodent origin (J82, RT4, 5637, MBT2, MB49, and NBT2). (B) Cisplatin sensitivity was assessed using clonogenic survival assays for all six bladder cancer cell lines. All survival curves were significantly different (p<0.05; with the exceptions of 5637 MBT2 = and MB49 = RT4). (C) Cisplatin LC50s are also plotted versus relative Ctr1 protein expression (R2 −0.71; correlation not significant; p = 0.111).
Ctr1 mulitmerisation and cisplatin cytotoxicity
The role of Ctr1 mulitmerisation in cisplatin uptake and cytotoxicity was assessed using Myc-tagged Ctr1 HEK293 cells. As shown previously by Guo et al. [15], 200 μM cisplatin treatment results in Ctr1 trimer formation in WT Myc-tagged cells, but not M1/M2 mutant cells. We confirmed this finding using WT or M1/M2 Ctr1-Myc-tagged cells with 200 μM cisplatin (Figure 3A). Two different concentrations of the M1/M2 plasmid were transfected into the cells (low and high) to confirm that we were comparing cells with similar levels of Myc-tagged Ctr1. To assess the role of multi-merisation of Ctr1 in cisplatin cytotoxicity, we compared clonogenic survival of these cell lines following 2 h of cisplatin treatment (Figure 3B). WT Myc-tagged cells were more sensitive to cisplatin relative to the M1/M2 Ctr1 mutant line. Using linear regression modelling, cell line and cisplatin concentration were significant predictors (p<0.05) of clonogenic survival.
Figure 3.

Role of Ctr1 mulitmerisation in cisplatin sensitivity. (A) Ctr1 multimerisation was confirmed in WT Myc-tagged Ctr1 cells following a 2-h treatment with 200 μM of cisplatin. HEK293 cells were transfected with either 2 μg (low) or 4 μg (high) of M1/M2 Ctr1. Multimerisation did not occur in the M1/M2 cells following cisplatin exposure. The high levels of M1/M2 was used for survival assays so that similar levels of basal Myc-tagged Ctr1 levels would be compared. • indicates the monomer, •• indicates the dimer, ••• and indicates the trimer (multimer). (B) Clonogenic survival of WT Myc-tagged Ctr1 cells versus M1/M2 Ctr1 cells. WT cells were significantly more sensitive to cisplatin. ** indicates statistical significance (p<0.001).
Effect of hyperthermia on cisplatin-induced Ctr1 multimerisation
We next investigated the effect of hyperthermia on Ctr1 multimerisation as a potential mechanism for increasing cisplatin uptake. Using HEK293 cells expressing WT Myc-tagged Ctr1 and the pcDNA3.1 vector control, we examined Ctr1 multimerisation following cisplatin treatment with and without hyperthermia treatment. Cells were treated with cycloheximide 30 min prior to cisplatin treatment to prevent new protein synthesis. As shown in Figure 4(A) and quantified in Figure 4(B), hyperthermia treatment increased the rate and level of Ctr1 multimerisation compared to cells treated at 37 °C. The rate of multimerisation increased overtime for both treatment groups, but was more pronounced in the hyperthermia-treated group. There was an increase in multimer formation over time, with a reduction in monomer levels.
Figure 4.

Hyperthermia treatment increases cisplatin-induced Ctr1 multimerisation. (A) Western blot analysis of Myc-tagged Ctr1 HEK293 cells treated with 100 μM cisplatin at 37°C or 41°C. Cells were treated with hyperthermia over the course of 15 to 60 min. Cells were harvested at the indicated time point, with 60→60 indicating 60 min post-hyperthermia treatment (or 120 min of normothermia). •indicates the monomer, •• indicates the dimer and •••, indicates the trimer (multimer). (B) Quantification of relative Myc-tagged Ctr1 expression (normalised to Actin expression) for monomer and trimer formation. These results are representative of three independent experiments.
In addition to the time course, Ctr1 multimerisation was assessed over a range of temperatures from 37 °C to 43 °C. Cells were treated for 60 min with concurrent hyperthermia and cisplatin (200 μM) and harvested at 60 min or 60 min post-hyperthermia. As shown in Figure 5, we observed higher levels of multimerisation following hyperthermia treatment compared to normothermia at both time points.
Figure 5.

Hyperthermic temperature treatment increases cisplatin-induced Ctr1 multimerisation. (A and C) Western blot analysis of Myc-tagged Ctr1 HEK293 cells treated with 200 μM cisplatin over a range of temperatures from 37°C to 43°C. Cells were treated with hyperthermia for 60 min and then harvested at either 60 min or 60 min post-heating. • indicates the monomer, • • indicates the dimer, and ••• indicates the trimer (multimer). (B and D) Quantification of relative Myc-tagged Ctr1 expression (normalised to actin expression) for trimer formation. These results are representative of three independent experiments.
Cisplatin accumulation in WT and Ctr1−/− cells
Cisplatin accumulation was measured in WT and Ctr1−/− MEFs following concurrent treatment with 0, 2, 6, and 10 μM cisplatin at 37°, 41°, and 42 °C. Raw data and data that were normalised to the lowest cisplatin dose at 37 °C for each cell line were analysed separately to distinguish between the cisplatin and temperature effects between cell lines. We observed a significant dose-dependent increase in platinum accumulation in both cell lines (p<0.001). Further, there was a significant difference in accumulation levels between cell lines (p<0.001), with higher levels of platinum measured in the WT cells relative to the Ctr1−/− cells. A temperature-dependent increase in platinum accumulation was observed in the WT cells (p=0.04), but not Ctr1−/− cells (p=0.951) (Figure 6B). Normalising the data removed the cell line effect. This revealed significant effects of cisplatin dose and temperature response (p<0.001). When the normalised data were analysed for each cell line (Figures 6C and D), a statistically significant increase in cisplatin due to increased temperature was observed following analyses with a linear regression (p<0.0001 for WT and p=0.028 for Ctr1−/−); however, this increase was approximately 2-fold higher in the WT versus Ctr1−/− cells. Additionally, using Tukey's post-hoc analyses, we detected significant differences between the 37 °C and 42 °C treatments at the 6 and 10 μM doses for the WT cells (p<0.05), whereas significant differences were not detected for the Ctr1−/−cells.
Figure 6.

Ctr1 expression and cisplatin accumulation in WT and Ctr1−/−MEF cell lines following hyperthermia treatment. (A) Ctr1 expression was assessed western blot analysis. (B, C, and D) Platinum levels were measured in WT and Ctr1−/−MEFs following concurrent cisplatin (2, 6, and 10 μM) and 1 h of hyperthermia treatment (41°and 42°C). (B) Raw platinum accumulation data are shown for the two cell lines. Cisplatin accumulation was significantly higher in the WT versus the Ctr1−/−cells (p < 0.001). Increasing temperature resulted in significantly enhanced cisplatin uptake in WT cells (p = 0.04), but not in the Ctr1−/− cells (p > 0.05; NS). (C and D) Platinum accumulation data were normalised to the 37°C 2 μM cisplatin treatment for the WT (C) and Ctr1−/−(D) cell lines. Significant differences in accumulation following heat treatment were observed in the WT cell line. *statistical significance at p < 0.05, and **significance at p < 0.001.
Cytotoxicity of WT versus Ctr1−/− cells following concurrent cisplatin and hyperthermia treatment
Although increased resistance to cisplatin has been observed in Ctr1−/− MEFs using trypan blue exclusion assays [6], we confirmed this differential sensitivity to cisplatin in WT and Ctr1−/− MEF by assessing clonogenic survival (Figure 7). WT cells were significantly more sensitive to cisplatin than Ctr1−/− cells (LC50s of 2.2 μM versus 4.6 μM in WT and Ctr1−/− cells, respectively). Survival was assessed at 37° and 41 °C. The effect of heat was statistically significant for both cell lines (p<0.01). We compared the survival difference due to temperature between cell lines by calculating the dose modifying factor (DMF) of hyperthermia for each cell line. The DMF for WT cells was 1.77, which was higher than the DMF the Ctr1−/− cells at 1.4. All survival curves were significantly different from each other (p<0.01), indicating that the DMF for the WT cells was significantly higher than for the Ctr1−/− cells. The corresponding LC50 and LC90 for each cell line and treatment is provided in Table I. It is important to note that the survival curves for each cell line were normalised to the vehicle-treated control for each temperature. Treatment at 41 °C for 1 h did not increase cell death relative to the 37 °C control.
Figure 7.
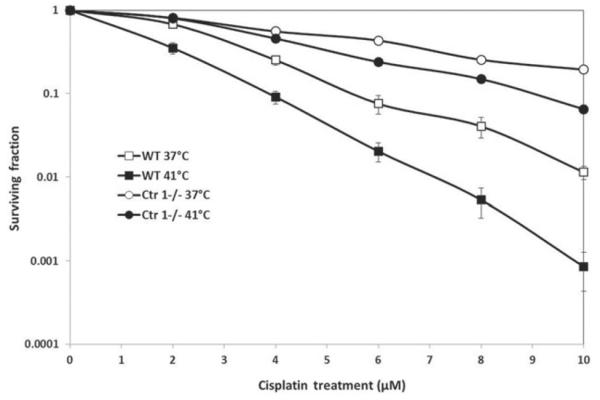
WT and Ctr1−/−MEF sensitivity to concurrent cisplatin and hyperthermia treatment. Cisplatin sensitivity was determined with clonogenic survival assays. Cells were treated with concurrent cisplatin (0–10 μM) and normothermia (37°C) or hyperthermia (41°C). The cell lines showed differential sensitivity to cisplatin (all survival curves were significantly different; p < 0.01). The effect of hyperthermia was significant for both cell lines (p < 0.01). All experiments were done independently three times.
Table I.
Survival curve data for MEF cells following cisplatin and hyperthermia treatment. A single exponential fit was used to analyse the data and calculate the rate, LC50, LC90, and dose modifying factor (DMF).
| Cell line and treatment | Survival curve rate (b) | LC50 (μM) | LC90 (μM) | DMF |
|---|---|---|---|---|
| WT 37 °C | 0.31 | 2.24 | 7.43 | 1.77 |
| WT 41 °C | 0.55 | 1.26 | 4.19 | |
| Ctrl−/− 37°C | 0.15 | 4.62 | 15.35 | 1.4 |
| Ctrl−/− 41 °C | 0.21 | 3.30 | 10.96 |
Discussion
Our data suggest that Ctr1 is involved in the synergistic interaction between hyperthermia and cisplatin. Hyperthermia significantly increased cisplatin uptake in WT but not Ctr1−/− cells. Furthermore, a higher DMF was observed for the WT cells following concurrent heat and cisplatin treatment relative to Ctr1−/− cells (1.77 versus 1.4, respectively). These data suggest that the response of Ctr1 to hyperthermia may, in part, increase cisplatin uptake, resulting in increased platinum-DNA adduct formation, and subsequent cell death.
The effects of hyperthermia are multi-factorial. We hypothesise that Ctr1 is involved in the synergism between hyperthermia and cisplatin therapies, but it is important to note that hyperthermia induces a wide variety of cellular responses/changes, depending on temperature and duration. One known effect of hyperthermia is increased membrane fluidity and permeability. A thorough review by Yatvin and Cramp discussed the cellular membrane changes induced by heat treatment, including physical and compositional alterations [26]. Specifically, heat treatment increases membrane-associated cholesterol, phospholipid, and protein levels, potentially altering fluidity and membrane function. Many of these earlier studies focused on temperatures higher (>43 °C) than the clinically relevant mild hyperthermia temperatures. Hayat and Friedberg assessed cell membrane permeability in mouse fibroblasts (3T3 and 3T6) over a range of temperatures from 37–45 °C for 5 to 30 min [27]. Permeability was assessed by measuring efflux of radioactively labelled, intracellular metabolites. Increased efflux was observed following heat treatment (starting at 41 °C) over time, but the effect was more pronounced at 43 °C. Because we wanted to assess the role of Ctr1 with minimal hyperthermic effects on membrane permeability, we used temperatures of 41° and 42 °C to assess cisplatin accumulation (Figure 6) and a range of 39° to 43 °C to assess Ctr1 multimerisation (Figure 5).
Using the WT versus Ctr1−/− MEF cell lines, we showed that Ctr1 is responsible for cisplatin uptake. Our studies demonstrate that Ctr1 function is involved in increased cellular cisplatin accumulation and cytotoxicity following hyperthermia. Guo et al. showed that copper exposure results in Ctr1 internalisation, whereas cisplatin treatment causes Ctr1 multimerisation [15]. The authors suggested that this multimerisation results in the formation of pores that allow cisplatin uptake. We observed decreased platinum sensitivity in the M1/M2 mutant cell line that does not trimerise relative to WT Myc-tagged cells, indicating that trimerisation is involved in cisplatin uptake. We also observed increased multimerisation following heat treatment in the WT cells. These data suggest that the underlying mechanism for both increased platinum accumulation and decreased survival following hyperthermia and cisplatin treatments is increased Ctr1 multimerisation. We confirmed that these cell lines expressed similar levels of the Myc-tagged Ctr1. Although multimerisation likely increased active cisplatin import into cells, it is important to note that the M1/M2 cells used in this study also express endogenous Ctr1, which could permit some cisplatin accumulation and cytotoxicity despite absence of Myc-tagged Ctr1 multimerisation.
Bladder cancer is the fifth most common type of cancer in the western world [28]. Depending on staging, this disease may be treated with a wide range of chemotherapeutic agents, (e.g. cisplatin, mitomycin C, epirubicin) immumotherapy (e.g. Bacillus Calmette-Guerin (BCG)), radiotherapy, and/or surgical resection [29]. Bladder cancer patients have also shown clinical benefits from hyperthermia treatment. Colombo et al. reported significantly fewer recurrences when primary or recurrent superficial transitional cell carcinoma of the bladder were treated with intravesical mitomycin C and hyperthermia compared to mitomycin C treatment alone [30]. A similar study using this same intravesical system combined with mitomycin C in patients with multiple or recurrent transitional cell carcinoma of the bladder exhibited comparable results showing a high percentage of recurrence-free patients [31]. A trial treating high-grade superficial bladder cancer with mild hyperthermia (goal temperature of 42 °C) and mitomycin C was also beneficial with 62.5% of patients being recurrence-free [32].
In the human bladder cancer samples, we observed a wide range of Ctr1 expression levels. Similar work in the ovarian cancer field showed the importance of these expression levels for platinum-based therapy [16–18]. Our data suggest that Ctr1 expression may also be an important prognostic marker for bladder cancer patients receiving platinum-based therapy. For example, assessing Ctr1 expression levels prior to treatment may provide information as to whether cisplatin-based therapy would be effective for patients. Using six different bladder cancer cell lines, we observed increased cisplatin sensitivity in the cell lines expressing higher levels of Ctr1. More importantly, our data show that Ctr1 expression levels are detectable in the bladder, suggesting that the combined treatment of cisplatin and local/regional hyperthermia may be beneficial in the clinical setting. Although superficial lesions seem to be an ideal target for adjuvant hyperthermia treatment because of accessibility, improvements in heating devices now allow for targeting deep-seated tumours. Heating devices, such as the Synergo® System (Synergo SB-TS:101-1; Medical Enterprises, Amsterdam, the Netherlands) [33], used specifically for bladder heating, and the BSD-2000 (BSD Medical, Salt Lake City, UT, USA), have been developed to accommodate these deep-seated lesions (e.g. cervical, gastrointestinal, bladder) [34].
Adjuvant hyperthermia treatment may also have a potential benefit for other cancer types commonly treated with cisplatin (testicular, ovarian, bladder, cervical, head and neck, and small cell lung cancers). In 1986, Kapp authored a review on the types of cancer that, based on lesion location and potential progression, would benefit from hyperthermia [35]. Based on mortality statistics, the author reported that local failures contribute to death in a high proportion of patients with brain, ovarian, prostate, cervical, oesophageal, bladder, and head and neck cancers. Lesions for which local-regional recurrence and metastases are problematic, such as breast cancer, head and neck cancer (lymph node metastases), colorectal cancer (nodal metastases), bladder cancer (muscle invasive disease), and malignant melanomas (symptomatic cutaneous, subcutaneous, or superficial lymph node metastases), were identified as potential candidates for hyperthermia. Biochemical evaluation of Ctr1 protein levels in biopsies on a per-patient basis may demonstrate elevated Ctr1 and serve as an excellent biomarker for treatment with cisplatin/hyperthermia therapy.
Our work suggests a potential role for Ctr1 in the synergistic interaction between hyperthermia and cisplatin treatment. These data raise additional questions at the mechanistic level, but also in a clinical setting. There may be therapeutic benefit by maximising the effect of this combination based on the role of Ctr1.
At the mechanistic level it may be of interest to analyse Ctr1 multimers in more detail. Assessment of the content of the multimer complex may be useful, as it is unclear if Ctr1 multimerises with itself or if the multimers represent Ctr1 complexed with other proteins or protein modifications. Hyperthermia is known to induce glycosyltransferase activities involved in O-linked glycoproteins [36] and ubiquitin expression [37]. Interestingly, these modifications are important for the function and degradation of Ctr1 [38–40].
Additionally, it may be interesting to determine whether lipid raft dynamics are involved in the process of Ctr1 multimerisation. The structure and function of lipid raft membrane microdomains are temperature sensitive [41,42], as are certain signalling pathways associated with lipid rafts [43]. Mace et al. demonstrated that fever-range hyperthermia (39.5 °C) results in increased GM1+ lipid microdomain clustering in CD8+ T cells [21,22]. A recent study conducted by Ashino et al. found that Ctr1 was predominantly localised in caveolae/lipid rafts in vascular smooth muscle cells [19]. It may be possible that the increased membrane fluidity and lipid raft clustering induced by hyperthermia allow for enhanced Ctr1 multimerisation, but further studies are necessary to confirm this hypothesis.
Future work should be conducted to investigate the possibility of combining cisplatin and hyperthermia treatment. It could potentially be beneficial to increase Ctr1 expression on the tumour cell surface to allow for increased multimerisation and cisplatin uptake. A recent review by Gupte and Mumper examined studies assessing copper levels in cancer patients [44]. Interestingly, elevated copper levels have been reported in a variety of cancer types, including breast, cervical, ovarian, lung, prostate, stomach, reticulo-endothelial system, and leukaemia. Additionally, they have been shown to correlate with cancer stage and/or progression. Copper is known to be an endogenous stimulator of angiogenesis by both promoting the motility of endothelial cells and inducing the synthesis of fibronectin (a matrix glycoprotein associated with angiogenesis) [44].
Due to observations of elevated copper levels in cancer patients and involvement of copper in promoting angiogenesis, there has been interest in using copper chelators for anti-angiogenic treatment. Several animal studies using copper chelators alone or in combination with anticancer drugs or radiotherapy have shown encouraging anti-tumour and anti-angiogenic results [45–49]. Based on these results, clinical trials using copper chelators, such as penicillamine and tetrathiomolybdate, as anti-angiogenic treatment have been completed but with mixed results [44]. Brewer et al. conducted a phase I clinical trial attempting to reduce angiogenesis with tetrathiomolybdate in patients with meta-static solid tumours [50]. They successfully induced and maintained copper deficiency in a non-toxic manner and observed stable disease in several of the patients. In a phase II trial treating patients with advanced renal cancer with tetrathiomolybdate, the drug was well-tolerated and it successfully reduced serum copper levels, but the clinical activity was limited to stable disease [51]. A phase II trial for patients with hormone-refractory prostate cancer concluded that tetrathiomolybdate was not an effective treatment strategy [52]. Brem et al. used penicillamine in a phase II clinical trial in combination with radiation therapy in patients with glioblastoma multiforme [53]. Serum copper levels were reduced and tolerated for months, but this antiangiogenic treatment did not improve patient survival. Although survival benefits were not observed in these clinical trials, copper chelators were able to effectively reduce copper levels without added toxicity and result in disease stabilisation in several studies.
Ishida et al. conducted a study using copper chelation combined with cisplatin to treat cervical cancer in a mouse model [16]. This treatment combination resulted in increased platinum uptake in the tumours, inhibition of angiogenesis, and anti-tumour effect. The treatment combination was also effective at increasing cisplatin sensitivity in vitro with several different human ovarian and cervical cancer cell lines. Although an exact mechanism for the improved treatment was not elucidated, the authors identified Ctr1 as the target. Liang et al. demonstrated a similar anti-tumour effect using this treatment combination in a xenograft mouse model with small cell lung cancer tumours [18]. Based on our results, the combination of cisplatin and copper chelation may benefit from adjuvant hyperthermia treatment. Future studies with this combination are warranted and may have potential as an effective treatment modality with clinical applicability.
Acknowledgements
The authors would like to thank Kim Hutchison at the North Carolina State University Department of Soils Science for her help with ICP-MS analysis, as well as Greg Palmer, Ph.D. and Peter Scarbrough, Ph.D., for help with statistical analyses. Additionally, the authors thank Dennis J. Thiele, Ph.D. for guidance with the Ctr1 work, Brant Inman, M.D. for supplying the human bladder RNA, and Kenneth Young for technical assistance.
Declaration of interest Financial support for this work was provided by NIH/NCCAM Grant 1F31 AT006644-01 and NIH CA42745. The authors alone are responsible for the content and writing of the paper.
References
- 1.Rosenberg B, Vancamp L, Krigas T. Inhibition of cell division in Escherichia coli by electrolysis products from a platinum electrode. Nature. 1965;205:698–9. doi: 10.1038/205698a0. [DOI] [PubMed] [Google Scholar]
- 2.Hahn GM. Hyperthermia and Cancer. Plenum Press; New York: 1982. [Google Scholar]
- 3.Hettinga JV, Konings AW, Kampinga HH. Reduction of cellular cisplatin resistance by hyperthermia – A review. Int J Hyperthermia. 1997;13:439–57. doi: 10.3109/02656739709023545. [DOI] [PubMed] [Google Scholar]
- 4.van de Vaart PJ, van der Vange N, Zoetmulder FA, van Goethem AR, van Tellingen O, ten Bokkel Huinink WW, et al. Intraperitoneal cisplatin with regional hyperthermia in advanced ovarian cancer: Pharmacokinetics and cisplatin-DNA adduct formation in patients and ovarian cancer cell lines. Eur J Cancer. 1998;34:148–54. doi: 10.1016/s0959-8049(97)00370-5. [DOI] [PubMed] [Google Scholar]
- 5.Kusumoto T, Holden SA, Ara G, Teicher BA. Hyperthermia and platinum complexes: Time between treatments and synergy in vitro and in vivo. Int J Hyperthermia. 1995;11:575–86. doi: 10.3109/02656739509022491. [DOI] [PubMed] [Google Scholar]
- 6.Ishida S, Lee J, Thiele DJ, Herskowitz I. Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals. Proc Natl Acad Sci USA. 2002;99:14298–302. doi: 10.1073/pnas.162491399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou B, Gitschier J. hCTR1: A human gene for copper uptake identified by complementation in yeast. Proc Natl Acad Sci USA. 1997;94:7481–6. doi: 10.1073/pnas.94.14.7481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Feo CJ, Aller SG, Siluvai GS, Blackburn NJ, Unger VM. Three-dimensional structure of the human copper transporter hCTR1. Proc Natl Acad Sci USA. 2009;106:4237–42. doi: 10.1073/pnas.0810286106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eisses JF, Kaplan JH. The mechanism of copper uptake mediated by human CTR1: A mutational analysis. J Biol Chem. 2005;280:37159–68. doi: 10.1074/jbc.M508822200. [DOI] [PubMed] [Google Scholar]
- 10.Dancis A, Haile D, Yuan DS, Klausner RD. The Saccharomyces cerevisiae copper transport protein (Ctr1p). Biochemical characterization, regulation by copper, and physiologic role in copper uptake. J Biol Chem. 1994;269:25660–7. [PubMed] [Google Scholar]
- 11.Petris MJ, Smith K, Lee J, Thiele DJ. Copper-stimulated endocytosis and degradation of the human copper transporter, hCtr1. J Biol Chem. 2003;278:9639–46. doi: 10.1074/jbc.M209455200. [DOI] [PubMed] [Google Scholar]
- 12.Larson CA, Blair BG, Safaei R, Howell SB. The role of the mammalian copper transporter 1 in the cellular accumulation of platinum-based drugs. Mol Pharmacol. 2009;75:324–30. doi: 10.1124/mol.108.052381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Song IS, Savaraj N, Siddik ZH, Liu P, Wei Y, Wu CJ, et al. Role of human copper transporter Ctr1 in the transport of platinum-based antitumor agents in cisplatin-sensitive and cisplatin-resistant cells. Mol Cancer Ther. 2004;3:1543–9. [PubMed] [Google Scholar]
- 14.Sinani D, Adle DJ, Kim H, Lee J. Distinct mechanisms for Ctr1-mediated copper and cisplatin transport. J Biol Chem. 2007;282:26775–85. doi: 10.1074/jbc.M703973200. [DOI] [PubMed] [Google Scholar]
- 15.Guo Y, Smith K, Petris MJ. Cisplatin stabilizes a multimeric complex of the human Ctr1 copper transporter: Requirement for the extracellular methionine-rich clusters. J Biol Chem. 2004;279:46393–9. doi: 10.1074/jbc.M407777200. [DOI] [PubMed] [Google Scholar]
- 16.Ishida S, McCormick F, Smith-McCune K, Hanahan D. Enhancing tumor-specific uptake of the anticancer drug cisplatin with a copper chelator. Cancer Cell. 2010;17:574–83. doi: 10.1016/j.ccr.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee YY, Choi CH, Do IG, Song SY, Lee W, Park HS, et al. Prognostic value of the copper transporters, CTR1 and CTR2, in patients with ovarian carcinoma receiving platinum-based chemo-therapy. Gynecol Oncol. 2011;122:361–5. doi: 10.1016/j.ygyno.2011.04.025. [DOI] [PubMed] [Google Scholar]
- 18.Liang ZD, Long Y, Tsai WB, Fu S, Kurzrock R, Gagea-Iurascu M, et al. Mechanistic basis for overcoming platinum resistance using copper chelating agents. Mol Cancer Ther. 2012;11:2483–94. doi: 10.1158/1535-7163.MCT-12-0580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ashino T, Sudhahar V, Urao N, Oshikawa J, Chen GF, Wang H, et al. Unexpected role of the copper transporter ATP7A in PDGF-induced vascular smooth muscle cell migration. Circ Res. 2010;107:787–99. doi: 10.1161/CIRCRESAHA.110.225334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moreno-Altamirano MM, Aguilar-Carmona I, Sanchez-Garcia FJ. Expression of GM1, a marker of lipid rafts, defines two subsets of human monocytes with differential endocytic capacity and lipopolysaccharide responsiveness. Immunology. 2007;120:536–43. doi: 10.1111/j.1365-2567.2006.02531.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mace TA, Zhong L, Kilpatrick C, Zynda E, Lee CT, Capitano M, et al. Differentiation of CD8 + T cells into effector cells is enhanced by physiological range hyperthermia. J Leukoc Biol. 2011;90:951–62. doi: 10.1189/jlb.0511229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mace TA, Zhong L, Kokolus KM, Repasky EA. Effector CD8 + T cell IFN-gamma production and cytotoxicity are enhanced by mild hyperthermia. Int J Hyperthermia. 2012;28:9–18. doi: 10.3109/02656736.2011.616182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kitada N, Takara K, Minegaki T, Itoh C, Tsujimoto M, Sakaeda T, et al. Factors affecting sensitivity to antitumor platinum derivatives of human colorectal tumor cell lines. Cancer Chemother Pharmacol. 2008;62:577–84. doi: 10.1007/s00280-007-0640-3. [DOI] [PubMed] [Google Scholar]
- 24.Dyrskjot L, Kruhoffer M, Thykjaer T, Marcussen N, Jensen JL, Moller K, et al. Gene expression in the urinary bladder: A common carcinoma in situ gene expression signature exists disregarding histopathological classification. Cancer Res. 2004;64:4040–8. doi: 10.1158/0008-5472.CAN-03-3620. [DOI] [PubMed] [Google Scholar]
- 25.Minami T, Ichii M, Okazaki Y. Comparison of three different methods for measurement of tissue platinum level. Biol Trace Elem Res. 1995;48:37–44. doi: 10.1007/BF02789077. [DOI] [PubMed] [Google Scholar]
- 26.Yatvin MB, Cramp WA. Role of cellular membranes in hyperthermia: Some observations and theories reviewed. Int J Hyperthermia. 1993;9:165–85. doi: 10.3109/02656739309022533. [DOI] [PubMed] [Google Scholar]
- 27.Hayat H, Friedberg I. Heat-induced alterations in cell membrane permeability and cell inactivation of transformed mouse fibroblasts. Int J Hyperthermia. 1986;2:369–78. doi: 10.3109/02656738609004967. [DOI] [PubMed] [Google Scholar]
- 28.Wu XR. Urothelial tumorigenesis: A tale of divergent pathways. Nat Rev Cancer. 2005;5:713–25. doi: 10.1038/nrc1697. [DOI] [PubMed] [Google Scholar]
- 29.Griffiths TR. Current perspectives in bladder cancer management. Int J Clin Pract. 2012;67:435–48. doi: 10.1111/ijcp.12075. [DOI] [PubMed] [Google Scholar]
- 30.Colombo R, Da Pozzo LF, Salonia A, Rigatti P, Leib Z, Baniel J, et al. Multicentric study comparing intravesical chemotherapy alone and with local microwave hyperthermia for prophylaxis of recurrence of superficial transitional cell carcinoma. J Clin Oncol. 2003;21:4270–6. doi: 10.1200/JCO.2003.01.089. [DOI] [PubMed] [Google Scholar]
- 31.Moskovitz B, Meyer G, Kravtzov A, Gross M, Kastin A, Biton K, et al. Thermo-chemotherapy for intermediate or high-risk recurrent superficial bladder cancer patients. Ann Oncol. 2005;16:585–9. doi: 10.1093/annonc/mdi124. [DOI] [PubMed] [Google Scholar]
- 32.Gofrit ON, Shapiro A, Pode D, Sidi A, Nativ O, Leib Z, et al. Combined local bladder hyperthermia and intravesical chemotherapy for the treatment of high-grade superficial bladder cancer. Urology. 2004;63:466–71. doi: 10.1016/j.urology.2003.10.036. [DOI] [PubMed] [Google Scholar]
- 33.Colombo R, Salonia A, Da Pozzo LF, Naspro R, Freschi M, Paroni R, et al. Combination of intravesical chemotherapy and hyperthermia for the treatment of superficial bladder cancer: Preliminary clinical experience. Crit Rev Oncol Hematol. 2003;47:127–39. doi: 10.1016/s1040-8428(03)00076-3. [DOI] [PubMed] [Google Scholar]
- 34.Fatehi D, van der Zee J, van der Wal E, Van Wieringen WN, Van Rhoon GC. Temperature data analysis for 22 patients with advanced cervical carcinoma treated in Rotterdam using radiotherapy, hyperthermia and chemotherapy: A reference point is needed. Int J Hyperthermia. 2006;22:353–63. doi: 10.1080/02656730600715796. [DOI] [PubMed] [Google Scholar]
- 35.Kapp DS. Site and disease selection for hyperthermia clinical trials. Int J Hyperthermia. 1986;2:139–56. doi: 10.3109/02656738609012390. [DOI] [PubMed] [Google Scholar]
- 36.Henle KJ, Stone A, Chatterjee SK. Effect of hyperthermia on activity of three glycosyltransferases in Chinese hamster ovary cells. Cancer Res. 1988;48:5717–21. [PubMed] [Google Scholar]
- 37.Bond U, Schlesinger MJ. Ubiquitin is a heat shock protein in chicken embryo fibroblasts. Mol Cell Biol. 1985;5:949–56. doi: 10.1128/mcb.5.5.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maryon EB, Molloy SA, Kaplan JH. O-linked glycosylation at threonine 27 protects the copper transporter hCTR1 from proteolytic cleavage in mammalian cells. J Biol Chem. 2007;282:20376–87. doi: 10.1074/jbc.M701806200. [DOI] [PubMed] [Google Scholar]
- 39.Maryon EB, Zhang J, Jellison JW, Kaplan JH. Human copper transporter 1 lacking O-linked glycosylation is proteolytically cleaved in a Rab9-positive endosomal compartment. J Biol Chem. 2009;284:28104–14. doi: 10.1074/jbc.M109.044925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu J, Sitaram A, Burd CG. Regulation of copper-dependent endocytosis and vacuolar degradation of the yeast copper transporter, Ctr1p, by the Rsp5 ubiquitin ligase. Traffic. 2007;8:1375–84. doi: 10.1111/j.1600-0854.2007.00616.x. [DOI] [PubMed] [Google Scholar]
- 41.Garcia-Saez AJ, Chiantia S, Schwille P. Effect of line tension on the lateral organization of lipid membranes. J Biol Chem. 2007;282:33537–44. doi: 10.1074/jbc.M706162200. [DOI] [PubMed] [Google Scholar]
- 42.Weise K, Triola G, Janosch S, Waldmann H, Winter R. Visualizing association of lipidated signaling proteins in heterogeneous membranes - Partitioning into subdomains, lipid sorting, interfacial adsorption, and protein association. Biochim Biophys Acta. 2010;1798:1409–17. doi: 10.1016/j.bbamem.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 43.Katkere B, Rosa S, Caballero A, Repasky EA, Drake JR. Physiological-range temperature changes modulate cognate antigen processing and presentation mediated by lipid raft-restricted ubiquitinated B cell receptor molecules. J Immunol. 2010;185:5032–9. doi: 10.4049/jimmunol.1001653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gupte A, Mumper RJ. Elevated copper and oxidative stress in cancer cells as a target for cancer treatment. Cancer Treat Rev. 2009;35:32–46. doi: 10.1016/j.ctrv.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 45.Pan Q, Kleer CG, van Golen KL, Irani J, Bottema KM, Bias C, et al. Copper deficiency induced by tetrathiomolybdate suppresses tumor growth and angiogenesis. Cancer Res. 2002;62:4854–9. [PubMed] [Google Scholar]
- 46.Khan MK, Miller MW, Taylor J, Gill NK, Dick RD, Van Golen K, et al. Radiotherapy and antiangiogenic TM in lung cancer. Neoplasia. 2002;4:164–70. doi: 10.1038/sj.neo.7900218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moriguchi M, Nakajima T, Kimura H, Watanabe T, Takashima H, Mitsumoto Y, et al. The copper chelator trientine has an antiangiogenic effect against hepatocellular carcinoma, possibly through inhibition of interleukin-8 production. Int J Cancer. 2002;102:445–52. doi: 10.1002/ijc.10740. [DOI] [PubMed] [Google Scholar]
- 48.Yoshida D, Ikeda Y, Nakazawa S. Suppression of tumor growth in experimental 9L gliosarcoma model by copper depletion. Neurol Med Chir (Tokyo) 1995;35:133–5. doi: 10.2176/nmc.35.133. [DOI] [PubMed] [Google Scholar]
- 49.Brem SS, Zagzag D, Tsanaclis AM, Gately S, Elkouby MP, Brien SE. Inhibition of angiogenesis and tumor growth in the brain. Suppression of endothelial cell turnover by penicillamine and the depletion of copper, an angiogenic cofactor. Am J Pathol. 1990;137:1121–42. [PMC free article] [PubMed] [Google Scholar]
- 50.Brewer GJ, Dick RD, Grover DK, LeClaire V, Tseng M, Wicha M, et al. Treatment of metastatic cancer with tetrathiomolybdate, an anticopper, antiangiogenic agent: Phase I study. Clin Cancer Res. 2000;6:1–10. [PubMed] [Google Scholar]
- 51.Redman BG, Esper P, Pan Q, Dunn RL, Hussain HK, Chenevert T, et al. Phase II trial of tetrathiomolybdate in patients with advanced kidney cancer. Clin Cancer Res. 2003;9:1666–72. [PubMed] [Google Scholar]
- 52.Henry NL, Dunn R, Merjaver S, Pan Q, Pienta KJ, Brewer G, et al. Phase II trial of copper depletion with tetrathiomolybdate as an antiangiogenesis strategy in patients with hormone-refractory prostate cancer. Oncology. 2006;71:168–75. doi: 10.1159/000106066. [DOI] [PubMed] [Google Scholar]
- 53.Brem S, Grossman SA, Carson KA, New P, Phuphanich S, Alavi JB, et al. Phase 2 trial of copper depletion and penicillamine as antiangiogenesis therapy of glioblastoma. Neuro Oncol. 2005;7:246–53. doi: 10.1215/S1152851704000869. [DOI] [PMC free article] [PubMed] [Google Scholar]