

Abstract
Mitochondria have a myriad of essential functions including metabolism and apoptosis. These chief functions are reliant on electron transfer reactions and the production of ATP and reactive oxygen species (ROS). The production of ATP and ROS are intimately linked to the electron transport chain (ETC). Electrons from nutrients are passed through the ETC via a series of acceptor and donor molecules to the terminal electron acceptor molecular oxygen (O2) which ultimately drives the synthesis of ATP. Electron transfer through the respiratory chain and nutrient oxidation also produces ROS. At high enough concentrations ROS can activate mitochondrial apoptotic machinery which ultimately leads to cell death. However, if maintained at low enough concentrations ROS can serve as important signaling molecules. Various regulatory mechanisms converge upon mitochondria to modulate ATP synthesis and ROS production. Given that mitochondrial function depends on redox reactions, it is important to consider how redox signals modulate mitochondrial processes. Here, we provide the first comprehensive review on how redox signals mediated through cysteine oxidation, namely S-oxidation (sulfenylation, sulfinylation), S-glutathionylation, and S-nitrosylation, regulate key mitochondrial functions including nutrient oxidation, oxidative phosphorylation, ROS production, mitochondrial permeability transition (MPT), apoptosis, and mitochondrial fission and fusion. We also consider the chemistry behind these reactions and how they are modulated in mitochondria. In addition, we also discuss emerging knowledge on disorders and disease states that are associated with deregulated redox signaling in mitochondria and how mitochondria-targeted medicines can be utilized to restore mitochondrial redox signaling.
Keywords: Redox, S-glutathionylation, S-oxidation, S-nitrosylation, Mitochondria
Highlights
-
•
Mitochondria are essential for various cellular processes.
-
•
Mitochondria are major sites for regulation by redox signaling.
-
•
Redox signaling regulates mitochondrial metabolism and function.
-
•
Redox signaling in mitochondria is deregulated in various disease states.
Introduction
Mitochondria can be a major source of cellular ROS which inherently depends on the metabolic “state” of mitochondria [1]. However, in order to understand mitochondrial ROS production, it is important to examine aerobic metabolism in detail. Nutrients (e.g. glucose, fatty acids, amino acids) are converted into metabolic intermediates (e.g. pyruvate, acetyl-CoA, oxaloacetate, 2-oxoglutarate) which are then systematically metabolized and decarboxylated by eight different enzymes in the tricarboxylic acid (TCA) cycle (Fig. 1). The decarboxylation steps are coupled to the transfer of electrons to NAD+ producing NADH [2]. Complex I (NADH:ubiquinone oxidoreductase) subsequently oxidizes NADH and the liberated electrons are passed to ubiquinone (Q) generating ubiquinol (QH2) [3]. Electrons from Complex II (succinate dehydrogenase; SDH), electron transfer flavoprotein oxidoreductase (ETF-QO), dihydroorotate dehydrogenase, and FAD-linked glycerol-3-phosphate dehydrogenase can also feed electrons into the Q pool [4], [5], [6], [7]. The electrons are then passed to Complex III through cytochrome C and on to Complex IV which then reduces the terminal electron acceptor O2 into H2O (Fig. 1) [8]. The most important aspect of this process is the redox potentials of NADH (Eo'=−340 mV), and O2 (Eo'=+810 mV). Due to this redox difference electron flow from NADH through the well-ordered prosthetic groups located within the individual respiratory complexes to O2 at Complex IV is an energetically favorable process. According to Peter Mitchell's Chemiosmotic Theory, the energetically favorable transfer of electrons from NADH to O2 through the respiratory complexes is coupled to the pumping of protons through Complexes I, III, and IV into the intermembrane space. This creates a temporary form of stored energy, namely, the protonmotive force (PMF) which is used by Complex V to produce ATP from ADP and Pi. Hence, the production of ATP from nutrient oxidation in mitochondria depends on a multitude of redox reactions.
Fig. 1.
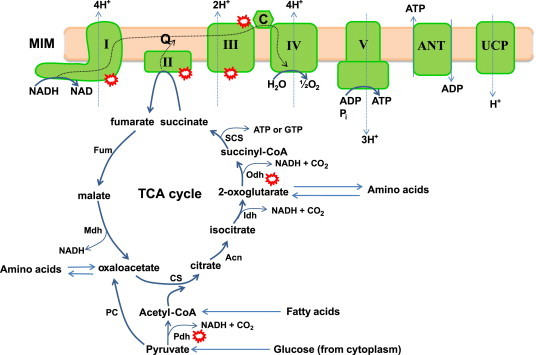
Aerobic respiration, oxidative phosphorylation, and chemiosmotic theory: nutrients in the form of pyruvate (generated from glucose via glycolysis in the cytoplasm), acetyl-CoA (produced from pyruvate or generated by fatty acid oxidation), or amino acids enter the tricarboxylic acid (TCA) and are systematically oxidized by the concerted action of 8 different enzymes. Carbon oxidation is coupled to decarboxylation reactions which yield the necessary electrons required to drive oxidative phosphorylation (OXPHOS). Electrons from the TCA cycle are transferred to NAD generating the electron carrier NADH which is then oxidized by Complex I. Electrons can also be provided by Complex II which oxidizes succinate to fumarate in the TCA cycle. Electrons then flow through a series of redox active prosthetic groups and molecules to the terminal electron O2 at Complex IV. The favorable energy change associated with electron flow is coupled to the pumping of protons through Complexes I, III, and IV into the intermembrane space. The protons are then transported back into the matrix by ATP synthase which couples the favorable energy change of proton transfer to the production of ATP from ADP and Pi. This is referred to as “coupled” respiration or OXPHOS. ATP is then exported into the cytoplasm to do “work” in the cell. The antiporter ANT is responsible for exporting ATP in exchange for ADP. The potential difference created by nutrient oxidation can be “uncoupled” from OXPHOS by MIM proteins ANT and UCP for the purpose of thermogenesis or controlling mitochondrial ROS production. Dotted line: represents flow of electrons and protons. Red circles: represent major sites for ROS production in mitochondria. Note that the ROS species is not defined in the figure and that other relevant sources of ROS, dihydroorotate dehydrogenase, sn-glycerol-3-phosphate dehydrogenase (mGPDH), electron transfer flavoprotein:ubiquinol oxidoreductase, p66shc/Cytochrome C, Mia40p/Erv1p, have been excluded for simplicity. Pdh; pyruvate dehydrogenase, PC; pyruvate carboxylase, CS; citrate synthase, Acn; aconitase, Idh; isocitrate dehydrogenase, Odh; 2-oxoglutarate dehydrogenase, SCS; succinyl-CoA synthase, Fum; fumarase, Mdh; malate dehydrogenase, I; Complex I, II; Complex II, III; Complex III, IV; Complex IV, V; Complex V, ANT; adenine nucleotide translocase, UCP; uncoupling protein, Q; quinone, C; cytochrome C.
Oxygen can only accept one electron at a time during its reduction to H2O. Electrons from Complexes I and III (and Complex II depending on experimental conditions) can “spin-off” prematurely, univalently reducing O2 to produce the proximal ROS superoxide anion radical (O2−) [9]. O2− is then quickly dismutated to H2O2 which is considered to be a chief ROS signaling molecule due to its longer half-life and capacity to diffuse through membranes. Complexes I and III remain the most well characterized sources of O2− in mitochondria [10]. Other mitochondrial sources of O2− and H2O2 include 2-oxoglutarate dehydrogenase (Odh), pyruvate dehydrogenase (Pdh) [11], dihydroorotate dehydrogenase, sn-glycerol-3-phosphate dehydrogenase (mGPDH), electron transfer flavoprotein:ubiquinol oxidoreductase, p66shc/Cytochrome C, Mia40p/Erv1p, and succinate dehydrogenase (Complex II) (reviewed in [10]). These enzymes can contribute significantly to total mitochondrial ROS production and can produce O2− or H2O2 or both which depends on experimental conditions and energetic substrates used [10], [12]. H2O2 emitted from mitochondria modulates insulin release, insulin signaling, the hypoxic response, adipocyte differentiation, regulation of cell cycle, and T cell receptor-initiated cell signaling [13], [14], [15], [16], [17]. H2O2 exerts its effects by reversibly oxidizing protein cysteine thiols (–SH) to sulfenic acid (–SOH). A protein cysteine thiol that can be reversibly oxidized by H2O2 is referred to as a “redox switch”. However, the signaling functions of ROS are not limited to just H2O2. Even O2− has also been shown to have a signaling role in cells due to its capacity to dismantle Fe–S clusters [18], [19]. It has also recently been shown that O2− increases and decreases “stochastically” with a periodicity of ~20–40 s. The periodicity of O2− “flashes” depends on the metabolic state of mitochondria and has been suggested to play an important role in mitochondrial permeability transition and induction of apoptosis [20]. It is important to note that the existence of periodic O2− “flashes” from mitochondria has recently been challenged since the cyclic permuted YFP (cpYFP) probe used to detect stochastic changes in O2− production by mitochondria is also highly sensitive to changes in pH [21]. Thus, it has been questioned whether the periodic changes in cpYFP are due to changes in O2− production or changes in pH [22]. Indeed, the actual mechanism by which cpYFP interacts with O2− has not been resolved. In addition, it has been suggested that cpYFP relies on protein cysteine thiols to interact with O2− which is unlikely given the slow rate of reaction between O2− and protein cysteine thiols. Thus, the existence of O2− “flashes” and their potential role in signaling needs to be carefully re-examined.
Using oxyradicals as signaling molecules can be dangerous since their accumulation can lead to production of the dreaded hydroxyl radical (OH), a highly reactive species that indiscriminately oxidizes biological macromolecules. But even mitochondrial O2− and H2O2 can have harmful effects if they accumulate to a high enough concentration. At high enough levels, H2O2 can oxidize –SOH to sulfinic (–SO2H) and sulfonic (–SO3H) acids which renders proteins irreversibly deactivated [23]. Similarly, O2− disassembles Fe–S clusters in a variety of TCA cycle enzymes and in respiratory complexes [24] and can also combine with nitric oxide (NO) to form the highly toxic species peroxynitrite (ONOO−) [25]. Oxidative stress due to H2O2 or O2− is associated with a number of pathologies, neurological disorders, and metabolic diseases. However, despite the ever present threat of oxidative stress, damage, and death, mitochondria continue to employ ROS for intra and intercellular signaling.
As illustrated above, it is very clear that mitochondrial ATP and ROS production are intimately linked [26]. The production of ATP over ROS (or vice versa) ultimately depends on electron transfer through the respiratory chain, nutrient oxidation, nutrient availability, the electronchemical proton gradient across the mitochondrial inner membrane (MIM), the availability of ADP, and even the structure and morphology of mitochondria [1], [27], [28], [29], [30]. A number of different control mechanisms converge upon mitochondria to adjust the output of ATP and ROS. These include allosteric control, covalent modification, protein synthesis, protein degradation, and formation of respirasomes [31], [32], [33], [34], [35]. However, over the past decade, there has been a surge in the number of articles published on the topic of redox control of mitochondrial proteins. Various types of redox modifications and switches, including S-oxidation (sulfenylation and sulfinylation), S-glutathionylation, and S-nitrosylation converge upon mitochondria to regulate a number of processes ranging from OXPHOS to mitochondrial morphology to cell death. However, very few reviews have actually covered the role of redox switches in the control of mitochondrial function in detail [23], [36], [37]. This is surprising as mitochondria use redox reactions to drive all of its functions. Mitochondria also harbor a microenvironment that favors redox signaling via cysteine oxidation reactions. Thus, mitochondria are also a “hotbed” for regulation by redox. In the following review, what is known about how mitochondrial biology can be regulated by redox switches will be surveyed. We will cover a broad range of subjects including the different types of reactions, why redox signaling is so prevalent in mitochondria, the role of redox switches in controlling mitochondrial function and apoptosis, and the association of deregulated mitochondrial redox signaling in pathologies and how these pathways can be targeted for therapeutic intervention. It is important to note that sources of mitochondrial O2− and H2O2, antioxidant defense systems, and mechanisms that control mitochondrial O2− and H2O2 production will not be discussed since a number of reviews have already covered these very important topics [9], [38], [39], [40].
Protein cysteine modifications in mitochondria
Cysteine accounts for ~2% of the amino acid content of cells (protein-bound, peptide-bound, free) making it the least utilized amino acid in biology [41]. Despite its limited use, it is one of the most conserved amino acids in proteins. This can be attributed to its unique physical properties which allow cysteine to fulfill very important functions in cells. Sulfur is large, polarizable, and electron rich and is thus very reactive and able to adopt multiple oxidation states [42], [43]. Cysteine residues are used in enzyme active sites and play structural roles which include iron–sulfur (Fe–S) assembly, heme, and zinc finger motifs [44], [45], [46]. Cysteine SH can also react with neighboring SH groups to form a disulfide bridge, an important structural component for secreted proteins [47]. Cysteine SH groups are also found on the surface of proteins making contact with the surrounding aqueous environment. This particular group of solvent exposed or “free” cysteine residues can be subjected to a range of redox-sensitive modifications (outlined below). Modification of a solvent exposed SH group depends on its ability to ionize and form a very nucleophilic thiolate anion (S−) [48]. Formation of S− is governed by the pKa of cysteine SH which is influenced heavily by the surrounding microenvironment. At neutral pH, cysteines have a pKa of ~8.3 meaning it will be in a protonated and unreactive state [49]. Exposure to a more alkaline environment can substantially increase the probability that a cysteine SH will adopt a deprotonated state [50]. Adjacent positively charged amino acids can also substantially decrease the pKa of cysteine thiols, rendering them more reactive. The presence of other adjacent positive charges such as the partial positive charge of an α-helix dipole or the nitrogen in the peptide backbone can also decrease the pKa of cysteines [51]. For instance, the catalytic cysteines in glutaredoxin (Grx) isoforms 1 and 2 adopt a very low pKa (Grx1, cytosolic isoform, pKa~3.5 and Grx2, mitochondrial matrix isoform, pKa~4.6) due to close proximity of a lysine residue and the N-terminus of an α-helix [52], [53]. Likewise, neighbouring negative charges can increase the pKa which is the case for reduced glutathione (GSH) which has a pKa of ~8.8 [49]. As mentioned SH reactivity also influences whether or not it can adopt other oxidation states; e.g. be reactive enough to sense changes in redox through interactions with H2O2. An excellent example of this is peroxiredoxin (Prx) which uses a catalytic or “peroxidatic” cysteine (Cysp), which has an estimated pKa of 5–6, to metabolize H2O2 with exquisite efficiency [49], [54]. Steric hindrance also plays a part in cysteine thiol reactivity since cysteine oxidation reactions are nucleophilic substitution (SN2) reactions [55], [56]. Thus, it is clear that the surrounding microenvironment will dictate whether or not a thiolate will be reactive enough to undergo modification which may also aid in dictating the selectivity for thiol modification.
These basic principles in SH chemistry and reactivity are highly relevant to mitochondria. This is in light of the unique physical properties of mitochondria. As outlined in the introduction, the chief functions of mitochondria are dependent on the pumping of protons from the matrix to the intermembrane space. This effectively alkalinizes the matrix environment which has a tremendous impact on mitochondrial redox potential and thiol ionization. For example, the calculated midpoint potential for GSH to glutathione disulfide (GSSG) ratio in the cytosol is −240 mV [57]. Since redox potentials are influenced by pH the alkalinity of the matrix environment lowers the potential even further (2GSH/GSSGmitochondria=−280 to −340 mV) [58]. Based on this it is very probable that protein cysteine residues in the mitochondrial matrix will exist in an ionized state. The mitochondrial proteome is also very rich in protein thiols. The estimated concentration of exposed protein thiols in mitochondria is 60–90 mM [59]. This actually makes protein cysteine residues the most concentrated thiol in mitochondria. In conjunction with these key properties mitochondria are also a major source for O2− and H2O2 and contain high amounts of GSH (~5 mM) which are both required for redox signaling. Mitochondria are also a major target for nitric oxide (NO) signaling and S-nitrosylation [37], [60]. Notably mitochondria have been reported to harbor NO synthase (NOS; specifically neuronal NOS isoform) however; this remains highly contentious (discussed below) [37]. Thus, it can be concluded that mitochondria harbor a unique environment that promotes thiol modification.
S-oxidation (sulfenylation and sulfinylation)
In ROS-mediated signaling, the oxidation of protein cysteine SH groups plays a central part in modulating protein function in response to local changes in redox environment. It is important, however, to note that all protein cysteine SH modifications are S-oxidation reactions since they involve changes in valency. Thus, cysteine SH oxidation by ROS is most often described as S-oxidation or thiol oxidation. For our purposes here, we will define S-oxidation specifically as sulfenylation and sulfinylation. It is also important to identify which oxyradical species are responsible for mediating these reactions. H2O2 is the most relevant radical species in redox signaling. This is by virtue of its reactivity with cysteine, capacity to diffuse through membranes, and its prolonged half-life (relative to other oxyradicals). H2O2 signaling is mediated through the formation of SOH [61]. In this reaction, the highly nucleophilic S− attacks H2O2, which results in formation of SOH and OH− [48]. Most protein cysteine SH groups react slowly with H2O2 (~20 M−1 s−1) [49]. This rate can be increased to as high as ~108 M−1 s1 which is dependent on protein microenvironment, pH, the pKa of the SH group, and presence of bulky groups surrounding SH groups [62], [63]. Due to their reactivity it has been very difficult to ascertain the pKa of SOH groups formed in proteins. However, studies with small molecule sulfenic acids have revealed that SOH groups have much lower pKa values than SH groups [64], [65]. Thus due to their lower pKa, it could be assumed that SOH adopt an ionized state (SO−) which makes them far more reactive. This could account for the labile nature of SOH groups in proteins explaining why they may be so difficult to detect and quantify. In addition, this also explains why SOH groups are amenable to undergo a variety of different modifications as outlined in Fig. 2a [66].
Fig. 2.
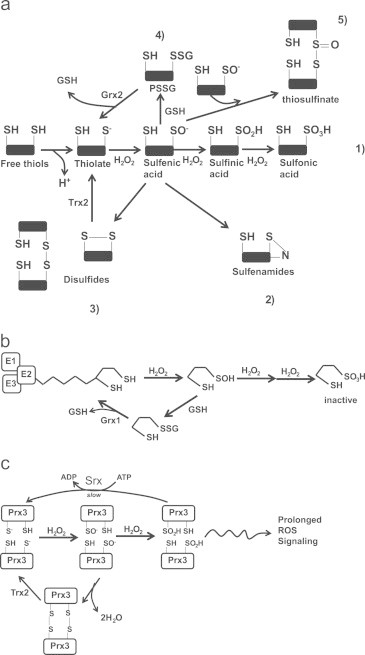
S-oxidation reactions and H2O2 signaling: (a) the various thiol oxidation reactions. Thiols are deprotonated generating a reactive thiolate anion which can then be oxidized by H2O2 to produce a SO−. Due to their highly reactive nature, SO− undergoes a range of other reactions. Depending on the conditions, environment, and proximity of the SO− to other reactive groups SO− can be (1) further oxidized by H2O2 to SO2H and SO3H, (2) react with amides on peptide backbones to generate sulfenamides, (3) cross react with neighboring thiols to form intra or intermolecular disulfide bonds which are reversed by Trx2, (4) undergo S-glutathionylation which is reversed by Grx2, and (5) react with a sulfenic acid to generate a thiosulfinate. (b) Lipoic acid residue on the E2 subunit of Odh can be protected from further oxidation by S-glutathionylation. H2O2 oxidizes a sulfhydryl moiety on the lipoate molecule generating SO− which can be oxidized further rendering Odh inactive. To protect from further oxidation, the SO− is S-glutathionylated which is reversed by Grx to regenerate an active enzyme. (c) Prx3 can adopt multiple oxidation states. The peroxidatic Cys of Prx3 is oxidized by H2O2 to yield SO− which then reacts with a neighboring resolving Cys to produce a disulfide bridge. The Cys residues are then reduced by Trx2 to regenerate active Prx3. SO− can also be further oxidized to SO2H which maintains Prx3 in an inactive state for a longer period which allows H2O2 signaling to persist. Prx3 is reduced back to an active enzyme complex by Srx. H2O2; hydrogen peroxide, SO−; sulfenic acid, SO2H; sulfinic acid, SO3H sulfonic acid, PSSG; protein glutathione disulfide mixture, Odh; 2-oxoglutarate dehydrogenase, Grx; glutaredoxin, Prx3; peroxiredoxin-3, Trx2; thioredoxin-2.
SOH groups can participate in a plethora of side reactions (Fig. 2a). For instance, SOH groups can quickly undergo S-glutathionylation if GSSG are in the vicinity. S-glutathionylation of SOH groups is very likely to proceed in mitochondria, given the small volume and high concentration of GSH. Further, S-glutathionylation of SOH has an important biological function in mitochondria, namely, the protection of cysteine residues from further oxidation when H2O2 levels are higher than usual. For example, the sulfur residues on lipoate in the E2 subunit of 2-oxoglutarate dehydrogenase (Odh), a TCA cycle enzyme that couples 2-oxoglutarate decarboxylation to NADH production, can be oxidized rapidly by H2O2 [67], [68]. To avoid the irreversible oxidation of SOH to SO3H, GSH is conjugated to SOH forming a protein glutathione mixed disulfide (PSSG) which essentially protects lipoate from further oxidation (Fig. 2b) [68]. Grx then catalyzes the deglutathionylation of Odh reactivating the enzyme [68]. Similar observations have been made for other mitochondrial proteins including Complex I and adenine nucleotide translocator (ANT) [69], [70]. The absolute number of mitochondrial protein SH groups that can form SOH has yet to be quantified. This may be difficult given that mitochondrial isolation protocols can result in unwanted oxidation of cysteine residues. Oxidation of cysteines during isolation can be prevented or minimized by the addition of N-ethylmaleimide (NEM), an alkylating agent that forms a Michael adduct with SH. Unfortunately, SOH groups are also very labile which also makes them difficult to quantify. Several methods have been used to properly trap and quantify SOH groups. These methods typically involve using dimedone molecules which react selectively with SOH groups tagging them for quantification [71]. Dimedone probes have also been used to measure SH oxidation to SOH in mitochondria [72]. However, it is possible to improve the efficacy of dimedone probes for quantification of mitochondrial SOH levels. For instance, triphenylphosphonium-tagged dimedone (TPP-dimedone) molecules can potentially be developed and used to trap and quantify mitochondrial SOH. TPP-dimedone could be administered in vivo allowing more accurate quantification of mitochondrial SOH. TPP is a lipophilic cation that preferentially accumulates in mitochondria due to large charge difference between the intermembrane space (positive) and matrix (negative) [73]. It is important to point out though that TPP probes can potentially hinder mitochondrial metabolism by uncoupling the proton gradient [74]. It is also important to acknowledge that SOH can form from one electron reduction reactions. These reactions involve the formation of a thiyl radical following exposure to radiation [75]. O2− has also been shown to catalyze the one electron reduction of low molecular weight thiols with a rate constant of 103 M−1 s−1 however; this may be insignificant given the rapid kinetics of superoxide dismutase (SOD; 108 M−1 s1) [10].
SOH groups can also react with neighboring SH groups to form intra or intermolecular disulfide bonds (Fig. 2a). This type of reaction is particularly important for antioxidant enzymes like Prx. In mitochondria, Prx3 is a homodimer with both subunits aligned in a head-to-tail fashion with Cysp on the N-terminus and the resolving cysteine (Cysr) on the C-terminus [54] (Fig. 2c). H2O2 quickly oxidizes Cysp producing SOH which then reacts with the neighbouring Cysr forming an intermolecular disulfide bridge. The catalytic cycle is completed by thioredoxin-2 (Trx2) and NADPH-mediated reduction of the intermolecular disulfide bridge. SOH groups can also react with other SOH groups to form thiosulfinates or react with neighboring nitrogens to form sulfenamides [75] (Fig. 2a). Thus, due to their highly reactive nature and nucleophilic and electrophilic properties, SOH can participate in a broad range of reactions. It is important to note though that it remains to be determined if thiosulfinates or sulfenamides are actually formed in mitochondria. Finally it is important to point out that a vast majority of these reactions are reversible either through Trx or Grx.
SOH can also undergo further oxidation by H2O2 (Fig. 2a). Since SOH can participate in a number of side reactions, further oxidation by H2O2 depends on protein microenvironment, presence of GSH, and quantity of H2O2. Oxidation of SOH generates SO2H. This reaction depends on the ionization of SOH and the nucleophilic attack of H2O2 [48]. Hyperoxidation of SOH to SO2H is often associated with oxidative stress [76]. However, it was recently observed that sulfiredoxin (Srx) can reduce SO2H back to SH in Prx (Fig. 2c) [77]. Although Srx has only been shown to reduce SO2H in Prx it would be important to ascertain if Srx has other targets. Indeed, the reversible nature of SO2H would suggest that sulfinic acid formation may be important for the redox regulation of proteins. Tt has been found that 5% of the cysteines in soluble rat liver proteins exist as SO2H [78]. In mitochondria SO2H formation is crucial for the modulation of Prx3 and DJ-1 [79], [80]. In the case of the former, SO2H formation is required to render Prx3 inactive for extended periods of time (Fig. 2c). This allows H2O2 to accumulate to sufficient amounts to exert regulatory effects on various proteins, either within or external to mitochondria [81]. Srx has recently been identified in mitochondria indicating mitochondria are equipped with the necessary enzymatic complement to reduce SO2H back to SH [82]. It is important to point out though that unlike cytoplasmic Prx2, Prx3 is more resistant to hyperoxidation [83]. Very little information exists on the importance of SO2H groups in the redox control of proteins, particularly in the mitochondria and which proteins and enzymes might be involved in reduction of SO2H. New tools to properly trap and measure SO2H in cells have been proposed including aryl-nitroso compounds which react with SO2H with high efficiency and specificity [84]. It would be intriguing to determine what percentage of the mitochondrial proteome forms SO2H.
S-glutathionylation
Another important redox switch required to control mitochondrial protein function is S-glutathionylation. In these reactions, GSH is conjugated to an exposed cysteine protein SH generating PSSG. The S-glutathionylation of proteins in mitochondria has been found to be highly sensitive to local changes in redox, specifically through fluctuations in 2GSH/GSSG [16], [85], [86]. As indicated above, the potential of the 2GSH/GSSG pair in the matrix of mitochondria is estimated to be between −280 to −340 mV [58]. This range has been attributed to the different methodologies used to measure and calculate absolute GSH and GSSG levels. Most measurements of GSH and GSSG levels have been performed using enzyme coupled assays. These types of assays should only be used to measure total glutathione levels and not for determination of 2GSH/GSSG since it is very difficult to accurately quantify the absolute amounts of GSSG. HPLC can also be used which does provide a far more accurate read of 2GSH/GSSG [86]. This is due to clean separation and resolution of GSH from GSSG which allows easy integration and calculation of absolute GSH and GSSG levels. Using an assumed mitochondrial volume of 0.75 µL (considered to vary between 0.5–1 μL [8]) the molar concentration can be calculated and then subsequently plugged into the Nernst equation to give the potential of the pair. However, isolation of mitochondria and treatment with acids for glutathione extraction can artificially increase GSSG levels decreasing the accuracy of the calculated potential. It is also important to extract mitochondria in the presence of 25 mM NEM to prevent any spontaneous reactions of GSH or GSSG with protein thiols. Samples should also be injected immediately after mitochondrial isolation since GSH can spontaneously oxidize over time. Control experiments should also be performed with mitochondria treated with reducing (DTT) or oxidizing agents (H2O2) [86]. For cells in culture, glutathione pools can be depleted using buthionine sulfoximine (BSO) [87]. Finally, a class of Grx1-GFP probes has been developed that allow the sensitive real-time measurement of 2GSH/GSSG in intact cells or in vivo [88]. These probes are targeted to the mitochondrial matrix using mitochondrial localization sequences. Briefly, the probe works through a series of cysteine disulfide exchange reactions between GSSG and probe itself which ultimately leads to the formation of a disulfide bond on GFP which alters its fluorescence at 408 nm and 488 nm. The calculated fluorescent ratio can then be used to determine the potential of 2GSH/GSSG. A very small increase in GSSG (10 µM) or H2O2 (12.5 µM), which produces GSSG, can induce a substantial increase in the ratiometric fluorescence of the probe [88]. The fluorescence can then be brought back down to base line with DTT. By virtue of its extreme sensitivity and non-invasiveness (e.g. no need to isolate mitochondria) this stands as the most sensitive and accurate measure for 2GSH/GSSG. It is important to accurately measure the absolute concentrations of GSH and GSSG since GSSG can S-glutathionylate cysteine residues via a simple thiol disulfide exchange reaction. Inaccurate measures of GSSG can lead to false conclusions on the state of the redox environment and whether or not S-glutathionylation reactions will proceed spontaneously. Thus, it is crucial to utilize methods that provide accurate measures of both GSH and GSSG and in this respect, spectrophotometric assays should be avoided.
The S-glutathionylation of proteins can either proceed non-enzymatically or enzymatically and it is thus important to distinguish between the two types. Non-enzymatic S-glutathionylation can proceed via a number of different pathways (Fig. 3a). Reaction 1 simply involves a thiol disulfide exchange reaction between protein cysteine S− and GSSG. The result is the formation of PSSG and GSH [89]. Reaction 1 can only proceed if GSSG has accumulated to a sufficient concentration and the 2GSH/GSSG is ~1 [90]. GSSG reaches this concentration only under oxidative stress. In reaction 2, a protein S− is oxidized to SOH which then reacts with GSSG to yield PSSG. For reaction 3, PSSG formation is initiated by the formation of a thiyl radical which interacts with GSH to form a glutathione anion protein thiyl radical intermediate [91]. This structure is stabilized by O2 which subsequently yields PSSG. Non-enzymatic glutathionylation reactions are very nonspecific and mostly proceed under oxidative stress. Under oxidative stress proteins in mitochondria can become hyper-glutathionylated which is often considered a marker for oxidative damage [92].
Fig. 3.
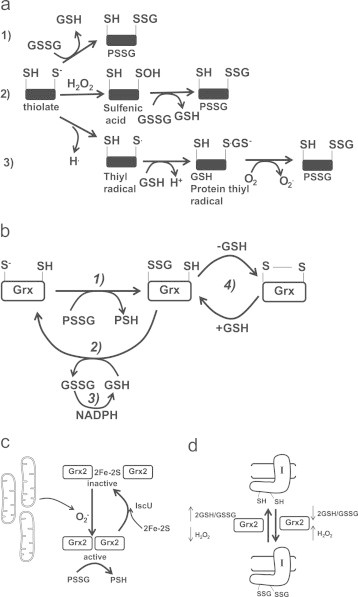
Non-enzymatic and enzymatic S-glutathionylation reactions and the modulation of Grx2. (a) Non-enzymatic glutathionylation reactions. (1) Solvent-exposed thiolate is S-glutathionylated by GSSG via a thiol disulfide exchange reaction. (2) Solvent-exposed thiolate are oxidized to a sulfenic acid residue by H2O2 which then undergoes S-glutathionylation. (3) one electron reduction of a thiol forms a thiyl radical which interacts with glutathione to form a glutathione anion thiyl radical intermediate. A disulfide bond then forms between the glutathione anion and the thiyl radical following the one electron reduction of O2 to O2−.(b) Catalytic cycle of Grx1 and Grx2 (in diagram it is simply referred to as Grx). In step 1 Grx catalyzes the rapid transfer of the gluthionyl moiety via a disulfide exchange reaction to its catalytic cysteine which produces a Grx-SSG intermediate and a deglutathionylated protein. Step 2 involves the GSH-mediated removal of the glutathionyl moiety from Grx-SSG which generates a fully reduced Grx enzyme and GSSG. For step 3 the GSSG is reduced by NADPH and glutathione reductase to regenerate GSH. Note the side reaction for Grx-SSG in step 4 an intramolecular disulfide can form in Grx. GSH is required to return Grx-SSG to its catalytic cycle. (c) Grx2 is modulated by 2Fe–2S cluster coordination and O2− mediated dissembly of the cluster. Grx2 is maintained is inactive as a homodimer. A subsequent burst of O2− results in the release of active Grx2 monomers which subsequently deglutathionylate target proteins. (d) Grx2 catalyzes the reversible S-glutathionylation of Complex I. When the 2GSH/GSSG ratio is low and H2O2 is high, Grx2 glutathionylase is activated. A high 2GSH/GSSG ratio and low H2O2 levels induce Grx2-mediated deglutathionylation of Complex I.
A number of mitochondrial proteins are S-glutathionylated under normal conditions, depending upon the tissue type [86], [93]. Further, S-glutathionylation of mitochondrial proteins under normal conditions is cysteine thiol specific, reversible, sensitive to changes in redox environment, and most importantly, enzyme driven [36], [90]. Enzyme-driven S-glutathionylation reactions are mostly investigated in the cytosol where Grx1 has been identified as the chief enzyme responsible for catalyzing the deglutathionylation of PSSG. The enzymatic mechanism involves two steps; (1) N-terminal cysteine on Grx deglutathionylates PSSG via a thiol disulfide exchange reaction yielding PSH and a Grx-SSG intermediate, (2) Grx-SSG binds GSH and the glutathionyl moiety is removed regenerating Grx1 and producing GSSG, (3) GSSG is reduced back to 2GSH by glutathione reductase and NADPH (Fig. 3b) [52]. Step 1 of the enzyme mechanism is very rapid with step 2 occurring much more slowly and is thus rate limiting [52]. It is also notable that Grx also has a C-terminal SH that can cross react with Grx-SSG to generate an intramolecular disulfide bridge and GSH. Reversal of the disulfide requires GSH to regenerate Grx1-SSG in the catalytic cycle. Formation of the intramolecular disulfide bridge has been suggested to serve as a site of regulation since it may allow an S-glutathionylation on a protein to persist [10]. The mechanistic details of the catalytic reaction still require clarification however; several catalytic models have been proposed including glutathione scaffold, glutathione activator, and an oxidative half-reaction (reviewed in [94]).
Discussion of the catalytic cycle of Grx1 is critical since mitochondria harbor a Grx1 isozyme, Grx2. Grx2 was only recently identified and is classified as a dithiol Grx since it conforms to the thioredoxin fold (four mixed β-sheets surrounded by α-helices) and contains the CXXC catalytic motif. Intriguingly, Grx2 only has ~34% sequence identity to Grx1 [53], [95], [96]. However, despite this clear lack of homology, Grx2 has been found to use the precise same catalytic mechanism as Grx1 [52]. The sequence differences between Grx1 and Grx2 are important to the regulation of both proteins. For instance, unlike Grx1, Grx2 has no exposed thiol residues on its surface making it less likely to be deactivated by ROS. Grx2 also forms a homodimer through coordination of a 2Fe-2S cluster which has been shown to be important for its regulation [18]. Grx2 is inactive when complexed with 2Fe–2S. Disassembly of the 2Fe–2S cluster releases active monomeric Grx2 which is mediated by O2−. This illustrates that there is a tight link between mitochondrial ROS, redox, and S-glutathionylation reactions (Fig. 3c) [97]. Reassembly of the 2Fe–2S cluster and the Grx2 homodimer requires iron sulfur cluster chaperone IscU [98]. It is also interesting that Grx2 has been found to also catalyze protein S-glutathionylation (Fig. 3d) [85]. This occurs when the 2GSH/GSSG is sufficiently oxidizing [85]. It is important to realize though that precisely how the glutaredoxins, namely Grx1 and Grx2, catalyze S-glutathionylation reactions is unresolved. There is though sufficient evidence to indicate that Grx2 harbors glutathionylase activity. For instance Complex I has been documented to be S-glutathionylated and deglutathionyated by Grx2 [85]. Grx2 has also been shown to be required for the S-glutathionylation of uncoupling protein-3 (UCP3) [86].The significance of Grx2-mediated modulation of Complex I and UCP3 by S-glutathionylation is discussed below. In addition, catalysis of S-glutathionylation by Grx2 is feasible from a thermodynamic and redox chemistry perspective given that thiol disulfide exchange reactions are generally reversible. Hypothetically, Grx2 may only harbor glutathionylase activity when Grx2-SSG has accumulated to a sufficient concentration. Indeed, step 2 of the catalytic cycle of Grx2 is rate limiting and, under certain conditions, this may prompt accumulation of a Grx2-SSG intermediate. It is also important to note that glutathione S-transferase (GST; Pi and Mu-family) has recently been found to catalyze the glutathionylation of proteins in the cytosol [99]. Mitochondria have also been reported to contain several GST isozymes including Alpha, Kappa, Mu, Pi and Zeta have been reported suggesting a similar function in mitochondria [100].
S-nitrosylation
Nitric oxide (NO) was among the first gaseous second messengers identified in mammals and plays a central role in vasodilation [101]. Following its production by NOS, NO rapidly diffuses through the endothelial cells, where it was produced, into smooth muscle cells where it activates soluble guanylate cyclase which prompts cyclic guanosine monophosphate (cGMP) production and smooth muscle relaxation. NO is also able to interact with mitochondria. NO binds cytochrome a3 and Cu2+B prosthetic groups of Complex IV [60]. By binding to the heme or Cu group, NO inhibits Complex IV activity which diminishes OXPHOS. Binding prosthetic groups is not the only way NO can modulate mitochondria. The S-nitrosylation (SNO) of free SH residues in proteins have also been shown to be an important component of NO signaling. A number of different mitochondrial proteins have been shown to be modulated by SNO. The means by which SNO is formed, however, remains elusive. NO is not an oxidant under biological conditions and thus cannot form a SNO via a direct reaction with S− [102]. However, NO can form a thionitroxide group (SNHO) which, following a one-electron oxidation, can possibly generate SNO [75]. Another mechanism for SNO involves the initial reaction of 2 mols of NO with 1 mol of O2 to form nitrogen dioxide (NO2). NO2 is capable of reacting with S− to form a thiyl radical which then reacts with NO to form SNO [103]. Finally, NO2 can react with NO to form dinitrogen trioxide (N2O3) which is able to react directly with S− to form SNO [104].
Where mitochondrial NO comes from is still enthusiastically debated. First, there have been several reports that NOS isoforms localize to mitochondria [37]. Specifically, it has been frequently reported that nNOS is found in liver and rat brain mitochondria where it actively produces NO following stimulation by calcium [105]. Unfortunately, the presence of mitochondrial NOS remains contentious since other groups have been unable to detect any NOS isoforms in mitochondria [37]. Other sources of mitochondrial NO include NO generated in the cytoplasm or neighboring cells. NO diffuses rapidly through membranes and thus can promptly gain access to the mitochondrial matrix. The issue surrounding external NO is the quenching of NO before it reaches mitochondria. For instance, in normoxia, NO can be scavenged by oxymyoglobin which produces nitrate (NO3−) which limits the amount of NO that actually reaches mitochondria [106]. Inside the cell, NO can react to form peroxynitrite (ONOO−) or, following NO2 formation, can react with unsaturated fatty acids to produce nitro-fatty acids [107].
The debate surrounding the source of NO, and which species actually generates SNO, does not diminish the importance of NO in the regulation of mitochondria. SNO has been shown be important for ischemic preconditioning and it has been shown that mitochondrial SNO (mSNO) can be used to greatly reduce cardiac infarct size through reversible SNO modification of Complex I [108]. SNO also modulates ATP synthase, several TCA cycle enzymes, and enzymes involved in β-oxidation [37], [109]. NO has also been shown to limit mitochondrial membrane permeability transition (MPT) through SNO formation on cyclophillin D [110]. A number of excellent reviews have been published on the topic NO signaling, SNO, and control of mitochondrial function [37], [111], [112]. Thus, SNO and the control of mitochondrial functions will not be considered any further in this review.
Regulation of mitochondrial biology by redox switches
From the above section it is clear that; (1) redox signaling is very complex, (2) we still understand very little about redox reactions, which can be attributed the lack of sensitive techniques, (3) the importance for regulation of mitochondrial function by redox switches. Mitochondrial ROS production, and how rapidly it is quenched, depends on the tissue and cell type, the energetic state of mitochondria (e.g. respiring under phosphorylating or non-phosphorylating conditions), and which substrate is being metabolized [93], [113], [114]. Regulation of mitochondria by redox switches would also be dependent upon mitochondrial density, protein content (e.g. thiol availability), antioxidant defense, and glutathione availability. The 2GSH/GSSG ratio is usually in a highly reduced state (in such tissues as heart, muscle, liver, and brain) but the state of this redox pair can change spatially and temporally depending upon ROS concentrations, transport of GSH from the cytosol, export of GSSG, and reduction of GSSG back to GSH by the efficient enzymatic activity of glutathione reductase [86], [115], [116]. In the following section we will discuss the various mitochondrial proteins and enzymes that are regulated by cysteine oxidation reactions and the impact cysteine oxidation has on enzyme function and activity. We will also highlight the conditions under which proteins in mitochondria are modulated by cysteine oxidation and the impact it has on mitochondrial metabolism and function.
Regulation of the TCA cycle
The TCA cycle is the most central metabolic pathway for anaerobic and aerobic organisms. For aerobes, the TCA cycle either serves as an engine that strips electrons from nutrients for production of ATP or provider of the necessary building blocks for the production of biological macromolecules. A number of enzymes in the TCA cycle have been shown to be either sulfenylated or S-glutathionylated. The impact of these modifications on mitochondrial function, and the circumstances under which the modification occurs, is enzyme dependent. Aconitase (Acn) can be reversibly deactivated by H2O2 through sulfydryl group modification [117]. This reversible inactivation has been shown to occur during ischemia-reperfusion injury. Intriguingly, Acn does not lose its 4Fe–4S cluster due to interactions with Frataxin protein [117]. However, if the stress persists, Acn can be irreversibly deactivated due to 4Fe–4S cluster disassembly [118]. NADP-dependent isocitrate dehydrogenase (Idh) has been reported to be inactivated by S-glutathionylation in HEK293 cells and rabbit heart mitochondria [119]. This is intriguing since this Idh isoform is required to contribute to the NADPH pool in mitochondria, which provides the reducing power necessary to maintain an active antioxidant defense system. The authors reported that half-maximal inhibition occurred at ~5 mM GSSG and can be reversed by 10 mM DTT [119]. This would set the 2GSH/GSSG ratio to ~1 and represents a system that is under oxidative stress. It would be important to discern if this inhibition occurs under normal conditions and how rapidly Grx2 (or another enzyme) reverses the inhibition. As mentioned Odh is a major target for redox regulation. Odh has been shown to be targeted by sulfenylation, sulfinylation, and S-glutathionylation; the latter being important in protecting key thiol residues from further oxidation [120]. Odh sits at a major metabolic junction that links carbon catabolism to amino acid production in mitochondria. It has been suggested that reversible oxidation of Odh may be required to modulate the flow and fluxes of metabolites through amino acid metabolism and the TCA cycle. Oxidative deactivation of Odh also has the added benefit of prompting the accumulation of 2-oxoglutarate, which has the capacity to quench H2O2 and would also aid in limiting H2O2 production [121]. Pyruvate dehydrogenase (Pdh), which bears dihydrolipoamide dehydrogenase subunits like Odh, can also be modulated by mitochondrial H2O2 through thiol oxidation [122]. In addition, like Odh, Pdh activity can be recovered with reducing agents, illustrating that this enzyme complex is regulated in manner similar to Odh [122]. SDH has also been found to be controlled by S-glutathionylation [123]. However, the circumstances of its regulation are quite intriguing. In Sprague-Dawley rat heart mitochondria, SDH is maintained in a glutathionylated state [123]. Following ischemia-reperfusion, SDH is deglutathionylated which not only decreases its activity but also induces a burst of O2− production from SDH [123]. L-carnitine/acyl-carnitine carrier (CAC) has also been found to be S-glutathionylated on Cys136 and Cys155, respectively [124]. S-glutathionylation state of CAC is sensitive to the potential of 2GSH/GSSG where high GSH leads to deglutathionylation and activation of CAC and high GSSG induces S-glutathionylation and deactivation of the transporter [124]. It was also found that exogenously added Grx1 can reverse S-glutathionylation indicating that CAC may also be targeted by either Grx1 in the intermembrane space or Grx2 in the matrix [124]. This illustrates the complexities surrounding S-glutathionylation and the regulation of protein function in mitochondria.
In the above examples, S-glutathionylation has mostly been shown to proceed under conditions of oxidative stress when GSSG levels are high (Fig. 3a). However, it is also clear that S-glutathionylation is reversible and occurs under normal conditions. For instance, Acn can be reversibly deactivated by S-glutathionylation [117]. It has also been shown that Odh can be deglutathionylated in vitro by purified Grx1 [68]. Also, SDH maintains an S-glutathionylated state in heart mitochondria under normal conditions [123]. The enzyme(s) that actually mediate S-glutathionylation and deglutathionylation under these conditions are still yet-to-be identified. However, Grx2 is a strong candidate for mediating these reactions. In regard to sulfenylation, it is doubtful that SOH formation plays any important regulatory role in mitochondria due to the reactivity of this moiety, especially in an environment rich in ROS, thiols, and glutathione. SOH is most likely a precursor in mitochondria for other reactions such as S-glutathionylation or sulfinylation.
Regulation of OXPHOS
Most of the information generated in terms of how redox switches work to control mitochondrial proteins has been accrued by studying the impact of S-oxidation reactions on OXPHOS. Most of these studies have been conducted using heart mitochondria of either bovine or mouse origin. Important information concerning the role of redox signaling in the control of mitochondrial bioenergetics has also been generated using cultured cells (lens epithelia, vascular smooth muscle cells, myotubes), isolated liver mitochondria, and isolated skeletal muscle mitochondria [86], [125], [126]. The impact of chemical-induced S-glutathionylation on mitochondrial bioenergetics has variable effects which are dependent on cell line. Acute treatment of vascular smooth muscle cells (VSMC) with 0.1 mM diamide, an S-glutathionylation catalyst, induces a transient decrease in resting respiration (basal respiration; respiration required to maintain cellular ATP levels under resting conditions) which then subsequently recovers over time [126]. Hill and colleagues also showed that 0.1 mM diamide induced a massive increase in proton leaks in VSMC. Higher diamide concentrations (>0.25 mM) induce an irreversible decrease in respiration [126]. These observations indicate that aerobic respiration can indeed be modulated by reversible S-glutathionylation reactions. Different results were obtained with primary mouse myotubes. Treatment with ≤0.2 mM diamide did not have any effect on resting respiration; however diamide treatment did induce an increase in PMF [87]. Assessment of state 4 respiratory parameters (proton leaks; non-phosphorylating respiration) revealed that diamide-induced S-glutathionylation inhibited proton leaks through UCP3 [87]. Leaks through UCP3 can be reactivated by a small increase in H2O2 which is required to decrease PMF and diminish mitochondrial ROS emission [87]. In addition, it has recently been shown that Grx2 is required to S-glutathionylate UCP3 [86]. Indeed, UCP3 is in a deglutathionylated and active state in skeletal muscle mitochondria isolated from Grx2 knock-out (Grx2−/−) mice [86]. Similar results have been obtained with Min6 insulinoma cells [16]. In this case, chemical induction of S-glutathionylation deactivates leaks through UCP2, a homolog of UCP3, which enhances glucose-stimulated insulin release (GSIS) [16]. The enhancement of GSIS is due to the maintenance of the mitochondrial protonic potential which increases aerobic ATP production for stimulation of insulin release. The progressive oxidation of glucose eventually leads to an increase in mitochondrial O2− and H2O2 which deglutathionylates UCP2, activates leaks, and desensitizes the GSIS [16]. Chemical induction of UCP2 glutathionylation can also be used to sensitize drug resistant cancer cells to chemotherapeutics [127]. This is due to the fact that certain drug resistant cancer cells overexpress UCP2 which prevents anthracycline-mediated ROS production [128]. Induction of UCP2 S-glutathionylation deactivates proton leaks which enhances drug toxicity through oxidative stress. Thus, reversible S-glutathionylation can control OXPHOS, proton leaks, and mitochondrial ROS emission which can have profound signaling effects in the cell.
Complex I was the first OXPHOS component identified to undergo either S-glutathionylation or sulfenylation. The mechanism of Complex I sulfenylation and S-glutathionylation has mostly been explored in heart mitochondria. In terms of sulfenylation, SOH formation was shown to undergo further oxidation to SO2H and SO3H which irreversibly deactivates Complex I [69]. However, cysteine residues that are oxidized to SOH in Complex I can also be protected from further oxidation by S-glutathionylation [69]. Although S-glutathionylation decreases Complex I activity, the modification can be reversed [85]. Complex I S-glutathionylation is also important since it can diminish O2− production. In bovine heart mitochondria, Cys531 and Cys704 on the 75 KDa subunit NDUSF1, were identified as S-glutathionylation sites on Complex I [69]. The NDUSF1 subunit sits in the hydrophilic arm of Complex I in proximity of FMN NADH binding site. S-glutathionylation of the solvent exposed Cys531 and Cys704 most likely induces conformational changes in the hydrophilic arm of Complex I which may sterically block the NADH binding site. Thus, preventing NADH oxidation can limit electron flow through the respiratory chain and diminish O2− production [129]. However, other studies have shown that S-glutathionylation of Complex I actually increases mitochondrial O2− emission [130]. This could be related to maintenance of Complex I in a prolonged S-glutathionylated state. It is important to connect Complex I S-glutathionylation to the S-glutathionylation of Odh since Odh generates NADH for oxidation by Complex I and both protein complexes produce mitochondrial ROS (Fig. 4). Notably, the S-glytathionylation of these two protein complexes diminishes the activity of both protein complexes when H2O2 is high. S-glutathionylation may serve as a mechanism that limits further O2− and H2O2 production by a mitochondrion that is already faced with the potential threat of oxidative stress (Fig. 4) [131], [132]. Once O2− and H2O2 levels are normal, Odh and Complex I are deglutathionylated by Grx2 and OXPHOS resumes unhindered (Fig. 4).
Fig. 4.
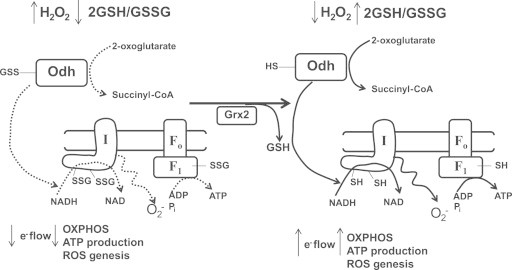
Hypothetical scheme depicting the regulation of mitochondrial O2− production and OXPHOS by S-glutathionylation. When H2O2 is high and 2GSH/GSSG is low in mitochondria, Odh, Complex I, and ATP synthase are temporarily S-glutathionylated to diminish NADH production and limit electron flow through the respiratory chain. Although this may diminish mitochondrial ATP production S-glutathionylation will limit O2− and H2O2 production by mitochondria. In addition, S-glutathionylation of ATP synthase may also prevent ATP hydrolysis and the pumping of protons back to the intermembrane space. This would effectively maintain a lower membrane potential and limit ROS genesis. Once ROS levels have decreased and the 2GSH/GSSG ratio is restored Grx2 deglutathionylates Complex I and potentially Odh and ATP synthase which restores mitochondrial ATP production.
Reversible S-glutathionylation of Complex I is mediated by Grx2. Complex I has been shown to be deglutathionylated by Grx2 in heart mitochondria, lens epithelia, and liver mitochondria [85], [86], [125]. Grx2 is required to maintain Complex I in an active deglutathionylated state in cardiac mitochondria. For instance, mice treated with doxorubicin display increased Complex I S-glutathionylation and decreased activity which compromises mitochondrial ATP production [129]. Selective overexpression of Grx2 in cardiac tissue maintains Complex I in an active deglutathionylated and active state [129]. Intriguingly, Complex I is not targeted by Grx2 in skeletal muscle mitochondria indicating that regulation of Complex I by Grx2 is tissue-specific [86]. Grx2 is able to catalyze the reversible glutathionylation of Complex I which is dependent on 2GSH/GSSG [85]. A more oxidized 2GSH/GSSG ratio induces Complex I S-glutathionylation whereas a more reduced glutathione pool activates the deglutathionylase activity of Grx2 [85]. Thus, the S-glutathionylation serves various important roles in the control of Complex I which include (1) protection from irreversible oxidation and (2) control of mitochondrial ROS genesis in response to alterations in local redox environment. The fact that Complex I can be controlled by redox switches has made it a pharmacological target for ischemic preconditioning and prevention of heart disease [108].
OXPHOS components have been shown to be modulated by redox switches. ATP synthase (Complex V) has been shown to be S-glutathionylated on Cys294 on its α-subunit which is located in the F1 hydrophilic part of the Complex [133]. This has been shown in heart mitochondria and occurs in dyssynchronous heart failure. Cys294 has also been shown to form SOH which reacts with a neighboring Cys103 residue on the γ subunit to form a disulfide bridge [133]. Formation of the disulfide bridge occurs only under chronic heart disease. S-glutathionylation blocks nucleotide binding which diminishes the production of ATP. Garcia et al. also showed that the α-subunit of the F1 complex of ATP synthase is S-glutathionylated in rat brain and liver [134]. Similar to heart mitochondria, S-glutathionylation of ATP synthase in these two tissues diminishes ATP production. Indeed, S-glutathionylation of Complex I and Odh not only limits NADH production but also lowers electron flow through the respiratory chain which would effectively diminish ROS genesis. Temporary S-glutathionylation of Complex I and Odh protects both enzyme complexes from irreversible oxidation when ROS is high but may also hypothetically serve as a mechanism to diminish mitochondrial ROS production (Fig. 4). Temporary S-glutathionylation of the F1 α-subunit of ATP synthase may also be part of this adaptive response to a burst of H2O2 genesis vis a vis the decrease in electron flow would limit ATP production. In addition S-glutathionylation mediated decrease in ATP synthase activity would also limit ATP hydrolysis and the pumping of protons back into the intermembrane space thus preventing the repolarization of the mitochondrial inner membrane (Fig. 4). The PMF is tightly associated with mitochondrial ROS production [27]. A high protonic potential nonlinearly increases mitochondrial ROS production by the respiratory chain while decoupling the MIM has the opposite effect. Intriguingly, when ROS is high in mitochondria UCP2 and UCP3 become deglutathionylated which activates proton leaks through these two proteins. Proton leaks lower the PMF across the MIM which subsequently diminishes ROS production by mitochondria. Thus, when ROS is high Complex I and Odh are S-glutathionylated to protect from irreversible oxidation which also lowers there activity to diminish electron flow through the respiratory chain which limits ROS production (Fig. 4). At the same time ATP synthase is S-glutathionylated to lower ATP production and prevent ATP hydrolysis and MIM recoupling. Notably, S-glutathionylation of ATP synthase may also protect cysteine residues from irreversible oxidation. In addition, if UCP2 or UCP3 are present in the tissue, high ROS deglutathionylates these two proteins resulting in their activation and a lowering of mitochondrial PMF which also diminishes ROS production. Thus, reversible S-glutathionylation may serve as an important mechanism for the control of mitochondrial ROS output which can have a profound effect on cellular ROS signaling.
Mitochondrial morphology
Mitochondrial structure and morphology plays a key role in dictating mitochondrial function [135]. The shape of mitochondria has a tremendous impact on mitochondrial metabolism and cell death. Likewise the nutrients being metabolized, the concentration of nutrients in the cell, and the amount of ROS play significant roles in dictating mitochondrial shape and morphology [135]. In mammalian cells, the proteins involved in mitochondrial fusion, are the integral membrane GTPases Mitofusin 1 (Mfn1), Mitofusin 2 (Mfn2) and autosomal dominant Optical Atrophy 1 (Opa1) [136]. Conversely fission is mediated by the dynamin family of GTPases Drp-1. Chronic oxidative stress, high glucose or fatty acids (which increase ROS), or gluco-lipotoxicity are known to induce mitochondrial fragmentation [135]. However, treatment with sub-lethal amounts of H2O2 and acute stresses induce a hyperfused state [137]. Co-treatment with antioxidants like Tempol prevents hyperfusion [137]. Recently, Shutt et al. provided evidence that S-glutathionylation of Mfn proteins is required to induce mitochondrial hyperfusion [137]. In vitro determinations showed that addition of GSSG to reaction mixtures induced a hyperfused state which was associated with S-glutathionylation of Mfn2 and formation of Opa1 oligomers. Intriguingly, performing the same reactions in the presence of cystosol fractions from HeLa cells reversed Mfn2 S-glutathionylation. Redpath et al. made similar observations showing that mitochondria in C2C12 cells adopt a hyperfused state when treated with diamide [138]. These results demonstrate S-glutathionylation also plays a key role in mitochondrial morphology and that induction of fusion is sensitive to changes in the 2GSH/GSSG ratio. It has been proposed that mitochondrial hyperfusion may increase resistant towards cell stress and cell death. Hyperfusion may promote protection through the sharing of mitochondrial resources, specifically, antioxidant defense enzymes and molecules which could provide more long term protection of mitochondrial proteins, enzymes, DNA, and lipids from acute stressors. Effectively, this protective response would also prevent oxidative stress and induction of intrinsic apoptotic cascades via preservation of mitochondrial redox balance.
MTP and redox switches
Oxyradicals are maintained at low concentrations by antioxidant defense systems. But what happens if oxyradical production cannot be controlled and antioxidant defense systems are overwhelmed? In this scenario, mitochondria induce cell death pathways. The mitochondrial intermembrane space contains several factors, including Smac/DIABLO, apoptosis inducing factor, and cytochrome C, that upon being released into the cytoplasm, activate programmed cell death pathways [139]. The use of cytochrome C as an apoptogenic signaling molecule is especially interesting since it is a key component of OXPHOS machinery. This illustrates the tight relationship between the efficiency of electron flow through the ETC and the production of either ATP or ROS.
Release of these apoptogenic factors in the cytoplasm can be mediated by MPT. The mitochondrial permeability transition pore (MPTP) is a non-selective pore that allows the free diffusion of molecules <1.5 KDa in size [140]. Once open, the MPTP dissipates ion and metabolite gradients and collapses the PMF, which can induce massive swelling of mitochondria and the rupture of the mitochondrial outer membrane [70]. The MPTP is normally closed but can be opened by a number of factors including elevated matrix [Ca2+], adenine nucleotide depletion, and increased inorganic phosphate concentration [141]. Notably for the purpose of redox switches, MPT can also be induced by ROS, hydroperoxides (t-butyl hydroperoxide), and chemical toxins (which can induce ROS production) [141]. Antioxidant supplementation prevents MPT. Interestingly, MPT can also be modulated by SH blockers and S-glutathionylation catalysts. For instance, induction of MPT can be prevented by incubating mitochondria in N-ethylmaleimide (NEM), a Michael acceptor that covalently binds to SH [70]. Alternatively, MPT can also be induced by diamide [70]. Studies using semiquinone, which actively produces O2−, have found that mitochondrial produced ROS can activate MPT [142], [143]. In addition, it has also been shown that H2O2 is the oxyradical species that most likely induces MPT since inclusion of catalase and thiol peroxidases to reaction mixtures prevents MPT [143], [144]. Thus, the capacity of mitochondria to induce programmed cell death depends on redox switches which, upon sulfenylation and/or S-glutathionylation, induces pore opening.
The function of redox switches in pore opening has been reviewed recently in [145]. Redox switches most likely serve as the connection between regulation of mitochondrial function and induction of death. The composition of MPTP still remains a contentious issue. MPTP has been shown to be composed of ANT, voltage dependent anion channel (VDAC), Pi carrier, and cyclophilin D (Cyp-D) [146]. For pore opening to occur, Cyp-D needs to bind to the protein complex. The role of ANT in MPT is enthusiastically debated however; several reports have conclusively shown that oxidation of key cysteine residues on ANT is required for pore opening. Pore opening was shown to require the cross-linking of Cys160 and Cys257 (rat) or Cys256 (bovine) in ANT [70]. This type of cross-linking only occurs if one of the SH groups is oxidized to SOH. Intriguingly, blocking Cys160 with adenine nucleotides actually prevents pore opening; most likely through prevention of disulfide bond formation [70]. Prolonged exposure to high (>100 µM) concentrations of H2O2 can decrease mitochondrial ATP production which could lead to a decreased amount of adenine nucleotide binding on ANT which would then exposed Cys160 for oxidation and subsequent disulfide bond formation. It is also important to point out that mitochondrial solute anion carriers share a high degree of homology including a Cys residue (Cys256) in the last loop region that contacts the matrix. It was recently reported that ANT can also be S-glutathionylated and maintains an S-glutathionylated state in mitochondria from primary astrocytes and rat cortex [147]. Following oxidative stress, ANT is deglutathionylated which leads to MPT and induction of cell death [147]. Thus, it is possible that S-glutathionylation of ANT may prevent cysteine oxidation and cross-linking of Cys160 and Cys256/257. However, several key aspects surrounding ANT, cysteine oxidation, and pore opening need to be resolved such as (1) S-glutathionylation can also potentiate Cys cross-linking and (2) does ANT actually form part of MPT. Indeed, the actual composition of MPTP is still enthusiastically debated and remains a subject of heavy scrutiny. It has been shown that ANT and VDAC are not required for pore formation. Various reports however have shown that Pi carrier may be a key part of the MPTP [148] but this has also been refuted [149]. It was also recently reported that MPTP is actually composed of ATP synthase dimers [150]. Since ATP synthase is amenable to cysteine oxidation when ROS is high it would be intriguing to determine if the oxidation of cysteines on ATP synthase is required to potentiate dimer formation.
Cyp-D has also been found to be modulated by S-oxidation reactions [151]. In this case, S-glutathionylation prevents Cyp-D binding to ANT which blocks MPT [151]. This modification was found to occur on Cys203 [110]. It is intriguing that S-glutathionylation of Cyp-D actually prevents MPTP. As mentioned above the actual composition of the MPTP is still enthusiastically debated however; Cyp-D has so far been the only component of MPTP found to be indispensable for pore opening. Cyp-D knockout or inhibition with cyclosporine A prevents MPTP and apoptosis [152], [153]. Since S-glutathionylation of Cyp-D prevents pore opening it is possible that blocking Cys residues on Cyp-D may be a protective response, e.g. when ROS production is transiently highly S-glutathionylation of Cyp-D prevents pore opening. Indeed, a transient burst of H2O2 production by mitochondria also S-glutathionylates Odh and Complex I which may effectively limit mitochondrial ROS production (Fig. 4). In conjunction with this, S-glutathionylation of Cyp-D may be required to prevent pore opening while mitochondria adapt to the transient increase in mitochondrial ROS production [110]. The involvement of ANT in pore opening is more contentious since some studies have shown that knockout of ANT does not diminish or prevent MPT [154]. However, it is possible that other proposed components of MPTP may be required to sense surrounding changes in redox and participate in pore formation following cysteine oxidation.
Deregulation of mitochondrial thiol reactions in disease
Cardiovascular diseases
An increasing number of studies have found that mitochondrial thiol oxidation is involved in cardiac arrest and stroke. Cessation of blood flow to a portion of the heart or brain results in localized hypoxia and often injury is observed during the reperfusion of oxygenated blood (ischemia/reperfusion injury or IRI). Injury mediated through the reperfusion stage is a result of the generation of ROS and RNS, although the sources of these are currently debated [155], [156], [157]. A key feature of IRI is mitochondrial energetic dysfunction [158]. ROS and RNS, generated during IRI, have been shown to damage components in Complexes I, III, IV and V of the respiratory chain, as well as many Kreb Cycle enzymes [159], [160]. Short ischemic periods precondition tissues to hypoxic exposure and provide cardio- and neuronal-protective effects (ischemic preconditioning). These effects are mediated through the expression of protective genes and proteins that remain in the cell long after the cessation of ischemic stress. Thiol modifications, particularly of Complex I subunits, have been identified during IRI and results in further ROS generation by this Complex [158]. It was also found that cysteine 90 of the 70 kDa FAD-binding protein of mitochondrial succinate ubiquinone reductase, or Complex II, was glutathionylated and that this modification (as well as electron transfer activity of the respiratory chain itself) was markedly decreased in post-ischemic heart during IRI [123]. A complete assessment of mitochondrial thiol-containing proteins that become oxidized during IRI in Langendorff perfused mouse heart has recently been carried out using mass spectrometry with isotope-coded affinity tags (ICAT) [161]. S-glutathionylation of Complex I increases when GSSG levels are high which subsequently decreases the activity of this respiratory complex [10]. In addition, a recent report has also shown that high GSSG induces S-glutathionylation and deactivation of CAC [124]. Since IRI also increases GSSG it would be important to determine if IRI also leads to the aberrant S-glutathionylation and deactivation of Complex I and CAC.
Enhancement of protein S-nitrosylation in the myocardium is marked in the post-ischemic heart [162], [163], [164]. RNS have been found to have both damaging and protective effects on myocardial tissues. Peroxynitrite-mediated tyrosine S-nitrosylation of the 70 kDa FAD-binding protein of Complex II occurs in the post-ischemic myocardium [165]. S-nitrosylation of Tyr56 and Tyr142 occurred while Cys267, Cys476, and Cys537 were involved in peroxynitrite-mediated S-sulfonation. Peroxynitrite-mediated disulfide formation was also found to occur between Cys306–Cys312, Cys439–Cys444, and Cys288–Cys575.
In other reports, a wide variety of nitric oxide donors and S-nitrosylating agents have been found to protect the ischemic myocardium from infarction. In this respect, S-nitrosylation of mitochondrial proteins during ischemia-reperfusion has cardioprotective effects. S-nitrosylation of Cys39 on the NADH-ubiquinone oxidoreductase chain 3 (ND3) subunit of Complex I occurs only after ischemia [108]. Reversible D-nitrosylation of this protein slows the reactivation of mitochondria during the crucial first minutes of reperfusion of heart tissue, thereby decreasing ROS production. Likewise, S-nitrosylation of Cys137 of the 75 kDa Complex I subunit was found to occur [166]. The implications of this are that ROS will decrease upon reperfusion. It has been previously shown that inhibition of Complex IV by NO can cause the back-up of electrons along the electron transport chain and increase ROS generation at Complex III [167], [168]. Since Complex I is the entry point for electrons into the chain, reversible inhibition of Complex I by S-nitrosylation would lower electron flux into the chain and thus lower ROS generation from the chain itself. Although inhibition of Complex I may increase generation of ROS from the complex itself [130], ROS generated from Complex I has been found to be much less than that generated from Complex III [169].
Mitochondrial thiol modification may also alter the timing, or synchrony, of contractions in the heart. Large differences in the timing of contractions can reduce cardiac output and efficiency and result in dyssynchronous heart failure (DHF). Mixed disulfide bond formation of mitochondrial F1FO-ATP synthase subunits have been found to be involved in DHF [133]. Two cysteine disulfide bond oxidations were identified to be involved in DHF. The first was a disulfide linkage between cysteine Cys294 of the α-subunit and Cys103 of the γ-subunit. The second was an oxidation of Cys294 to sulfenic acid. In addition, Cys294 of the α-subunit can also be S-glutathionylated and S-nitrosylated [170]. Either thiol modification led to a significant decrease in ATP synthase activity, resulting in the development of DHF. Glutathionylation of Cys294 of the α-subunit also has the potential to block nucleotide binding and thus decrease activity [171].
Metabolic syndrome, insulin resistance, obesity and diabetes
Diabetes is a major health risk, with the global population of diabetics estimated to reach 300 million by the year 2025. Mortality risk is complicated by the association of diabetes mellitus and myocardial infarction. Diabetes is associated with hyperglycemia and this has been shown to have a direct effect on mitochondrial function [172]. The mitochondria from diabetic hearts have been found to be sensitized to mitochondrial permeability transition pore opening, which leads to a propensity for cardiac injury in diabetic patients and in animal models [173], [174]. Hyperglycemia leads to high glucose-dependent production of electron transfer donors (NADH and FADH2) and thereby increases electron flux through the mitochondrial electron transport chain. Consequently there is an increase in the ATP/ADP ratio and hyperpolarization of the mitochondrial membrane potential results. This high electrochemical potential leads to a partial inhibition of electron transport at Complex III, leading to an accumulation of electrons on Q. This, in turn, drives the partial reduction of O2 to generate O2−. The generation of O2− from the highly reduced Q, and from Complex III in general, is believed to be the primary source of mitochondrial dysfunction during diabetes. Two recent reviews have addressed the role of redox signaling and S-glutathionylation in glucose-stimulated insulin release and how deregulated redox signaling could potentially be related to development of diabetes [175]. However, very few studies have actually addressed the role of redox signaling in modulation mitochondrial function in pancreatic β-cells. A recent report has shown that reversible S-glutathionylation is required to control proton leaks through UCP2 in mitochondria which plays a role in regulating glucose-stimulated insulin release in Min6 cells and pancreatic islets [16].
Nutrient excess leads to excessive levels of energy substrates utilized by the mitochondria with consequential effects on lipid and glucose metabolism. This, in turn, leads to mitochondrial dysfunction and to the development of obesity and obesity-related pathologies such as non-alcoholic fatty acid liver disease (NAFLD) [176]. Few studies have addressed the relationship between deregulated redox signaling and the development of metabolic disease. A recent report has shown that the S-glutathionylated proteome in hepatic and visceral adipose tissue changes drastically upon the onset of obesity in mice [177]. In addition, knock-out of Grx2 leads to a significant decrease in total adiposity in C57Bl6J mice [86]. Intriguingly the decrease in adiposity in Grx2−/− mice correlates strongly with an increase in mitochondrial respiration, nutrient oxidation, and proton leaks through UCP3 in skeletal muscle [86]. It has been suggested that increasing leaks through UCP3 may have a weight loss effect since an increase in leaks would inevitably increase respiration and nutrient oxidation [178]. Thus, the weight loss effect observed in Grx2−/− mice may be due to the maintenance of UCP3 in a deglutathionylated and active state which ultimately leads to increases proton leaks and nutrient metabolism. Previously, it was found that mitochondria from obese rats show an increased capacity for oxidizing succinate, likely due to increased activity of succinate dehydrogenase, the mitochondrial enzyme that oxidizes succinate and reduces Q [179]. Oxidized succinate, or fumarate, can form a Michael addition with thiol groups in proteins [180] and this post-translational modification of proteins has been found to inactivate glyceraldehyde-3-phosphate dehydrogenase. Increased succination of protein thiols in observed during obesity and diabetes [181]. Succination of protein thiols in adipocytes has been found to occur during mitochondrial stress in adipocytes during adipogenesis [182]. S-(2-succinyl)cysteine (2SC), the product of chemical modification of proteins by the Krebs cycle intermediate, fumarate, was found to be significantly increased during maturation of 3T3-L1 fibroblasts to adipocytes. Treatment of 3T3 cells with high glucose (30 mM) caused a significant increase in cellular ATP/ADP ratio, NADH/NAD+ ratio and mitochondrial membrane potential [183]. It also increased total cellular protein succination, but mitochondrial proteins alone were not examined in this study. Recently, mitochondrial Acn was found to be inhibited by succination in fumarate hydratase deficiency occurring in hereditary leiomyomatosis and renal cell cancer [184]. It remains to be determined whether a similar inhibition occurs during diabetes and obesity.
Neurodegenerative diseases
The major neurodegenerative diseases, including Alzheimer's (AD), Friedreich's (FD), amyotrophic lateral sclerosis (ALS or Lou Gehrig's), Huntington's (HD), Parkinson's (PD) and multiple sclerosis (MS) diseases, are characterized by a progressive loss of neurons and glutathionylation of proteins have been shown to have a role in this process [185]. All are characterized by either misfolded (e.g. tau), misprocessed (eg. amyloid precursor protein) or mutated (eg. α-synuclein) proteins. These proteins (or peptides from proteins) form insoluble aggregates and mitochondrial dysfunction, with corresponding generation of ROS and RNS, has been implicated in the process. The role of mitochondrial protein thiols in the development of neurodegenerative diseases is not fully understood and is an area that requires active investigation.
The levels of GSH have been reported to be reduced in AD [186]. Likewise, similar changes were observed in PD where GSH was reduced and GSSG was increased [187], [188]. PD is characterized by early GSH depletion in dopaminergic cells in the substantia nigra (SN). The reasons for depletion of GSH in neurodegenerative diseases are not always clear. In PD, there is no corresponding increase in GSSG [189], so GSH depletion cannot be explained by oxidative stress alone. In this study, there was no increase in glutathione synthase activity in PD, but a large increase in γ-glutamyltranspeptidase, an enzyme that catalyzes the transfer of the γ-glutamyl moiety from GSH or a glutathione conjugate onto an acceptor molecule, was observed. As mentioned previously, mitochondrial GSH is a substrate for mitochondrial Grx. Grx2 has been shown to be involved in mitochondrial iron–sulfur (Fe–S) center biogenesis [190]. Increased cellular iron levels are a hallmark of PD, and decreased levels of GSH in PD results in a depletion of substrate for Grx2. The result is decreased iron incorporation into mitochondrial Complex I and aconitase (Acn) [190]. It also results in significant increases in iron-regulatory protein (IRP) binding, transferrin receptor, decreased ferritin, and subsequent increases in mitochondrial iron levels. Decreases in GSH, and increased levels of GSSG and nitrosothiols, were also observed in MS patients [191].
Glutathionylation of mitochondrial proteins has been shown to be involved in neurodegenerative disorders. Glutaredoxin (Grx) is responsible for the deglutathionylation of proteins and has been shown to be decreased in post-mortem AD brains [192]. Loss of Grx would mean loss of the cell's ability to deglutathionylate proteins as well as its ability to bind to apoptosis signal-regulating kinase 1 (ASK1) (also known as mitogen-activated protein kinase kinase kinase 5 (MAP3K5)). In a mouse model of PD (acute treatment with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) which targets mitochondrial Complex I), Grx was observed to increase in brains [193]. When Grx1 was knocked down by antisense oligonucleotides, inactivation of Complex I by MPTP was not observed. It is also interesting to note that glutathionylation of the tau protein causes it to polymerize and form the neurofibrillary tangles [194] found in AD.
S-nitrosylation of mitochondrial proteins has been related to the development of neurodegenerative disorders. S-nitrosylation of mitochondrial Complex I has previously been implicated in the development of PD [195], [196]. Protein disulfide isomerase (PDI), a thioredoxin-like domain protein closely related to Grx and Trx, has also been implicated in PD [197]. PDI can become S-nitrosylated (forming SNO-PDI) and this modification disrupts both its chaperonin and neuroprotective functions in PD [198] and in AD [199]. The S-nitrosylation of dynamin-related protein 1 (Drp1) may be related to the pathological fragmentation of mitochondria and consequent loss of synapses in neurodegenerative disorders [197]. Drp1 controls mitochondrial fission, and is responsible for proper neuronal development. Drp1 knockout mice exhibit abnormal brain development and die around embryonic day 12. Mitochondrial fusion/fission is essential for maintaining mitochondrial integrity and disruption of these processes leads to mitochondrial dysfunction and the development of PD and AD [200], [201]. NO, produced in an inflammatory response to the Aβ peptide, nitrosylates a key cysteine on Drp1 [199], resulting in mitochondrial fission, synaptic loss, and neuronal damage. Prevention of S-nitrosylation of Drp1 by cysteine mutation abrogates these neurotoxic events. However, the role of Drp1 S-nitrosylation in its GTPase activity, mitochondrial fission and neuronal injury in AD remains debated as other reports have found that S-nitrosylation of Cys644 of Drp1 does not affect its GTPase activity or cause dimerization of the protein [202].
Aging
All organisms undergo aging and mitochondrial protein thiol group content may play a role in this process [203], [204]. Aging has been associated with more oxidized forms of biomacromolecules, particularly in the mitochondria where the majority of ROS/RNS are produced within the cell [205], [206], [207]. Previously, it has been shown that mitochondrial Complex I activity decreases with age and that protection of thiol groups with N-acetylcysteine prevented such decrease [208]. In an interesting study done on one short-lived and one long-lived strain (48% longer) of mouse, mitochondrial thiols (glutathione, reactive protein thiols) were found to be lower in short-lived mice (with corresponding higher levels of GSSG) than in long-lived mice [209]. Peroxiredoxin III, present in the mitochondria, was found to be present in it more oxidized, inactive form in aged rats [210]. Cysteine 109 in the catalytic site was found to be more sulfonated in aged animals. Not all modifications, associated with age, are viewed to be detrimental, with some being beneficial to the aging cell [211].
Age-related declines in GSH result in more oxidizing conditions within plasma[212]. It also means that there may be less available for glutathionylation within mitochondria, and thus proteins would be less glutathionylated during oxidizing conditions. Glutathionylation has been found to have both inactivating effects on its function [213], [214], but may also protect the modified protein (due to the reversibility of glutathionylation) from irreversible and permanent damage [68].
The S-nitrosylation of proteins with age has been well studied, but a lack of a focus has been made on the mitochondria. S-nitrosylation of general cellular proteins linked to neurodegenerative diseases [215], cardiovascular diseases [216], obesity and diabetes [217] have all been related to aging. Likewise, general glutathionylation of cellular proteins has been associated with age-related diseases [218]. Succination and disease has not been studied in great detail, but mitochondrial sirtuin 5 has been previously shown to desuccinylate mitochondrial proteins [219]. Such modifications could be elucidated by performing proteomic work on pure isolated mitochondrial preparations. The challenge is that current protocols for such preparations do not yield fully pure mitochondria due to the nature in the cell (mitochondria form a syncytium throughout the cell and not a distinct organelle).
The regulation of mitochondrial abundance and function by thiol modification in disease is an unexplored realm of research that is open for investigation. Nitrosylation, sulfonylation, glutathionylation and succination of protein thiols represent rapid posttranslational “switches” controlling the function of mitochondrial proteins and providing a “fast” response to environmental stimuli. Discovery of additional thiol modification targets in the mitochondria should help us attain a more complete understanding of the molecular processes by which these modifications occur. Further elucidation of the mechanism by which these controls alter function opens up new avenues for the development of thiol-based therapeutics and antioxidants for the treatment of mitochondrial dysfunction in aging and disease.
Conclusions and perspectives
Mitochondria fulfill a multitude of essential cellular functions which are ultimately dependent on the production of ATP and ROS. ATP is required to drive most cellular processes while ROS can serve as an important signaling molecule. Genesis of either molecule is dependent on the transfer of electrons from nutrients to O2 in the ETC. Mitochondrial dysfunction is related to wide range of diseases and some cases is actually the cause of disease. In the present review we have outlined the importance of redox signals in the modulation of mitochondrial function and how deregulation of the signals can lead to development of various disease states. Key enzymes and proteins involved in nutrient oxidation, metabolism, OXPHOS, solute transport, ROS production, ROS quenching, mitochondrial morphology, and even apoptosis are modulated by S-oxidation, S-glutathionylation, and S-nitrosylation. We illustrated the complexity of these reactions since ROS, GSH, and NO converge on protein cysteine thiols resulting in oxidation and change in protein function. We also showed that these modifications occur under to scenarios (1) controlled reactions which are site specific, rapid, and readily reversed by enzymes and (2) uncontrolled reactions which are nonspecific and associated with oxidative stress and deregulated redox signaling. We also discussed some emerging concepts in terms of mitochondrial medicine and how the concept of mitochondria-targeted pharmaceuticals can potentially be utilized to restore deregulated redox signaling through replenishment of mitochondrial GSH and/or restoring mitochondrial ROS levels. Notably, we overlooked some other important reactions such as formation of persulfides (Cys–S–S−) which are the result of H2S signaling. Indeed, mitochondria are not only sensitive to H2S signaling but also important sites for H2S metabolism [220], [221]. Much remains to be investigated in terms of redox signaling and the modulation of mitochondrial function. However, based on present knowledge it is clear that redox signaling plays a central role in modulating mitochondrial biology and physiology.
Acknowledgements
This work was supported by a Discovery Grant from the Natural Sciences and Engineering Research Council (NSERC) of Canada to WGW.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Brown G.C., Borutaite V. There is no evidence that mitochondria are the main source of reactive oxygen species in mammalian cells. Mitochondrion. 2012;12:1–4. doi: 10.1016/j.mito.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 2.Fernie A.R., Carrari F., Sweetlove L.J. Respiratory metabolism: glycolysis, the TCA cycle and mitochondrial electron transport. Curr. Opin. Plant Biol. 2004;7:254–261. doi: 10.1016/j.pbi.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 3.Baradaran R., Berrisford J.M., Minhas G.S., Sazanov L.A. Crystal structure of the entire respiratory complex I. Nature. 2013;494:443–448. doi: 10.1038/nature11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Quinlan C.L., Orr A.L., Perevoshchikova I.V., Treberg J.R., Ackrell B.A., Brand M.D. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J. Biol. Chem. 2012;287:27255–27264. doi: 10.1074/jbc.M112.374629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khutornenko A.A., Roudko V.V., Chernyak B.V., Vartapetian A.B., Chumakov P.M., Evstafieva A.G. Pyrimidine biosynthesis links mitochondrial respiration to the p53 pathway. Proc. Natl. Acad. Sci. USA. 2010;107:12828–12833. doi: 10.1073/pnas.0910885107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang J., Frerman F.E., Kim J.J. Structure of electron transfer flavoprotein-ubiquinone oxidoreductase and electron transfer to the mitochondrial ubiquinone pool. Proc. Natl. Acad. Sci. USA. 2006;103:16212–16217. doi: 10.1073/pnas.0604567103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Orr A.L., Quinlan C.L., Perevoshchikova I.V., Brand M.D. A refined analysis of superoxide production by mitochondrial sn-glycerol 3-phosphate dehydrogenase. J. Biol. Chem. 2012;287:42921–42935. doi: 10.1074/jbc.M112.397828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brand M.D., Nicholls D.G. Assessing mitochondrial dysfunction in cells. Biochem. J. 2011;435:297–312. doi: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mailloux R.J., Harper M.E. Uncoupling proteins and the control of mitochondrial reactive oxygen species production. Free Radic. Biol. Med. 2011;51:1106–1115. doi: 10.1016/j.freeradbiomed.2011.06.022. [DOI] [PubMed] [Google Scholar]
- 10.Mailloux R.J., McBride S.L., Harper M.E. Unearthing the secrets of mitochondrial ROS and glutathione in bioenergetics. Trends Biochem. Sci. 2013;38:592–632. doi: 10.1016/j.tibs.2013.09.001. [DOI] [PubMed] [Google Scholar]
- 11.Starkov A.A., Fiskum G., Chinopoulos C., Lorenzo B.J., Browne S.E., Patel M.S., Beal M.F. Mitochondrial alpha-ketoglutarate dehydrogenase complex generates reactive oxygen species. J. Neurosci.: Off. J. Soc. Neurosci. 2004;24:7779–7788. doi: 10.1523/JNEUROSCI.1899-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Quinlan C.L., Perevoshchikova I.V., Hey-Mogensen M., Orr A.L., Brand M.D. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013;1:304–312. doi: 10.1016/j.redox.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guzy R.D., Hoyos B., Robin E., Chen H., Liu L., Mansfield K.D., Simon M.C., Hammerling U., Schumacker P.T. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005;1:401–408. doi: 10.1016/j.cmet.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 14.Tormos K.V., Anso E., Hamanaka R.B., Eisenbart J., Joseph J., Kalyanaraman B., Chandel N.S. Mitochondrial complex III ROS regulate adipocyte differentiation. Cell Metab. 2011;14:537–544. doi: 10.1016/j.cmet.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Loh K., Deng H., Fukushima A., Cai X., Boivin B., Galic S., Bruce C., Shields B.J., Skiba B., Ooms L.M., Stepto N., Wu B., Mitchell C.A., Tonks N.K., Watt M.J., Febbraio M.A., Crack P.J., Andrikopoulos S., Tiganis T. Reactive oxygen species enhance insulin sensitivity. Cell Metab. 2009;10:260–272. doi: 10.1016/j.cmet.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mailloux R.J., Fu A., Robson-Doucette C., Allister E.M., Wheeler M.B., Screaton R., Harper M.E. Glutathionylation state of uncoupling protein-2 and the control of glucose-stimulated insulin secretion. J. Biol. Chem. 2012;287:39673–39685. doi: 10.1074/jbc.M112.393538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hamanaka R.B., Chandel N.S. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 2010;35:505–513. doi: 10.1016/j.tibs.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lillig C.H., Berndt C., Vergnolle O., Lonn M.E., Hudemann C., Bill E., Holmgren A. Characterization of human glutaredoxin 2 as iron-sulfur protein: a possible role as redox sensor. Proc. Natl. Acad. Sci. USA. 2005;102:8168–8173. doi: 10.1073/pnas.0500735102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.James A.M., Collins Y., Logan A., Murphy M.P. Mitochondrial oxidative stress and the metabolic syndrome. Trends Endocrinol. Metab. 2012;23:429–434. doi: 10.1016/j.tem.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 20.Wang W., Fang H., Groom L., Cheng A., Zhang W., Liu J., Wang X., Li K., Han P., Zheng M., Yin J., Mattson M.P., Kao J.P., Lakatta E.G., Sheu S.S., Ouyang K., Chen J., Dirksen R.T., Cheng H. Superoxide flashes in single mitochondria. Cell. 2008;134:279–290. doi: 10.1016/j.cell.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schwarzlander M., Logan D.C., Fricker M.D., Sweetlove L.J. The circularly permuted yellow fluorescent protein cpYFP that has been used as a superoxide probe is highly responsive to pH but not superoxide in mitochondria: implications for the existence of superoxide ‘flashes’. Biochem. J. 2011;437:381–387. doi: 10.1042/BJ20110883. [DOI] [PubMed] [Google Scholar]
- 22.Schwarzlander M., Murphy M.P., Duchen M.R., Logan D.C., Fricker M.D., Halestrap A.P., Muller F.L., Rizzuto R., Dick T.P., Meyer A.J., Sweetlove L.J. Mitochondrial ‘flashes’: a radical concept repHined. Trends Cell Biol. 2012;22:503–508. doi: 10.1016/j.tcb.2012.07.007. [DOI] [PubMed] [Google Scholar]
- 23.Murphy M.P. Mitochondrial thiols in antioxidant protection and redox signaling: distinct roles for glutathionylation and other thiol modifications. Antioxid. Redox Signal. 2012;16:476–495. doi: 10.1089/ars.2011.4289. [DOI] [PubMed] [Google Scholar]
- 24.Mailloux R.J., Lemire J., Appanna V.D. Hepatic response to aluminum toxicity: dyslipidemia and liver diseases. Exp. Cell Res. 2011;317:2231–2238. doi: 10.1016/j.yexcr.2011.07.009. [DOI] [PubMed] [Google Scholar]
- 25.Pacher P., Schulz R., Liaudet L., Szabo C. Nitrosative stress and pharmacological modulation of heart failure. Trends Pharmacol. Sci. 2005;26:302–310. doi: 10.1016/j.tips.2005.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lane N. Evolution. The costs of breathing. Science. 2011;334:184–185. doi: 10.1126/science.1214012. [DOI] [PubMed] [Google Scholar]
- 27.Korshunov S.S., Skulachev V.P., Starkov A.A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997;416:15–18. doi: 10.1016/s0014-5793(97)01159-9. [DOI] [PubMed] [Google Scholar]
- 28.Mailloux R.J., Dumouchel T., Aguer C., Dekemp R., Beanlands R., Harper M.E. Hexokinase II acts through UCP3 to suppress mitochondrial reactive oxygen species production and maintain aerobic respiration. Biochem. J. 2011;437:301–311. doi: 10.1042/BJ20110571. [DOI] [PubMed] [Google Scholar]
- 29.Lenaz G. The mitochondrial production of reactive oxygen species: mechanisms and implications in human pathology. IUBMB Life. 2001;52:159–164. doi: 10.1080/15216540152845957. [DOI] [PubMed] [Google Scholar]
- 30.Pouvreau S. Superoxide flashes in mouse skeletal muscle are produced by discrete arrays of active mitochondria operating coherently. PLoS One. 2010;5:e13035. doi: 10.1371/journal.pone.0013035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee I., Bender E., Kadenbach B. Control of mitochondrial membrane potential and ROS formation by reversible phosphorylation of cytochrome c oxidase. Mol. Cell. Biochem. 2002;234–235:63–70. [PubMed] [Google Scholar]
- 32.Hue L., Taegtmeyer H. The Randle cycle revisited: a new head for an old hat. Am. J. Physiol. Endocrinol. Metab. 2009;297:E578–E591. doi: 10.1152/ajpendo.00093.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scherz-Shouval R., Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem. Sci. 2011;36:30–38. doi: 10.1016/j.tibs.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 34.St-Pierre J., Drori S., Uldry M., Silvaggi J.M., Rhee J., Jager S., Handschin C., Zheng K., Lin J., Yang W., Simon D.K., Bachoo R., Spiegelman B.M. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 35.Maranzana E., Barbero G., Falasca A.I., Lenaz G., Genova M.L. Mitochondrial respiratory supercomplex association limits production of reactive oxygen species from Complex I. Antioxid. Redox Signal. 2013;19:1469–1480. doi: 10.1089/ars.2012.4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hurd T.R., Costa N.J., Dahm C.C., Beer S.M., Brown S.E., Filipovska A., Murphy M.P. Glutathionylation of mitochondrial proteins. Antioxid. Redox Signal. 2005;7:999–1010. doi: 10.1089/ars.2005.7.999. [DOI] [PubMed] [Google Scholar]
- 37.Piantadosi C.A. Regulation of mitochondrial processes by protein S-nitrosylation. Biochim. Biophys. Acta. 2012;1820:712–721. doi: 10.1016/j.bbagen.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murphy M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mailloux R.J., Harper M.E. Mitochondrial proticity and ROS signaling: lessons from the uncoupling proteins. Trends Endocrinol. Metab. 2012;23:451–458. doi: 10.1016/j.tem.2012.04.004. [DOI] [PubMed] [Google Scholar]
- 40.Sena L.A., Chandel N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell. 2012;48:158–167. doi: 10.1016/j.molcel.2012.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang B., Chen S.C., Wang D.L. Shear flow increases S-nitrosylation of proteins in endothelial cells. Cardiovasc. Res. 2009;83:536–546. doi: 10.1093/cvr/cvp154. [DOI] [PubMed] [Google Scholar]
- 42.Lindahl M., Mata-Cabana A., Kieselbach T. The disulfide proteome and other reactive cysteine proteomes: analysis and functional significance. Antioxid. Redox Signal. 2011;14:2581–2642. doi: 10.1089/ars.2010.3551. [DOI] [PubMed] [Google Scholar]
- 43.Grek C., Zhang J., Manevich Y., Townsend D.M., Tew K.D. Causes and consequences of cysteine S-glutathionylation. J. Biol. Chem. 2013;72:2383–2393. doi: 10.1074/jbc.R113.461368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chepelev N.L., Willmore W.G. Regulation of iron pathways in response to hypoxia. Free Radic. Biol. Med. 2011;50:645–666. doi: 10.1016/j.freeradbiomed.2010.12.023. [DOI] [PubMed] [Google Scholar]
- 45.Stewart M.D., Igumenova T.I. Reactive cysteine in the structural Zn(2+) site of the C1B domain from PKCalpha. Biochemistry. 2012;51:7263–7277. doi: 10.1021/bi300750w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lill R. Function and biogenesis of iron–sulphur proteins. Nature. 2009;460:831–838. doi: 10.1038/nature08301. [DOI] [PubMed] [Google Scholar]
- 47.Velan B., Grosfeld H., Kronman C., Leitner M., Gozes Y., Lazar A., Flashner Y., Marcus D., Cohen S., Shafferman A. The effect of elimination of intersubunit disulfide bonds on the activity, assembly, and secretion of recombinant human acetylcholinesterase. Expression of acetylcholinesterase Cys-580-Ala mutant. J. Biol. Chem. 1991;266:23977–23984. [PubMed] [Google Scholar]
- 48.Lo Conte M., Carroll K.S. The redox biochemistry of protein sulfenylation and sulfinylation. J. Biol. Chem. 2013;288:26480–26488. doi: 10.1074/jbc.R113.467738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Winterbourn C.C., Hampton M.B. Thiol chemistry and specificity in redox signaling. Free Radic. Biol. Med. 2008;45:549–561. doi: 10.1016/j.freeradbiomed.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 50.Smith R.E., MacQuarrie R. The role of cysteine residues in the catalytic activity of glycerol-3-phosphate dehydrogenase. Biochim. Biophys. Acta. 1979;567:269–277. doi: 10.1016/0005-2744(79)90112-8. [DOI] [PubMed] [Google Scholar]
- 51.Kortemme T., Creighton T.E. Ionisation of cysteine residues at the termini of model alpha-helical peptides. Relevance to unusual thiol pKa values in proteins of the thioredoxin family. J. Mol. Biol. 1995;253:799–812. doi: 10.1006/jmbi.1995.0592. [DOI] [PubMed] [Google Scholar]
- 52.Gallogly M.M., Starke D.W., Mieyal J.J. Mechanistic and kinetic details of catalysis of thiol-disulfide exchange by glutaredoxins and potential mechanisms of regulation. Antioxid. Redox Signal. 2009;11:1059–1081. doi: 10.1089/ars.2008.2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stroher E., Millar A.H. The biological roles of glutaredoxins. Biochem. J. 2012;446:333–348. doi: 10.1042/BJ20112131. [DOI] [PubMed] [Google Scholar]
- 54.Cox A.G., Winterbourn C.C., Hampton M.B. Mitochondrial peroxiredoxin involvement in antioxidant defence and redox signalling. Biochem. J. 2010;425:313–325. doi: 10.1042/BJ20091541. [DOI] [PubMed] [Google Scholar]
- 55.Lo Conte M., Carroll K.S. The redox biochemistry of protein sulfenylation and sulfinylation. J. Biol. Chem. 2013;288:26480–26488. doi: 10.1074/jbc.R113.467738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Paulsen C.E., Carroll K.S. Cysteine-mediated redox signaling: chemistry, biology, and tools for discovery. Chem. Rev. 2013;113:4633–4679. doi: 10.1021/cr300163e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Go Y.M., Jones D.P. Redox compartmentalization in eukaryotic cells. Biochim. Biophys. Acta. 2008;1780:1273–1290. doi: 10.1016/j.bbagen.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kemp M., Go Y.M., Jones D.P. Nonequilibrium thermodynamics of thiol/disulfide redox systems: a perspective on redox systems biology. Free Radic. Biol. Med. 2008;44:921–937. doi: 10.1016/j.freeradbiomed.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Requejo R., Hurd T.R., Costa N.J., Murphy M.P. Cysteine residues exposed on protein surfaces are the dominant intramitochondrial thiol and may protect against oxidative damage. FEBS J. 2010;277:1465–1480. doi: 10.1111/j.1742-4658.2010.07576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brown G.C. Nitric oxide and mitochondria. Front. Biosci.: J. Virtual Lib. 2007;12:1024–1033. doi: 10.2741/2122. [DOI] [PubMed] [Google Scholar]
- 61.Roos G., Messens J. Protein sulfenic acid formation: from cellular damage to redox regulation. Free Radic. Biol. Med. 2011;51:314–326. doi: 10.1016/j.freeradbiomed.2011.04.031. [DOI] [PubMed] [Google Scholar]
- 62.Peskin A.V., Low F.M., Paton L.N., Maghzal G.J., Hampton M.B., Winterbourn C.C. The high reactivity of peroxiredoxin 2 with H(2)O(2) is not reflected in its reaction with other oxidants and thiol reagents. J. Biol. Chem. 2007;282:11885–11892. doi: 10.1074/jbc.M700339200. [DOI] [PubMed] [Google Scholar]
- 63.Hall A., Parsonage D., Poole L.B., Karplus P.A. Structural evidence that peroxiredoxin catalytic power is based on transition-state stabilization. J. Mol. Biol. 2010;402:194–209. doi: 10.1016/j.jmb.2010.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Enami S., Hoffmann M.R., Colussi A.J. Simultaneous detection of cysteine sulfenate, sulfinate, and sulfonate during cysteine interfacial ozonolysis. J. Phys. Chem. B. 2009;113:9356–9358. doi: 10.1021/jp904316n. [DOI] [PubMed] [Google Scholar]
- 65.McGrath A.J., Garrett G.E., Valgimigli L., Pratt D.A. The redox chemistry of sulfenic acids. J. Am. Chem. Soc. 2010;132:16759–16761. doi: 10.1021/ja1083046. [DOI] [PubMed] [Google Scholar]
- 66.Hugo M., Turell L., Manta B., Botti H., Monteiro G., Netto L.E., Alvarez B., Radi R., Trujillo M. Thiol and sulfenic acid oxidation of AhpE, the one-cysteine peroxiredoxin from Mycobacterium tuberculosis: kinetics, acidity constants, and conformational dynamics. Biochemistry. 2009;48:9416–9426. doi: 10.1021/bi901221s. [DOI] [PubMed] [Google Scholar]
- 67.Tretter L., Adam-Vizi V. Inhibition of alpha-ketoglutarate dehydrogenase due to H2O2-induced oxidative stress in nerve terminals. Ann. N. Y. Acad. Sci. 1999;893:412–416. doi: 10.1111/j.1749-6632.1999.tb07867.x. [DOI] [PubMed] [Google Scholar]
- 68.Applegate M.A., Humphries K.M., Szweda L.I. Reversible inhibition of alpha-ketoglutarate dehydrogenase by hydrogen peroxide: glutathionylation and protection of lipoic acid. Biochemistry. 2008;47:473–478. doi: 10.1021/bi7017464. [DOI] [PubMed] [Google Scholar]
- 69.Hurd T.R., Requejo R., Filipovska A., Brown S., Prime T.A., Robinson A.J., Fearnley I.M., Murphy M.P. Complex I within oxidatively stressed bovine heart mitochondria is glutathionylated on Cys-531 and Cys-704 of the 75-kDa subunit: potential role of CYS residues in decreasing oxidative damage. J. Biol. Chem. 2008;283:24801–24815. doi: 10.1074/jbc.M803432200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McStay G.P., Clarke S.J., Halestrap A.P. Role of critical thiol groups on the matrix surface of the adenine nucleotide translocase in the mechanism of the mitochondrial permeability transition pore. Biochem. J. 2002;367:541–548. doi: 10.1042/BJ20011672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nelson K.J., Klomsiri C., Codreanu S.G., Soito L., Liebler D.C., Rogers L.C., Daniel L.W., Poole L.B. Use of dimedone-based chemical probes for sulfenic acid detection methods to visualize and identify labeled proteins. Methods Enzymol. 2010;473:95–115. doi: 10.1016/S0076-6879(10)73004-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hutson S.M., Poole L.B., Coles S., Conway M.E. Redox regulation and trapping sulfenic acid in the peroxide-sensitive human mitochondrial branched chain aminotransferase. Methods Mol. Biol. 2008;476:139–152. doi: 10.1007/978-1-59745-129-1_10. [DOI] [PubMed] [Google Scholar]
- 73.Cocheme H.M., Kelso G.F., James A.M., Ross M.F., Trnka J., Mahendiran T., Asin-Cayuela J., Blaikie F.H., Manas A.R., Porteous C.M., Adlam V.J., Smith R.A., Murphy M.P. Mitochondrial targeting of quinones: therapeutic implications. Mitochondrion. 2007;7(Suppl):S94–S102. doi: 10.1016/j.mito.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 74.Reily C., Mitchell T., Chacko B.K., Benavides G., Murphy M.P., Darley-Usmar V. Mitochondrially targeted compounds and their impact on cellular bioenergetics. Redox Biol. 2013;1:86–93. doi: 10.1016/j.redox.2012.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bindoli A., Fukuto J.M., Forman H.J. Thiol chemistry in peroxidase catalysis and redox signaling. Antioxid. Redox Signal. 2008;10:1549–1564. doi: 10.1089/ars.2008.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cho C.S., Lee S., Lee G.T., Woo H.A., Choi E.J., Rhee S.G. Irreversible inactivation of glutathione peroxidase 1 and reversible inactivation of peroxiredoxin II by H2O2 in red blood cells. Antioxid. Redox Signal. 2010;12:1235–1246. doi: 10.1089/ars.2009.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lowther W.T., Haynes A.C. Reduction of cysteine sulfinic acid in eukaryotic, typical 2-Cys peroxiredoxins by sulfiredoxin. Antioxid. Redox Signal. 2011;15:99–109. doi: 10.1089/ars.2010.3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hamann M., Zhang T., Hendrich S., Thomas J.A. Quantitation of protein sulfinic and sulfonic acid, irreversibly oxidized protein cysteine sites in cellular proteins. Methods Enzymol. 2002;348:146–156. doi: 10.1016/s0076-6879(02)48634-x. [DOI] [PubMed] [Google Scholar]
- 79.Chevallet M., Wagner E., Luche S., van Dorsselaer A., Leize-Wagner E., Rabilloud T. Regeneration of peroxiredoxins during recovery after oxidative stress: only some overoxidized peroxiredoxins can be reduced during recovery after oxidative stress. J. Biol. Chem. 2003;278:37146–37153. doi: 10.1074/jbc.M305161200. [DOI] [PubMed] [Google Scholar]
- 80.Canet-Aviles R.M., Wilson M.A., Miller D.W., Ahmad R., McLendon C., Bandyopadhyay S., Baptista M.J., Ringe D., Petsko G.A., Cookson M.R. The Parkinson's disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc. Natl. Acad. Sci. USA. 2004;101:9103–9108. doi: 10.1073/pnas.0402959101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Woo H.A., Yim S.H., Shin D.H., Kang D., Yu D.Y., Rhee S.G. Inactivation of peroxiredoxin I by phosphorylation allows localized H(2)O(2) accumulation for cell signaling. Cell. 2010;140:517–528. doi: 10.1016/j.cell.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 82.Noh Y.H., Baek J.Y., Jeong W., Rhee S.G., Chang T.S. Sulfiredoxin translocation into mitochondria plays a crucial role in reducing hyperoxidized peroxiredoxin III. J. Biol. Chem. 2009;284:8470–8477. doi: 10.1074/jbc.M808981200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Peskin A.V., Dickerhof N., Poynton R.A., Paton L.N., Pace P.E., Hampton M.B., Winterbourn C.C. Hyperoxidation of peroxiredoxins 2 and 3: rate constants for the reactions of the sulfenic acid of the peroxidatic cysteine. J. Biol. Chem. 2013;288:14170–14177. doi: 10.1074/jbc.M113.460881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lo Conte M., Carroll K.S. Chemoselective ligation of sulfinic acids with aryl-nitroso compounds. Angew. Chem. Int. Ed. Engl. 2012;51:6502–6505. doi: 10.1002/anie.201201812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Beer S.M., Taylor E.R., Brown S.E., Dahm C.C., Costa N.J., Runswick M.J., Murphy M.P. Glutaredoxin 2 catalyzes the reversible oxidation and glutathionylation of mitochondrial membrane thiol proteins: implications for mitochondrial redox regulation and antioxidant DEFENSE. J. Biol. Chem. 2004;279:47939–47951. doi: 10.1074/jbc.M408011200. [DOI] [PubMed] [Google Scholar]
- 86.Mailloux R.J., Xuan J.Y., Beauchamp B., Jui L., Lou M., Harper M.E. Glutaredoxin-2 is required to control proton leak through uncoupling protein-3. J. Biol. Chem. 2013;288:8365–8379. doi: 10.1074/jbc.M112.442905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mailloux R.J., Seifert E.L., Bouillaud F., Aguer C., Collins S., Harper M.E. Glutathionylation acts as a control switch for uncoupling proteins UCP2 and UCP3. J. Biol. Chem. 2011;286:21865–21875. doi: 10.1074/jbc.M111.240242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gutscher M., Pauleau A.L., Marty L., Brach T., Wabnitz G.H., Samstag Y., Meyer A.J., Dick T.P. Real-time imaging of the intracellular glutathione redox potential. Nat. Methods. 2008;5:553–559. doi: 10.1038/nmeth.1212. [DOI] [PubMed] [Google Scholar]
- 89.Dalle-Donne I., Rossi R., Giustarini D., Colombo R., Milzani A. S-glutathionylation in protein redox regulation. Free Radic. Biol. Med. 2007;43:883–898. doi: 10.1016/j.freeradbiomed.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 90.Gallogly M.M., Mieyal J.J. Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Curr. Opin. Pharmacol. 2007;7:381–391. doi: 10.1016/j.coph.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 91.Kang P.T., Zhang L., Chen C.L., Chen J., Green K.B., Chen Y.R. Protein thiyl radical mediates S-glutathionylation of complex I. Free Radic. Biol. Med. 2012;53:962–973. doi: 10.1016/j.freeradbiomed.2012.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ziegler D.M. Role of reversible oxidation–reduction of enzyme thiols-disulfides in metabolic regulation. Annu. Rev. Biochem. 1985;54:305–329. doi: 10.1146/annurev.bi.54.070185.001513. [DOI] [PubMed] [Google Scholar]
- 93.Mailloux R.J., Adjeitey C.N., Xuan J.Y., Harper M.E. Crucial yet divergent roles of mitochondrial redox state in skeletal muscle vs. brown adipose tissue energetics. Faseb J. 2012;26:363–375. doi: 10.1096/fj.11-189639. [DOI] [PubMed] [Google Scholar]
- 94.Deponte M. Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochim. Biophys. Acta. 2013;1830:3217–3266. doi: 10.1016/j.bbagen.2012.09.018. [DOI] [PubMed] [Google Scholar]
- 95.Gladyshev V.N., Liu A., Novoselov S.V., Krysan K., Sun Q.A., Kryukov V.M., Kryukov G.V., Lou M.F. Identification and characterization of a new mammalian glutaredoxin (thioltransferase), Grx2. J. Biol. Chem. 2001;276:30374–30380. doi: 10.1074/jbc.M100020200. [DOI] [PubMed] [Google Scholar]
- 96.Lundberg M., Johansson C., Chandra J., Enoksson M., Jacobsson G., Ljung J., Johansson M., Holmgren A. Cloning and expression of a novel human glutaredoxin (Grx2) with mitochondrial and nuclear isoforms. J. Biol. Chem. 2001;276:26269–26275. doi: 10.1074/jbc.M011605200. [DOI] [PubMed] [Google Scholar]
- 97.Mitra S., Elliott S.J. Oxidative disassembly of the [2Fe–2S] cluster of human Grx2 and redox regulation in the mitochondria. Biochemistry. 2009;48:3813–3815. doi: 10.1021/bi900112m. [DOI] [PubMed] [Google Scholar]
- 98.Qi W., Cowan J.A. Mechanism of glutaredoxin-ISU [2Fe–2S] cluster exchange. Chem. Commun. (Camb) 2011;47:4989–4991. doi: 10.1039/c0cc05079b. [DOI] [PubMed] [Google Scholar]
- 99.Klaus A., Zorman S., Berthier A., Polge C., Ramirez S., Michelland S., Seve M., Vertommen D., Rider M., Lentze N., Auerbach D., Schlattner U. Glutathione S-transferases interact with AMP-activated protein kinase: evidence for S-glutathionylation and activation in vitro. PLoS One. 2013;8:e62497. doi: 10.1371/journal.pone.0062497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Raza H. Dual localization of glutathione S-transferase in the cytosol and mitochondria: implications in oxidative stress, toxicity and disease. FEBS J. 2011;278:4243–4251. doi: 10.1111/j.1742-4658.2011.08358.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Knowles R.G., Moncada S. Nitric oxide as a signal in blood vessels. Trends Biochem. Sci. 1992;17:399–402. doi: 10.1016/0968-0004(92)90008-w. [DOI] [PubMed] [Google Scholar]
- 102.Zhao Y.L., Houk K.N. Thionitroxides, RSNHO⁎: the structure of the SNO moiety in “S-Nitrosohemoglobin”, a possible NO reservoir and transporter. J. Am. Chem. Soc. 2006;128:1422–1423. doi: 10.1021/ja057097f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ford E., Hughes M.N., Wardman P. Kinetics of the reactions of nitrogen dioxide with glutathione, cysteine, and uric acid at physiological pH. Free Radic. Biol. Med. 2002;32:1314–1323. doi: 10.1016/s0891-5849(02)00850-x. [DOI] [PubMed] [Google Scholar]
- 104.Sun J., Steenbergen C., Murphy E. S-nitrosylation: NO-related redox signaling to protect against oxidative stress. Antioxid. Redox Signal. 2006;8:1693–1705. doi: 10.1089/ars.2006.8.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bates T.E., Loesch A., Burnstock G., Clark J.B. Immunocytochemical evidence for a mitochondrially located nitric oxide synthase in brain and liver. Biochem. Biophys. Res. Commun. 1995;213:896–900. doi: 10.1006/bbrc.1995.2213. [DOI] [PubMed] [Google Scholar]
- 106.Brown G.C., Borutaite V. Nitric oxide and mitochondrial respiration in the heart. Cardiovasc. Res. 2007;75:283–290. doi: 10.1016/j.cardiores.2007.03.022. [DOI] [PubMed] [Google Scholar]
- 107.Freeman B.A., Baker P.R., Schopfer F.J., Woodcock S.R., Napolitano A., d’Ischia M. Nitro-fatty acid formation and signaling. J. Biol. Chem. 2008;283:15515–15519. doi: 10.1074/jbc.R800004200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chouchani E.T., Methner C., Nadtochiy S.M., Logan A., Pell V.R., Ding S., James A.M., Cocheme H.M., Reinhold J., Lilley K.S., Partridge L., Fearnley I.M., Robinson A.J., Hartley R.C., Smith R.A., Krieg T., Brookes P.S., Murphy M.P. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat. Med. 2013;19:753–759. doi: 10.1038/nm.3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Doulias P.T., Tenopoulou M., Greene J.L., Raju K., Ischiropoulos H. Nitric oxide regulates mitochondrial fatty acid metabolism through reversible protein S-nitrosylation. Sci. Signal. 2013;6:rs1. doi: 10.1126/scisignal.2003252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nguyen T.T., Stevens M.V., Kohr M., Steenbergen C., Sack M.N., Murphy E. Cysteine 203 of cyclophilin D is critical for cyclophilin D activation of the mitochondrial permeability transition pore. J. Biol. Chem. 2011;286:40184–40192. doi: 10.1074/jbc.M111.243469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Piantadosi C.A., Tatro L.G., Whorton A.R. Nitric oxide and differential effects of ATP on mitochondrial permeability transition. Nitric Oxide: Biol. Chem./Off. J. Nitric Oxide Soc. 2002;6:45–60. doi: 10.1006/niox.2001.0368. [DOI] [PubMed] [Google Scholar]
- 112.Chinta S.J., Andersen J.K. Nitrosylation and nitration of mitochondrial complex I in Parkinson's disease. Free Radic. Res. 2011;45:53–58. doi: 10.3109/10715762.2010.509398. [DOI] [PubMed] [Google Scholar]
- 113.Kwong L.K., Sohal R.S. Substrate and site specificity of hydrogen peroxide generation in mouse mitochondria. Arch. Biochem. Biophys. 1998;350:118–126. doi: 10.1006/abbi.1997.0489. [DOI] [PubMed] [Google Scholar]
- 114.Kushnareva Y., Murphy A.N., Andreyev A. Complex I-mediated reactive oxygen species generation: modulation by cytochrome c and NAD(P)+ oxidation-reduction state. Biochem. J. 2002;368:545–553. doi: 10.1042/BJ20021121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Aon M.A., Cortassa S., Maack C., O’Rourke B. Sequential opening of mitochondrial ion channels as a function of glutathione redox thiol status. J. Biol. Chem. 2007;282:21889–21900. doi: 10.1074/jbc.M702841200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.de la Asuncion J.G., Millan A., Pla R., Bruseghini L., Esteras A., Pallardo F.V., Sastre J., Vina J. Mitochondrial glutathione oxidation correlates with age-associated oxidative damage to mitochondrial DNA. Faseb J. 1996;10:333–338. doi: 10.1096/fasebj.10.2.8641567. [DOI] [PubMed] [Google Scholar]
- 117.Bulteau A.L., Lundberg K.C., Ikeda-Saito M., Isaya G., Szweda L.I. Reversible redox-dependent modulation of mitochondrial aconitase and proteolytic activity during in vivo cardiac ischemia/reperfusion. Proc. Natl. Acad. Sci. USA. 2005;102:5987–5991. doi: 10.1073/pnas.0501519102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mailloux R.J., Hamel R., Appanna V.D. Aluminum toxicity elicits a dysfunctional TCA cycle and succinate accumulation in hepatocytes. J. Biochem. Mol. Toxicol. 2006;20:198–208. doi: 10.1002/jbt.20137. [DOI] [PubMed] [Google Scholar]
- 119.Kil I.S., Park J.W. Regulation of mitochondrial NADP+-dependent isocitrate dehydrogenase activity by glutathionylation. J. Biol. Chem. 2005;280:10846–10854. doi: 10.1074/jbc.M411306200. [DOI] [PubMed] [Google Scholar]
- 120.McLain A.L., Cormier P.J., Kinter M., Szweda L.I. Glutathionylation of alpha-ketoglutarate dehydrogenase: the chemical nature and relative susceptibility of the cofactor lipoic acid to modification. Free Radic. Biol. Med. 2013;61C:161–169. doi: 10.1016/j.freeradbiomed.2013.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.McLain A.L., Szweda P.A., Szweda L.I. alpha-Ketoglutarate dehydrogenase: a mitochondrial redox sensor. Free Radic. Res. 2011;45:29–36. doi: 10.3109/10715762.2010.534163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yan L.J., Sumien N., Thangthaeng N., Forster M.J. Reversible inactivation of dihydrolipoamide dehydrogenase by mitochondrial hydrogen peroxide. Free Radic. Res. 2013;47:123–133. doi: 10.3109/10715762.2012.752078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chen Y.R., Chen C.L., Pfeiffer D.R., Zweier J.L. Mitochondrial complex II in the post-ischemic heart: oxidative injury and the role of protein S-glutathionylation. J. Biol. Chem. 2007;282:32640–32654. doi: 10.1074/jbc.M702294200. [DOI] [PubMed] [Google Scholar]
- 124.Giangregorio N., Palmieri F., Indiveri C. Glutathione controls the redox state of the mitochondrial carnitine/acylcarnitine carrier Cys residues by glutathionylation. Biochim. Biophys. Acta. 2013;1830:5299–5304. doi: 10.1016/j.bbagen.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 125.Wu H., Lin L., Giblin F., Ho Y.S., Lou M.F. Glutaredoxin 2 knockout increases sensitivity to oxidative stress in mouse lens epithelial cells. Free Radic. Biol. Med. 2011;51:2108–2117. doi: 10.1016/j.freeradbiomed.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hill B.G., Higdon A.N., Dranka B.P., Darley-Usmar V.M. Regulation of vascular smooth muscle cell bioenergetic function by protein glutathiolation. Biochim. Biophys. Acta. 2010;1797:285–295. doi: 10.1016/j.bbabio.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Pfefferle A., Mailloux R.J., Adjeitey C.N., Harper M.E. Glutathionylation of UCP2 sensitizes drug resistant leukemia cells to chemotherapeutics. Biochim. Biophys. Acta. 2013;1833:80–89. doi: 10.1016/j.bbamcr.2012.10.006. [DOI] [PubMed] [Google Scholar]
- 128.Baffy G., Derdak Z., Robson S.C. Mitochondrial recoupling: a novel therapeutic strategy for cancer? Br. J. Cancer. 2011;105:469–474. doi: 10.1038/bjc.2011.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Diotte N.M., Xiong Y., Gao J., Chua B.H., Ho Y.S. Attenuation of doxorubicin-induced cardiac injury by mitochondrial glutaredoxin 2. Biochim. Biophys. Acta. 2009;1793:427–438. doi: 10.1016/j.bbamcr.2008.10.014. [DOI] [PubMed] [Google Scholar]
- 130.Taylor E.R., Hurrell F., Shannon R.J., Lin T.K., Hirst J., Murphy M.P. Reversible glutathionylation of complex I increases mitochondrial superoxide formation. J. Biol. Chem. 2003;278:19603–19610. doi: 10.1074/jbc.M209359200. [DOI] [PubMed] [Google Scholar]
- 131.Tretter L., Adam-Vizi V. Inhibition of Krebs cycle enzymes by hydrogen peroxide: a key role of [alpha]-ketoglutarate dehydrogenase in limiting NADH production under oxidative stress. J. Neurosci.: Off. J. Soc. Neurosci. 2000;20:8972–8979. doi: 10.1523/JNEUROSCI.20-24-08972.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Gibson G.E., Park L.C., Sheu K.F., Blass J.P., Calingasan N.Y. The alpha-ketoglutarate dehydrogenase complex in neurodegeneration. Neurochem. Int. 2000;36:97–112. doi: 10.1016/s0197-0186(99)00114-x. [DOI] [PubMed] [Google Scholar]
- 133.Wang S.B., Murray C.I., Chung H.S., Van Eyk J.E. Redox regulation of mitochondrial ATP synthase. Trends Cardiovasc. Med. 2013;23:14–18. doi: 10.1016/j.tcm.2012.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Garcia J., Han D., Sancheti H., Yap L.P., Kaplowitz N., Cadenas E. Regulation of mitochondrial glutathione redox status and protein glutathionylation by respiratory substrates. J. Biol. Chem. 2010;285:39646–39654. doi: 10.1074/jbc.M110.164160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Liesa M., Shirihai O.S. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013;17:491–506. doi: 10.1016/j.cmet.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chan D.C. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006;125:1241–1252. doi: 10.1016/j.cell.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 137.Shutt T., Geoffrion M., Milne R., McBride H.M. The intracellular redox state is a core determinant of mitochondrial fusion. EMBO Rep. 2012;13:909–915. doi: 10.1038/embor.2012.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Redpath C.J., Bou Khalil M., Drozdzal G., Radisic M., McBride H.M. Mitochondrial hyperfusion during oxidative stress is coupled to a dysregulation in calcium handling within a C2C12 cell model. PLoS One. 2013;8:e69165. doi: 10.1371/journal.pone.0069165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lin M.T., Beal M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 140.Kowaltowski A.J., Castilho R.F., Vercesi A.E. Mitochondrial permeability transition and oxidative stress. FEBS Lett. 2001;495:12–15. doi: 10.1016/s0014-5793(01)02316-x. [DOI] [PubMed] [Google Scholar]
- 141.Halestrap A.P., Brenner C. The adenine nucleotide translocase: a central component of the mitochondrial permeability transition pore and key player in cell death. Curr. Med. Chem. 2003;10:1507–1525. doi: 10.2174/0929867033457278. [DOI] [PubMed] [Google Scholar]
- 142.Kowaltowski A.J., Castilho R.F., Vercesi A.E. Ca(2+)-induced mitochondrial membrane permeabilization: role of coenzyme Q redox state. Am. J. Physiol. 1995;269:C141–C147. doi: 10.1152/ajpcell.1995.269.1.C141. [DOI] [PubMed] [Google Scholar]
- 143.Kowaltowski A.J., Castilho R.F., Grijalba M.T., Bechara E.J., Vercesi A.E. Effect of inorganic phosphate concentration on the nature of inner mitochondrial membrane alterations mediated by Ca2+ ions. A proposed model for phosphate-stimulated lipid peroxidation. J. Biol. Chem. 1996;271:2929–2934. doi: 10.1074/jbc.271.6.2929. [DOI] [PubMed] [Google Scholar]
- 144.Kowaltowski A.J., Netto L.E., Vercesi A.E. The thiol-specific antioxidant enzyme prevents mitochondrial permeability transition. Evidence for the participation of reactive oxygen species in this mechanism. J. Biol. Chem. 1998;273:12766–12769. doi: 10.1074/jbc.273.21.12766. [DOI] [PubMed] [Google Scholar]
- 145.Yin F., Sancheti H., Cadenas E. Mitochondrial thiols in the regulation of cell death pathways. Antioxid. Redox Signal. 2012;17:1714–1727. doi: 10.1089/ars.2012.4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Halestrap A.P. A pore way to die: the role of mitochondria in reperfusion injury and cardioprotection. Biochem. Soc. Trans. 2010;38:841–860. doi: 10.1042/BST0380841. [DOI] [PubMed] [Google Scholar]
- 147.Queiroga C.S., Almeida A.S., Martel C., Brenner C., Alves P.M., Vieira H.L. Glutathionylation of adenine nucleotide translocase induced by carbon monoxide prevents mitochondrial membrane permeabilization and apoptosis. J. Biol. Chem. 2010;285:17077–17088. doi: 10.1074/jbc.M109.065052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Leung A.W., Varanyuwatana P., Halestrap A.P. The mitochondrial phosphate carrier interacts with cyclophilin D and may play a key role in the permeability transition. J. Biol. Chem. 2008;283:26312–26323. doi: 10.1074/jbc.M805235200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Varanyuwatana P., Halestrap A.P. The roles of phosphate and the phosphate carrier in the mitochondrial permeability transition pore. Mitochondrion. 2012;12:120–125. doi: 10.1016/j.mito.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Giorgio V., von Stockum S., Antoniel M., Fabbro A., Fogolari F., Forte M., Glick G.D., Petronilli V., Zoratti M., Szabo I., Lippe G., Bernardi P. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc. Natl. Acad. Sci. USA. 2013;110:5887–5892. doi: 10.1073/pnas.1217823110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Sanchez G., Fernandez C., Montecinos L., Domenech R.J., Donoso P. Preconditioning tachycardia decreases the activity of the mitochondrial permeability transition pore in the dog heart. Biochem. Biophys. Res. Commun. 2011;410:916–921. doi: 10.1016/j.bbrc.2011.06.095. [DOI] [PubMed] [Google Scholar]
- 152.Baines C.P., Kaiser R.A., Purcell N.H., Blair N.S., Osinska H., Hambleton M.A., Brunskill E.W., Sayen M.R., Gottlieb R.A., Dorn G.W., Robbins J., Molkentin J.D. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 153.Nakagawa T., Shimizu S., Watanabe T., Yamaguchi O., Otsu K., Yamagata H., Inohara H., Kubo T., Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–658. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 154.Kokoszka J.E., Waymire K.G., Levy S.E., Sligh J.E., Cai J., Jones D.P., MacGregor G.R., Wallace D.C. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature. 2004;427:461–465. doi: 10.1038/nature02229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ambrosio G., Zweier J.L., Flaherty J.T. The relationship between oxygen radical generation and impairment of myocardial energy metabolism following post-ischemic reperfusion. J. Mol. Cell. Cardiol. 1991;23:1359–1374. doi: 10.1016/0022-2828(91)90183-m. [DOI] [PubMed] [Google Scholar]
- 156.Zweier J.L., Flaherty J.T., Weisfeldt M.L. Direct measurement of free radical generation following reperfusion of ischemic myocardium. Proc. Natl. Acad. Sci. USA. 1987;84:1404–1407. doi: 10.1073/pnas.84.5.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Bolli R., Marban E. Molecular and cellular mechanisms of myocardial stunning. Physiol. Rev. 1999;79:609–634. doi: 10.1152/physrev.1999.79.2.609. [DOI] [PubMed] [Google Scholar]
- 158.Tompkins A.J., Burwell L.S., Digerness S.B., Zaragoza C., Holman W.L., Brookes P.S. Mitochondrial dysfunction in cardiac ischemia-reperfusion injury: ROS from complex I, without inhibition. Biochim. Biophys. Acta. 2006;1762:223–231. doi: 10.1016/j.bbadis.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 159.Lesnefsky E.J., Moghaddas S., Tandler B., Kerner J., Hoppel C.L. Mitochondrial dysfunction in cardiac disease: ischemia—reperfusion, aging, and heart failure. J. Mol. Cell. Cardiol. 2001;33:1065–1089. doi: 10.1006/jmcc.2001.1378. [DOI] [PubMed] [Google Scholar]
- 160.Rouslin W. Mitochondrial complexes I, II, III, IV, and V in myocardial ischemia and autolysis. Am. J. Physiol. 1983;244:H743–H748. doi: 10.1152/ajpheart.1983.244.6.H743. [DOI] [PubMed] [Google Scholar]
- 161.Kumar V., Kleffmann T., Hampton M.B., Cannell M.B., Winterbourn C.C. Redox proteomics of thiol proteins in mouse heart during ischemia/reperfusion using ICAT reagents and mass spectrometry. Free Radic. Biol. Med. 2013;58:109–117. doi: 10.1016/j.freeradbiomed.2013.01.021. [DOI] [PubMed] [Google Scholar]
- 162.Liu B., Tewari A.K., Zhang L., Green-Church K.B., Zweier J.L., Chen Y.R., He G. Proteomic analysis of protein tyrosine nitration after ischemia reperfusion injury: mitochondria as the major target. Biochim. Biophys. Acta. 2009;1794:476–485. doi: 10.1016/j.bbapap.2008.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Zhao X., Chen Y.R., He G., Zhang A., Druhan L.J., Strauch A.R., Zweier J.L. Endothelial nitric oxide synthase (NOS3) knockout decreases NOS2 induction, limiting hyperoxygenation and conferring protection in the postischemic heart. Am. J. Physiol. Heart Circ. Physiol. 2007;292:H1541–H1550. doi: 10.1152/ajpheart.00264.2006. [DOI] [PubMed] [Google Scholar]
- 164.Zhao X., He G., Chen Y.R., Pandian R.P., Kuppusamy P., Zweier J.L. Endothelium-derived nitric oxide regulates postischemic myocardial oxygenation and oxygen consumption by modulation of mitochondrial electron transport. Circulation. 2005;111:2966–2972. doi: 10.1161/CIRCULATIONAHA.104.527226. [DOI] [PubMed] [Google Scholar]
- 165.Zhang L., Chen C.L., Kang P.T., Garg V., Hu K., Green-Church K.B., Chen Y.R. Peroxynitrite-mediated oxidative modifications of complex II: relevance in myocardial infarction. Biochemistry. 2010;49:2529–2539. doi: 10.1021/bi9018237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Burwell L.S., Nadtochiy S.M., Tompkins A.J., Young S., Brookes P.S. Direct evidence for S-nitrosation of mitochondrial complex I. Biochem. J. 2006;394:627–634. doi: 10.1042/BJ20051435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Poderoso J.J., Carreras M.C., Lisdero C., Riobo N., Schopfer F., Boveris A. Nitric oxide inhibits electron transfer and increases superoxide radical production in rat heart mitochondria and submitochondrial particles. Arch. Biochem. Biophys. 1996;328:85–92. doi: 10.1006/abbi.1996.0146. [DOI] [PubMed] [Google Scholar]
- 168.Brookes P., Darley-Usmar V.M. Hypothesis: the mitochondrial NO(⁎) signaling pathway, and the transduction of nitrosative to oxidative cell signals: an alternative function for cytochrome C oxidase. Free Radic. Biol. Med. 2002;32:370–374. doi: 10.1016/s0891-5849(01)00805-x. [DOI] [PubMed] [Google Scholar]
- 169.Chen Q., Vazquez E.J., Moghaddas S., Hoppel C.L., Lesnefsky E.J. Production of reactive oxygen species by mitochondria: central role of complex III. J. Biol. Chem. 2003;278:36027–36031. doi: 10.1074/jbc.M304854200. [DOI] [PubMed] [Google Scholar]
- 170.Zweier J.L., Chen C.A., Talukder M.A. Cardiac resynchronization therapy and reverse molecular remodeling: importance of mitochondrial redox signaling. Circ. Res. 2011;109:716–719. doi: 10.1161/CIRCRESAHA.111.253864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Chen C.A., Wang T.Y., Varadharaj S., Reyes L.A., Hemann C., Talukder M.A., Chen Y.R., Druhan L.J., Zweier J.L. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature. 2010;468:1115–1118. doi: 10.1038/nature09599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Rolo A.P., Palmeira C.M. Diabetes and mitochondrial function: role of hyperglycemia and oxidative stress. Toxicol. Appl. Pharmacol. 2006;212:167–178. doi: 10.1016/j.taap.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 173.Anderson E.J., Rodriguez E., Anderson C.A., Thayne K., Chitwood W.R., Kypson A.P. Increased propensity for cell death in diabetic human heart is mediated by mitochondrial-dependent pathways. Am. J. Physiol. Heart Circ. Physiol. 2011;300:H118–H124. doi: 10.1152/ajpheart.00932.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Williamson C.L., Dabkowski E.R., Baseler W.A., Croston T.L., Alway S.E., Hollander J.M. Enhanced apoptotic propensity in diabetic cardiac mitochondria: influence of subcellular spatial location. Am. J. Physiol. Heart Circ. Physiol. 2010;298:H633–H642. doi: 10.1152/ajpheart.00668.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Sanchez-Gomez F.J., Espinosa-Diez C., Dubey M., Dikshit M., Lamas S. S-glutathionylation: relevance in diabetes and potential role as a biomarker. Biol. Chem. 2013;394:1263–1280. doi: 10.1515/hsz-2013-0150. [DOI] [PubMed] [Google Scholar]
- 176.Bournat J.C., Brown C.W. Mitochondrial dysfunction in obesity. Curr. Opin. Endocrinol. Diabetes Obes. 2010;17:446–452. doi: 10.1097/MED.0b013e32833c3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Picklo M.J., Sr., Idso J.P., Jackson M.I. S-Glutathionylation of hepatic and visceral adipose proteins decreases in obese rats. Obesity (Silver Spring) 2013;21:297–305. doi: 10.1002/oby.20002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Harper M.E., Green K., Brand M.D. The efficiency of cellular energy transduction and its implications for obesity. Annu. Rev. Nutr. 2008;28:13–33. doi: 10.1146/annurev.nutr.28.061807.155357. [DOI] [PubMed] [Google Scholar]
- 179.Lazarin Mde O., Ishii-Iwamoto E.L., Yamamoto N.S., Constantin R.P., Garcia R.F., da Costa C.E., Vitoriano Ade S., de Oliveira M.C., Salgueiro-Pagadigorria C.L. Liver mitochondrial function and redox status in an experimental model of non-alcoholic fatty liver disease induced by monosodium L-glutamate in rats. Exp. Mol. Pathol. 2011;91:687–694. doi: 10.1016/j.yexmp.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 180.Blatnik M., Thorpe S.R., Baynes J.W. Succination of proteins by fumarate: mechanism of inactivation of glyceraldehyde-3-phosphate dehydrogenase in diabetes. Ann. N. Y. Acad. Sci. 2008;1126:272–275. doi: 10.1196/annals.1433.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Frizzell N., Lima M., Baynes J.W. Succination of proteins in diabetes. Free Radic. Res. 2011;45:101–109. doi: 10.3109/10715762.2010.524643. [DOI] [PubMed] [Google Scholar]
- 182.Nagai R., Brock J.W., Blatnik M., Baatz J.E., Bethard J., Walla M.D., Thorpe S.R., Baynes J.W., Frizzell N. Succination of protein thiols during adipocyte maturation: a biomarker of mitochondrial stress. J. Biol. Chem. 2007;282:34219–34228. doi: 10.1074/jbc.M703551200. [DOI] [PubMed] [Google Scholar]
- 183.Frizzell N., Thomas S.A., Carson J.A., Baynes J.W. Mitochondrial stress causes increased succination of proteins in adipocytes in response to glucotoxicity. Biochem. J. 2012;445:247–254. doi: 10.1042/BJ20112142. [DOI] [PubMed] [Google Scholar]
- 184.Ternette N., Yang M., Laroyia M., Kitagawa M., O’Flaherty L., Wolhulter K., Igarashi K., Saito K., Kato K., Fischer R., Berquand A., Kessler B.M., Lappin T., Frizzell N., Soga T., Adam J., Pollard P.J. Inhibition of mitochondrial aconitase by succination in fumarate hydratase deficiency. Cell Rep. 2013;3:689–700. doi: 10.1016/j.celrep.2013.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Sabens Liedhegner E.A., Gao X.H., Mieyal J.J. Mechanisms of altered redox regulation in neurodegenerative diseases—focus on S-glutathionylation. Antioxid. Redox Signal. 2012;16:543–566. doi: 10.1089/ars.2011.4119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Ansari M.A., Scheff S.W. Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J. Neuropathol. Exp. Neurol. 2010;69:155–167. doi: 10.1097/NEN.0b013e3181cb5af4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Jenner P., Dexter D.T., Sian J., Schapira A.H., Marsden C.D. Oxidative stress as a cause of nigral cell death in Parkinson's disease and incidental Lewy body disease. The Royal Kings and Queens Parkinson's Disease Research Group. Ann. Neurol. 1992;32(Suppl:):S82–S87. doi: 10.1002/ana.410320714. [DOI] [PubMed] [Google Scholar]
- 188.Sian J., Dexter D.T., Lees A.J., Daniel S., Agid Y., Javoy-Agid F., Jenner P., Marsden C.D. Alterations in glutathione levels in Parkinson's disease and other neurodegenerative disorders affecting basal ganglia. Ann. Neurol. 1994;36:348–355. doi: 10.1002/ana.410360305. [DOI] [PubMed] [Google Scholar]
- 189.Schulz J.B., Lindenau J., Seyfried J., Dichgans J. Glutathione, oxidative stress and neurodegeneration. Eur. J. Biochem./FEBS. 2000;267:4904–4911. doi: 10.1046/j.1432-1327.2000.01595.x. [DOI] [PubMed] [Google Scholar]
- 190.Lee D.W., Kaur D., Chinta S.J., Rajagopalan S., Andersen J.K. A disruption in iron–sulfur center biogenesis via inhibition of mitochondrial dithiol glutaredoxin 2 may contribute to mitochondrial and cellular iron dysregulation in mammalian glutathione-depleted dopaminergic cells: implications for Parkinson's disease. Antioxid. Redox Signal. 2009;11:2083–2094. doi: 10.1089/ars.2009.2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Calabrese V., Scapagnini G., Ravagna A., Bella R., Butterfield D.A., Calvani M., Pennisi G., Giuffrida Stella A.M. Disruption of thiol homeostasis and nitrosative stress in the cerebrospinal fluid of patients with active multiple sclerosis: evidence for a protective role of acetylcarnitine. Neurochem. Res. 2003;28:1321–1328. doi: 10.1023/a:1024984013069. [DOI] [PubMed] [Google Scholar]
- 192.Akterin S., Cowburn R.F., Miranda-Vizuete A., Jimenez A., Bogdanovic N., Winblad B., Cedazo-Minguez A. Involvement of glutaredoxin-1 and thioredoxin-1 in beta-amyloid toxicity and Alzheimer's disease. Cell Death Differ. 2006;13:1454–1465. doi: 10.1038/sj.cdd.4401818. [DOI] [PubMed] [Google Scholar]
- 193.Kenchappa R.S., Ravindranath V. Glutaredoxin is essential for maintenance of brain mitochondrial complex I: studies with MPTP. Faseb J. 2003;17:717–719. doi: 10.1096/fj.02-0771fje. [DOI] [PubMed] [Google Scholar]
- 194.Dinoto L., Deture M.A., Purich D.L. Structural insights into Alzheimer filament assembly pathways based on site-directed mutagenesis and S-glutathionylation of three-repeat neuronal Tau protein. Microsc. Res. Tech. 2005;67:156–163. doi: 10.1002/jemt.20195. [DOI] [PubMed] [Google Scholar]
- 195.Chinta S.J., Andersen J.K. Reversible inhibition of mitochondrial complex I activity following chronic dopaminergic glutathione depletion in vitro: implications for Parkinson's disease. Free Radic. Biol. Med. 2006;41:1442–1448. doi: 10.1016/j.freeradbiomed.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 196.Hsu M., Srinivas B., Kumar J., Subramanian R., Andersen J. Glutathione depletion resulting in selective mitochondrial complex I inhibition in dopaminergic cells is via an NO-mediated pathway not involving peroxynitrite: implications for Parkinson's disease. J. Neurochem. 2005;92:1091–1103. doi: 10.1111/j.1471-4159.2004.02929.x. [DOI] [PubMed] [Google Scholar]
- 197.Nakamura T., Lipton S.A. Redox modulation by S-nitrosylation contributes to protein misfolding, mitochondrial dynamics, and neuronal synaptic damage in neurodegenerative diseases. Cell Death Differ. 2011;18:1478–1486. doi: 10.1038/cdd.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Uehara T., Nakamura T., Yao D., Shi Z.Q., Gu Z., Ma Y., Masliah E., Nomura Y., Lipton S.A. S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature. 2006;441:513–517. doi: 10.1038/nature04782. [DOI] [PubMed] [Google Scholar]
- 199.Cho D.H., Nakamura T., Fang J., Cieplak P., Godzik A., Gu Z., Lipton S.A. S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science. 2009;324:102–105. doi: 10.1126/science.1171091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Van Laar V.S., Berman S.B. Mitochondrial dynamics in Parkinson's disease. Exp. Neurol. 2009;218:247–256. doi: 10.1016/j.expneurol.2009.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Chen H., Chan D.C. Mitochondrial dynamics—fusion, fission, movement, and mitophagy--in neurodegenerative diseases. Hum. Mol. Genet. 2009;18:R169–R176. doi: 10.1093/hmg/ddp326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Bossy B., Petrilli A., Klinglmayr E., Chen J., Lutz-Meindl U., Knott A.B., Masliah E., Schwarzenbacher R., Bossy-Wetzel E. S-Nitrosylation of DRP1 does not affect enzymatic activity and is not specific to Alzheimer's disease. J. Alzheimer's Dis.: JAD. 2010;20(Suppl 2):S513–S526. doi: 10.3233/JAD-2010-100552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Moosmann B. Respiratory chain cysteine and methionine usage indicate a causal role for thiyl radicals in aging. Exp. Gerontol. 2011;46:164–169. doi: 10.1016/j.exger.2010.08.034. [DOI] [PubMed] [Google Scholar]
- 204.Schindeldecker M., Stark M., Behl C., Moosmann B. Differential cysteine depletion in respiratory chain complexes enables the distinction of longevity from aerobicity. Mech. Ageing Dev. 2011;132:171–179. doi: 10.1016/j.mad.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 205.Held J.M., Gibson B.W. Regulatory control or oxidative damage? Proteomic approaches to interrogate the role of cysteine oxidation status in biological processes. Mol. Cell. Proteomics: MCP. 2012;11(R111):013037. doi: 10.1074/mcp.R111.013037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Butterfield D.A., Perluigi M., Reed T., Muharib T., Hughes C.P., Robinson R.A., Sultana R. Redox proteomics in selected neurodegenerative disorders: from its infancy to future applications. Antioxid. Redox Signal. 2012;17:1610–1655. doi: 10.1089/ars.2011.4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Go Y.M., Jones D.P. Cysteine/cystine redox signaling in cardiovascular disease. Free Radic. Biol. Med. 2011;50:495–509. doi: 10.1016/j.freeradbiomed.2010.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Martinez Banaclocha M. N-acetylcysteine elicited increase in complex I activity in synaptic mitochondria from aged mice: implications for treatment of Parkinson's disease. Brain Res. 2000;859:173–175. doi: 10.1016/s0006-8993(00)02005-9. [DOI] [PubMed] [Google Scholar]
- 209.Rebrin I., Sohal R.S. Comparison of thiol redox state of mitochondria and homogenates of various tissues between two strains of mice with different longevities. Exp. Gerontol. 2004;39:1513–1519. doi: 10.1016/j.exger.2004.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Musicco C., Capelli V., Pesce V., Timperio A.M., Calvani M., Mosconi L., Zolla L., Cantatore P., Gadaleta M.N. Accumulation of overoxidized Peroxiredoxin III in aged rat liver mitochondria. Biochim. Biophys. Acta. 2009;1787:890–896. doi: 10.1016/j.bbabio.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 211.Cai Z., Yan L.J. Protein oxidative modifications: beneficial roles in disease and health. J. Biochem. Pharmacol. Res. 2013;1:15–26. [PMC free article] [PubMed] [Google Scholar]
- 212.Droge W. The plasma redox state and ageing. Ageing Res. Rev. 2002;1:257–278. doi: 10.1016/s1568-1637(01)00008-3. [DOI] [PubMed] [Google Scholar]
- 213.Velu C.S., Niture S.K., Doneanu C.E., Pattabiraman N., Srivenugopal K.S. Human p53 is inhibited by glutathionylation of cysteines present in the proximal DNA-binding domain during oxidative stress. Biochemistry. 2007;46:7765–7780. doi: 10.1021/bi700425y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Kambe T., Song T., Takata T., Hatano N., Miyamoto Y., Nozaki N., Naito Y., Tokumitsu H., Watanabe Y. Inactivation of Ca2+/calmodulin-dependent protein kinase I by S-glutathionylation of the active-site cysteine residue. FEBS Lett. 2010;584:2478–2484. doi: 10.1016/j.febslet.2010.04.059. [DOI] [PubMed] [Google Scholar]
- 215.Nakamura T., Tu S., Akhtar M.W., Sunico C.R., Okamoto S., Lipton S.A. Aberrant protein s-nitrosylation in neurodegenerative diseases. Neuron. 2013;78:596–614. doi: 10.1016/j.neuron.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Schulman I.H., Hare J.M. Regulation of cardiovascular cellular processes by S-nitrosylation. Biochim. Biophys. Acta. 2012;1820:752–762. doi: 10.1016/j.bbagen.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Kaneki M., Shimizu N., Yamada D., Chang K. Nitrosative stress and pathogenesis of insulin resistance. Antioxid. Redox Signal. 2007;9:319–329. doi: 10.1089/ars.2006.1464. [DOI] [PubMed] [Google Scholar]
- 218.Cooper A.J., Pinto J.T., Callery P.S. Reversible and irreversible protein glutathionylation: biological and clinical aspects. Expert Opin. Drug Metab. Toxicol. 2011;7:891–910. doi: 10.1517/17425255.2011.577738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Newman J.C., He W., Verdin E. Mitochondrial protein acylation and intermediary metabolism: regulation by sirtuins and implications for metabolic disease. J. Biol. Chem. 2012;287:42436–42443. doi: 10.1074/jbc.R112.404863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Bouillaud F., Blachier F. Mitochondria and sulfide: a very old story of poisoning, feeding, and signaling? Antioxid. Redox Signal. 2011;15:379–391. doi: 10.1089/ars.2010.3678. [DOI] [PubMed] [Google Scholar]
- 221.Szabo C., Ransy C., Modis K., Andriamihaja M., Murghes B., Coletta C., Olah G., Yanagi K., Bouillaud F. Regulation of mitochondrial bioenergetic function by hydrogen sulfide. Part I. Biochemical and physiological mechanisms. Br. J. Pharmacol. 2013;221 doi: 10.1111/bph.12369. [DOI] [PMC free article] [PubMed] [Google Scholar]